
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox-active ligand promoted multielectron reactivity at earth-abundant transition metal complexes
Minzhu
Zou
 and
Kate M.
Waldie
*
and
Kate M.
Waldie
*
Department of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, USA. E-mail: kate.waldie@rutgers.edu
First published on 5th August 2024
Abstract
The introduction of redox-active ligands into transition metal complexes can lead to novel redox behavior due to the ability of these ligands to serve as electron reservoirs. This feature is especially attractive in earth-abundant transition metal systems that typically favor one-electron redox processes and radical reactivity, as opposed to the two-electron redox cycles common with the noble metals. The redox flexibility afforded by redox-active ligands can enable substrate activation and/or bond forming and breaking processes that would otherwise be inaccessible with traditional redox-innocent ligands. This review discusses key examples of stoichiometric substrate activation and organic transformations facilitated by redox-active ligand-promoted multielectron reactivity at earth-abundant metal complexes. We highlight the electrochemical properties of these systems in relation to their substrate reactivity, where, in many cases, the complexes exhibit sequential one-electron redox events. Only a few examples have achieved electrocatalytic reactivity based on two-electron redox features, which underscores the untapped potential for further development of redox-active ligand systems to expand the capabilities of earth-abundant metal complexes in electrocatalysis.
 Minzhu Zou | Minzhu Zou received his B.Sc. in Chemistry from Wuhan University, China in 2018, and completed his Ph.D. in Chemistry at Rutgers, The State University of New Jersey (New Brunswick) in 2024 under the guidance of Prof. Kate M. Waldie. His Ph.D. dissertation focused on the development of earth-abundant cobalt complexes with redox-active ligands to facilitate multi-electron transformations. Minzhu is currently Chemist II, Extractables & Leachables at SGS North America Inc. |
 Kate M. Waldie | Kate M. Waldie earned her Ph.D. with Prof. Robert M. Waymouth at Stanford University in 2016. After her postdoctoral research with Prof. Clifford P. Kubiak at the University of California San Diego, she started her independent career as Assistant Professor in the Department of Chemistry and Chemical Biology at Rutgers, The State University of New Jersey (New Brunswick) in 2018. Her research group focuses on the use of molecular transition metal complexes as electrocatalysts for renewable energy conversion and small-molecule activation. |
1. Introduction
The design of ancillary ligands is a central tenet in organometallic chemistry. By tuning the ligand properties, the reactivity of molecular transition metal complexes can be directed to achieve selective transformations with organic substrates, toward the development of (electro)catalytic systems. While our understanding of how the properties of traditional redox-innocent ligands can be exploited to tune the structural and electronic properties of the metal center are well established, such ligands typically serve as redox spectators and do not directly participate in the redox processes needed for substrate activation or the catalytic mechanism (Fig. 1a).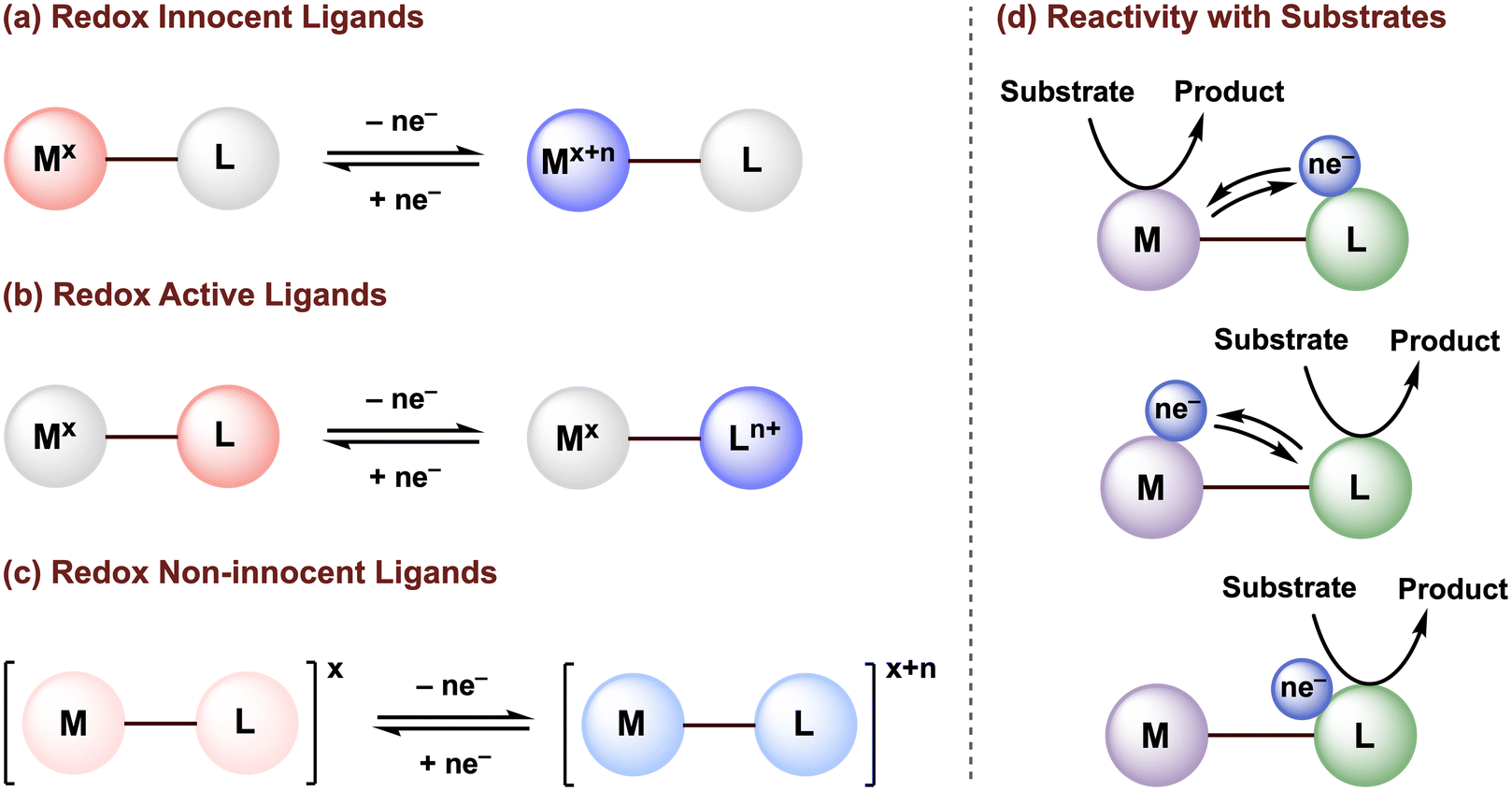 | ||
| Fig. 1 Classification of (a) redox innocent, (b) redox-active, and (c) redox non-innocent ligands. (d) Possible metal–ligand cooperativity for substrate activation. | ||
The activity of transition metal catalysts is typically governed by the redox events occurring at the metal center. However, over the past two decades, greater attention has been given to redox-active (or redox non-innocent) ligands (Fig. 1b and c). The terms “redox non-innocent” and “redox-active” ligands are both commonly found in the literature. The concept of ligand non-innocence was originally introduced by Jørgensen to indicate the oxidation state ambiguity of certain redox-active metal complexes: “ligands are innocent when they allow oxidation states of the central atoms to be defined”.1 This definition subtly emphasizes that non-innocent behavior depends on both the metal and the ligand.2 In these cases, the difficulty in assigning the metal and ligand oxidation states is usually attributed to strong mixing of ligand and metal frontier orbitals due to their effective energy level matching. On the other hand, the designation of a redox-active ligand system is reserved for metal complexes in which the oxidation states can be established through experimental methods, typically high-resolution X-ray crystallography and/or various spectroscopic characterizations.3,4
Since redox-active ligands possess multiple accessible redox states, not only can these ligands tune the electronic properties (i.e., Lewis acidity/basicity) of the metal, but can also maintain a more stable oxidation state of the metal center by accepting and/or releasing electrons at milder potentials, and/or can facilitate bond-breaking or bond-forming reactions via ligand-to-substrate or substrate-to-ligand electron transfers (Fig. 1d).5–9 van der Vlugt, de Bruin, and their co-workers have summarized in detail how the coordination of redox-active ligand(s) to a transition metal center can create an extended π-conjugated network and provide enough stabilization to ligand-based radicals generated through single electron transfer, enabling high control over radical-type one-electron (1e−) reactivity.10–12 Nevertheless, an in-depth understanding of the electronic structures in paramagnetic non-innocent species can be challenging and often requires advanced techniques, such as electron paramagnetic resonance (EPR) and X-ray absorption (XAS) spectroscopy. Furthermore, off-cycle radical reactions may still occur. Thus, the development of new earth-abundant transition metal complexes that display multielectron reactivity centered at the ligand scaffold or promoted via metal–ligand multielectron cooperativity is of great significance to advance sustainable methodologies for chemical transformations.
In nature, a wide variety of multielectron processes are promoted with high selectivity and efficiency by metalloenzymes based on earth-abundant transition metal cations. The reactions occurring in the active site of these enzymes rely on close cooperation of both the earth-abundant metal center and surrounding organic redox-active cofactors (Fig. 2).13,14 Much inspiration has been taken from these biological systems in the context of organometallic chemistry, resulting in numerous investigations into the reactivity of transition metal complexes bearing redox-active ligands.9,10,15 However, it should be stressed that the presence of redox-active ligands does not guarantee preferential multielectron behavior and radical 1e− reactivity may still be observed, as is the case for first-row metalloenzymes that operate by single electron or coupled electron–proton transfer.
 | ||
| Fig. 2 Select metalloenzyme active sites with redox-active organic cofactors.14 | ||
This frontier article discusses key examples from the literature on the application of redox-active ligands to achieve multielectron reactivity at earth-abundant transition metal complexes where the electrons are supplied by either the redox-active ligand(s) or by both the ligand and metal center. We largely focus our discussion on first-row systems, and highlight examples of stoichiometric and catalytic reactions for multielectron substrate activation facilitated by redox-active ligand(s). When available, the electrochemical properties of each system are noted to encourage greater consideration of the relevant redox potentials of the metal complex and substrate on activity, toward the rational design of redox-active ligand systems for organic transformations.
2. Ligand-promoted stoichiometric reactivity
2.1. Multielectron reactivity at early transition metal complexes
Two-electron processes are ubiquitous in homogenous catalysis mediated by transition metal complexes. For example, oxidative addition and its microscopic reverse, reductive elimination, are representative elementary steps for substrate activation and bond formation reactions. This reactivity has been well-studied for palladium catalysts, where these key steps are achieved through the two-electron Pd0/II redox cycle (Fig. 3).16 | ||
| Fig. 3 General catalytic cycle for Pd-catalyzed cross-coupling reactions.16 | ||
Beyond palladium, tremendous progress has been made in this area with early transition metal complexes. Seminal studies by Heyduk and co-workers focused on halogen oxidative addition at ZrIV and TiIV complexes bearing dianionic amidophenolate (ap2−) or o-phenylenediamide (opda2−) redox-active ligand(s).17–20 Since these early transition metal complexes contain formal d0 metal centers, the metal oxidation state cannot be increased further and the ap2− or opda2− ligand(s) must supply the necessary redox equivalents required for the overall 2e− transformation. However, the authors suggest this process may be viewed as a molecular oxidative addition due to the strong σ and π donation from the electron-rich ap2− ligands to the metal.17 For example, [Zr(ap)2(THF)2] reacted rapidly with 1 equiv. PhICl2 to afford the oxidative addition product [ZrCl2(isq˙−)2], where 1e− oxidation of both ap2− ligands to the imino-semiquinonate (isq˙−) form has occurred (Scheme 1a).18 Other halogen-based oxidants (e.g., Br2, XeF2, PCl2Ph3, PBr2Ph3, and ClCPh3) also reacted with [Zr(ap)2L2] in a similar fashion, affording the singlet diradical product [ZrX2(isq˙−)2] (X = F, Cl, Br). Notably, [ZrIVF2(isq˙−)2] was also obtained by reaction of [Zr(ap)2(py)2] (py = pyridine) with 2 equiv. ferrocenium hexafluorophosphate [Cp2Fe][PF6], where the only fluoride source was the hexafluorophosphate anion. Similarly, [(N2O2red)TiIVL2] (L = THF or py) also reacted with 1 equiv. PhICl2 to afford oxidative addition products (Scheme 1b).20 Depending on the axial ligands in the parent complex, a six- or seven-coordinate TiIVCl2 was generated with an oxidized [N2O2]2− redox-active ligand backbone.
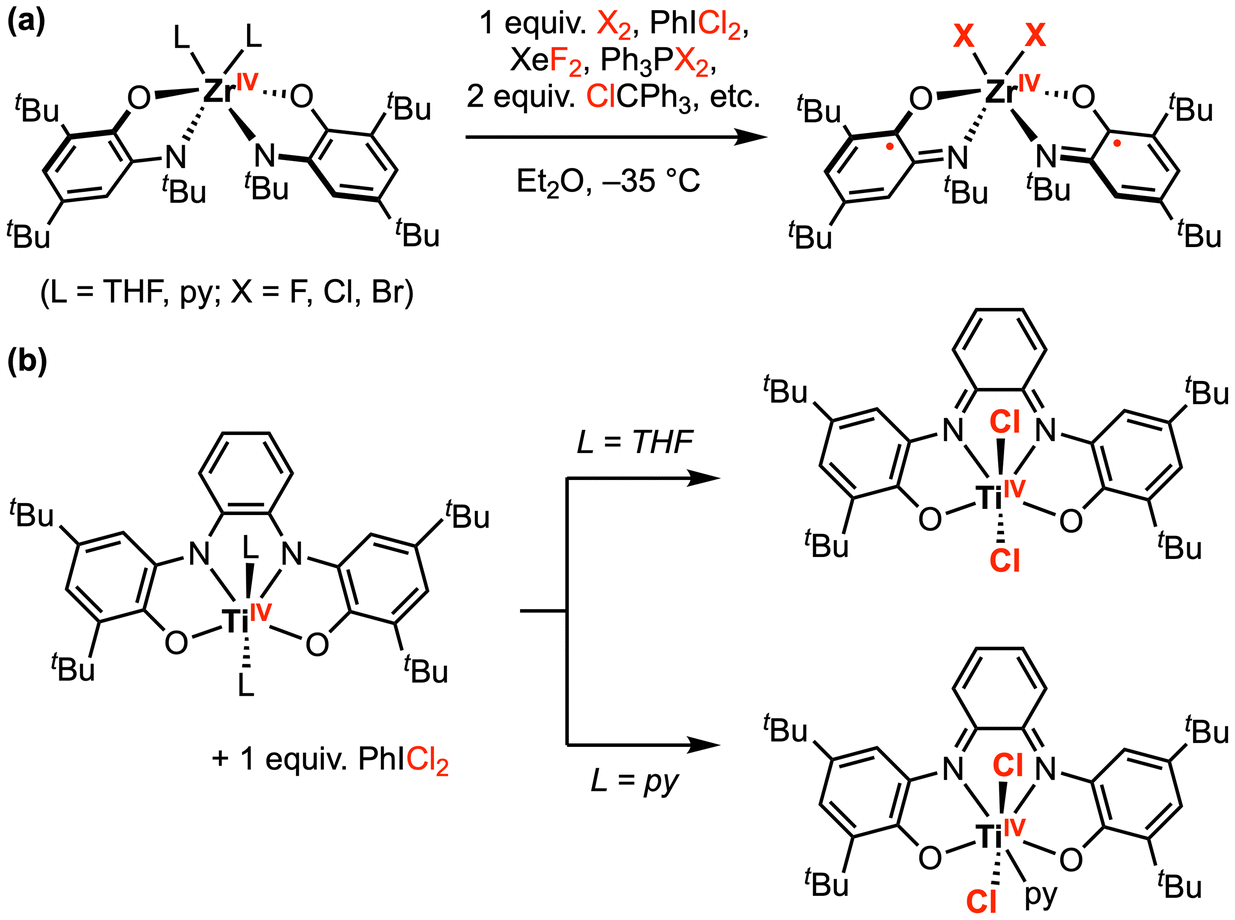 | ||
| Scheme 1 Representative halogen oxidative addition at (a) ZrIV, and (b) TiIV amindophenolate complexes.18,20 | ||
In complementary reactivity studies, carbon–carbon bond formation via reductive elimination from the ZrIV and TiIV complexes has also been reported.21,22 When the dianionic complexes [ZrR2(ap)2]2− (R = Ph, p-tolyl, or Me) were treated with 2 equiv. [Cp2Fe][PF6] in THF, the homocoupling product (R–R) was obtained (Scheme 2a).21 The proposed pathway for this reaction involves an initial oxidation of both redox-active ap2− ligands by the decamethylferrocenium oxidant, from which reductive elimination is accessible. The putative ZrIV diradical intermediate was detected by low-temperature UV-vis studies, supporting the proposed mechanism. In another example, treatment of a TiIV bis-phenylacetylide complex with 1 equiv. PhICl2 afforded 1,4-diphenylbutadiyne and the corresponding TiIVCl2 complex (Scheme 2b), where the latter was reported to undergo ligand-centered quasi-reversible 1e− oxidations at E1/2 = −0.52 V and −0.19 V vs. Fc+/0 in THF with 0.1 M [nBu4N][PF6] (Scheme 3).22 Two possible intermediates were suggested with respect to the fluxionality of the amine arms, depending on whether the C–C bond formation via reductive elimination occurred before or after chloride coordination to the titanium center.
 | ||
| Scheme 2 Representative carbon–carbon bond formation via reductive elimination at (a) ZrIV amindophenolate, and (b) TiIV phenylenediamide complexes.21,22 | ||
 | ||
| Scheme 3 Sequential ligand-centered one-electron oxidations of TiIV-dichloride.22 | ||
More recently, Roberts and co-workers reported catalytic alkyl-alkyl cross coupling reactions mediated by early transition metal complexes with a tris(amido) redox-active ligand.23–26 For example, the addition of (1-bromoethyl)benzene (PhMeCH–Br) to a mixture of [(NNN)ScIII(THF)3] and benzylzinc bromide (BnZnBr) resulted in a mixture of the alkyl-alkyl cross-coupling (PhMeCH–Bn) and homocoupling [(PhMeCH)2] products. Formation of the homocoupling side product could be minimized by using catalytic amounts of the Sc complex. A radical coupling mechanism was proposed involving an organic alkyl radical (ArMeCH˙), partnered with the ligand-centered radical intermediate [(NNN˙)ScIIIBr(THF)2]− in the solvent cage (Fig. 4).25 The presence of an alkyl radical was supported by radical trapping studies with TEMPO, as well as by the racemization of the stereocenter in the cross-coupled product. Unlike classic palladium-catalyzed cross-coupling reactions, β-hydride elimination was not observed in this system as the organic alkyl radical is never bound to the metal. Moreover, the substrate scope for alkyl–alkyl cross coupling extended to include coupling partners with functional groups (e.g., Br, CF3) that are incompatible with late-transition-metal catalysis. In previous work by Heyduk and co-workers, a series of tantalum complexes featuring the tris(amido) ligand were prepared in which the electronic properties of the ligand phenylene backbone and amide donors were systematically varied.27 While derivatization on the ligand backbone significantly affected the ligand-centered redox potentials, enabling redox tuning over more than 270 mV, changes to the amido substituents had little impact on the electronic profile of the complex.
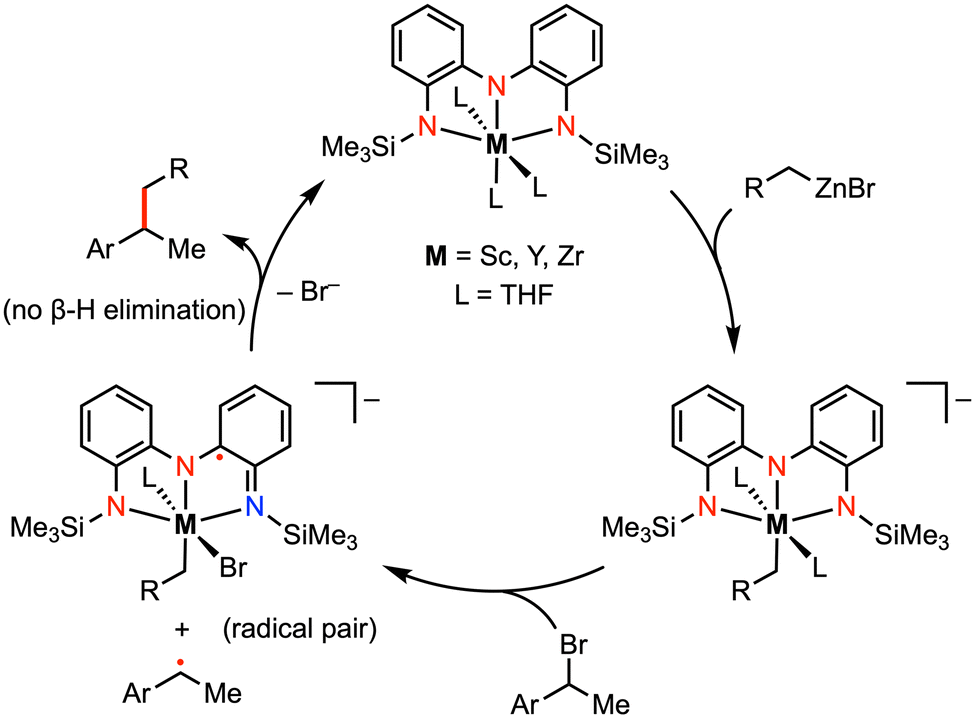 | ||
| Fig. 4 Representative carbon–carbon bond formation via reductive elimination at Sc, Y, or Zr complexes.25 | ||
2.2 Multielectron reactivity at late transition metal complexes
Compared to the early transition metals, first-row late transition metals typically favor lower oxidation states and thus have a non-zero d electron count. Traditionally, these first-row transition metal complexes undergo sequential 1e− processes due to metal-centered redox events. However, the introduction of redox active ligands into these complexes can introduce additional redox events at various potentials, and in select cases, can facilitate multielectron reactivity with organic substrates. For example, the aminophenol-derived ligand architecture has been applied to achieve novel 2e− reactivity at copper and cobalt complexes.28–30 In 2010, the Soper group reported square planar, intermediate spin (S = 1) CoIII complexes [CoIII(Arap)2]− (Ar = phenyl or 2,6-diisopropylphenyl) bearing two amidophenolate redox-active ligands.29 The phenyl derivative was shown to undergo two quasi-reversible ligand-based 1e− oxidations to the neutral [CoIII(Arap)(Arisq˙−)] and monocationic [CoIII(Arisq˙−)2]+ species at −0.77 V and −0.35 V vs. Fc+/0, respectively, in MeCN. The neutral complex [CoIII(Arap)(Arisq˙−)] was isolated and characterized as an S = 1/2 ground state. Treatment of [CoIII(Arap)(Arisq˙−)] with 1 equiv. [Cl˙] radical donor, such as CCl4 or N-chlorosuccinimide (NCS), in THF resulted in a quick color change to blue-green. The product was identified as square pyramidal [CoIII(Cl)(Arisq˙−)2] through UV-vis studies (Scheme 4). Interestingly, the same CoIII–Cl complex was also obtained by the reaction of [CoIII(Arap)(Arisq˙−)] with 1 equiv. electrophilic [Cl+] source ([Cl+] = 2,3,4,5,6,6-hexachloro-2,4-cyclohexadien-1-one) in MeCN, which was proposed to proceed via a net 2e− redox process promoted entirely by the redox-active ligands.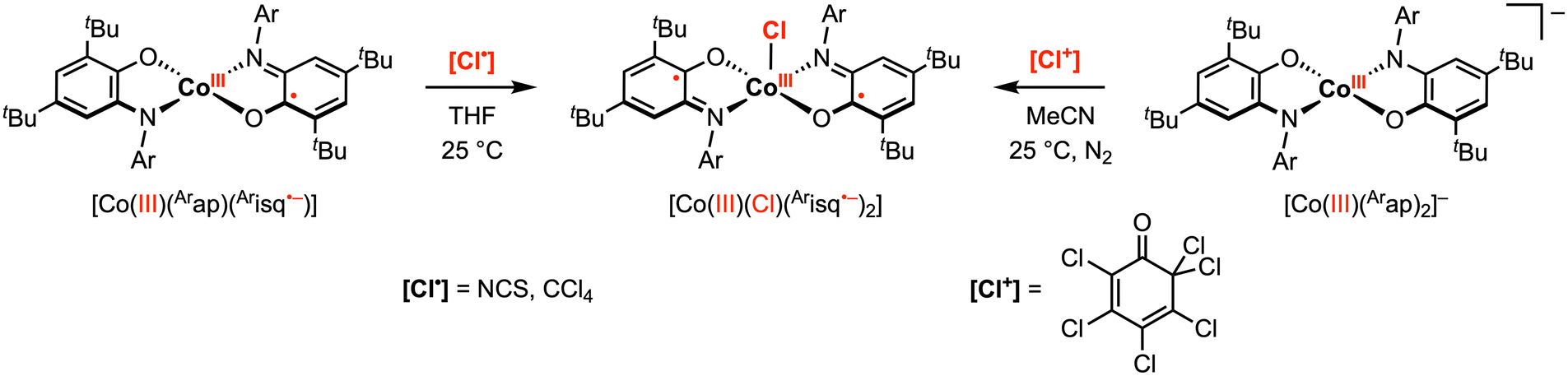 | ||
| Scheme 4 Cobalt(III)–C bond formation promoted by one or two amidophenolate ligands.29 | ||
In a related study, Soper and co-workers showed that [CoIII(Phap)2]− reacts with electrophilic alkyl reagents (R–X) to form a CoIII–R bond (Fig. 5),30 resembling their previous work with the Cl+ reagent. The solid-state structures of square pyramidal complexes [CoIII(CH2Cl)(Phisq˙−)2] and [CoIII(Et)(Phisq˙−)2] revealed contracted C–O and C–N bond distances, consistent with the [Phisq]˙− ligand backbone. This reactivity was explored with various alkyl halides (and MeOTf), in which the reaction rate followed the order of OTf− ≈ I− > Br− > Cl−. The CoIII–R bond formation was also highly sensitive to steric hinderance at the cobalt center, and no addition reaction was observed over weeks with iodobenzene or vinyl bromide, both bearing a sp2-hybridized carbon center. Based on the abovementioned results, an SN2 mechanism was proposed for the oxidative addition of organohalide to the cobalt center, where the two electrons required for bond formation originated from the two amidophenolate redox-active ligands.
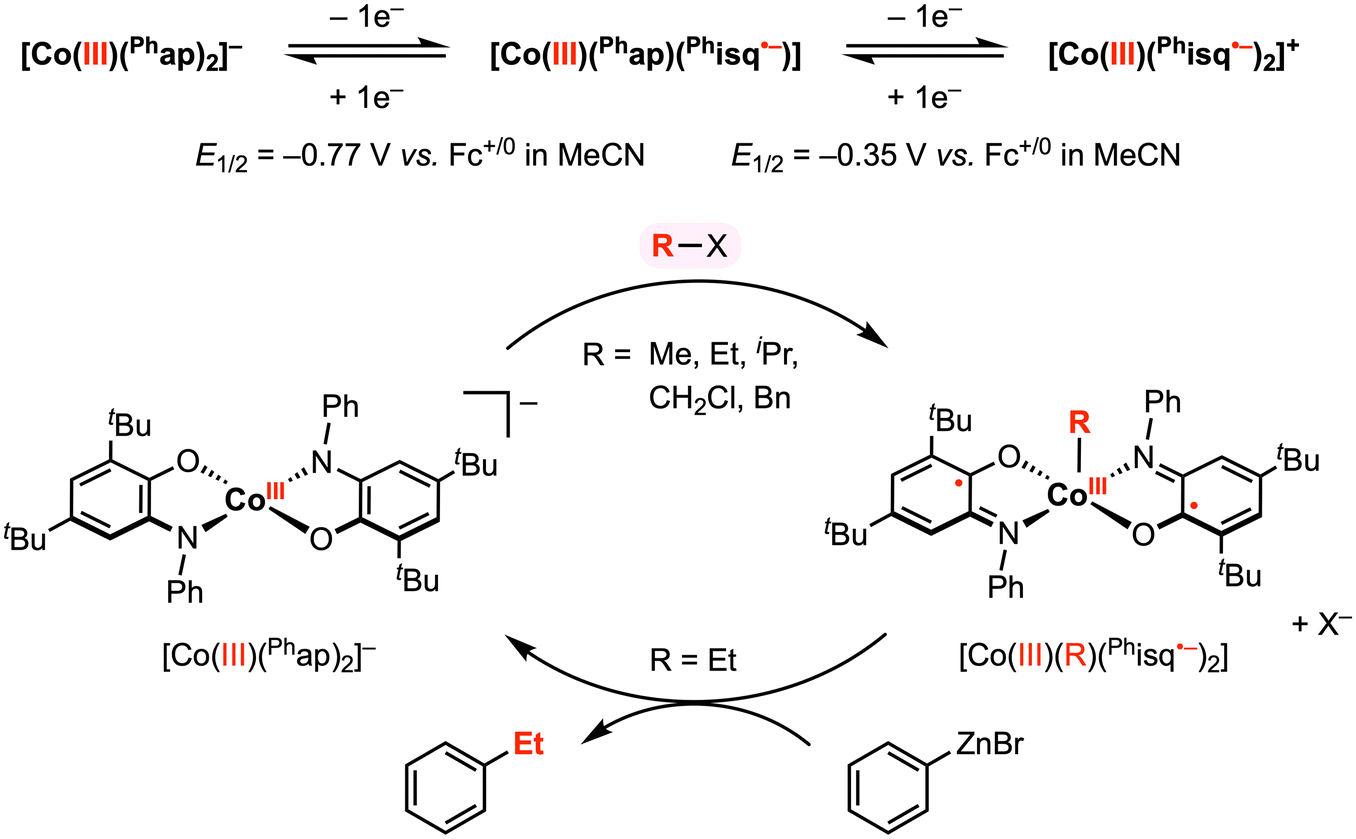 | ||
| Fig. 5 Stoichiometric CoIII–R and carbon–carbon bond formation reactions promoted by the amidophenolate redox-active ligands.30 | ||
The authors reported that the CoIII–Et diradical complex is relatively inert to β-hydride elimination; however, this complex was shown to exhibit further reactivity with phenyl- or hexylzinc bromide to give ethylbenzene and n-octane, respectively, in 5–15% yield. This result demonstrated a remarkable oxidative addition and reductive elimination mediated by a 2e− redox cycle at the cobalt center. Although the precise mechanism of reductive elimination remains unclear and the reaction conditions have yet to be optimized, these stoichiometric studies revealed key insights into the use of redox-active ligands to promote carbon–carbon bond formation at a first-row late transition metal center.
Beyond electrophilic alkyl addition at the first-row transition metals, [M]–CF3 bond formation has also been achieved. The diradical CuII complex [CuII(Phisq˙−)2], originally reported by Wieghardt and co-workers in 2001,3 was found to undergo ligand-based reversible 1e− oxidations at E1/2 = −0.26 V and 0.37 V vs. Fc+/0 in CH2Cl2 with 0.1 M [nBu4N][PF6]. Later, Desage-El Murr, Fensterbank, and co-workers31 discovered that [CuII(Phisq˙−)2] reacts with Umemoto's reagent ([DBT–CF3]+, Ep,c = −0.75 V vs. Fc+/0 in MeCN)32 to yield the first example of a CuII–CF3 complex (Scheme 5). The incorporation of the CF3 group in the product was confirmed by ESI mass spectrometry. The UV-vis, EPR, and cyclic voltammetry (CV) studies of the paramagnetic CuII–CF3 complex displayed similar features as the CuII–Br2 analogue reported by Chaudhuri and co-workers, in which both ligands are in the neutral iminobenzoquinone form (Phbq).28 Further investigations into the stability and reactivity of [CuII(CF3)(Phbq)2] revealed an intramolecular CF3− group transfer to the electrophilic sites on the ligand backbone. Nucleophilic trifluoromethylation of external electrophilic substrates was unsuccessful, likely due to the inability to access ligand exchange at the copper center. Despite a limited substrate scope, the formal umpolung of the electrophilic CF3+ source to a nucleophilic CF3− anion was demonstrated. The mechanism of Cu–CF3 bond formation was not discussed; however, in their following study, single electron transfer (SET) was proposed between the CF3+ reagent and the redox-active ligand in [CuII(Phisq˙−)2] to generate a CF3˙ radical, which could be trapped by TEMPO or other organic substrates (e.g., heteroaromatics, silyl enol ethers, and alkynes) in CH2Cl2 or N-methyl-2-pyrrolidone (NMP) (Fig. 6).33 This catalytic protocol of radical trifluoromethylation circumvents changes in the oxidation state of the copper center, thus limiting uncontrolled radical reactivities. It is worth mentioning that [CuII(CF3)(Phbq)2] is inactive for radical trifluoromethylation, meaning this is a non-productive species for trifluoromethyl transfer protocols.
 | ||
| Scheme 5 Stoichiometric CuII–CF3 bond formation promoted by the amidophenolate redox-active ligands and subsequent CF3 group transfer to the ligand backbone.31 | ||
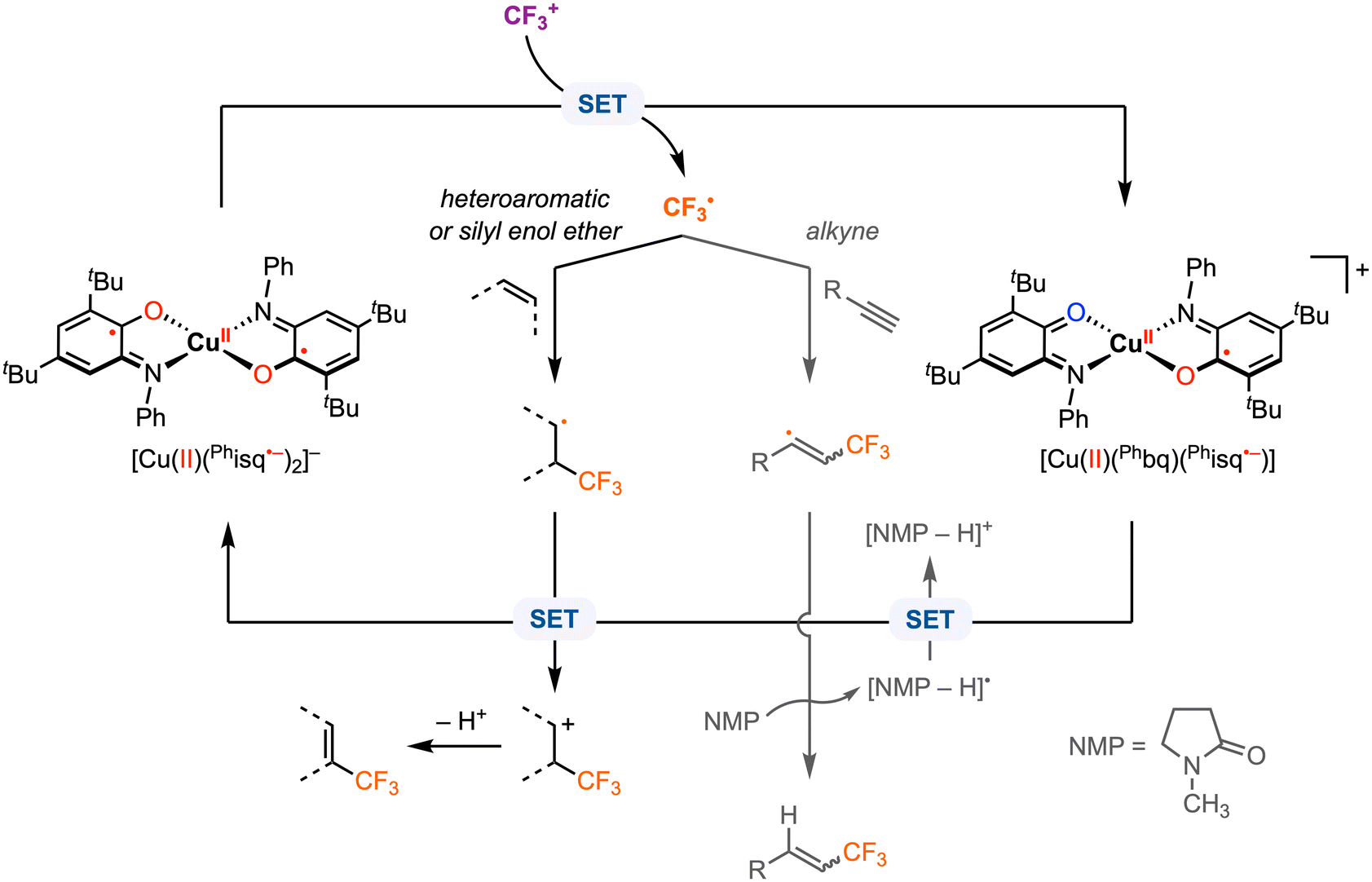 | ||
| Fig. 6 Proposed mechanism for radical trifluoromethylation with [CuII(Phisq˙−)2].33 | ||
We recently reported a family of [CpCo(Ropda)] complexes (Cp = cyclopentadienyl) that exhibit a reversible 2e− oxidation feature by CV.34 The redox potential for the isopropyl derivative [CpCo(iPropda)] was observed at E1/2 = −0.17 vs. Fc+/0 in acetonitrile: by varying the electronic properties of the ligand substituents, this potential could be tuned by over 0.5 V. Structural and spectroscopic characterization of the 2e− oxidation product, obtained by treatment of the neutral species with 2 equiv. chemical oxidant, established this species as [CpCoIII(Rbqdi)(MeCN)]2+ with the neutral benzoquinonediimime (bqdi) ligand (Scheme 6). DFT computational studies were used to probe the pathway for 2e− oxidation: initial loss of 1e− from [CpCo(Ropda)] occurs with redox-induced electron transfer to form a CoII intermediate, where the decrease in delocalization in the Co-ligand metallocycle enables acetonitrile coordination and promotes the loss of the second 1e− at a more negative potential. This family is one of only a handful of examples exhibiting a two-electron redox feature at first-row metals.3,35–48 Such multielectron behavior may be advantageous for developing electrocatalysts that operate at minimal overpotential (vide infra).
 | ||
| Scheme 6 Stoichiometric CoIII–X bond formation (X = CF3 or F) promoted by the phenylenediamide redox-active ligand.49 | ||
In a following report, we applied this 2e− oxidation behavior to achieve reactivity with electrophilic reagents.49 The [CpCo(iPropda)] was shown to react with Umemoto's reagent ([DBT–CF3]+ and NFSI (N-fluorobenzenesulfonimide)) as electrophilic CF3+ and F+ sources, respectively, resulting in formation of a new Co–X bond in [CpCoIII(iPrbqdi)(X)]+ (X = CF3, F) via ligand-to-substrate two-electron transfer (Scheme 6). The CoIII–CF3 complex can also be accessed using other electrophilic CF3 sources, which yielded a clear correlation between the redox potential of the cobalt complex and the electrophile reagent (Fig. 7). Moving forward, redox potentials may prove to be a useful guide for selection of other appropriate substrates and reaction partners. We further showed that [CpCoIII(iPrbqdi)(F)]+ can deliver nucleophilic fluoride to the trityl cation by formal umpolung of the original F+ source. In contrast, the CoIII–CF3 bond in [CpCoIII(iPrbqdi)(CF3)]+ is robust and does not act as a CF3− source. Instead, trapping experiments in the presence of TEMPO or 3-methyl-1H-indole suggested a SET mechanism between Umemoto's reagent and the cobalt complex, similar to the behavior of [CuII(Phisq˙−)2] in Fig. 6.33
 | ||
| Fig. 7 Reactivity of [CpCo(iPropda)] (1) with CF3+ reagents. 2 = [CpCoIII(iPrbqdi)(CF3)]+. Reproduced with permission from ref. 49. Copyright 2023 Royal Society of Chemistry. | ||
Other redox-active ligand frameworks have also been explored for promoting metal–carbon bond formation. In 2018, Hisaeda and co-workers50 described a CoII porphycene (Pc) complex that undergoes two reversible reduction processes at E1/2 = −0.93 and −1.25 V vs. Ag/AgCl in THF with 0.1 M [nBu4N][PF6] (Scheme 7a). Electrolysis of CoIIPc at −1.20 V vs. Ag/AgCl resulted in reduction of the Pc ligand to the radical anion form (Pc˙−), confirmed by their and previously reported UV-vis spectroelectrochemistry results. The EPR spectrum for the 1e− reduced species in THF at room temperature was also consistent with a ligand-centered radical that is delocalized throughout the porphycene skeleton. In contrast, the cyclic voltammogram of CoIIPc in CH2Cl2 under N2 showed a chemically irreversible reduction feature, indicating that a chemical reaction occurs after 1e− reduction. Combining NMR, UV-vis, and mass spectra analysis, the reduction product in CH2Cl2 was confirmed as the CoIII complex, CH2Cl–CoIIIPc (Scheme 7b), thus resembling the reactivity of [CoIII(Phap)2]− with CH2Cl2 reported by Soper and co-workers in 2010.30 However, in this case, the two electrons required for cobalt–carbon bond formation are provided by both the Pc ligand and the cobalt center. Similarly, CoIII–CH3 bond formation was observed with CH3I under reductive conditions in DMF, where the 1H NMR signal of the axial methyl group was found at −5.0 ppm. Based on radical-trapping studies with α-phenyl-N-tert-butylnitrone (PBN) and DFT calculations, the CoIII–R bond forming reaction likely proceeds via an SN2 mechanism.
 | ||
| Scheme 7 Stoichiometric Co–R bond formation promoted by both the Pc redox-active ligand and the cobalt center.50 | ||
The Chirik group has demonstrated the reactivity of iron complexes featuring a bis(imino)pyridine (PDI) redox-active ligand for two-electron oxidative addition51 and catalytic cyclization reactions,52–54 achieved via metal–ligand cooperativity. In their 2013 report,54 the isolation of catalytically relevant iron metallacycles was explored. The oxidative addition of enyne or diyne substrates to [(iPrPDI)FeII(N2)] yielded a metallocycle complex formulated as an intermediate-spin FeIII with antiferromagnetic coupling to the PDI˙− radical anion (overall S = 1), based on magnetic susceptibility measurements and 57Fe Mössbauer spectroscopy. Subsequent H2 hydrogenation and reductive elimination led to release of the cyclized product and regeneration of the starting FeII catalyst (Fig. 8). This study revised their previously proposed mechanism for iron-catalyzed cycloaddition in which the PDI ligand was thought to serve as the electron reservoir to maintain the FeII oxidation state.52,55 While electrochemical characterization data for [(iPrPDI)Fe(N2)] is unavailable, electrochemical studies for 4-substituted PDI ligands and related iron dicarbonyl complexes [(4-R-iPrPDI)Fe(CO)2] (R = H, CF3, tBu, Bn, NMe2) have been reported.56,57 For the iron dicarbonyl complex [(4-H-iPrPDI)Fe(CO)2], reversible 1e− oxidation and 1e− reduction waves are observed at −0.49 V and −2.46 V vs. Fc+/0, respectively, in THF with 0.1 M [nBu4N][PF6].56
 | ||
| Fig. 8 Proposed catalytic cycle for the hydrogenative cyclization of enynes by redox-active bis(imino)pyridine iron complexes.54 | ||
3. Electrocatalytic multielectron reactions
The abovementioned proof-of-principle studies demonstrating [M]–R and C–C bond formation at first-row transition metals laid the foundation for the development of electrocatalytic transformations promoted by redox-active ligand(s).58,59 In 2014, Sarkar and co-workers reported a (pseudo)tetrahedral CoII diradical complex [CoII(MesL˙−)2] (Mes = mesityl), in which two semi-benzoquinonediimine radicals are strongly antiferromagnetically coupled to the central CoII ion, yielding an S = 1/2 ground state (confirmed by magnetic susceptibility measurements and EPR studies).58 This complex [CoII(MesL˙−)2] displayed two reversible 1e− reductions at E1/2 = –1.23 V and −2.10 V vs. Fc+/0 in THF with 0.1 M [nBu4N][PF6]. In contrast, CV studies in CH2Cl2 exhibited a significant current increase near the second reduction potential, indicating an electrocatalytic reaction mediated by the doubly reduced species [CoII(MesL2−)2]2−. In particular, the authors proposed that [CoII(MesL2−)2]2−, generated at the working electrode surface, reacts with the CH2Cl2 solvent via C–Cl bond activation.Inspired by this result, the authors attempted catalytic C–C bond formation using [CoII(MesL˙−)2] as the pre-catalyst. Indeed, using a benzyl halide as a substrate, electrocatalytic current (icat) was observed in THF at −2.10 V. At 100 mV s−1 scan rate, the observed rate constant using benzyl bromide or benzyl chloride was estimated to be 10 and 2.8 s−1, respectively. The slower reaction rate with benzyl chloride suggests that C–X cleavage may be the rate determining step. The ESI mass spectrum recorded for the mixture of in situ generated [CoII(MesL2−)2]2− and benzyl chloride showed [CoII(Bn)(MesL˙−)2]− as the main product, while dibenzyl was isolated as the exclusive organic product in this reaction mixture. Taken together, an electrocatalytic mechanism was proposed (Fig. 9): electrochemical reduction of [CoII(MesL˙−)2] by two electrons generates [CoII(MesL2−)2]2−, which activates the benzyl halide to yield the CoII–Bn intermediate, followed by homocoupling of CoII–Bn with a second equivalent of CoII–Bn to yield the dibenzyl product with concomitant formation of [CoII(L2−)(L˙−)]−. This 1e− reduced intermediate can be further reduced at the working electrode to regenerate the active dianionic form for benzyl halide activation and subsequent bond formation.
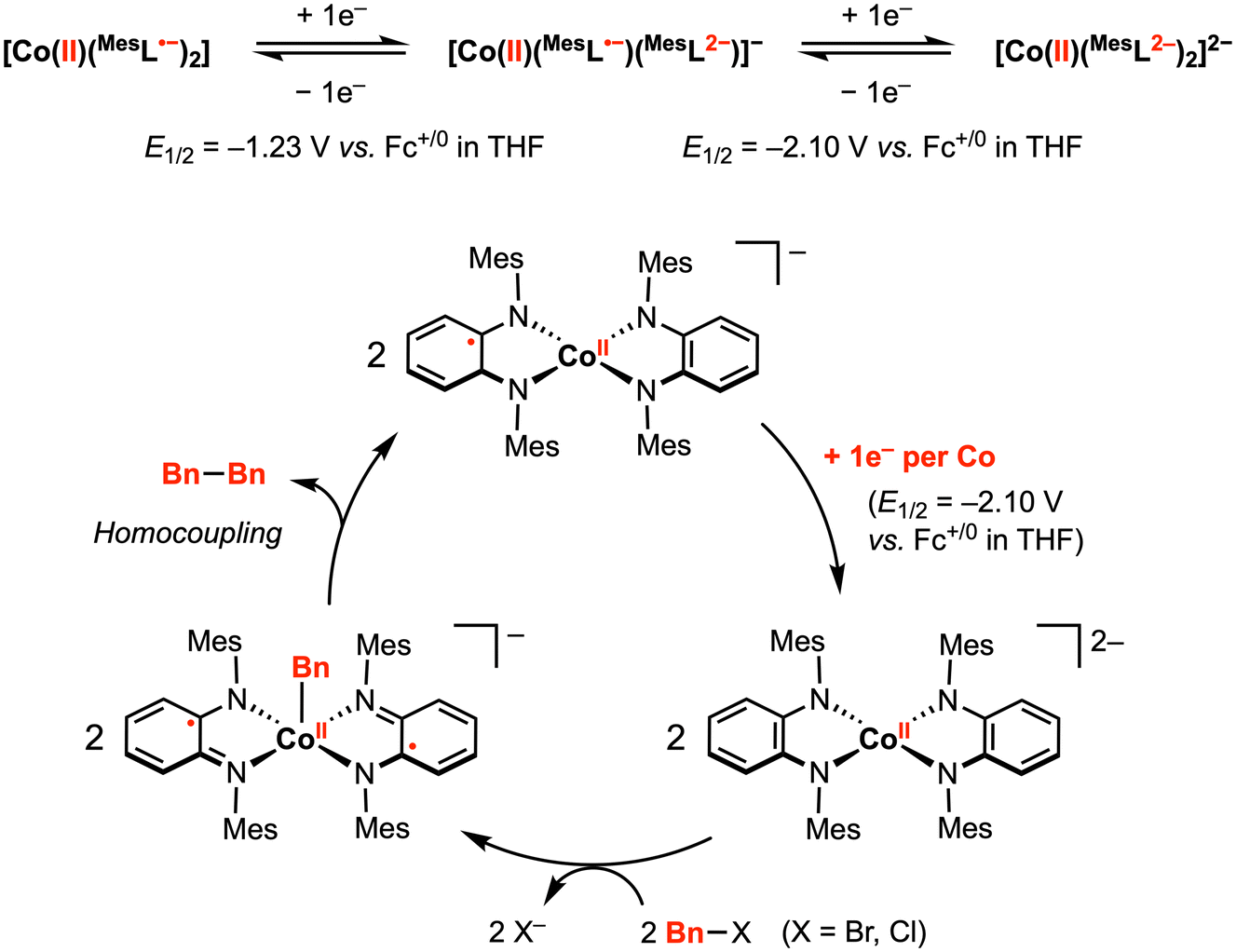 | ||
| Fig. 9 Proposed electrocatalytic C–C bond formation promoted by two diimine-type redox-active ligands.58 | ||
The above study is an elegant example of electrocatalysis at the first-row transition metal facilitated by redox-active ligands. However, we note that this cobalt electrocatalyst, as well as most of the stoichiometric systems discussed in Sections 2.1 and 2.1 (all except [CpCo(Ropda)]), operate by two sequential 1e− redox processes, and thus the two-electron reactivity only occurs after the second electron transfer at the E2 potential to generate the active species (Fig. 10a). Sequential, well-separated 1e− redox events (often referred to as normal ordering of potentials) are typically observed due to electrostatic repulsion.60 For electrochemical or electrocatalytic reactions mediated by molecular metal complexes, overpotential is defined as the difference between the standard thermodynamic potential for the reaction and the potential at which the electrocatalyst operates.61 Thus, if the potential difference (ΔE = E2 − E1) between the first and second redox events at the metal complex is decreased, the overpotential for a two-electron transformation will also be lowered. In an ideal case, the overpotential will be minimized when the potential difference (ΔE) is zero or even a negative (for oxidations) or positive (for reductions) value. This situation is known as potential inversion: the second electron transfer is as favorable or easier than the first electron transfer, leading to an overall two-electron redox process (Fig. 10b).
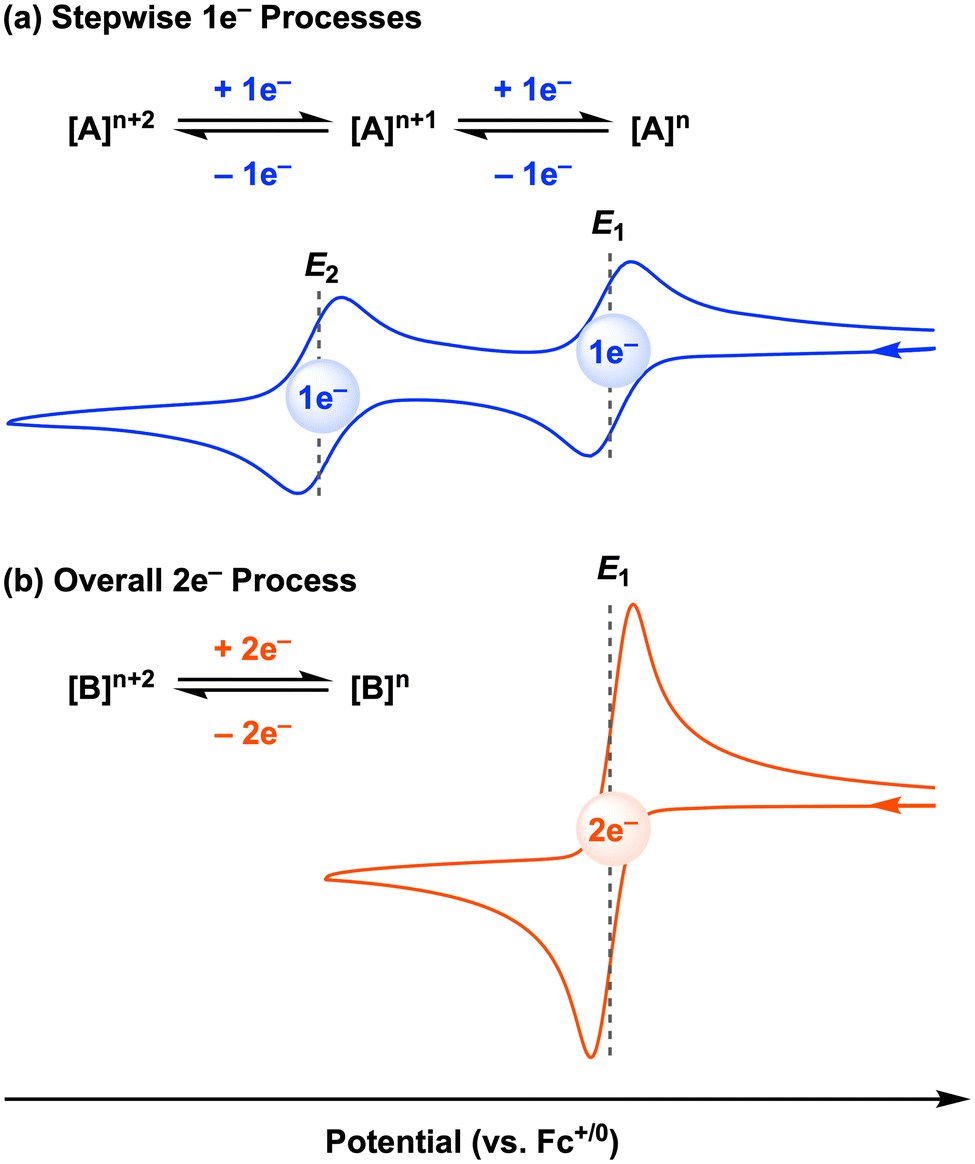 | ||
| Fig. 10 (a) Normal ordering of redox potentials. (b) Potential inversion. | ||
The ordering of potentials can be affected by several factors, including coupled proton transfer, structural reorganization (e.g., ligand binding/release), and ion solvation energies that approximately depend on the square of the charge number.60,62 Nevertheless, as mentioned above, only a limited number of first-row transition metal complexes have been shown to exhibit overall two-electron redox cycles in a single CV feature.3,35–48 Among them, only one example has shown multielectron reactivity for electrocatalysis (CO2 reduction).43
From the CO2 reduction literature, we highlight select examples in which the overpotential for electrocatalytic CO2 reduction is decreased due to the presence of a redox-active ligand that causes the second redox potential (E2) of the metal complex to shift to a more positive value. In 2020, the Chang group63 reported an iron polypyridyl complex, [FeII(ppy0)(MeCN)]2+, that exhibited two closely spaced 1e− reductions, centered at −1.43 V vs. Fc+/0 in MeCN (Fig. 11), both of which were assigned as ligand-centered events. Two-electron reduction with decamethylcobaltocene (Cp*2Co) in THF afforded a neutral diamagnetic complex. Experimental and DFT computational characterization of this species led to its formulation as [FeII(ppy˙˙)2−]0 with an open-shell singlet ground state where an intermediate-spin iron(II) center (SFe = 1) is antiferromagnetically coupled to a doubly-reduced polypyridyl (Sppy = 1) ligand. The strong metal–ligand exchange coupling shifts the second reduction of the polypyridyl ligand to a more positive potential relative to the zinc(II)63 and cobalt(II)64 analogues by 640 mV and 770 mV, respectively. The authors also showed that [FeII(ppy0)(MeCN)]2+ is highly selective for CO2 reduction. With phenol (3.5 M) as a proton source, two electrocatalytic waves were observed at ca. −1.50 and −2.0 V vs. Fc+/0, the former being near the polypyridyl reduction potential. The appearance of a second catalytic wave was attributed to a change in the electrocatalytic mechanism, according to their follow-up studies.65 The faradaic efficiency for CO production (FECO) reached 94% at −1.46 V vs. Fc+/0, with a turnover frequency (TOF) of 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1 and overpotential (η) of only 190 mV. This result marked a substantial decrease in the energetic requirements for CO2 reduction compared to previously reported molecular catalysts.
000 s−1 and overpotential (η) of only 190 mV. This result marked a substantial decrease in the energetic requirements for CO2 reduction compared to previously reported molecular catalysts.
 | ||
| Fig. 11 Cyclic voltammograms of FeII and CoII polypyridyl complexes. Reproduced with permission from ref. 64. Copyright 2022 American Chemical Society. | ||
More recently, Zhang and co-workers examined the electrocatalytic CO2 reduction reaction at a series of CoII tetraphenylporphyrin complexes in which different numbers of redox-active nitro groups (NO2) were incorporated into the ligand backbone.66 The parent complex [TPPCoII] displayed two reversible reductions at E1/2 = −1.28 V and −2.41 V vs. Fc+/0 in DMF with 0.1 M [nBu4N][PF6], for which the first reduction was assigned as cobalt-based.67 The CoII/CoI couple in the nitro-substituted complexes was shifted slightly to more positive potentials by 40–130 mV due to the electron-withdrawing effect of the nitro group(s). A second reversible reduction for all nitro-substituted complexes was observed at E1/2 = −1.59 V vs. Fc+/0. This redox feature was replicated at the [BNPPZnII] analogue and is thus assigned as a ligand-based process. Interestingly, differential pulse voltammetry (DPV) revealed that the n2/n1 ratio (number of electrons being transferred in the second/first reduction) are associated with the number of nitro groups on the porphyrin ligand (Fig. 12). All complexes in this series were shown to be active electrocatalysts for CO2 reduction, with onset potentials observed at −1.50 V for all nitro-substituted complexes and at −2.32 V for the parent [TPPCoII]. Thus, by introducing the redox-active nitro groups, the overpotential (η) for catalytic CO2-to-CO conversion was lowered to merely 270 mV (compared to η = ca. 1090 mV for [TPPCo]). The foot-of-the-wave analysis (FOWA) of the catalytic wave yielded the maximum TOF value of 4.9 × 104 s−1 for [TNPPCoII] bearing four nitro substituents.
 | ||
| Fig. 12 Cobalt(II) tetraphenylporphyrin complexes and their differential pulse voltammograms in DMF with 0.1 M [nBu4N][PF6]. Reproduced from ref. 66 under a Creative Commons License CC-BY-NC-ND 4.0. | ||
In another case, the introduction of bulky mesityl groups on the bipyridine ligand in [Mn(Mesbpy)(CO)3Br] (Mesbpy = 6,6′-dimesityl-2,2′-bipyridine) was shown to have a significant effect on the electrochemical reduction behavior, preventing dimerization after bromide dissociation.43 The parent complex [Mn(bpy)(CO)3Br] exhibited two irreversible 1e− reduction waves separated by ca. 300 mV, while [Mn(Mesbpy)(CO)3Br] undergoes a reversible 2e− reduction at E1/2 = −1.55 V vs. Fc+/0 in MeCN to yield the anionic complex [Mn(Mesbpy)(CO)3]−. This anion was synthesized independently via reduction with potassium graphite (KC8) in THF – its crystal structure and DFT calculations indicated significant electron density is present on the bpy ligand. The authors showed clear evidence for the binding and activation of CO2 by [Mn(Mesbpy)(CO)3]−, generating a Mn(I)–CO2H intermediate; however, electrocatalytic current enhancement was not observed until another 1e− reduction of the Mn(I)–CO2H complex at ca. −2.0 V vs. Fc+/0. Consequently, despite the ability of [Mn(Mesbpy)(CO)3Br] to achieve two-electron behavior in a single redox feature, the overpotential for CO2 reduction was found to be comparable to the parent system. This example highlights an issue that may arise when attempting to decrease the catalytic overpotential by potential inversion without knowledge or consideration of the reactivity of the resulting species – if the multielectron redox feature does not generate an active species to initiate electrocatalytic turnover, the potential inversion may not benefit electrocatalytic performance and additional considerations must be accounted for to optimize reactivity. Thus, it is critical to balance all aspects of the catalytic system when considering electrocatalyst design toward the development of systems that operate via multielectron reactions near the thermodynamic potential.
4. Conclusions and outlook
This article provides an overview of recent developments in the field of multielectron reactivity facilitated by redox-active ligand(s) at earth-abundant transition metal complexes. Through these various examples, we highlight how redox-active ligand scaffolds can serve as efficient electron reservoirs to maintain the metal oxidation state and to activate substrates through ligand-based oxidation or reduction processes. The redox flexibility of these ligands has emerged as a valuable strategy for modulating the behavior of first-row transition metal centers to access novel stoichiometric, and in some instances, electrocatalytic multielectron reactivity with organic substrates. While the progress in this field is exciting, the potential of redox-active ligands for multielectron transformations may yet to be fully realized.Throughout this review, we noted the relevant redox potentials for each system, where available. These examples largely focused on select ligand structures rather than a systematic exploration of ligand substituent effects. While the influence of ligand electronic properties on the electrochemical behavior of transition metal complexes has been thoroughly established, limited work has been done to demonstrate how the redox potentials of the metal complex affect the favorability, rate, selectivity, and/or mechanism for multielectron reactions with organic substrates. Further investigations in this area are warranted to establish structure–activity relationships with a variety of redox-active ligand scaffolds and metal centers, and to push the boundaries of inductive ligand alterations to better understand the interplay between metal and ligand properties for multielectron reactivity. This knowledge will be invaluable as the field moves toward rational redox-active ligand design for targeted synthetic applications.
The prospect of accessing multielectron behavior at a single redox feature with earth-abundant metal complexes is of great interest for electrocatalytic applications. Achieving two electron transfers in a single redox feature, as observed by CV or other electrochemical technique, eliminates the need for additional applied potential to reach the second oxidation state, which in principle would minimize the overpotential required for electrocatalytic substrate conversion. To date, this strategy has only been successfully demonstrated in a few reports. A key challenge with this approach is that the two-electron redox process may not directly generate an intermediate active for electrocatalysis, as observed with [Mn(Mesbpy)(CO)3Br] for CO2 reduction. Further work is needed to better understand how to strategically design redox-active ligand complexes that minimize electrocatalytic overpotentials while maintaining the necessary properties at the metal for substrate reactivity. Furthermore, as highlighted in this review, most earth-abundant metal complexes showing multielectron substrate reactivity display sequential 1e− electrochemical processes rather than 2e− redox features. As more first-row metal complexes exhibiting 2e− behavior are developed, greater attention should be paid to understanding how to effectively exploit this reactivity for electrocatalysis. Advancements in this area will further broader the opportunities for redox-active ligands in sustainable catalysis.
Author contributions
Conceptualization, M.Z. and K.M.W.; writing – original draft, M.Z. and K.M.W.; writing – review and editing, M.Z. and K.M.W.; supervision, K.M.W.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported the ACS Petroleum Research Fund (65171-DNI3) and Rutgers, The State University of New Jersey.References
- C. K. Jørgensen, Differences between the four halide ligands, and discussion remarks on trigonal-bipyramidal complexes, on oxidation states, and on diagonal elements of one-electron energy, Coord. Chem. Rev., 1966, 1, 164–178 CrossRef.
- M. D. Ward and J. A. McCleverty, Non-innocent behaviour in mononuclear and polynuclear complexes: consequences for redox and electronic spectroscopic properties, J. Chem. Soc., Dalton Trans., 2002, 275–288 RSC.
- P. Chaudhuri, C. N. Verani, E. Bill, E. Bothe, T. Weyhermüller and K. Wieghardt, Electronic Structure of Bis(o-iminobenzosemiquinonato)metal Complexes (Cu, Ni, Pd). The Art of Establishing Physical Oxidation States in Transition-Metal Complexes Containing Radical Ligands, J. Am. Chem. Soc., 2001, 123, 2213–2223 CrossRef CAS PubMed.
- P. J. Chirik, Preface: Forum on Redox-Active Ligands, Inorg. Chem., 2011, 50, 9737–9740 CrossRef CAS PubMed.
- P. J. Chirik and K. Wieghardt, Radical Ligands Confer Nobility on Base-Metal Catalysts, Science, 2010, 327, 794–795 CrossRef CAS PubMed.
- W. Kaim, Manifestations of Noninnocent Ligand Behavior, Inorg. Chem., 2011, 50, 9752–9765 CrossRef CAS.
- V. Lyaskovskyy and B. de Bruin, Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- F. F. Khan, A. D. Chowdhury and G. K. Lahiri, Bond Activations Assisted by Redox Active Ligand Scaffolds, Eur. J. Inorg. Chem., 2020, 1138–1146 CrossRef CAS.
- K. Singh, A. Kundu and D. Adhikari, Ligand-Based Redox: Catalytic Applications and Mechanistic Aspects, ACS Catal., 2022, 12, 13075–13107 CrossRef CAS.
- N. P. van Leest, F. J. de Zwart, M. Zhou and B. de Bruin, Controlling Radical-Type Single-Electron Elementary Steps in Catalysis with Redox-Active Ligands and Substrates, JACS Au, 2021, 1, 1101–1115 CrossRef CAS.
- J. I. van der Vlugt, Radical-Type Reactivity and Catalysis by Single-Electron Transfer to or from Redox-Active Ligands, Chem. – Eur. J., 2019, 25, 2651–2662 CrossRef.
- A. I. O. Suarez, V. Lyaskovskyy, J. N. H. Reek, J. I. van der Vlugt and B. deBruin, Complexes with Nitrogen-Centered Radical Ligands: Classification, Spectroscopic Features, Reactivity, and Catalytic Applications, Angew. Chem., Int. Ed., 2013, 52, 12510–12529 CrossRef.
- W. Kaim and B. Schwederski, Non-innocent ligands in bioinorganic chemistry - An overview, Coord. Chem. Rev., 2010, 254, 1580–1588 CrossRef.
- J. Stubbe and W. A. van der Donk, Protein Radicals in Enzyme Catalysis, Chem. Rev., 1998, 98, 705–762 CrossRef PubMed.
- D. L. J. Broere, R. Plessius and J. I. van der Vlugt, New avenues for ligand-mediated processes – expanding metal reactivity by the use of redox-active catechol, o-aminophenol and o-phenylenediamine ligands, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 Nobel Prize, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed.
- K. J. Blackmore, J. W. Ziller and A. F. Heyduk, “Oxidative Addition” to a Zirconium(IV) Redox-Active Ligand Complex, Inorg. Chem., 2005, 44, 5559–5561 CrossRef PubMed.
- K. J. Blackmore, M. B. Sly, M. R. Haneline, J. W. Ziller and A. F. Heyduk, Group IV Imino-Semiquinone Complexes Obtained by Oxidative Addition of Halogens, Inorg. Chem., 2008, 47, 10522–10532 CrossRef CAS.
- N. A. Ketterer, H. Fan, K. J. Blackmore, X. Yang, J. W. Ziller, M.-H. Baik and A. F. Heyduk, π˙–π˙ Bonding Interactions Generated by Halogen Oxidation of Zirconium(IV) Redox-Active Ligand Complexes, J. Am. Chem. Soc., 2008, 130, 4364–4374 CrossRef CAS.
- K. J. Blackmore, N. Lal, J. W. Ziller and A. F. Heyduk, Group IV Coordination Chemistry of a Tetradentate Redox-Active Ligand in Two Oxidation States, Eur. J. Inorg. Chem., 2009, 735–743 CrossRef CAS.
- M. R. Haneline and A. F. Heyduk, C−C Bond-Forming Reductive Elimination from a Zirconium(IV) Redox-Active Ligand Complex, J. Am. Chem. Soc., 2006, 128, 8410–8411 CrossRef CAS.
- G. Szigethy and A. F. Heyduk, Steric and Electronic Consequences of Flexibility in a Tetradentate Redox-Active Ligand: Ti(IV) and Zr(IV) Complexes, Inorg. Chem., 2011, 50, 125–135 CrossRef CAS.
- R. G. Belli, V. C. Tafuri, N. A. Garcia and C. C. Roberts, One- and Two-Electron Redox Catalysis with Lutetium Enabled by a Tris(Amido) Redox-Active Ligand, Organometallics, 2023, 42, 1059–1064 CrossRef CAS.
- R. G. Belli, V. C. Tafuri and C. C. Roberts, Improving Alkyl–Alkyl Cross-Coupling Catalysis with Early Transition Metals through Mechanistic Understanding and Metal Tuning, ACS Catal., 2022, 12, 9430–9436 CrossRef CAS.
- R. G. Belli, V. C. Tafuri, M. V. Joannou and C. C. Roberts, d0 Metal-Catalyzed Alkyl–Alkyl Cross-Coupling Enabled by a Redox-Active Ligand, ACS Catal., 2022, 12, 3094–3099 CrossRef CAS.
- J. T. Gavin, R. G. Belli and C. C. Roberts, Radical-Polar Crossover Catalysis with a d0 Metal Enabled by a Redox-Active Ligand, J. Am. Chem. Soc., 2022, 144, 21431–21436 CrossRef CAS PubMed.
- R. F. Munhá, R. A. Zarkesh and A. F. Heyduk, Tuning the Electronic and Steric Parameters of a Redox-Active Tris(amido) Ligand, Inorg. Chem., 2013, 52, 11244–11255 CrossRef.
- C. Mukherjee, T. Weyhermüller, E. Bothe and P. Chaudhuri, Oxidation of an o-Iminobenzosemiquinone Radical Ligand by Molecular Bromine: Structural, Spectroscopic, and Reactivity Studies of a Copper(II) o-Iminobenzoquinone Complex, Inorg. Chem., 2008, 47, 2740–2746 CrossRef CAS PubMed.
- A. L. Smith, L. A. Clapp, K. I. Hardcastle and J. D. Soper, Redox-active ligand-mediated Co–Cl bond-forming reactions at reducing square planar cobalt(III) centers, Polyhedron, 2010, 29, 164–169 CrossRef CAS.
- A. L. Smith, K. I. Hardcastle and J. D. Soper, Redox-Active Ligand-Mediated Oxidative Addition and Reductive Elimination at Square Planar Cobalt(III): Multielectron Reactions for Cross-Coupling, J. Am. Chem. Soc., 2010, 132, 14358–14360 CrossRef CAS.
- J. Jacquet, E. Salanouve, M. Orio, H. Vezin, S. Blanchard, E. Derat, M. Desage-El Murr and L. Fensterbank, Iminosemiquinone radical ligands enable access to a well-defined redox-active CuII–CF3 complex, Chem. Commun., 2014, 50, 10394–10397 RSC.
- Y. Yasu, T. Koike and M. Akita, Three-component Oxytrifluoromethylation of Alkenes: Highly Efficient and Regioselective Difunctionalization of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C Bonds Mediated by Photoredox Catalysts, Angew. Chem., Int. Ed., 2012, 51, 9567–9571 CrossRef CAS.
C Bonds Mediated by Photoredox Catalysts, Angew. Chem., Int. Ed., 2012, 51, 9567–9571 CrossRef CAS. - J. Jacquet, S. Blanchard, E. Derat, M. Desage-El Murr and L. Fensterbank, Redox-ligand sustains controlled generation of CF3 radicals by well-defined copper complex, Chem. Sci., 2016, 7, 2030–2036 RSC.
- M. Zou, T. J. Emge and K. M. Waldie, Two-Electron Redox Tuning of Cyclopentadienyl Cobalt Complexes Enabled by the Phenylenediamide Ligand, Inorg. Chem., 2023, 62, 10397–10407 CrossRef CAS PubMed.
- S. A. Richert, P. K. S. Tsang and D. T. Sawyer, Ligand-Centered Redox Processes for MnL3, FeL3, and CoL3 Complexes (L = Acetylacetonate, 8-Quinolinate, Picolinate, 2,2′-Bipyridyl, 1,10-Phenanthroline) and for Their Tetrakis(2,6-dichlorophenyl)porphinato Complexes [M(Por)], Inorg. Chem., 1989, 28, 2471–2475 CrossRef CAS.
- P. N. Bartlett and V. Eastwick-Field, A reinvestigation of the electrochemistry of [Ni(II)(bpy)3(ClO4)2] in acetonitrile using rotating disc and rotating ring-disc electrodes, Electrochim. Acta, 1993, 38, 2515–2523 CrossRef CAS.
- E. Bill, E. Bothe, P. Chaudhuri, K. Chlopek, D. Herebian, S. Kokatam, K. Ray, T. Weyhermüller, F. Neese and K. Wieghardt, Molecular and Electronic Structure of Four- and Five-Coordinate Cobalt Complexes Containing Two o-Phenylenediamine- or Two o-Aminophenol-Type Ligands at Various Oxidation Levels: An Experimental, Density Functional, and Correlated ab initio Study, Chem. – Eur. J., 2005, 11, 204–224 CrossRef.
- K. Chlopek, E. Bothe, F. Neese, T. Weyhermuller and K. Wieghardt, Molecular and Electronic Structures of Tetrahedral Complexes of Nickel and Cobalt Containing N,N’-Disubstituted, Bulky o-Diiminobenzosemiquinonate(1−) π-Radical Ligands, Inorg. Chem., 2006, 45, 6298–6307 CrossRef CAS PubMed.
- C. Mukherjee, T. Weyhermüller, E. Bothe and P. Chaudhuri, Targeted Oxidase Reactivity with a New Redox-Active Ligand Incorporating N2O2 Donor Atoms. Complexes of Cu(II), Ni(II), Pd(II), Fe(III), and V(V), Inorg. Chem., 2008, 47, 11620–11632 CrossRef CAS PubMed.
- S. K. Sharma, P. S. May, M. B. Jones, S. Lense, K. I. Hardcastle and C. E. MacBeth, Catalytic dioxygen activation by Co(II) complexes employing a coordinatively versatile ligand scaffold, Chem. Commun., 2011, 47, 1827–1829 RSC.
- P. Ghosh, S. Samanta, S. K. Roy, S. Joy, T. Krämer, J. E. McGrady and S. Goswami, Redox Noninnocence in Coordinated 2-(Arylazo)pyridines: Steric Control of Ligand-Based Redox Processes in Cobalt Complexes, Inorg. Chem., 2013, 52, 14040–14049 CrossRef CAS.
- D. Sengupta, P. Ghosh, T. Chatterjee, H. Datta, N. D. Paul and S. Goswami, Ligand-Centered Redox in Nickel(II) Complexes of 2-(Arylazo)pyridine and Isolation of 2-Pyridyl-Substituted Triaryl Hydrazines via Catalytic N-Arylation of Azo-Function, Inorg. Chem., 2014, 53, 12002–12013 CrossRef CAS PubMed.
- M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, Manganese Catalysts with Bulky Bipyridine Ligands for the Electrocatalytic Reduction of Carbon Dioxide: Eliminating Dimerization and Altering Catalysis, J. Am. Chem. Soc., 2014, 136, 5460–5471 CrossRef CAS.
- K. M. Waldie, S. Ramakrishnan, S.-K. Kim, J. K. Maclaren, C. E. D. Chidsey and R. M. Waymouth, Multielectron Transfer at Cobalt: Influence of the Phenylazopyridine Ligand, J. Am. Chem. Soc., 2017, 139, 4540–4550 CrossRef CAS.
- B. D. Matson, E. A. McLoughlin, K. C. Armstrong, R. M. Waymouth and R. Sarangi, Effect of Redox Active Ligands on the Electrochemical Properties of Manganese Tricarbonyl Complexes, Inorg. Chem., 2019, 58, 7453–7465 CrossRef CAS PubMed.
- C. S. Richburg and B. H. Farnum, Influence of Pyridine on the Multielectron Redox Cycle of Nickel Diethyldithiocarbamate, Inorg. Chem., 2019, 58, 15371–15384 CrossRef CAS.
- M. M. R. Mazumder, A. Burton, C. S. Richburg, S. Saha, B. Cronin, E. Duin and B. H. Farnum, Controlling One-Electron vs Two-Electron Pathways in the Multi-Electron Redox Cycle of Nickel Diethyldithiocarbamate, Inorg. Chem., 2021, 60, 13388–13399 CrossRef CAS PubMed.
- H. Bamberger, U. Albold, J. Dubnická Midlíková, C.-Y. Su, N. Deibel, D. Hunger, P. P. Hallmen, P. Neugebauer, J. Beerhues, S. Demeshko, F. Meyer, B. Sarkar and J. van Slageren, Iron(II), Cobalt(II), and Nickel(II) Complexes of Bis(sulfonamido)benzenes: Redox Properties, Large Zero-Field Splittings, and Single-Ion Magnets, Inorg. Chem., 2021, 60, 2953–2963 CrossRef CAS PubMed.
- M. Zou and K. M. Waldie, Redox-active ligand promoted electrophile addition at cobalt, Chem. Commun., 2023, 59, 14693–14696 RSC.
- T. Koide, I. Aritome, T. Saeki, Y. Morita, Y. Shiota, K. Yoshizawa, H. Shimakoshi and Y. Hisaeda, Cobalt–Carbon Bond Formation Reaction via Ligand Reduction of Porphycene–Cobalt(II) Complex and Its Noninnocent Reactivity, ACS Omega, 2018, 3, 4027–4034 CrossRef CAS.
- J. M. Darmon, S. C. E. Stieber, K. T. Sylvester, I. Fernández, E. Lobkovsky, S. P. Semproni, E. Bill, K. Wieghardt, S. DeBeer and P. J. Chirik, Oxidative Addition of Carbon–Carbon Bonds with a Redox-Active Bis(imino)pyridine Iron Complex, J. Am. Chem. Soc., 2012, 134, 17125–17137 CrossRef CAS.
- M. W. Bouwkamp, A. C. Bowman, E. Lobkovsky and P. J. Chirik, Iron-Catalyzed [2π + 2π] Cycloaddition of α,ω-Dienes: The Importance of Redox-Active Supporting Ligands, J. Am. Chem. Soc., 2006, 128, 13340–13341 CrossRef CAS PubMed.
- K. T. Sylvester and P. J. Chirik, Iron-Catalyzed, Hydrogen-Mediated Reductive Cyclization of 1,6-Enynes and Diynes: Evidence for Bis(imino)pyridine Ligand Participation, J. Am. Chem. Soc., 2009, 131, 8772–8774 CrossRef CAS.
- J. M. Hoyt, K. T. Sylvester, S. P. Semproni and P. J. Chirik, Synthesis and Electronic Structure of Bis(imino)pyridine Iron Metallacyclic Intermediates in Iron-Catalyzed Cyclization Reactions, J. Am. Chem. Soc., 2013, 135, 4862–4877 CrossRef CAS.
- K. T. Sylvester and P. J. Chirik, Iron-Catalyzed, Hydrogen-Mediated Reductive Cyclization of 1,6-Enynes and Diynes: Evidence for Bis(imino)pyridine Ligand Participation, J. Am. Chem. Soc., 2009, 131, 8772–8774 CrossRef CAS.
- A. M. Tondreau, C. Milsmann, E. Lobkovsky and P. J. Chirik, Oxidation and Reduction of Bis(imino)pyridine Iron Dicarbonyl Complexes, Inorg. Chem., 2011, 50, 9888–9895 CrossRef CAS.
- J. M. Darmon, Z. R. Turner, E. Lobkovsky and P. J. Chirik, Electronic Effects in 4-Substituted Bis(imino)pyridines and the Corresponding Reduced Iron Compounds, Organometallics, 2012, 31, 2275–2285 CrossRef CAS.
- M. van der Meer, Y. Rechkemmer, I. Peremykin, S. Hohloch, J. van Slageren and B. Sarkar, (Electro)catalytic C–C bond formation reaction with a redox-active cobalt complex, Chem. Commun., 2014, 50, 11104–11106 RSC.
- D. L. J. Broere, D. K. Modder, E. Blokker, M. A. Siegler and J. I. vanderVlugt, Metal–Metal Interactions in Heterobimetallic Complexes with Dinucleating Redox-Active Ligands, Angew. Chem., Int. Ed., 2016, 55, 2406–2410 CrossRef CAS PubMed.
- D. H. Evans, One-Electron and Two-Electron Transfers in Electrochemistry and Homogeneous Solution Reactions, Chem. Rev., 2008, 108, 2113–2144 CrossRef CAS.
- A. M. Appel and M. L. Helm, Determining the Overpotential for a Molecular Electrocatalyst, ACS Catal., 2014, 4, 630–633 CrossRef CAS.
- C. Hessin, J. Schleinitz, N. Le Breton, S. Choua, L. Grimaud, V. Fourmond, M. Desage-El Murr and C. Léger, Assessing the Extent of Potential Inversion by Cyclic Voltammetry: Theory, Pitfalls, and Application to a Nickel Complex with Redox-Active Iminosemiquinone Ligands, Inorg. Chem., 2023, 62, 3321–3332 CrossRef CAS PubMed.
- J. S. Derrick, M. Loipersberger, R. Chatterjee, D. A. Iovan, P. T. Smith, K. Chakarawet, J. Yano, J. R. Long, M. Head-Gordon and C. J. Chang, Metal–Ligand Cooperativity via Exchange Coupling Promotes Iron- Catalyzed Electrochemical CO2 Reduction at Low Overpotentials, J. Am. Chem. Soc., 2020, 142, 20489–20501 CrossRef CAS PubMed.
- P. De La Torre, J. S. Derrick, A. Snider, P. T. Smith, M. Loipersberger, M. Head-Gordon and C. J. Chang, Exchange Coupling Determines Metal-Dependent Efficiency for Iron- and Cobalt-Catalyzed Photochemical CO2 Reduction, ACS Catal., 2022, 12, 8484–8493 CrossRef CAS.
- M. Loipersberger, J. S. Derrick, C. J. Chang and M. Head-Gordon, Deciphering Distinct Overpotential-Dependent Pathways for Electrochemical CO2 Reduction Catalyzed by an Iron–Terpyridine Complex, Inorg. Chem., 2022, 61, 6919–6933 CrossRef CAS.
- W.-W. Yong, H.-T. Zhang, Y.-H. Guo, F. Xie and M.-T. Zhang, Redox-Active Ligand Assisted Multielectron Catalysis: A Case of Electrocatalyzed CO2-to-CO Conversion, ACS Org. Inorg. Au, 2023, 3, 384–392 CrossRef CAS PubMed.
- L. A. Truxillo and D. G. Davis, Electrochemistry of cobalt tetraphenylporphyrin in aprotic media, Anal. Chem., 1975, 47, 2260–2267 CrossRef CAS.
| This journal is © the Partner Organisations 2024 |