
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient access to hexaaryl-substituted borazines in batch and continuous-flow†
Alireza
Nazari Khodadadi
,
Ejdi
Cela
,
Dario
Marchionni
,
Fan
Huang
,
Francesco
Ferlin
 and
Luigi
Vaccaro
*
and
Luigi
Vaccaro
*
Laboratory of Green S.O.C. – Dipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, Via Elce di Sotto 8, 06123 – Perugia, Italy. E-mail: luigi.vaccaro@unipg.it; Web: https://greensoc.chm.unipg.it/
First published on 23rd May 2024
Abstract
Borazine-doped π-conjugated frameworks have significantly contributed to advancing the application of boron–nitrogen-doped organic materials. However, the challenging synthetic procedure has imposed limits on progress. In the case of hexaaryl-substituted borazines (HABs), the instability of B,B′,B′′-trichloro-N,N′,N′′-triarylborazine (TCB) due to the high reactivity of the boron site necessitates the synthesis and nucleophilic substitution of the chlorinated borazine under strict avoidance of moisture and oxygen to obtain HABs. Moving toward an ideal more sustainable synthesis, the first continuous flow process for the synthesis of HABs has been developed, enabling fast and safe boron arylation. The new process facilitates the neutralization and cleanup of the TCB solution from acidic by-products by utilizing an inorganic scavenger. BY making use of bio-derived 2-MeTHF as reaction medium, the common wasteful isolation procedure of HABs can be avoided, thus allowing not only simplifying the preparation of substituted borazines in high yields but also minimizing its environmental impact and improving its safety profile.
Introduction
Borazine (H3B3N3H3), discovered by Stock and Pohland in 1926,1 is the isoelectronic and (partially) isostructural inorganic analogue of benzene in which C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds are replaced by boron–nitrogen (B–N) bonds.2,3 The B–N bond causes weaker cyclic π-electron delocalization in borazine4 and stronger polarity, thus increasing the HOMO–LUMO gap, delivering unique optoelectronic properties in this class of molecules.5–9 These properties make borazine and its derivatives valuable molecular frameworks that can be incorporated as dopants into organic materials to modify their electronic and optical properties.10–14
C bonds are replaced by boron–nitrogen (B–N) bonds.2,3 The B–N bond causes weaker cyclic π-electron delocalization in borazine4 and stronger polarity, thus increasing the HOMO–LUMO gap, delivering unique optoelectronic properties in this class of molecules.5–9 These properties make borazine and its derivatives valuable molecular frameworks that can be incorporated as dopants into organic materials to modify their electronic and optical properties.10–14
In addition to hexagonal boron–nitrogen–carbon (h-BNC) and hexagonal boron–nitrogen (h-BN) sheets, the synthesis of borazine-doped organic architectures with acceptable thermal and hydrolytic stability has given rise to a new class of materials in organic electronics that is growing rapidly.15–18 Significant examples of hexaaryl-substituted borazine (HAB) are shown in Fig. 1, where borazine is substituted by aromatic moieties. To explore the optoelectronic properties of HABs, hexaphenylborazine and B-trimesityl-N-triphenylborazine (Fig. 1, HAB1 and HAB3 respectively) were incorporated into light-emitting diodes (LEDs) and light-emitting electrochemical cells (LECs).
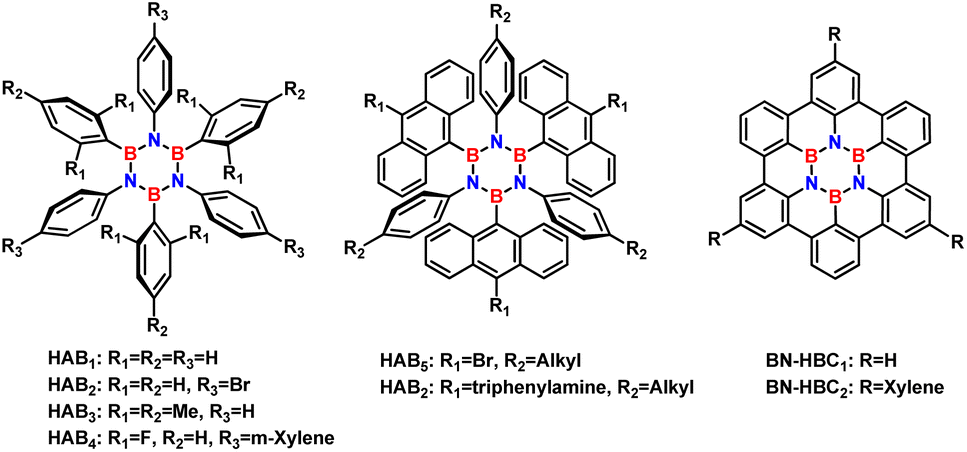 | ||
| Fig. 1 Aryl-substituted borazines with improved photophysical properties compared to their all-carbon analogues. | ||
Development of a facile synthetic methodology to access organic borazines in an efficient and potentially larger scale, may help the progress of their application in different fields of fundamental and applied interest.
From the synthetic point of view, dehydrogenation19,20 using diborane or metal borohydride, and (cyclo)condensation10,21 with BX3, are among the most widely relevant strategies for the preparation of N-aryl substituted borazines. Subsequently, access to HABs can be achieved by functionalizing chlorinated boron atoms using arylstannanes22via metal-catalysis or nucleophilic substitutions with organolithium or organomagnesium.23
This latter approach for the synthesis of N-substituted borazine derivatives is the most fundamental method that consists in the condensation reaction in which an amine and usually a boron halide are used to generate B,B′,B′′-trichloro-N,N′,N′′-triphenylborazole (TCB) intermediate (Fig. 2) with an active B-halide site ready for further derivatization.24 The use of boron trichloride does not only facilitate dehydrohalogenation (in comparison with boron trifluoride),24 but also improves selectivity for the ring closure compared to the corresponding boron tribromide.25
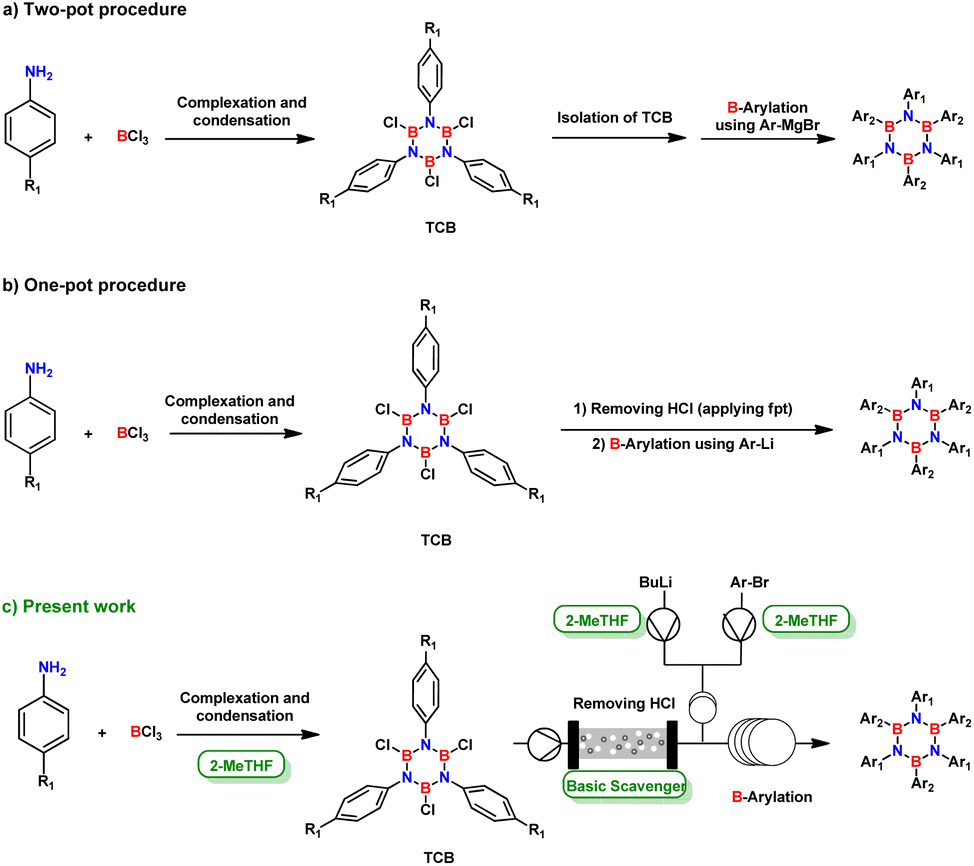 | ||
| Fig. 2 Evolution of common synthetic routes for obtaining HABs by condensation/nucleophilic substitution. | ||
In 1958, Stafiej first reported a two-pot synthetic procedure to obtain hexaphenylborazine via the TCB prepared in 73% yield by reacting boron trichloride and aniline in toluene.21 The procedure proceeded after the isolation of TCB from the reaction mixture and its reaction with Ph–Li or Ph–MgBr.21 The yields of the B-arylation step were 18% or 65%, respectively (Fig. 2a). Despite the low productivity and difficulties in handling sensitive intermediates, this procedure was the most versatile tool for the preparation of alkyl/aryl substituted borazines until 2005, when Yamaguchi and coworkers developed a one-pot protocol, which was further optimized by Bonifazi.18,26 In this protocol, TCB was treated without isolation from the mixture14 and the main impurity, hydrochloric acid, was removed by freeze–pump–thaw procedure (Fig. 2b).13,26
Although this approach improved the initial procedure, it still required strictly controlled conditions to achieve moderate or satisfactory yields.
Therefore, for these reasons, we have decided to define a protocol for the safe, faster, and productive synthesis of HABs.
Our final goal is also to disclose a protocol that could possibly simplify access to a larger-scale production of HABs libraries.
The protocol should be able to eliminate the risks and difficulties associated with handling sensitive intermediates, low productivity, long reaction time, the use of petrol-based reaction media, and potentially facilitate scaling up the process.
At this aim, we have directed our attention and efforts toward implementing current batch syntheses into an inherently more sustainable continuous flow protocol.27–33
Efficient heat and mass transfer, advanced mixing efficiency, unique scalability, and reaction control, along with the ability to operate under safer conditions are some of the useful advantages of continuous flow processing34–39 that we intended to exploit within this work.
Besides, to the best of our knowledge, there is no example on the use of flow conditions in the fundamental processes based on boron arylation. Therefore, the present study represents the first approach in this direction (Fig. 2c).
We believe that a significant advance in sustainability for the specific synthesis of HABs can be achieved by combining the advantages of flow chemistry with the possibility of using an environmentally benign reaction medium.40–45
Results and discussion
We have initially studied the batch-to-flow translation by studying in detail the two-pot consecutive batch procedure (Fig. 2b). The synthesis of TCB using BCl3 has been clearly explained by Bettinger and co-workers.46 Briefly, it involves complexation and condensation steps in which the combination of the amine and boron sources, at low temperature, leads to the formation of a RNH2·BCl3 complex. Then, the process proceeds via the unimolecular ring closure47 or dehydrochlorination48 of the complex at high temperature towards the (cyclo)condensation step. Evidently, the formation of TCB is accompanied by the formation of large amounts of HCl, which must be removed before its reaction with a nucleophile (e.g. organolithium or organomagnesium).In the above-mentioned optimized procedures,14,18,26 repetitive freeze pump thawing is used to remove the acid but this approach cannot be effective in a continuous-flow reactor.
Another critical point for batch-to-flow transition is the solubility of TCB intermediate as it strongly depends on the substituted aniline utilized. Indeed, most of the differently substituted TCBs herein synthesized are insoluble at the low temperatures required for the second step, which is the addition of nucleophile. This condition must be avoided when working in flow, as TCB precipitation will inevitably clog the system. To solve these issues, we planned to study both the adoption of a heterogeneous scavenger capable of removing the HCl formed in flow and the efficiency of the process in the presence of an adequate co-solvent capable of dissolving TCB to pass it through the flow system.
Screening of co-solvent
The solubility of TCB derivatives in nonpolar solvents is generally lower compared to polar or moderately polar solvents like ethereal solvents. However, the key reason for synthesizing TCB in a nonpolar solvent such as toluene is to promote the easy thermal dehydrochlorination of the Lewis acid–base adduct, leading to an enhanced yield of TCB formation.49Therefore, we decided that the best approach is to add the co-solvent only after the formation of TCB to eliminate its negative impact on its formation. Type and quantity of co-solvent has been carefully studied to control its influence on the subsequent aryllithium functionalization step.
Results are summarized in Table 1. Nevertheless, acknowledging that a higher concentration can have a positive impact on the TCB functionalization step, opting for the smallest volume of co-solvent (determined through stepwise cooling down and solubilization) was deemed preferable. This choice was evident in the case of DME, where an increase in the amount was observed to negatively affect the reaction's productivity.
|
|
||
|---|---|---|
| Entry | Reaction medium | Yieldb (%) |
| a Reaction conditions: aniline (3.3 mmol, 1.0 equiv.); BCl3 (1.2 equiv.), Mes–Li prepared with treatment of mesitylbromide (5.28 mmol) and n-BuLi (8.0 mmol). b Isolated yield over two steps. Further details are explained in ESI.† | ||
| 1 | – (Control) | 49 |
| 2 | Tetrahydrofuran | 31 |
| 3 | Cyclopentyl methyl ether | 27 |
| 4 | Anisole | 38 |
| 5 | 2-MeTHF | 45 |
| 6 | 1,2-Dimethoxyethane (DME) | 45 |
| 7 | Diethyl ether | 19 |
| 8 | o-Dichlorobenzene | <10 |
| 9 | Dichloromethane | Trace |
| 10 | 1,2-Dichloroethane | <10 |

Considering the acid stability of the co-solvent as well as moisture content, CPME, and 2-MeTHF are the best candidates for our purpose (compared to DME, THF, and DEE). Interestingly, 2-MeTHF was selected as the very promising choice as giving almost comparable yields against the control experiment while unfortunately CPME negatively affected the final yield of HAB.
Screening of heterogeneous basic scavengers for HCl elimination
The presence of HCl can terminate the functionalization of TCB with organolithium. From a practical standpoint, installing a cartridge filled with a basic scavenger can remove the unwanted acid. What limits scavenger screening is the sensitivity of the intermediate, where an active boron site can be attacked by various nucleophiles. In addition, the insolubility of protonated scavengers was considered since the reactivity of the organolithium can be negatively affected by these impurities. Bearing in mind sensitivity of TCB and organolithium species, different classes of basic scavengers were investigated, including polymer-supported organic bases, clay minerals, and inorganic antiacids (Table 2).|
|
||
|---|---|---|
| Entry | Reaction medium | Yieldb (%) |
| a Reaction conditions: aniline (3.3 mmol, 1.0 equiv.); BCl3 (1.2 equiv.), Mes–Li prepared with treatment of mesitylbromide (5.28 mmol) and n-BuLi (8.0 mmol), 2-MeTHF (10.0 mL), scavenger (1.0 g dispersed in quartz beads 1 mm). b Isolated yield over two steps. Further details are explained in ESI.† | ||
| 1 | – (Control) | 49 |
| 2 | Diethylaminomethyl–polystyrene | Trace |
| 3 | Amberlyst A21 free base | <10 |
| 4 | PS-tetraalkylammonium carbonate | Trace |
| 5 | Di(1H-imidazol-1-yl) butane (BisIm) | 0 |
| 6 | Montmorillonite K10 | <10 |
| 7 | Montmorillonite, surface modified | 0 |
| 8 | Hydrotalcite | <5 |
| 9 | Basic aluminum oxide | Trace |
| 10 | Calcium carbonate | 52 |
| 11 | Sodium carbonate | Trace |

Through this experimentation, we experienced that inert quartz beads can be used to disperse the scavenger and regulate the movement and residence time of the solution. To investigate the compatibility of TCB with the scavenger, the sensitive intermediate was analyzed by 11B NMR before and after the scavenging stage.
The quest for an efficient scavenger commenced with the screening of polymeric organic bases (Table 2, entries 1–4), including diethylaminomethyl polystyrene, Amberlyst A21 free base, and PS–tetraalkylammonium carbonate. However, the performance of the selected amine-based polymeric bases proved unimpressive, with issues such as leaching of the base from the support and trapping of TCB. Alternatively, the use of an organic base that is not soluble in the reaction media was explored (Table 2, entry 5). Di(1H-imidazol-1-yl) butane (BisIm) was synthesized ad-hoc for investigation.50 We speculated that, the absence of polymer support and the presence of the imidazole moiety could positively impact HCl removal compared to PS-NR2. However, a non-compatibility with TCB was observed which led to a negative result.
Then, the use of commercial inorganic basic materials was explored (Table 2, entries 6–11). The main problem with the use of these scavengers is the water content. Although the moisture content was minimized by long-term drying, TCB was degraded after passing through these scavengers. Among all the bases tested, calcium carbonate, due to its compatibility with TCB, insolubility in the reaction media, and efficient HCl removal, was found to be the best candidate to function as an acid scavenger leading to good results even in comparison with control experiments. The improvement of porosity by dispersing dry CaCO3 in quartz beads lead to a 52% yield in the synthesis of (Mes–B–N–Ph)3. To be accurate, 0.5 and 1.0 mm quartz beads were tested in different portions to control the residence time of the solution in the scavenging medium, and the amount of scavenger was also minimized by designing a reaction model in which the HCl concentration and the solvent mixture were the same as the TCB mixture.
Development of the continuous flow process
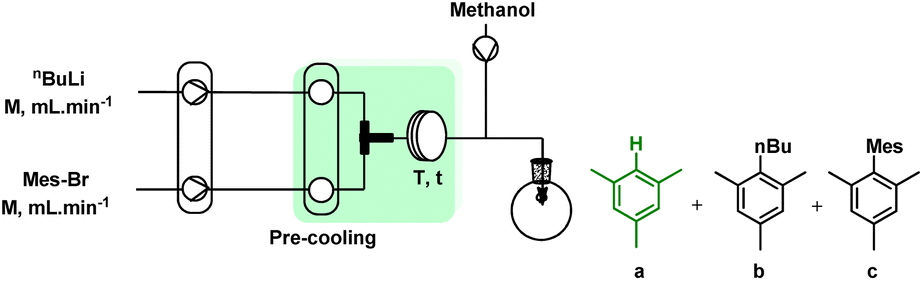
To improve the productivity of the reaction and minimize the waste associated, we focused on the development of the continuous flow protocol. Initially, we proposed a modular flow system to produce both TCB and organolithium in two separate reactors. The target compound will be eventually obtained by merging these two flow procedures. Despite the adoption of different flow conditions and reactor setups, yield of TCB did not exceed 15%. This confirmed again that the synthesis of TCB in a closed system cannot be satisfactory due to the inefficient removal of HCl which can substantially hamper the condensation of RNH2·BCl3 complex.46The subsequent development started with an examination of the bromine–lithium exchange reaction in a flow (labelled R-A). First, a Br/Li exchange reaction model between n-BuLi and mesityl bromide followed by protonation with MeOH was considered to optimise the influencing parameters such as residence time, molar ratio of reagents, concentration, and flow rates. In addition to the conversion of mesityl bromide, the formation of mesitylene was also considered to confirm the minimum formation of homocoupling and n-butyl mesityl formation. The reactor type, mixer, and concentration were chosen to achieve the highest possible flow rate, and concentration for a high throughput system. Regarding the lithiation agent, there was no significant difference between n-BuLi and tBuLi, while the molar ratio (changes in concentration and flow rate) significantly changed the exchange results.
In addition, the optimal molar ratio of n-BuLi to Mes–Br was found to be 1.0, whereas in batch systems the use of 2.1 equivalents of tBuLi or 1.2 equivalents of n-BuLi at −78 °C was reported under strictly controlled conditions for a limited scale.13,14 Concerning both bromine/lithium exchange and aryl lithium decomposition of mesityl bromide, the residence time was optimized, and as shown in Table 3, less mesitylene formation was observed with longer residence times.
|
|
|||||
|---|---|---|---|---|---|
| # | F n-BuLi (mL min−1) | F Mes-Br (mL min−1) | Molar ratio | Residence time (s) | Yield of ab (%) |
| a Reaction conditions: solution of mesityl bromide (0.18 M in THF) and n-BuLi (0.5 M in hexane) were introduced to the flow system fabricated by PTFE tube (∅i.d.: 1 mm) at 0 °C using high-pressure syringe pump. b Yield of mesitylene determined by GC. c Using t-BuLi instead of n-BuLi. Further details are explained in ESI.† | |||||
| 1 | 1.0 | 3.47 | 0.8 | 10.54 | 28 |
| 2 | 1.2 | 3.33 | 1.0 | 10.39 | 84.4 |
| 3 | 1.2 | 3.33 | 1.0 | 10.39 | 81.4c |
| 4 | 1.4 | 3.24 | 1.2 | 10.15 | 71.8 |
| 5 | 1.6 | 3.17 | 1.4 | 9.87 | 46 |
| 6 | 1.2 | 3.33 | 1.0 | 8.32 | 58.9 |
| 7 | 1.2 | 3.33 | 1.0 | 12.47 | 38.4 |
| 8 | 1.5 | 4.16 | 1.0 | 8.32 | 93 |
| 9 | 1.8 | 5.0 | 1.0 | 6.92 | >99 |
| 10 | 2.0 | 5.56 | 1.0 | 6.23 | >99 |
After the production of aryl lithium in reactor-A was optimized, this was intently delivered to the second reactor (labelled R-B), where the TCB solution was subjected to arylation and the formation of HAB. At that time, with the intention to optimize the final isolation of the product while preserving the waste minimization, we reasonably thought that a good strategy could simplify as much as possible the composition of the mixture in terms of reaction media.
Considering waste minimization, we replaced the THF used to dilute the aryl-bromide with 2-MeTHF while keeping the same final molar concentration (0.18 M). Within this refinement, as 2-MeTHF is almost insoluble in water, we could also avoid wasteful extraction procedures.
Continuing with the process optimization, the TCB solution was cleaned up by passing it through an Omnifit column filled with calcium carbonate dispersed in quartz beads. To control the flow rate, a backpressure regulator was installed upstream of the second micromixer (M2). Treatment of TCB with aryl lithium under a continuous flow regime was more complicated, as triple arylation of TCB without degradation appeared to be difficult. Initial studies showed low product formation at residence times below 32 seconds (Table 4, entries 1–4) using a T-mixer with 2.0 mm of internal diameter, and unlike batch process arylation of TCB, where the reaction should start at a low temperature, the reaction in the continuous flow process at low temperature was found to furnish HAB in low yield (obtained 17% and 18% at −15 and 0 °C, respectively).
|
|
|||||
|---|---|---|---|---|---|
| # | F TCB (mL min−1) | [TCB] (mol L−1) | Temp. (°C) | Residence time(s) | Yieldb (%) |
| a Reaction conditions: solution of TCB(B,B′,B′′-trichloro-N,N′,N′′-tri(4-tertbutylphenyl), 0.05 M in toluene/heptane/2-MeTHF) produced from aniline derivatives (1.0 equiv.); BCl3 (1.2 equiv.) was introduced using high-pressure syringe pumps to the flow system fabricated by PEEK Tee (ID: 0.5 mm) and PTFE tube (∅i.d.: 1 mm) at r.t. and Mes–Li preparation reactor was same as Table 3. b Isolated yield over two steps. c Using T-mixer with ID: 2.0 mm, residence time was altered by changing tube length, tube diameter, or flow rate. | |||||
| 1 | 2.0 | 0.050 | −15 | 32 | 17c |
| 2 | 2.0 | 0.050 | 0 | 32 | 18c |
| 3 | 2.0 | 0.050 | r. t. | 19 | 19c |
| 4 | 2.0 | 0.050 | r. t. | 26 | 39c |
| 5 | 2.0 | 0.050 | r. t. | 42 | 64 |
| 6 | 1.8 | 0.050 | r. t. | 80 | 74 |
| 7 | 2.0 | 0.044 | r. t. | 42 | 35 |
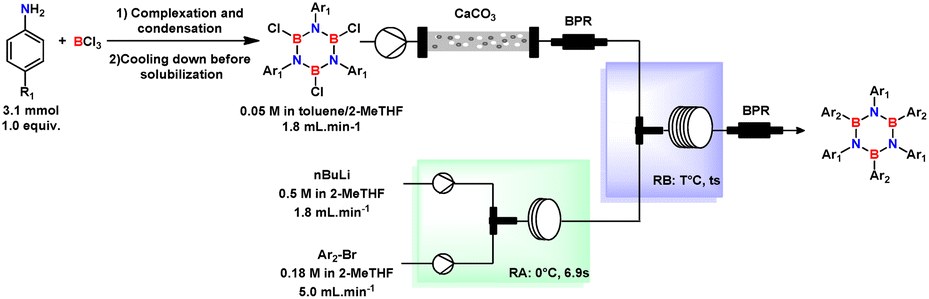
By increasing mixing efficiency with a T-shaped mixer with a smaller inner diameter (∅i.d. = 0.5 mm) and the molar ratio of Ar–Li/TCB with a concomitant increasing in the temperature for the B-arylation stage (R-B) to room temperature (23 °C) improved the reaction efficiency of HAB formation (Table 4, entry 5), indicating that the substitution of boron by the aryl lithium intermediate was enhanced by more efficient mixing of the solutions. Not surprisingly, we also confirmed that lowering the concentration of TCB led to a lower yield of HAB (entry 6 compared to 7).
Having established the feasibility and soundness of the flow system, the scope was broadened by synthesizing various TCBs (B,B′,B′′- trichloro-N,N′,N′′-tri(4-substituted phenyl)) and aryl bromides (Fig. 3).
 | ||
| Fig. 3 HABs synthetized under continuous flow process. Reaction conditions: Ar–Br (0.18 M in 2-MeTHF), n-BuLi (0.5 M in hexane/2-MeTHF), and TCB solution (0.05 M in toluene/heptane/2-MeTHF) produced from aniline derivatives (1.0 equiv.); BCl3 (1.2 equiv.) were introduced to the flow system fabricated by PEEK Tee (ID: 0.5 mm) and PTFE tube (∅i.d.: 1 mm) using high-pressure syringe pump, isolated yield over two steps. | ||
1,3 dimethyl aryl bromides were mainly selected for the synthesis of hydrolytically stable HABs in which the empty p orbital of B is shielded by the presence of methyl groups.23 The system is well-suited for producing various substituted borazines, encompassing not only alkylated aryl rings. Remarkably, the continuous flow system was used for the synthesis of (B,B′,B′′-tri(2,6-difluorophenyl)-N,N′,N′′-tri(4-substituted phenyl)), which serves as a precursor for the synthesis of BN–HBCs. Slight modifications were made to prevent the formation of benzyne and to increase the boron arylation step, as shown in Fig. 4. Benzyne formation was minimized by increasing the mixing efficiency and shorter residence time in both Br–Li exchange and B-substitution reactors. Independent of the residence time, this reaction is predominantly affected by the reactor tube diameter. Therefore, implementing the usage of a PTFE tube with a smaller diameter (0.8 mm) proved essential for achieving the desired product.
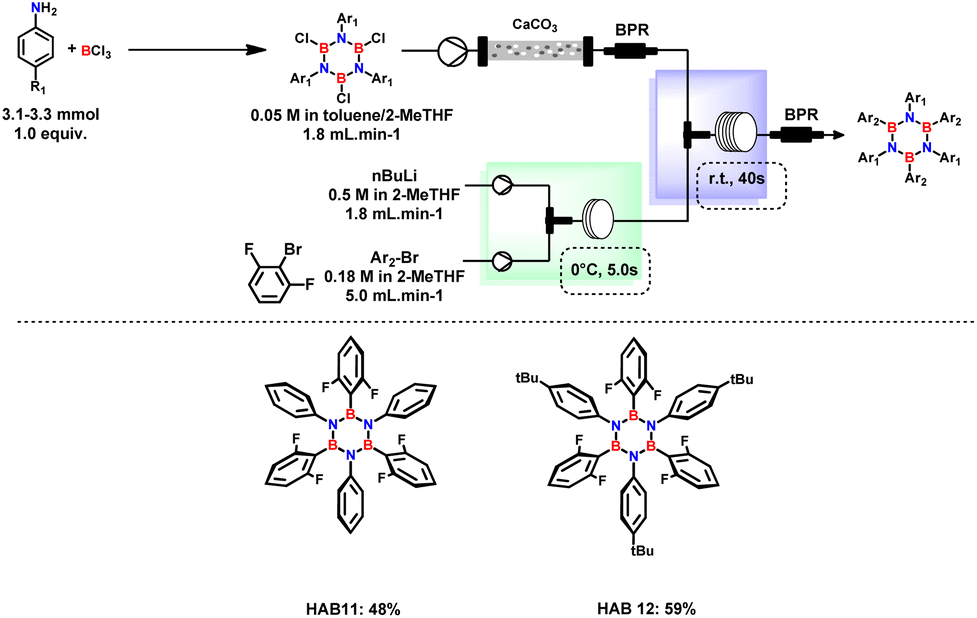 | ||
| Fig. 4 Orthofluoro-substituted HABs for the synthesis of BN–HBCs. Reaction conditions: Ar–Br (0.18 M in 2-MeTHF), n-BuLi (0.5 M in hexane/2-MeTHF), and TCB solution (0.05 M in toluene/heptane/2-MeTHF) produced from aniline derivatives (1.0 equiv.); BCl3 (1.2 equiv.) were introduced to the flow system fabricated by SS-micromixer and PTFE tube (∅i.d.: 0.8 mm) using high-pressure syringe pump, isolated yield over two steps. | ||
Aminated HABs are substrates of high interest because they display peculiar optical properties.7,14 In this context, by replacing n-BuLi with t-BuLi and with almost identical conditions (Fig. 5), aminated HABs were successfully synthesized, thus encompassing the usual problems encountered with the preparation of such compounds.
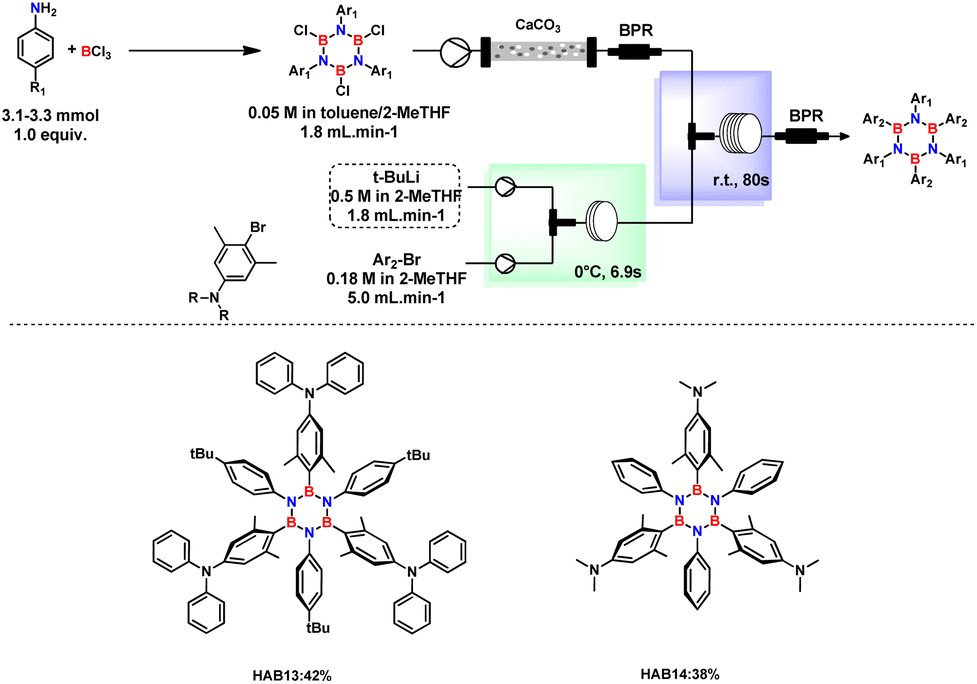 | ||
| Fig. 5 Synthesis of aminated HABs under continuous flow conditions. Reaction conditions: Ar–Br (0.18 M in 2-MeTHF) t-BuLi (0.5 M in pentane/2-MeTHF), and TCB solution (0.05 M in toluene/heptane/2-MeTHF) produced from aniline derivatives (1.0 equiv.); BCl3 (1.2 equiv.) were introduced to the flow system fabricated by PEEK Tee (ID: 0.5 mm) and PTFE tube (∅i.d.: 1 mm) using a high-pressure syringe pump. | ||
Importantly, the final work-up of all the HABs synthesized was very simple due to the presence of 2-MeTHF, hexane (from n-BuLi stock solution) heptane (from the BCl3 stock solution), and toluene (from the initial synthesis of TCB). The reaction mixture at the end of the process was quenched with a small amount of water (to remove salts formed during the process) and after separation, the mixture of solvent can be fractionally separated by both simple distillation or reduced pressure distillation allowing the recovery and reuse of the 2-MeTHF (up to 90% of mass recovered). Final addition of methanol led HAB to precipitate.
To provide quantitative insight into the improvement in terms of scalability and overall sustainability assessed by our continuous flow procedure, we also developed a fast and simple large-scale production by running the reaction for a long period with the same concentration and the same flow parameters. Using both small- and large-scale data we calculated the E-factor51 and safety/benign index52,53 (for the output solvent waste) for our procedures and we compared these results with the other batch literature processes (Fig. 6) (see ESI† for details on calculation).
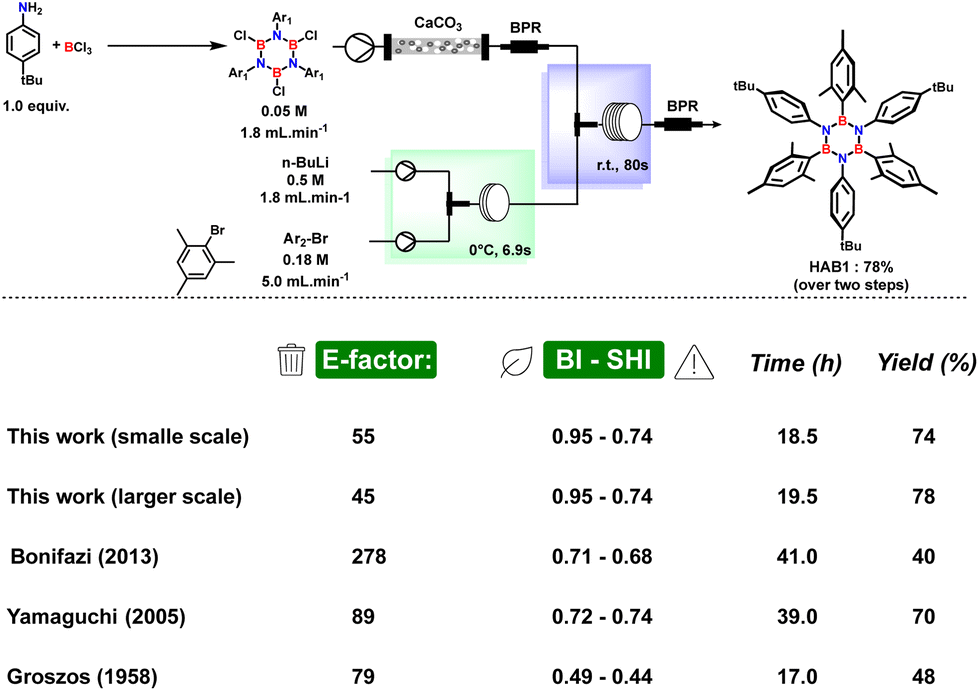 | ||
| Fig. 6 Comparison of E-factor, benign–safety/hazard index, reaction time and yields for this work in flow and other procedures in batch conditions. | ||
Although batch methods provide moderate to good yields, the environmental impact of such methods is significant due to the use of petrol-based solvents and wasteful work-up procedures leading to E-factor values in the range 79–278. On the other hand, the capabilities, and advantages of our method, which have been explained in detail, are undeniable allowing an E-factor of 55 for the small-scale process which can be further reduced to 45 in the large-scale with a safety and environmental profile far better than other known methods to synthesize HABs mainly ascribed to the stepwise utilization of bio-derived 2-MeTHF as reaction medium. A further qualitative comparison can be made considering the energy efficiency. In fact, by adopting our newly developed flow procedure, it is possible to completely avoid the energy-demanding freeze–pump–thaw cycles based on the use of liquid nitrogen, which is necessary to efficiently remove hydrochloric acid. The freeze–pump–thaw cycles are generally used in all the batch procedures in the literature. The avoidance of freeze–pump–thaw cycles is a crucial advantage when considering a scaling-up of a common batch procedure. The efficiency of this procedure can be more difficult at a larger scale and almost impossible. In contrast, the flow procedure is scalable just by multiplying or increasing the size of the reactor.
Conclusion
In summary, we have demonstrated a flow-based boron arylation and synthesis of aryl-substituted borazines using a procedure that requires limited manual handling of reagents or intermediates. In addition, the use of scavenger-filled cartridges allows reaction media to be cleaned as part of the continuous process, which plays a critical role in reaction productivity as well as large-scale productions. This represents a significant improvement over existing protocols that require strict precautions in both handling and safety to produce the same products with lower productivity. The outlined synthetic route also has the potential to be used for the lithiation of 2,6-difluorinated and 4-aminated aryl bromides to highlight the role of the continuous flow system in the preparation of HABs by nucleophilic substitution of boron chloride by organolithium nucleophiles.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
L. V. gratefully acknowledged for Università degli Studi di Perugia and EU through European Union's Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie entitled STiBNite (N_ 956923). D. M. and L. V. wish also to thank INPS for the Ph.D. grant and training. This work has been received funding also by the National Innovation Ecosystem grant ECS00000041 – VITALITY. The University of Perugia is acknowledged for financial support to the university project “Fondo Ricerca di Ateneo, edizione 2022”.References
- A. Stock and E. Pohland, Ber. Dtsch. Chem. Ges. B, 1926, 59, 2215–2223 CrossRef.
- S. W. Yang, H. Zhang, J. M. Soon, C. W. Lim, P. Wu and K. P. Loh, Diamond Relat. Mater., 2003, 12, 1194–1200 CrossRef CAS.
- H. F. Bettinger, T. Kar and E. Sánchez-García, J. Phys. Chem. A, 2009, 113, 3353–3359 CrossRef CAS PubMed.
- M. D. R. Merino-García, L. A. Soriano-Agueda, J. D. D. Guzmán-Hernández, D. Martínez-Otero, B. Landeros Rivera, F. Cortés-Guzmán, J. E. Barquera-Lozada and V. Jancik, Inorg. Chem., 2022, 61, 6785–6798 CrossRef PubMed.
- R. Ghiasi and S. Akbari, J. Chil. Chem. Soc., 2014, 59, 2666–2673 CrossRef CAS.
- D. Benker, T. M. Klapötke, G. Kuhn, J. Li and C. Miller, Heteroat. Chem., 2005, 16, 311–315 CrossRef CAS.
- I. H. T. Sham, C. C. Kwok, C. M. Che and N. Zhu, Chem. Commun., 2005, 3547–3549 RSC.
- S. Limberti, L. Emmett, A. Trandafir, G. Kociok-Köhna and G. D. Pantoş, Chem. Sci., 2019, 10, 9565–9570 RSC.
- J. K. Parker and S. R. Davis, J. Phys. Chem. A, 1997, 101, 9410–9414 CrossRef CAS.
- M. Maier, J. Klopf, C. Glasmacher, F. Fantuzzi, J. Bachmann, O. Ayhan, A. Koner, B. Engels and H. Helten, Chem. Commun., 2022, 58, 4464–4467 RSC.
- D. Marchionni, S. Basak, A. Nazari Khodadadi, A. Marrocchi and L. Vaccaro, Adv. Funct. Mater., 2023, 33, 2303635 CrossRef CAS.
- K. T. Jackson, M. G. Rabbani, T. E. Reich and H. M. El-Kaderi, Polym. Chem., 2011, 2, 2775–2777 RSC.
- S. Kervyn, O. Fenwick, F. Di Stasio, Y. S. Shin, J. Wouters, G. Accorsi, S. Osella, D. Beljonne, F. Cacialli and D. Bonifazi, Chem. – Eur. J., 2013, 19, 7771–7779 CrossRef CAS PubMed.
- A. Wakamiya, T. Ide and S. Yamaguchi, J. Am. Chem. Soc., 2005, 127, 14859–14866 CrossRef CAS PubMed.
- G. Imamura, C. W. Chang, Y. Nabae, M. A. Kakimoto, S. Miyata and K. Saiki, J. Phys. Chem. C, 2012, 116, 16305–16310 CrossRef CAS.
- C. Sánchez-Sánchez, S. Brüller, H. Sachdev, K. Müllen, M. Krieg, H. F. Bettinger, A. Nicolaï, V. Meunier, L. Talirz, R. Fasel and P. Ruffieux, ACS Nano, 2015, 9, 9228–9235 CrossRef PubMed.
- M. Krieg, F. Reicherter, P. Haiss, M. Ströbele, K. Eichele, M. J. Treanor, R. Schaub and H. F. Bettinger, Angew. Chem., Int. Ed., 2015, 54, 8284–8286 CrossRef CAS PubMed.
- J. Dosso, J. Tasseroul, F. Fasano, D. Marinelli, N. Biot, A. Fermi and D. Bonifazi, Angew. Chem., Int. Ed., 2017, 56, 4483–4487 CrossRef CAS PubMed.
- J. S. Li, C. R. Zhang, B. Li, F. Cao and S. Q. Wang, Eur. J. Inorg. Chem., 2010, 1763–1766 CrossRef CAS.
- M. Hasenbeck, J. Becker and U. Gellrich, Angew. Chem., Int. Ed., 2020, 59, 1590–1594 CrossRef CAS PubMed.
- S. J. Groszos and S. F. Stafiej, J. Am. Chem. Soc., 1958, 80, 1357–1360 CrossRef CAS.
- G. E. Rudebusch, L. N. Zakharov and S. Y. Liu, Angew. Chem., Int. Ed., 2013, 52, 9316–9319 CrossRef CAS PubMed.
- K. Xagasawa, Inorg. Chem., 1966, 5, 442–445 CrossRef.
- C. A. Brown and A. W. Laubengayer, J. Am. Chem. Soc., 1955, 77, 3699–3700 CrossRef CAS.
- S. Allaoud and B. Frange, Inorg. Chem., 1985, 24, 2520–2523 CrossRef CAS.
- D. Marinelli, F. Fasano, B. Najjari, N. Demitri and D. Bonifazi, J. Am. Chem. Soc., 2017, 139, 5503–5519 CrossRef CAS PubMed.
- V. Hessel, M. Escribà-Gelonch, J. Bricout, N. N. Tran, A. Anastasopoulou, F. Ferlin, F. Valentini, D. Lanari and L. Vaccaro, ACS Sustainable Chem. Eng., 2021, 9, 9508–9540 CrossRef CAS.
- D. Dallinger, B. Gutmann and C. O. Kappe, Acc. Chem. Res., 2020, 53, 1330–1341 CrossRef CAS PubMed.
- F. Ferlin, D. Lanari and L. Vaccaro, Green Chem., 2020, 22, 5937–5955 RSC.
- L. Rogers and K. F. Jensen, Green Chem., 2019, 21, 3481–3498 RSC.
- L. Vaccaro, Sustainable Flow Chemistry: Methods and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017 Search PubMed.
- J. A. M. Lummiss, P. D. Morse, R. L. Beingessner and T. F. Jamison, Chem. Rec., 2017, 17, 667–680 CrossRef CAS PubMed.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed.
- M. Guidi, P. H. Seeberger and K. Gilmore, Chem. Soc. Rev., 2020, 49, 8910–8932 RSC.
- A.-C. Bédard, A. Adamo, K. C. Aroh, M. G. Russell, A. A. Bedermann, J. Torosian, B. Yue, K. F. Jensen and T. F. Jamison, Science, 2018, 361, 1220–1225 CrossRef PubMed.
- M. B. Plutscack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef PubMed.
- L. Vaccaro, D. Lanari, A. Marrocchi and G. Strappaveccia, Green Chem., 2014, 16, 3680–3704 RSC.
- V. Hessel, D. Kralisch, N. Kockmann, T. Noël and Q. Wang, ChemSusChem, 2013, 6, 746–789 CrossRef CAS PubMed.
- F. Valentini, B. Di Erasmo, M. Ciani, S. Chen, Y. Gu and L. Vaccaro, Green Chem., 2024, 26, 4871–4879 RSC.
- G. Brufani, F. Valentini, G. Rossini, L. Rosignoli, Y. Gu, P. Liu and L. Vaccaro, Green Synth. Catal., 2023, 4, 154–159 CrossRef CAS.
- N. Salameh, F. Ferlin, F. Valentini, I. Anastasiou and L. Vaccaro, ACS Sustainable Chem. Eng., 2022, 10, 3766–3776 CrossRef CAS.
- J. Osorio-Tejada, F. Ferlin, L. Vaccaro and V. Hessel, Green Chem., 2022, 24, 325–337 RSC.
- F. Ferlin, D. Sciosci, F. Valentini, G. Cravotto, J. Menzio and K. Martina, Green Chem., 2021, 23, 7210–7218 RSC.
- F. Ferlin, I. Anastasiou, L. Carpisassi and L. Vaccaro, Green Chem., 2021, 23, 6576–6582 RSC.
- J. Hahn, M. Krieg, C. Keck, C. Maichle-Mössmer, R. F. Fink and H. F. Bettinger, Dalton Trans., 2018, 47, 17304–17316 RSC.
- R. K. Bartlett, H. S. Turner, R. J. Warne, M. A. Young and I. J. Lawrenson, J. Chem. Soc. A, 1966, 479–500 RSC.
- W. Gerrard and E. F. Mooney, J. Chem. Soc., 1960, 4028–4036 RSC.
- T. E. Reich, K. T. Jackson, S. Li, P. Jena and H. M. El-Kaderi, J. Mater. Chem., 2011, 21, 10629 RSC.
- P. Langer, L. Yang, C. R. Pfeiffer, W. Lewis and N. R. Champness, Dalton Trans., 2018, 48, 58–64 RSC.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- J. Andraos, Org. Process Res. Dev., 2012, 16, 1482–1506 CrossRef CAS.
- J. Andraos, Org. Process Res. Dev., 2013, 17, 175–192 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc00830h |
| This journal is © The Royal Society of Chemistry 2024 |