
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Construction of a BiVO4/VS-MoS2 S-scheme heterojunction for efficient photocatalytic nitrogen fixation†
Han-Ying
Luo‡
a,
Zhao-Lei
Liu‡
a,
Meng-Ran
Zhang
a,
Yan-Fei
Mu
 *b and
Min
Zhang
*a
*b and
Min
Zhang
*a
aMOE International Joint Laboratory of Materials Microstructure, Institute for New Energy Materials and Low Carbon Technologies, School of Materials Science and Engineering, School of Chemistry and Chemical Engineering, Tianjin University of Technology, Tianjin 300384, China. E-mail: zm2016@email.tjut.edu.cn
bSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, Jiangsu 225009, China. E-mail: 007916@yzu.edu.cn
First published on 20th February 2024
Abstract
Photocatalytic nitrogen (N2) reduction to ammonia (NH3), adopting H2O as the electron source, suffers from low efficiency owing to the sluggish kinetics of N2 reduction and the requirement of a substantial thermodynamic driving force. Herein, we present a straightforward approach for the construction of an S-scheme heterojunction of BiVO4/VS-MoS2 to successfully achieve photocatalytic N2 fixation, which is manufactured by coupling an N2-activation component (VS-MoS2 nanosheet) and water-oxidation module (BiVO4 nanocrystal) through electrostatic self-assembly. The VS-MoS2 nanosheet, enriched with sulfur vacancies, plays a pivotal role in facilitating N2 adsorption and activation. Additionally, the construction of the S-scheme heterojunction enhances the driving force for water oxidation and improves charge separation. Under simulated sunlight irradiation (100 mW cm−2), BiVO4/VS-MoS2 exhibits efficient photocatalytic N2 reduction activity with H2O as the proton source, yielding NH3 at a rate of 132.8 μmol g−1 h−1, nearly 7 times higher than that of pure VS-MoS2. This study serves as a noteworthy example of efficient N2 reduction to NH3 under mild conditions.
1. Introduction
Ammonia (NH3) serves as both a promising energy storage intermediary and an essential raw material affecting agricultural and industrial production.1,2 However, given the molecular inertness of N2 (dissociation energy of 945 kJ mol−1), the current industrial synthesis of NH3 predominantly relies on the Haber–Bosch process.3,4 This traditional method requires high temperature and pressure (300–550 °C and 15–25 MPa), resulting in significant investment costs and high energy consumption.5,6 Therefore, the pursuit of synthetic processes under mild conditions has become a paramount objective. Photocatalytic N2 fixation, employing semiconductor photocatalysts that harness sunlight as the energy source while utilizing N2 and H2O as reactants, stands out as a sustainable approach for NH3 production.7–10 Nonetheless, because of the poor N2 adsorption–activation and carrier separation in semiconductor photocatalysts, achieving efficient photocatalytic conversion of N2 to NH3 remains a scientific challenge.11–13Efficient N2 adsorption–activation and good light absorption of photocatalysts are the fundamental prerequisites for realizing the efficient photoreduction of N2 to produce NH3.14 Consequently, the initial step involves the selection of potential semiconductor materials to reasonably design photocatalysts with suitable energy levels for N2 reduction and favorable properties for N2 adsorption–activation.15–17 Schrauzer and co-workers conducted the pioneering systematic investigation into the photocatalytic NH3 synthesis using TiO2-based materials.18 In recent years, numerous studies have explored traditional metal oxides, carbonaceous materials, and layered double hydroxide semiconductors as photocatalysts for N2 photofixation.19–22 However, these semiconductor materials often exhibit wide band gaps and narrow spectral absorption characteristics, which are not conducive for efficient photon utilization in photocatalytic reactions. Inspired by the MoFe-cofactor in natural nitrogenase, molybdenum disulfide (MoS2) has garnered significant attention in the field of N2 fixation due to its structural resemblance to nitrogenase with a Mo–S configuration.23,24 As a two-dimensional direct bandgap semiconductor material, MoS2 not only possesses good photon capture ability but also can be used as a carrier to construct composite catalysts to achieve more efficient catalytic conversion. Nonetheless, MoS2 exhibits poor activity in photocatalytic N2 reduction systems using water as the electron source due to its lack of water oxidation capacity.25 In addition, akin to other semiconductor photocatalysts, its weak photogenerated carrier separation ability also hinders the progress of photocatalytic N2 reduction.26,27 To improve the photocatalytic activity of semiconductor photocatalysts, extensive research efforts have focused on bolstering charge separation through the construction of p–n, II-type, and S-scheme heterojunctions.28–31 Mimicking natural photosynthesis, the S-scheme charge transfer system has received special attention, which can simultaneously facilitate spatial carrier separation and improve redox ability.32,33 Additionally, monoclinic BiVO4 as a “star” material has a broad range of visible light utilization and excellent photoelectric stability, and has been widely used in the study of photoanodic water oxidation.34 Based on this, BiVO4 has potential to serve as a water oxidation unit for MoS2 to construct heterojunctions, further enhancing the efficiency of photocatalytic N2 fixation.
Herein, we present an innovative S-scheme heterojunction prepared by coupling sulfur vacancy enriched MoS2 nanosheets (VS-MoS2) with BiVO4 nanocrystals through electrostatic self-assembly for photocatalytic N2 reduction. Under simulated sunlight irradiation (100 mW cm−2), BiVO4/VS-MoS2 exhibits efficient photocatalytic N2 reduction activity, with a NH3 yield of 132.8 μmol g−1 h−1, nearly 7 times that of pure VS-MoS2 (20 μmol g−1 h−1). In addition, the experimental results from in situ X-ray photoelectron spectroscopy and electron spin resonance spectroscopy verify the presence of sulfur-rich vacancies on the surface of VS-MoS2 nanosheets and the successful formation of an S-scheme heterojunction, which are conducive to the improvement of N2 adsorption–activation and driving force of water oxidation as well as charge separation. The mechanism of the N2 reduction reaction is speculated using in situ diffuse reflectance infrared Fourier transform spectroscopy.
2. Experimental
2.1. Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 mg, 10 mg:20 mg, 10 mg:40 mg, denoted as BiVO4/VS-MoS2 (1/1), BiVO4/VS-MoS2 (1/2) and BiVO4/VS-MoS2 (1/4), respectively.
:10 mg, 10 mg:20 mg, 10 mg:40 mg, denoted as BiVO4/VS-MoS2 (1/1), BiVO4/VS-MoS2 (1/2) and BiVO4/VS-MoS2 (1/4), respectively.
2.2. Photocatalytic N2 reduction experiment
The photocatalytic N2 reduction reactions were carried out in a gas–solid reaction system (25 °C). 20 mg of photocatalyst was placed on the sample table of the reactor. 200 μL of water was injected into the bottom of the reactor as the proton source. Before initiating the photocatalytic reactions, the system was heated to produce water vapor. The system was meticulously degassed to remove air with N2 (99.999%), where N2 was bubbled through 1 M HCl as well as a mixed solution of potassium permanganate and KOH to ensure the removal of potential contaminants. A 300 W xenon lamp equipped with a UVVISCUT400 filter was employed as the light source (light intensity of 100 mW cm−2). For detection, 2 mL of H2O was injected into the system to dissolve the product to form the product solution.2.3. Product detection
2.4. In situ diffuse reflectance infrared Fourier transform spectra (DRIFTS)
In situ diffuse reflectance infrared Fourier transform spectra were recorded using a Bruker IFS 66v Fourier transform spectrometer. The samples were mixed with KBr in a quartz mortar and then placed in an infrared reaction chamber. Pure N2 (99.999%) containing water vapor was continuously introduced into the experimental chamber during in situ characterization. The IR spectrum of pure KBr was first collected as a background spectrum. The final spectra were obtained by subtracting the background spectrum from the spectrum of the sample.3. Results and discussion
3.1. Preparation and structure of BiVO4/VS-MoS2
The synthetic process of BiVO4/VS-MoS2 is illustrated in Scheme 1. Briefly, VS-MoS2 and BiVO4 were initially prepared through the hydrothermal method and thermal injection method, respectively. The measured zeta potential (ζ) of VS-MoS2 is negative (−45 mV), opposited with that of BiVO4 (33 mV) in toluene (Fig. S1†). This opposite surface charging characteristic of VS-MoS2 and BiVO4 should facilitate the spontaneous assembly to form BiVO4/VS-MoS2 in solution. X-ray diffraction (XRD) patterns (Fig. 1a) show that the as-prepared VS-MoS2 and BiVO4 can be indexed to the P63/mmc(194) hexagonal space group (PDF card no. 01-075-1539) and I2/a(15) monoclinic space group (PDF card no. 00-014-0688), respectively. The characteristic diffraction peaks at 14.39°, 32.8°, 39.65° and 58.56° of VS-MoS2 correspond to the (002), (100), (103), and (110) crystal planes. The diffraction peaks of BiVO4 are located at 18.67°, 28.95°, 35.22°, 39.78°, 46.71°, 49.96°, and 59.26°, corresponding to the (110), (121), (002), (211), (240), (−202), and (123) crystal planes. Notably, the diffraction pattern of BiVO4/VS-MoS2 contains the characteristic peaks of both VS-MoS2 and BiVO4, indicating the successful loading of BiVO4 nanocrystals onto VS-MoS2 nanosheets. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) measurements revealed that VS-MoS2 exhibits a nanoflower morphology (Fig. 1b), which is formed by the gathering of nanosheets (Fig. 1c). Atomic force microscopy (AFM) analysis in Fig. S2† further confirmed that VS-MoS2 has a nanosheet morphology with a mean thickness of ∼3 nm. TEM images of pure BiVO4 and BiVO4/VS-MoS2 (Fig. 1d and e) show that BiVO4 nanocrystals are interspersed on VS-MoS2 nanosheets, with an average size of ∼5 nm, which is almost identical to that of the pure BiVO4 nanocrystal (Fig. S3†). Further high-resolution TEM (HRTEM) analysis (Fig. 1f) showed clear lattice fringes of 2.72 Å and 3.08 Å, corresponding to the (100) plane of VS-MoS2 (ref. 35) and the (121) plane of monoclinic BiVO4 (ref. 36) (Fig. S4†), respectively, confirming the presence of BiVO4 nanocrystals on the surface of VS-MoS2 nanosheets. The corresponding energy-dispersive X-ray spectroscopy (EDS) mapping analysis (Fig. 1g) demonstrated the homogeneous distribution of Mo, S, Bi, V, and O elements on BiVO4/VS-MoS2, further verifying the construction of BiVO4/VS-MoS2.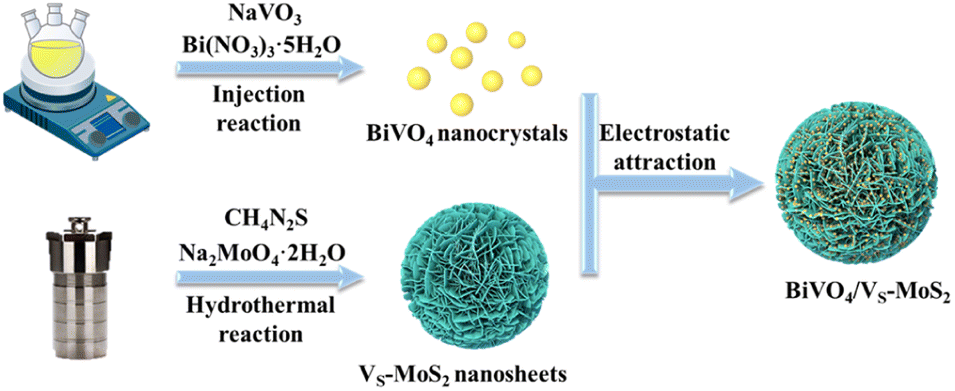 | ||
| Scheme 1 Schematic illustration of the preparation process of BiVO4/VS-MoS2. | ||
 | ||
| Fig. 1 (a) XRD patterns of VS-MoS2, BiVO4, and BiVO4/VS-MoS2. (b and c) SEM and TEM images of a VS-MoS2 nanosheet. (d) TEM image of a BiVO4 nanocrystal. (e–g) TEM, HRTEM, and corresponding EDS images of BiVO4/VS-MoS2. (h) EPR spectra of VS-MoS2 and BiVO4/VS-MoS2. | ||
To gain insights into the structural characteristics of the as-prepared photocatalysts, electron paramagnetic resonance (EPR) measurements were performed initially. As depicted in Fig. 1h, a distinct EPR signal at g = 2.003 can be recognized for VS-MoS2, signifying the formation of the sulfur vacancies on the surface of VS-MoS2.37 In the case of BiVO4/VS-MoS2, a stronger EPR signal is detected, providing evidence for the formation of heterojunction interfaces, which can turn spin-coupled electron pairs into unpaired electron states.38 Sulfur vacancies can serve as active sites to promote carrier capture and N2 adsorption and activation.39 The X-ray photoelectron spectra (XPS) were further analyzed to investigate the potential interfacial interaction and charge transfer processes within the BiVO4/VS-MoS2 heterojunction. Doublet XPS characteristic peaks can be identified at 229.3 eV and 232.4 eV in pure VS-MoS2, attributed to Mo 3d5/2 and Mo 3d3/2 (Fig. S5a†), which can confirm the reduction of Mo6+ to Mo4+ during the growth of VS-MoS2 nanosheets.40 In addition, a comparison between pure VS-MoS2, BiVO4, and BiVO4/VS-MoS2 reveals noticeable shifts in binding energy for Mo 3d, S 2p, Bi 4f, and V 2p. Specifically, Mo 3d and S 2p exhibit positive shifts of 0.50 eV and 0.20 eV, while Bi 4f and V 2p display negative shifts of 0.20 eV and 0.20 eV, respectively (Fig. S5b–d†). These charge redistributions within the BiVO4/VS-MoS2 heterojunction reflect a strong chemical interaction at the interface of the BiVO4/VS-MoS2 heterojunction. This robust interfacial electronic coupling in BiVO4/VS-MoS2 is expected to facilitate the charge transfer between the VS-MoS2 nanosheets and BiVO4 nanocrystals.
3.2. S-scheme mechanism of the BiVO4/VS-MoS2 heterojunction
The energy levels and interfacial charge transfer processes of BiVO4/VS-MoS2 were explored to evaluate the photocatalytic N2 reduction potential of the as-prepared photocatalysts. Fig. 2a and b show the UV-vis diffuse reflectance spectra (DRS), revealing a good light response for both the VS-MoS2 nanosheets and BiVO4 nanocrystals. From the corresponding Tauc plots (insets of Fig. 2a and b), the band gaps (Eg) of VS-MoS2 and BiVO4 can be deduced to be 1.71 eV and 2.22 eV, respectively. To ascertain the energy band structures of VS-MoS2 and BiVO4, the onset edge (Ei) and the secondary electron cutoff (Ecutoff) were determined by carrying out ultraviolet photoelectron spectroscopy (UPS) (Fig. 2c). According to the equation EVB = 21.22 − (Ecutoff − Ei), the valence band edge potentials (EVB) can be calculated to be 1.35 V and 2.23 V versus the standard hydrogen electrode (vs. NHE) for VS-MoS2 and BiVO4, respectively. By combining the values from Eg and EVB analysis, the corresponding conduction band edge potentials (ECB) can be calculated to be −0.36 V and 0.01 V (vs. NHE) for VS-MoS2 and BiVO4, respectively. The resultant energy band structures of VS-MoS2 and BiVO4 are presented in Fig. 2d. The photogenerated electrons in VS-MoS2 possess sufficient energy to drive the photoreduction of N2 to NH3 (−0.13 V vs. NHE),41 while their driving force for H2O oxidation is relatively weak. Meanwhile, the photogenerated holes in BiVO4 can trigger H2O photooxidation to O2 (0.82 V vs. NHE, pH = 7), while they are insufficient for the photoreduction of N2. In addition, the Fermi levels were also estimated according to the UPS spectra, to be −4.62 eV and −5.27 eV (vs. vacuum) for VS-MoS2 and BiVO4, respectively. These results reveal that both VS-MoS2 and BiVO4 are n-type semiconductors, consistent with the results from the Mott–Schottky tests, where the slopes of Mott–Schottky curves for both BiVO4 and VS-MoS2 are positive (Fig. S6†). From the above results and analysis, we can preliminarily deduce that, given the higher Fermi level of VS-MoS2 compared to that of BiVO4, the free electrons in VS-MoS2 would spontaneously flow to BiVO4 to establish a Fermi level equilibrium upon contact (Fig. S7(I–II)†). A built-in electric field and band bending of the interface in BiVO4/VS-MoS2 would be formed. Driven by the interfacial built-in electric field, photogenerated electrons in BiVO4 would recombine with the photogenerated holes in VS-MoS2 (Fig. S7(III)†), facilitating the formation of the S-scheme charge transfer pathway.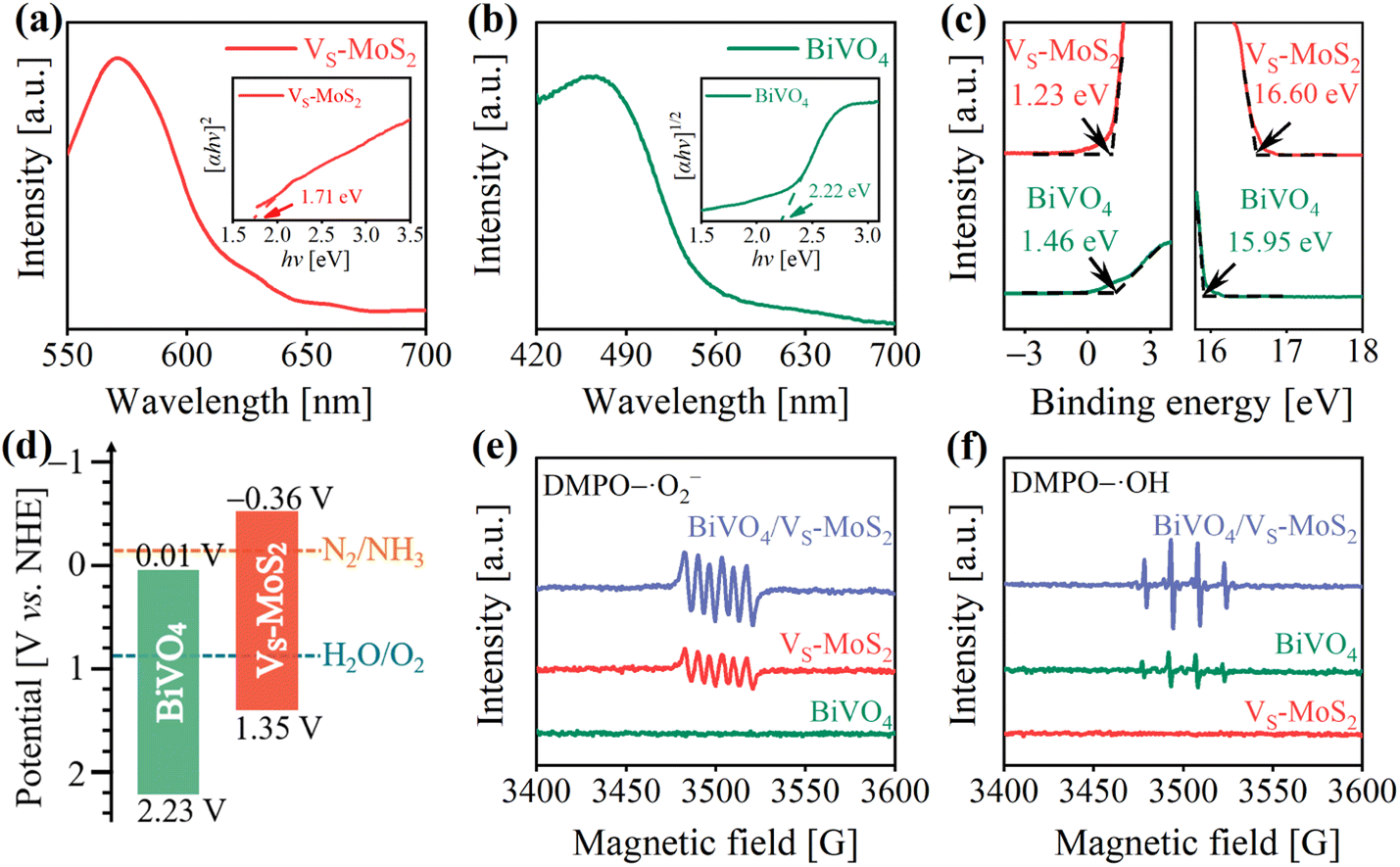 | ||
| Fig. 2 (a and b) UV-vis DRS spectra of VS-MoS2 and BiVO4. The insets show the corresponding Tauc plots. (c) UPS spectra of VS-MoS2 and BiVO4. (d) Energy band structures of VS-MoS2 and BiVO4 (pH = 7). EPR signals of (e) DMPO–·O2− and (f) DMPO–·OH over VS-MoS2, BiVO4, and BiVO4/VS-MoS2 under illumination. | ||
To scrutinize the S-scheme charge transfer pathway in the BiVO4/VS-MoS2 heterojunction, in situ irradiated XPS (ISI-XPS) spectra were first measured to detect photoinduced changes in the electron cloud density around the nuclei of elements. As depicted in Fig. S8a and b,† the binding energies of Mo 3d and V 2p in BiVO4/VS-MoS2 demonstrate a negative displacement (∼0.40 eV) and positive movement (∼0.30 eV) under illumination, respectively, compared with those in the dark. These binding energy changes indicate the accumulation of photogenerated electrons on VS-MoS2 and photogenerated holes on BiVO4, revealing the occurrence of S-scheme interfacial charge transfer within the BiVO4/VS-MoS2 heterojunction. To further confirm this S-scheme interfacial charge transfer mechanism in the BiVO4/VS-MoS2 heterojunction, the EPR spectra were recorded using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the trapping agent.42 In the case of pure VS-MoS2, only the DMPO–·O2− characteristic signal can be identified (Fig. 2e), as its EVB potential is insufficient to drive the oxidation of H2O to ·OH. Conversely, in pure BiVO4, only the DMPO–·OH characteristic signal can be recognized (Fig. 2f), due to the insufficient driving force of photogenerated electrons in ECB to trigger the reduction of O2 to ·O2− (−0.33 V vs. NHE). For the BiVO4/VS-MoS2 heterojunction, more obvious DMPO–·O2− and DMPO–·OH signals can be simultaneously identified, providing compelling evidence for the establishment of an interfacial S-scheme charge transport channel. That is to say, the photogenerated electrons in BiVO4 and the photogenerated holes in VS-MoS2 initially recombine through the interfacial S-scheme channel, leading to the accumulation of photogenerated holes on BiVO4 and photogenerated electrons on VS-MoS2. This spatial separation of charge carriers in the BiVO4/VS-MoS2 heterojunction would more effectively promote their participation in the redox reaction.
3.3. Photogenerated carrier evolution
In general, the evolution of photogenerated charges within photocatalysts plays a pivotal role in determining their photocatalytic activity.43,44 To evaluate the behavior of photogenerated carriers in BiVO4/VS-MoS2, steady-state photoluminescence (PL) spectrum measurements were first performed. As presented in Fig. 3a, the PL spectrum of BiVO4 displays an intrinsic emission band at ∼490 nm under 365 nm excitation, which is related to photogenerated electron–hole recombination. Upon the construction of the BiVO4/VS-MoS2 heterojunction, the PL spectrum exhibits significant quenching, indicating the effective transfer of photogenerated carriers between the BiVO4 nanocrystals and VS-MoS2 nanosheets. In addition, the photoelectrochemical properties of the samples were characterized to assess the potential positive influence of the construction of a heterostructure on charge evolution behavior. Notably, under light irradiation, BiVO4/VS-MoS2 exhibits a smaller charge-transport resistance compared with both pure BiVO4 and VS-MoS2 (Fig. 3b and Table S1†), indicating that the construction of the electric field in the heterojunction improves the charge transport characteristics. From the transient photocurrent (I–t) measurements (Fig. 3c), it is evident that BiVO4/VS-MoS2 has a higher current density than both BiVO4 and VS-MoS2, providing further evidence of expedited charge separation kinetics within the BiVO4/VS-MoS2 heterojunction. Moreover, to more intuitively reveal the space charge separation, Kelvin probe force microscopy (KPFM) was utilized to measure the surface photovoltage (SPV) response of photocatalysts under light. Atomic force microscope (AFM) images (Fig. 3d and f) present the morphology outline of VS-MoS2 and BiVO4/VS-MoS2 nanosheet clusters, which are basically consistent with the SEM results (Fig. 1b). Under illumination, the SPV response zone (Fig. 3e and g) emerges, aligning with the morphology outlines of the samples. These observations also indicate the spatial charge separation and redistribution after light irradiation.45 As shown in Fig. 3h, pure VS-MoS2 presents a feeble SPV signal under light irradiation, primarily due to rapid photogenerated carrier recombination. Benefiting from the construction of heterojunctions, BiVO4/VS-MoS2 displays a noticeable enhancement of the SPV response, of ∼100 mV, far surpassing that of pure VS-MoS2. Ultimately, these characterization studies underscore that the construction of heterojunctions can promote charge separation, and enhance the accumulation of electrons/holes on VS-MoS2 and BiVO4, respectively, which is beneficial for these carriers to participate in the subsequent multi-electron transfer processes during the photoreduction N2 reaction (Fig. 3i). | ||
| Fig. 3 (a) Steady-state photoluminescence spectra of BiVO4 and BiVO4/VS-MoS2. (b and c) EIS plots and I–t curves of BiVO4, VS-MoS2, and BiVO4/VS-MoS2 plotted at a bias potential of −0.4 V vs. Ag/AgCl. AFM height images of (d) VS-MoS2 and (f) BiVO4/VS-MoS2. SPV images of (e) VS-MoS2 and (g) BiVO4/VS-MoS2 under illumination. (h) Potential difference changes (ΔCPD) of VS-MoS2 and BiVO4/VS-MoS2 by subtracting the potential under dark conditions from that under illumination. (i) Schematic illustration of the photogenerated carrier separation. | ||
3.4. Photocatalytic N2 reduction activity
The photocatalytic N2 reduction reactions were conducted in a gas–solid reaction apparatus (Fig. S9†) containing N2 and H2O vapor under simulated sunlight irradiation (100 mW cm−2). To ensure the accuracy of our experiments, the samples were pretreated with a DMSO solution of KI. This step is crucial to remove any residual ligands introduced during the synthesis process, preventing any interference with the experimental results (Fig. S10,† details are described in the Experimental section). The photocatalytic activities were evaluated by evaluating the NH4+ production using the indophenol blue method (Fig. S11,† details are described in the Experimental section).46 As illustrated in Fig. S12,† a prominent absorption signal at ∼655 nm can be observed, indicating that N2 is effectively photoreduced to NH3 in the presence of light for BiVO4/VS-MoS2. The NH3 generation performances for the as-prepared photocatalysts are presented in Fig. 4a and S13.† For pure BiVO4, there is virtually no NH3 synthesis activity due to the insufficient driving force of electron energy in its conduction band (Fig. 2d). Meanwhile, VS-MoS2 exhibits a weak N2 photoreduction to NH3 activity, with a rate of 20.0 ± 1.1 μmol g−1 h−1, resulting from its insufficient thermodynamic driving force for H2O oxidation and serious charge recombination. Upon combining the VS-MoS2 nanosheets with the BiVO4 nanocrystals, the corresponding photocatalytic N2 reduction activity improves significantly, reaching 132.8 ± 4.2 μmol g−1 h−1 within 4 hours, which is nearly 7-fold that of pure VS-MoS2. The apparent quantum efficiency at 575 nm can reach 0.3% (Fig. S14†). Certainly, we also analyzed the NH4+ production by ion chromatography (Fig. S15†), and the results are in agreement with those obtained by the indophenol blue method. Obviously, this improvement of NH3 evolution activity can be attributed to the increased driving force for water oxidation and the enhanced separation of photogenerated carriers in the BiVO4/VS-MoS2 heterojunction, compared with pure VS-MoS2 nanosheets. By adjusting the mass ratio of BiVO4 and VS-MoS2 (Fig. S16†), the NH3 evolution activities of the samples were screened, in which BiVO4/VS-MoS2 with a mass ratio (BiVO4:VS-MoS2) of 1:3 exhibits the best performance (Fig. 4a).
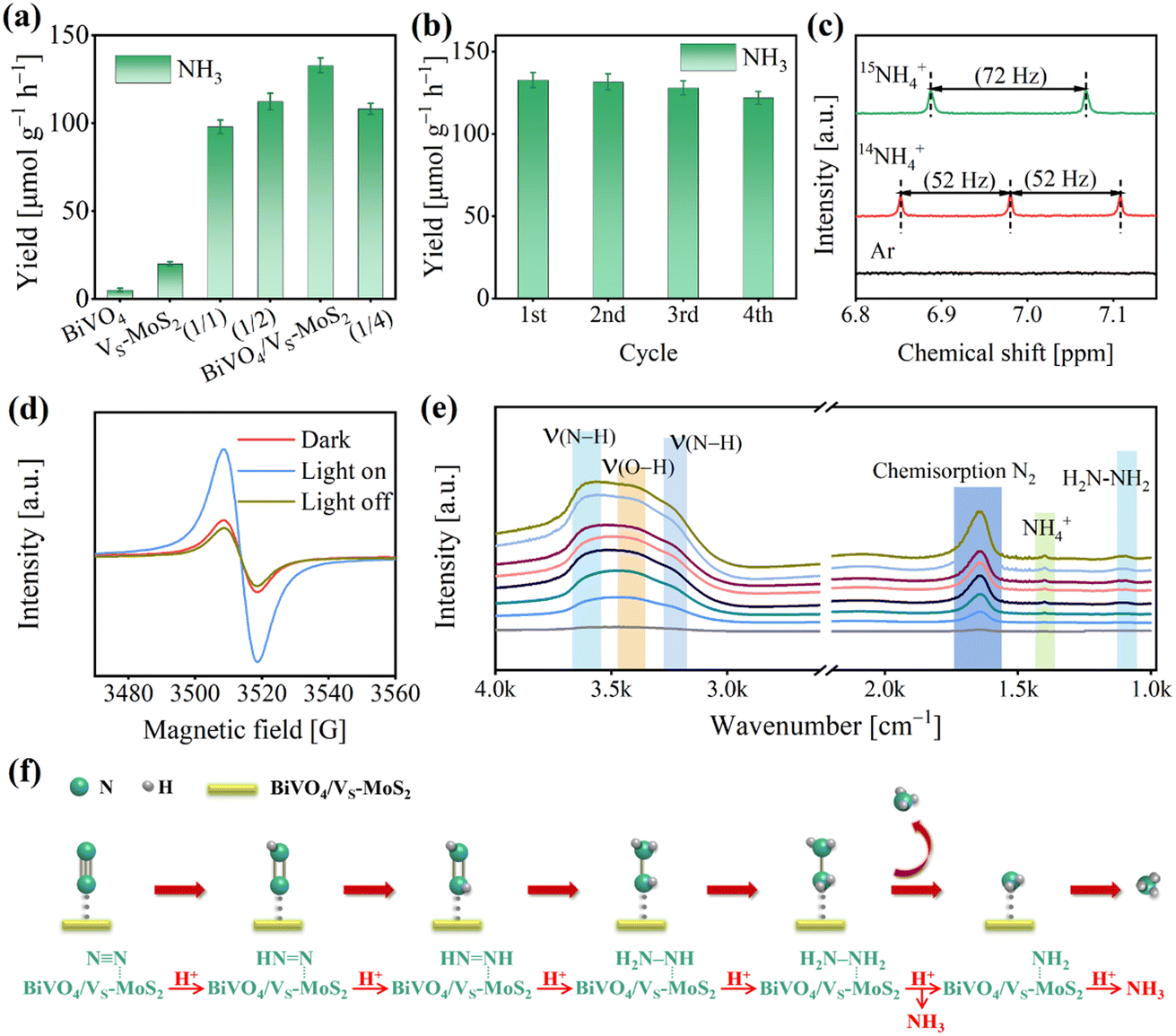 | ||
| Fig. 4 (a) Photocatalytic NH3 production rates for BiVO4, VS-MoS2, BiVO4/VS-MoS2, BiVO4/VS-MoS2 (1/1), BiVO4/VS-MoS2 (1/2), and BiVO4/VS-MoS2 (1/4) under 4 h light irradiation. (b) NH3 synthesis for four cyclic tests of BiVO4/VS-MoS2. (c) The 1H NMR spectra of products with 14N2 or 15N2 as the reaction atmosphere. (d) In situ EPR spectra of BiVO4/VS-MoS2 in the dark or under light irradiation in a N2 atmosphere or light off in a N2 atmosphere. (e) In situ DRIFTS spectra of the photoreduction N2 reaction. (f) The proposed reaction pathway of the photoreduction N2 reaction. | ||
Moreover, the O2 evolution activity of BiVO4/VS-MoS2 was also measured through a gas chromatograph, to be 110.0 ± 4.5 μmol g−1 h−1 (Fig. S17†). This result suggests that the rate of consumed electrons (Relectron) for N2 reduction to NH3 is nearly equal to the rate of consumed holes (Rhole) for water oxidation to O2, in accordance with the formulae of Relecreon = 3 × RNH3 and Rhole = 4 × RO2. This balance signifies the effective utilization of the photogenerated electrons and holes. Based on the above analysis, the whole photocatalytic reaction including N2 reduction and H2O oxidation by BiVO4/VS-MoS2 can be described as 2N2 + 6H2O → 4NH3 + 3O2. Additionally, the robustness of BiVO4/VS-MoS2 was evaluated by reemploying the photocatalyst in four cycles of tests (Fig. 4b), which reveals little decrement in activity. In addition, the XRD patterns (Fig. S18†) and TEM images (Fig. S19†) of BiVO4/VS-MoS2 after the photocatalytic reaction exhibit no obvious changes, confirming the good stability of the photocatalyst in this gas–solid photocatalytic system. The Mo 3d and Bi 4f XPS spectra of BiVO4/VS-MoS2 after catalysis show almost no significant changes, further suggesting the maintenance of the local coordination environment of the Bi and Mo sites.
To elucidate the origin of NH3 and O2 during the photocatalytic reaction, several controlled experiments were carried out using BiVO4/VS-MoS2 as the photocatalyst (Fig. S21†). In the absence of a photocatalyst (BiVO4/VS-MoS2), N2, light, or H2O, there is nearly no NH3 evolution, unequivocally demonstrating that the N2 reduction reaction is indeed a photocatalytic reaction using BiVO4/VS-MoS2 as the photocatalyst with H2O as the proton source. According to the characterization by the colorimetric method and ion chromatography (the details are described in the Experimental section), nearly no NO2− and NO3− signals in the product system can be detected before and after the photocatalytic reaction (Fig. S22 and S23†). In addition, we have further measured the N content in the obtained photocatalyst by EDS analysis (Fig. S24 and Table S2†), and no N atoms are present. These results confirm that NH3 remained unoxidized, and substantiate that NH3 originated solely from N2 reduction, excluding impurity conversion as a source. To further verify the origin of NH3 in the product system, isotope labeling experiments were performed using BiVO4/VS-MoS2 as the photocatalyst in atmospheres containing 15N2, 14N2, or Ar, respectively (Fig. S25†). More specifically, no NH4+ signal is detected under a Ar atmosphere in the nuclear magnetic resonance (NMR) spectra, while distinct signals belonging to 15NH4+ or 14NH4+ can be observed in a 15N2 or 14N2 atmosphere, respectively (Fig. 4c). These results directly confirm that NH4+ is indeed generated from the N2 in the system. In addition, the mass spectrometry analysis of the product (Fig. S26†) was also performed, revealing an 18O2 signal at m/z = 36, which confirms that O2 is produced from H2O oxidation.
To gain insight into the photocatalytic N2 reduction process of the BiVO4/VS-MoS2 heterojunction, we conducted further characterization to explore the active sites and monitor the evolution of intermediates during the photocatalytic reaction, by using in situ EPR spectra and in situ diffuse reflectance infrared Fourier transform spectra (DRIFTS), respectively. As presented in Fig. 4d, the intensity of the sulfur vacancy signal for BiVO4/VS-MoS2 exhibits an increment when exposed to light irradiation for 15 min, and decreases once the light was turned off for 10 min, suggesting that the sulfur vacancies facilitate the trapping of photogenerated electrons from the conduction band.47,48 The exposed Mo site at the sulfur vacancy is in a coordination unsaturated state, and can act as the photocatalytic active site bonding with N2. The trapped photogenerated electrons at sulfur vacancies can transfer to N2 molecules to achieve N2 reduction and transformation. As the photocatalytic N2 reduction reaction proceeded, a series of absorption signals were detected belonging to intermediates adsorbed on BiVO4/VS-MoS2 (Fig. 4e). Specifically, an obvious peak at 1648 cm−1 belonging to the chemisorbed N2 (–N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) appeared,49 which can be attributed to the efficient adsorption and activation of N2 by the Mo sites at the vacancies of the nanosheets.50 The peak at 3432 cm−1 is attributed to ν(O–H) of H2O, which serves as the proton source for N2 activation. The overlapped signal bands at 3230 cm−1 and 3555 cm−1 can be attributed to ν(N–H) stretching modes of NH3, and the peak at 1403 cm−1 can be attributed to NH4+ adsorption. The enhancement of these characteristic signals implies the progression of N2 activation and conversion to NH3via a multistep proton-coupled electron transfer (PCET) process over BiVO4/VS-MoS2. In addition, a peak at 1102 cm−1 can be identified, originating from the H2N–NH2 characteristic signal.51 Given that there is no hydrazine formation in the product (Fig. S27†), we thus speculate that the photocatalytic N2 fixation process on BiVO4/VS-MoS2 follows an associative alternating pathway (Fig. 4f). In this pathway, hydrogenation occurs alternatively on two N atoms, with the final step involving the cleavage of the N–N bond to generate the first NH3 molecule, followed by the last hydrogenation and another NH3 desorption.
N) appeared,49 which can be attributed to the efficient adsorption and activation of N2 by the Mo sites at the vacancies of the nanosheets.50 The peak at 3432 cm−1 is attributed to ν(O–H) of H2O, which serves as the proton source for N2 activation. The overlapped signal bands at 3230 cm−1 and 3555 cm−1 can be attributed to ν(N–H) stretching modes of NH3, and the peak at 1403 cm−1 can be attributed to NH4+ adsorption. The enhancement of these characteristic signals implies the progression of N2 activation and conversion to NH3via a multistep proton-coupled electron transfer (PCET) process over BiVO4/VS-MoS2. In addition, a peak at 1102 cm−1 can be identified, originating from the H2N–NH2 characteristic signal.51 Given that there is no hydrazine formation in the product (Fig. S27†), we thus speculate that the photocatalytic N2 fixation process on BiVO4/VS-MoS2 follows an associative alternating pathway (Fig. 4f). In this pathway, hydrogenation occurs alternatively on two N atoms, with the final step involving the cleavage of the N–N bond to generate the first NH3 molecule, followed by the last hydrogenation and another NH3 desorption.
4. Conclusions
In summary, we have successfully prepared an S-scheme heterojunction of BiVO4/VS-MoS2 by coupling the N2-activated component (VS-MoS2) and the water-oxidized module (BiVO4) to facilitate the process of photocatalytic N2 reduction. BiVO4/VS-MoS2 exhibits efficient NH3 synthesis activity using H2O as a proton source under illumination (100 mW cm−2), with an NH3 yield of 132.8 μmol g−1 h−1, nearly 7 times higher than that of pure VS-MoS2. Our experimental findings underscore the pivotal roles played by the VS-MoS2 nanosheet with sulfur-rich vacancies in enhancing N2 adsorption and activation. Furthermore, the construction of the S-scheme heterojunction substantially elevates the driving force for H2O oxidation and significantly improves charge separation within the system. Moreover, an alternate association pathway for N2 photoreduction reaction is proposed according to the in situ DRIFTS characterization. This study presents an efficient example for achieving the N2 conversion process under mild conditions.Author contributions
Han-Ying Luo: data curation, formal analysis, validation. Zhao-Lei Liu: data curation, formal analysis, validation. Meng-Ran Zhang: formal analysis, supervision. Yan-Fei Mu: validation, writing – original draft preparation, funding acquisition. Min Zhang: conceptualization, funding acquisition, supervision, validation, writing – reviewing and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (U21A20286 and 22305214), the National Key R&D Program of China (2022YFA1502902), the Natural Science Foundation of Tianjin City (17JCJQJC43800), the Jiangsu Funding Program for Excellent Postdoctoral Talent, the 111 Project of China (D17003), and the Tianjin Research Innovation Project for Postgraduate Students (2022SKY178).References
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- X.-B. Li, Z.-K. Xin, S.-G. Xia, X.-Y. Gao, C.-H. Tung and L.-Z. Wu, Chem. Soc. Rev., 2020, 49, 9028–9056 RSC.
- Y. Yang, S.-Q. Wang, H. Wen, T. Ye, J. Chen, C.-P. Li and M. Du, Angew. Chem., Int. Ed., 2019, 58, 15362–15366 CrossRef CAS.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- X. Zheng, Y. Yan, X. Li, Y. Liu and Y. Yao, J. Hazard. Mater., 2023, 446, 130679 CrossRef CAS PubMed.
- S. Wang, Y. Wang, T. C. Zhang, X. Jia and S. Yuan, Nanoscale, 2023, 15, 16219–16226 RSC.
- Y. Yu, Y. Li, Y. Fang, L. Wen, B. Tu and Y. Huang, Appl. Catal., B, 2024, 340, 123161 CrossRef CAS.
- Z. Zhao, H. Ren, D. Yang, Y. Han, J. Shi, K. An, Y. Chen, Y. Shi, W. Wang, J. Tan, X. Xin, Y. Zhang and Z. Jiang, ACS Catal., 2021, 11, 9986–9995 CrossRef CAS.
- S. H. W. Kok, J. Lee, W.-K. Chong, B.-J. Ng, X. Y. Kong, W.-J. Ong, S.-P. Chai and L.-L. Tan, J. Alloys Compd., 2023, 952, 170015 CrossRef CAS.
- J. Di, C. Chen, Y. Wu, Y. Zhao, C. Zhu, Y. Zhang, C. Wang, H. Chen, J. Xiong, M. Xu, J. Xia, J. Zhou, Y. Weng, L. Song, S. Li, W. Jiang and Z. Liu, Adv. Mater., 2022, 34, 2204959 CrossRef CAS.
- S. Zhang, Y. Zhao, R. Shi, C. Zhou, G. I. N. Waterhouse, Z. Wang, Y. Weng and T. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2554–2560 CrossRef CAS.
- S. Wang, X. Hai, X. Ding, K. Chang, Y. Xiang, X. Meng, Z. Yang, H. Chen and J. Ye, Adv. Mater., 2017, 29, 1701774 CrossRef.
- Z. Lu, H. Wang, Y. Tao, S. Zhu, W. Hao, X. Liu, Y. Min and J. Fan, Nanoscale, 2023, 15, 14847–14857 RSC.
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- Y. Xiong, B. Li, Y. Gu, T. Yan, Z. Ni, S. Li, J.-L. Zuo, J. Ma and Z. Jin, Nat. Chem., 2023, 15, 286–293 CrossRef CAS PubMed.
- S. Liu, Z. Teng, H. Liu, T. Wang, G. Wang, Q. Xu, X. Zhang, M. Jiang, C. Wang, W. Huang and H. Pang, Angew. Chem., Int. Ed., 2022, 61, e202207026 CrossRef CAS PubMed.
- Y. Lv, S.-W. Ke, Y. Gu, B. Tian, L. Tang, P. Ran, Y. Zhao, J. Ma, J.-L. Zuo and M. Ding, Angew. Chem., Int. Ed., 2023, 62, e202305246 CrossRef CAS.
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 1977, 99, 7189–7193 CrossRef CAS.
- S. Cao, N. Zhou, F. Gao, H. Chen and F. Jiang, Appl. Catal., B, 2017, 218, 600–610 CrossRef CAS.
- L. Zhang, S. Hou, T. Wang, S. Liu, X. Gao, C. Wang and G. Wang, Small, 2022, 18, 2202252 CrossRef CAS PubMed.
- S. Hu, Y. Li, F. Li, Z. Fan, H. Ma, W. Li and X. Kang, ACS Sustainable Chem. Eng., 2016, 4, 2269–2278 CrossRef CAS.
- Y. Zhao, Y. Zhao, G. I. N. Waterhouse, L. Zheng, X. Cao, F. Teng, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, Adv. Mater., 2017, 29, 1703828 CrossRef.
- H. Su, L. Chen, Y. Chen, R. Si, Y. Wu, X. Wu, Z. Geng, W. Zhang and J. Zeng, Angew. Chem., Int. Ed., 2020, 59, 20411–20416 CrossRef CAS PubMed.
- G. Zhan, F. Quan, Y. Yao, S. Zhao, X. Liu, H. Gu, Y. Huang, X. Liu, F. Jia and L. Zhang, Appl. Catal., B, 2023, 323, 122186 CrossRef CAS.
- Q. Li, Y. Shi, Z. Wang, C. Liu, J. Bi, J. C. Yu and L. Wu, J. Colloid Interface Sci., 2023, 652, 1568–1577 CrossRef CAS.
- T. He, Z. Zhao, R. Liu, X. Liu, B. Ni, Y. Wei, Y. Wu, W. Yuan, H. Peng, Z. Jiang and Y. Zhao, J. Am. Chem. Soc., 2023, 145, 6057–6066 CrossRef CAS.
- X. Dong, K. Wang, Z. Cui, X. Shi, Z. Wang and F. Dong, Nano Res., 2023, 16, 6679–6686 CrossRef CAS.
- Z. Ai, M. Huang, D. Shi, M. Yang, H. Hu, B. Zhang, Y. Shao, J. Shen, Y. Wu and X. Hao, Appl. Catal., B, 2022, 315, 121577 CrossRef CAS.
- Y. Chen, M. Yu, G. Huang, Q. Chen and J. Bi, Small, 2022, 18, 2205388 CrossRef CAS.
- C. Cheng, J. Zhang, B. Zhu, G. Liang, L. Zhang and J. Yu, Angew. Chem., Int. Ed., 2023, 62, e202218688 CrossRef CAS.
- E. Aslan, M. K. Gonce, M. Z. Yigit, A. Sarilmaz, E. Stathatos, F. Ozel, M. Can and I. H. Patir, Appl. Catal., B, 2017, 210, 320–327 CrossRef CAS.
- Y. Zhang, J. Di, X. Zhu, M. Ji, C. Chen, Y. Liu, L. Li, T. Wei, H. Li and J. Xia, Appl. Catal., B, 2023, 323, 122148 CrossRef CAS.
- Y. Wang, S. Wang, J. Gan, J. Shen, Z. Zhang, H. Zheng and X. Wang, ACS Sustainable Chem. Eng., 2023, 11, 1962–1973 CrossRef CAS.
- Y. Lu, Y. Yang, X. Fan, Y. Li, D. Zhou, B. Cai, L. Wang, K. Fan and K. Zhang, Adv. Mater., 2022, 34, 2108178 CrossRef CAS.
- A. Shan, X. Teng, Y. Zhang, P. Zhang, Y. Xu, C. Liu, H. Li, H. Ye and R. Wang, Nano Energy, 2022, 94, 106913 CrossRef CAS.
- C. Liu, S. Mao, M. Shi, X. Hong, D. Wang, F. Wang, M. Xia and Q. Chen, Chem. Eng. J., 2022, 449, 137757 CrossRef CAS.
- X. Guo, E. Song, W. Zhao, S. Xu, W. Zhao, Y. Lei, Y. Fang, J. Liu and F. Huang, Nat. Commun., 2022, 13, 5954 CrossRef PubMed.
- P. Xia, X. Pan, S. Jiang, J. Yu, B. He, P. M. Ismail, W. Bai, J. Yang, L. Yang, H. Zhang, M. Cheng, H. Li, Q. Zhang, C. Xiao and Y. Xie, Adv. Mater., 2022, 34, 2200563 CrossRef CAS.
- Z. Li, G. Gu, S. Hu, X. Zou and G. Wu, Chin. J. Catal., 2019, 40, 1178–1186 CrossRef CAS.
- X. Xu, X. Liu, J. Zhao, D. Wu, Y. Du, T. Yan, N. Zhang, X. Ren and Q. Wei, J. Colloid Interface Sci., 2022, 606, 1374–1379 CrossRef CAS PubMed.
- C. Zhao, X. Li, L. Yue, X. Ren, S. Yuan, Z. Zeng, X. Hu, Y. Wu and Y. He, ACS Appl. Nano Mater., 2023, 6, 15709–15720 CrossRef CAS.
- Y.-F. Mu, C. Zhang, M.-R. Zhang, W. Zhang, M. Zhang and T.-B. Lu, ACS Appl. Mater. Interfaces, 2021, 13, 22314–22322 CrossRef CAS PubMed.
- R. Chen, D. Zhang, Z. Wang, D. Li, L. Zhang, X. Wang, F. Fan and C. Li, J. Am. Chem. Soc., 2023, 145, 4667–4674 CrossRef CAS.
- A. Kumar, M. Sharma, S. Sheoran, S. Jaiswal, A. Patra, S. Bhattacharya and V. Krishnan, Nanoscale, 2023, 15, 11667–11680 RSC.
- R. Chen, Z. Ren, Y. Liang, G. Zhang, T. Dittrich, R. Liu, Y. Liu, Y. Zhao, S. Pang, H. An, C. Ni, P. Zhou, K. Han, F. Fan and C. Li, Nature, 2022, 610, 296–301 CrossRef CAS PubMed.
- M.-M. Shi, D. Bao, B.-R. Wulan, Y.-H. Li, Y.-F. Zhang, J.-M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1606550 CrossRef PubMed.
- Z. Wang, J. Zhu, X. Zu, Y. Wu, S. Shang, P. Ling, P. Qiao, C. Liu, J. Hu, Y. Pan, J. Zhu, Y. Sun and Y. Xie, Angew. Chem., Int. Ed., 2022, 61, e202203249 CrossRef CAS.
- L. Luo, X. Han, K. Wang, Y. Xu, L. Xiong, J. Ma, Z. Guo and J. Tang, Nat. Commun., 2023, 14, 2690 CrossRef CAS.
- Y. Zhang, J. Di, X. Qian, M. Ji, Z. Tian, L. Ye, J. Zhao, S. Yin, H. Li and J. Xia, Appl. Catal., B, 2021, 299, 120680 CrossRef CAS.
- Y. Bo, H. Wang, Y. Lin, T. Yang, R. Ye, Y. Li, C. Hu, P. Du, Y. Hu, Z. Liu, R. Long, C. Gao, B. Ye, L. Song, X. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2021, 60, 16085–16092 CrossRef CAS.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, J. Am. Chem. Soc., 2018, 140, 1496–1501 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, characterization approaches, and some supporting data. See DOI: https://doi.org/10.1039/d3na01091k |
| ‡ Han-Ying Luo and Zhao-Lei Liu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |