
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRing-opening copolymerization of (dihydro)levoglucosenone-derived spiro-epoxides and cyclic anhydrides
Tianle Gao†
a,
Hiroto Ayakawa†b,
Tatsuya Nishimura c,
Ayano Yoshidab,
Moeno Sugiyamab,
Takuya Yamamotoa,
Kenji Tajimaa,
Kenji Takahashid,
Feng Li*a,
Takuya Isonoa,
Katsuhiro Maedae and
Toshifumi Satoh*afg
c,
Ayano Yoshidab,
Moeno Sugiyamab,
Takuya Yamamotoa,
Kenji Tajimaa,
Kenji Takahashid,
Feng Li*a,
Takuya Isonoa,
Katsuhiro Maedae and
Toshifumi Satoh*afg
aDivision of Applied Chemistry, Faculty of Engineering, Hokkaido University, Sapporo 060-8628, Japan. E-mail: feng.li@eng.hokudai.ac.jp; satoh@eng.hokudai.ac.jp
bGraduate School of Chemical Sciences and Engineering, Hokkaido University, Sapporo 060-8628, Japan
cGraduate School of Natural Science and Technology, Kanazawa University, Kakuma-machi, Kanazawa, 920-1192 Japan
dFaculty of Biological Science and Technology, Institute of Science and Engineering, Kanazawa University, Kanazawa, 920-1192, Japan
eNano Life Science Institute (WPI-NanoLSI), Kanazawa University, Kakuma-machi, Kanazawa, 920-1192 Japan
fList Sustainable Digital Transformation Catalyst Collaboration Research Platform (ICReDD List-PF), Institute for Chemical Reaction Design and Discovery, Hokkaido University, Sapporo 001-0021, Japan
gDepartment of Chemical & Materials Engineering, National Central University, Taoyuan 320317, Taiwan
First published on 4th June 2026
Abstract
Ring-opening copolymerization (ROCOP) of epoxides with cyclic anhydrides is a powerful strategy for synthesis of structure-controlled polyesters with tunable properties. Although considerable efforts have been devoted to expanding the monomer scope by introducing specific side groups or utilizing new biomass-derived feedstocks, the influence of monomer substitution patterns has received far less attention. In particular, in contrast to the well-studied mono- and 1,2-disubstituted epoxides, 1,1-disubstituted epoxides have been much less explored. Herein, we report cellulose-derived spiro-epoxides DBOO and DBOOPh as a new class of 1,1-disubstituted epoxide monomers for ROCOP with cyclic anhydrides. ROCOP of DBOO with phthalic anhydride proceeds in a living/controlled manner, affording polyesters with predominantly alternating structures, as confirmed by NMR and MALDI-TOF MS analyses. By varying the monomer combinations, a series of polyesters bearing spirocyclic backbones were synthesized. Thermal analyses revealed sufficient thermal stability and widely tunable glass-transition temperatures ranging from 29 to 130 °C. Moreover, the chirality of DBOO and DBOOPh induced chiral environments along the polymer chain, as evidenced by the circular dichroism spectra of the resulting polyesters. It may further induce the polymer chains to adopt a biased one-handed helical conformation. This work not only expands the monomer scope of ROCOP to biomass-derived spiro-epoxides, thus providing access to polyesters with unique rigid spirocyclic backbones, but also demonstrates the potential of polyesters synthesized via ROCOP as new chiral materials.
1. Introduction
Polyesters constitute one of the most important classes of polymeric materials owing to their structural diversity, broad utility, and intrinsic degradability. Owing to the presence of hydrolyzable ester linkages, polyesters have long been regarded as attractive alternatives to persistent polyolefin-based plastics and have found widespread applications in the packaging, commodity materials, engineering plastics, and biomedical fields.1–3 Conventionally, polyesters are synthesized either by the polycondensation of diols and dicarboxylic acids or by the ring-opening polymerization (ROP) of cyclic esters.4,5 In addition, the ring-opening copolymerization (ROCOP) of epoxides with cyclic anhydrides has emerged as an attractive approach for polyester synthesis.6–8 This approach offers good control of polymerization through a chain-growth mechanism as well as remarkable versatility in structural design by varying the combination of the epoxide and cyclic anhydride monomers. As a result, ROCOP provides a powerful platform for tuning polymer composition, functionality, and material properties and has developed into an important alternative to classical polyester synthesis.Over the past several decades, numerous studies have focused on the development of new monomers. However, most of these efforts have primarily involved either introduction of specific side groups or utilization of new biomass-derived feedstocks.9–13 By contrast, relatively little attention has been devoted to innovations in the fundamental substitution patterns of monomers. Taking epoxides as an example, monosubstituted epoxides and 1,2-disubstituted epoxides represent the most common substitution patterns employed in ROCOP with cyclic anhydrides (Fig. 1A). For 1,1-disubstituted epoxides, ROCOP using cyclic anhydrides and isobutylene oxide (IBO) has attracted research attention only recently.14–18 The bulky gem-dimethyl groups of IBO directly nucleophilically attack the less hindered methylene carbon, thereby favoring highly regular head-to-tail linkages. In addition, the symmetric structure of the polyesters synthesized from IBO facilitates regular chain packing, leading to semi-crystalline materials that are difficult to obtain using racemic monosubstituted epoxides or meso-1,2-disubstituted epoxides. Although these studies highlight the potential of 1,1-disubstituted epoxides in ROCOP, the reported examples are limited to IBO.
 | ||
| Fig. 1 (A) Ring-opening copolymerization of cyclic anhydrides and epoxides bearing different substitution patterns; (B) schematic illustration of this work. | ||
Spiro-epoxides, a special subclass of 1,1-disubstituted epoxides, have not been explored in ROP prior to our recent report describing the successful ROP of spiro-epoxide DBOO which was synthesized from cellulose-derived dihydrolevoglucosenone (cyrene) in a single step.19,20 This polymerization afforded a biobased aliphatic polyether with an unexpectedly high glass-transition temperature (Tg = 140 °C). This success encouraged us to further explore the potential of spiro-epoxides in other polymerization processes, particularly in the ROCOP with cyclic anhydrides (Fig. 1B). Given the unique structure of DBOO, ROCOP of its structurally related spiro-epoxides with cyclic anhydrides are expected to produce polyesters with previously unexplored backbone structures, thereby expanding the scope of ROCOP chemistry. This strategy represents a new approach for the utilization of cellulose-derived LGO/cyrene in polymer synthesis.21–24 Furthermore, the bulky and optically active nature of these epoxides may endow the resulting polyesters with regular chiral secondary structures.
In this study, we report the ROCOP of (dihydro)levoglucosenone-derived spiro-epoxides, including DBOO and DBOOPh, with cyclic anhydrides. The resulting polyesters were investigated with regard to their material properties, including thermal properties and chiral secondary structures.
2. Results and discussion
2.1. ROCOP of DBOO and phthalic anhydride
Following a previously reported procedure, DBOO monomers were synthesized via the Corey–Chaykovsky reaction using Cyrene™. Depending on the type of ylide agent, DBOO with two different diastereomeric ratios (dr = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 and 87:13) were obtained.19,20 They are referred to as DBOO(50:50) and DBOO(87:13), respectively.
:50 and 87:13) were obtained.19,20 They are referred to as DBOO(50:50) and DBOO(87:13), respectively.
ROCOP was initially performed using DBOO(50:50) and phthalic anhydride (PA). 1,4-Benzenedimethanol (BDM) was used as an initiator. The investigation began with catalyst screening which was performed in bulk at 60 °C (Table 1, runs 1–4; Scheme S1; Fig. S1).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Run | Catalyst | [PA]0/[DBOO]0/[I]0/[Cat.] | Temp. (°C) | Time (h) | Conv.PAb (%) |
Mn,theo.c |
Mn,NMRb |
Mn,SECd |
Đd |
| a Polymerization conditions: Ar atmosphere; BDM initiator.b Determined from 1H NMR spectrum of the obtained polymer in CDCl3.c Calculated from [PA]0/[BDM]0 × conv.PA/100 × ((M.W. of PA) + (M.W. of DBOO)) + (M.W. of BDM).d Determined by SEC of the polymer obtained in THF using a polystyrene standard.e n(PA) = 2.60 mmol, Vol(toluene) = 520 µL.f DBOO(87:13) was used.g Determined by SEC (CHCl3, polystyrene standard). | |||||||||
| 1 | t-BuP2 | 70/280/1/1 | 60 | 20 | 22 | 4500 | n.d. | 1100 | 1.31 |
| 2 | MTBD | 70/280/1/1 | 60 | 20 | 6 | 1400 | n.d. | n.d. | n.d |
| 3 | MTBD + TEB | 70/280/1/1 | 60 | 20 | 10 | 2200 | n.d. | n.d. | n.d. |
| 4 | CsOPiv | 70/280/1/1 | 60 | 20 | 30 | 6100 | n.d. | 2000 | 1.28 |
| 5 | CsOPiv | 70/200/1/1 | 60 | 120 | 79.6 | 16000 |
10700 |
4400 | 1.22 |
| 6e | CsOPiv | 60/60/1/1 | 100 | 24 | 74.7 | 13000 |
12900 |
5200 | 1.16 |
| 7e | CsOPiv | 30/30/1/1 | 100 | 8 | 72.0 | 6200 | 6500 | 3300 | 1.14 |
| 8e | CsOPiv | 90/90/1/1 | 100 | 72 | 75.1 | 19600 |
21900 |
7500 | 1.14 |
| 9e,f | CsOPiv | 90/90/1/1 | 100 | 22.5 | 72.0 | 18900 |
17800 |
5700g | 1.31g |
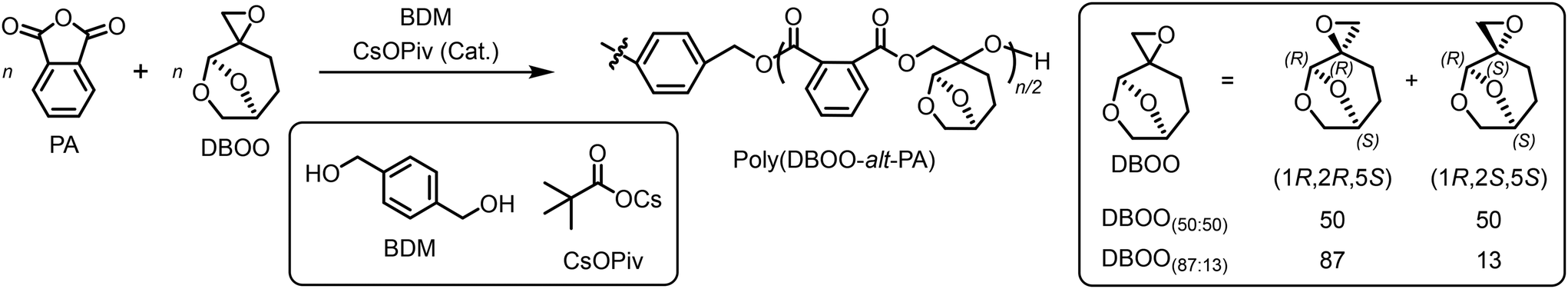
First, several different catalyst systems including t-BuP2, 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD) organobase catalysts, MTBD/triethylborane (TEB) acid/base binary catalysts, and cesium pivalate (CsOPiv), were tested within the same period of 20 h.25–27 The systems using t-BuP2 and CsOPiv catalysts gave monomer conversions higher than 20%, while the MTBD and MTBD/TEB catalytic systems did not show sufficient reactivity, with a monomer conversion of no more than 10%. t-BuP2 showed superior reactivity and controllability in the ROCOP of PA and isobutylene oxide (IBO), the most representative 1,1-disubstituted epoxide, in our previous report.17 However, t-BuP2 does not outperform CsOPiv with regard to the reactivity. Moreover, when using t-BuP2 as the catalyst, the polymerization system rapidly changed from colorless to black. Small unidentified peaks were observed in the crude 1H NMR spectrum in contrast to that obtained using CsOPiv (Fig. S1). After identifying CsOPiv as the most suitable catalyst, the polymerization time was extended to obtain higher monomer conversion. As a result, after 120 h, PA conversion reached 79.6%, and the corresponding polymer was obtained with relatively narrow dispersity (Đ = 1.22) (Table 1, run 5; Fig. S2). The poly(DBOO(50:50)-alt-PA) structure was confirmed by 1H NMR spectroscopy and MALDI-TOF MS, indicating a perfectly alternating structure. No poly(DBOO) homopolymer or segments were observed (Fig. 2A and B). Although a small shoulder peak was observed in the SEC trace in the low-molecular-weight region, the molecular weight distribution remained relatively narrow. The low-molecular-weight shoulder peak was assigned to poly(DBOO(50:50)-alt-PA), which was initiated by CsOPiv. As CsOPiv is a monofunctional initiator, it should result in a polymer with a molecular weight that is half of that of the polymer initiated by the difunctional initiator BDM. This is in agreement with the SEC trace observations.
 | ||
| Fig. 2 (A) 1H NMR spectra of poly(DBOO(50:50)-alt-PA) (Table 1, run 5); (B) MALDI-TOF MS of poly(DBOO(50:50)-alt-PA) (Table 1, run 5); (C) SEC trace of synthesized poly(DBOO(50:50)-alt-PA) with different molecular weight (Table 1, run 6–8); (D) monomer conversion monitoring during ROCOP of PA and DBOO (Table S1). | ||
Following the success of the DBOO/PA ROCOP in bulk, the solution polymerization in toluene with [PA]0 = [DBOO]0 was examined to fully exploit DBOO. Because the reduced monomer concentration resulting from the addition of solvent decreases the polymerization rate, the reaction temperature was increased to 100 °C to allow the reaction to reach completion within a reasonable time. To our delight, the polymerization proceeded well. The monomer conversion reached 74.7% after 24 h, and the theoretical molecular weight (Mn,theo) and NMR molecular weight (Mn,NMR) matched well (Table 1, run 6). The obtained polymer exhibited a relatively narrow dispersity (Đ = 1.16). Polymerization with different target degrees of polymerization (DP) was examined by varying the [monomer]0/[initiator]0 ratio (Table 1, runs 7, 8). Mn increased with the feed ratio, and the dispersity remained narrow, further demonstrating the well-controlled nature of the polymerization (Fig. 2C).
When using DBOO(87:13), which is a DBOO monomer with a different diastereomeric ratio, in the same solution polymerization conditions (Table 1, run 9), the obtained result was similar to that obtained using DBOO(50:50). However, the obtained poly(DBOO(87:13)-alt-PA) exhibited much lower solubility in THF than poly(DBOO(50:50)-alt-PA), and its Mn,SEC (5.7 kDa) was measured in chloroform rather than in THF (Fig. S3). The 1H NMR spectrum of poly(DBOO(87:13)-alt-PA) exhibited different relative peak intensities from those of poly(DBOO(50:50)-alt-PA) due to the different diastereomeric ratios of the monomers (Fig. S4 and S5).
In our previous study on the homopolymerization of DBOO, the two diastereomers exhibited different reaction rates, with the (1R,2S,5S)-diastereomer consumed approximately 1.6 times faster than the (1R,2R,5S)-diastereomer. To understand the reactivities of the two ROCOP diastereomers with PA, a kinetic monitoring experiment was conducted (Table S1; Fig. 2D and S6). It was found that the difference in the consumption rate between the two diastereomers of DBOO was much smaller than that in the ROP of DBOO (Fig. S7). This result demonstrated the nearly ideal random distribution of DBOO diastereomers along the poly(DBOO(50:50)-alt-PA) polymer chain.
2.2. Monomer scope
Following the successful ROCOP of DBOO and PA, other monomer combinations were examined (Table 2). Based on our previous experience in the ROCOP of IBO and cyclic anhydrides, we selected glutaric anhydride (GA), diglycolic anhydride (DGA), and norbornene anhydride (NA) as the cyclic anhydride monomers to test the copolymerization with DBOO(50:50), with the [cyclic anhydride]0/[DBOO(50:50)]0/[BDM]/[CsOPiv] feed ratio fixed at 100/100/1/1 (Table 2, runs 1–3). The polymerizations using GA and DGA were performed in toluene at 100 °C. Similar to the reaction using PA which has a high target DP (Table 1, run 8), these two polymerizations required a comparably long reaction time to obtain high monomer conversion. In both cases, the Mn,theo and Mn,NMR values showed reasonable agreement. Although the dispersity values of the polymer were relatively narrow (Đ = 1.18 and 1.26), Mn,SEC values were drastically lower than Mn,NMR. The alternating structures of the resulting polymers were confirmed by 1H NMR and MALDI-TOF MS (Fig. S8–S13). Multiple unidentified small peaks were observed in the MALDI-TOF MS spectra of both poly(DBOO(50:50)-alt-GA) and poly(DBOO(50:50)-alt-DGA), indicating the occurrence of undesired side reactions during polymerization.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Run | Cyclic anhydride/epoxide | Temp. (°C) | Time (h) | Conv.b (%) | Mn, theo.c |
Mn, NMRb |
Mn, SECd |
Đd |
| a Polymerization conditions: [cyclic anhydride]0/[epoxide]0/[BDM]/[CsOPiv] = 100/100/1/1; Ar atmosphere; n(cyclic anhydride) = 4.00 mmol, Vol(toluene) = 800 µL.b Determined from 1H NMR spectrum of the obtained polymer in CDCl3.c Calculated from [cyclic anhydride]0/[BDM]0 × conv.DBOO/100 × ((M.W. of cyclic anhydride) + (M.W. of DBOO)) + (M.W. of BDM).d Determined by SEC of the polymer obtained in THF using a polystyrene standard. | ||||||||
| 1 | GA/DBOO(50:50) | 100 | 72 | 84.0 | 21800 |
26700 |
3000 | 1.18 |
| 2 | DGA/DBOO(50:50) | 100 | 96 | >99.9 | 26100 |
27200 |
3600 | 1.26 |
| 3 | NA/DBOO(50:50) | 80 | 620 | n.d. | n.d. | n.d. | 2300 | 1.45 |
| 4 | PA/DBOOPh | 110 | 228 | 98.0 | 25100 |
n.d. | 3600 | 1.32 |
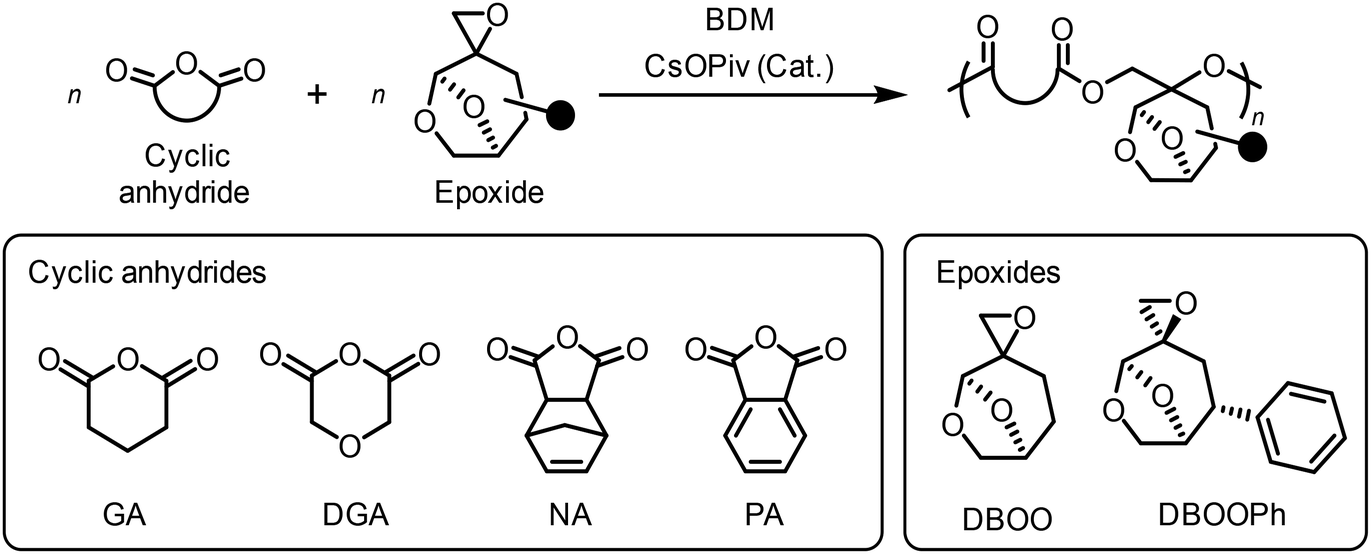
After examining GA and DGA, which are monocyclic anhydrides, tricyclic anhydride NA was tested in ROCOP with DBOO (Table 2, run 3). However, polymerization at 100 °C resulted in a polymer product with very broad dispersity (Đ = 1.74, Table S2, run 1). The product was improved by reducing the polymerization temperature to 80 °C (Đ = 1.48, Table S2, run 2). In contrast to the aforementioned polymerization systems, it was difficult to identify monomer conversion in NA/DBOO ROCOP using 1H NMR because of peak overlap. Therefore, polymerization was performed for a long time (620 h) to ensure sufficient monomer conversion. As a result, the poly(DBOO(50:50)-alt-NA) polymer was obtained with a Mn,SEC = 2.3 kDa, and a slightly broader dispersity (Đ = 1.45, Table 2, run 3; Fig. S14 and S15). In the MALDI-TOF MS spectrum, although multiple peak series were observed, and some of these could not be identified, the periodic peak interval (306.3 Da) matched the sum of the molecular weights of NA and DBOO, supporting the formation of an alternating copolymer (Fig. S16).
After examining different cyclic anhydrides, we expanded the scope of cellulose-derived spiro epoxide monomers. In LGO, the α,β-unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond provides some opportunities to introduce an additional substituent. As a proof-of-concept, we synthesized DBOOPh via the introduction of a phenyl group using the well-known 1,4-addition of LGO, followed by the Corey–Chaykovsky reaction (Schemes S2 and S3). DBOOPh was obtained as a single diastereomer (Fig. S17–S20).
C double bond provides some opportunities to introduce an additional substituent. As a proof-of-concept, we synthesized DBOOPh via the introduction of a phenyl group using the well-known 1,4-addition of LGO, followed by the Corey–Chaykovsky reaction (Schemes S2 and S3). DBOOPh was obtained as a single diastereomer (Fig. S17–S20).
ROCOP of DBOOPh with PA was carried out at 110 °C due to the increased steric hindrance by the additional phenyl group (Table S3; Table 2, run 4). The monomer conversion reached 98% after 228 h. Despite low reactivity, it afforded poly(DBOOPh-alt-PA) with Mn,SEC = 3.6 kDa and moderate dispersity (Đ = 1.32) (Fig. S21). The 1H NMR spectrum and MALDI-TOF MS of the polymer further supported the alternating structure (Fig. S22 and S23).
2.3. Physical properties investigation
The thermal properties of the polymers obtained by the aforementioned polymerization were evaluated. First, thermogravimetric analysis (TGA) of the synthesized polyesters was performed. The 5% weight-loss decomposition temperatures (Td,5%) were determined to be in the range of 224–297 °C, demonstrating sufficient thermal stability (Fig. S24–S29). Subsequently, the obtained polyesters were examined by differential scanning calorimetry (DSC) analysis (Fig. S30–S35). As summarized in Fig. 3, the glass-transition temperatures (Tg) of the polyesters ranged from 29 °C to 130 °C. In particular, poly(DBOO(50:50)-alt-GA) and poly(DBOO(50:50)-alt-DGA) exhibited relatively low Tg values of 29 °C and 64 °C, respectively, consistent with their incorporation of relatively flexible aliphatic segments in the backbone. The higher Tg of poly(DBOO(50:50)-alt-DGA) compared to that of poly(DBOO(50:50)-alt-GA) can be attributed to the presence of additional aliphatic ether moieties which result in additional interactions between the polymer chains. Introduction of bulky and rigid cyclic anhydride units restricted segmental motion, resulting in a much higher Tg of 130 °C for both poly(DBOO(50:50)-alt-PA) and poly(DBOO(50:50)-alt-NA). | ||
| Fig. 3 DSC traces of synthesized polyesters. | ||
For poly(DBOO(87:13)-alt-PA) and poly(DBOOPh-alt-PA), more ordered structures were expected owing to the use of diastereomerically enriched and even optically pure monomers. However, no crystallization (Tc) or melting (Tm) transitions were detected in the DSC traces. The Tg of poly(DBOO(87:13)-alt-PA) was found to be 127 °C, which is similar to that of poly(DBOO(50:50)-alt-PA). The Tg for poly(DBOOPh-alt-PA) was measured as 123 °C, which is even slightly lower than that of poly(DBOO-alt-PA), despite the additional introduction of a rigid phenyl substituent. The amorphous nature of the obtained polyesters was further confirmed by X-ray diffraction (XRD) analysis (Fig. S36–S41).
2.4 Investigation of the secondary structure via circular dichroism
In our previous study on the ROP of DBOO, the resulting PDBOO polyether was expected to adopt a helical conformation when a single diastereomer or a diastereomerically enriched monomer was used.20 However, experimental validation of this conformation proved difficult owing to the lack of the ultraviolet-visible (UV-VIS) absorption and poor solubility of poly(DBOO(87:13)).In the present study, for ROCOP with PA, the rigid benzene ring of PA together with the bulky spiro-epoxide monomers afforded polyesters with high glass transition temperatures, indicating relatively rigid backbone structures. Moreover, the chirality of DBOO or DBOOPh may induce the polymer chains to adopt chiral secondary conformations such as helices.
Taking advantage of the UV absorption arising from the aromatic ring, the UV-VIS absorption and circular dichroism (CD) spectra of poly(DBOO(50:50)-alt-PA), poly(DBOO(87:13)-alt-PA), and poly(DBOOPh-alt-PA) were measured in chloroform (Fig. 4 and S42). An absorption band attributed to the α-band of the aromatic rings in the polymer backbone was observed in the region of 260–300 nm. For all polymers, the molar absorptivity (ε) at 270 nm was approximately 1200 L mol−1 cm−1, confirming that the absorption originated from the aromatic backbone.
 | ||
| Fig. 4 (A) Chemical structures of poly(DBOO-alt-PA) and poly(DBOOPh-alt-PA) with highlighted benzene rings; (B) CD (top) and UV–vis absorption (bottom) spectra of poly(DBOO(50:50)-alt-PA) (red), poly(DBOO(87:13)-alt-PA) (green), and poly(DBOOPh-alt-PA) (blue) measured in chloroform at −20 °C ([polymer] = 10 mM, based on repeating units). | ||
The CD spectra of these three polymers exhibited a similar split Cotton effect in the region of 250–300 nm: the negative 1st Cotton, positive 2nd Cotton, and the cross point at approximately 270 nm. These results indicate that, regardless of the diastereomeric ratio of DBOO or the use of the DBOOPh monomer, the exciton coupling between aromatic units along the polymer chain preferentially adopts a counterclockwise arrangement, which may induce a biased left-handed helical conformation. Among the three samples, the most distinct CD signal was observed for poly(DBOOPh-alt-PA), presumably due to the use of an optically pure epoxide monomer which further increased the rigidity by introducing a phenyl side group.
3. Conclusions
In summary, we report the synthesis of polyesters with spirocyclic backbones via the ROCOP of cellulose-derived spiro-epoxides with cyclic anhydrides. Using a CsOPiv catalyst in combination with an alcohol initiator, this polymerization system exhibited living/controlled characteristics and afforded polyesters with alternating structures. This strategy is applicable to various cyclic anhydrides, including PA, GA, DGA, and NA, and was successfully extended to the structurally modified spiroepoxide DBOOPh. The resulting polyesters exhibited sufficient thermal stability and broadly tunable glass-transition temperatures (29–130 °C). Although no crystallization behavior was observed by DSC and XRD, the chirality of the spiro-epoxides together with their rigid spirocyclic backbones induced chiral environments along the polymer chains, as confirmed by CD measurements. It may further induce the polymer chains to adopt a biased one-handed helical conformation. This study expands the structural diversity of polyesters and broadens the scope of ROCOP chemistry by employing novel biomass-derived spiroepoxides. Moreover, the chiral secondary structure of the obtained polymers demonstrates the potential of the polyesters synthesized via ROCOP as novel chiral materials.Author contributions
T. Gao and H. Ayakawa contributed equally to this study. Tianle Gao: methodology, visualization, data curation, writing – original draft, funding acquisition. Hiroto Ayakawa: methodology, visualization, data curation, writing – original draft; Tatsuya Nishimura: methodology, visualization, data curation, funding acquisition; Ayano Yoshida: methodology; Moeno Sugiyama: methodology; Takuya Yamamoto: supervision; Kenji Tajima: supervision; Kenji Takahashi: supervision, funding acquisition; Feng Li: supervision, conceptualization, methodology, writing – original draft, writing – review and editing, and funding acquisition. Takuya Isono: supervision, writing – review & editing, funding acquisition; Katsuhiro Maeda: supervision, funding acquisition; Toshifumi Satoh: supervision, conceptualization, writing – review and editing, and funding acquisition.Conflicts of interest
Hokkaido University filed a patent (JP 2025-109705) based on this work. T. S., T. L., F. L., T. G., and H. A. are listed as inventors for this application. The authors declare no conflicts of interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6py00397d.Acknowledgements
This work was financially supported by the Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research(A) 24H00766 (T. S.), Grant-in-Aid for Scientific Research(B) JP25K01815 (K. M., T. N.) and JP24K01466 (T. N.), Grant-in-Aid for Challenging Research (Exploratory) JP25K22281 (T. N.), Grant-in-Aid for Early-Career Scientists 24K17719 (F. L.), the JST COI-NEXT Program (no. JPMJPF2102) (K. T.), the Frontier Chemistry Center (Hokkaido University) (T. S.), the Photo-Excitonic Project (Hokkaido University) (T. S.), the List Sustainable Digital Transformation Catalyst Collaboration Research Platform (List-PF, Hokkaido University) (T. S.), the Project of Junior Scientist Promotion (Hokkaido University) (T. S.), Cocreation Core for Soft Materials Aspiring Research & Translation (C3-SMART) (T. I.), the JSPS Program for Forming Japan's Peak Research Universities (J-PEAKS; No. JPJS00420230001) “Soft Materials Platform Aspiring the Unique Properties from Natural Polymers” (T. I.), and ENEOS TONENGENERAL RESEARCH/DEVELOPMENT ENCOURAGEMENT & SCHOLARSHIP FOUNDATION (F. L.). T. G. gratefully acknowledges the JSPS Fellowship for Young Scientists.References
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- G. Odian, Principles of polymerization, John Wiley & Sons, 2004 Search PubMed.
- S. M. Guillaume, E. Kirillov, Y. Sarazin and J. F. Carpentier, Chem. – Eur. J., 2015, 21, 7988–8003 CrossRef CAS.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- D. Ryzhakov, G. Printz, B. Jacques, S. Messaoudi, F. Dumas, S. Dagorne and F. Le Bideau, Polym. Chem., 2021, 12, 2932–2946 RSC.
- T. Aida and S. Inoue, J. Am. Chem. Soc., 1985, 107, 1358–1364 CrossRef CAS.
- M. Sengoden, V. Satheesh, S. Sarkar and D. J. Darensbourg, Polym. Chem., 2025, 16, 4548–4556 RSC.
- T. M. McGuire, E. F. Clark and A. Buchard, Macromolecules, 2021, 54, 5094–5105 CrossRef CAS.
- F. D. Monica and A. W. Kleij, ACS Sustainable Chem. Eng., 2021, 9, 2619–2625 Search PubMed.
- J. Xu, J. Liu and N. Hadjichristidis, J. Am. Chem. Soc., 2024, 147, 945–956 CrossRef.
- A. Brandolese, F. Della Monica, M. A. Pericàs and A. W. Kleij, Macromolecules, 2022, 55, 2566–2573 CrossRef CAS.
- R. C. Jeske, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11330–11331 CrossRef CAS.
- A. K. Hubbell, J. R. Lamb, K. Klimovica, M. Mulzer, T. D. Shaffer, S. N. MacMillan and G. W. Coates, ACS Catal., 2020, 10, 12537–12543 CrossRef CAS.
- L. Hu, X. Zhang, X. Cao, D. Chen, Y. Sun, C. Zhang and X. Zhang, Macromolecules, 2021, 54, 6182–6190 CrossRef CAS.
- M. Sugiyama, R. Suzuki, H. Ayakawa, T. Gao, F. Li, T. Yamamoto, T. Isono and T. Satoh, Polym. J., 2025, 57, 1153–1163 CrossRef CAS.
- Z. Xie, Z. Yang, C. Hu, M. Niu, Y. Zhang, T. Yang, X. Pang and X. Chen, Nat. Commun., 2026, 17, 2668 CrossRef CAS.
- M. M. Zanardi, A. G. Suárez and A. M. Sarotti, J. Org. Chem., 2017, 82, 1873–1879 CrossRef CAS PubMed.
- H. Ayakawa, T. Nishimura, F. Li, T. Gao, T. Yamamoto, K. Tajima, T. Isono, K. Maeda and T. Satoh, ACS Macro Lett., 2026, 15, 725–731 CrossRef CAS.
- M. K. Stanfield, R. S. Terry, J. A. Smith and S. C. Thickett, Polym. Chem., 2023, 14, 4949–4956 RSC.
- P. Ray, T. Hughes, C. Smith, M. Hibbert, K. Saito and G. P. Simon, Polym. Chem., 2019, 10, 3334–3341 RSC.
- G. Wang, S. Ji, Z.-C. Li and C. Shi, Chin. Chem. Lett., 2026, 112581 CrossRef.
- Y. Mizukami, Y. Kakehi, F. Li, T. Yamamoto, K. Tajima, T. Isono and T. Satoh, ACS Macro Lett., 2024, 13, 252–259 CrossRef CAS.
- H. Li, J. Zhao and G. Zhang, ACS Macro Lett., 2017, 6, 1094–1098 CrossRef CAS PubMed.
- L.-F. Hu, C.-J. Zhang, H.-L. Wu, J.-L. Yang, B. Liu, H.-Y. Duan and X.-H. Zhang, Macromolecules, 2018, 51, 3126–3134 CrossRef CAS.
- X. Xia, R. Suzuki, K. Takojima, D.-H. Jiang, T. Isono and T. Satoh, ACS Catal., 2021, 11, 5999–6009 CrossRef CAS.
Footnote |
| † T. Gao and H. Ayakawa contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2026 |