
Coupling doped halogen sites in copper(I)–organic frameworks with cuprous oxide for high selectivity CO2 photoreduction with H2O†
Jing
Liang
a,
Shengfu
Huang
a,
Yuan
Chang
b,
Andreas
Terfort
 c,
Junfeng
Gao
*b and
Jinxuan
Liu
*ad
c,
Junfeng
Gao
*b and
Jinxuan
Liu
*ad
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, 116024 Dalian, P. R. China. E-mail: Jinxuan.liu@dlut.edu.cn
bLaboratory of Materials Modification by Laser, Ion and Electron Beams, Ministry of Education, Dalian University of Technology, 116024 Dalian, P. R. China. E-mail: gaojf@dlut.edu.cn
cDepartment of Chemistry, Institute of Inorganic and Analytical Chemistry, Goethe University Frankfurt, Max-von-Laue-Street 7, 60438 Frankfurt, Germany
dLeicester International Institute, Dalian University of Technology Panjin Campous, 124221 Panjin, P. R. China
First published on 26th April 2024
Abstract
Photoreduction catalysis offers an eco-friendly approach to convert CO2 into valuable chemicals. However, understanding the influence of halide active sites in metal–organic framework (MOF) catalysts on photoreduction and selectivity remains limited. We report different halogen (X) sites (X = Cl, Br and I) in copper(I)–organic frameworks (CuX–bpy) grown on Cu2O for photoreduction of CO2 without the assistance of additional sacrificial agents and photosensitizers. Impressively, the Cu2O@CuCl–bpy-MOF efficiently photo-reduces CO2 into formate with a high yield of 1520 μmol g−1 after 12 hours of reaction. Density functional theory calculation results reveal that the effective coordination coupling between the MOF and semiconductor facilitates rapid electron transfer leading to efficient CO2 reduction and H2O oxidation.
Introduction
Mimicking natural photosynthesis to convert CO2 with H2O into high-value products offers a promising sustainable solution for decreasing anthropogenic CO2 emissions and producing renewable chemicals.1 However, it faces the challenge of coupling the CO2 reduction reaction (CO2RR) and water oxidation reaction into one photosynthetic system and achieving high selectivity of the overall reaction.2 To address these challenges, combining two components with independent functions to construct catalysts has been considered to be a significant method.3 Nevertheless, numerous vital aspects are challenging to address simultaneously in a majority of composite systems including efficient light absorption, the separation and rapid migration of photo-generated carriers to redox sites and the rapid adsorption and desorption of CO2.4–8 Consequently, the design and fabrication of composite photocatalysts with well-defined structures to achieve highly selective CO2 photoreduction is of great significance.Metal–organic frameworks (MOFs) with excellent structural designability and the presence of both adsorption and active sites have emerged as a promising candidate for the capture and conversion of CO2.9–11 By adjusting appropriate metal nodes and functional organic linkers, MOFs are capable of possessing efficient light absorption and fast separation and migration of photogenerated carriers.12,13 A variety of MOF-based photocatalysts have been demonstrated for the photocatalytic reduction of CO2.14–16 Inorganic semiconductors such as Cu2O,17 TiO2,18 and α-Fe2O3 (ref. 19) have been reported to be able to achieve highly efficient water photooxidation. On this basis, a feasible strategy is to combine CO2 reduction MOFs with water oxidation inorganic semiconductors to create composite catalysts to enhance the overall selective reaction in artificial photosynthesis.20–23
MOF derived composites, such as the heterojunction of TTCOF and NH2-UiO-66 (TTCOF/NUZ),24 Ni-doped CdS nanoparticles@bimetallic MOF (Ni@CdS⊂Zn/Co-ZIF),25 MOF-808 loaded with a molecular photosensitizer (Zr-MBA-Ru/Re-MOF),26 doped nano-gold@MOF (Au-NC@UiO-68-NHC)27 and ligand modified Zr-MOF (BUT-110),28 have been demonstrated to be capable of reduction of CO2 under light illumination. However, most of these materials primarily yield low-value CO products and require additional photosensitizers and sacrificial agents, which can be economically unfriendly and harmful to the healthy development of the environment. We hypothesize that incorporating a MOF into photosensitive semiconductors may reduce the reliance on photosensitizers and sacrificial agents in the photocatalytic system.29,30
Halogens plays a pivotal role in the field of photocatalysis. A substantial body of research has revealed their significance, particularly in metal halide perovskite photocatalysts, where halogens profoundly affect the electrolytic transport properties and overall performance.31 Zhao et al. demonstrated that in CsCuCl2Br, the substitution of Cl− with Br− effectively narrows the bandgap, harnessing a wider range of solar energy and enhancing the transport of photoelectrons and holes.32 Guo et al. also demonstrated the influence of halogens by regulating the ratio of Br− and Cl−, leading to notable changes in the yield of CO and CH4 (ref. 33). The efficient charge separation and favorable stability in mixed-halide perovskites contribute to their exceptional performance in the CO2RR. However, in MOF composite materials, relevant research on the impact of halogens on their photocatalytic CO2RR performance has not yet been reported.
Herein, different halogen (X) sites (X = Cl, Br and I) in copper(I)–organic frameworks in situ compounded on cuprous oxide (Cu2O@CuX–bpy; bpy = 4,4′-bipyridine) have been synthesized towards selectively photocatalytic CO2RR integrated with water photooxidation. Cu2O@CuCl–bpy achieves efficient photo-reduction of CO2 into formate (HCOOH) with a high yield of 1520 μmol g−1 in a 12 h photoreduction process without sacrificial agents and photosensitizers. In situ spectroscopic results suggest that the prominent photocatalytic performance arises from increased electronegativity, enhancing CO2 adsorption and stabilizing HCOOH intermediates, and it has been confirmed by density functional theory (DFT) calculation results that the photogenerated electrons rapidly migrate from the semiconductor to the MOF through coordination coupling, accumulating at MOF active sites for the CO2RR, while holes in the semiconductor enable H2O oxidation, mimicking natural photosynthesis. Cu2O@CuCl–bpy serves as a rational platform for investigating the correlation between halogen sites and photocatalytic efficiency.
Results and discussion
The synthesis of powdery doped halogen sites in copper(I)–organic frameworks loaded on Cu2O (Cu2O@CuX–bpy) is schematically illustrated in Fig. 1(a). Cu2O nanocubes were synthesized by reducing copper oxide nanocubes with ascorbic acid.34 Cu2O@CuCl–bpy was obtained through in situ recombination of CuCl–bpy on the Cu2O nanocube surface. The Cu2O@CuBr–bpy and Cu2O@CuI–bpy were synthesized by changing the added cuprous chloride into cuprous bromide and cuprous iodide, respectively.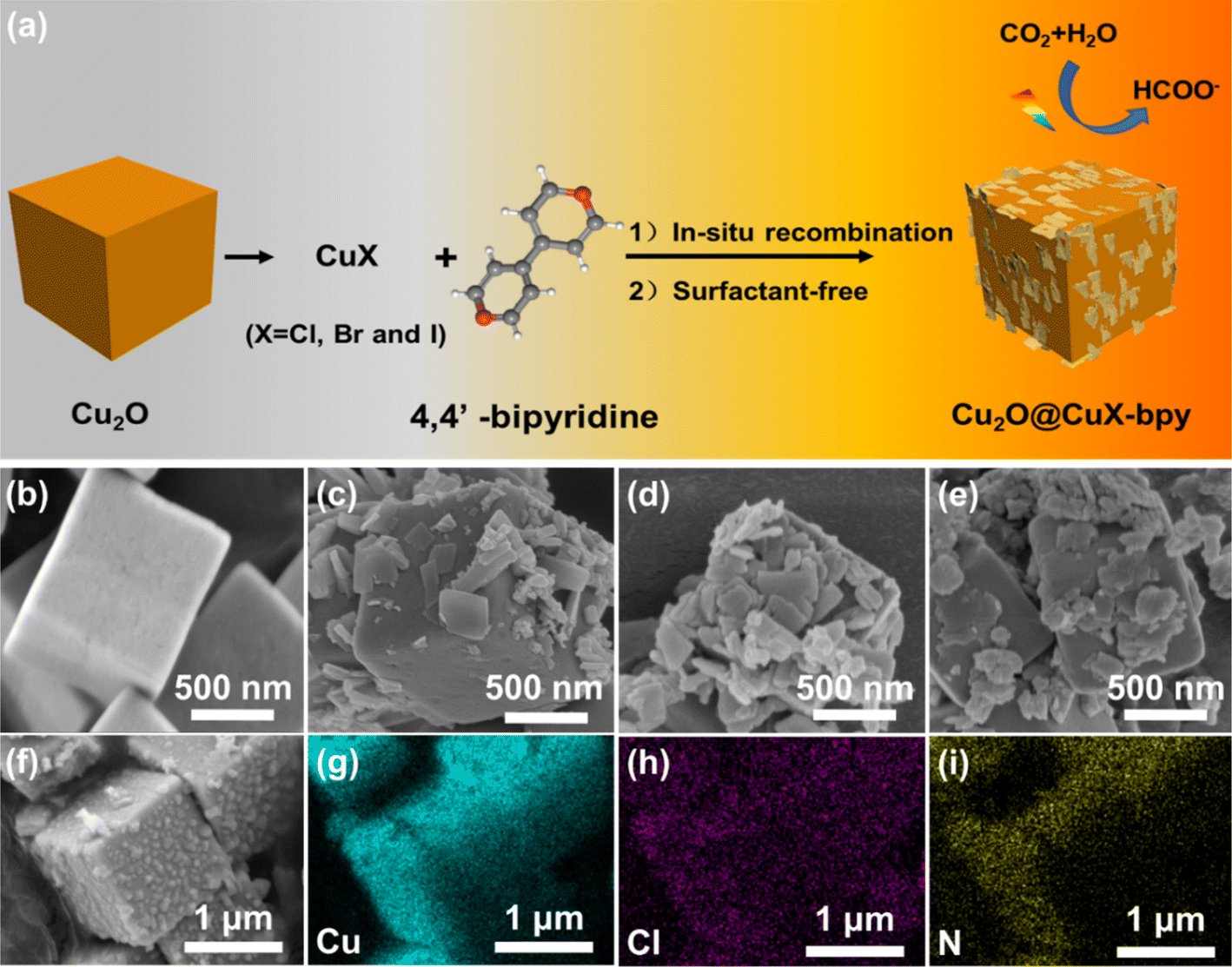 | ||
| Fig. 1 (a) Schematic illustration of the fabrication method of Cu2O@CuX–bpy. SEM images of (b) Cu2O nanocubes, (c) Cu2O@CuCl–bpy, (d) Cu2O@CuBr–bpy and (e) Cu2O@CuI–bpy. (f–i) SEM image of the Cu2O@CuCl–bpy and the corresponding element mapping images of Cu, Cl, and N. | ||
The X-ray diffraction (XRD) patterns of Cu2O@CuX–bpy are shown in Fig. S1.† In Fig. S1(a),† the pronounced diffraction peaks at 29.5°, 36.4°, 42.3°, 61.4° and 73.5° can be indexed to the planes of Cu2O (JCPDS card no. 05-0667). Taking Cu2O@CuCl–bpy as an example, the PXRD pattern of the product highly matches with that of the simulated CuCl–bpy, indicating the synthesis of the Cu2O@CuCl–bpy compound (Fig. S1(b)†). To detect the morphology, field emission scanning electron microscopy (SEM) was employed. The SEM image in Fig. 1(b) revealed the formation of a square-shaped morphology for the as-prepared Cu2O with an average size of 1 μm. As shown in Fig. 1(c)–(e), in situ recombination of a two-dimensional sheet-like nanostructure on Cu2O was observed in Cu2O@CuX–bpy as well as distribution at the surface of Cu2O nanocubes. From Fig. 1(f)–(i) and S2 and S3,† the homogeneous distribution of each element was observed by energy-dispersive spectra (EDS) for Cu2O@CuX–bpy, indicating successful combination of CuX–bpy on the Cu2O surface.
The chemical composition and valence state of Cu2O@CuCl–bpy were determined by X-ray photoelectron spectroscopy (XP spectra) as shown in Fig. 2. The XP spectra results clearly demonstrate the existence of Cu, O, N, and Cl elements within the Cu2O@CuCl–bpy. The XP spectra of Cu 2p (Fig. 2(a)) exhibit two main spin–orbit doublets for Cu2O and Cu2O@CuCl–bpy. The peaks due to Cu2O located at 932.4 eV and 952.3 eV can be assigned to Cu+ 2p3/2 and Cu+ 2p1/2, respectively.35,36 In the Cu LMM Auger spectrum presented in Fig. S4,† the peak at 569 eV indicates the absence of Cu(0). Compared to Cu2O, the slight position shift of Cu+ for Cu2O@CuCl–bpy located at 932.1 eV and 952 eV to lower binding energy can be ascribed to the change of local environment charge caused by the presence of halogen element. The chlorine element in CuCl–bpy can coordinate with Cu on the Cu2O surface, which made Cu+ move slightly towards lower binding energy. The detailed peak assignments and fitted results are listed in Table S1.†
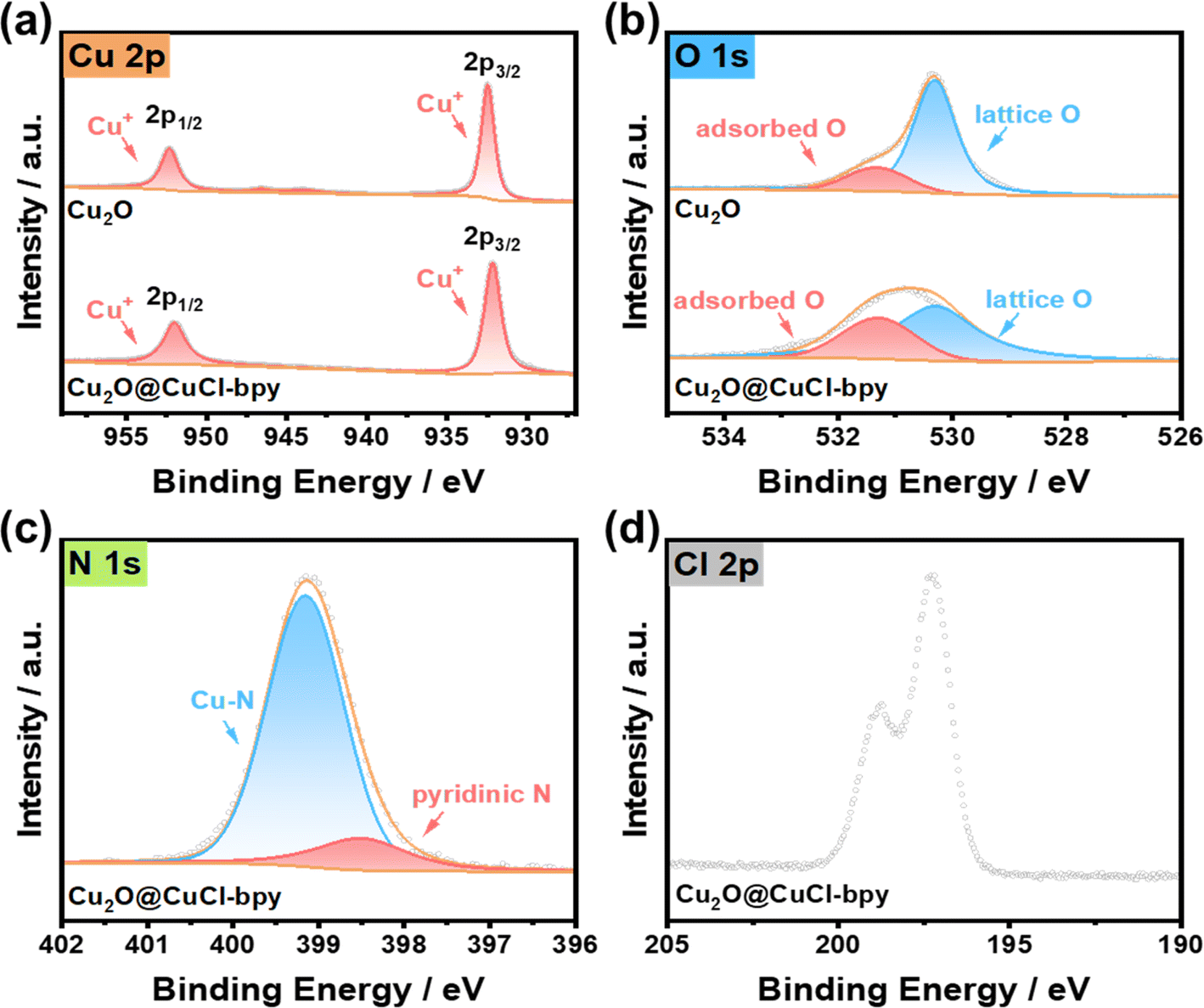 | ||
| Fig. 2 XP spectra of Cu2O and Cu2O@CuCl–bpy. (a) Cu 2p, (b) O 1s, (c) N 1s and (d) Cl 2p. | ||
Two types of oxygen are observed for Cu2O (Fig. 2(b)), i.e., the peaks at 531.3 eV and 530.3 eV, which can be attributed to the adsorbed O and lattice O, respectively.37 The adsorbed O slightly increased and lattice O significantly decreased for Cu2O@CuCl–bpy as compared to the adsorbed O and the lattice O for Cu2O. This results from the successful integration of CuCl–bpy with rich adsorption sites onto the Cu2O surface, and thus decreases the lattice O and increases the adsorbed O within Cu2O@CuCl–bpy. For the XPS N 1s spectrum of Cu2O@CuCl–bpy in Fig. 2(c), a main peak at 399.1 eV can be observed, assigned to Cu–N bonding.38 The extra weak peak at 398.5 eV can be assigned to pyridinic N, which did not successfully coordinate with the metal ions during synthesis. The XPS chlorine 2p spectrum of Cu2O@CuCl–bpy displayed peaks at 198.7 eV and 197.2 eV that could be assigned to Cl− in chloride (Fig. 2(d)).39
For photo-reduction of CO2, the separation behavior of photogenerated charge carriers is one of the vital factors. The optical properties of the samples were analyzed with the UV-vis diffuse reflectance spectra (UV-vis-DRS). They show broad and strong absorption in the region of 400–600 nm (Fig. 3(a) and S5†), which indicates the suitable light absorption range and potential photo-response properties of Cu2O@CuX–bpy. Using Tauc's relation, the band-gap of Cu2O@CuCl–bpy, Cu2O@CuBr–bpy and Cu2O@CuI–bpy can be determined to be 1.86 eV, 1.95 eV and 1.97 eV, respectively. The conduction band (CB) positions are estimated by measuring the flat band potential (Efb) according to Mott–Schottky (M–S) curves (Fig. S6†). Thus, based on the combined UV-vis-DRS and M–S analyses, the energy band structures of Cu2O@CuX–bpy are shown in Fig. 3(b). The appropriate energy band structure ensures that they are suitable for photocatalytic reduction of CO2 together with oxidation of water to various fuels.
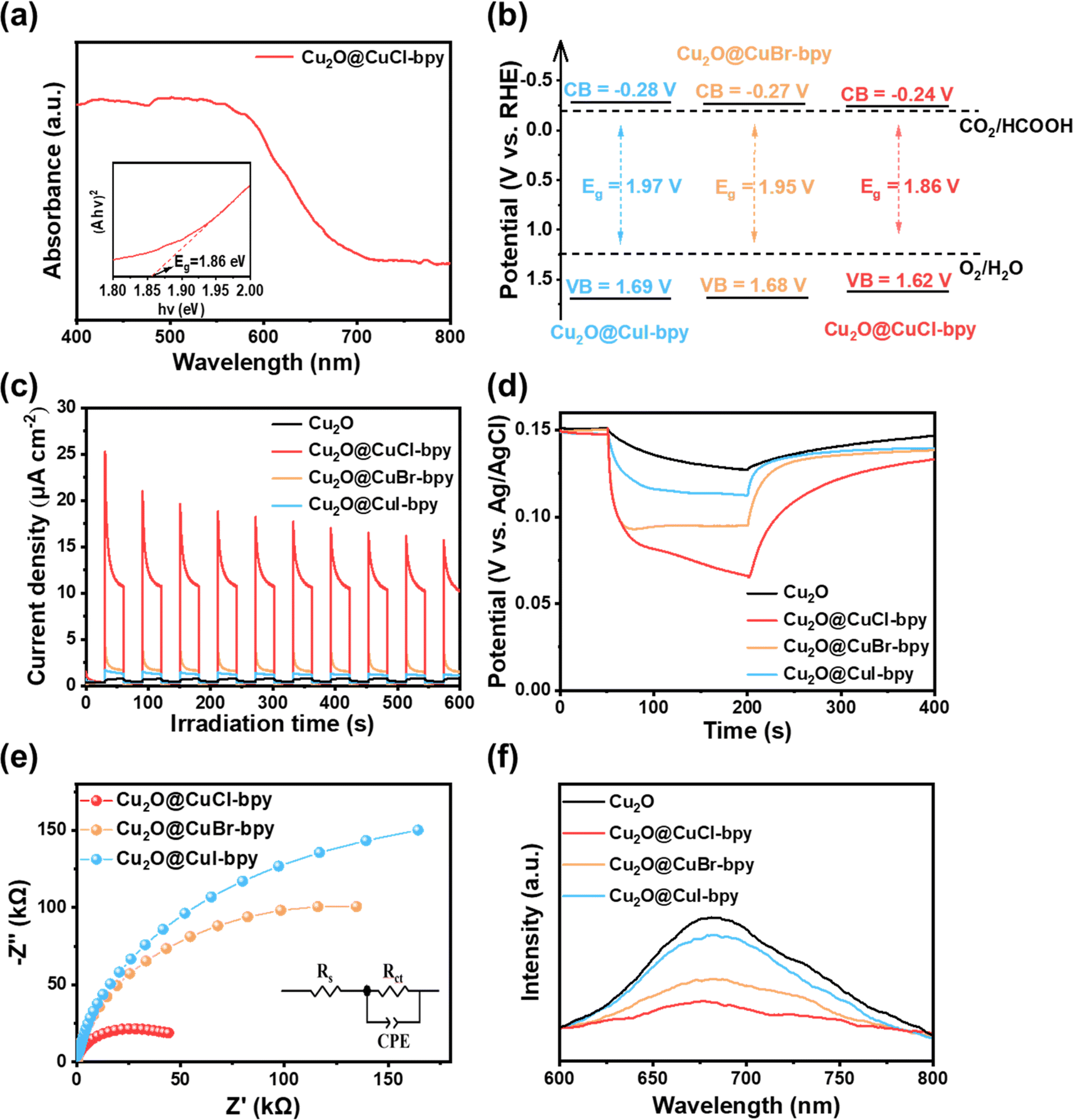 | ||
| Fig. 3 (a) UV-vis DRS spectra (inset: Tauc plots) of Cu2O@CuCl–bpy. (b) The energy band structure of Cu2O@CuX–bpy. (c) Transient photocurrent responses. (d) Open-circuit potential decay curves of Cu2O and Cu2O@CuX–bpy. (e) EIS spectra of Cu2O@CuX–bpy. (f) Steady-state PL spectra of Cu2O and Cu2O@CuX–bpy. | ||
In order to have efficient performance in photocatalytic reactions, the suitable photocatalyst also needs to have prominent charge migration ability. According to the photoelectrochemical (PEC) tests, the Cu2O@CuX–bpy samples all display more remarkable photocurrent density in comparison to Cu2O (Fig. 3(c)). Especially, Cu2O@CuCl–bpy reaches the largest photocurrent density, suggesting efficient carrier transfer and separation. Furthermore, the photoelectron lifetimes of Cu2O and Cu2O@CuX–bpy were determined by the decay of open circuit potentials (Fig. 3(d)). Compared with Cu2O, the Cu2O@CuX–bpy samples (especially Cu2O@CuCl–bpy) present a much more prolonged photoelectron lifetime and a higher photovoltage under irradiation, confirming the more efficient separation of photogenerated electron–hole pairs. On the basis of electrochemical impedance spectroscopy (EIS) spectra and the equivalent circuit (Fig. 3(e)), the EIS spectrum of Cu2O@CuCl–bpy presents the smallest semicircle diameter, indicating the lowest charge transfer resistance.
The photochemical properties of compound materials are studied with the help of steady-state photoluminescence (PL) spectra to display charge migration dynamics. As shown in Fig. 3(f), the Cu2O@CuX–bpy samples all show a lower emission peak in comparison to Cu2O, implying weaker recombination of photoinduced carriers. Particularly, Cu2O@CuCl–bpy exhibits the lowest PL intensity, suggesting enhanced charge separation.
Time-resolved transient photoluminescence (TRPL) spectra were further investigated to track the charge dynamics in real time (Fig. S7†). After being fitted by a bi-exponential equation, a long average lifetime is achieved for Cu2O@CuCl–bpy (τCl = 48.28 ns) compared to Cu2O (τO = 1.62 ns), indicating the suppressed recombination of the photogenerated electrons and holes of Cu2O@CuCl–bpy. The efficient charge separation is ascribed to CuCl–bpy loaded on Cu2O as the charge capture and enrichment center. Combining the analysis results of PL and TRPL, the appropriate introduction of doped halogen sites in copper(I)–organic frameworks loaded on Cu2O can shorten the electron-transfer distance, accelerate the transfer dynamics and slow down the photogenerated carrier recombination.
Considering the above discussions of all the characterization results, the Cu2O@CuX–bpy samples have an appropriate energy band structure, photoresponse capability and photogenerated carrier separation performance, meeting the needs of photocatalytic reduction of CO2 with oxidation of water, which have optimistic application prospects in the field of overall reaction of artificial photosynthesis. Therefore, the photocatalytic assay of the as-prepared Cu2O and Cu2O@CuX–bpy toward photocatalytic reduction of CO2 coupled with oxidation of water was evaluated by using visible-light (λ > 400 nm, 200 mW cm−2) illumination in CO2-saturated water under mild reaction conditions (25 °C). No additional sacrificial agent and photosensitizer are needed in the reaction process.
After photocatalytic reaction, only HCOOH as a liquid reduction product could be detected by ion chromatography (Fig. S8†) and 1HNMR (Fig. S9†). The evolution of HCOOH from Cu2O@CuX–bpy as a function of reaction time is shown in Fig. 4(a). The evolution yield of HCOOH exhibits a linear increase behaviour during the initial 12 hour reaction, while no other gaseous and liquid products of the CO2RR were detected, resulting in a high selectivity. We found that the evolution yield of HCOOH in the photocatalytic CO2RR over the Cu2O@CuX–bpy increased significantly with an increase in the electronegativity of the halogen, suggesting that the halogen modification promotes the reaction steps of HCOOH formation. Notably, it was found that Cu2O@CuCl–bpy is the optimum photocatalyst to achieve the highest evolution yield of HCOOH and the total yield is 1520 μmol g−1 after 12 hours of reaction, which is higher than that of the majority of MOF-based photocatalysts (Table S2†). This could be ascribed to the appropriate energy band structure, the best photoresponse performance and charge separation ability of Cu2O@CuCl–bpy, which is consistent with the above characterization results.
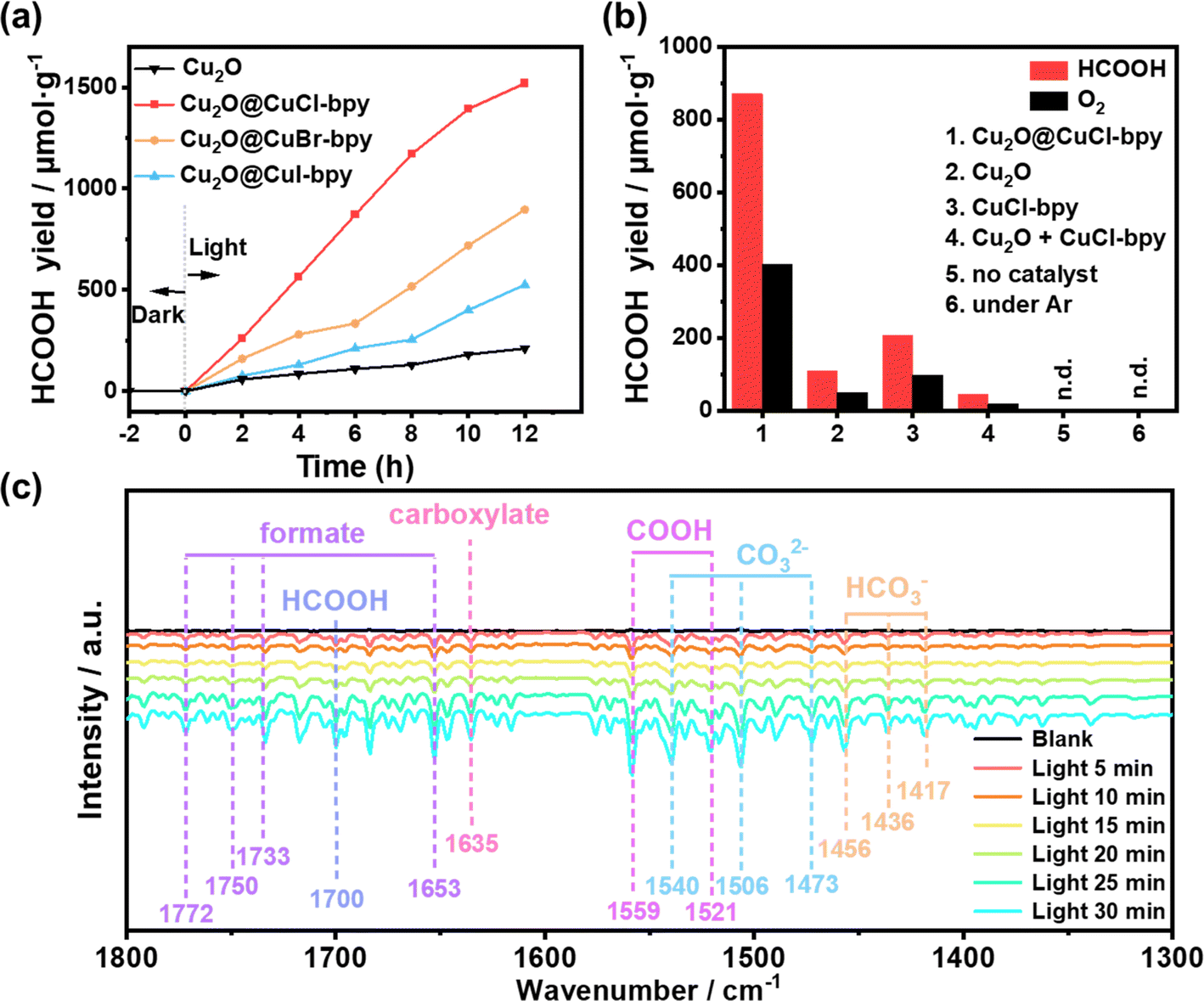 | ||
| Fig. 4 (a) The yield of HCOOH as a function of the time of visible light illumination over Cu2O and Cu2O@CuX–bpy. (b) The yield of HCOOH produced with different catalyst and reaction conditions after 6 h of visible light illumination. (c) In situ FTIR spectra for the simulated photo-driven CO2 reduction process over Cu2O@CuCl–bpy. | ||
For comparison, control experiments were conducted under various reaction conditions as displayed in Fig. 4(b). The evolution yield of O2 produced by photocatalytic oxidation of water was detected and the HCOOH/O2 ratio is close to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as expected, which is consistent with the assumption of the photocatalytic reduction of CO2 integrated with oxidation of water in one photoredox cycle. Remarkably, the HCOOH evolution yield with Cu2O@CuCl–bpy can reach as high as 870.6 μmol g−1 in 6 h, over 7 times higher than that with Cu2O (109.4 μmol g−1) and over 4 times higher than that with CuCl–bpy (208.1 μmol g−1). In addition, physically mixed control experiments with Cu2O + CuCl–bpy composites were performed. The evolution yield of HCOOH is only about five percent that of Cu2O@CuCl–bpy, which demonstrates the necessity of the in situ recombination of CuCl–bpy on the Cu2O nanocube surface. Additional control experiments demonstrated that negligible reduction products could be detected without the photocatalyst, CO2, or light illumination. These results suggested that those factors were all indispensable for photocatalytic CO2 reduction and the generated HCOOH production does indeed originate from the reduction of CO2 molecules rather than the photolysis from other carbon-containing species in this photocatalytic system.
:1 as expected, which is consistent with the assumption of the photocatalytic reduction of CO2 integrated with oxidation of water in one photoredox cycle. Remarkably, the HCOOH evolution yield with Cu2O@CuCl–bpy can reach as high as 870.6 μmol g−1 in 6 h, over 7 times higher than that with Cu2O (109.4 μmol g−1) and over 4 times higher than that with CuCl–bpy (208.1 μmol g−1). In addition, physically mixed control experiments with Cu2O + CuCl–bpy composites were performed. The evolution yield of HCOOH is only about five percent that of Cu2O@CuCl–bpy, which demonstrates the necessity of the in situ recombination of CuCl–bpy on the Cu2O nanocube surface. Additional control experiments demonstrated that negligible reduction products could be detected without the photocatalyst, CO2, or light illumination. These results suggested that those factors were all indispensable for photocatalytic CO2 reduction and the generated HCOOH production does indeed originate from the reduction of CO2 molecules rather than the photolysis from other carbon-containing species in this photocatalytic system.
In order to further explore the photocatalytic stability of Cu2O@CuCl–bpy, the cyclic catalytic performance was examined by running the experiment for five consecutive cycles (Fig. S10†). The Cu2O@CuCl–bpy retained the original photocatalytic efficiency for the HCOOH production during three consecutive cycles, indicating the remarkable stability and activity of the photocatalyst. However, in the last two cycles, the performance of Cu2O@CuCl–bpy decreases gradually, which may be due to the partial desquamation of CuCl–bpy on the Cu2O surface. The structure, morphology, and chemical states of Cu2O@CuX–bpy after the photocatalytic reaction were characterized using PXRD, SEM and XP spectra as shown in Fig. S11–S15.† No noticeable change in PXRD between the initial and 12 h catalyzed Cu2O@CuX–bpy can be observed, demonstrating that the structure of Cu2O@CuX–bpy remains unchanged during the photocatalytic reaction process (Fig. S11†). Furthermore, SEM images show that Cu2O@CuX–bpy maintains its basic morphology after photocatalysis and the partial desquamation of CuX–bpy on the Cu2O surface can be observed, which is consistent with the previous conjecture (Fig. S12–S14†). After 12 hours of photoreaction, the existence of Cu, O, N and Cl is observed, while adsorbed O slightly increased due to the oxygen generation during the photoreaction process (Fig. S15†). Thus, the above analysis demonstrates the outstanding photocatalytic performance and stability of Cu2O@CuX–bpy for application.
To further clarify the reasons for the outstanding photocatalytic performance and HCOOH conversion selectivity, in situ FTIR spectroscopy was performed to probe the intermediates of Cu2O@CuCl–bpy during visible light illumination. As shown in Fig. 4(c), new peaks obviously appeared and their intensities gradually increased with the illumination time from 0 to 30 min. The peaks at 1540, 1506 and 1473 cm−1 are attributed to monodentate carbonate (CO32−) and the peaks at 1456, 1436, and 1417 cm−1 are ascribed to bicarbonate (HCO3−).40–42 The peak intensities of these carbonates increase significantly, indicating that Cu2O@CuCl–bpy can chemically adsorb and interact with CO2 and H2O molecules. The emerging peaks at 1653, 1733, 1750, and 1772 cm−1 may result from formate and the peak at 1635 cm−1 could be attributed to carboxylate. Moreover, the intensities of the peaks at 1559 and 1521 corresponding to *COOH increased with the extension of visible light illumination time, which might be possibly caused by the favorable proton capture capability of CO2 radicals. Notably, the intensity of the peak at 1700 cm−1 gradually increased with light irradiation and this can be ascribed to *HCOOH, which is an important intermediate for the formation of HCOOH.40 For the purpose of comparison, in situ FTIR spectroscopy was performed to analyze the intermediates of Cu2O@CuBr–bpy and Cu2O@CuI–bpy under visible light illumination (Fig. S16†). During the illumination time from 0 to 30 min, the peak intensities of Cu2O@CuBr–bpy and Cu2O@CuI–bpy obviously appeared and their intensities gradually increased, which is the same as what was observed for Cu2O@CuCl–bpy. It is worth noting that the peak intensity of Cu2O@CuCl–bpy is the highest while that of Cu2O@CuI–bpy is the lowest. We speculate that the increase in electronegativity would increase the interaction between halide anions and intermediates of HCOOH, thus enhancing CO2 reduction.
Density Functional Theory (DFT) computational methods were used to explain the photoexcitation process and the catalytic reaction mechanism. The model structures established in the calculation and the reaction mechanism are shown in Fig. S17–S22† (details in the Experimental section). To investigate the connection mode between CuX–bpy and the semiconductor Cu2O, we investigated the adsorption of CuX–bpy on the Cu2O(100) facets through the charge density difference. As shown in Fig. 5(a) and (b), one CuX–bpy molecule is put on the Cu2O(100) surface. The halogen element X will bond to two surface Cu atoms to form a tetra-coordinated [ClCu4]3+ structure. There is obvious charge transfer at the interface between CuX–bpy and Cu2O(100), where a photoelectron would potentially transfer from Cu2O to the CuX–bpy layer.
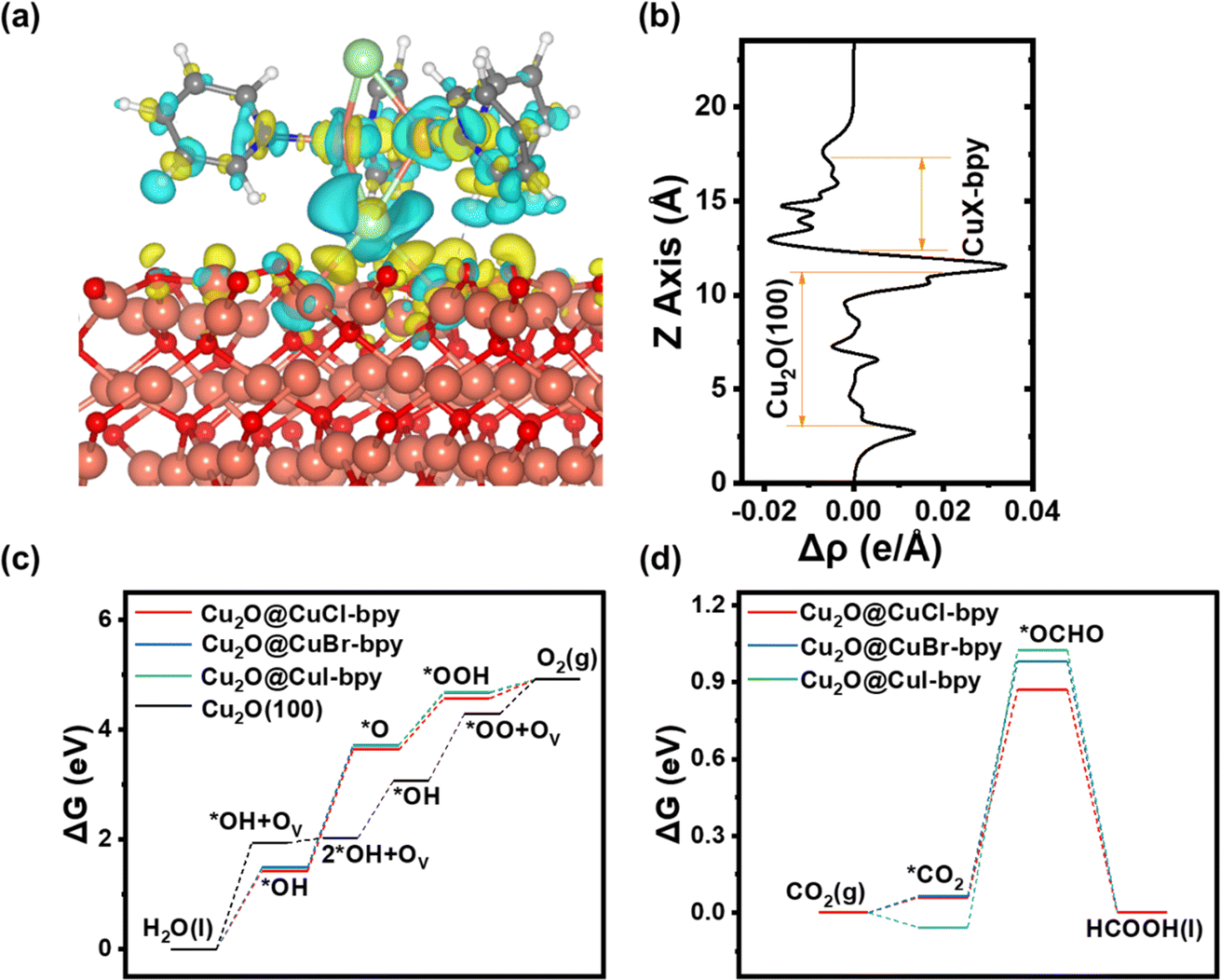 | ||
| Fig. 5 (a) Charge density difference of Cu2O@CuX–bpy (X = Cl, Br, and I, isosurface level = 0.0015 |e| per bohr3). Yellow and blue indicate electron accumulation and depletion. (b) Projected charge density difference along the Z axis. Catalytic performance of (c) the OER and (d) CO2RR processes. | ||
A comparison graph about the OER and CO2RR catalytic performance of CuX–bpy molecules and the Cu2O(100) surface is given in Fig. 5(c) and (d). More detailed information is available in Tables S3–S7.† As for the OER process, CuCl–bpy, CuBr–bpy and CuI–bpy all follow the adsorbate evolution mechanism, but exhibit very high free energies of 2.211 eV, 2.222 eV and 2.23 eV, respectively. At U = 1.23 V, their corresponding overpotentials are 0.981 V, 0.992 V and 1 V, respectively. The bare Cu2O(100) surface also does not perform well for the OER due to the 3.075 eV free energy. Meanwhile, following the lattice oxygen oxidation mechanism, the Cu2O(100) surface exhibits a lower free energy of 1.943 eV, whose corresponding overpotential is 0.713 V. Therefore, the bare Cu2O(100) surface plays a role in water oxidation. As for the CO2RR process, the *OCHO adsorbate is more energetically stable than the *COOH adsorbate in CuX–bpy molecules. Thus, CuCl–bpy, CuBr–bpy and CuI–bpy all follow the formic acid pathway. Their free energies are 0.872 eV, 0.982 eV and 1.083 eV, respectively. In other words, the catalytic performance of CuX–bpy increases with the increase in electronegativity of halogen element X. The adsorption energies of *CO2 are all negative in CuX–bpy, which indicates the effortless adsorption beginning of the CO2RR. For comparison, the bare Cu2O(100) surface can hardly adsorb the *CO2 adsorbate due to its 2.058 eV adsorption energy, leading to ultrahigh free energy as well as making it unsuitable for the CO2RR. Hence, CuX–bpy molecules play a role in carbon dioxide reduction.
According to the above analysis, we propose a probable photocatalytic mechanism in Cu2O@CuX–bpy. Upon visible-light illumination, abundant photo-generated electron–hole pairs are formed in the Cu2O@CuX–bpy system. The introduction of halogen sites enables faster migration of photo-induced carriers, achieving long-life carrier separation and stabilizing the formate intermediate to promote the photoredox cycle. Cu2O presented excellent visible-light adsorption capacity, which means it can serve as a sensor to absorb light to form photo-generated carriers. The photogenerated electrons in Cu2O were further transferred to CuX–bpy loaded on its surface through Cu–Cl coordination bonds. Subsequently, the photogenerated electrons in CuX–bpy were transferred to the Cu active sites, promoting the reduction of CO2 to HCOOH assisted by halogen sites, whereas the photogenerated holes in Cu2O were consumed for the oxidation of water.
Conclusion
In summary, different halogen (X) sites (M = Cl, Br and I) in copper(I)–organic frameworks in situ compounded on cuprous oxide were constructed for outstanding photocatalytic reduction of CO2 integrated with oxidation of water. The introduction of halogen sites makes the MOF-based catalysts have better photoresponse capability, photo-generated carrier separation and migration capability and photoredox stability. Remarkably, Cu2O@CuCl–bpy achieves efficient photo-reduction of CO2 into formate with a high yield of 1520 μmol g−1 after 12 hours of reaction. From in situ FTIR spectroscopy analysis, the superior photocatalytic performance is attributed to the increase in electronegativity, which would increase the interaction between halide anions and the intermediates of HCOOH, enhancing CO2 adsorption and stabilizing the intermediates of HCOOH to promote the photoredox cycle. Through comprehensive characterization and DFT calculations, we confirmed that the effective coordination coupling between the MOF and semiconductor enables photo-generated electrons to transfer rapidly between the active sites of the MOF and the semiconductor. This feature results in the photoexcited electrons and holes accumulating in the CuX–bpy and Cu2O, respectively, which can be efficiently used for CO2 reduction and H2O oxidation, thus simulating artificial photosynthesis. This work not only affords the rational design strategy of halide active sites in functional MOF catalysts, but also provides fundamental insights into the mechanism of CO2 photoreduction reaction systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was conducted by the Fundamental Research Center of Artificial Photosynthesis (FReCAP), National Key R&D Program of China (2022YFA0911904), financially supported by the National Natural Science Foundation of China (NSFC) under grand no. 22088102. Financial support from the State Key Laboratory of Fine Chemicals, Dalian University of Technology (KF 2002) is gratefully acknowledged.References
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- M. Liras, M. Barawi and V. A. de la Peña O’Shea, Chem. Soc. Rev., 2019, 48, 5454–5487 RSC.
- N. Y. Huang, J. Q. Shen, X. W. Zhang, P. Q. Liao, J. P. Zhang and X. M. Chen, J. Am. Chem. Soc., 2022, 144, 8676–8682 CrossRef CAS PubMed.
- M. Lu, M. Zhang, J. Liu, T. Y. Yu, J. N. Chang, L. J. Shang, S. L. Li and Y. Q. Lan, J. Am. Chem. Soc., 2022, 144, 1861–1871 CrossRef CAS PubMed.
- H. Ou, S. Ning, P. Zhu, S. Chen, A. Han, Q. Kang, Z. Hu, J. Ye, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202206579 CrossRef CAS PubMed.
- R. Chen, Z. Ren, Y. Liang, G. Zhang, T. Dittrich, R. Liu, Y. Liu, Y. Zhao, S. Pang, H. An, C. Ni, P. Zhou, K. Han, F. Fan and C. Li, Nature, 2022, 610, 296–301 CrossRef CAS PubMed.
- S. Huang, M. Wang, D. J. Su, J. Liang, F. Sun, W. Tian, L. B. Zhao and J. Liu, ChemSusChem, 2022, 15, e202200704 CrossRef CAS PubMed.
- J. R. Li, H. Huang, W. Xue, K. Sun, X. Song, C. Wu, L. Nie, Y. Li, C. Liu, Y. Pan, H. Jiang, D. Mei and C. Zhong, Nat. Catal., 2021, 4, 719–729 CrossRef CAS.
- J. Zhang, Y. Wang, H. Wang, D. Zhong and T. Lu, Chin. Chem. Lett., 2022, 33, 2065–2068 CrossRef CAS.
- H. Wang, C. Zhang, B. Dong, D. Zhong and T. Lu, Sci. China Mater., 2023, 66, 839–858 CrossRef.
- X. Ma, H. Liu, W. Yang, G. Mao, L. Zheng and H. L. Jiang, J. Am. Chem. Soc., 2021, 143, 12220–12229 CrossRef CAS PubMed.
- J. K. Jin, K. Wu, X. Y. Liu, G. Q. Huang, Y. L. Huang, D. Luo, M. Xie, Y. Zhao, W. Lu, X. P. Zhou, J. He and D. Li, J. Am. Chem. Soc., 2021, 143, 21340–21349 CrossRef CAS PubMed.
- X. Meng, J. Yang, C. Zhang, Y. Fu, K. Li, M. Sun, X. Wang, C. Dong, B. Ma and Y. Ding, ACS Catal., 2021, 12, 89–100 CrossRef.
- X. He, Z. Gan, S. Fisenko, D. Wang, H. M. El-Kaderi and W. N. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 9688–9698 CrossRef CAS PubMed.
- S. Wang, X. Wang, X. Cheng, J. G. Ma and W. Sun, J. Mater. Chem. A, 2022, 10, 16396–16402 RSC.
- L. Yu, X. Ba, M. Qiu, Y. Li, L. Shuai, W. Zhang, Z. Ren and Y. Yu, Nano Energy, 2019, 60, 576–582 CrossRef CAS.
- H. Li, S. Wang, M. Wang, Y. Gao, J. Tang, S. Zhao, H. Chi, P. Zhang, J. Qu, F. Fan and C. Li, Angew. Chem., Int. Ed., 2022, 61, e202204272 CrossRef CAS PubMed.
- J. Rong, Z. Wang, J. Lv, M. Fan, R. Chong and Z. Chang, Chin. J. Catal., 2021, 42, 1999–2009 CrossRef CAS.
- D. Li, M. Kassymova, X. Cai, S. Zang and H. L. Jiang, Coord. Chem. Rev., 2020, 412, 213262 CrossRef CAS.
- W. Dong, J. Jia, Y. Wang, J. An, O. Yang, X. Gao, Y. Liu, J. Zhao and D. Li, Chem. Eng. J., 2022, 438, 135622 CrossRef CAS.
- C. Zheng, X. Qiu, J. Han, Y. Wu and S. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 42243–42249 CrossRef CAS PubMed.
- C. Wang, X. Yi and P. Wang, Appl. Catal., B, 2019, 247, 24–48 CrossRef CAS.
- Q. Niu, S. Dong, J. Tian, G. Q. Huang, J. Bi and L. Wu, ACS Appl. Mater. Interfaces, 2022, 14, 24299–24308 CrossRef CAS PubMed.
- H. Z. Liu, X. Liu, B. Li, H. Luo, J. G. Ma and P. Cheng, ACS Appl. Mater. Interfaces, 2022, 14, 28123–28132 CrossRef CAS PubMed.
- S. Karmakar, S. Barman, F. A. Rahimi and T. K. Maji, Energy Environ. Sci., 2021, 14, 2429–2440 RSC.
- Y. Jiang, Y. Yu, X. Zhang, M. Weinert, X. Song, J. Ai, L. Han and H. Fei, Angew. Chem., Int. Ed., 2021, 60, 17388–17393 CrossRef CAS PubMed.
- X. J. Kong, T. He, J. Zhou, C. Zhao, T. C. Li, X. Q. Wu, K. Wang and J. R. Li, Small, 2021, 17, e2005357 CrossRef PubMed.
- L. Z. Dong, L. Zhang, J. Liu, Q. Huang, M. Lu, W. X. Ji and Y. Q. Lan, Angew. Chem., Int. Ed., 2020, 59, 2659–2663 CrossRef CAS PubMed.
- X. Zu, Y. Zhao, X. Li, R. Chen, W. Shao, L. Li, P. Qiao, W. Yan, Y. Pan, Q. Xu, J. Zhu, Y. Sun and Y. Xie, Angew. Chem., Int. Ed., 2023, 62, e202215247 CrossRef CAS PubMed.
- J. Wang, Y. Shi, Y. Wang and Z. Li, ACS Energy Lett., 2022, 7, 2043–2059 CrossRef CAS.
- H. B. Zhao, J. F. Liao, Y. Teng, H. Y. Chen and D. B. Kuang, ACS Appl. Mater. Interfaces, 2022, 14, 43354–43361 CrossRef CAS PubMed.
- S.-H. Guo, J. Zhou, X. Zhao, C.-Y. Sun, S.-Q. You, X.-L. Wang and Z.-M. Su, J. Catal., 2019, 369, 201–208 CrossRef CAS.
- Y. Fang, D. Luan, Y. Chen, S. Gao and X. W. D. Lou, Angew. Chem., Int. Ed., 2020, 59, 7178–7183 CrossRef CAS PubMed.
- C. Bai, S. Fan, X. Li, Z. Niu, J. Wang, Z. Liu and D. Zhang, Adv. Funct. Mater., 2022, 32, 2205569 CrossRef CAS.
- X. Zhou, B. Fu, L. Li, Z. Tian, X. Xu, Z. Wu, J. Yang and Z. Zhang, Nat. Commun., 2022, 13, 5770 CrossRef CAS PubMed.
- H. Luo, B. Li, J. G. Ma and P. Cheng, Angew. Chem., Int. Ed., 2022, 61, e202116736 CrossRef CAS PubMed.
- Q. Lai, J. Zhu, Y. Zhao, Y. Liang, J. He and J. Chen, Small, 2017, 13, 1700740 CrossRef PubMed.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- J. Zhou, J. Li, L. Kan, L. Zhang, Q. Huang, Y. Yan, Y. Chen, J. Liu, S. L. Li and Y. Q. Lan, Nat. Commun., 2022, 13, 4681 CrossRef CAS PubMed.
- M. Kou, W. Liu, Y. Wang, J. Huang, Y. Chen, Y. Zhou, Y. Chen, M. Ma, K. Lei, H. Xie, P. K. Wong and L. Ye, Appl. Catal., B, 2021, 291, 120146 CrossRef CAS.
- Y. Xi, X. Zhang, Y. Shen, W. Dong, Z. Fan, K. Wang, S. Zhong and S. Bai, Appl. Catal., B, 2021, 297, 120411 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00259h |
| This journal is © The Royal Society of Chemistry 2024 |