
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevisiting group 4–7 transition metals for heterogeneous ammonia synthesis
Wenbo
Gao
 ab,
Yawei
Wang
ab,
Qianru
Wang
ab,
Zhaolong
Sun
ab,
Jianping
Guo
*ab and
Ping
Chen
ab
ab,
Yawei
Wang
ab,
Qianru
Wang
ab,
Zhaolong
Sun
ab,
Jianping
Guo
*ab and
Ping
Chen
ab
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: guojianping@dicp.ac.cn
bCenter of Materials and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing, 100049, China
First published on 2nd February 2024
Abstract
Ammonia is a key small molecule for manufacturing nitrogen-based fertilizers and organic chemicals and equally important for renewable energy storage and conversion. The available Haber–Bosch ammonia synthesis process using fused iron catalysts operated under harsh conditions is, however, unsustainable. The development of alternative and more efficient approaches to sustainable ammonia production has garnered much attention recently. Most of the prior work has been devoted to the investigation of Fe, Ru or Co-based metal catalysts for ammonia synthesis. In comparison, there are very limited studies on group 4–7 transition metals, because they are prone to form metal nitrides, which are difficult to hydrogenate to ammonia. This mini-review summarizes recent advances in activating these metals for heterogeneous ammonia synthesis. We show that the potential properties of group 4–7 transition metals for ammonia synthesis should be revisited, which may lead to the development of more efficient materials or chemical processes for ammonia production under mild conditions.
Broader contextAmmonia has been not only a key molecule for manufacturing nitrogen-based fertilizers, but also a ‘star molecule’ for energy storage, conversion and utilization in recent years. Sustainable ammonia production is the most important part of the future ammonia-powered clean energy systems. Although the Haber–Bosch ammonia synthesis process is available and well defined, it can only be operated under very harsh conditions. The development of more energy-efficient approaches and advanced materials for sustainable ammonia synthesis under mild conditions is highly required. The group 4–7 transition metals have the advantage of high affinity to N2, but the activated N is difficult to convert further to ammonia and thus exhibits low catalytic activities. It is of both scientific significance and practical relevance to make these transition metals active for ammonia synthesis typically under mild conditions. In this mini-review, the recent advances in employing group 4–7 metals (nitrides) for heterogeneous ammonia synthesis are summarized, emphasizing the opportunities in activating these group 4–7 metals for ammonia production. |
1. Introduction
There has been recently considerable interest in dinitrogen fixation to ammonia (N2-to-NH3), because ammonia is not only an indispensable chemical intermediate for producing nitrogenous fertilizers and organic chemicals, but also a promising carbon-free energy (hydrogen) carrier and fuel.1–3 Ammonia has the metrics of high hydrogen content (17.8 wt%), high energy density (22.5 MJ kg−1 or 15.6 MJ L−1), facile storage and transportation, etc. The synthesis, separation, storage, transportation, and utilization of ammonia are important for the forthcoming ‘ammonia energy systems’.4 The artificial ammonia synthesis, the most difficult process, relies highly on the well-defined and centralized Haber–Bosch (H–B) ammonia synthesis process nowadays, which is operated under high-temperature and high-pressure reaction conditions (673–823 K and 10–30 MPa) using an iron-based catalyst. In the 1990s, a carbon supported ruthenium catalyst was used industrially in the KBR advanced ammonia process, known as KAAP. The development of alternative methods and advanced materials for ammonia synthesis typically under mild conditions is a long sought-after goal. This task is further motivated by the requirements of decentralized, distributed, flexible, and high-efficiency ammonia production processes that could be coupled with the isolated and intermittent renewable energy resources.5However, ammonia synthesis under mild conditions is never an easy task. From the viewpoint of thermodynamics, ammonia synthesis from N2 and H2 is an exergonic reaction (ΔG° = −32.9 kJ mol−1). The equilibrium ammonia concentration reaches 91.5% at 298 K and 1 bar N2/H2 pressure. This is, unfortunately, unattainable under normal thermal reaction conditions because of large kinetic barriers. To obtain an exit NH3 concentration of practical interest (i.e., >10 mol%), high temperatures and pressures are essential even for the commercialized fused iron or Ru catalyst. This contradiction between thermodynamic and kinetic properties of ammonia synthesis reaction lies in at least two aspects.6 On one hand, the chemical inertness of the N2 molecule is basically due to the strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond (942 kJ mol−1), high HOMO–LUMO gap (10.82 eV), and nonpolarity.7 On the other hand, it is more difficult to maintain a high N hydrogenation rate with a high N2 dissociation rate, which is restricted by the energy scaling relationship among different adsorbed species and transition states on a specific active site.8 This relationship gives rise to a classic volcano-type plot for a series of transition metal surfaces between catalytic activity and the adsorption energy of N as illustrated in Fig. 1a. Metals such as Fe and Ru have moderate N adsorption energies and high intrinsic activities and have thus been applied in the ammonia synthesis industry. For elements at the left- or right-hand side of the volcano plot, a lower activity is found because of their either higher or lower N adsorption energy. Specifically, it is hard for the right-hand side metals to activate N2, although the hydrogenation of N is facile. For the left-hand side metals, it turns out that a high N adsorption energy leads to a low activation energy for N2 dissociation but a high energy barrier for N hydrogenation to NHx (x = 1–3) species. As an example, the clean Cr metal surface has a strong N adsorption energy (EN = −1.8 eV for the Cr(110) surface),9 and surface science studies showed that the dissociative adsorption of N2 and formation of surface Cr nitride could take place at 300 K.10 The formed surface Cr nitride, however, is too stable to be hydrogenated to NH3 under moderate conditions. These left-hand side metals have high affinity toward N2, and hence have advantages of N2 activation and/or dissociation, which has been generally accepted as the rate-determining step in ammonia synthesis reaction. It is thus of both scientific and practical interest to find out whether or how to enhance the catalytic activities of these left-hand side metals.
N triple bond (942 kJ mol−1), high HOMO–LUMO gap (10.82 eV), and nonpolarity.7 On the other hand, it is more difficult to maintain a high N hydrogenation rate with a high N2 dissociation rate, which is restricted by the energy scaling relationship among different adsorbed species and transition states on a specific active site.8 This relationship gives rise to a classic volcano-type plot for a series of transition metal surfaces between catalytic activity and the adsorption energy of N as illustrated in Fig. 1a. Metals such as Fe and Ru have moderate N adsorption energies and high intrinsic activities and have thus been applied in the ammonia synthesis industry. For elements at the left- or right-hand side of the volcano plot, a lower activity is found because of their either higher or lower N adsorption energy. Specifically, it is hard for the right-hand side metals to activate N2, although the hydrogenation of N is facile. For the left-hand side metals, it turns out that a high N adsorption energy leads to a low activation energy for N2 dissociation but a high energy barrier for N hydrogenation to NHx (x = 1–3) species. As an example, the clean Cr metal surface has a strong N adsorption energy (EN = −1.8 eV for the Cr(110) surface),9 and surface science studies showed that the dissociative adsorption of N2 and formation of surface Cr nitride could take place at 300 K.10 The formed surface Cr nitride, however, is too stable to be hydrogenated to NH3 under moderate conditions. These left-hand side metals have high affinity toward N2, and hence have advantages of N2 activation and/or dissociation, which has been generally accepted as the rate-determining step in ammonia synthesis reaction. It is thus of both scientific and practical interest to find out whether or how to enhance the catalytic activities of these left-hand side metals.
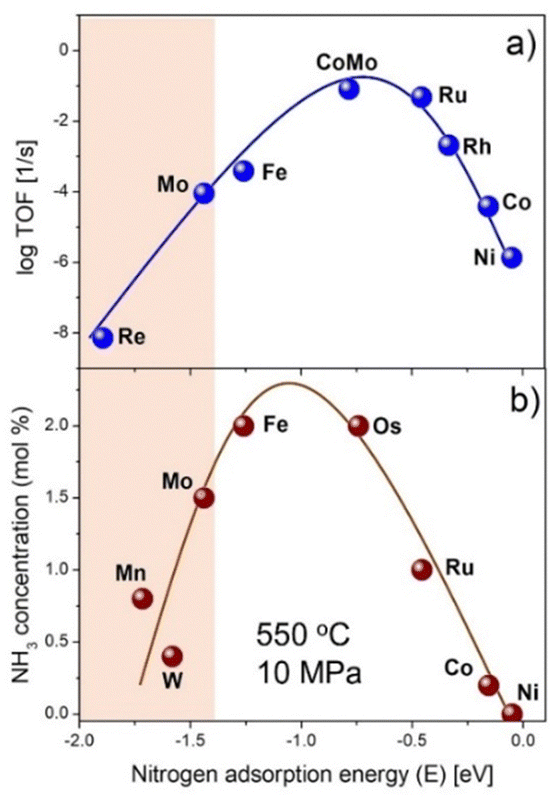 | ||
| Fig. 1 The classic volcano plot for thermal-catalytic ammonia synthesis. (a) Calculated rate as a function of EN. (b) The NH3 concentration data were taken from ref. 28. The N adsorption energies of Mn and W were taken from ref. 9. | ||
Group 4–7 metals such as Ti, V, Cr, Mn, Mo, Nb, etc. are located at the left-hand side of the volcano plot and most of them have been rarely used as catalytic materials for thermo-catalytic ammonia synthesis relative to Fe, Ru or Co. However, it is well known that Mo and V are key elements in the cofactors of MoFe and VFe nitrogenases, respectively. The nitrogenase can catalyze N2 reduction and protonation to NH3 (NH4+) at room temperature and atmospheric pressure although this biological process is also energy-intensive (ca. 500 kJ mol−1 N2). There are a large number of soluble transition metal–dinitrogen complexes based on the group 4–7 metals. Among them, a couple of Mo- and V-based complexes have been synthesized and shown to be catalytically active for homogeneous ammonia synthesis.11 The knowledge on the role of group 4–7 metals in biological or homogenous N2 fixation would be helpful for the design and development of novel catalysts or functional materials for heterogenous ammonia synthesis. In one early monograph, Aika and Tamaru gave a comprehensive review on the non-iron catalysts including group 4–7 metals for ammonia synthesis before 1995.12 In this minireview, we would like to summarize the recent advances in the group 4–7 metals for heterogeneous ammonia synthesis, typically heterogeneous thermal catalysis and the chemical looping route. Some strategies to activate the group 4–7 metal nitride surfaces are discussed. We hope this minireview could arouse the interest of the researchers to rethink the role of these transition metals in dinitrogen fixation and ammonia synthesis, which have received far less attention compared with Fe, Ru, and Co metals.
2. Thermo-catalytic ammonia synthesis
Before the invention of the fused iron-based ammonia synthesis catalyst, Mittasch et al. had explored various transition metals for ammonia synthesis.28 They found some of the group 4–7 transition metals such as Mo, W, and Mn are catalytically active for ammonia synthesis at high temperature and pressure (Fig. 1b). With the addition of promoters, the multicomponent iron catalyst was found to be more active than others. Moreover, Fe is much cheaper and easily available, which render it attractive for practical applications. Since then, very little attention has been given to other metals including the group 4–7 metals for ammonia synthesis.The group 4–7 metals are prone to form thermodynamically stable metal nitrides under the ammonia synthesis reaction conditions. Aika et al. studied the mechanism of ammonia synthesis over a molybdenum nitride (Mo2N) catalyst and proposed that the rate-determining step (RDS) is the chemisorption of N2, which is retarded by nitrogen atoms on the nitride surface.29 Boudart et al. found the structure sensitivity and strong ammonia inhibition effects on the Mo2N catalyst for ammonia synthesis.30 However, the catalytic activity of Mo-based catalysts is commonly lower than those of Fe-based and Ru-based catalysts. With the advancement of materials science and preparation technology, some active ammonia synthesis catalysts employing the group 4–7 metals have been developed. As shown in Table 1, these catalysts could be divided into three types: alloy or metal nitride catalysts, transition metal hydride-based catalysts, and composite catalysts of alkali (alkaline earth) hydride and transition metals.
| Samples | NH3 synthesis rate (μmol g−1 h−1) | Reaction conditions | WHSV (ml g−1 h−1) | Ref. |
|---|---|---|---|---|
| V | Undetected | 673 K, 50 bar | 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
13 |
| VN | 10 | 673 K, 50 bar | 60000 |
13 |
| VN | Undetected | 573 K, 10 bar | 60000 |
14 |
| CrN | Undetected | 573 K, 10 bar | 60000 |
14 |
| Mn4N | Undetected | 573 K, 10 bar | 60000 |
14 |
| Mo2N | 68 | 673 K, 1 bar | 9000 | 15 |
| Co3Mo3N | 652 | 673 K, 1 bar | 9000 | 15 |
| Cs–Co3Mo3N | 986 | 673 K, 1 bar | 9000 | 15 |
| Cs–Co3Mo3N | 5000 | 673 K, 11 bar | 9000 | 15 |
| Fe–Mo–N | 143 | 673 K, 1 bar | 9000 | 15 |
| Ni2Mo3N | 383 | 673 K, 1 bar | 12000 |
16 |
| Re3N | 430 | 673 K, 1 bar | 9000 | 17 |
| CoRe4 | 943 | 673 K, 1 bar | 12000 |
18 |
| ZrCr2 | 580 | 673 K, 30 bar | 60000 |
19 |
| ZrMn2 | 400 | 673 K, 30 bar | 60000 |
19 |
| ZrV2 | 200 | 673 K, 30 bar | 60000 |
19 |
| TaV2 | Undetected | 673 K, 30 bar | 60000 |
19 |
| Zr(Cr0.8Fe0.2)2 | 200 | 673 K, 30 bar | 60000 |
19 |
| Zr(Cr0.8Co0.2)2 | 130 | 673 K, 30 bar | 60000 |
19 |
| Zr(Cr0.8Ni0.2)2 | 170 | 673 K, 30 bar | 60000 |
19 |
| Zr(Cr0.8Cu0.2)2 | 280 | 673 K, 30 bar | 60000 |
19 |
| LaMnSi | 120 | 673 K, 1 bar | 36000 |
20 |
| TiH2 | 2800 | 673 K, 50 bar | 60000 |
13 |
| VH0.39 | 3200 | 673 K, 50 bar | 60000 |
13 |
| NbH0.6 | 400 | 673 K, 50 bar | 60000 |
13 |
| ZrH2 | Undetected | 673 K, 50 bar | 60000 |
13 |
| BaTiO2.5H0.5 | 1400 | 673 K, 50 bar | 60000 |
21 |
| Ru/BaTiO2.5H0.5 | 28200 |
673 K, 50 bar | 60000 |
22 |
| Fe/BaTiO2.4H0.6 | 14000 |
673 K, 50 bar | 60000 |
22 |
| Co/BaTiO2.5H0.5 | 4000 | 673 K, 50 bar | 60000 |
22 |
| LaN–Ru/ZrH2 | 5600 | 623 K, 10 bar | 60000 |
23 |
| Mn–5LiH | 3120 | 573 K, 10 bar | 60000 |
14 |
| Mn4N–LiH | 2253 | 573 K, 10 bar | 60000 |
14 |
| Mn4N–NaH | 509 | 573 K, 10 bar | 60000 |
14 |
| Mn4N–KH | 70 | 573 K, 10 bar | 60000 |
14 |
| Mn4N–CaH2 | 224 | 573 K, 10 bar | 60000 |
14 |
| Mn4N–BaH2 | 1320 | 573 K, 10 bar | 60000 |
14 |
| V–5LiH | 273 | 573 K, 10 bar | 60000 |
14 |
| Cr–5LiH | 3604 | 573 K, 10 bar | 60000 |
14 |
| ZrCr2–LiH | 750 | 673 K, 30 bar | 60000 |
19 |
| CrN–BaH2 | 1190 | 573 K, 10 bar | 60000 |
24 |
| K2[Mn(NH2)4] | 1124 | 573 K, 10 bar | 60000 |
25 |
| Rb2[Mn(NH2)4] | 1281 | 573 K, 10 bar | 60000 |
25 |
| [BaCrHN] | 6860 | 573 K, 10 bar | 60000 |
24 |
| Ca3CrN3H | 3800 | 673 K, 50 bar | 60000 |
26 |
| Ru/MgO | 310 | 573 K, 10 bar | 60000 |
14 |
| Cs–Ru/MgO | 1390 | 573 K, 10 bar | 60000 |
14 |
| Fe cat. | 5400 | 573 K, 9 bar | 36000 |
27 |
2.1 Alloy or metal nitride catalysts
Based on the understanding of the Sabatier principle and concept of scaling relations, it appears that improved ammonia synthesis catalysts with optimum N adsorption energies could be found by alloying group 4–7 metals having high N binding energies with those metals having low N adsorption energies. One typical example is the CoMo catalyst developed by Aika and Jacobsen parallelly in 2000,31,32 which exhibits a higher activity comparable to those of Fe- and Ru-based catalysts. It should be noted that the CoMo alloy transforms into CoMo nitride (Co3Mo3N) under the conditions of ammonia synthesis. Alternatively, it has been suggested that the lattice nitrogen may participate in the catalytic cycle via a Mars–van Krevelen-like mechanism.33,34 This reaction pathway involves the synthesis of ammonia by the hydrogenation of lattice nitrogen leading to nitrogen vacancies, which can be replenished by gaseous N2. Except for the CoMo nitride catalyst, NiMo and FeMo nitride catalysts were also tested, which are less active than CoMo. The addition of Co can also improve the activity of Re or Re3N but the formation of a ternary metal nitride phase was not observed.17This approach for catalyst design could also be generalized to other transition metals. For example, rare earth metals can combine with Co, Ru, or Fe forming intermetallic compounds such as CeCo3, CeRu2, and CeFe2 showing high specific activities for ammonia synthesis.12 Ni metal alone is generally inactive for ammonia synthesis, but recent studies showed that combining Ni with rare earth metals, which transformed into nitrides, can significantly enhance its catalytic activity.35 Some ternary intermetallic compounds such as LaRuSi, LaCoSi, and LaMnSi have been reported as active catalysts for ammonia synthesis.20,36,37 The intermetallic compounds containing group 4–7 metals, such as ZrCr2, ZrMn2, and ZrV2, can absorb both H2 and N2, and they have been tested for ammonia synthesis recently and form the intermetallic nitride hydride phase, although their catalytic activities are not high.19 Wang et al. reported that a CoMoFeNiCu high-entropy alloy is active for ammonia decomposition, the reverse reaction of ammonia synthesis.38 There has been so far little work on high-entropy alloys or high-entropy metal hydrides and nitrides for ammonia synthesis catalysis, which is worthy of future study. The artificial intelligence and machine learning would help guide the design and construction of active alloy, hydride or nitride catalysts. Based on the available data sets of adsorption energies of NHx species on a number of metal, alloy, or nitride surfaces from the databases such as Open Catalyst, a machine learning model could be trained and used to predict N adsorption energies of other different sites. Combining DFT calculations and machine learning could produce an automated framework that is able to search for active sites approaching the optimum N adsorption energies. If the controlled synthesis of alloy or intermetallic compound materials with specific composition, morphology, and size can be achieved, the N adsorption energy could be finely tuned, which may lead to the development of more active alloy or metal nitride catalysts for ammonia synthesis. However, as discussed before, the catalytic activity will be still limited by the scaling relationships on the transition metal surfaces,8 and hence it remains unclear whether we could achieve high-efficiency ammonia synthesis under mild conditions by using transition metal-only catalysts.
2.2 Transition metal hydrides
As mentioned above, it is known that early transition metals such as Ti, V, or Cr are inactive for ammonia synthesis at low temperatures and pressures (Table 1) because of their strong metal–N bonds. In organometallic chemistry, the reactivity of transition metals can be manipulated by the selection of different kinds of ligands.11 A series of transition metal (Ti, V, Cr, Nb etc.) hydride complexes have been shown to activate or even cleave N2via reductive elimination and/or reductive protonation mechanisms.39 For example, a soluble trititanium hydride cluster cleaves N2 to form N–H bonds under mild conditions (Fig. 2a).40 Some heterogeneous metal hydride-based materials for N2 activation have also been reported. Basset et al. reported an isolated silica supported tantalum hydride complex ([(Si–O)2Ta–Hx, x = 1 or 3]) capable of cleaving N2 at 523 K forming a Ta amido imido product [(SiO)2Ta(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH)(NH2)] (Fig. 2b).41 However, the hydrogenation to ammonia was not obtained. A single site Mo hydride supported on silica was shown to be active for ammonia synthesis catalysis at 673 K.42
NH)(NH2)] (Fig. 2b).41 However, the hydrogenation to ammonia was not obtained. A single site Mo hydride supported on silica was shown to be active for ammonia synthesis catalysis at 673 K.42
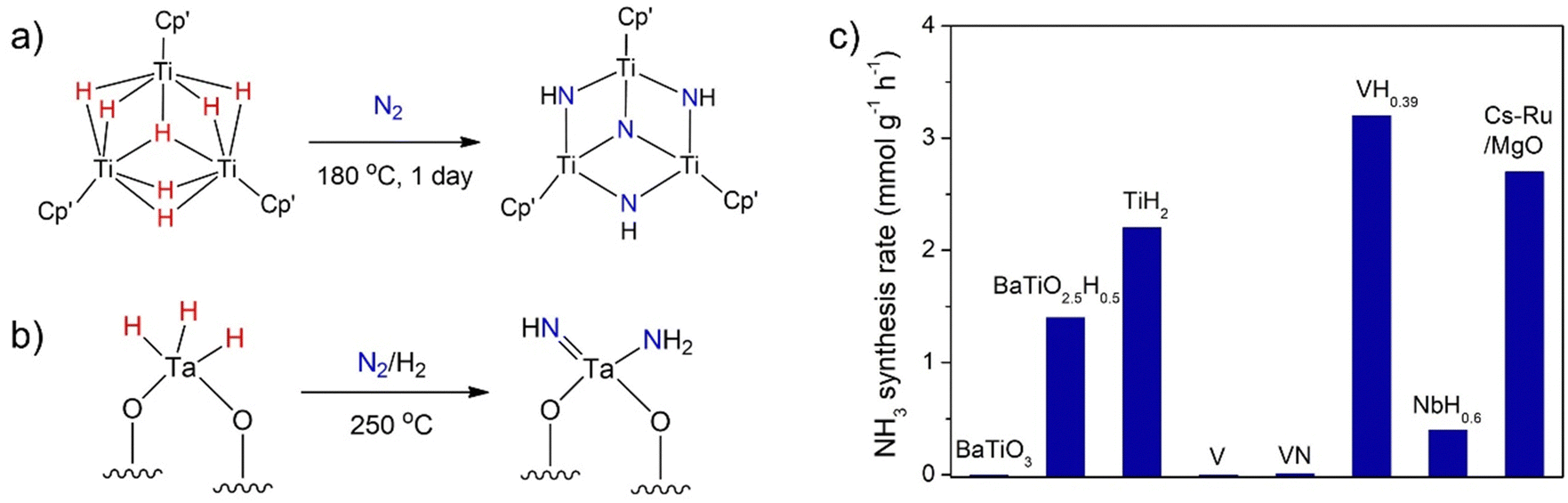 | ||
| Fig. 2 (a) N2 cleavage by a soluble trititanium hydride cluster. (b) N2 activation over an isolated silica supported tantalum hydride complex. (c) NH3 synthesis rates over a series of early transition metal hydride precatalysts. Reaction conditions: 673 K, 5 MPa. | ||
The inclusion of hydrogen into the lattice of transition metals forming metal hydrides has been found to counteract the negative effects of overly strong TM–N bonds and make them more active for ammonia synthesis (Fig. 2c). For example, Kageyama et al. found that although BaTiO3 is inactive, the titanium-based hydrides (TiH2 and BaTiO2.5H0.5) could catalyze ammonia synthesis via a hydrogen-based Mars–van Krevelen mechanism.21 After the test, TiH2 and BaTiO2.5H0.5 may change to surface TiN and BaTiO2.5N0.2H0.3 oxyhydride–nitride structures, respectively. The presence of hydride in the catalyst phase is demonstrated to be crucial. It has been shown later that vanadium hydride (VH0.39) and niobium hydride (NbH0.6) are also active for ammonia synthesis, whereas V metal and VN are inactive.13 However, VH0.39 finally turns into a nitride–hydride composition of VH0.44N0.16, which should be the active phase. These (oxy)hydrides have also been shown as effective supports for loading Ru, Fe, or Co metals for ammonia synthesis catalysis.22 The reactions of N2 with gas-phase transition metal atoms and clusters have been studied and the knowledge may be transferred into the real catalytic systems.43,44 Ma et al. reported that N2 can be activated by the well-defined Ta3N3H− and Ta3N3− gas-phase clusters yielding Ta3N5H− and Ta3N5−, respectively.45 The presence of hydrogen in Ta3N3H− was suggested to modify the charge distribution and the geometry of Ta3N3H−, which is crucial to increase the reactivity. Although these transition metal hydrides or nitride–hydride pre-catalysts exhibit insufficient activities in ammonia synthesis reaction, these results encourage that other group 4–7 metal hydrides or multinary metal hydrides may be worthy of further study as (pre)catalysts for ammonia synthesis.
2.3 Alkali (alkaline earth) hydride-transition metal composite catalysts
It has been proposed that the dissociation of N2 is retarded by the strong bonded N atoms on transition metal surfaces. Provided the N atoms can be removed by a substance, the reactive sites would be open to N2 activation, and the substance should be regenerated. The alkali and alkaline earth metal hydrides (denoted hereafter as AH) containing negatively charged hydrogen atoms (H−) possess this functionality. As an example, lithium hydride (LiH) is able to remove nitrogen atoms from the transition metal nitrides (denoted as TMN) to clean the metal surface. It is also important that LiH can bind N atoms to form LiNH2/Li2NH species, which can further split H2 heterolytically to give NH3 and regenerate LiH.46 This synergy between TMN and LiH creates a favourable pathway that allows 3d TM–LiH composites to exhibit unexpectedly high catalytic activities typically at lower temperatures. As shown in Fig. 3a, the addition of LiH can increase the activity of neat 3d metals (from V to Ni) by nearly 1–4 orders of magnitude at 573 K and 10 bar pressure.14 Recent work showed that the addition of LiH can improve the activity of Zr-based intermetallic compounds for ammonia synthesis (Table 1). This promoting effect is more remarkable on the early transition metals V, Cr, and Mn. A more prominent feature is that the catalytic activities of Cr–LiH, Mn–LiH, Fe–LiH, and Co–LiH composite catalysts are quite similar and outperform that of the reference Cs–Ru/MgO catalyst. This activity trend is distinctly different from the volcano plot for the neat transition metals (Fig. 1), suggesting that the presence of LiH weakens the dependence of activity on the identity of transition metals, and the scaling relationships on transition metal surfaces have been interfered by the presence of alkali metal hydrides, which are non-transition metal components.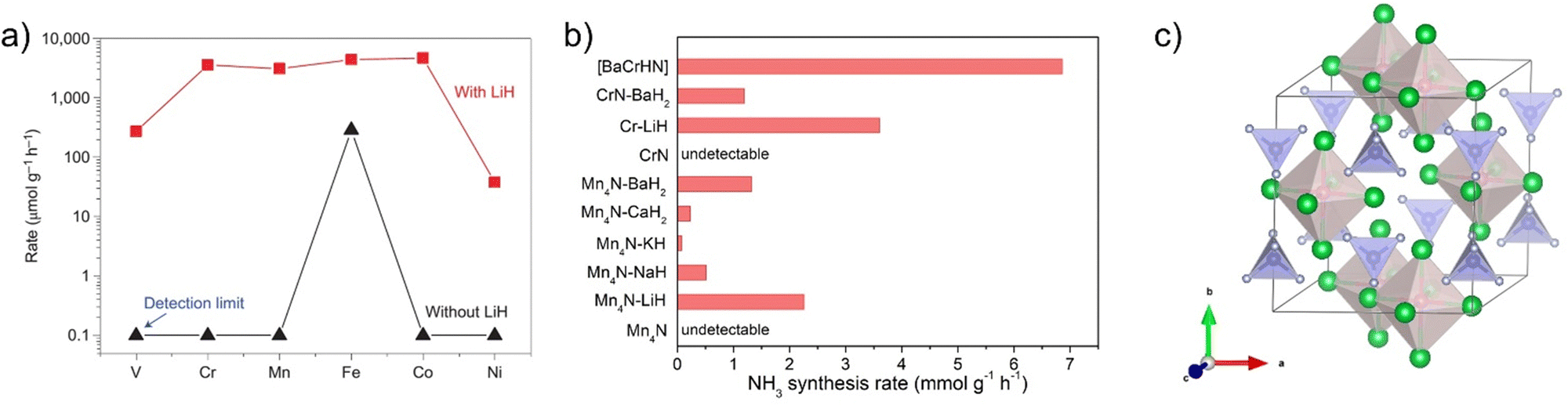 | ||
| Fig. 3 (a) Impact of LiH on the catalytic activity of 3d transition metals for ammonia synthesis. Reproduced with permission from ref 14. Copyright (2017) Springer Nature. (b) NH3 production rates of a series of Cr-based and Ru-based catalysts. (c) Crystal structure of Ba5CrN4H (Ba, green; H, red; Cr, blue; N, gray). | ||
In addition to LiH, other alkali or alkaline earth metal hydrides have also been demonstrated to be able to enhance the catalytic activity of early transition metals. For instance, the ammonia synthesis rate of Mn4N can be improved significantly by the addition of NaH, KH, CaH2, or BaH2 at 573 K and 1 MPa. The Mn4N–BaH2 catalyst has an activity of 1320 μmol gcat−1 h−1, which is comparable to that of the active Cs–Ru/MgO catalyst. Another important observation is that the activities of Mn4N–AHs are ranked in the order of Mn4N–BaH2 > Mn4N–LiH > Mn4N–KH > Mn4N–CaH2 > Mn4N–NaH on the basis of per gram of Mn.47 Such an order of promoting capability is different from that of the conventional alkali or alkaline earth oxide or hydroxide promoters, i.e., Cs2O > K2O > Na2O > BaO > CaO.48 Such a difference in promoting order suggests that the function mechanism of alkali and alkaline earth metals depends strongly on their chemical forms. Similarly, CrN has a negligible activity, while the CrN–BaH2 sample prepared simply by ball-milling CrN and BaH2 powders can achieve an ammonia synthesis rate of 1190 μmol gcat−1 h−1.24 It is also interesting to see that the phase structures of transition metal nitrides vary with the changes in temperature, pressure, and space velocity, showing a complex dynamic behavior in catalysis.47 Besides these alkali or alkaline earth metal hydrides, substances with similar functionalities need further study. It would be useful to mention here that the combination of MnN, CrN, VN, etc. with alkali or alkaline earth metal amides/imides leads to high catalytic activities for the ammonia decomposition reaction.49
Regarding the nature of the active sites of the alkali hydride–transition metal composite catalysts, we found that the alkali or alkaline earth metal hydrides can combine with late transition metals such as Ru or Ir forming complex metal hydrides (Li4RuH6, Ba2RuH6, K3RuH7, Li3IrH7etc.).50,51 Based on this understanding, we supposed that an alkali/alkaline earth-transition metal nitride–hydride species may be formed at the interface of alkali hydrides and transition metal nitrides. To verify this hypothesis, an amorphous barium chromium nitride–hydride catalyst, i.e., [BaCrNH], was synthesized and tested for ammonia synthesis reaction. Experimental results showed that the [BaCrNH] nitride–hydride catalyst exhibits excellent catalytic performance typically under milder conditions (Fig. 3b).24 The ammonia synthesis rate is ca. 4 times that of the reference Cs–Ru/MgO catalyst at 573 K and 1 MPa. The co-existence of Ba and H− helps to create more reactive N species in the lattice, which is facilely hydrogenated to ammonia and simultaneously leaves a vacant site for N2 adsorption and activation. The active phase has a Ba5CrN4H-like structure containing Ba6H octahedral and CrN4 tetrahedral units (Fig. 3c). These reactive H and N species are involved in the ammonia formation process. It has been suggested that N2 is adsorbed either on the Cr or Ba site of the nitride–hydride catalyst. By heating an orthorhombic nitride Ca3CrN3 under hydrogen at 673 K, a hexagonal nitride hydride, Ca3CrN3H, was synthesized recently, which exhibited catalytic performance for ammonia synthesis.26 Because of its well-defined crystal structure, it is beneficial to correlate the surface structure and catalytic performance. It has been suggested that N2 activation and hydrogenation involves the Ca cations, rather than the Cr sites.52 Although there is a lack of strong experimental evidence, the catalytic role of transition metals in activating alkali hydrides for N2 activation is open. Only a handful of nitride–hydride compounds of transition metals have been reported up to now such as Ca6Cr2N6H, Ba3CrN3H, etc.53,54 There will be much room to develop new nitride–hydride compounds containing various group 4–7 transition metals and alkali or alkaline earth metals for ammonia synthesis as well as other chemical transformations.
3. Chemical looping ammonia synthesis
Chemical looping is a reaction intensification technology by splitting a reaction into two or multiple sub-reactions mediated by certain solid carrier materials. In the chemical looping ammonia synthesis (abbreviated as CLAS) process, N2 is first fixed by a suitable nitrogen carrier and NH3 is then harvested by the reactions of N carriers and the hydrogen source. Potential advantages of CLAS include the ability to circumvent the competitive adsorption of N2 and the H source in the catalytic process and to control process conditions for N2 fixation and NH3 production steps independently.55 In this sense, CLAS provides an alternative route to circumvent the scaling relations in ammonia synthesis catalysis. Chemical looping ammonia synthesis using early transition metal nitrides as nitrogen carriers could date back to the late 19th century when Tessie du Motay attempted ammonia synthesis via a chemical loop based on the interconversion of TiN (or Ti3N2) and Ti5N3.56 Haber studied ammonia formation through the stepwise formation and hydrogenation of Mn nitride (Mn3N2–Mn), but the ammonia yields were too low for practical applications.57 Since the advent of the Haber–Bosch catalytic ammonia synthesis process, the development of CLAS has fallen behind. Until the beginning of this century, there was renewed interest in CLAS because it has the potential advantage of utilizing the intermittent renewable energy.58The challenge of CLAS is to find efficient nitrogen carriers and process conditions that produce ammonia at economically acceptable rates. Group 4–7 transition metals enable facile N2 activation and their nitrides have high and variable nitrogen contents and thus are the main candidate N carrier materials.55 As shown in Fig. 4, there are basically two types of CLAS processes using transition metal nitrides as nitrogen carriers. The first type uses H2O as the hydrogen source (Fig. 4a). For example, Pfromm et al. designed Cr/CrN/Cr2O3 mediated CLAS, which contains three steps: metallic Cr reacts with N2 to form CrN; CrN hydrolyzes to produce NH3 and generate Cr2O3; and Cr2O3 is reduced by CO or H2 to regenerate metallic Cr.59 Analogously, Mn/Mn nitride/Mn oxide and Mo/Mo nitride/Mo oxide mediated CLAS processes were also studied.60,61 Musgrave et al. screened more than a thousand metal nitride/metal oxide pairs for CLAS by using high-throughput equilibrium analysis.62 Many early transition metals had been identified to be promising candidates. The endothermic reduction step generally requires high temperatures (>1000 K) and could use the concentrated solar energy. The second type uses H2 as the hydrogen source. In this process, NH3 is produced through the interconversion between N-rich and N-poor nitrides. Michalsky et al. studied the Mn6N2.58/Mn4N mediated CLAS, but the NH3 production rate is only 55 μmol g−1 h−1 at 823 K and 0.1 MPa H2.63 Metal doping has been shown to alter the geometrical and electrical structures and chemical properties of N carriers, and thus could boost NH3 production.64 Hargreaves et al. reported that doping Mn nitride (Mn3N2) with low levels of lithium resulted in enhanced reactivity toward hydrogenation at low temperature.65 With the addition of Co, the conversion rate of lattice N in CrN can be increased by one order of magnitude at 973 K and 0.1 MPa H2.66 Gao et al. showed that alkali and alkaline earth metal hydrides-imides can be employed as N carriers in a two-step NH3 loop (eqn (1) and (2)).55 On this basis, composite N carriers containing both early transition metal nitrides and alkali or alkaline earth metal imides, e.g., Mn4N–LiH/Mn2N–Li2NH and Mn4N–BaH2/Mn2N–BaNH, have been proposed.67 The Mn nitride not only acts as a nitrogen carrier but also functions as a catalyst enhancing the kinetics of N2 fixation by alkali hydride to form imide (eqn (1)) and the subsequent hydrogenation of the imide to NH3 (eqn (2)). The hydrides also facilitate the kinetics of N2 fixation and subsequent hydrogenation of the Mn nitride. These features make the loop feasible at ambient pressure and temperatures below 300 °C (Fig. 4c). The CLAS performances could be potentially further improved by selecting different couples of transition metal nitrides and alkali or alkaline earth metal imides to optimize the thermodynamic and kinetic properties. On the other hand, the advancement of preparation methods to increase the number of active sites on the surfaces of N carriers could also provide an opportunity to enhance the energy efficiency of CLAS.
| 4LiH + N2 → 2Li2NH + H2 | (1) |
| 2Li2NH + 4H2 → 4LiH + 2NH3 | (2) |
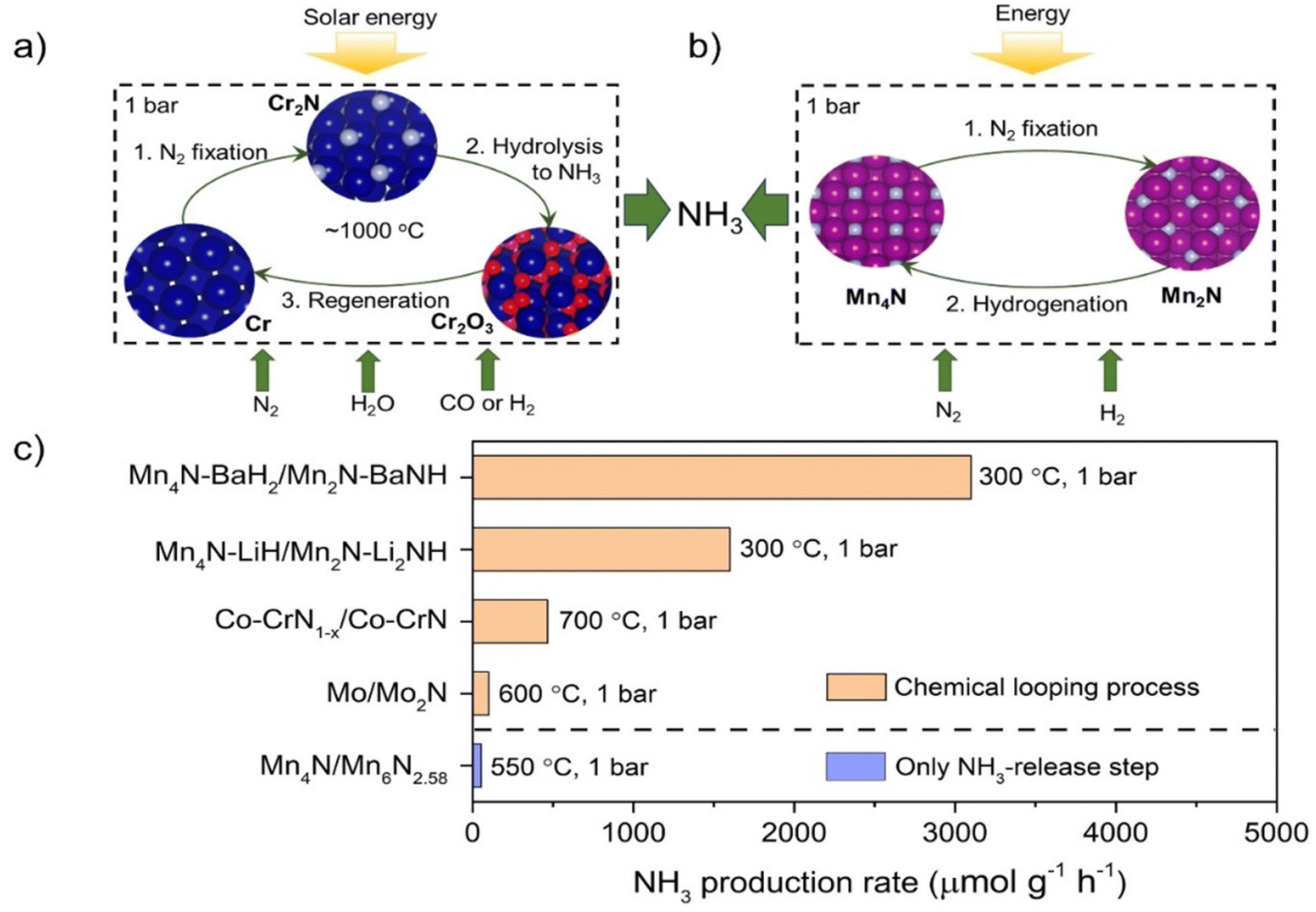 | ||
| Fig. 4 Two types of chemical looping ammonia synthesis processes using transition metal nitrides as nitrogen carriers. (a) Metal–metal nitride–metal oxide loop (Cr, blue; N, light blue; O, red). (b) N-rich metal nitride–N–lean nitride loop (Mn, purple; N, light blue). (c) Comparison of chemical looping ammonia synthesis rates for selected N carriers. | ||
4. Beyond thermal ammonia synthesis
Besides the thermal activation routes, the interactions of transition metal nitrides with external fields such as electricity, photon, microwave, and plasma would increase freedom to manipulate the properties of metal nitrides toward N2-to-NH3.Electrocatalytic N2 reduction to NH3 (eNRR) using group 4–7 transition metals such as ZrN, VN, NbN, CrN, and Mo2N has been studied both theoretically and experimentally. Theoretical calculation results showed that NH3 forms on these nitrides by way of a Mars–van Krevelen mechanism.68–70 The free energy required for the endergonic protonation of surface N to NH3 can be overcome with an applied bias. On the other hand, since the early transition metals bind N stronger than H, they could be more active toward N2 reduction than toward the competing hydrogen evolution reaction. However, later studies reported that Mo2N undergoes fast chemical decomposition in aqueous electrolytes and shows no catalytic activity for the NRR.71 These controversial results leave open room for the careful study of these transition metal nitrides for the eNRR reaction.
Photo-chemical ammonia synthesis has also received increasing attention recently.72 Photon-driven nitrogen fixation by titania catalysts was reported decades ago by Schrauzer and Guth, but the efficiency is very low.73 It has been suggested that oxygen defects and/or Ti3+ are the active sites for nitrogen adsorption and activation.74 Recent studies, however, reported that surface carbon has a strong interaction with N2, which may assist the N2 reduction process.75 Many other oxides, nitrides, and sulfides of group 4–7 metals are semiconductors, which could potentially be used as light-absorbing materials and photocatalysts. For example, Tsang et al. reported an Fe-decorated 2D MoS2 photocatalyst for ammonia synthesis,76 where the main role of MoS2 is to generate excited electrons upon light illumination transferring to Fe sites for N2 activation. Some nitrides such as TiN are plasmonic and they can turn illumination into photothermal heating, which may assist in reactions.77 Tantalum nitride (Ta3N5) has a small band gap (2.1 eV) and is considered to be a promising photocatalyst for solar water splitting under visible light.78 It is interesting to find out whether Ta3N5 and other group 4–7 transition metal nitrides could function both as light absorbing materials and photocatalysts for N2 reduction to NH3. On the other hand, the influence of photon illumination on weakening the metal nitrogen bonding is also worthy of future investigation.
5. Outlook
Although the studies of ammonia synthesis have continued for more than one century, the goal of ammonia synthesis under mild conditions has not been achieved. It is not only a scientific task, but also of practical relevance, as ammonia has been considered to be a key energy molecule for the sustainable development of human society. The success of biological N2 fixation suggests that this goal is not unattainable and would continue to provide valuable inspirations for the future studies on artificial nitrogen conversion processes. We should continue to find ways to tackle the contradictions of thermodynamics and kinetics of ammonia synthesis reaction, which may also have impacts on other chemical reactions. Because of the high affinity of group 4–7 transition metals for nitrogen, they are potential candidate materials. Unfortunately, these metals have not received much attention because there is a lack of powerful strategies to weaken the strong metal–N bond and facilitate the hydrogenation of N species to ammonia. Metal alloying and introduction of non-transition metal elements especially hydrogen (hydride) and alkali (or alkaline earth) metals have been demonstrated to be efficient methods. In addition, the introduction of external fields provides greater flexibility for the development of efficient materials or catalysts to achieve ammonia synthesis under mild conditions.Artificial intelligence (AI) and machine learning have been believed to be able to accelerate catalyst design by identifying key descriptors correlated with the catalytic performances in recent years.79–81 For example, an active Cu–Al electrocatalyst for CO2 reduction to ethylene was predicted using density functional theory calculations in combination with active machine learning.79 This approach may be used for the studies of catalysts or nitrogen carriers for ammonia production. Applying these approaches is challenging since they rely on large data sets with high accuracy and high reliability, which are often not available typically in the fields of chemical looping, plasma, and electro- and photo-chemical ammonia synthesis processes. Extensive and intensive experimental and theoretical work is still needed at this stage.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Key R&D Program of China (2021YFB4000400), National Natural Science Foundation of China (Grants No. 21988101, 22279131, 22379139), and Youth Innovation Promotion Association CAS (No. Y2022060 and 2022180).Notes and references
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- J. Guo and P. Chen, Chem, 2017, 3, 709–712 CAS.
- L. Ye, R. Nayak-Luke, R. Bañares-Alcántara and E. Tsang, Chem, 2017, 3, 712–714 CAS.
- F. Chang, W. Gao, J. Guo and P. Chen, Adv. Mater., 2021, 33, 2005721 CrossRef CAS PubMed.
- W. David, Ammonia: zero-carbon fertiliser, fuel and energy store, 2020 Search PubMed.
- J. Guo and P. Chen, Acc. Chem. Res., 2021, 54, 2434–2444 CrossRef CAS PubMed.
- H.-P. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547–564 RSC.
- A. Vojvodic, A. J. Medford, F. Studt, F. Abild-Pedersen, T. S. Khan, T. Bligaard and J. K. Nørskov, Chem. Phys. Lett., 2014, 598, 108–112 CrossRef CAS.
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- P. A. Dowben, A. Miller, H. J. Ruppender and M. Grunze, Surf. Sci., 1988, 193, 336–352 CrossRef CAS.
- K. C. MacLeod and P. L. Holland, Nat. Chem., 2013, 5, 559–565 CrossRef CAS PubMed.
- K.-I. Aika and K. Tamara, in Ammonia: Catalysis and Manufacture, ed. A. Nielsen, Springer Berlin Heidelberg, Berlin, Heidelberg, 1995, pp. 103–148 DOI:10.1007/978-3-642-79197-0_3.
- Y. Cao, A. Saito, Y. Kobayashi, H. Ubukata, Y. Tang and H. Kageyama, ChemCatChem, 2021, 13, 191–195 CrossRef CAS.
- P. Wang, F. Chang, W. Gao, J. Guo, G. Wu, T. He and P. Chen, Nat. Chem., 2017, 9, 64–70 CrossRef CAS PubMed.
- R. Kojima and K.-i Aika, Appl. Catal., A, 2001, 218, 121–128 CrossRef CAS.
- B. Dragoi, A. Ungureanu, A. Chirieac, C. Ciotonea, C. Rudolf, S. Royer and E. Dumitriu, Appl. Catal., A, 2015, 504, 92–102 CrossRef CAS.
- R. Kojima and K.-I. Aika, Appl. Catal., A, 2001, 209, 317–325 CrossRef CAS.
- K. McAulay, J. S. J. Hargreaves, A. R. McFarlane, D. J. Price, N. A. Spencer, N. Bion, F. Can, M. Richard, H. F. Greer and W. Z. Zhou, Catal. Commun., 2015, 68, 53–57 CrossRef CAS.
- Y. Cao, Z. Wei, W. Al Maksoud, R. Rai, Y. Kobayashi and H. Kageyama, Solid State Sci., 2023, 145, 107331 CrossRef CAS.
- H. Li, Y. Gong, H. Yang, X. Yang, K. Li, J. Wang and H. Hosono, ChemSusChem, 2023, 16, e202301016 CrossRef CAS PubMed.
- Y. Kobayashi, Y. Tang, T. Kageyama, H. Yamashita, N. Masuda, S. Hosokawa and H. Kageyama, J. Am. Chem. Soc., 2017, 139, 18240–18246 CrossRef CAS PubMed.
- Y. Tang, Y. Kobayashi, N. Masuda, Y. Uchida, H. Okamoto, T. Kageyama, S. Hosokawa, F. Loyer, K. Mitsuhara, K. Yamanaka, Y. Tamenori, C. Tassel, T. Yamamoto, T. Tanaka and H. Kageyama, Adv. Energy Mater., 2018, 8, 1801772 CrossRef.
- L. Li, T. Zhang, J. Cai, H. Cai, J. Ni, B. Lin, J. Lin, X. Wang, L. Zheng, C.-T. Au and L. Jiang, J. Catal., 2020, 389, 218–228 CrossRef CAS.
- Y. Guan, W. Zhang, Q. Wang, C. Weidenthaler, A. Wu, W. Gao, Q. Pei, H. Yan, J. Cui, H. Wu, S. Feng, R. Wang, H. Cao, X. Ju, L. Liu, T. He, J. Guo and P. Chen, Chem. Catal., 2021, 1, 1042–1054 CrossRef CAS.
- H. Cao, J. Guo, F. Chang, C. Pistidda, W. Zhou, X. Zhang, A. Santoru, H. Wu, N. Schell, R. Niewa, P. Chen, T. Klassen and M. Dornheim, Chem. – Eur. J., 2017, 23, 9766–9771 CrossRef CAS PubMed.
- Y. Cao, M. A. Kirsanova, M. Ochi, W. Al Maksoud, T. Zhu, R. Rai, S. Gao, T. Tsumori, S. Kobayashi, S. Kawaguchi, E. Abou-Hamad, K. Kuroki, C. Tassel, A. M. Abakumov, Y. Kobayashi and H. Kageyama, Angew. Chem., Int. Ed., 2022, 61, e202209187 CrossRef CAS PubMed.
- M. Kitano, Y. Inoue, M. Sasase, K. Kishida, Y. Kobayashi, K. Nishiyama, T. Tada, S. Kawamura, T. Yokoyama, M. Hara and H. Hosono, Angew. Chem., Int. Ed., 2018, 57, 2648–2652 CrossRef CAS PubMed.
- A. Mittasch and W. Frankenburg, in Adv. Catal., ed. W. G. Frankenburg, V. I. Komarewsky and E. K. Rideal, Academic Press, 1950, vol. 2, pp. 81–104 Search PubMed.
- K.-i Aika and A. Ozaki, J. Catal., 1969, 14, 311–321 CrossRef CAS.
- L. Volpe and M. Boudart, J. Phys. Chem., 1986, 90, 4874–4877 CrossRef CAS.
- C. J. H. Jacobsen, Chem. Commun., 2000, 1057–1058, 10.1039/b002930k.
- K. Ryoichi and A. Ken-ichi, Chem. Lett., 2000, 514–515 Search PubMed.
- D. McKay, D. H. Gregory, J. S. J. Hargreaves, S. M. Hunter and X. Sun, Chem. Commun., 2007, 3051–3053 RSC.
- S. M. Hunter, D. H. Gregory, J. S. Hargreaves, M. Richard, D. Duprez and N. Bion, ACS Catal., 2013, 3, 1719–1725 CrossRef CAS PubMed.
- T.-N. Ye, S.-W. Park, Y. Lu, J. Li, M. Sasase, M. Kitano, T. Tada and H. Hosono, Nature, 2020, 583, 391–395 CrossRef CAS PubMed.
- J. Z. Wu, J. Li, Y. T. Gong, M. Kitano, T. Inoshita and H. Hosono, Angew. Chem., Int. Ed., 2019, 58, 825–829 CrossRef CAS PubMed.
- Y. T. Gong, H. C. Li, J. Z. Wu, X. Y. Song, X. Q. Yang, X. B. Bao, X. Han, M. Kitano, J. J. Wang and H. Hosono, J. Am. Chem. Soc., 2022, 144, 8683–8692 CrossRef CAS PubMed.
- P. F. Xie, Y. G. Yao, Z. N. Huang, Z. Y. Liu, J. L. Zhang, T. Y. Li, G. F. Wang, R. Shahbazian-Yassar, L. B. Hu and C. Wang, Nat. Commun., 2019, 10, 12 CrossRef PubMed.
- Q. Wang, Y. Guan, J. Guo and P. Chen, Cell Rep. Phys. Sci., 2022, 3, 100779 CrossRef CAS.
- T. Shima, S. Hu, G. Luo, X. Kang, Y. Luo and Z. Hou, Science, 2013, 340, 1549–1552 CrossRef CAS PubMed.
- P. Avenier, M. Taoufik, A. Lesage, X. Solans-Monfort, A. Baudouin, A. de Mallmann, L. Veyre, J.-M. Basset, O. Eisenstein, L. Emsley and E. A. Quadrelli, Science, 2007, 317, 1056–1060 CrossRef CAS PubMed.
- L. M. Azofra, N. Morlanés, A. Poater, M. K. Samantaray, B. Vidjayacoumar, K. Albahily, L. Cavallo and J.-M. Basset, Angew. Chem., Int. Ed., 2018, 57, 15812–15816 CrossRef CAS PubMed.
- Z. Luo, A. W. Castleman, Jr. and S. N. Khanna, Chem. Rev., 2016, 116, 14456–14492 CrossRef CAS PubMed.
- P. Wang, H. Xie, J. Guo, Z. Zhao, X. Kong, W. Gao, F. Chang, T. He, G. Wu, M. Chen, L. Jiang and P. Chen, Angew. Chem., Int. Ed., 2017, 56, 8716–8720 CrossRef CAS PubMed.
- Y. Zhao, J. T. Cui, M. Wang, D. Y. Valdivielso, A. Fielicke, L. R. Hu, X. Cheng, Q. Y. Liu, Z. Y. Li, S. G. He and J. B. Ma, J. Am. Chem. Soc., 2019, 141, 12592–12600 CrossRef CAS PubMed.
- D. H. Gregory, J. Mater. Chem., 2008, 18, 2321–2330 RSC.
- F. Chang, Y. Guan, X. Chang, J. Guo, P. Wang, W. Gao, G. Wu, J. Zheng, X. Li and P. Chen, J. Am. Chem. Soc., 2018, 140, 14799–14806 CrossRef CAS PubMed.
- K.-i Aika, T. Takano and S. Murata, J. Catal., 1992, 136, 126–140 CrossRef CAS.
- J. Guo, P. Wang, G. Wu, A. Wu, D. Hu, Z. Xiong, J. Wang, P. Yu, F. Chang, Z. Chen and P. Chen, Angew. Chem., Int. Ed., 2015, 54, 2950–2954 CrossRef CAS PubMed.
- Q. Wang, H. Wen, Y. Guan, S. Zhang, W. Gao, J. Guo and P. Chen, ACS Catal., 2023, 13, 9882–9890 CrossRef CAS.
- H. Yan, W. Gao, Q. Wang, J. Guo and P. Chen, Faraday Discuss., 2023, 243, 55–64 RSC.
- Y. Cao, E. Toshcheva, W. Almaksoud, R. Ahmad, T. Tsumori, R. Rai, Y. Tang, L. Cavallo, H. Kageyama and Y. Kobayashi, ChemSusChem, 2023, 16, e202300234 CrossRef CAS PubMed.
- M. S. Bailey, M. N. Obrovac, E. Baillet, T. K. Reynolds, D. B. Zax and F. J. DiSalvo, Inorg. Chem., 2003, 42, 5572–5578 CrossRef CAS PubMed.
- N. W. Falb, J. N. Neu, T. Besara, J. B. Whalen, D. J. Singh and T. Siegrist, Inorg. Chem., 2019, 58, 3302–3307 CrossRef CAS PubMed.
- W. B. Gao, J. P. Guo, P. K. Wang, Q. R. Wang, F. Chang, Q. J. Pei, W. J. Zhang, L. Liu and P. Chen, Nat. Energy, 2018, 3, 1067–1075 CrossRef CAS.
- J. R. Jennings, Catalytic ammonia synthesis: fundamentals and practice, Springer Science & Business Media, 1991 Search PubMed.
- F. Haber and G. Van Oordt, Z. Anorg. Allg. Chem., 1905, 44, 341–378 CrossRef.
- M. E. Gálvez, M. Halmann and A. Steinfeld, Ind. Eng. Chem. Res., 2007, 46, 2042–2046 CrossRef.
- R. Michalsky and P. H. Pfromm, Sol. Energy, 2011, 85, 2642–2654 CrossRef CAS.
- R. Michalsky, P. H. Pfromm and A. Steinfeld, Interface Focus, 2015, 5, 20140084 CrossRef PubMed.
- R. Michalsky, B. J. Parman, V. Amanor-Boadu and P. H. Pfromm, Energy, 2012, 42, 251–260 CrossRef CAS.
- C. J. Bartel, J. R. Rumptz, A. W. Weimer, A. M. Holder and C. B. Musgrave, ACS Appl. Mater. Interfaces, 2019, 11, 24850–24858 CrossRef CAS PubMed.
- R. Michalsky, A. M. Avram, B. A. Peterson, P. H. Pfromm and A. A. Peterson, Chem. Sci., 2015, 6, 3965–3974 RSC.
- N. Shan, C. Huang, R. T. Lee, N. Manavi, L. Xu, V. Chikan, P. H. Pfromm and B. Liu, ChemCatChem, 2020, 12, 2233–2244 CrossRef CAS.
- S. Laassiri, C. D. Zeinalipour-Yazdi, C. R. A. Catlow and J. S. J. Hargreaves, Appl. Catal., B, 2018, 223, 60–66 CrossRef CAS.
- S. Wang, F. Gong, Q. Zhou, Y. Xie, H. Li, M. Li, E. Fu, P. Yang, Y. Jing and R. Xiao, Appl. Catal., B, 2023, 339, 123134 CrossRef CAS.
- S. Feng, W. Gao, Q. Wang, Y. Guan, H. Yan, H. Wu, H. Cao, J. Guo and P. Chen, J. Mater. Chem. A, 2021, 9, 1039–1047 RSC.
- Y. Abghoui and E. Skúlason, J. Phys. Chem. C, 2017, 121, 6141–6151 CrossRef CAS.
- X. Ren, G. Cui, L. Chen, F. Xie, Q. Wei, Z. Tian and X. Sun, Chem. Commun., 2018, 54, 8474–8477 RSC.
- X. Yang, J. Nash, J. Anibal, M. Dunwell, S. Kattel, E. Stavitski, K. Attenkofer, J. G. Chen, Y. Yan and B. Xu, J. Am. Chem. Soc., 2018, 140, 13387–13391 CrossRef CAS PubMed.
- B. Hu, M. Hu, L. Seefeldt and T. L. Liu, ACS Energy Lett., 2019, 4, 1053–1054 CrossRef CAS.
- A. J. Medford and M. C. Hatzell, ACS Catal., 2017, 7, 2624–2643 CrossRef CAS.
- G. N. Schrauzer and T. Guth, J. Am. Chem. Soc., 1977, 99, 7189–7193 CrossRef CAS.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS PubMed.
- B. M. Comer, Y.-H. Liu, M. B. Dixit, K. B. Hatzell, Y. Ye, E. J. Crumlin, M. C. Hatzell and A. J. Medford, J. Am. Chem. Soc., 2018, 140, 15157–15160 CrossRef CAS PubMed.
- J. Zheng, L. Lu, K. Lebedev, S. Wu, P. Zhao, I. J. McPherson, T. S. Wu, R. Kato, Y. Li, P.-L. Ho, G. Li, L. Bai, J. Sun, D. Prabhakaran, R. A. Taylor, Y. L. Soo, K. Suenaga and S. C. E. Tsang, Chem Catal., 2021, 1, 162–182 CrossRef CAS.
- D. F. Swearer, N. R. Knowles, H. O. Everitt and N. J. Halas, ACS Energy Lett., 2019, 4, 1505–1512 CrossRef CAS.
- J. Eichhorn, S. P. Lechner, C. M. Jiang, G. Folchi Heunecke, F. Munnik and I. D. Sharp, J. Mater. Chem. A, 2021, 9, 20653–20663 RSC.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C.-S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S.-C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- Y. Guo, X. He, Y. Su, Y. Dai, M. Xie, S. Yang, J. Chen, K. Wang, D. Zhou and C. Wang, J. Am. Chem. Soc., 2021, 143, 5755–5762 CrossRef CAS PubMed.
- F. Göltl, J. Phys. Chem. C, 2022, 126, 3305–3313 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |