
Tailoring metal–support interaction over faceted TiO2 and copper nanoparticles for electrocatalytic nitrate reduction to ammonia†
Wahyu Prasetyo
Utomo
a,
Hao
Wu
*b,
Rui
Liu
 a and
Yun Hau
Ng
*a
a and
Yun Hau
Ng
*a
aLow-Carbon and Climate Impact Research Centre, School of Energy and Environment, City University of Hong Kong, Kowloon, Hong Kong SAR 999077, China. E-mail: yunhau.ng@cityu.edu.hk
bMacao Institute of Materials Science and Engineering (MIMSE), Faculty of Innovation Engineering, Macau University of Science and Technology, Taipa, Macau SAR 999078, China. E-mail: wuhao@must.edu.mo
First published on 8th December 2023
Abstract
The electrocatalytic nitrate reduction reaction (NO3−RR) provides a sustainable route for ammonia production while mitigating nitrate pollutants in the environment. Metal–support interaction has a significant influence on this electrocatalytic process. However, the mechanism of the facet-dependent metal–support interaction in the NO3−RR is still unknown. Herein, we report the modulation of the metal–support interaction by depositing copper nanoparticles on anatase TiO2 with different facet exposures, i.e., (001) and (101) facets. The result of copper nanoparticles being deposited on TiO2 with dominant (101) facet exposure is an enhanced ammonia yield rate of 447.5 μg mgcat−1 h−1 at −0.9 V vs. reversible hydrogen electrode (RHE), which is 4.2 times higher than the pristine TiO2 counterpart. The strong interaction between copper nanoparticles and TiO2 with dominant (101) facet exposure contributes to a greater increase in catalytic performance than TiO2 with dominant (001) facet exposure. The strong interaction leads to electron-deficient copper nanoparticles, efficient electron transfer, and stronger binding of the *NO2 intermediate, promoting the hydrogenation process in the NO3− reduction reaction for selective NH3 synthesis.
Introduction
Ammonia (NH3) is an essential feedstock chemical for the fertilizer, pharmaceutical, and nitrogen-containing chemical industries, as well as a potential energy carrier.1–5 Nevertheless, the production of NH3 still heavily relies on the Haber–Bosch process, which requires high temperature (400–500 °C) and high pressure (100–200 atm).6,7 Annually, this process needs an energy supply of up to ∼2.5 exajoule and is responsible for 1.4% of carbon dioxide emissions.8,9 Therefore, the development of a more energy-saving and environmentally friendly process is important.The electrocatalytic nitrate reduction reaction (NO3−RR) to NH3 has attracted increasing attention as an alternative method for sustainable NH3 production since it can be performed under ambient conditions. Moreover, NO3− is highly soluble in water and is known as one of the most widespread pollutants in the environment, especially in water bodies. NO3− mainly comes from industrial wastewater, liquid nuclear waste, livestock excrement, and chemical fertilizers, with a wide range of concentrations up to 2 mol L−1.10,11 The increased concentration of NO3− could lead to eutrophication, which decreases the oxygen level in water and destroys aquatic ecosystems.12–16 From a thermodynamic point of view, NO3− has a lower dissociation energy of the N=O bond (204 kJ mol−1), which could be more readily reduced with a lower thermodynamic barrier compared to bonds in other nitrogen-containing sources, such as N2.17 Therefore, the process of converting NO3− to NH3via the NO3−RR could potentially provide a sustainable route for green NH3 synthesis and address current issues with environmental water pollution.
Several electrocatalysts have been developed for the NO3−RR, such as titanium dioxide (TiO2), copper(II) oxide (CuO), copper(I) oxide (Cu2O), and cobalt oxide (CoO).18–21 However, the exhibited performance is typically still limited and remains far below the practical performances as mentioned by the US Department of Energy (300 mA cm−2 with 90% faradaic efficiency for NH3, FENH3).22 Continuous studies and investigations are still needed to acquire a better understanding of the catalytic system; hence, the catalytic performance can be further improved. In this regard, exposing specific facets of the catalyst is a promising strategy to modulate catalytic activity as different facet exposures reveal different atomic arrangements and electronic properties.23–28 Typically, the (001) facet of anatase TiO2 was reported to have higher reactivity due to the dominance of undercoordinated Ti atoms. An ideal (001) surface of TiO2 is occupied by five-coordinated Ti (Ti5c), i.e., undercoordinated Ti, accounting for 100% of coverage. On the other hand, the (101) surface is occupied by 50% Ti5c and 50% six-coordinated Ti (Ti6c).29–31 Therefore, the exposure of more (001) facets can, theoretically, lead to higher activity due to the more reactive Ti5c species on the surface. Other factors, such as electronic properties, band energy, or electron trapping states, can also contribute to the different catalytic performances induced by the facet effect.29,32–36 As reported by Pan et al., TiO2 with (010) facet exposure (TiO2-010) showed the highest catalytic activity, followed by TiO2-101 and TiO2-001.29 The performances exhibited were attributed to the cooperative effects of the undercoordinated Ti (Ti5c) on the (010) surface of TiO2-(010), and a more energetic conduction band (CB) position resulting in stronger reducing electrons in the CB. Besides, Mikrut et al. reported that the TiO2-(100) catalyst showed the highest activity toward the catalytic oxidation reaction compared to its TiO2-(001) and the TiO2-(101) counterparts.32 The superior performance was contributed to by the promoted separation of charge carriers in the TiO2-(100) catalyst, which facilitated hole transfer to the active sites. Moreover, Sun et al. reported a silver (Ag)-loaded TiO2 for a catalytic NO3−RR.33 Benefitting from the Ag nanoparticles as trap centres for electron transfer, the removal efficiency of NO3− reached ∼95% with high selectivity of 90% towards N2 production.
In addition to facet control, metal loading is another effective way to improve the catalytic activity of the NO3−RR. For instance, Kim et al. reported the strong metal–support interaction between the Pd–Cu cocatalyst and oxygen-deficient TiO2, promoting interfacial electron transfer and enhancing the NO3−RR performance.37 Nevertheless, NO2− is still the dominant product instead of the desired NH3 using the reported catalysts. Following this study, Li et al. reported using Pd–In supported on an FeOx substrate as the catalyst.38 The intrinsic electron transfer from Pd to the support led to the formation of positively charged Pd sites, which facilitates the adsorption of NO3− for better activation. A similar result was also observed in Cu–Pd-loaded nitrogen-doped carbon (Cu–Pd/CN). The interaction between Cu–Pd nanoparticles and the CN support stimulated the interfacial charge polarization and resulted in the electron-deficient Cu as the active site, which is favourable for NO3− adsorption and activation.39
The aforementioned studies indicate the significance of facet exposure and metal–support interaction on the catalytic performance for the NO3−RR. However, the determining factor in the electrocatalytic activity for the faceted TiO2 is still debatable since many factors could contribute to the apparent catalytic activity.29,30,33,34 Moreover, the contribution of the exposed TiO2 facet to the metal–support interaction in metal-loaded faceted TiO2 in correlation with their electrocatalytic performance for the NO3−RR remains unclear. The deposition of metal on the specific facet of TiO2 can lead to different levels of metal–support interaction due to the differing natures of the specific facets of TiO2. It can also lead to different reactivities during the catalytic process. In this context, Cu nanoparticles are deposited on the surface of TiO2 with different facet exposures, i.e., (001) and (101) facets, and catalytic tests toward the NO3−RR as a proof of concept of tailoring the interaction between metal and support are performed. The strong metal–support interaction between the Cu nanoparticles and (101) facets lead to effective electron transfer from the Cu nanoparticles to the TiO2 support resulting in electron-deficient Cu. In addition to the oxygen vacancies (OVs) in the TiO2 support, the electron-deficient Cu nanoparticles then serve as active sites for the NO3− adsorption and may also supply the NO3− reactants to the nearby OVs of TiO2. As a result, the production of NH3 by the (101)-dominant Cu-TiO2 electrocatalyst reaches the highest yield rate of 447.5 μg mgcat−1 h−1 at −0.9 V vs. reversible hydrogen electrode (RHE) among its prepared counterparts, which is 4.2 times higher than that of the pristine (101)-dominant TiO2 (NH3 yield: 106.7 μg mgcat−1 h−1). The increase is more exaggerated compared to the enhancement exhibited by the (001)-dominant Cu-TiO2 with respect to the pristine (001)-dominant TiO2. Moreover, time-dependent experiments and electrochemical analysis disclose that strong metal–support interaction results in a more strongly binding *NO2 intermediate, which promotes the electron transfer, facilitating the subsequent hydrogenation process for the selective NH3 synthesis.
Results and discussion
Material characterization
The TiO2 catalyst with (001) facet or (101) facet as the dominant facet is denoted as (001)-dominant TiO2 and (101)-dominant TiO2, respectively, which was prepared by a hydrothermal process using NaF as a capping agent (details in the Experimental section).32,40 Subsequently, Cu nanoparticles were evenly deposited on the surface of (001)-dominant TiO2 and (101)-dominant TiO2, resulting in (001)-dominant Cu-TiO2 and (101)-dominant Cu-TiO2 (details in the Experimental section). X-ray diffraction (XRD) patterns of the prepared catalysts are presented in Fig. 1a. All XRD peaks can be indexed to the anatase TiO2 (JCPDS 98-105-4604) without the rutile phase being observed.31,41 The peak intensities of the (001)-dominant Cu-TiO2 and (101)-dominant Cu-TiO2 are slightly lower than those of the pristine (001)-dominant TiO2 and (101)-dominant TiO2, which indicate slight decreases in crystallinity. The decline in the crystallinity can be correlated with the slight reduction of pristine TiO2 by NaBH4 during the Cu deposition process.42 However, no additional peaks from the deposited Cu can be observed in the XRD patterns because of the low amount of deposited Cu and its high dispersion.43 Inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis shows that the amounts of loaded Cu are 0.86% w/w and 0.82% w/w for the (001)-dominant Cu-TiO2 and (101)-dominant Cu-TiO2, respectively. | ||
| Fig. 1 (a) XRD patterns of the (001)-dominant TiO2, (101)-dominant TiO2, (001)-dominant Cu-TiO2, and (101)-dominant Cu-TiO2. SEM images of (b) the (001)-dominant TiO2, (c) the (101)-dominant TiO2, (d) the (001)-dominant Cu-TiO2, and (e) the (101)-dominant Cu-TiO2. HRTEM images of (f) the (001)-dominant Cu-TiO2 with Cu nanoparticles deposited on the (001) facet and (g) the (101)-dominant Cu-TiO2 with Cu nanoparticles deposited on the (101) facet. | ||
Scanning electron microscopye (SEM) analysis was performed to verify the morphology of the faceted TiO2 catalysts. As presented in Fig. 1b and c, the (001)-dominant TiO2 catalysts show a flat sheet morphology, while the (101)-dominant TiO2 catalysts are truncated octahedral particles. The exposures of (001) facets relative to all facets in the (001)-dominant TiO2 and (101)-dominant TiO2 were calculated to be ∼70.2% and ∼33.2%, respectively (details in Fig. S1a–c in ESI†). This means that the (001)-dominant TiO2 is dominated by (001) facets with an exposure of ∼70.2% from its total facets, while the (101)-dominant TiO2 is dominated by (101) facets with an exposure of ∼66.8%. The Cu nanoparticles are deposited on both (001) and (101) facets of the (001)-dominant TiO2 and (101)-dominant TiO2, as shown in Fig. 1d and e. The dispersion of the Cu nanoparticles on the surface of the (001)-dominant TiO2 and the (101)-dominant TiO2 is further verified using transmission electron microscopy (TEM) analysis. The TEM images with the corresponding energy dispersive X-ray (EDX) mapping show the uniform dispersion of the Cu nanoparticles on the (001)-dominant Cu-TiO2 and the (101)-dominant Cu-TiO2 (Fig. S2a–d and Fig. S3a–d in the ESI†). The average size of the Cu nanoparticle was estimated to be 6.65 ± 3.51 nm and 6.81 ± 2.34 nm for the (001)-dominant Cu-TiO2 and the (101)-dominant Cu-TiO2, respectively (Fig. S4a–c and Fig. S5a–d in ESI†). Moreover, the high-resolution TEM (HRTEM) image of the (001)-dominant Cu-TiO2 shows a Cu nanoparticle anchored on the surface of the (001) facet of the (001)-dominant TiO2 (Fig. 1f). The lattice fringe of 0.19 nm can be attributed to the (200) and (020) crystal planes of anatase TiO2, which are typically correlated with the (001) facet as both (200) and (020) facets have an interfacial angle of 90° between each other on the (001) zone axis diffraction.31,44 Meanwhile, the attachment of the Cu nanoparticle is identified by the lattice fringe of 0.20 nm coming from the (111) plane of Cu. We note that a lattice fringe of 0.24 nm coming from Cu2O (111) is also observed,45,46 which indicates the superficial oxidation of Cu metal under an ambient atmosphere.47 In addition to the (001) plane, the Cu nanoparticles can also be found on the (101) surface of the (101)-dominant TiO2, as presented in Fig. 1g. The lattice fringe of 0.35 nm refers to the (101) plane of the (101)-dominant TiO2. Similar to the (001)-dominant Cu-TiO2, the crystal planes of both Cu and Cu2O are also present on the (101)-dominant TiO2 catalyst.
The chemical states of all catalysts were investigated using X-ray photoelectron spectroscopy (XPS) analysis. As presented in Fig. 2a, the Ti 2p XPS spectra of the (001)-dominant TiO2 and (001)-dominant Cu-TiO2 samples show pronounced peaks at 457.3 eV and 462.9 eV, which refer to Ti4+ 2p3/2 and Ti4+ 2p1/2, respectively.48 The same position of the Ti4+ 2p3/2 and Ti4+ 2p1/2 peaks between the pristine (001)-dominant TiO2 and (001)-dominant Cu-TiO2 indicates that there is no significant change in the TiO2 phase of the (001)-dominant TiO2 upon depositing Cu nanoparticles. In contrast, the (101)-dominant Cu-TiO2 shows a shift of the Ti4+ 2p3/2 and Ti4+ 2p1/2 peaks to lower binding energies (457.4 eV and 463.1 eV, respectively) compared to the pristine (101)-dominant TiO2 (457.5 eV and 463.2 eV, respectively), as shown in Fig. 2b. The shift of binding energy to lower values suggests the tuned electronic structure of the (101)-dominant TiO2 upon Cu deposition.41,48
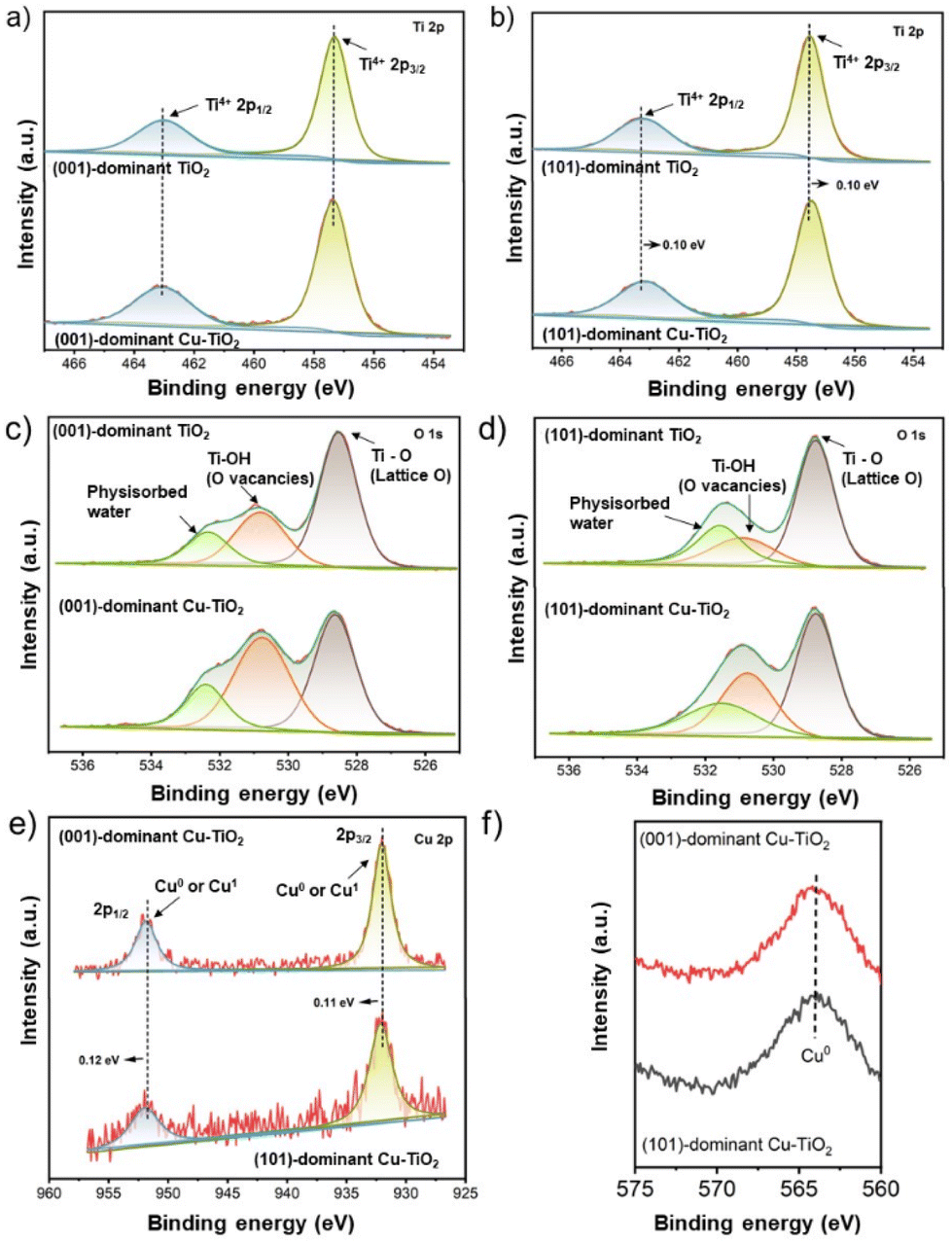 | ||
| Fig. 2 The Ti 2p XPS spectra of (a) the (001)-dominant TiO2 and (001)-dominant Cu-TiO2, and (b) the (101)-dominant TiO2 and (101)-dominant Cu-TiO2. O 1s XPS spectra of (c) the (001)-dominant TiO2 and (001)-dominant Cu-TiO2, and (d) the (101)-dominant TiO2 and (101)-dominant Cu-TiO2. (e) The Cu 2p XPS spectra of the (001)-dominant Cu-TiO2 and (101)-dominant Cu-TiO2. (f) The Cu LMM AES spectra of the (001)-dominant Cu-TiO2 and (101)-dominant Cu-TiO2. | ||
The O 1s XPS spectra are presented in Fig. 2c and d. All catalysts show three deconvoluted peaks at 528.5 eV, 530.8 eV, and 532.3 eV, which can be assigned to the lattice O of TiO2, the O ions associated with bridging Ti-OH (OVs), and physisorbed water, respectively.41,48 Upon the deposition of the Cu nanoparticles on the surface of the pristine (001)-dominant TiO2 and (101)-dominant TiO2, the intensity of OVs increased, indicating the generation of more OVs. The formation of more OVs in the (001)-dominant Cu-TiO2 and (101)-dominant Cu-TiO2 can be correlated with the Cu deposition process, which involves NaBH4 as a reducing agent.42
The chemical states analysis of the Cu nanoparticles was performed using argon-etched depth profiling XPS to avoid surface oxidation of Cu in air. As presented in Fig. 2e, two pronounced peaks can be observed at 932.03 eV and 951.85 eV in the (001)-dominant Cu-TiO2, which can be ascribed to the 2p3/2 and 2p1/2 peaks of Cu+ or Cu0.41 Further analysis using Cu LMM Auger electron spectroscopy (Fig. 2f) confirmed the presence of Cu0 species in both the (001)-dominant Cu-TiO2 and (101)-dominant Cu-TiO2 as indicated by a single peak at 565.2 eV. The presence of single Cu0 species was also supported by cyclic voltammetry (CV) measurement over freshly prepared (001)-dominant Cu-TiO2 and (101)-dominant Cu-TiO2 samples (Fig. S6 in the ESI†), in which both materials do not show reduction peaks correlated to the reduction of Cu2+ and Cu+, indicating the presence of Cu metal (Cu0) on the surface of faceted TiO2. However, it is worth noting that in the Cu 2p XPS spectrum of the (101)-dominant Cu-TiO2 (Fig. 2e), the peaks shifted to the higher binding energies of 932.14 eV and 951.97 eV compared to the (001)-dominant Cu-TiO2, suggesting lower electron density in the (101)-dominant Cu-TiO2 resulting from the electron transfer from the Cu nanoparticles to the (101)-dominant TiO2 support.37,41,49
The above XPS results imply that the electron transfer occurred from the Cu nanoparticles to the (101)-dominant TiO2 support, which decreased the electron density in the Cu nanoparticles and increased the electron density in the OVs and hence the surrounding Ti atoms. On the other hand, such an electron transfer phenomenon was hardly observed in the (001)-dominant Cu-TiO2, which suggests that the Cu nanoparticles have a stronger interaction with (101) facets compared to (001) facets.50–52 The electron transfer from Cu to TiO2 correlates with the presence of the OVs on the surface of TiO2. In this regard, OVs can serve as electron trapping centres.53,54 Due to the strong interaction between (101) facets of TiO2 and the Cu nanoparticles, the electrons are transferred from Cu to TiO2 and pair with singlet electrons in the OVs near the interface.55 Additionally, it is also acknowledged that electrons are shallowly trapped in Ti5c on the surface of (101) facets, while electrons are also deeply trapped in Ti6c on the subsurface of (001) facets.34 Therefore, the shallow traps on the surface of Ti5c in (101) facets can provide a more conducive environment for electron mobility, which result in stronger interactions with the Cu nanoparticles.34 To sum up, arising from the strong interaction between the Cu nanoparticles and (101)-dominant TiO2 support, the electrons transfer from Cu nanoparticles to the OVs in (101) facets, leading to electron-deficient Cu nanoparticles.39,41,49
Electrocatalytic NO3−RR evaluation
An electrocatalytic activity test of faceted-TiO2-based catalysts was performed in an H-type cell with a typical three-electrode configuration using a Nafion 117 membrane to separate the cathode and the anode chambers. The prepared catalysts were dropcast on carbon paper substrates as the working electrodes (1.0 mgcat cm−2, details in the Experimental section). Prior to the NO3−RR test, the prepared electrodes were initially examined using linear sweep voltammetry (LSV) from +0.1 V to −1.2 V vs. RHE in 0.5 M Na2SO4 solution with and without the addition of 50 ppm NO3−–N. The LSV curves of both the pristine (001)-dominant TiO2 and (101)-dominant TiO2 electrodes showed a slight increase in current density upon the addition of NO3− (Fig. S7 in the ESI†), while the increase in current density was apparently exaggerated with the deposition of Cu nanoparticles (Fig. 3a and b).19 Besides, the corresponding onset potential of the LSV curves for both faceted-TiO2 electrodes also shifted to the less negative value (−0.3 V vs. RHE) with the deposition of Cu nanoparticles. Note that the increase of the current density for the (101)-dominant Cu-TiO2 is greater than that of the (001)-dominant Cu-TiO2, suggesting the stronger promoting effect in the NO3−RR upon Cu deposition on the (101) facet of the (101)-dominant TiO2.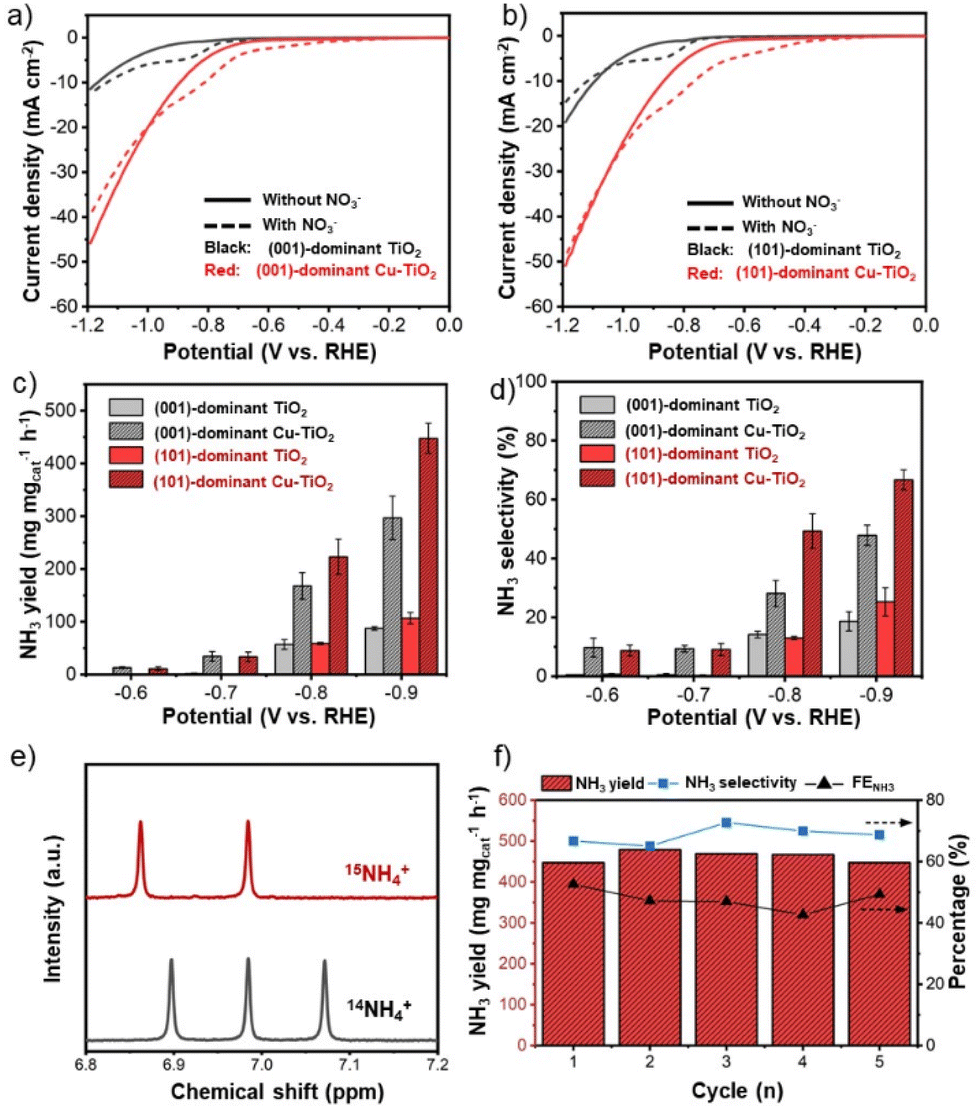 | ||
| Fig. 3 LSV curves of (a) the (001)-dominant TiO2 and (001)-dominant Cu-TiO2, and (b) the (101)-dominant TiO2 and (101)-dominant Cu-TiO2 in 0.5 M Na2SO4 without and with the addition of 50 ppm NO3−–N. (c) NH3 yield and (d) NH3 selectivity. All catalytic activity experiments were performed in 0.5 M Na2SO4 containing 50 ppm NO3−–N. (e) 1H NMR spectra of the electrolyte after the electrocatalytic reduction reaction using 15NO3− and 14NO3− as the nitrogen sources. The reaction for isotope labelling was performed for the (101)-dominant Cu-TiO2 at −0.9 V vs. RHE in 0.5 M Na2SO4 containing 50 ppm NO3−–N. (f) Stability test of the (101)-dominant Cu-TiO2 was performed at −0.9 V vs. RHE in 0.5 M Na2SO4 containing 50 ppm NO3−–N. | ||
The electrocatalytic NO3−RR performances were further examined using chronoamperometry (i–t) tests at different applied potentials in 0.5 M NaSO4 containing 50 ppm NO3−–N. The concentration levels of NO3−, NH3, and NO2− were determined using spectrophotometric methods (Fig. S8a–c in the ESI†).56 The NO3− conversion rate increased along with the more negative potentials (Fig. S9 in the ESI†). The effect of Cu deposition on the NO3− conversion can be observed clearly at the potentials of −0.8 and −0.9 V vs. RHE, where both the (001)-dominant Cu-TiO2 and (101)-dominant Cu-TiO2 show higher NO3− conversion rates compared to the pristine TiO2 counterparts.
Moreover, an obvious trend related to the effect of Cu deposition on the different facets of TiO2 can be observed in the NH3 yield rate (Fig. 3c). Within all the applied potentials, both the (001)-dominant Cu-TiO2 and the (101)-dominant Cu-TiO2 showed significantly higher NH3 yield rates compared to the pristine (001)-dominant TiO2 and (101)-dominant TiO2. The activity increased more significantly along with the more negative potentials, reaching the highest value at −0.9 V vs. RHE with NH3 yields of 296.6 μg mgcat−1 h−1 and 447.5 μg mgcat−1 h−1 for the (001)-dominant Cu-TiO2 and the (101)-dominant Cu-TiO2, respectively. The NH3 yield rate of the (101)-dominant Cu-TiO2 is 4.2 times higher than that of the pristine (101)-dominant TiO2 (106.7 μg mgcat−1 h−1), while the NH3 yield of the (001)-dominant Cu-TiO2 is only 3.3 times higher than the pristine counterpart (87.9 μg mgcat−1 h−1). Therefore, the (101)-dominant Cu-TiO2 exhibits an exaggerated NH3 yield increase compared to the (001)-dominant Cu-TiO2. Similar trends can also be observed in the corresponding NH3 selectivity (Fig. 3d) and FENH3 (Fig. S10 in the ESI†). The highest NH3 selectivity value was achieved at −0.9 V with values of 66.7% and 47.8% for the (101)-dominant Cu-TiO2 and the (001)-dominant Cu-TiO2, respectively. Additionally, it is worth noting that FENH3 shows a volcano plot with the highest values being reached at −0.8 V vs. RHE with values of 67.1% and 47.0% for the (101)-dominant Cu-TiO2 and the (001)-dominant Cu-TiO2, respectively. Note that the NH3 selectivity value was calculated based on the concentration change of NO3− only with the corresponding products, while the FE was calculated based on the total current regardless of the reactions (details in the ESI†). Therefore, the calculation of the NH3 selectivity value excluded the contribution of the hydrogen evolution reaction (HER), while the FENH3 took it into consideration.19,57 The decrease of the FENH3 for all electrodes at −0.9 V vs. RHE is attributed to the intensified HER, as evidenced by the significant increase of the FEH2 (Fig. S11 in the ESI†).14,19 Accordingly, both the NO2− selectivity and the FENO2 also decreased at −0.9 V vs. RHE (Fig. S12 and Fig. S13 in the ESI†). Another possible byproduct, i.e., hydrazine (N2H4), was also detected using the Watt and Chrisp method (detailed in the Experimental section in the ESI†). In all catalytic reactions, no hydrazine (N2H4) was detected as presented in Fig. S14a–c in the ESI,† excluding the possibility of N2H4 as a byproduct. We note that the greatest performance exhibited by the (101)-dominant Cu-TiO2 catalyst in this work is, in fact, on a par with previous studies reported in the literature, specifically for Ti-based and Cu0-based catalysts in neutral media (Table S1 in the ESI†). However, it is worth noting that the exhibited performances in this work resulted from lower external potentials (−0.9 V vs. RHE) compared to the other Ti-based catalysts (−1.0 to −1.6 V vs. RHE), which is also one of the important parameters for evaluating the electrocatalytic performance.58,59
Control experiments were then performed to verify the origin of the produced NH3. As presented in Fig. 3e, the electrolyte taken from the electrocatalytic reduction of Na15NO3 shows typical doublet peaks at δ = 6.86 and δ = 6.98 ppm, while the triplet peak was observed in the spectra when employing Na14NO3. These 15N isotope-labelling results strongly confirm that the produced NH3 originated from the electroreduction of NO3−.19,56 Moreover, a negligible amount of NH3 produced in the absence of NO3− and at the open circuit potential (OCP) further supports the results from 15N isotope-labelling experiments (Fig. S15 in the ESI†). No accumulation of NH3 was observed upon exposing the electrolyte to the air, excluding possible NH3 contamination from the environment (Fig. S16 in the ESI†).
The exaggerated improvement in the NH3 yield rate and NH3 selectivity can be correlated with the strong metal–support interaction exhibited by the (101)-dominant Cu-TiO2 catalyst. On the other hand, we note that the concentration of OV also increases in both the (001)-dominant TiO2 and (101)-dominant TiO2 upon Cu deposition as indicated by the O 1s XPS spectra in Fig. 2c and d. One of the oxygen (O) atoms in NO3− can occupy the OV and coordinate with the surrounding Ti3+ of TiO2, which weakens the N–O bonding for better activation.18 Therefore, to investigate the contribution of OVs, we also performed the electrocatalytic test on the reduced-(001)-dominant TiO2 and reduced-(101)-dominant TiO2 prepared by the NaBH4 reduction method on the pristine faceted TiO2 without the addition of Cu salts. As verified by the electron paramagnetic resonance (EPR) spectra of the pristine (101)-dominant TiO2 and the reduced (101)-dominant TiO2 (Fig. S17 in the ESI†), the reduced (101)-dominant TiO2 shows a slightly higher peak intensity at g = 2.003, indicating a higher concentration of OVs upon implementing the NaBH4 reduction process.48 Moreover, as presented in Fig. S18a–d in the ESI,† the reduced-(001)-dominant TiO2 and the reduced-(101)-dominant TiO2 also show slight increases in NH3 yield rate and selectivity, suggesting the benefits of OVs generation in the NO3−RR to a certain degree.18 However, the improved performances by the reduced-faceted-TiO2 electrodes are much less significant compared to the enhancements exhibited by the Cu-loaded faceted TiO2 electrodes.
To further investigate the contribution from the Cu nanoparticles, which serve as the co-catalyst in the Cu-loaded TiO2 electrode, we also performed electrocatalytic tests over Cu nanoparticles prepared using the NaBH4 reduction method. For this purpose, we deposited an equal amount of Cu nanoparticles contained in the (001)-dominant Cu-TiO2 and (101)-dominant Cu-TiO2 electrodes. As presented in Fig. S19 in the ESI,† Cu nanoparticles show activity for NO3− reduction to NH3.60 NH3 yield rates increase along with the more negative potentials. However, the magnitude of the NH3 yield rate is much inferior compared to that of the Cu-loaded faceted TiO2 catalysts. These control experiments provide evidence that significant enhancement of the NH3 yield rate, selectivity, and FENH3 over the (001)-dominant Cu-TiO2 and the (101)-dominant Cu-TiO2 is mainly determined by the interaction between the Cu nanoparticles and the faceted TiO2. Moreover, the stronger metal–support interaction between the Cu nanoparticles and the (101) facets of TiO2 further enhance the performance. The presumption can be derived from the fact that the increase in NH3 yield rate and selectivity is exaggerated for the (101)-dominant Cu-TiO2 compared to the (001)-dominant Cu-TiO2. Moreover, the (101)-dominant Cu-TiO2 shows a stable NH3 yield rate, NH3 selectivity, and FENH3 in five consecutive reactions (Fig. 3f), suggesting the appreciable stability of the designed system for the NO3−RR.
Mechanism study
Time-dependent experiments were performed to understand the kinetics and reaction mechanism of the NO3−RR (Fig. 4a–d). For all prepared catalysts, the NO3− concentration decreases over time accompanied by the increase in NH3 and NO2− concentrations. The apparent reduction rate constant values of NO3− calculated based on the Langmuir–Hinshelwood model for the (101)-dominant Cu-TiO2 (kap = 0.00551 min−1) and the (001)-dominant Cu-TiO2 (kap = 0.00476 min−1) are higher than those of the pristine (101)-dominant TiO2 (kap = 0.00368 min−1) and (001)-dominant TiO2 (kap = 0.00322 min−1) counterparts, suggesting the positive role of Cu deposition in promoting the NO3−RR (Fig. S20a and b in the ESI†).38,61,62 The apparent activation energy (Ea) also decreases upon Cu loading on the surface of faceted TiO2. The Ea for the (101)-dominant Cu-TiO2 is significantly lower (Ea = 4.61 kJ mol−1) than that of the pristine (101)-dominant TiO2 (Ea = 15.79 kJ mol−1). The same trend can be observed in the (001)-dominant Cu-TiO2 (Ea = 13.90 kJ mol−1) and the (001)-dominant TiO2 (Ea = 24.03 kJ mol−1) (Fig. S21a and b in the ESI†).63,64 It is noteworthy that among the four samples, the (101)-faceted TiO2-based catalysts typically show lower apparent Ea values than the (001)-faceted TiO2-based catalysts, indicating the advantage of (101) facet exposure and Cu loading on these facets for reducing the Ea. These Ea values agree well with the Tafel plots (Fig. S22 in the ESI†), in which the (101)-dominant Cu-TiO2 shows a lower Tafel slope (178.7 mV dec−1) than the (001)-dominant Cu-TiO2 (203.7 mV), indicating the higher kinetics rate of the former for the electrocatalytic NO3−RR.65,66 These apparent Ea values and the Tafel slopes are in good agreement with the reduction rate (Fig. 4a–d and Fig. S20a, b in the ESI†), as mentioned before. In addition to the kinetics information, the higher reduction rate of the (101)-dominant Cu-TiO2 suggests a higher adsorption strength between the NO3− ions with the catalyst surface. This assumption can be rationalized based on the presence of the electron-deficient Cu nanoparticles in the (101)-dominant Cu-TiO2, which are more conducive to attracting the negatively charged NO3− ions.38,39 This attraction could also lead to the accumulation of NO3− near the catalyst surface, which facilitates the contact between the NO3− ions and the surface OVs in the surrounding Cu nanoparticles.39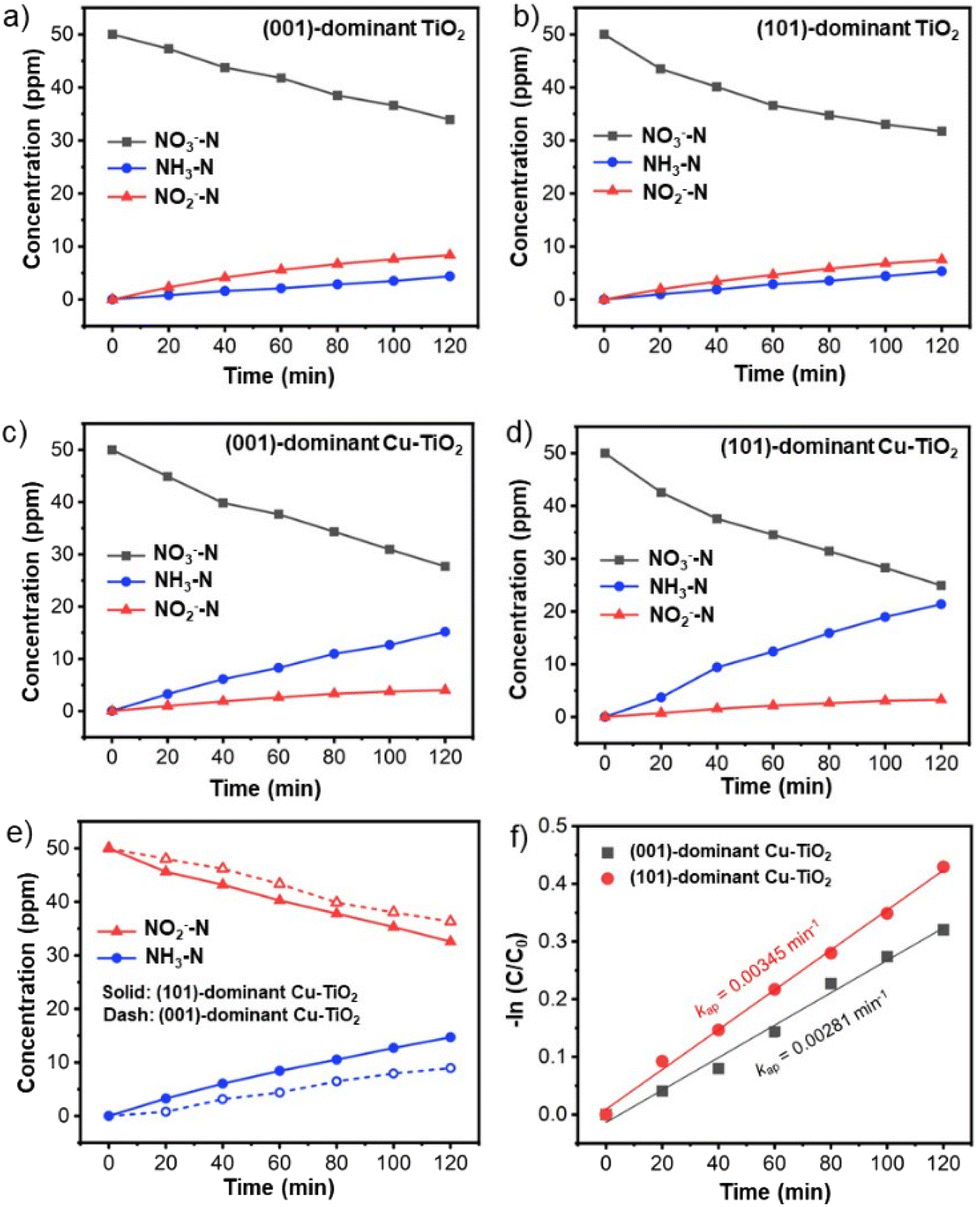 | ||
| Fig. 4 NO3−, NH3, and NO2− concentration changes over (a) the (001)-dominant TiO2, (b) the (101)-dominant TiO2, (c) the (001)-dominant Cu-TiO2, and (d) the (101)-dominant Cu-TiO2. All experiments were performed at −0.9 V vs. RHE in 0.5 M Na2SO4 containing 50 ppm of NO3—N for 2 h. € NO2− and NH3 concentration changes over the (001)-dominant Cu-TiO2 and the (101)-dominant Cu-TiO2, and (f) the corresponding Langmuir–Hinshelwood plots. All the experiments were performed at −0.9 V vs. RHE in 0.5 M Na2SO4 containing 50 ppm of NO2−–N for 2 h. | ||
The influence of the strong metal–support interaction on the catalysis process can be observed by the product distribution, specifically the evolution of NH3 and NO2− as the two major products. As shown in Fig. 4a and b, the (001)-dominant TiO2 and the (101)-dominant TiO2 electrodes show that NO2− is the dominant product at −0.9 V vs. RHE. On the other hand, upon the deposition of Cu on the faceted TiO2, NH3 emerges as the dominant product from the beginning of the reaction (Fig. 4c and d). Notably, the increase in the NH3 concentration of the (101)-dominant Cu-TiO2 is much higher than that of the (001)-dominant Cu-TiO2, which is in good agreement with the trends of NH3 yield rate and NH3 selectivity in the catalytic test. The time-dependent experiments in a prolonged reaction period show similar trends for the concentration changes of the NO3− reactant and the reduction products (Fig. S23a and b in the ESI†). The greater increase in NH3 production of the (101)-dominant Cu-TiO2 indicates the benefit of the strong metal–support interaction between the Cu nanoparticles and (101) facets of TiO2 in promoting the selective NO3−RR to NH3.
We note that the detection of NO2− in the electrolyte during the NO3−RR indicates that the production rate of NO3− to *NO2 is faster than that of NO2− to NH3, and thus leads to the desorption of *NO2 from the catalyst surface into the electrolyte.60 The detection of higher amounts of NO2− in the electrolyte may also indicate that the binding strength of *NO2 to the catalyst surface is weaker, hence it can easily be desorbed from the catalyst surface into the electrolyte. Therefore, to further investigate the role of the *NO2 intermediate in the NO3−RR, time-dependent NO2− reduction reactions (NO2−RRs) over the (001)-dominant Cu-TiO2 and the (101)-dominant Cu-TiO2 catalysts were performed. As presented in Fig. 4e, the decrease in the NO2− concentration using the (001)-dominant Cu-TiO2 is slower than that using the (101)-dominant Cu-TiO2. The corresponding apparent rate constant values based on the Langmuir–Hinshelwood model of the (101)-dominant Cu-TiO2 and the (001)-dominant Cu-TiO2 are 0.00345 min−1 and 0.00281 min−1 (Fig. 4f), respectively.61,67 The higher rate constant of NO2− reduction may indicate the higher binding strength between NO2− and the surface of the (101)-dominant Cu-TiO2 when forming *NO2. These results agree well with the lower amount of NO2− produced using the (101)-dominant Cu-TiO2 compared with that produced using the (001)-dominant Cu-TiO2 in the NO3−RR (Fig. S24 in the ESI†). Based on these findings, the exaggerated enhancement in the NH3 production by the (101)-dominant Cu-TiO2 can also be attributed to the stronger binding strength of *NO2, which suppresses the desorption of *NO2 into the electrolyte and subsequently promotes the subsequent hydrogenation process.
The intrinsic electrochemical properties of the prepared electrodes are important factors in influencing catalytic performance. Therefore, electrochemical impedance spectroscopy (EIS), electrochemical active surface area (ECSA) analysis, and Mott–Schottky analysis were performed for the pristine and Cu-deposited TiO2 electrodes. The Nyquist plots of the Cu-deposited TiO2 electrodes show smaller arc radii than the pristine counterparts (Fig. 5a and b). The series resistance (Rs), which is correlated with the resistance from the solution, is comparable among all the electrodes (Table S2 in the ESI†).61,68,69 On the other hand, the charge transfer resistance (RCT) decreases significantly upon Cu deposition. The RCT value of the (001)-dominant Cu-TiO2 (27.20 Ω) is 2.7 times lower than that of the pristine (001)-dominant TiO2 (74.38 Ω). Meanwhile, the (101)-dominant Cu-TiO2 shows a more significant decrease in the RCT value (40.28 Ω), which is 3.8 times lower than that of the pristine (101)-dominant TiO2 (152.50 Ω). The more efficient electron transfer as evidenced by the higher decrease in the RCT value on the (101)-dominant Cu-TiO2 than on the (001)-dominant Cu-TiO2 is likely contributed to by the stronger interaction between Cu nanoparticles and (101) facets of TiO2 in the (101)-dominant Cu-TiO2.41,70 The ECSA was further investigated by measuring the electrochemical double-layer capacitance (Cdl), which is proportional to the ECSA. The linear fitting of the charging current density with various scan rates (Fig. 5c and d and Fig. S25a–d in the ESI†) reveals that the Cdl values of the (001)-dominant Cu-TiO2 (Cdl = 0.0601 mF cm−2) and the (101)-dominant Cu-TiO2 (Cdl = 0.0886 mF cm−2) are higher than those of the pristine (001)-dominant TiO2 (Cdl = 0.0546 mF cm−2) and the (101)-dominant TiO2 (Cdl = 0.0685 mF cm−2), suggesting that more active sites are present in the Cu-deposited TiO2 catalysts.41 Moreover, the Mott–Schottky plots show positive slopes for all the electrodes, indicating the n-type nature of the faceted-TiO2 electrodes (Fig. S26a and b in the ESI†).41,71 The (001)-dominant Cu-TiO2 and the (101)-dominant Cu-TiO2 show smaller slopes compared with those of the pristine (001)-dominant TiO2 and (101)-dominant TiO2, also indicating the higher donor density in the Cu-deposited TiO2.72 These results suggest that the significant enhancement in NH3 production exhibited by the (101)-dominant Cu-TiO2 is likely contributed to by the promoted electron transfer. In this regard, the nature of (101) facets, which possess shallow traps on the surface of Ti5c in (101) facets, can provide a more conducive environment for electron mobility as reported in the literature.34
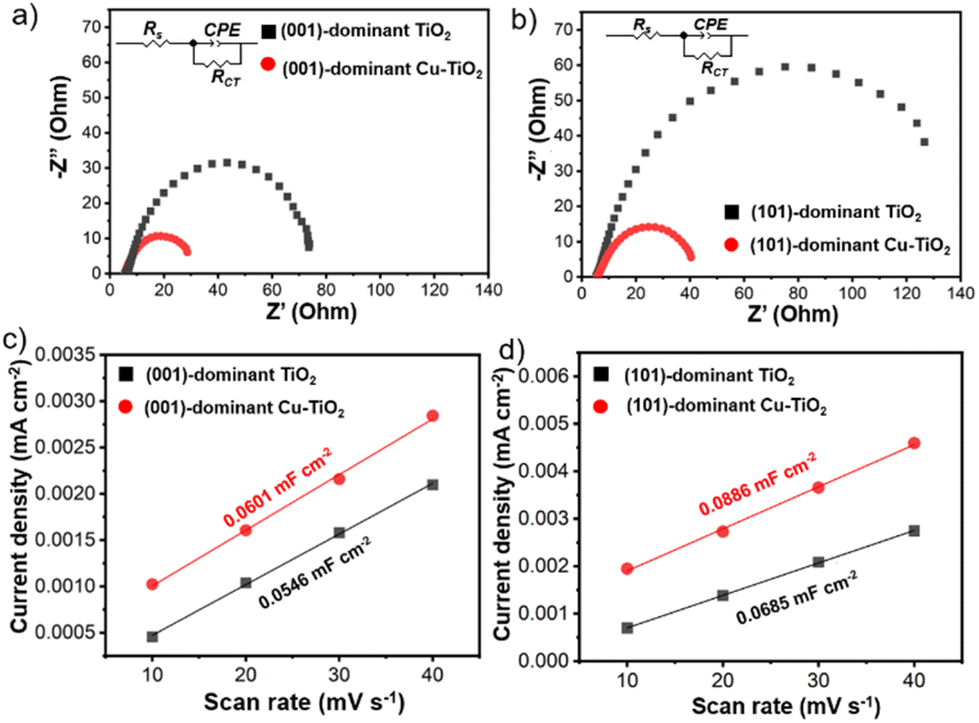 | ||
| Fig. 5 Nyquist plots of (a) the (001)-dominant TiO2 and (001)-dominant Cu-TiO2 and (b) the (101)-dominant TiO2 and (101)-dominant Cu-TiO2. The insets are the equivalent electrical circuits. Charging current density plotted against the scan rate at 0.655 V vs. RHE for (c) the (001)-dominant TiO2 and (001)-dominant Cu-TiO2, and (d) the (101)-dominant TiO2 and (101)-dominant Cu-TiO2. | ||
The above electrocatalytic NO3−RR activities, time-dependent experiments, and electrochemical analysis have collectively suggested that (1) the Cu deposition on the faceted-TiO2 surface leads to higher NH3 production, NH3 selectivity, and FENH3 and (2) the deposition of Cu on the (101) facet is more beneficial for promoting NH3 production compared to Cu deposition on the (001) facet due to the stronger interaction between the Cu nanoparticles and the dominant (101) facet in the (101)-dominant Cu-TiO2. The Cu metal was previously reported as an active catalyst for the NO3−RR in the literature.62,73–75 The activity originates from the d-orbital electronic configuration of Cu, where the NO3− reactant binds with Cu by electron donation from the highest occupied molecular orbital (HOMO) of NO3− to the empty orbital of Cu accompanied by the backdonation from the fully occupied d orbital to the lowest unoccupied molecular orbital (LUMO) of NO3−.60 Therefore, in addition to OVs as active sites, the deposited Cu nanoparticles can also serve as active sites in the (001)-dominant Cu-TiO2 and the (101)-dominant Cu-TiO2 catalysts, resulting in significantly higher NH3 production than that from the pristine counterparts. Additionally, due to the strong interaction between the Cu nanoparticles and (101) facets of TiO2, the electrons are transferred from the Cu nanoparticles to the (101) facets of TiO2 resulting in electron-deficient Cu, which can strongly attract and accumulate the negatively charged NO3−.39 The stronger absorption strength is also found in the NO2− (and *NO2), which suppresses the desorption of *NO2 to the electrolyte. Finally, the exposure of (101) facets of TiO2, possessing shallow traps for electrons on the Ti5c surface could provide a more conducive environment for electron mobility, thus promoting the electron transfer in the (101)-dominant Cu-TiO2 resulting in significant enhancements in NH3 production and NH3 selectivity over the (101)-dominant Cu-TiO2 catalyst.
Conclusions
In conclusion, the strong interaction between the deposited Cu nanoparticles and the TiO2 support with the dominant (101) facet exposure promoted selective NH3 production from the NO3−RR. The (101)-dominant Cu-TiO2 catalyst demonstrated the highest NH3 yield rate of 447.5 μg mgcat−1 h−1 at −0.9 V vs. RHE, which is 4.2 times higher than that of the pristine (101)-dominant TiO2 catalyst with an NH3 yield of 106.7 μg mgcat−1 h−1. In comparison, the (001)-dominant Cu-TiO2 counterpart showed NH3 yield rate of 296.6 μg mgcat−1 h−1 under identical reaction conditions, which is 3.3 times higher than that of the pristine (001)-dominant TiO2 with an NH3 yield of 87.9 μg mgcat−1 h−1. The greater increase in catalytic performance for the (101)-dominant TiO2 with Cu deposition was ascribed to the formation of electron-deficient Cu nanoparticles, the stronger binding energy of *NO2, and the promoted electron transfer originating from the strong interaction between the Cu nanoparticles and (101) facets of TiO2. Collectively, these factors suppressed the *NO2 desorption and promoted the subsequent hydrogenation process, leading to direct NO3− reduction to NH3. These findings can stimulate the future surface design over other oxide supports toward the efficient electrocatalytic NO3−RR to produce NH3.Experimental
The experimental details are provided in the ESI.† This section summarizes the material synthesis, characterization, and electrochemical measurements.Preparation of the (001)-dominant TiO2 (flat sheet) and the (101)-dominant TiO2 (truncated octahedral)
The TiO2 samples with different dominant facets were prepared using the hydrothermal method with NaF as a capping agent, as reported by Mikrut et al.40 In a typical procedure for preparing (001)-dominant TiO2, 532 mg NaF was mixed with 160 mL of HCl (12.8%). The mixture was then stirred to form a clear solution. Subsequently, 781.6 mg of TiOSO4 was added to the solution, followed by constant stirring for 1 h to form a clear solution. The resulting solution was then placed into a 200 mL Teflon-lined stainless-steel autoclave and was heated at 180 °C for 12 h. After naturally cooling down to room temperature, the powders were collected and centrifuged in water and ethanol several times. The obtained powders were then dried at 60 °C in a vacuum oven for 12 h and calcined at 600 °C for 2 h at a heating rate of 2 °C min−1 under an argon atmosphere (20 mL min−1). The (101)-dominant TiO2 (truncated octahedral) was prepared using the same method as that of the (001)-dominant TiO2 by adjusting the concentration of HCl (6.7%), the mass of NaF (279.6 mg), and the amount of TiOSO4 (390.8 mg).Copper (Cu) deposition on (001)-dominant TiO2 and (101)-dominant TiO2
The Cu deposition was performed using NaBH4 as a reducing agent.41 20 mg of the (001)-dominant TiO2 or the (101)-dominant TiO2 was dispersed in 15 mL of DI water, followed by sonication for 30 min. After sonication, 315 μL of 20 mM Cu(NO3)2·3H2O solution was added slowly under vigorous stirring. The stirring was continued for 30 min. 30 mg of NaBH4 was then added, and the stirring was continued for another 20 min. The powders were subsequently separated by centrifugation and washed using excess deionized water and ethanol. The powders were finally dried in a vacuum oven at 60 °C for 12 h.Characterization
The crystal phase of each prepared sample was analyzed using an X-ray diffraction (XRD) PANalytical X'pert3 powder X-ray diffractometer operated at 40 kV and 100 mA with Cu Kα radiation. The morphology and the composition of the catalyst were observed using a transmission electron microscope (TEM) FEI Tecnai F20 and scanning electron microscope (SEM) Zeiss EVO 10. The oxidation states of Ti, Cu, and O elements were characterized using X-ray photoelectron spectroscopy (XPS, Thermo Scientific NEXSA) conducted with an Al Kα X-ray excitation source. All binding energies were referenced to the C 1s peak at 283.67 eV. Electron paramagnetic resonance (EPR) analysis was performed using a Bruker EMXPLUS EPR instrument (Germany). The amount of deposited metal was measured using inductively coupled plasma optical emission spectroscopy (ICP-OES, Optima 8000). The ultraviolet–visible (UV–vis) absorbance data were collected on a Shimadzu UV-3600 UV spectrophotometer.Electrochemical measurements
The prepared samples were dropcast on pieces of carbon paper as the working electrodes. Before the drop-casting, the pieces of carbon paper were washed in a mixture of methanol and acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; v/v) and ultrasonicated for 10 min. Subsequently, the pieces of carbon paper were washed again in water and ultrasonicated for another 10 min. The pieces of carbon paper were then dried at room temperature. Meanwhile, 10 mg of TiO2 powders, e.g., the (001)-dominant TiO2, were mixed with 0.24 mL of water and 0.72 mL of ethanol. After that, 0.04 mL of Nafion solution (5%) was added. The mixture was then ultrasonicated for 1 h to form a homogeneous suspension. 0.1 mL of the prepared homogeneous suspension was then dropped onto 1.0 × 1.0 cm2 carbon paper (1.0 mgcat cm−2). The same procedure was applied to prepare the electrodes loaded with the other catalysts (i.e., the (101)-dominant TiO2, (001)-dominant Cu-TiO2, and (101)-dominant Cu-TiO2).
:1; v/v) and ultrasonicated for 10 min. Subsequently, the pieces of carbon paper were washed again in water and ultrasonicated for another 10 min. The pieces of carbon paper were then dried at room temperature. Meanwhile, 10 mg of TiO2 powders, e.g., the (001)-dominant TiO2, were mixed with 0.24 mL of water and 0.72 mL of ethanol. After that, 0.04 mL of Nafion solution (5%) was added. The mixture was then ultrasonicated for 1 h to form a homogeneous suspension. 0.1 mL of the prepared homogeneous suspension was then dropped onto 1.0 × 1.0 cm2 carbon paper (1.0 mgcat cm−2). The same procedure was applied to prepare the electrodes loaded with the other catalysts (i.e., the (101)-dominant TiO2, (001)-dominant Cu-TiO2, and (101)-dominant Cu-TiO2).
The electrochemical measurements were carried out using a CHI 660 instrument in an H-type cell using the TiO2 based-thin film electrode as the working electrode, saturated calomel electrode (SCE) as the reference electrode, and platinum foil as the counter electrode. The surface area of the working electrode was controlled within 1.0 × 1.0 cm2. 0.5 M Na2SO4 was used as the electrolyte. The electrolyte volumes in the anodic and cathodic chambers were 30 and 40 mL, respectively. A Nafion 117 membrane was placed between the anodic and cathodic chambers. The catholyte was purged with Ar at a flow rate of 50 mL min−1 for 15 min under vigorous stirring. Before the NO3−RR measurement, cyclic voltammetry (CV) was performed from +0.1 V to −1.2 V vs. RHE to reach a stable curve. After the CV, NaNO3 (50 ppm NO3−–N) was added to the cathode chamber. The chronoamperometry (i–t) test was carried out at different potentials for 2 h at a stirring rate of 300 rpm. All potentials were referenced to reversible hydrogen electrode (RHE) by the Nernst equation (ERHE = ESCE + 0.059 × pH + 0.241).
To investigate the electrochemically active surface area (ECSA), the working electrode was cycled in the non-faradaic potential region at various scan rates from 10 to 40 mV s−1 in 0.5 M Na2SO4. By plotting the charging current density against the scan rate, the double-layer capacitance (Cdl) was calculated by determining the slope value. Electrochemical impedance spectroscopy (EIS) was performed at the potential of −0.7 V vs. RHE at a frequency of 10−1–105 Hz and amplitude of 5 mV. Mott–Schottky plots were recorded at a frequency of 0.5 kHz and amplitude of 0.01 V.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was financially supported by the Hong Kong Research Grant Council (RGC) General Research Fund (GRF) CityU 11306920, CityU 11308721, and CityU 11316522. The authors acknowledge the financial support by the Science and Technology Development Fund, Macao SAR (File No. FDCT-0125/2022/A), and the National Natural Science Foundation of China (22305009).References
- L. Wang, M. Xia, H. Wang, K. Huang, C. Qian, C. T. Maravelias and G. A. Ozin, Joule, 2018, 2, 1055–1074 CrossRef CAS.
- B. M. Comer, P. Fuentes, C. O. Dimkpa, Y.-H. Liu, C. A. Fernandez, P. Arora, M. Realff, U. Singh, M. C. Hatzell and A. J. Medford, Joule, 2019, 3, 1578–1605 CrossRef CAS.
- L. Hollevoet, F. Jardali, Y. Gorbanev, J. Creel, A. Bogaerts and J. A. Martens, Angew. Chem., Int. Ed., 2020, 59, 23825–23829 CrossRef CAS PubMed.
- D. W. Kim, D. W. Kang, M. Kang, J.-H. Lee, J. H. Choe, Y. S. Chae, D. S. Choi, H. Yun and C. S. Hong, Angew. Chem., Int. Ed., 2020, 59, 22531–22536 CrossRef CAS PubMed.
- W. P. Utomo, M. K. H. Leung, Z. Yin, H. Wu and Y. H. Ng, Adv. Funct. Mater., 2022, 32, 2106713 CrossRef CAS.
- A. J. Martín, T. Shinagawa and J. Pérez-Ramírez, Chem, 2019, 5, 263–283 Search PubMed.
- S. Lin, X. Zhang, L. Chen, Q. Zhang, L. Ma and J. Liu, Green Chem., 2022, 24, 9003–9026 RSC.
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- O. Elishav, B. M. Lis, E. M. Miller, D. J. Arent, A. Valera-Medina, A. G. Dana, G. E. Shter and G. S. Grader, Chem. Rev., 2020, 120, 5352–5436 CrossRef CAS PubMed.
- L. Barrera and R. Bala Chandran, ACS Sustainable Chem. Eng., 2021, 9, 3688–3701 CrossRef CAS.
- T. A. Belay, F. M. Lin, C. Y. Lin, H. M. Hsiao, M. F. Chang and J. C. Liu, Water Sci. Technol., 2015, 72, 960–965 CrossRef CAS PubMed.
- X. Zhang, Y. Wang, C. Liu, Y. Yu, S. Lu and B. Zhang, Chem. Eng. J., 2021, 403, 126269 CrossRef CAS.
- Y. Li, Y. K. Go, H. Ooka, D. He, F. Jin, S. H. Kim and R. Nakamura, Angew. Chem., Int. Ed., 2020, 59, 9744–9750 CrossRef CAS PubMed.
- X. Wan, W. Guo, X. Dong, H. Wu, X. Sun, M. Chu, S. Han, J. Zhai, W. Xia, S. Jia, M. He and B. Han, Green Chem., 2022, 24, 1090–1095 RSC.
- Y. Wang, L. Zhang, Y. Niu, D. Fang, J. Wang, Q. Su and C. Wang, Green Chem., 2021, 23, 7594–7608 RSC.
- Y.-T. Xu, K.-C. Ren, Z.-M. Tao, D. K. Sam, E. Feng, X. Wang, G. Zhang, J. Wu and Y. Cao, Green Chem., 2023, 25, 589–595 RSC.
- A. Stirling, I. Pápai, J. Mink and D. R. Salahub, J. Chem. Phys., 1994, 100, 2910–2923 CrossRef CAS.
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS.
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef CAS.
- R. Daiyan, T. Tran-Phu, P. Kumar, K. Iputera, Z. Tong, J. Leverett, M. H. A. Khan, A. A. Esmailpour, A. Jalili, M. Lim, A. Tricoli, R.-S. Liu, X. Lu, E. Lovell and R. Amal, Energy Environ. Sci., 2021, 14, 3588–3598 RSC.
- J. Fu, F. Yao, T. Xie, Y. Zhong, Z. Tao, S. Chen, L. He, Z. Pi, K. Hou, D. Wang, X. Li and Q. Yang, Sep. Purif. Technol., 2021, 276, 119329 CrossRef CAS.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. F. Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- G. Liu, H. G. Yang, J. Pan, Y. Q. Yang, G. Q. (Max) Lu and H.-M. Cheng, Chem. Rev., 2014, 114, 9559–9612 CrossRef CAS.
- Z. Xiong, Z. Lei, Y. Li, L. Dong, Y. Zhao and J. Zhang, J. Photochem. Photobiol., C, 2018, 36, 24–47 CrossRef CAS.
- B. Shao, W. Zhao, S. Miao, J. Huang, L. Wang, G. Li and W. Shen, Chin. J. Catal., 2019, 40, 1534–1539 CrossRef CAS.
- Z. Xie, H. L. Tan, H. Wu, R. Amal, J. Scott and Y. H. Ng, Mater. Today Energy, 2022, 26, 100986 CrossRef CAS.
- H. L. Tan, X. Wen, R. Amal and Y. H. Ng, J. Phys. Chem. Lett., 2016, 7, 1400–1405 CrossRef CAS PubMed.
- J. Chen, X. Li, B. Lei, L. Zhou and S. Wang, Green Chem., 2019, 21, 483–490 RSC.
- J. Pan, G. Liu, G. Q. (Max) Lu and H.-M. Cheng, Angew. Chem., Int. Ed., 2011, 50, 2133–2137 CrossRef CAS PubMed.
- W. Wang, J. Fang, Y. Zhou, W. Zhang and C. Lu, RSC Adv., 2016, 6, 67556–67564 RSC.
- N. Sutradhar, A. K. Biswas, S. K. Pahari, B. Ganguly and A. B. Panda, Chem. Commun., 2014, 50, 11529–11532 RSC.
- P. Mikrut, D. Mitoraj, R. Beranek and W. Macyk, Appl. Surf. Sci., 2021, 566, 150662 CrossRef CAS.
- D. Sun, W. Yang, L. Zhou, W. Sun, Q. Li and J. K. Shang, Appl. Catal., B, 2016, 182, 85–93 CrossRef CAS.
- H. Zhou, M. Wang, F. Kong, Z. Chen, Z. Dou and F. Wang, J. Am. Chem. Soc., 2022, 144, 21224–21231 CrossRef CAS PubMed.
- H. Wu, L. Zhang, A. Du, R. Irani, R. van de Krol, F. F. Abdi and Y. H. Ng, Nat. Commun., 2022, 13, 6231 CrossRef CAS PubMed.
- H. Wu, R. Irani, K. Zhang, L. Jing, H. Dai, H. Y. Chung, F. F. Abdi and Y. H. Ng, ACS Energy Lett., 2021, 6, 3400–3407 CrossRef CAS.
- M.-S. Kim, S.-H. Chung, C.-J. Yoo, M. S. Lee, I.-H. Cho, D.-W. Lee and K.-Y. Lee, Appl. Catal., B, 2013, 142–143, 354–361 CrossRef CAS.
- J. Li, M. Li, X. Yang, S. Wang, Y. Zhang, F. Liu and X. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 33859–33867 CrossRef CAS PubMed.
- Y. Xu, K. Shi, T. Ren, H. Yu, K. Deng, X. Wang, Z. Wang, H. Wang and L. Wang, Small, 2022, 18, 2203335 CrossRef CAS PubMed.
- P. Mikrut, M. Kobielusz and W. Macyk, Electrochim. Acta, 2019, 310, 256–265 CrossRef CAS.
- W. P. Utomo, H. Wu and Y. H. Ng, Small, 2022, 18, 2200996 CrossRef CAS PubMed.
- X. Zhang, L. Luo, R. Yun, M. Pu, B. Zhang and X. Xiang, ACS Sustainable Chem. Eng., 2019, 7, 13856–13864 CrossRef CAS.
- Y. Zhang, J. Hu, C. Zhang, Y. Liu, M. Xu, Y. Xue, L. Liu and M. K. H. Leung, J. Mater. Chem. A, 2020, 8, 9091–9098 RSC.
- G. Liu, H. G. Yang, X. Wang, L. Cheng, H. Lu, L. Wang, G. Q. (Max) Lu and H.-M. Cheng, J. Phys. Chem. C, 2009, 113, 21784–21788 CrossRef CAS.
- Y. Zhai, Y. Ji, G. Wang, Y. Zhu, H. Liu, Z. Zhong and F. Su, RSC Adv., 2015, 5, 73011–73019 RSC.
- M. Fernández-Arias, M. Boutinguiza, J. del Val, A. Riveiro, D. Rodríguez, F. Arias-González, J. Gil and J. Pou, Nanomaterials, 2020, 10, 300 CrossRef PubMed.
- F. Yao, M. Jia, Q. Yang, F. Chen, Y. Zhong, S. Chen, L. He, Z. Pi, K. Hou, D. Wang and X. Li, Water Res., 2021, 193, 116881 CrossRef CAS PubMed.
- Z. Han, C. Choi, S. Hong, T.-S. Wu, Y.-L. Soo, Y. Jung, J. Qiu and Z. Sun, Appl. Catal., B, 2019, 257, 117896 CrossRef CAS.
- H. Wang, L. Wang, D. Lin, X. Feng, Y. Niu, B. Zhang and F.-S. Xiao, Nat. Catal., 2021, 4, 418–424 CrossRef CAS.
- Z. Song, M. N. Banis, L. Zhang, B. Wang, L. Yang, D. Banham, Y. Zhao, J. Liang, M. Zheng, R. Li, S. Ye and X. Sun, Nano Energy, 2018, 53, 716–725 CrossRef CAS.
- I. Jiménez-Morales, S. Cavaliere, D. Jones and J. Rozière, Phys. Chem. Chem. Phys., 2018, 20, 8765–8772 RSC.
- L. Lin, S. Yao, R. Gao, X. Liang, Q. Yu, Y. Deng, J. Liu, M. Peng, Z. Jiang, S. Li, Y.-W. Li, X.-D. Wen, W. Zhou and D. Ma, Nat. Nanotechnol., 2019, 14, 354–361 CrossRef CAS PubMed.
- K. Zhu, F. Shi, X. Zhu and W. Yang, Nano Energy, 2020, 73, 104761 CrossRef CAS.
- G. Wu, G. Zhao, J. Sun, X. Cao, Y. He, J. Feng and D. Li, J. Catal., 2019, 377, 271–282 CrossRef CAS.
- Y. Zhang, F. Du, R. Wang, X. Ling, X. Wang, Q. Shen, Y. Xiong, T. Li, Y. Zhou and Z. Zou, J. Mater. Chem. A, 2021, 9, 17442–17450 RSC.
- W. P. Utomo, H. Wu and Y. H. Ng, Energies, 2023, 16, 27 CrossRef CAS.
- C. Wang, Y. Zhang, H. Luo, H. Zhang, W. Li, W. Zhang and J. Yang, Small Methods, 2022, 6, 2200790 CrossRef CAS PubMed.
- O. Schmidt, A. Gambhir, I. Staffell, A. Hawkes, J. Nelson and S. Few, Int. J. Hydrog. Energy, 2017, 42, 30470–30492 CrossRef CAS.
- C. A. Fernandez, N. M. Hortance, Y.-H. Liu, J. Lim, K. B. Hatzell and M. C. Hatzell, J. Mater. Chem. A, 2020, 8, 15591–15606 RSC.
- G.-F. Chen, Y. Yuan, H. Jiang, S.-Y. Ren, L.-X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- D. Hartanto, G. Yuhaneka, W. P. Utomo, A. I. Rozafia, Y. Kusumawati, W. Dahani and A. Iryani, RSC Adv., 2022, 12, 5665–5676 RSC.
- X. Wang, M. Zhu, G. Zeng, X. Liu, C. Fang and C. Li, Nanoscale, 2020, 12, 9385–9391 RSC.
- P. Wang, L. Yang, Y. Q. Gao and X. S. Zhao, Nucleic Acids Res., 2015, 43, 7207–7216 CrossRef CAS PubMed.
- X. Niu, X. Nie, C. Yang and J. G. Chen, Catal. Sci. Technol., 2020, 10, 1881–1888 RSC.
- Y. Tang, C. Yang, M. Sheng, X. Yin and W. Que, ACS Sustainable Chem. Eng., 2020, 8, 12990–12998 CrossRef CAS.
- X. Teng, J. Wang, L. Ji, Y. Lv and Z. Chen, Nanoscale, 2018, 10, 9276–9285 RSC.
- C. Zhao, Y. Liang, W. Li, Y. Tian, X. Chen, D. Yin and Q. Zhang, RSC Adv., 2017, 7, 52614–52620 RSC.
- K. Ao, Q. Wei and W. A. Daoud, ACS Appl. Mater. Interfaces, 2020, 12, 33595–33602 CrossRef CAS PubMed.
- L. L. Zulfa, R. Ediati, A. R. P. Hidayat, R. Subagyo, N. Faaizatunnisa, Y. Kusumawati, D. Hartanto, N. Widiastuti, W. P. Utomo and M. Santoso, RSC Adv., 2023, 13, 3818–3834 RSC.
- H. Wu, S. Qu, Z. Xie and Y. H. Ng, ACS Appl. Energy Mater., 2022, 5, 8419–8427 CrossRef CAS.
- G. Yuhaneka, A. I. Rozafia, W. P. Utomo, A. Iryani and D. Hartanto, Malays. J. Fundam. Appl. Sci., 2022, 18, 463–472 CrossRef.
- H. Y. Chung, R. J. Wong, R. Amal and Y. H. Ng, Energy Fuels, 2022, 36, 11550–11558 CrossRef CAS.
- X. Fu, X. Zhao, X. Hu, K. He, Y. Yu, T. Li, Q. Tu, X. Qian, Q. Yue, M. R. Wasielewski and Y. Kang, Appl. Mater. Today, 2020, 19, 100620 CrossRef.
- E. Pérez-Gallent, M. C. Figueiredo, I. Katsounaros and M. T. M. Koper, Electrochim. Acta, 2017, 227, 77–84 CrossRef.
- S.-E. Bae and A. A. Gewirth, Faraday Discuss., 2008, 140, 113–123 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, Fig. S1–S19 and Tables S1 and S2. See DOI: https://doi.org/10.1039/d3gc02018e |
| This journal is © The Royal Society of Chemistry 2024 |