
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Shining light on hybrid perovskites for photoelectrochemical solar to fuel conversion
Sudhanshu
Shukla
 *abc,
Vishal
Jose
abc and
Nripan
Mathews
*de
*abc,
Vishal
Jose
abc and
Nripan
Mathews
*de
aImec, imo-imomec, Thor Park 8320, 3600 Genk, Belgium. E-mail: Sudhanshu.shukla@imec.be
bEnergyVille, imo-imomec, Thor Park 8320, 3600 Genk, Belgium
cHasselt University, imo-imomec, Martelarenlaan 42, 3500 Hasselt, Belgium
dSchool of Materials Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore. E-mail: Nripan@ntu.edu.sg
eEnergy Research Institute @ NTU, Nanyang Technological University, Research Techno Plaza, 50 Nanyang Drive, Singapore 637553, Singapore
First published on 17th July 2024
Abstract
Hybrid halide perovskites (HaPs) represent a class of material with excellent optoelectronic properties providing distinct avenues for disruptive photo(-electro) catalytic technologies. However, their photocatalytic activity, selectivity and stability remains a scientific and technological hurdle. In this perspective, we discuss fundamental aspects of perovskite based photocatalytic systems, specifically for CO2 conversion and high value oxidation reactions, and highlight critical limiting factors and on-going challenges in the field. We critically assess the recent advances in designing halide perovskite hetero-interfaces and characterization methodologies which are often used to define the performance metrics. Furthermore, we outline important questions and identify emerging trends in relation to the remediation strategy towards improved photocatalytic performance and stability from halide perovskite semiconductors.
 Sudhanshu Shukla | Sudhanshu Shukla is a senior researcher and Marie Skłodowska–Curie fellow at Interuniversity Microelectronics Centre (IMEC), Belgium. He obtained his PhD from Nanyang Technological University, Singapore in 2017. He was a visiting research scholar at Lawrence Berkeley National Laboratory (LBNL) and the University of California, Berkeley, USA (2016). His research interest includes fundamental understanding and application of novel compound semiconductors for photovoltaics and photoelectrochemical solar fuels generation. |
 Vishal Jose | Vishal Jose is a senior researcher at the IMOMEC division of IMEC. He also holds an MSCA fellowship that will commence in September 2024. He received his PhD from Nanyang Technological University, Singapore, in 2022. Dr Vishal's key research interests include electrocatalysis, solar fuels, and electrochemical recycling technologies. |
 Nripan Mathews | Nripan Mathews is an associate professor and the Provost's Chair in Materials Science and Engineering at the School of Materials Science and Engineering at Nanyang Technological University (NTU), Singapore. His interest lies in the development of novel and inexpensive electronic materials through cost-effective techniques for electronics and energy conversion. These include organic–inorganic halide perovskites, metal oxides, and organic thin films. He is interested both in the fundamental properties of the electronic materials as well as their applications in practical devices such as solar cells, thin-film transistors, and memory devices. |
Broader contextFossil fuel-dependent growth has led to an unprecedented rise in atmospheric CO2 levels. This has triggered climate change which is one of the biggest global challenges of our time. A paradigm shift to renewable energy is urgently required to decarbonize the economy and ensure carbon-neutral and sustainable growth. Artificial photosynthetic systems, inspired by natural photosynthesis, have garnered tremendous interest in the spontaneous generation of value-added chemical fuels from CO2 and water entirely from solar energy. Halide perovskites have emerged as a promising material system for photoelectrocatalysis, after their resounding success in photovoltaics. The versatile properties of halide perovskites unlock the key to facilitating many important catalytic reactions, beyond CO2 conversion. This perspective provides a comprehensive and critical assessment of the potential of cost-competitive halide perovskite-based photocatalytic systems. We intend to focus the research community's interest on driving value-added catalytic reactions using halide perovskites. By doing so, we identify fundamental issues that require immediate attention and provide clear future directions that must be considered for this technology to become a commercial reality and have tangible impact. |
1. Introduction
Fossil fuel dependent growth has led to an unprecedented rise in atmospheric CO2 levels. This has triggered a climate emergency which has become one of the biggest global challenges of our time.1 Therefore, a paradigm shift to renewable energy is urgently required to meet global energy demands and ensure sustainable growth in a cost-effective manner.2 Two dominant factors are fueling the rapidly transforming renewable energy landscape – (i) a decrease in the levelized cost of electricity (LCOE) of photovoltaics,3 and (ii) heavy reliance on fossil-based resources to generate liquid fuels for transportation and chemical feedstocks for high value synthetic chemicals in fertilizer and pharmaceutical industries.4 While falling PV prices are favorable for ever increasing energy demands, the associated intermittency related to diurnal changes and climatic variations pose a limit to its applicability, especially in hard to decarbonize sectors such as transportation.5 Thus, renewable technologies aiming to convert CO2 and store energy in the form of synthetic fuels and value-added chemicals are highly desirable not only for developing alterative sustainable fuels but also for closing the carbon cycle.Solar-driven photo(electro)catalysis systems, inspired from natural photosynthesis, enable spontaneous generation of high energy density and value-added chemical fuels from CO2 and water, also known as artificial photosynthesis, solar fuels, and carbon-capture, and utilization (CCU).6 The energy density (per unit weight or volume) of photoelectrochemically produced molecules is higher than those of conventionally used lithium batteries, making them suitable for long-term storage and mobility.7 Thus, a clean, cost-competitive, and flexible alternative to current fossil-fuel based technologies is offered for sustainable intersectoral societal growth.
The idea of renewably sourced chemicals from CO2 recycling has intrigued researchers for decades, since the first demonstration of photocatalytic water splitting on a semiconducting TiO2 surface by Honda and Fujishima8 and later, photoelectrochemical CO2 reduction using a p-GaP semiconductor by Halmann.9 Since then, strides have been made in photocatalytic water splitting in terms of efficiency and scale.10 In contrast, the progress in photoelectrochemical CO2 reduction remained limited due to complexities associated with solvation dynamics, activation barrier, reaction kinetics (multiple electron and proton transfer processes), preferential dimerization, material stability in aqueous media and difficulties related to up-scaling. Moreover, the interference of impurities and spurious carbon signals renders unreliable quantification of product distribution from photocatalytic CO2 reduction.11
Halide perovskites (HaPs) have emerged as low-cost, easy processable semiconductors with intriguing properties like long charge-carrier diffusion lengths and bandgap tunability and direct optical excitation with strong absorption coefficients (>105 cm−1). With 26.54% certified power conversion efficiency,12 HaP based solar cells have outcompeted many established thin film photovoltaic technologies in just 10 years. In parallel, the versatile properties of HaPs place them at the forefront of photocatalysis. For the first time, a HaP was exploited for conducting photocatalytic CO2 reduction by Y.-F. Xu et al., in 2017.13 The authors utilized CsPbBr3 QDs and their composites with graphene oxide for artificial CO2 reduction in ethyl acetate solvent, where the pristine QDs demonstrated an average electron consumption rate of 23.7 μmol g−1 h−1. Since then multiple studies have been conducted to improve the efficiency, selectivity, and stability of various HaPs in organic and inorganic solvents along with shedding light on reaction mechanisms and material degradation processes. For instance, recently, L. Ding and co-workers revealed the CO2 reduction potential of Cs2AgBiBr6 QDs encapsulated in a metal organic framework by attaining CO production rates of 309.01 μmol g−1 h−1.14 However, the selectivity of these materials is still primarily limited to CO and CH4, unlike materials such as carbon nitride that can selectively generate higher order carbon products. For example, the C2H6 evolution rate was 616.6 μmol g−1 h−1,15 and the CH3OH evolution rate was 13.9 μmol g−1 h−1.16 Profound advancements have been made in tailoring and employing HaPs for various redox reactions and chemical valorisation.
2. Considerations and fundamental challenges for driving key photoelectrochemical reactions: there is plenty of room for perovskites
Several high energy density and value-added compounds can be targeted through photoelectrochemical reduction of environmental feedstocks such as water, CO2 (hydrocarbons), and N2 to obtain hydrogen, hydrocarbons and ammonia, respectively. For instance, CO2 can be reduced and converted to obtain several high energy densities and value-added gaseous and liquid C1 (such as CO, methane, methanol, formic acid) and C2+ (such as ethylene, ethanol, propanol, ethylene glycol) chemical products.17 Thermodynamically, CO2 reduction is an energy demanding reaction. The energy required to break the C–O bond is ∼750 kJ mol−1. This is slightly higher than the electrochemical potential required for the hydrogen evolution reaction (HER) (∼237 kJ mol−1). However, kinetic factors related to CO2 activation barrier, and complex multi-step reaction pathways push the energetics well above the thermodynamic limit, giving rise to electrochemical overpotential.18 Similarly N2 can be converted to NH3, although it poses a higher challenge due to the high energy required to break the strong N–N bond. Standard reduction potentials (vs. RHE) and typical products are given below for various reactions involving electron and proton transfers:19,20| 2H+ + 2e− → H2 (ERHE = 0.00 V) | (1) |
| CO2 + 2H+ + 2e− → HCO2H (ERHE = −0.17 V) | (2) |
| CO2 + 2H+ + 2e− → CO + H2O (ERHE = −0.10 V) | (3) |
| CO2 + 4H+ + 4e− → HCHO + H2O (ERHE = −0.07 V) | (4) |
| CO2 + 6H+ + 6e− → CH3OH + H2O (ERHE = +0.02 V) | (5) |
| CO2 + 8H+ + 8e− → CH4 + 2H2O (ERHE = +0.17 V) | (6) |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O (ERHE = +0.08 V) | (7) |
| 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O (ERHE = +0.09 V) | (8) |
| 2CO2 + 14H+ + 14e− → C2H6 + 4H2O (ERHE = +14 V) | (9) |
| N2 + 6H+ + 6e− → 2NH3 (ERHE = 0.00 V) | (10) |
Oxidation reduction:
| 2H2O + 4H+ → O2 + 4H+ (ERHE = +1.23 V) | (11) |
Therefore, the fundamental limitation arises from the overall voltage requirement imposed by thermodynamics and kinetics. Typically, the other half of the reduction reaction is generally coupled to the oxidation reaction, namely the oxygen evolution reaction (OER). From the above mentioned overpotential losses, the cumulative voltage requirement for the coupled HER–OER or the CO2R–OER reaction exceeds beyond 1.8 V. Fig. 1a presents the typical reduction (in blue) and oxidation (in black) reactions performed at the photocathode and photoanode, respectively. Thus, the choice of anodic reaction is not merely limited to the OER. There are alternative oxidation reactions (AOR) such as – iodide oxidation, glycerol oxidation, plastic and biomass oxidation, degradation of organic pollutants, and selective organic transformation.21 These are useful to obtain high value chemical compounds with overall less voltage requirement.
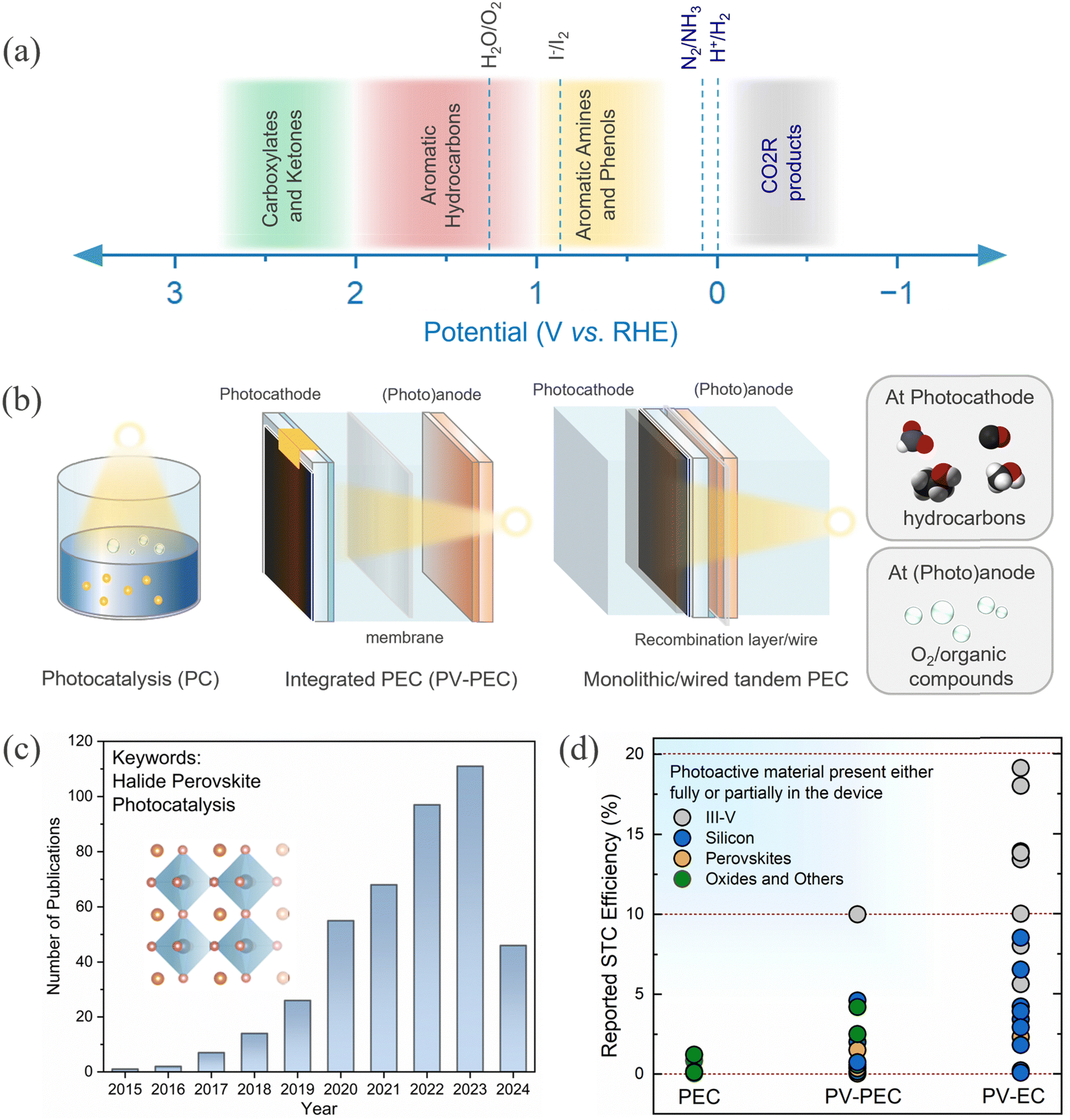 | ||
| Fig. 1 (a) Common reduction and organic oxidation reactions are depicted over potential ranges.20–23 (b) Different architecture and corresponding components of a PC, integrated PV-PEC and monolithic/wired tandem PEC device. PEC devices are configured with at least one photoelectrode (photocathode with anode or photoanode with cathode) and the integrated PEC may have a wired connected PV component. (c) Trends in publications on halide perovskite related papers on photoelectrocatalysis for the period 2015 to 2024 (source: Scopus; keyword: halide perovskite and photocatalysis, assessed on 25 April 2024). (d) Reported solar-to-carbon (STC) efficiency for different device architectures using various photoactive materials.24–55 | ||
The input energy for endergonic reduction can be provided by the photocatalyst (an irradiated semiconductor in direct contact with the reactants) in the form of photovoltage. The fundamental principle behind a photo(electro)catalytic process consists of (i) efficient absorption of photons followed by the generation of electron hole pairs, (ii) carrier separation and transport to the surface, and (iii) electrochemical reduction or oxidation reaction involving electrons from the conduction band or holes from the valence band.56 The overall photocurrent of the process can be expressed as:
| J = Jabs × ηtrans × ηFE | (12) |
Hence, the photocurrent and photovoltage are two critical parameters that determine the overall efficiency. Subsequent sections in the article describe how these two parameters can be optimized.
It is essential to consider different routes57 that are used to accomplish HaP driven photoelectrochemical CO2R: (1) photocatalysis (PC), (2) photoelectrocatalysis (PEC), and photovoltaic integrated PEC (PV-PEC) or buried junction PEC, and (3) tandem PEC scheme as depicted in Fig. 1b. Alternatively, PV powered electrolysis (PV-EC) is also a viable way to accomplish CO2R through electrolysis. However, we will keep our focus on direct and integrated approaches in this perspective. A good account of the latest summary on perovskite-based PV-EC can be found in ref. 58. Recently, there has been a surge in research activities on HaPs for photo(electro)catalysis, evident from the rapidly growing trend of the publications in this field (Fig. 1c). Fig. 1d depicts a comparative view of the solar to carbon (STC) efficiency for different device architectures and material systems. It becomes clear that STC efficiencies for PEC and PV-PECs are well below 10% for most of the materials. It is straightforward to envisage above 10% STC from HaP devices, considering that comparable optoelectronic properties can be achieved for HaPs as for the III–V semiconductor (GaAs).
Inorganic metal oxides such as TiO2, WO3, ZnO, and α-Fe2O3 appeared as early adopters for photocatalytic systems, benefiting from the high electronegativity of oxygen, versatile metal–oxygen chemistry, and economic viability.59–61 The ionic character of the bonds between the metal and oxygen atomic orbitals results in stable compounds with sufficiently large bandgaps, that typically straddle the redox potentials of various reduction (CO2, H+) and oxidation reactions (water, ethylene glycol, and 5-hydroxymethylfurfural). However, the wide bandgap of these materials limits the spectral range of optical absorption, and they suffer from high recombination losses due to a plethora of point defects and polaron formation, severely limiting the availability of carriers for photocatalysis.62 Beyond oxides, transition metal dichalcogenides (TMDs) have garnered significant attention for photocatalysis, exhibiting superior optoelectronic properties such as optical absorption, charge transport and rich defect chemistry.63–66 However, the number of chalcogenides with sufficiently high bandgap is relatively small and, in addition, there are aggravated instability concerns related to photocorrosion in aqueous media.65,67
Halide perovskites (HaPs) have emerged as a promising class of materials for a wide variety of optoelectronic applications; solar cells,68 photodetectors,69 light-emitting diodes (LED),70 lasers,71 memristors,72 and photoelectrocatalysis.73 The rapid evolution of HaP based devices and their performance is underpinned by their exceptional optoelectronic properties such as defect tolerance, direct bandgap with high optical absorption and ambipolar transport with long-range balanced diffusion lengths. Carrier diffusion lengths, given by 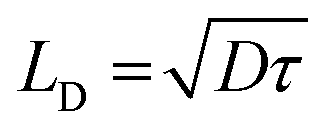 , of up to 1 μm have been observed, manifesting the low defect density in HaPs. Another feature associated with the defect or disorder induced band tailing is the Urbach energy (Eu). The significantly low value of Urbach energy (∼15–30 meV) is a manifestation of the superior optoelectronic quality of HaPs, compared to conventional semiconductors (>30 meV).74 A gamut of unique properties of HaPs stems from its high degree of compositional flexibility and tuneability of structure, dimension, and electronic properties. The structure of perovskites can be easily engineered, beyond archetypal MAPbI3, to yield a variety of inorganic–organic hybrids and purely inorganic systems, and facile dimensional tailoring enables realization of 3D, 2D, 1D, 0D, and even mixed dimensional HaPs.75,76 This allows band gap tuneability in a wide spectral range (from ultraviolet to near infrared) and provides control over energy band alignment for favorable energetics. Considering the above properties, HaPs seemingly satisfy the key requirements for photocatalytic devices by ensuring efficient optical absorption of photons, electron–hole pair generation, loss-less carrier transport, and seamless utilization of carriers at the interface to drive redox reactions with sufficiently high rates. However, one of the major limitations that has kept its potential untapped is their aqueous stability, and achieving product selectivity beyond C1 (CO, CH4) compounds. This requires more understanding of several unexplored elements related to composition, morphology, charge separation, passivation, and catalytically active sites in HaPs. The development of a HaP based photocatalyst relies on improving the optoelectronic as well as photocatalytic properties and mechanistic insights into the underlying descriptors. We discuss important factors governing the photocatalytic activity of HaPs and present methodologies to gain fundamental understanding, as shown in Fig. 2.
, of up to 1 μm have been observed, manifesting the low defect density in HaPs. Another feature associated with the defect or disorder induced band tailing is the Urbach energy (Eu). The significantly low value of Urbach energy (∼15–30 meV) is a manifestation of the superior optoelectronic quality of HaPs, compared to conventional semiconductors (>30 meV).74 A gamut of unique properties of HaPs stems from its high degree of compositional flexibility and tuneability of structure, dimension, and electronic properties. The structure of perovskites can be easily engineered, beyond archetypal MAPbI3, to yield a variety of inorganic–organic hybrids and purely inorganic systems, and facile dimensional tailoring enables realization of 3D, 2D, 1D, 0D, and even mixed dimensional HaPs.75,76 This allows band gap tuneability in a wide spectral range (from ultraviolet to near infrared) and provides control over energy band alignment for favorable energetics. Considering the above properties, HaPs seemingly satisfy the key requirements for photocatalytic devices by ensuring efficient optical absorption of photons, electron–hole pair generation, loss-less carrier transport, and seamless utilization of carriers at the interface to drive redox reactions with sufficiently high rates. However, one of the major limitations that has kept its potential untapped is their aqueous stability, and achieving product selectivity beyond C1 (CO, CH4) compounds. This requires more understanding of several unexplored elements related to composition, morphology, charge separation, passivation, and catalytically active sites in HaPs. The development of a HaP based photocatalyst relies on improving the optoelectronic as well as photocatalytic properties and mechanistic insights into the underlying descriptors. We discuss important factors governing the photocatalytic activity of HaPs and present methodologies to gain fundamental understanding, as shown in Fig. 2.
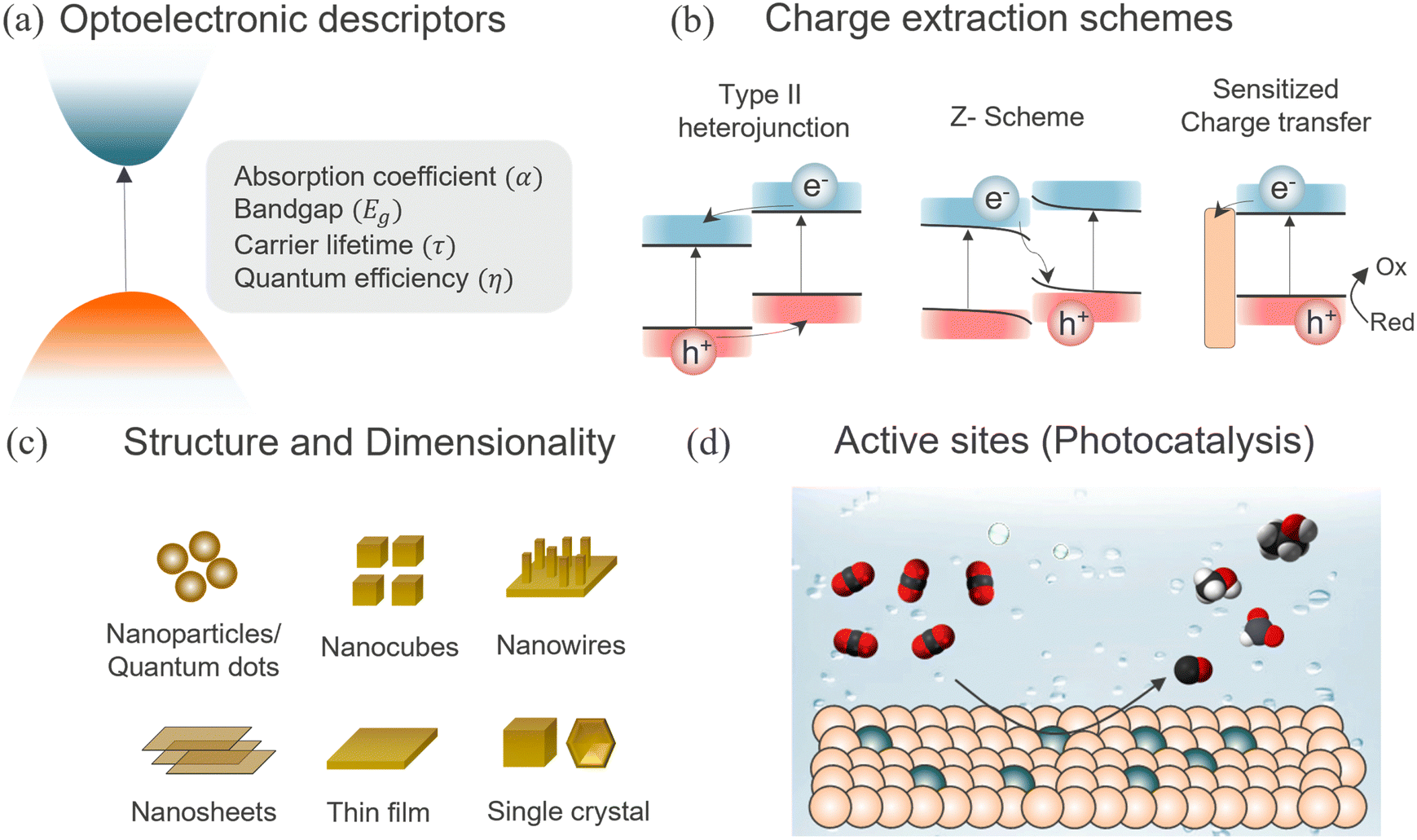 | ||
| Fig. 2 Overview of the themes covered in this perspective. (a) Perovskite band structure and key optoelectronic properties, (b) different heterojunction schemes for efficient charge transfer, (c) influence of different morphology and dimensionality and (d) the role of surface chemistry in defining the catalytically active site. | ||
In this perspective, we identify the mechanistic challenges that need to be addressed and discuss potential routes to advance the field and rationalize the role that perovskites can play in the abatement of CO2 emissions and facilitate its solar driven conversion to value-added chemicals and sustainable fuels.
3. Optoelectronic aspects of a halide perovskite based photocatalytic system
The excellent optoelectronic properties of HaPs are due to their peculiar crystallographic structure and electronic structure. The general formula for perovskites is ABX3, where the A site is typically occupied by a monovalent organic/inorganic cation (CH3NH3+ or Cs+), B is a divalent metal (Pb2+, Sn2+), and X is a single or mixed halide anion (I−, Br−, Cl−). The structure remains stable for a wide range of small (Cs+) and large cation sizes (CH3NH3+), governed by an empirical Goldschmidt tolerance factor.77 The bandgap is formed primarily due to the hybridization between metal (B) and halide (X) orbitals.The valence band maximum (VBM) is dominated by the X p characteristics (with some B site contribution) and the conduction band minimum (CBM) is derived from π antibonding of B p and X p orbitals. A cation seems to primarily behave as a spacer and does not directly contribute to the electronic structure but influences the bandgap via lattice deformations. Thus, the direct bandgap along with favorable p → p transition are the key factors behind high optical absorption in HaPs.68 Notably, strong spin–orbit coupling (SOC), due to the presence of heavy metals (like Pb), has been shown to influence the optical transition and carrier lifetime due to Rashba splitting of the CBM.78–80
Broad spectral tunability and different bandgaps can be achieved by facile halide substitution and varying compositions of HaPs, as shown in Fig. 3a. The conduction band position for most of the perovskites is sufficiently negative relative to the redox potential of water splitting, CO2R and N2 reduction reaction (N2RR),81 providing the driving force to facilitate these reactions. The conduction band position can be varied through halide substitution to selectively match the redox potential. It is also evident that some HaPs have a valence band position positive enough to make them suitable for water oxidation and even for high value oxidation reactions like HMF and ethylene glycol oxidation.21 HaPs are mostly investigated as photocatalysts for reduction reactions which are commonly coupled to a water oxidation reaction. The photoexcited electrons in the conduction band reduce CO2, while the holes in the valence band are consumed in H2O oxidation generating oxygen. Water also acts as a source of protons for hydrogenation of photocatalyzed CO2, in addition to serving as a reducing agent.
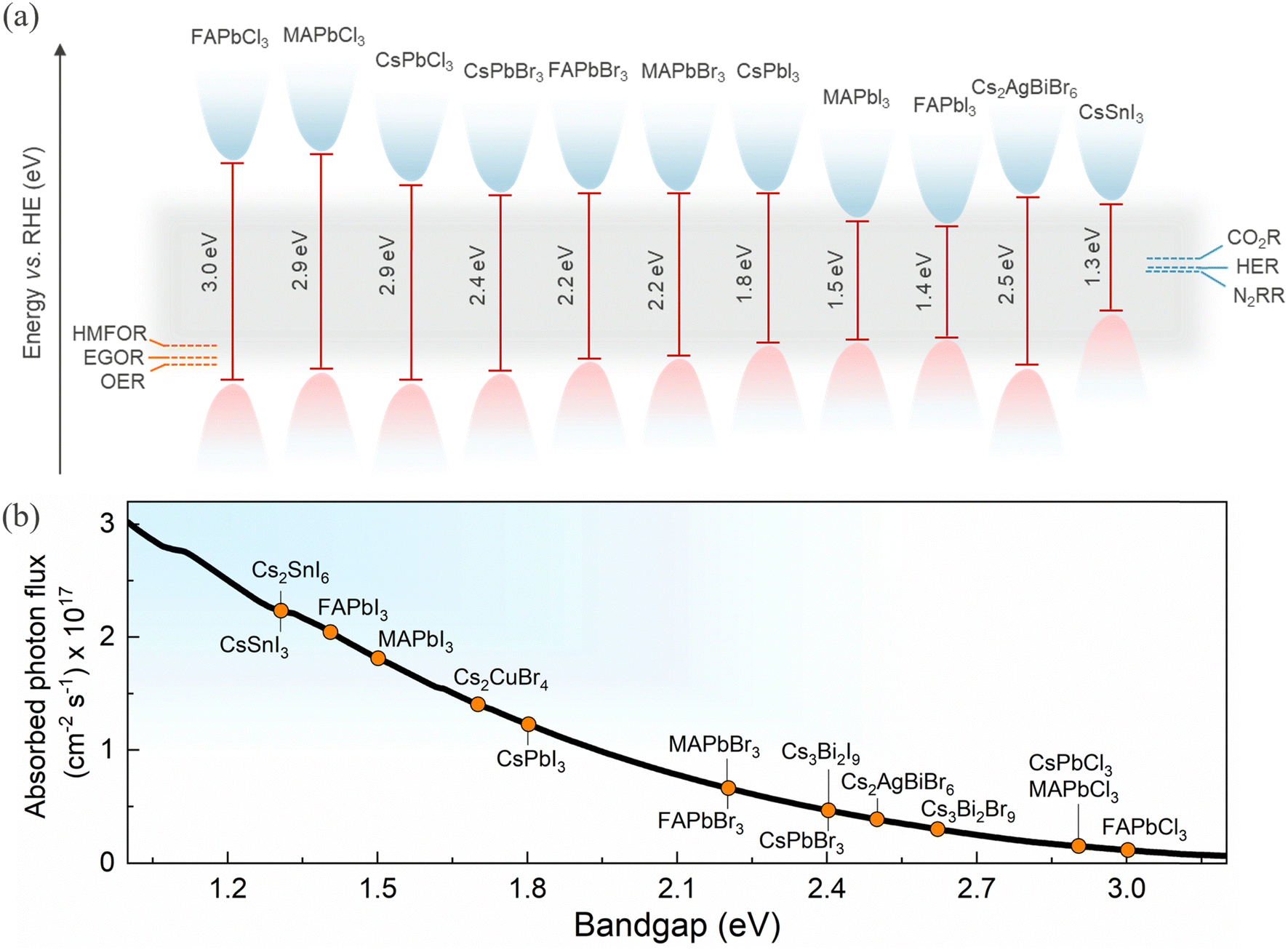 | ||
| Fig. 3 (a) Valence and conduction band positions of selected HaPs with respect to relevant redox cathodic and anodic reactions.82,83 (b) AM1.5G equivalent steady-state absorbed photon flux available for different bandgap HaPs (assuming step-function-like absorptivity) representing their light absorption ability. | ||
In practice, a combination of photocatalysts is more conducive for optimum light absorption, as shown in Fig. 3b, while maintaining the necessary photochemical potential higher than the redox potential. This is generally achieved by forming heterostructures.
3.1. Photophysical and charge transfer processes in halide perovskites
Upon irradiation of light above or equal to the bandgap, photoexcited electron–hole pairs are generated and migrate to the surface primarily through diffusion. Drift transport is believed to be less important in perovskites, as the electric fields are screened by the presence of moving ions.84 During the migration, carriers undergo various photophysical radiative and/or non-radiative recombination processes due to the presence of disorder, shallow traps, and deep defects. The lower defect density in HaPs ensures long carrier diffusion lengths, implying a reasonably high carrier recombination lifetime and mobilities. Since photoelectrochemical reactions are kinetically slow processes, the photoexcited carrier lifetime must be long enough to ensure the availability of carriers at the surface for redox reactions (Fig. 4a).85 Thus, charge carrier lifetime (τ) is the most essential descriptor to describe the photocatalytic ability associated with its optoelectronic quality. Determination of the carrier lifetime has mostly relied on the photoluminescence (PL) based measurements. Fig. 4a shows the carrier lifetime values reported in the literature and the corresponding regime for various HaPs, deduced from photoluminescence measurements. Apart from PL, other transient techniques such as – time resolved microwave conductivity (TRMC), optical pump THz probe (OPTP) spectroscopy, transient absorption spectroscopy (TAS), transient photovoltage (TPV) and photoconductivity measurements, have also proven to be quite useful in lifetime assessment.86–88 For instance, Chen et al. revealed a long carrier lifetime of up to 30 μs and 2.7 ms for MAPbI3 polycrystalline films and MAPbBr3 single crystals, respectively, from steady-state photoconductivity and Hall measurements.89 Brenes et al. observed a carrier lifetime of 32 μs in the passivated MAPbI3 thin films from TRMC measurements.90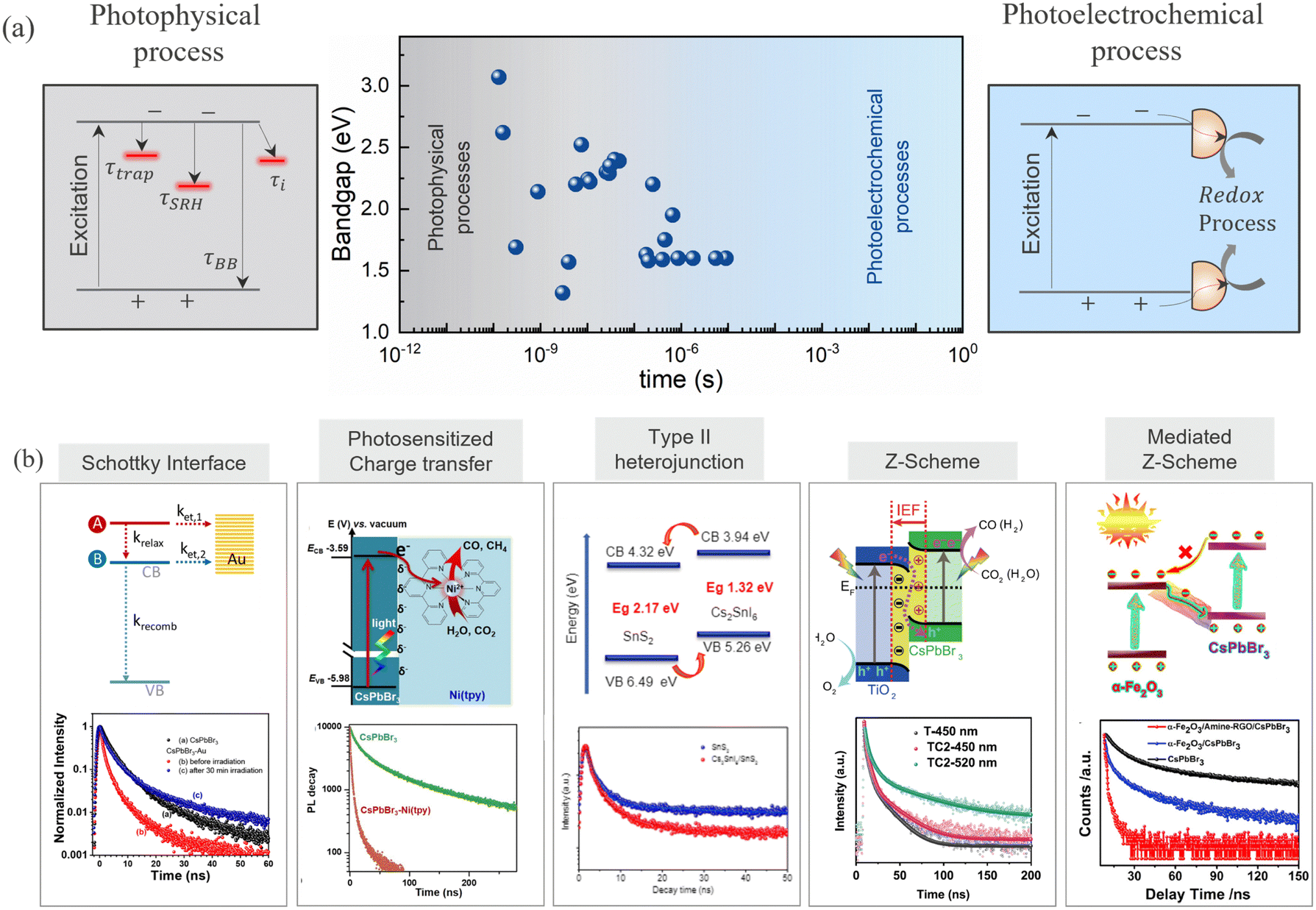 | ||
| Fig. 4 (a) Charge carrier lifetimes for various HaPs derived from transient photoluminescence spectroscopy. Photophysical and photoelectrochemical regimes are differentiated based on the different timescales. The data points are taken from the literature.91–113 (b) Heterojunction schemes utilized to separate the photogenerated charge carriers and drive photoelectrochemical CO2 reduction from HaPs. Schottky interface, reprinted with permission from ref. 114. Copyright 2021 American Chemical Society. Photosensitized charge transfer, reprinted with permission from ref. 92. Copyright 2021 American Chemical Society. Type II heterojunction, reprinted with permission from ref. 93. Copyright 2019 American Chemical Society. Z-Scheme, reproduced from ref. 94. Copyright 2020 Nature Publishing Group. Mediated Z-Scheme, reproduced from ref. 95. Copyright 2020 Cell Press. | ||
Photophysical processes like – trap assisted recombination, Shockley–Read–Hall (SRH) recombination, and surface or interface recombination reduce the carrier lifetime and manifest as a change in decay dynamics of transient PL. All these processes are usually concealed in the carrier decay dynamics. While it is straightforward to deduce lifetimes from transient profiles, it is hardly possible to discern underlying physical recombination processes without fluence dependent measurements and an appropriate model.115 The identification of the lifetime limiting process is imperative to develop focused passivation strategies and enhance the carrier lifetime. Several strategies ranging from doping and alloying,116 nanostructuring,117,118 bulk and passivation,91,119–122 and dimensional tailoring,123–125 have been demonstrated to enhance the lifetime of carriers in HaPs. Lessons from photovoltaics could be extremely useful to design passivation strategies and harness the photocatalytic activity from enhanced carrier lifetimes.
Next to long carrier lifetimes, charge carrier extraction is extremely important for efficient functioning of a photocatalytic device. Charge carrier separation is achieved through different heterojunction schemes, as shown in Fig. 4b. The common heterojunction charge transfer schemes are – Schottky junction, type II heterojunction, Z-scheme heterojunction (direct and mediated), molecular sensitization. Heterojunction layers commonly serve the dual role of passivation and carrier selective transport layer.126–128 It is shown that despite long carrier diffusion lengths, carrier collection can be limited by unoptimized, and low mobility charge transport layers.129 The charge transfer from perovskites reduces the carrier density within the perovskite absorber, therefore the concomitant drop in the PL intensity can act as a qualitative descriptor of the transfer kinetics. Consequently, transient PL has been exploited as a quantitative method to analyze recombination and charge transfer kinetics. This is evident from the reduction in PL lifetimes for CsPbBr3/Au (Schottky),114 CsPbBr3/[Ni(tertpy)2]2+ (molecular sensitizer),92 Cs2SnI6/SnS2 (type II),93 CsPbBr3/TiO2 (Z-scheme),94 FAPbBr3/α-Fe2O3 (Z-scheme),130 and CsPbBr3/rGO/α-Fe2O3 (mediated Z-scheme),95 heterojunctions studied in the literature, as shown in Fig. 4b. The enhanced charge separation correlates directly with the improvement in the respective photocatalytic activities. The trend holds across a wide range of heterojunction combinations – CsPbBr3 QDs/GO,13 CsPbBr3/g-C3N4,131 CsPbBr3/TiO–g-C3N4,132 CsPbBr3/MXene nanosheets (Ti3C2Tx),133 CsPbBr3/N-doped carbon dots,134 CsPbBr3 QDs/Bi2WO6 nanosheet,135 FAPbBr3/Bi2WO6,136 CsPbBr3/CdS,137 ZnSe nanorods/CsSnCl3,138 and Cs3Bi2Br9/g-C3N4.139 Some of these benefit from an interfacial electric field that further assists in charge separation. High surface area and porous metal organic frameworks (MOFs) have also been explicitly used as charge extracting layers which are advantageous against insulating SiO2,140 or an inorganic/polymer matrix.141 CsPbBr3–zeolite imidazolate (ZIF) core–shell composite,96 MAPbI3 QDs/PCN-221 (Fe-based porphyrin) encapsulated structure,97 and Cs3Bi2Br9 and Cs2AgBiBr6 nanodots/mesoporous titania facilitated enhanced electron transfer to promote photocatalytic CO2 reduction to CO and CH4 respectively.98 While lifetime changes help, the unambiguous determination of charge transfer from PL kinetics requires the knowledge of carrier injection levels. This is frequently overlooked in the measurements. Thus, fluence conditions are extremely critical to unambiguously discern decay components related to charge transfer and not due to recombination activity. Moreover, subjecting to similar light illumination conditions used during the device testing would be beneficial to gain mechanistic understanding.
Another concern is regarding the steady-state PL quenching, which is also described as a marker of the charge transfer process. However, PL quenching may also occur due to enhanced recombination at the interface and/or reabsorption effects from the heterojunction layer rather than charge transfer. Ideally, the heterojunction should maintain a high PL under open-circuit conditions (which is mostly the case) due to the passivating nature of the interface. This way steady-state PL is quite useful to screen the passivating interfaces. On the other hand, rapid PL quenching should occur when deviating from open circuit conditions i.e., when the charge carriers are efficiently extracted. Considerations on energy band alignments are necessary to ensure efficient transfer of charges.
3.2. Charge separation and its utilization for catalysis
Despite a plethora of experimental demonstrations, the fundamental understanding remains unclear regarding how separated charge carriers participate in the catalytic reaction. How many separated charge carriers indeed participate in the photocatalytic reaction and determine the faradaic efficiency? How do surface traps/defects influence the surface charge and photocatalytic activity? What is the effect of charge separation on stability? These questions can be partially addressed by in situ and operando spectroscopic techniques such as photoluminescence, transient absorption, and impedance spectroscopy, which can provide valuable mechanistic insights with high spatial resolution.The success of defect passivation and charge transport layers in HaPs based solar cells has not been translated to photoelectrochemical devices. In situ studies can provide mechanistic insights on charge transfer at the complex HaPs/electrolyte interface and determine the rate limiting step. Multilayered tandem architectures and selective co-catalyst integration should be explored to maximize charge transfer efficiency. Thus, minimization of energy loss should be considered at every interface. Recent demonstrations on the possibility of hot carrier extraction in particulate HaPs photocatalysts expand the capabilities of these materials.
4. Photocatalytic reactions on halide perovskites
4.1. Engineering morphology and dimensionality for enhanced photocatalytic performance
HaPs morphology and dimensional engineering have been shown to influence photocatalytic activity through a change in the local electronic structure induced by the surface coordination environment, catalytic reaction sites, surface charge density, binding energy of CO2 and intermediates, and surface area. In terms of morphology, quantum confined HaP structures have been explored in photocatalysis, such as QDs (0D), nanorods/nanowires (1D), nanosheets/nanoplatelets (2D), and nano/microcrystals crystals (>100 nm, 3D).142Nanocrystalline HaPs show enhanced photocatalytic activity due to their high surface-to-volume ratio and demonstrate more resilience to phase transformations. However, they tend to be less stable compared to their bulk counterparts. Specific advantages and opportunities for nanocrystalline HaPs include suppressed phase segregation, exploitation of hot carriers, and high surface area. On the other hand, charge carrier extraction is challenging in such nano dimensional systems due to the excitonic nature of the optical excitations as evidenced from high PL quantum yields (PLQY) and lower lifetimes. An additional challenge comes from charge transport limitations as many of the QDs are coated with insulating ligands. Hence, the efficiency of perovskite QD solar cells is far lower than perovskite thin film solar cells. A comparative view based on important metrics for photocatalysis for single crystals, thin films and nanoparticles is shown in Fig. 5.
 | ||
| Fig. 5 Radar chart depicting the comparison of different metrics for nanocrystals, thin films, and single crystals HaP-based photocatalytic systems. | ||
Lowering the dimensionality leads to a higher surface area which is beneficial for catalytic charge transfer. A study on nanocrystal size dependence showed higher photocatalytic activity and stability for a 8.5 nm quantum confined nanocrystal, leading to a longer PL lifetime of 9.7 ns.143 Two dimensional (2D) layered structures provide better conducting pathways for charge transfer due to the large interfacial area. For instance, Jiang et al. fabricated a heterojunction from CsPbBr3/Bi2WO6 2D sheets for CO2 reduction,144 where a higher interface area of 2D sheet led to a 5-fold increase in CO2 conversion yield compared with bare CsPbBr3 nanosheets (Fig. 6a–c). Functionalization of a co-catalyst can further help in boosting the photocatalytic activity. However, maintaining the structural integrity of the co-catalyst on the support and avoiding precipitation during the photocatalytic reaction is extremely daunting.145 The improvements from nano sizing, although seemingly obvious, need to be rationalized for long term performance. Research work from Zhu et al. critically assessed the viability of CsPbBr3 nanoparticles with different sizes (4 nm to 24 nm) for organic transformation, as shown in Fig. 6d and e. They observed that while faster photocatalytic activity is observed initially for smaller HaP nanocrystals, the overall yield remained less than larger nanocrystals in long term operation146 (Fig. 6f).
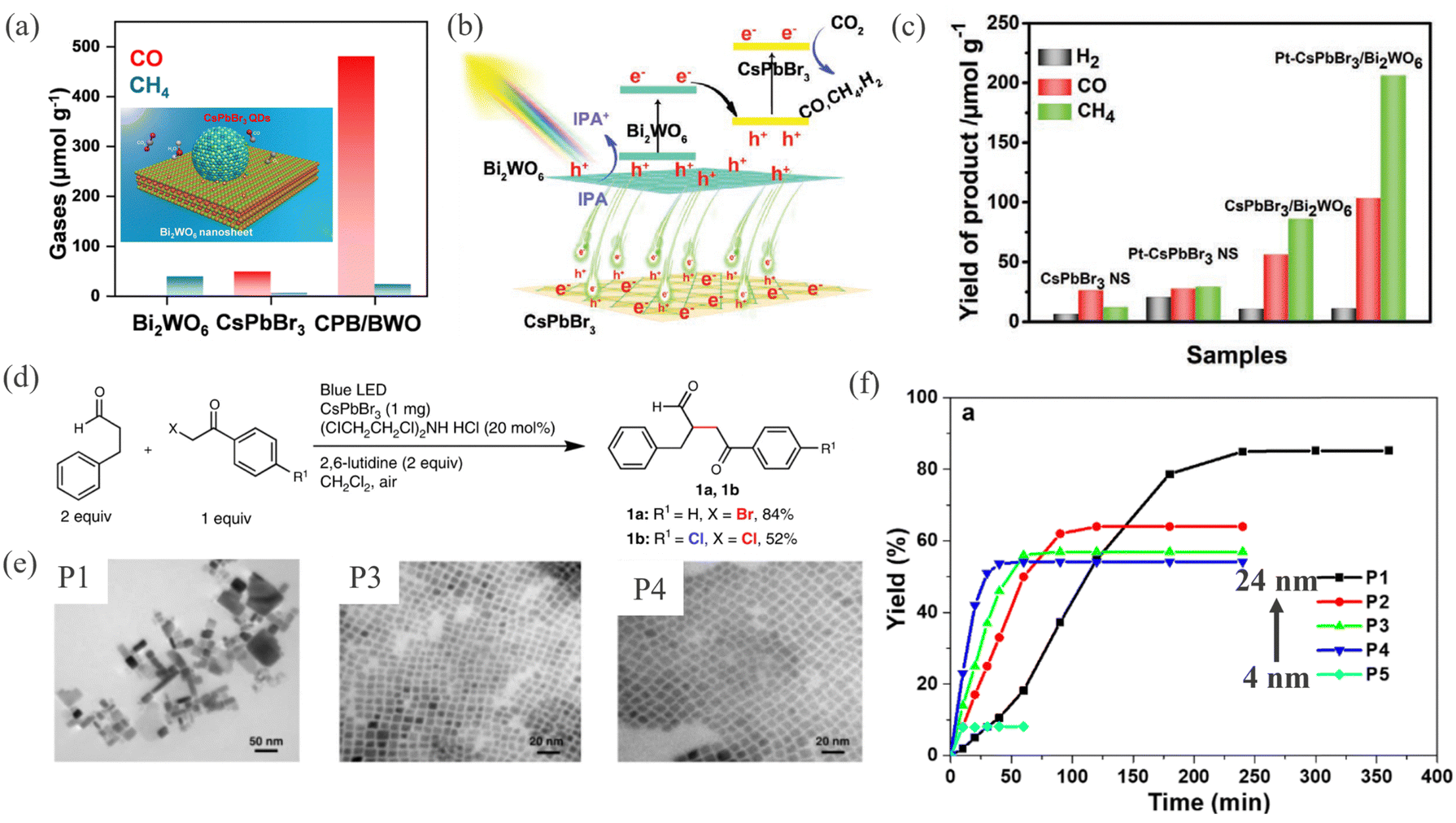 | ||
| Fig. 6 (a) Product yield during CO2 photoreduction/H2O photooxidation over 0D/2D CsPbBr3/Bi2WO4 and its individual components. The inset shows a schematic illustration of the CsPbBr3 zero-dimensional (0D) nanocrystals. Reprinted with permission from ref. 135. Copyright 2020 American Chemical Society. (b) Conceptual band diagram of the 2D/2D heterojunction depicting the Z-scheme and charge carrier dynamics. (c) Product yield rates during photocatalytic CO2 reduction/isopropyl alcohol oxidation over the 2D/2D heterostructure system and its individual components. Reproduced from ref. 144. Copyright 2020 Wiley. (d) Organic transformation reaction and the resulting product. (e) Transmission electron microscopy images of various sizes of CsPbBr3 nanocrystals as photocatalysts. (f) Percent yield of product over time for CsPbBr3 nanocrystals as a function of nanocrystal size (P1, P2, P3, P4 and P5 refer to 24 nm, 14 nm, 9 nm, 6 nm, and 4 nm respectively). Reproduced from ref. 146. Copyright 2019 Nature Publishing Group. | ||
4.2. Exploitation of different crystal shapes and facets
The synthesis of different shapes of halide perovskite nanoparticles offers new opportunities in photocatalysis. Reasonable success has been achieved in synthesizing shape-controlled nanoparticles, such as facile room temperature synthesis of CsPbX3 nanoparticles.147 Since halide perovskites have long electron and hole diffusion lengths, the anisotropy in the crystal structure can lead to different electron and hole migration pathways which can help in spatial decoupling of the reduction and oxidation reactions. Li et al. have showed migration of holes and electrons to the edge (100) and (006) basal facets, respectively, on Cs3Bi2I9 hexagonal prisms, using Co2+ (for oxidation) and Pt4+ (for reduction) as redox probes, as shown in Fig. 7a.148 Different atomic arrangements on crystal facets dictate the surface energy and the interaction with the reaction environment. Selective functionalization of co-catalysts over crystal planes that have reduced kinetic barriers for photocatalytic CO2 reduction is highly interesting. This strategy has recently showed quantum efficiency approaching 100% for photocatalytic water splitting reaction by selectively functionalizing Rh/Cr2O3 at (100) and CoOOH at the (110) facet of the SrTiO3-Al nanoparticle as a HER and OER co-catalyst, respectively.149 Similarly, taking advantage of the crystallographic anisotropy, pseudo type-II facet selective CsPbBr3–sulfobromide Pb4S3Br2 epitaxial heterostructures have been demonstrated to improve the catalytic activity (Fig. 7b). | ||
| Fig. 7 (a) Anisotropic charge transport pathways leading to distinct interaction at (100) and (006) facets of Cs3Bi2I9 hexagonal prisms. Reprinted with permission from ref. 148. Copyright 2022 American Chemical Society. (b) Atomic models of CsPbBr3–sulfobromide Pb4S3Br2 heterostructures of different shapes: rhombicuboctahedronin, hexapods, and dodecahedron nanostructures. (bottom) Product distribution after 2 h of photocatalytic reaction for different heterostructure shapes under CO2-saturated H2O vapor. Reprinted with permission from ref. 150. Copyright 2022 American Chemical Society. | ||
In a recent study, albeit for solar cells, it is shown that the (111) facet dominated FAPbI3 film is more stable against moisture and phase transition, which the authors attributed to the reduced chemisorption/interaction strength of water molecules on the (111) facet compared to the (100) facet that is predominant is conventional thin films.151 These studies clearly suggest the importance of facet engineering in achieving high performing and stable photocatalytic devices.
4.3. Activation and manipulation of catalytically active sites
Catalytically active sites are essential for the desired reduction and oxidation activity. The intrinsic activity of the active sites depends on inherent electronic structuring and surface atomic arrangements that affect the adsorption and desorption of reactive intermediates. Exposing active sites over the surface and enhancing their accessibility for the reactants could modulate photocatalytic performance. Apart from poor stability in electrolytes, HaP also suffers from a lack of highly active sites for the desired reactions.152 Various strategies, such as morphology engineering, surface/interface structuring, heterojunction construction, encapsulation methodologies, and pairing co-catalysts, are explored to enhance the activity of HaP photocatalysts. For instance, Y.-F. Xu et al., reported the development of CsPbBr3 nanocrystal/palladium nanosheet (CsPbBr3NC/Pd NS) composites for improved photocatalytic CO2 reduction in water vapor.153 This study revealed that even though the pristine CsPbBr3 nanocrystals displayed activity for CO2 reduction, the performance could be significantly enhanced by integrating with Pd nanosheets (photoelectron consumption rate increased from 9.86 to 33.9 μmol g−1 h−1). This performance enhancement could be stemmed in fact from the creation of metal/semiconductor Schottky contact between Pd NS and CsPbBr3 NCs that accelerates the charge separation and transfer properties along with exposure of catalytically active Pd sites.Using the binding sites on HaP to immobilize catalytically active species can assist in achieving a high reaction activity of HaP-based hybrid photocatalysts. For example, Z. Chen and co-workers stabilized a [Ni(terpy)2]2+(Ni(tpy)) metal complex on inorganic ligand-capped CsPbBr3 NCs to form a CsPbBr3–Ni(tpy) hybrid photocatalyst.92 Apart from providing active Ni(tpy) catalytic centers, the metal complexes also served as electron sinks by accepting photoexcited electrons from HaP nanocrystals and thus suppressing electron–hole recombination. Consequently, CsPbBr3–Ni(tpy) hybrid photocatalyst yielded 1724 μmol g−1 (CO/CH4) in the reduction of CO2, which is about 26 times higher than the yield achieved by pristine CsPbBr3 NCs. In another work, L.-Y. Wu et al., encapsulated MAPbI3 perovskite QDs in the pores of a Fe-porphyrin based metal organic framework (PCN-221(Fex)) through a sequential deposition procedure.97 Utilizing steady-state and time-resolved PL measurements, it was revealed that, due to close contact of absorber and catalysts, photogenerated electrons from MAPbI3 QDs can easily be transferred to catalytically active sites of Fe porphyrins and thus enhance the charge separation efficiencies and activity of the resulting hybrid photocatalyst. Furthermore, metal–organic framework structures were also found to improve the stability of MAPbI3 QDs in water-involved photocatalytic systems. Following these effects, the optimized MAPbI3@PCN-221(Fex) exhibited a CO2 reduction yield of 1559 mmol g−1 (CO (34%) and CH4 (66%)) with high stability (linear productions over 80 hours). The exploration of such strategies seems to improve the effectiveness of HaP-based photocatalysts in CO2 reduction. However, more efforts are required to enhance the activity-stability of these newly emerged photocatalysts.
Surface active sites should be optimized in terms of their density, accessibility, and intrinsic activities. Given that the fundamental understanding of catalytic mechanisms is still limited, in situ characterization techniques and DFT-based calculations on surface energies with considerations on intermediates should help to achieve better optimizations. For instance, the defective states in materials can be trap sites for carriers, leading to an increased rate of electron–hole pairs recombination. On the other hand, defective sites could come with increased intrinsic activity, facilitating better reaction rates. Therefore, systematic analysis and detailed investigations based on in situ and theoretical studies help exploit the materials to off-limits. In situ analysis also assists in revealing the dynamic surface reconstruction of catalysts during the actual testing. Further augmentation with advanced machine learning algorithms can help in rationalizing and elucidating the catalytically active sites via high-throughput complex calculations of site-specific reactant binding energies and reaction intermediates.
4.4. Product selectivity for CO2R products
The product selectivity primarily depends on the surface electronic states which govern the CO2 activation/adsorption, catalytically active sites, and intermediate adsorption/desorption properties. Also, the availability of surface charges dictates the reaction pathways. Evidently, it is difficult to achieve reduction products that involve a higher number of electrons at the surface, such as CH3OH and other C2+ products. CO and CH4 are the major CO2 reduction products for HaPs. The product yield for various HaPs is shown in Fig. 8a and b and compared against other photocatalysts for CO2 reduction. There is no report on methanol or higher order C2 product yet. Success in improving CO and CH4 production has been achieved through different heterojunction schemes, morphology, compositional and dimensional engineering, as discussed above. The relative distribution of CO and CH4 from different heterostructures is shown in Fig. 8c. Mechanistic and theoretical insights on CO2 activation and adsorption energy of intermediates, and proton coupled electron transfer reaction (PCET) is lacking for HaPs, contrary to oxide perovskites.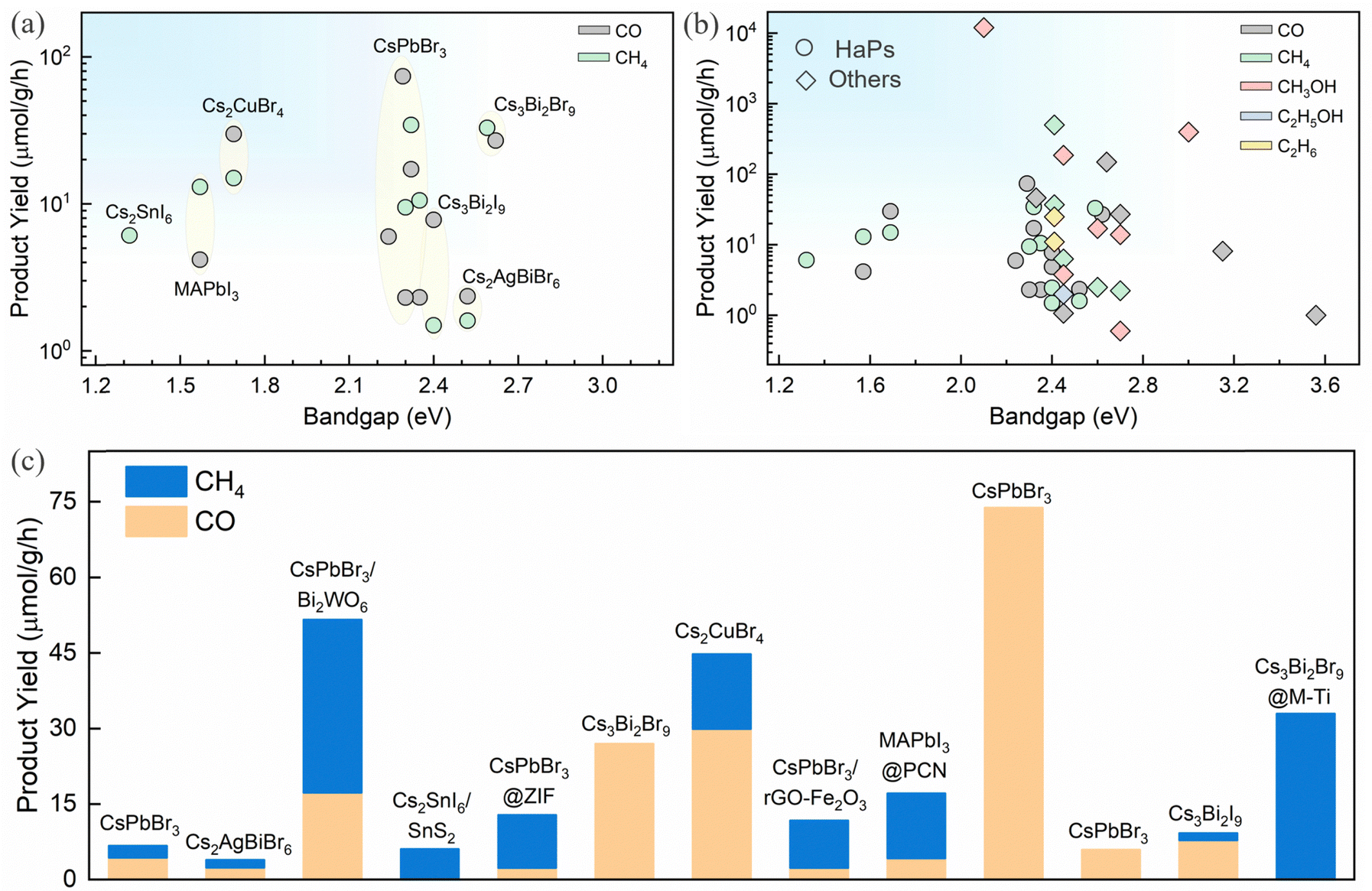 | ||
| Fig. 8 (a) Product yield during photocatalytic CO2 reduction for various perovskites, (b) comparison of products and their respective yield for HaPs and other semiconductors,16,131,154–166 and (c) product distribution from different perovskites and perovskite heterojunctions.13,93–101,109,111,144 | ||
Sheng et al. exploited the concept of frustrated Lewis pairs (FLP) to achieve efficient dissociation of H2 and CO2 reduction on Pb-free Cs3Bi2Br9 and Cs2CuBr4 quantum dots.99,101 Utilizing in situ diffuse reflectance infrared Fourier transform spectra (DRIFT) and density functional theory (DFT) calculations, the authors show that the surface catalytic sites can be regulated via bromine modulation and spontaneous polarization effect due to Cu-d band properties in Cs3Bi2Br9 and Cs2CuBr4 respectively.
The estimation of the energetic barrier and binding strengths of key intermediates such as CO*, COOH* and CH4* species on perovskite surfaces are critical to alter and design surfaces that can steer the reaction kinetics in different hydrogenation pathways. Studies combining in situ experiments and theory can be extremely valuable to achieve breakthroughs in the field.
5. Improvements in stability enabling advanced photoelectrochemical devices
Poor stability of perovskite based photoelectrodes in PEC type devices limits their potential due to the reduction in the photovoltage output over time. The photovoltage loss incurred due the degradation reduces the solar-to-fuel conversion efficiency. Both intrinsic (ion migration, defects and traps, phase instability) as well extrinsic environmental stresses act to trigger or accelerate degradation. Adequate bulk and surface passivation schemes through additive engineering, molecular passivation, and barrier layers have drastically improved the stability of perovskite devices with minimal photovoltage loss.167–169 Additionally, applying an external encapsulation layer and/or changing the chemical environment improves the operational stability of perovskite photoelectrodes in aqueous media, providing an extended window to drive various reduction and oxidation reactions.5.1. Perovskite based photocathodes for reduction reactions
The most widely explored CsPbBr3 had an average carrier lifetime in the range of 1–50 ns, which is significantly lower than the HaPs used in best performing solar cells. This is due to the aqueous instability issues associated with HaP compositions used in solar cells. The chemical bonding in HaPs is highly ionic in nature, which causes instability in polar solvents and liquid electrolytes. This is usually mitigated by exploiting dynamic precipitation-solubility equilibrium in HX acid solution,170 or using purely organic or mixed aqueous–organic solvents.135,144 Therefore, alternative oxidation of organic compounds is advantageous both with regards to stability and energetics. Ethyl acetate, isopropyl alcohol and benzyl alcohol have been used for oxidative half reactions. This provides an opportunity for synthesizing a wide range of value-added chemicals. However, the practical and economic benefit of utilizing these chemicals must be assessed in advance.Additive engineering and site-specific molecular passivation routes have also been quite successful in achieving long term stability of perovskites in PV devices while simultaneously addressing the ion/halide migration issues. Encouraging device modifications have been proposed to make them stable in different photoelectrochemical environments. As an example of an integrated system, Liang et al. demonstrated carbon encapsulated MAPbI3 perovskite solar cells sealed by an electrode integrated with a catalyst layer for photoelectrochemical water splitting.171 Recently, direct integration of a catalyst on photocathode/photoanodes has been possible, thanks to a passivating and conductive carbon layer.
Fehr et al. showed solar driven water splitting from a mixed cation Cs0.05FA0.85MA0.1Pb(I0.95Br0.05)3 (photocathode) and FA0.97MA0.03PbI3 (photoanode) perovskite solar cell directly integrated with a carbon electrode with an overall STH efficiency of 20.8%.172
For CO2 photoelectrochemical reduction, Andrei et al. demonstrated the success of a carbon encapsulation approach to fabricate a monolithic triple cation (CsFAMA)PbI3.2Br0.66 based perovskite–BiVO4 tandem PEC as a standalone system for CO2 reduction (see Fig. 9a).29,173,174 Carbon encapsulation provides a distinct avenue to extend the applicability of wider perovskite compositions in photocatalysis. In addition to carbon, Field's metal (FM) and metallic foils are also used to enhance electrical conduction and stability. For example, Tayyebi et al.175 and Choi et al.176 have demonstrated ammonia and hydrogen production respectively, from FM and metallic layer protected perovskite photocathodes, as shown in Fig. 9b and c. In this way, multi-layer protective layers, also serving as charge transport layers, have been successfully applied to drive different redox reactions (CO2, nitrate, and water reduction) at high photocurrent density (∼20 mA cm−2). The coupling of alternative oxidation reactions such as glycerol and lignocellulosic biomass has opened a new paradigm in this direction.
 | ||
| Fig. 9 (a) Schematic representation of a wireless standalone BiVO4–perovskite artificial leaf like device and corresponding faradaic efficiency of various products after photocatalytic CO2 reduction. Reproduced from ref. 173. Copyright 2023 Springer Nature Limited. (b) Schematic of the perovskite photocathode-based PEC cell used for NH3 production. The bottom graph shows the amount of NH3 generated for Ru@TiNS/Ni/perovskite photocathode – Pt@TiNS anode device for simultaneous nitrate reduction and glycerol oxidation. Reproduced from ref. 175. Copyright 2024 Springer Nature Limited. (c) Schematic of the perovskite PEC device for hydrogen generation at the photocathode and biomass oxidation at the anode. The bottom figure shows the production of vanillin and acetovanillone from lignocellulosic biomass, lignin, hemicellulose, and cellulose oxidation. Reproduced from ref. 176. Copyright 2022 Springer Nature Limited. | ||
5.2. Perovskite based photoanodes for oxidation reactions
Carbon encapsulation is not merely limited to reduction reactions but extended to perovskite photoanodes to drive oxidation chemistry. Excellent progress has been made in driving oxidation reactions from carbon encapsulated perovskite photoanodes. Mesoporous carbon protected FAPbBr3 based photoanode demonstrated 8.5% STH efficiency, surpassing the performance achieved from analogous photoanodes such as BiVO4, Fe2O3, TiO2, WO3, and Ta3N5.177 Poli et al. utilized an Ir-catalyst embedded bilayer of graphite sheet and mesoporous carbon electrode protected inorganic CsPbBr3 photoanode for water oxidation.178 Stability and activity can be further improved through modulating surface chemistry and catalyst engineering. Depending on the electrochemical response of organic ligands and metallic catalysts, the redox ability of HaP-based materials can be tuned to yield value-added compounds with reduced energy input.Recently, Zhu et al. have achieved remarkable stability of 210 h for water oxidation in aqueous electrolyte through encapsulating with electrocatalytically active glassy carbon and boron doped diamond sheets containing an earth abundant NiFeOOH catalyst.179 A similar catalyst has been integrated into a high performing FAPbI3 based photoanode to realize an impressive 8.5% solar-to-hydrogen (STH) efficiency on scaled-up 123 cm2 mini modules.180 Recent demonstrations on heterojunctions based on conjugated polymers with metal sulfide (Sb2S3),181 metal oxide (Mo:BiVO4),126 and metal nitride (Ta3N5)127 can be used as a basis to develop a perovskite photoanode with carrier extraction efficiency. Beyond water oxidation, few studies explore perovskite photoanodes for alternative oxidation reactions such as iodide oxidation and organic transformations for environmental remediation. The iodide oxidation reaction (IOR) has a thermodynamically lower energetic requirement and is kinetically more favorable than the OER. Yun et al. have developed a Co–Ni2S3 catalyst embedded in a carbon matrix and integrated this into a standard n–i–p perovskite solar cell stack to realize photoanodes for the IOR. Fig. 10b shows the photoanode device configuration and corresponding voltammogram with and without catalyst. The protected photoanode stack yielded a STH efficiency of 11.45% along with a stable 25 h of continuous operation.23 Climent et al. demonstrated a reversible photoelectrochemical transformation of benzyl alcohol (BzOH) to benzyl aldehyde (BzCHO) and vice versa using a selective charge transfer scheme, i.e. TiO2/CsPbBr3 for oxidation and NiO/CsPbBr3 for reduction.182 The same group later extended the approach to drive the oxidation of organic pollutant such as 2-mercaptobenzothiazol (MBA) using Al2O3 protected CsPbBr3 photoanode, as shown in Fig. 10c.183 The efficiency of the process was further enhanced by externally powering the PEC device with a perovskite PV module.
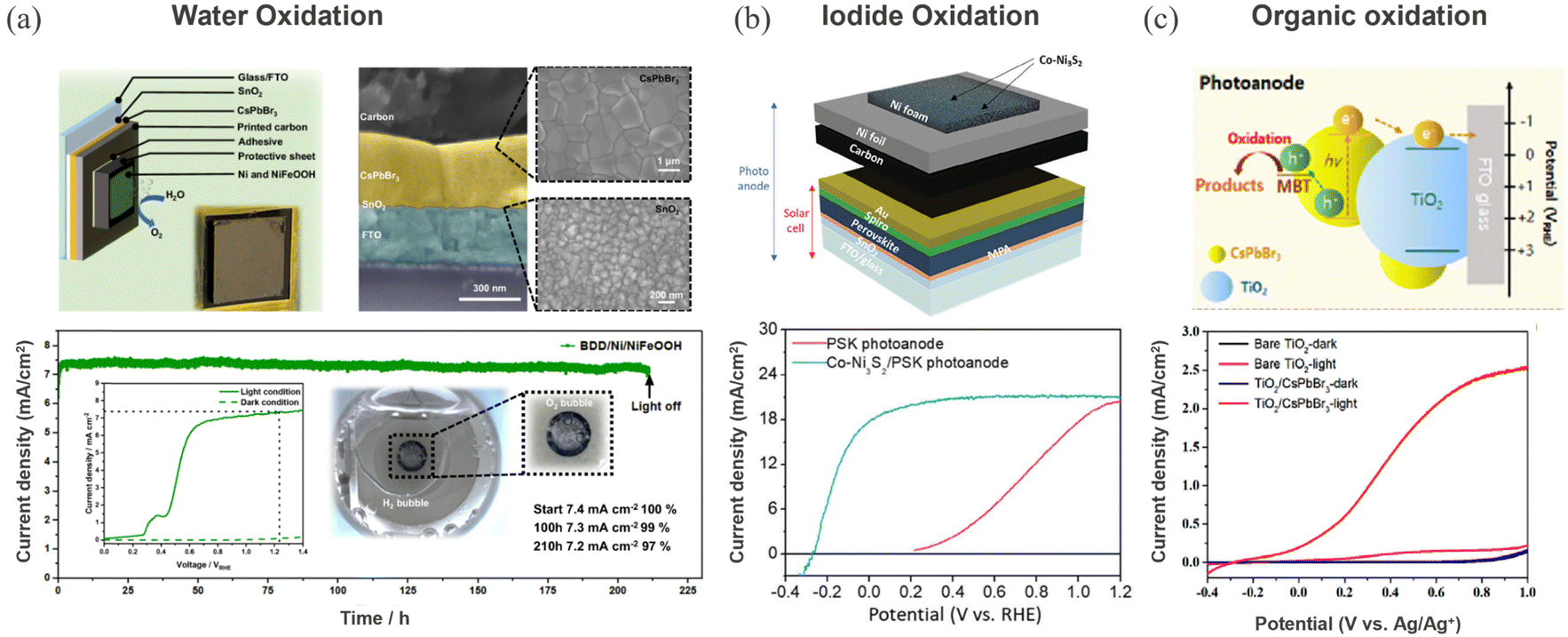 | ||
| Fig. 10 (a) Water oxidation: CsPbBr3 photoanode protected with GC/Ni/NiFeOOH and corresponding cross-sectional and top-view SEM micrographs of the device stack. The bottom shows the device stability testing at +1.23 VRHE under 1 sun illumination. The inset shows the voltammogram under 1 sun (solid line) and in dark (green dashed line) and a photograph of the photoanode under operation showing the evolved O2 bubbles. Reproduced from ref. 179. Copyright 2024 Springer Nature Limited. (b) Iodide oxidation: device structure of the perovskite photoanode showing Co–Ni3S2/Ni foam/Ni foil/carbon powder stacked on a solar cell structure and the corresponding voltammogram under 1-sun illumination in 0.5 M KPi electrolyte containing 0.5 M KI. Reproduced from ref. 23. Copyright 2024 Wiley. (c) Organic oxidation: perovskite photoanode based PEC cell for MBE oxidation and below shows the LSV scans of the photoanodes in 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) in dichloromethane (DCM) with 0.05 M MBT. Reprinted with permission from ref. 183. Copyright 2023 American Chemical Society. | ||
6. Emerging concepts
While the vast majority of perovskites remain unexplored for photocatalytic application, high-throughput combinatorial screening and DFT augmented machine learning provide immense opportunities to discover stable and catalytically active perovskite compositions. Notable efforts are already being made in this direction to discover unique perovskite compounds for specific optoelectronic devices.184–187 The optoelectronic and photocatalytic performance descriptors discussed above can guide in refining the compositional library. Advance computational methods can also help in understanding the complex and multistep photoelectrocatalysis process involving various interdependent parameters such as charge transfer, CO2 activation, catalytic sites, reaction intermediates and competing reactions. In a recent attempt, Caruso et al. demonstrated the importance of choosing the correct descriptor for optimizing photocatalytic property. They combined machine learning models with the DFT and found that Zn2+ metal substitution at the B-site in the Cs2AgBiBr6 perovskite enhances the photoactivity due to the optimum electronic structure, especially the d-orbital configuration (d10).188 The vast library of compositionally feasible perovskite compounds remained explored. The rapid rise in the computation capabilities to screen, predict and rationalize materials properties has opened new avenues to discover stable and non-toxic perovskite or perovskite inspired compounds targeted for specific photoelectrochemical processes. Moreover, the accelerated testing platforms, also known as the laboratory of the future, can be a powerful tool for rapid screening of compositionally and operationally stable compounds. Expanding the scope of reactions from reduction to alternative oxidation reaction is gaining interest to cover wider aspects of photoelectrochemical transformations. It is important to recognize that specific HaPs might be more suitable for certain reactions and testing conditions (pH, temperature, and irradiation etc.).7. Outlook and future directions
Despite significant milestones achieved for optoelectronic devices (photovoltaics, photodetectors, lasers, LEDs, etc.), the full potential of halide perovskites remains untapped for photocatalysis application. The photochemical performance and stability are far from its practical usage. In this perspective, we have emphasized fundamental concepts and examined key advancements propelling halide perovskites for photocatalysis. Promising paths are identified for future advancements and valorization of perovskite based photocatalytic systems.(i) Targeting stable compositions and measurement conditions
Stability of the photoelectrodes is one of the biggest challenges faced in a photoelectrochemical process. To date, archetypical MAPbI3 and wide bandgap CsPbX3 perovskite compositions are the most widely explored photocatalytic processes. Screening of a large number of compositions with suitable bandgaps for both reduction and oxidation reaction can accelerate the development of interesting candidates and help in identifying which composition modifications lead to higher stability. With rapidly advancing materials development initiatives, there is an urgent need for a perovskite materials database, combining theory and experiments, specific for photoelectrochemical processes. This will also lead to the discovery of more robust, stable, and Pb-free compositions suitable for photocatalysis. There is a need to critically define the optimum reaction conditions such as – pH, temperature, and consider the viability of driving complete reaction at lower overpotentials with high production rate. External strategies such as facet engineering, novel protective coatings (oxide/metal/organic) and structural engineering approaches (like core–shell structures, MOF-based encapsulation, etc.) must be developed and explored to stabilize perovskites in electrolytes. Apart from developing protective materials, their deposition/coating strategies, growth mechanisms, effects on perovskites, and photoelectrochemical activity should be investigated and understood in-depth.(ii) Better charge transfer schemes for enhanced photocurrent
Leveraging upon the successful passivation schemes used for aqueous water splitting and CO2 reduction, further improvement in performance can be achieved through better charge collection. In this regard, alternative heterojunctions beyond conventional electron and hole transport layers need to be developed, particularly Z-scheme configurations. Z-scheme with an optimum combination of bandgap (1.6 eV, top cell and 1.1 eV bottom cell) not only enhances the light absorption but also enhances the photo reduction/oxidation ability. Developing high surface area textured surfaces can further enhance the charge collection with better light management.(iii) Driving alternative reactions for value added chemicals
Due to the instability in water based aqueous media, perovskites are stabilized in alternative media, mainly, saturated hydrohalic acid (HX; HI, HBr) solutions. The resulting products from HX splitting, I2 and Br2, are useful products for hygiene and energy industries. Moreover, exploring alternative anodic oxidations to the OER is desirable to lower the PEC energy requirements. Halide oxidation is thermodynamically more favorable over the OER and requires less kinetic overpotential due to two electrons being involved in the reactions compared to four electrons for water oxidation. By coupling alternative reactions such as oxidation of biomass derived organic compounds (lignin, glucose, furfural, 5-hydroxymethyl furfural; HMF, glycerol, etc.) and chemical wastes (polyethylene terephthalate; PET, glyceric acid, wastewater remediation), several high value chemicals such as FDCA, dimethoxydihydrofuran, vanillin, and glycolic acid, etc. can be achieved as byproducts of these reactions. This approach has significant potential in reducing the energy input of the paired electrochemical process, enhancing the overall techno-economic viability, and maximizing the return of energy investments. The integration of high selectivity metal complex (Ni, Fe, Ru, and Ir) based molecular catalysts and (bi-) metallic (Au, Pt, and Pd) co-catalysts is a promising direction to enhance the functionality of the perovskite photocatalysts. The future endeavors must eventually go beyond the above and cover additional important reactions like seawater splitting and wastewater treatment.(iv) Towards higher order carbon compounds
A higher photovoltage is desirable to overcome overpotential losses associated with photoelectrochemical CO2 reduction. Inspiration from photovoltaic architectures might be useful to engineer novel integration methods enhancing charge extraction and stability. The high photovoltage from encapsulated and integrated perovskite PEC devices has shown promising results for unbiased standalone operation. Semitransparent perovskite PV devices can provide extra photovoltage without compromising the total photon flux. Next, overcoming mass transport limitations associated with CO2 electrolysis, an integrated perovskite device with flow reactor and catalyst loaded gas diffusion electrode (GDE) flow reactors will pave the way for a standalone PEC device at high current densities. Surface reaction kinetics can be tuned by changing adsorption strengths of the intermediates. Advanced in situ characterization tools such as synchrotron-based techniques, X-ray scattering and photoelectron spectroscopy are helpful in identifying surface adsorbates and catalytic sites. Moreover, observation of catalytic transient states and charge transfer processes using ultrafast infrared and pump–probe spectroscopy bring exciting opportunities to understand the physical origin of the electrochemical processes at the perovskite surface.(v) Scalability challenges
From a practical perspective, the scalability aspects should be considered in the beginning itself. The facile low temperature synthesis position perovskites in a favorable scenario to develop large scale processes compared to other photocatalysts, especially oxides. While nanoparticle photocatalysts show great promise, they are confronted with scaling issues. The development of thin film artificial leaf like devices is excellent in this regard while keeping the fundamental features of nanoparticles and even single crystals intact. Device scalability depends also on optimizing other important components such as reactant solubility limits, mass transport limitations and the design of the reactor.Further development lies in understanding the charge transfer processes in widely utilized heterojunction schemes, specifically the perovskite–electrolyte interface. To further expand the scope of halide perovskite based photocatalytic systems, utilization of other forms of CO2 and targeting alternative oxidation reactions is indispensable.
Author contributions
S. Shukla: conceptualization and writing – original draft. V. Jose and S. Shukla: reviewing and editing. N. Mathews: conceptualization, supervision, reviewing and editing.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. S. acknowledges funding from the European Union's Horizon Europe program under the Marie Skłodowska-Curie Grant Agreement No. 101067667. S. S. and V. J. acknowledge Catalisti VLAIO (Vlaanderen Agentschap Innoveren & Ondernemen) for their funding through the Moonshot SYN-CAT project (HBC.2020.2614), and the Belgian federal government through the Energy Transition Fund for T-REX project. NM would like to acknowledge the funding from the National Research Foundation (NRF), Singapore, under its Competitive Research Program (CRP) (NRF-CRP25-2020-0002). The authors would like to thank Dr Tom Aernouts and Prof. Bart Vermang for fruitful discussions.References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- B. Obama, Science, 2017, 355, 126–129 CrossRef CAS PubMed.
- M. Victoria, N. Haegel, I. M. Peters, R. Sinton, A. Jäger-Waldau, C. del Cañizo, C. Breyer, M. Stocks, A. Blakers, I. Kaizuka, K. Komoto and A. Smets, Joule, 2021, 5, 1041–1056 CrossRef CAS.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- E. Papadis and G. Tsatsaronis, Energy, 2020, 205, 118025 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- M. Halmann, Nature, 1978, 275, 115–116 CrossRef CAS.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- C.-C. Yang, Y.-H. Yu, B. van der Linden, J. C. S. Wu and G. Mul, J. Am. Chem. Soc., 2010, 132, 8398–8406 CrossRef CAS PubMed.
- S. Liu, J. Li, W. Xiao, R. Chen, Z. Sun, Y. Zhang, X. Lei, S. Hu, M. Kober-Czerny, J. Wang, F. Ren, Q. Zhou, H. Raza, Y. Gao, Y. Ji, S. Li, H. Li, L. Qiu, W. Huang, Y. Zhao, B. Xu, Z. Liu, H. J. Snaith, N.-G. Park and W. Chen, Nature, 2024 DOI:10.1038/s41586-024-07723-3.
- Y.-F. Xu, M.-Z. Yang, B.-X. Chen, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed.
- L. Ding, F. Bai, B. Borjigin, Y. Li, H. Li and X. Wang, Chem. Eng. J., 2022, 446, 137102 CrossRef CAS.
- G. Wang, Z. Chen, T. Wang, D. Wang and J. Mao, Angew. Chem., Int. Ed., 2022, 61, e202210789 CrossRef CAS PubMed.
- Y. Wang, X. Liu, X. Han, R. Godin, J. Chen, W. Zhou, C. Jiang, J. F. Thompson, K. B. Mustafa, S. A. Shevlin, J. R. Durrant, Z. Guo and J. Tang, Nat. Commun., 2020, 11, 2531 CrossRef CAS PubMed.
- T. Kunene, L. Xiong and J. Rosenthal, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9693–9695 CrossRef CAS PubMed.
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- D. Li, K. Yang, J. Lian, J. Yan and S. Liu, Adv. Energy Mater., 2022, 12, 2201070 CrossRef CAS.
- H. Wan, A. Bagger and J. Rossmeisl, J. Phys. Chem. Lett., 2022, 13, 8928–8934 CrossRef CAS PubMed.
- C. R. Lhermitte and K. Sivula, ACS Catal., 2019, 9, 2007–2017 CrossRef CAS.
- T. Fuchigami, S. Inagi and M. Atobe, Appendix B: Tables of Physical Data. Fundamentals and Applications of Organic Electrochemistry, John Wiley & Sons, 2014 Search PubMed.
- J. Yun, Y. S. Park, H. Lee, W. Jeong, C.-S. Jeong, C. U. Lee, J. Lee, S. Moon, E. Kwon, S. Lee, S. Kim, J. Kim, S. Yu and J. Moon, Adv. Energy Mater., 2024, 2401055 CrossRef.
- X. Deng, R. Li, S. Wu, L. Wang, J. Hu, J. Ma, W. Jiang, N. Zhang, X. Zheng, C. Gao, L. Wang, Q. Zhang, J. Zhu and Y. Xiong, J. Am. Chem. Soc., 2019, 141, 10924–10929 CrossRef CAS PubMed.
- U. Kang, S. K. Choi, D. J. Ham, S. M. Ji, W. Choi, D. S. Han, A. Abdel-Wahab and H. Park, Energy Environ. Sci., 2015, 8, 2638–2643 RSC.
- T. Arai, S. Sato, T. Kajino and T. Morikawa, Energy Environ. Sci., 2013, 6, 1274–1282 RSC.
- S. Y. Lee, S. Y. Lim, D. Seo, J.-Y. Lee and T. D. Chung, Adv. Energy Mater., 2016, 6, 1502207 CrossRef.
- K. Sekizawa, S. Sato, T. Arai and T. Morikawa, ACS Catal., 2018, 8, 1405–1416 CrossRef CAS.
- V. Andrei, B. Reuillard and E. Reisner, Nat. Mater., 2020, 19, 189–194 CrossRef CAS PubMed.
- C. Li, T. Wang, B. Liu, M. Chen, A. Li, G. Zhang, M. Du, H. Wang, S. F. Liu and J. Gong, Energy Environ. Sci., 2019, 12, 923–928 RSC.
- X. Zhou, R. Liu, K. Sun, Y. Chen, E. Verlage, S. A. Francis, N. S. Lewis and C. Xiang, ACS Energy Lett., 2016, 1, 764–770 CrossRef CAS.
- T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998–2002 RSC.
- Y. Sugano, A. Ono, R. Kitagawa, J. Tamura, M. Yamagiwa, Y. Kudo, E. Tsutsumi and S. Mikoshiba, RSC Adv., 2015, 5, 54246–54252 RSC.
- T. Sekimoto, H. Hashiba, S. Shinagawa, Y. Uetake, M. Deguchi, S. Yotsuhashi and K. Ohkawa, J. Phys. Chem. C, 2016, 120, 13970–13975 CrossRef CAS.
- Gurudayal, J. W. Beeman, J. Bullock, H. Wang, J. Eichhorn, C. Towle, A. Javey, F. M. Toma, N. Mathews and J. W. Ager, Energy Environ. Sci., 2019, 12, 1068–1077 RSC.
- N. C. Deb Nath, S. Y. Choi, H. W. Jeong, J.-J. Lee and H. Park, Nano Energy, 2016, 25, 51–59 CrossRef CAS.
- Y. J. Jang, I. Jeong, J. Lee, J. Lee, M. J. Ko and J. S. Lee, ACS Nano, 2016, 10, 6980–6987 CrossRef CAS PubMed.
- H. S. Jeon, J. H. Koh, S. J. Park, M. S. Jee, D.-H. Ko, Y. J. Hwang and B. K. Min, J. Mater. Chem. A, 2015, 3, 5835–5842 RSC.
- Q. Jia, S. Tanabe and I. Waki, Chem. Lett., 2018, 436–439 CrossRef CAS.
- T. Arai, S. Sato, K. Sekizawa, T. M. Suzuki and T. Morikawa, Chem. Commun., 2019, 55, 237–240 RSC.
- T. N. Huan, D. A. Dalla Corte, S. Lamaison, D. Karapinar, L. Lutz, N. Menguy, M. Foldyna, S.-H. Turren-Cruz, A. Hagfeldt, F. Bella, M. Fontecave and V. Mougel, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9735–9740 CrossRef CAS PubMed.
- M. A. Ghausi, J. Xie, Q. Li, X. Wang, R. Yang, M. Wu, Y. Wang and L. Dai, Angew. Chem., Int. Ed., 2018, 57, 13135–13139 CrossRef CAS PubMed.
- M. Schreier, F. Héroguel, L. Steier, S. Ahmad, J. S. Luterbacher, M. T. Mayer, J. Luo and M. Grätzel, Nat. Energy, 2017, 2, 17087 CrossRef CAS.
- Gurudayal, J. Bullock, D. F. Srankó, C. M. Towle, Y. Lum, M. Hettick, M. C. Scott, A. Javey and J. Ager, Energy Environ. Sci., 2017, 10, 2222–2230 RSC.
- F. Urbain, P. Tang, N. M. Carretero, T. Andreu, L. G. Gerling, C. Voz, J. Arbiol and J. R. Morante, Energy Environ. Sci., 2017, 10, 2256–2266 RSC.
- G. M. Sriramagiri, N. Ahmed, W. Luc, K. D. Dobson, S. S. Hegedus and F. Jiao, ACS Sustainable Chem. Eng., 2017, 5, 10959–10966 CrossRef CAS.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. Salehi-Khojin, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- M. Schreier, L. Curvat, F. Giordano, L. Steier, A. Abate, S. M. Zakeeruddin, J. Luo, M. T. Mayer and M. Grätzel, Nat. Commun., 2015, 6, 7326 CrossRef CAS PubMed.
- B. Kim, H. Seong, J. T. Song, K. Kwak, H. Song, Y. C. Tan, G. Park, D. Lee and J. Oh, ACS Energy Lett., 2020, 5, 749–757 CrossRef CAS.
- L. Q. Zhou, C. Ling, H. Zhou, X. Wang, J. Liao, G. K. Reddy, L. Deng, T. C. Peck, R. Zhang, M. S. Whittingham, C. Wang, C.-W. Chu, Y. Yao and H. Jia, Nat. Commun., 2019, 10, 4081 CrossRef PubMed.
- S. Y. Chae, S. Y. Lee, S. G. Han, H. Kim, J. Ko, S. Park, O.-S. Joo, D. Kim, Y. Kang, U. Lee, Y. J. Hwang and B. K. Min, Sustainable Energy Fuels, 2020, 4, 199–212 RSC.
- W. Deng, L. Zhang, H. Dong, X. Chang, T. Wang and J. Gong, Chem. Sci., 2018, 9, 6599–6604 RSC.
- G. Piao, S. H. Yoon, D. S. Han and H. Park, ChemSusChem, 2020, 13, 698–706 CrossRef CAS PubMed.
- Z. Chen, T. Wang, B. Liu, D. Cheng, C. Hu, G. Zhang, W. Zhu, H. Wang, Z.-J. Zhao and J. Gong, J. Am. Chem. Soc., 2020, 142, 6878–6883 CrossRef CAS PubMed.
- D. Ren, N. W. X. Loo, L. Gong and B. S. Yeo, ACS Sustainable Chem. Eng., 2017, 5, 9191–9199 CrossRef CAS.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2017, 16, 70–81 CrossRef PubMed.
- C. Moon and B. Shin, Discovery Mater., 2022, 2, 5 CrossRef.
- J. W. Yang, Y. J. Ahn, D. K. Cho, J. Y. Kim and H. W. Jang, Inorg. Chem. Front., 2023, 10, 3781–3807 RSC.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef CAS PubMed.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- E. Pastor, M. Sachs, S. Selim, J. R. Durrant, A. A. Bakulin and A. Walsh, Nat. Rev. Mater., 2022, 7, 503–521 CrossRef CAS.
- H. Li, X. Jia, Q. Zhang and X. Wang, Chem, 2018, 4, 1510–1537 CAS.
- A. Iwase, S. Yoshino, T. Takayama, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2016, 138, 10260–10264 CrossRef CAS PubMed.
- G. Giuffredi, T. Asset, Y. Liu, P. Atanassov and F. Di Fonzo, ACS Mater. Au, 2021, 1, 6–36 CrossRef CAS PubMed.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- Y. Liu, M. Xia, D. Ren, S. Nussbaum, J.-H. Yum, M. Grätzel, N. Guijarro and K. Sivula, ACS Energy Lett., 2023, 8, 1645–1651 CrossRef CAS PubMed.
- J. Y. Kim, J.-W. Lee, H. S. Jung, H. Shin and N.-G. Park, Chem. Rev., 2020, 120, 7867–7918 CrossRef CAS PubMed.
- M. Ahmadi, T. Wu and B. Hu, Adv. Mater., 2017, 29, 1605242 CrossRef PubMed.
- X. Y. Chin, A. Perumal, A. Bruno, N. Yantara, S. A. Veldhuis, L. Martínez-Sarti, B. Chandran, V. Chirvony, A. S.-Z. Lo, J. So, C. Soci, M. Grätzel, H. J. Bolink, N. Mathews and S. G. Mhaisalkar, Energy Environ. Sci., 2018, 11, 1770–1778 RSC.
- Q. Zhang, Q. Shang, R. Su, T. T. H. Do and Q. Xiong, Nano Lett., 2021, 21, 1903–1914 CrossRef CAS PubMed.
- R. A. John, N. Yantara, S. E. Ng, M. I. B. Patdillah, M. R. Kulkarni, N. F. Jamaludin, J. Basu, Ankit, S. G. Mhaisalkar, A. Basu and N. Mathews, Adv. Mater., 2021, 33, 2007851 CrossRef CAS.
- F. Temerov, Y. Baghdadi, E. Rattner and S. Eslava, ACS Appl. Energy Mater., 2022, 5, 14605–14637 CrossRef CAS PubMed.
- E. Ugur, M. Ledinský, T. G. Allen, J. Holovský, A. Vlk and S. De Wolf, J. Phys. Chem. Lett., 2022, 13, 7702–7711 CrossRef CAS PubMed.
- H. Lin, C. Zhou, Y. Tian, T. Siegrist and B. Ma, ACS Energy Lett., 2018, 3, 54–62 CrossRef CAS.
- T. M. Koh, K. Thirumal, H. S. Soo and N. Mathews, ChemSusChem, 2016, 9, 2541–2558 CrossRef CAS PubMed.
- Z. Li, M. Yang, J.-S. Park, S.-H. Wei, J. J. Berry and K. Zhu, Chem. Mater., 2016, 28, 284–292 CrossRef CAS.
- D. Niesner, M. Wilhelm, I. Levchuk, A. Osvet, S. Shrestha, M. Batentschuk, C. Brabec and T. Fauster, Phys. Rev. Lett., 2016, 117, 126401 CrossRef PubMed.
- T. Etienne, E. Mosconi and F. De Angelis, J. Phys. Chem. Lett., 2016, 7, 1638–1645 CrossRef CAS PubMed.
- E. M. Hutter, M. C. Gélvez-Rueda, A. Osherov, V. Bulović, F. C. Grozema, S. D. Stranks and T. J. Savenije, Nat. Mater., 2017, 16, 115–120 CrossRef CAS PubMed.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- H. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS.
- J. Wang, Y. Shi, Y. Wang and Z. Li, ACS Energy Lett., 2022, 7, 2043–2059 CrossRef CAS.
- P. Calado, A. M. Telford, D. Bryant, X. Li, J. Nelson, B. C. O’Regan and P. R. F. Barnes, Nat. Commun., 2016, 7, 13831 CrossRef CAS PubMed.
- K. Takanabe, ACS Catal., 2017, 7, 8006–8022 CrossRef CAS.
- H. Hempel, T. J. Savenjie, M. Stolterfoht, J. Neu, M. Failla, V. C. Paingad, P. Kužel, E. J. Heilweil, J. A. Spies, M. Schleuning, J. Zhao, D. Friedrich, K. Schwarzburg, L. D. A. Siebbeles, P. Dörflinger, V. Dyakonov, R. Katoh, M. J. Hong, J. G. Labram, M. Monti, E. Butler-Caddle, J. Lloyd-Hughes, M. M. Taheri, J. B. Baxter, T. J. Magnanelli, S. Luo, J. M. Cardon, S. Ardo and T. Unold, Adv. Energy Mater., 2022, 12, 2102776 CrossRef CAS.
- L. M. Herz, ACS Energy Lett., 2017, 2, 1539–1548 CrossRef CAS.
- L. Krückemeier, Z. Liu, T. Kirchartz and U. Rau, Adv. Mater., 2023, 35, 2300872 CrossRef PubMed.
- Y. Chen, H. T. Yi, X. Wu, R. Haroldson, Y. N. Gartstein, Y. I. Rodionov, K. S. Tikhonov, A. Zakhidov, X. Y. Zhu and V. Podzorov, Nat. Commun., 2016, 7, 12253 CrossRef CAS PubMed.
- R. Brenes, D. Guo, A. Osherov, N. K. Noel, C. Eames, E. M. Hutter, S. K. Pathak, F. Niroui, R. H. Friend, M. S. Islam, H. J. Snaith, V. Bulović, T. J. Savenije and S. D. Stranks, Joule, 2017, 1, 155–167 CrossRef CAS.
- M. Abdi-Jalebi, Z. Andaji-Garmaroudi, S. Cacovich, C. Stavrakas, B. Philippe, J. M. Richter, M. Alsari, E. P. Booker, E. M. Hutter, A. J. Pearson, S. Lilliu, T. J. Savenije, H. Rensmo, G. Divitini, C. Ducati, R. H. Friend and S. D. Stranks, Nature, 2018, 555, 497–501 CrossRef CAS PubMed.
- Z. Chen, Y. Hu, J. Wang, Q. Shen, Y. Zhang, C. Ding, Y. Bai, G. Jiang, Z. Li and N. Gaponik, Chem. Mater., 2020, 32, 1517–1525 CrossRef CAS.
- X.-D. Wang, Y.-H. Huang, J.-F. Liao, Y. Jiang, L. Zhou, X.-Y. Zhang, H.-Y. Chen and D.-B. Kuang, J. Am. Chem. Soc., 2019, 141, 13434–13441 CrossRef CAS PubMed.
- F. Xu, K. Meng, B. Cheng, S. Wang, J. Xu and J. Yu, Nat. Commun., 2020, 11, 4613 CrossRef CAS PubMed.
- Y. Jiang, J.-F. Liao, H.-Y. Chen, H.-H. Zhang, J.-Y. Li, X.-D. Wang and D.-B. Kuang, Chem, 2020, 6, 766–780 CAS.
- Z.-C. Kong, J.-F. Liao, Y.-J. Dong, Y.-F. Xu, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS.
- L.-Y. Wu, Y.-F. Mu, X.-X. Guo, W. Zhang, Z.-M. Zhang, M. Zhang and T.-B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed.
- Q.-M. Sun, J.-J. Xu, F.-F. Tao, W. Ye, C. Zhou, J.-H. He and J.-M. Lu, Angew. Chem., Int. Ed., 2022, 61, e202200872 CrossRef CAS PubMed.
- J. Sheng, Y. He, M. Huang, C. Yuan, S. Wang and F. Dong, ACS Catal., 2022, 12, 2915–2926 CrossRef CAS.
- Y.-F. Mu, W. Zhang, G.-X. Dong, K. Su, M. Zhang and T.-B. Lu, Small, 2020, 16, 2002140 CrossRef CAS PubMed.
- J. Sheng, Y. He, J. Li, C. Yuan, H. Huang, S. Wang, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 13103–13114 CrossRef CAS PubMed.
- Z. Liu, L. Krückemeier, B. Krogmeier, B. Klingebiel, J. A. Márquez, S. Levcenko, S. Öz, S. Mathur, U. Rau, T. Unold and T. Kirchartz, ACS Energy Lett., 2019, 4, 110–117 CrossRef CAS.
- M. Stolterfoht, C. M. Wolff, J. A. Márquez, S. Zhang, C. J. Hages, D. Rothhardt, S. Albrecht, P. L. Burn, P. Meredith, T. Unold and D. Neher, Nat. Energy, 2018, 3, 847–854 CrossRef CAS.
- D. W. deQuilettes, S. Koch, S. Burke, R. K. Paranji, A. J. Shropshire, M. E. Ziffer and D. S. Ginger, ACS Energy Lett., 2016, 1, 438–444 CrossRef CAS.
- I. L. Braly, D. W. deQuilettes, L. M. Pazos-Outón, S. Burke, M. E. Ziffer, D. S. Ginger and H. W. Hillhouse, Nat. Photonics, 2018, 12, 355–361 CrossRef CAS.
- A. Al-Ashouri, A. Magomedov, M. Roß, M. Jošt, M. Talaikis, G. Chistiakova, T. Bertram, J. A. Márquez, E. Köhnen, E. Kasparavičius, S. Levcenco, L. Gil-Escrig, C. J. Hages, R. Schlatmann, B. Rech, T. Malinauskas, T. Unold, C. A. Kaufmann, L. Korte, G. Niaura, V. Getautis and S. Albrecht, Energy Environ. Sci., 2019, 12, 3356–3369 RSC.
- M. Abdi-Jalebi, M. Ibrahim Dar, S. P. Senanayak, A. Sadhanala, Z. Andaji-Garmaroudi, L. M. Pazos-Outón, J. M. Richter, A. J. Pearson, H. Sirringhaus, M. Grätzel and R. H. Friend, Sci. Adv., 2019, 5, eaav2012 CrossRef CAS PubMed.
- M. Abdi-Jalebi, M. Pazoki, B. Philippe, M. I. Dar, M. Alsari, A. Sadhanala, G. Divitini, R. Imani, S. Lilliu, J. Kullgren, H. Rensmo, M. Grätzel and R. H. Friend, ACS Nano, 2018, 12, 7301–7311 CrossRef CAS PubMed.
- S. S. Bhosale, A. K. Kharade, E. Jokar, A. Fathi, S.-m Chang and E. W.-G. Diau, J. Am. Chem. Soc., 2019, 141, 20434–20442 CrossRef CAS PubMed.
- A. H. Slavney, T. Hu, A. M. Lindenberg and H. I. Karunadasa, J. Am. Chem. Soc., 2016, 138, 2138–2141 CrossRef CAS PubMed.
- L. Zhou, Y.-F. Xu, B.-X. Chen, D.-B. Kuang and C.-Y. Su, Small, 2018, 14, 1703762 CrossRef PubMed.
- H. Huang, H. Yuan, J. Zhao, G. Solís-Fernández, C. Zhou, J. W. Seo, J. Hendrix, E. Debroye, J. A. Steele, J. Hofkens, J. Long and M. B. J. Roeffaers, ACS Energy Lett., 2019, 4, 203–208 CrossRef CAS.
- H. Wang, X. Wang, R. Chen, H. Zhang, X. Wang, J. Wang, J. Zhang, L. Mu, K. Wu, F. Fan, X. Zong and C. Li, ACS Energy Lett., 2019, 4, 40–47 CrossRef CAS.
- J. Chakkamalayath, G. V. Hartland and P. V. Kamat, J. Phys. Chem. C, 2021, 125, 17881–17889 CrossRef CAS.
- T. Kirchartz, J. A. Márquez, M. Stolterfoht and T. Unold, Adv. Energy Mater., 2020, 10, 1904134 CrossRef CAS.
- O. Stroyuk, O. Raievska, J. Hauch and C. J. Brabec, Angew. Chem., Int. Ed., 2023, 62, e202212668 CrossRef CAS PubMed.
- M. Li, S. Bhaumik, T. W. Goh, M. S. Kumar, N. Yantara, M. Grätzel, S. Mhaisalkar, N. Mathews and T. C. Sum, Nat. Commun., 2017, 8, 14350 CrossRef CAS PubMed.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- D. W. de Quilettes, S. M. Vorpahl, S. D. Stranks, H. Nagaoka, G. E. Eperon, M. E. Ziffer, H. J. Snaith and D. S. Ginger, Science, 2015, 348, 683–686 CrossRef CAS PubMed.
- G. Han, T. M. Koh, J. Li, B. Febriansyah, Y. Fang, N. F. Jamaludin, Y. F. Ng, P. J. S. Rana, S. Mhaisalkar and N. Mathews, ACS Appl. Energy Mater., 2021, 4, 2716–2723 CrossRef CAS.
- G. H. Ahmed, J. K. El-Demellawi, J. Yin, J. Pan, D. B. Velusamy, M. N. Hedhili, E. Alarousu, O. M. Bakr, H. N. Alshareef and O. F. Mohammed, ACS Energy Lett., 2018, 3, 2301–2307 CrossRef CAS.
- P. J. S. Rana, B. Febriansyah, T. M. Koh, A. Kanwat, J. Xia, T. Salim, T. J. N. Hooper, M. Kovalev, D. Giovanni, Y. C. Aw, B. Chaudhary, Y. Cai, G. Xing, T. C. Sum, J. W. Ager, S. G. Mhaisalkar and N. Mathews, Adv. Mater., 2023, 35, 2210176 CrossRef CAS PubMed.
- G. Grancini and M. K. Nazeeruddin, Nat. Rev. Mater., 2019, 4, 4–22 CrossRef CAS.
- P. P. Boix, S. Agarwala, T. M. Koh, N. Mathews and S. G. Mhaisalkar, J. Phys. Chem. Lett., 2015, 6, 898–907 CrossRef CAS PubMed.
- T. M. Koh, V. Shanmugam, X. Guo, S. S. Lim, O. Filonik, E. M. Herzig, P. Müller-Buschbaum, V. Swamy, S. T. Chien, S. G. Mhaisalkar and N. Mathews, J. Mater. Chem. A, 2018, 6, 2122–2128 RSC.
- J. W. Yang, S. G. Ji, C.-S. Jeong, J. Kim, H. R. Kwon, T. H. Lee, S. A. Lee, W. S. Cheon, S. Lee, H. Lee, M. S. Kwon, J. Moon, J. Y. Kim and H. W. Jang, Energy Environ. Sci., 2024, 17, 2541–2553 RSC.
- J. W. Yang, H. R. Kwon, S. G. Ji, J. Kim, S. A. Lee, T. H. Lee, S. Choi, W. S. Cheon, Y. Kim, J. Park, J. Y. Kim and H. W. Jang, Adv. Funct. Mater., 2024, 2400806 CrossRef.
- H. R. Kwon, J. W. Yang, S. Choi, W. S. Cheon, I. H. Im, Y. Kim, J. Park, G.-H. Lee and H. W. Jang, Adv. Energy Mater., 2024, 14, 2303342 CrossRef CAS.
- S. Akel, A. Kulkarni, U. Rau and T. Kirchartz, Phys. Rev. X, 2023, 2, 013004 Search PubMed.
- Y.-F. Mu, C. Zhang, M.-R. Zhang, W. Zhang, M. Zhang and T.-B. Lu, ACS Appl. Mater. Interfaces, 2021, 13, 22314–22322 CrossRef CAS PubMed.
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed., 2018, 57, 13570–13574 CrossRef CAS PubMed.
- X.-X. Guo, S.-F. Tang, Y.-F. Mu, L.-Y. Wu, G.-X. Dong and M. Zhang, RSC Adv., 2019, 9, 34342–34348 RSC.
- A. Pan, X. Ma, S. Huang, Y. Wu, M. Jia, Y. Shi, Y. Liu, P. Wangyang, L. He and Y. Liu, J. Phys. Chem. Lett., 2019, 10, 6590–6597 CrossRef CAS PubMed.
- E. Rathore, K. Maji, D. Rao, B. Saha and K. Biswas, J. Phys. Chem. Lett., 2020, 11, 8002–8007 CrossRef CAS PubMed.
- J. Wang, J. Wang, N. Li, X. Du, J. Ma, C. He and Z. Li, ACS Appl. Mater. Interfaces, 2020, 12, 31477–31485 CrossRef CAS PubMed.
- H. Huang, J. Zhao, Y. Du, C. Zhou, M. Zhang, Z. Wang, Y. Weng, J. Long, J. Hofkens, J. A. Steele and M. B. J. Roeffaers, ACS Nano, 2020, 14, 16689–16697 CrossRef CAS PubMed.
- A. Kipkorir, J. DuBose, J. Cho and P. V. Kamat, Chem. Sci., 2021, 12, 14815–14825 RSC.
- N. Li, X. Chen, J. Wang, X. Liang, L. Ma, X. Jing, D.-L. Chen and Z. Li, ACS Nano, 2022, 16, 3332–3340 CrossRef CAS PubMed.
- L. Romani, A. Speltini, C. N. Dibenedetto, A. Listorti, F. Ambrosio, E. Mosconi, A. Simbula, M. Saba, A. Profumo, P. Quadrelli, F. De Angelis and L. Malavasi, Adv. Funct. Mater., 2021, 31, 2104428 CrossRef CAS.
- Q. Zhong, M. Cao, H. Hu, D. Yang, M. Chen, P. Li, L. Wu and Q. Zhang, ACS Nano, 2018, 12, 8579–8587 CrossRef CAS PubMed.
- K. Ma, X.-Y. Du, Y.-W. Zhang and S. Chen, J. Mater. Chem. C, 2017, 5, 9398–9404 RSC.
- A. F. Gualdrón-Reyes, C. A. Mesa, S. Giménez and I. Mora-Seró, Sol. RRL, 2022, 6, 2200012 CrossRef.
- J. Hou, S. Cao, Y. Wu, Z. Gao, F. Liang, Y. Sun, Z. Lin and L. Sun, Chem. – Eur. J., 2017, 23, 9481–9485 CrossRef CAS PubMed.
- Y. Jiang, H.-Y. Chen, J.-Y. Li, J.-F. Liao, H.-H. Zhang, X.-D. Wang and D.-B. Kuang, Adv. Funct. Mater., 2020, 30, 2004293 CrossRef CAS.
- J. T. DuBose and P. V. Kamat, ACS Energy Lett., 2022, 7, 1994–2011 CrossRef CAS.
- X. Zhu, Y. Lin, J. San Martin, Y. Sun, D. Zhu and Y. Yan, Nat. Commun., 2019, 10, 2843 CrossRef PubMed.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648–3657 CrossRef CAS PubMed.
- M. Li, S. Xu, L. Wu, H. Tang, B. Zhou, J. Xu, Q. Yang, T. Zhou, Y. Qiu, G. Chen, G. I. N. Waterhouse and K. Yan, ACS Energy Lett., 2022, 7, 3370–3377 CrossRef CAS.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- R. Das, A. Patra, S. K. Dutta, S. Shyamal and N. Pradhan, J. Am. Chem. Soc., 2022, 144, 18629–18641 CrossRef CAS PubMed.
- C. Ma, F. T. Eickemeyer, S.-H. Lee, D.-H. Kang, S. J. Kwon, M. Grätzel and N.-G. Park, Science, 2023, 379, 173–178 CrossRef CAS PubMed.
- Z.-Y. Chen, N.-Y. Huang and Q. Xu, Coord. Chem. Rev., 2023, 481, 215031 CrossRef CAS.
- Y.-F. Xu, M.-Z. Yang, H.-Y. Chen, J.-F. Liao, X.-D. Wang and D.-B. Kuang, ACS Appl. Energy Mater., 2018, 1, 5083–5089 CrossRef CAS.
- A. Li, T. Wang, C. Li, Z. Huang, Z. Luo and J. Gong, Angew. Chem., Int. Ed., 2019, 58, 3804–3808 CrossRef CAS PubMed.
- L. Wang, M. Ghoussoub, H. Wang, Y. Shao, W. Sun, A. A. Tountas, T. E. Wood, H. Li, J. Y. Y. Loh, Y. Dong, M. Xia, Y. Li, S. Wang, J. Jia, C. Qiu, C. Qian, N. P. Kherani, L. He, X. Zhang and G. A. Ozin, Joule, 2018, 2, 1369–1381 CrossRef CAS.
- W. Zhao, M. Ding, P. Yang, Q. Wang, K. Zhang, X. Zhan, Y. Yu, Q. Luo, S. Gao, J. Yang and Y. Xie, EES Catal., 2023, 1, 36–44 RSC.
- S. Gao, B. Gu, X. Jiao, Y. Sun, X. Zu, F. Yang, W. Zhu, C. Wang, Z. Feng, B. Ye and Y. Xie, J. Am. Chem. Soc., 2017, 139, 3438–3445 CrossRef CAS PubMed.
- Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C.-J. Sun, Z. Cai, J. R. Guest, Y. Ren, V. Stamenkovic, L. A. Curtiss, Y. Liu and T. Rajh, Nat. Energy, 2019, 4, 957–968 CrossRef CAS.
- L. Wang, J. Wan, Y. Zhao, N. Yang and D. Wang, J. Am. Chem. Soc., 2019, 141, 2238–2241 CrossRef CAS PubMed.
- L. Hao, L. Kang, H. Huang, L. Ye, K. Han, S. Yang, H. Yu, M. Batmunkh, Y. Zhang and T. Ma, Adv. Mater., 2019, 31, 1900546 CrossRef PubMed.
- P. Xia, M. Antonietti, B. Zhu, T. Heil, J. Yu and S. Cao, Adv. Funct. Mater., 2019, 29, 1900093 CrossRef.
- S. Sorcar, Y. Hwang, J. Lee, H. Kim, K. M. Grimes, C. A. Grimes, J.-W. Jung, C.-H. Cho, T. Majima, M. R. Hoffmann and S.-I. In, Energy Environ. Sci., 2019, 12, 2685–2696 RSC.
- S. Sorcar, J. Thompson, Y. Hwang, Y. H. Park, T. Majima, C. A. Grimes, J. R. Durrant and S.-I. In, Energy Environ. Sci., 2018, 11, 3183–3193 RSC.
- Z. Jiang, W. Wan, H. Li, S. Yuan, H. Zhao and P. K. Wong, Adv. Mater., 2018, 30, 1706108 CrossRef PubMed.
- T. Di, B. Zhu, B. Cheng, J. Yu and J. Xu, J. Catal., 2017, 352, 532–541 CrossRef CAS.
- G. Yin, M. Nishikawa, Y. Nosaka, N. Srinivasan, D. Atarashi, E. Sakai and M. Miyauchi, ACS Nano, 2015, 9, 2111–2119 CrossRef CAS PubMed.
- H. Zhu, S. Teale, M. N. Lintangpradipto, S. Mahesh, B. Chen, M. D. McGehee, E. H. Sargent and O. M. Bakr, Nat. Rev. Mater., 2023, 8, 569–586 CrossRef.
- S. Shukla, T. M. Koh, R. Patidar, J. H. Lew, P. Kajal, S. G. Mhaisalkar and N. Mathews, J. Phys. Chem. C, 2021, 125, 6585–6592 CrossRef CAS.
- A. Bashir, S. Shukla, J. H. Lew, S. Shukla, A. Bruno, D. Gupta, T. Baikie, R. Patidar, Z. Akhter, A. Priyadarshi, N. Mathews and S. G. Mhaisalkar, Nanoscale, 2018, 10, 2341–2350 RSC.
- S. Park, W. J. Chang, C. W. Lee, S. Park, H.-Y. Ahn and K. T. Nam, Nat. Energy, 2016, 2, 16185 CrossRef.
- J. Liang, X. Han, Y. Qiu, Q. Fang, B. Zhang, W. Wang, J. Zhang, P. M. Ajayan and J. Lou, ACS Nano, 2020, 14, 5426–5434 CrossRef CAS PubMed.
- A. M. K. Fehr, A. Agrawal, F. Mandani, C. L. Conrad, Q. Jiang, S. Y. Park, O. Alley, B. Li, S. Sidhik, I. Metcalf, C. Botello, J. L. Young, J. Even, J. C. Blancon, T. G. Deutsch, K. Zhu, S. Albrecht, F. M. Toma, M. Wong and A. D. Mohite, Nat. Commun., 2023, 14, 3797 CrossRef CAS PubMed.
- M. Rahaman, V. Andrei, D. Wright, E. Lam, C. Pornrungroj, S. Bhattacharjee, C. M. Pichler, H. F. Greer, J. J. Baumberg and E. Reisner, Nat. Energy, 2023, 8, 629–638 CrossRef CAS.
- V. Andrei, G. M. Ucoski, C. Pornrungroj, C. Uswachoke, Q. Wang, D. S. Achilleos, H. Kasap, K. P. Sokol, R. A. Jagt, H. Lu, T. Lawson, A. Wagner, S. D. Pike, D. S. Wright, R. L. Z. Hoye, J. L. MacManus-Driscoll, H. J. Joyce, R. H. Friend and E. Reisner, Nature, 2022, 608, 518–522 CrossRef CAS PubMed.
- A. Tayyebi, R. Mehrotra, M. A. Mubarok, J. Kim, M. Zafari, M. Tayebi, D. Oh, S.-h Lee, J. E. Matthews, S.-W. Lee, T. J. Shin, G. Lee, T. F. Jaramillo, S.-Y. Jang and J.-W. Jang, Nat. Catal., 2024, 7, 510–521 CrossRef CAS.
- Y. Choi, R. Mehrotra, S.-H. Lee, T. V. T. Nguyen, I. Lee, J. Kim, H.-Y. Yang, H. Oh, H. Kim, J.-W. Lee, Y. H. Kim, S.-Y. Jang, J.-W. Jang and J. Ryu, Nat. Commun., 2022, 13, 5709 CrossRef CAS PubMed.
- H. Yang, Y. Liu, Y. Ding, F. Li, L. Wang, B. Cai, F. Zhang, T. Liu, G. Boschloo, E. M. J. Johansson and L. Sun, Nat. Commun., 2023, 14, 5486 CrossRef CAS PubMed.
- I. Poli, U. Hintermair, M. Regue, S. Kumar, E. V. Sackville, J. Baker, T. M. Watson, S. Eslava and P. J. Cameron, Nat. Commun., 2019, 10, 2097 CrossRef PubMed.
- Z. Zhu, M. Daboczi, M. Chen, Y. Xuan, X. Liu and S. Eslava, Nat. Commun., 2024, 15, 2791 CrossRef CAS PubMed.
- D. Hansora, J. W. Yoo, R. Mehrotra, W. J. Byun, D. Lim, Y. K. Kim, E. Noh, H. Lim, J.-W. Jang, S. I. Seok and J. S. Lee, Nat. Energy, 2024, 9, 272–284 CrossRef CAS.
- L. Wang, W. Lian, B. Liu, H. Lv, Y. Zhang, X. Wu, T. Wang, J. Gong, T. Chen and H. Xu, Adv. Mater., 2022, 34, 2200723 CrossRef CAS PubMed.
- R. Fernández-Climent, A. F. Gualdrón-Reyes, M. García-Tecedor, C. A. Mesa, D. Cárdenas-Morcoso, L. Montañes, E. M. Barea, E. Mas-Marzá, B. Julián-López, I. Mora-Seró and S. Giménez, Sol. RRL, 2022, 6, 2100723 CrossRef.
- S.-Y. Lee, P. Serafini, S. Masi, A. F. Gualdrón-Reyes, C. A. Mesa, J. Rodríguez-Pereira, S. Giménez, H. J. Lee and I. Mora-Seró, ACS Energy Lett., 2023, 8, 4488–4495 CrossRef CAS PubMed.
- M. Saliba, Adv. Energy Mater., 2019, 9, 1803754 CrossRef.
- Q. Sun, W.-J. Yin and S.-H. Wei, J. Mater. Chem. C, 2020, 8, 12012–12035 RSC.
- M. Ahmadi, M. Ziatdinov, Y. Zhou, E. A. Lass and S. V. Kalinin, Joule, 2021, 5, 2797–2822 CrossRef CAS.
- S. Sun, A. Tiihonen, F. Oviedo, Z. Liu, J. Thapa, Y. Zhao, N. T. P. Hartono, A. Goyal, T. Heumueller, C. Batali, A. Encinas, J. J. Yoo, R. Li, Z. Ren, I. M. Peters, C. J. Brabec, M. G. Bawendi, V. Stevanovic, J. Fisher and T. Buonassisi, Matter, 2021, 4, 1305–1322 CrossRef CAS.
- X. Li, H. Mai, J. Lu, X. Wen, T. C. Le, S. P. Russo, D. A. Winkler, D. Chen and R. A. Caruso, Angew. Chem., Int. Ed., 2023, 62, e202315002 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |