
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Orange peel biochar/clay/titania composites: low cost, high performance, and easy-to-reuse photocatalysts for the degradation of tetracycline in water†
Morenike O.
Adesina
 abcd,
Moses O.
Alfred
bc,
Harald
Seitz
e,
Katlen
Brennenstuhl
a,
Harshadrai M.
Rawel
f,
Pablo
Wessig
a,
Jiyong
Kim
g,
Armin
Wedel
g,
Wouter
Koopman
h,
Christina
Günter
i,
Emmanuel I.
Unuabonah
bc and
Andreas
Taubert
*a
abcd,
Moses O.
Alfred
bc,
Harald
Seitz
e,
Katlen
Brennenstuhl
a,
Harshadrai M.
Rawel
f,
Pablo
Wessig
a,
Jiyong
Kim
g,
Armin
Wedel
g,
Wouter
Koopman
h,
Christina
Günter
i,
Emmanuel I.
Unuabonah
bc and
Andreas
Taubert
*a
aInstitute of Chemistry, University of Potsdam, D-14476 Potsdam, Germany. E-mail: ataubert@uni-potsdam.de
bAfrican Centre of Excellence for Water and Environmental Research (ACEWATER), Redeemer's University, PMB 230, Ede, Osun State, Nigeria
cDepartment of Chemical Sciences, Redeemer's University, PMB 230, Ede, Osun State, Nigeria
dLead City University, Ibadan, Oyo State, Nigeria
eFraunhofer Institute for Cell Therapy and Immunology, Branch Bioanalytics and Bioprocesses (IZI-BB), 14476 Potsdam, Germany
fInstitute of Nutritional Science, University of Potsdam, D-14558 Nuthetal, Potsdam, Germany
gFunctional Materials and Devices, Fraunhofer Institute for Applied Polymer Research IAP, Potsdam, 14476 Potsdam, Germany
hInstitute of Physics and Astronomy, University of Potsdam, D-14476 Potsdam, Germany
iInstitute of Geosciences, University of Potsdam, D-14476 Potsdam, Germany
First published on 10th April 2024
Abstract
New orange peel biochar/clay/titania nanocomposites (NCs) were studied for photocatalytic degradation of tetracycline (TET) under both UV and natural solar irradiation by variation of NC dose, initial TET concentration, ionic strength, and competing anions. Total organic carbon (TOC) reduction was used to assess mineralization. Intermediate product formation during TET degradation was characterized using liquid chromatography-mass spectrometry and agar-based diffusion assays. The as-synthesized material prepared with biochar obtained at 600 °C (C600KT) exhibits the best TET degradation performance under UV light exposure and solar irradiation with up to 92 and 89% after 2 h, respectively. Especially under UV exposure, C600KT exhibits the highest apparent rate constant of 2.9 × 10−2 min−1 and a half-life of 23.9 min. About 60 and 50% TOC are removed after 2 h under UV and solar irradiation, respectively. Quenching experiments confirm that superoxide and hydroxyl radicals are the major reactive species involved in the degradation process. Furthermore, the treated effluents are harmless to both Escherichia coli and Staphylococcus xylosus, indicating that no intermediate products with higher toxicity are produced during the photocatalytic degradation. Additionally, the results show that the main fraction of TET is degraded within the first 15 min of irradiation. The C600KT composite is recyclable and retains its performance over at least four cycles, proving its stability and reusability. Overall, the new NCs are therefore highly attractive for the remediation of TET pollution in water.
Water impactThe article focuses on a low cost-high impact technology for micropollutant removal from water. The water impact is very straightforward: the materials are cheap, highly effective, and the synthesis can be scaled to larger amounts. The material and the process described here are therefore of high interest to both the developing and industrialized countries for effectively treating (organic) micropollutant-contaminated waters. |
1. Introduction
Tetracycline (TET), an antibiotic commonly administered in human and veterinary medicine, is frequently detected in water environments due to its stable chemical structure, which hinders complete metabolization or full uptake by human or animal bodies.1,2 Consequently, a significant amount is released through feces or urine as persistent degradation by-products into the aquatic environment.3 Residual TET concentrations have been found in various water sources across several countries, including surface water, groundwater, drinking water, and wastewater.4–6 While trace amounts of TET ranging from 2 ng L−1–60 μg L−1 have been detected,3–6 their residues and metabolites contribute to the spread of antibiotic-resistance genes, disrupting ecosystem function and ultimately harming human health.7–10Therefore, an effective treatment for the removal or degradation of TET is of high interest and current relevance. Titanium dioxide (TiO2) is a suitable photocatalyst for water treatment due to its low toxicity, chemical and biological inertness, and photocatalytic stability.11 Its anatase phase has a slightly larger band gap (3.20 eV) than the rutile phase (3.03 eV).12,13 Because of the wide band gap, both titania phases only exhibit a strong photoactivity in the ultraviolet (UV) region of the electromagnetic spectrum and low efficiencies in the visible (vis) region.14,15
As such, a UV light-based device would be ideal for fully exploiting the power of titania-based water treatment materials. However, considering the situation in Africa and other developing regions, there are two main issues: (1) the cost of UV-based devices and (2) insufficient or intermittent power supply in large parts of these regions.16 Therefore, harnessing solar (visible) light instead of using UV lamps for photocatalytic water treatment is one of the major factors in reducing the cost and technical challenges of water treatment via (titania-based) photocatalysis.
Moreover, in addition to poor activity under visible light irradiation, any large-scale application of TiO2 nanoparticles (NPs) for water treatment is hampered by the tendency of titania NPs to agglomerate and by difficulties in NP separation after water treatment leading to poor catalyst recovery.17–20 Consequently, numerous efforts are underway to
(1) improve the photocatalytic response of TiO2 under visible light,21
(2) improve the ease of separation of the photocatalyst from a water body that has been treated,22 and
(3) maintain a good catalytic performance over many cycles of water treatment.23
Several studies have investigated strategies of immobilizing TiO2 onto supports like biochar, glass, activated carbon, or clay minerals to create composite materials with improved visible light photocatalytic activity and recovery properties.24–27 In particular, the addition of a carbonaceous component to a photocatalyst composite can improve the photocatalytic efficiency by improving pollutant adsorption, visible light absorption, and efficient charge separation.28–30 Carbon materials have remarkable adsorption properties allowing for the accumulation of pollutants on their surface; this facilitates rapid interaction between active species and pollutant molecules.28,31–34 Secondly, doping of TiO2 with carbonaceous material contributes to increased absorption of visible light.28,35 This doping interaction can occur through the substitution of carbon at titanium (C–O bond) or oxygen sites (Ti–C bond) in the TiO2 lattice, resulting in band gap narrowing and extended absorption edge.35–37 Moreover, carbon materials can form electron sinks which can enhance the charge separation within TiO2.38–40
On the other hand, clay minerals improve the photocatalyst stability and its recyclability over extended periods of use.16,41 The combination of titania, a carbonaceous species, and a (clay) mineral should therefore provide materials with a high application potential for photocatalytic degradation of contaminants in water bodies.
Indeed, Cheng et al. reported an improved photocatalytic efficiency of different TiO2-carbon hybrids for the removal of rhodamine B, which they assigned to (1) improved electron–hole separation and (2) a larger surface area of the composite compared to pure titania.42 Lu et al. prepared a TiO2-biochar composite by depositing TiO2 onto walnut shell biochar. The resulting composite showed a high photodegradation and mineralization efficiency, implying that the addition of biochar improved the photocatalytic process. At the same time, the addition of biochar also seems to simplify the recovery of the composite photocatalyst after use.43 Oseghe et al. described C-doped TiO2 with a higher photocatalytic efficiency under vis-LED light than undoped TiO2.15 Likewise, Alfred et al. reported the efficient degradation of pharmaceuticals in aqueous media using solar-active clay/TiO2 nanocomposites (NCs) prepared through a biomass-assisted synthetic route.16
Recently, we reported the development and performance of a new class of materials based on biochar derived from orange peel (OP), kaolin clay, and titania for the removal of the emerging contaminants (ECs); tetracycline (TET) and bisphenol A (BPA).44 The NCs have better adsorption capacities for TET (30 mg g−1) and BPA (23 mg g−1) than the unmodified TiO2 counterpart or any of the NC constituents (OP biochar, kaolin, titania) alone. Moreover, the composites show a performance that is comparable to some other efficient materials for TET and BPA removal via adsorption.44–47 However, adsorption is a phase transfer phenomenon, and once the active sites are occupied, no further removal of pollutants is possible. On the other hand, in the case of photocatalysis, the contaminants are continually degraded and the fragments are removed from the surface, extending the duration that the surface active sites are available for usein catalytic degradation. This reduces the cost of regeneration as well as the challenge of disposing of spent adsorbent. Therefore, an additional photoactivity of these NCs would offer a superior potential to remove contaminants.
In this current study, we evaluate OP biochar/kaolin clay/TiO2 NCs, but with respect to the photocatalytic degradation of TET under UV and visible light (rather than exploiting pure adsorption, which was described previously44). The choice of TET as a model case for this study is motivated by the extensive use of this drug in veterinary and human medicine around the world resulting in a significant accumulation of TET in freshwater systems.48
2. Experimental
2.1 Materials
Precursors and composites were obtained as described previously;44 details are available in the ESI.† In short, 1.0 g of biochar and 1.0 g of kaolin clay were dispersed in 30 mL of ethanol, sonicated, then 5 mL of titanium tetraisopropoxide (TTIP) was added and the mixture was vigorously stirred for 1 h. Subsequently, 70 mL of deionized water was added dropwise under continued agitation over the course of another hour. The slurry was aged overnight at room temperature, dried, and then pyrolyzed. The resulting products obtained were washed, dried to constant weight, and stored in a dry container. NCs are labelled C###KT, where C is carbon (biochar), ### = 300, 400, 500, 600 is the calcination temperature for biochar synthesis, K is kaolin clay, and T is titania. A pure TiO2 powder (P-TiO2) without the other components was also synthesized as a control.2.2 Characterization
The analytical characterization (surface chemistry, morphology, composition, point of zero charge, thermal properties, and surface areas) of the NCs has been reported previously.44 For the current study, optical and spectroscopic properties were determined to further characterize the NCs.Elemental maps were recorded on a JEOL JSM 6510 scanning electron microscope (JEOL, Freising, Germany) equipped with an Xplore EDX detector (Oxford Instruments, UK). The back scattered electron (BSE) detector was used for material contrast images at 20 kV. The secondary electron (SE) detector was used for topographic imaging at 7 kV.
Raman spectra were recorded with a Raman microscope (Witec alpha300 AR, Ulm, Germany) and a 10× objective (Nikon E Plan 10×/0.25). A 532 nm diode laser was used as the excitation source. To increase the signal-to-noise ratio, each Raman spectrum was accumulated over 10 individual measurements with an integration time of 1 s each.
UV-vis diffuse reflectance spectroscopy (UV-vis DRS) data were collected on a Perkin Elmer Lambda 750 UV-vis spectrophotometer with the solid material attachment Praying Mantis (Harrick Scientific Products Inc.). Magnesium sulfate (MgSO4) AnalaR NORMAPUR (VWR, Leuven, Belgium) was used as background material. The measuring range was λ = 250–850 nm with a resolution of 2 nm. The measured reflection data R were converted to the K/S ratio according to the Kubelka–Munk function, eqn (1).49
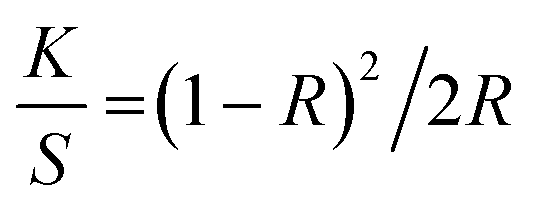 | (1) |
In eqn (1), K is the absorption coefficient, S is the scattering coefficient, and R is the reflectance. From these data, the optical bandgaps were graphically analyzed using the Tauc analysis50viaeqn (2).
| (αhν)2 = A(hν − Eg) | (2) |
Photoluminescence (PL) spectra were recorded using a homebuilt micro-spectrometer. In short, a 355 nm (Coherent Genesis) laser was directed to the sample through a 40× objective (Nikon Plan Fluor ELWD). The excitation power at the sample plane was 1.5 mW. The emission was collected through the same objective and separated from the excitation beam by a dichroic mirror (Chroma zt 375 RCD). Finally, the spectra were recorded by an imaging spectrograph (Andor Kymera 328i, equipped with an Andor Newton EMCCD). The spectra were averaged over 20 individual spectra each with an integration time of 1 second per spectrum.
The chemical composition and chemical states of the NC were obtained with an Axis Supra+ (Kratos Analytical, UK) X-ray photoelectron spectroscopy (XPS) setup using monochromatised Al Kα radiation for excitation (15 kV, typical 20 mA). CasaXPS software was used for data processing and interpretation; XPS signals were fitted using GL (30) line shapes combining Gaussian (70%) and Lorentzian (30%) line shapes.51
2.3 Photocatalysis study
Photodegradation studies were carried out under UVB (using a homebuilt photoreactor, see ESI,† Fig. S1) and sunlight (during Potsdam summer 2022 between 10:00 and 16:00) irradiation. The photoreactor has 8 sockets for the lamps (36 W each, UVB, λmax = 311 nm, Philips UV-B PL-L 36 W/01/4p 2G11) that are arranged circularly around a stir plate, where the reaction mixture can be placed in an Erlenmeyer flask or beaker. The lamp array is covered with an open cylindrical aluminum jacket to prevent light emission to the outside and to allow air circulation. In this study, six UV tubes were used, that is, two sockets were left unused.For photodegradation experiments, the photocatalyst composite (100 mg) was deposited in a 200 mL beaker containing 100 mL of an aqueous TET solution (20 mg L−1) and afterwards the flask was exposed to the light source (either the photoreactor or sunlight) with simultaneous agitation with a magnetic stirrer. Aliquots of 3.0 mL were withdrawn at specific times and immediately filtered through 0.45 μm PTFE syringe filters (VWR, Radnor, USA). The remaining TET concentration in the liquid phase was determined via UV-vis spectroscopy (Shimadzu UV-1900, Kyoto, Japan) using the TET absorption band at 358 nm. Solutions were placed in a quartz cuvette with a path length of 10 mm using water as reference. Calibration was done via a dilution series with TET in water. To ascertain that the PTFE filters do not adsorb TET and affect the measurements, calibrated TET solutions were directly filtered with the PTFE filters and no TET adsorption on the PTFE filter was found. A control photolysis study was conducted under identical experimental conditions as described above, but without the photocatalyst.
The TET degradation efficiency was calculated from eqn (3):
| R (%) = (C0 − Ct)/C0 × 100 | (3) |
The degree of mineralization was determined via the measurement of the total organic carbon (TOC) of the treated and untreated water using a Vario TOC analyzer (TOC/TNb Analyzer, vario TOC cube, Elementar Analysensysteme GmbH, Hanau, Germany). The percentage of mineralization was calculated using eqn (4),
| % Mineralization = (TOC0 − TOCe)/TOC0 × 100 | (4) |
The kinetics of the photodegradation reaction was evaluated according to the Langmuir–Hinshelwood (L–H) kinetics model (first-order kinetics model), eqn (5),
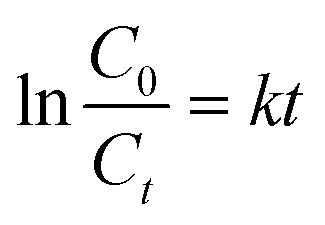 | (5) |
The influence of operating variables on TET photodegradation was studied using the best performing catalyst, C600KT. To that end, 50 mL of a 20 mg L−1 TET solution were irradiated for 60 min using C600KT for the effect of catalyst dose (10, 25, 50 mg), solution pH between 3.0 and 11.0 (adjusted with 0.1 M HCl or 0.1 M NaOH), ionic strength (0.1, 0.01 M, and 0.001 M NaCl), and anions (1.0 mM of SO42−, Cl−, HCO3−). For the effect of initial concentration, different concentrations of tetracycline (10, 20, 40, 80 mg L−1) were investigated.
To evaluate the recyclability of the NCs, again the best performing photocatalyst, C600KT, was selected. 200 mL of 20 mg L−1 of TET solution was mixed with 200 mg of C600KT for 120 min under UV irradiation. After each experiment, the NC was recovered, rinsed with water, and dried in the oven at 80 °C before reuse.
2.4 Identification of reactive species and degradation products
In order to identify the main reactive species (positive valence band holes h+, hydroxyl radicals ˙OH, or superoxide ˙O2−) generated upon irradiation, 1 mM sodium oxalate (Na2C2O4, a h+ scavenger), 0.2 mL isopropanol (IPA, an ˙OH scavenger), or 0.5 mM benzoquinone (BQ, a ˙O2− scavenger) were used.30,52 All measurements were performed in duplicate.Identification of the degradation products was done via high-performance liquid chromatography-mass spectrometry (HPLC-MS, Agilent Infinity HPLC 1260 System using Agilent G6470A Series Triple Quad LC/MS, Agilent Technologies Sales & Services GmbH & Co.KG, Waldbronn, Germany) using solutions that were irradiated for 120 min in the presence of the photocatalyst. A Poroshell 120 EC-C18 Column (3.0 × 50 mm, 2.7 μm, Agilent Technologies Sales & Services GmbH & Co.KG, Waldbronn, Germany) was employed for separation. The binary mobile phase consisted of 0.1% formic acid (A) and methanol (B) at a flow rate of 0.6 mL min−1. The column temperature was 25 °C. Analyses were performed in ESI† positive ion mode using the following settings: nebulizer pressure of 35.0 psi, fragmentor voltage of 80 V, and a desolvation nitrogen gas at a flow rate of 11.0 L min−1. MS scans were performed in the range = 100–1000 m/z at fragmentor voltage = 80 V with a scan time = 500 ms.
2.5 Toxicity assessment of treated water
Agar-disc diffusion assays were applied to determine whether the degradation products still retain antibiotic activity and to follow the effects of TET degradation over treatment (i.e. irradiation) time. In order to compare the potential toxicities of TET and aliquots from treated water at different time intervals, the change in toxicity against TET-sensitive bacteria (Gram-positive Staphylococcus xylosus and Gram-negative Escherichia coli SCS1) was monitored. One colony of each bacterial strain was cultured in 20 mL 2YT medium and incubated overnight at 37 °C while shaking at 100 rpm min−1 (called “overnight culture”). Subsequently, 20 mL of 45 °C warm 2YT agar was mixed with 10 μL of the overnight culture to prepare the plate. From each of the test samples (the parent compound and the aliquots taken at specific time intervals), 500 μL were taken and lyophilized down to 50 μL. The necessary number of paper discs (4.5 mm) was deposited on the bacteria agar plate and 25 μL of 10× concentrated process samples were pipetted on each disc. The antibiotic TET was used as a positive control and distilled water with 5% ethanol was used as a negative control. The plate was incubated over night at 37 °C and evaluated. Finally, the inhibition zone diameter was used as a criterion to determine the antimicrobial toxicity.3. Results and discussion
3.1 Characterization
A previous study44 describes the synthesis and the general characteristics of the NCs in detail, including scanning electron microscopy (SEM), elemental analysis (EA), energy dispersive X-ray (EDX) spectroscopy, X-ray diffraction (XRD), and nitrogen sorption (see ref. 44 for full details). SEM showed that the NCs are composed of well-dispersed nanometer-sized titania particles deposited on a porous kaolin clay/carbon support.44Furthermore, additional characterization of the NCs was done for the present study. Fig. 1 displays a representative SEM image with elemental mapping of one of the composites, C600KT. The X-ray map shows the presence of C, Ti, Al, Si, and O. This result matches well with the EDX profile and elemental analysis obtained for the NCs in the previous study.44 The Ti-, C-, and O-mapping shows that Ti, C, and O are uniformly distributed in the composites revealing the close contact between these elements. Si and Al stem from the clay (and hence must be closely associated) while C stems from the orange peel biochar.
 | ||
| Fig. 1 SEM image and elemental maps of all elements present in the C600KT NCs. Green, blue, yellow, red, and cyan represent carbon, aluminum, silicon, oxygen, and titanium, respectively. Scale bar applies to all images. | ||
Fig. 2a displays the Raman spectra of the NCs. The spectra show five bands that are characteristic of anatase at 147 (Eg, O–Ti–O symmetric bending), 198 (Eg, unassigned band from TiO2), 395 (B1g, Ti–O asymmetric bending), 513 (A1g + B1g, Ti–O symmetric stretching or asymmetric bending), and 636 (Eg, O–Ti–O symmetric stretching) cm−1. The spectra of all NCs show an intense band at 147 cm−1, suggesting that the crystallite sizes of the NCs are identical. All other bands, however, are broad, indicating the defective nature of TiO2 in the NCs.53,54
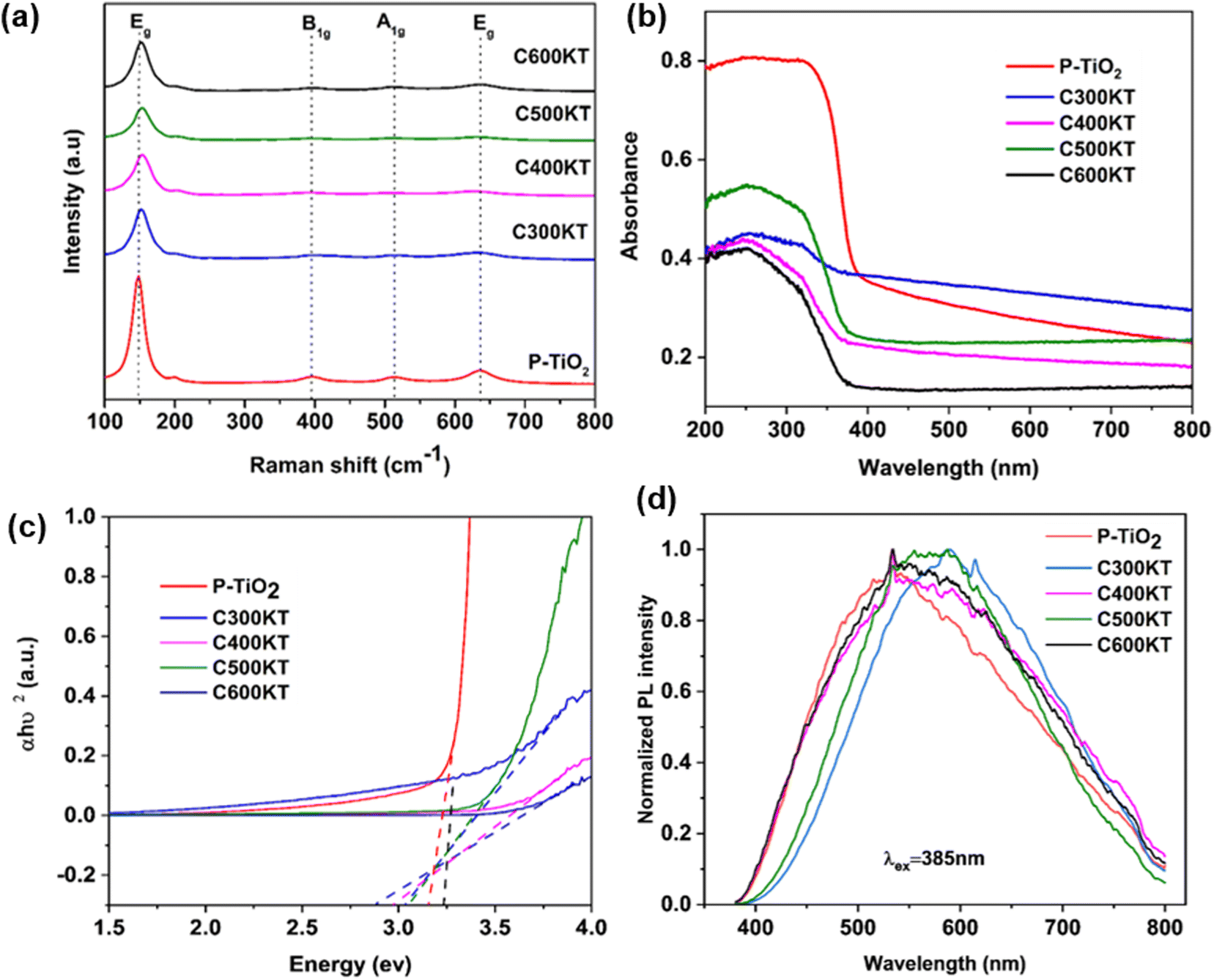 | ||
| Fig. 2 (a) Raman spectra, (b) UV-vis DRS spectra, (c) Tauc plot analysis, and (d) PL spectra of P-TiO2 and all NCs. Raman spectra in panel (a) are shifted vertically for clarity. Band gaps calculated from Fig. 1c are shown in Table 1. | ||
The bands observed in the Raman spectra of the NCs are less intense and exhibit a slight red shift (ca. 2–3 cm−1) when compared to those found in the spectra of P-TiO2. This suggests a slightly higher defect density in the TiO2 in the NCs.41,55 which has been linked to the presence of impurities such as carbon or oxygen vacancies in the TiO2 lattice.54 Surprisingly, even though about 28% of the NC are carbon44 there is no Raman signal that can be assigned to the presence of carbonaceous species. Similarly, from the EDX data,44 the Si content (indicative of kaolinite) is very low at ca. 3–4%, while the Ti content (indicative of the titania) is ca. 23%. This indicates that there is a rather high fraction of defect-rich54,56 titania which consequently dominates the Raman spectra of the NCs.
Fig. 2b shows the solid-state UV-vis diffuse reflectance spectroscopy (UV-vis DRS) data of P-TiO2 and the NCs. They all show similar absorption edges at around 380 nm. P-TiO2 has the strongest absorption band around 200–380 nm in the UV region. The NCs show a much lower absorption in the UV region in the order C600KT > C300KT > C500KT > C400KT. Previous studies indeed report that the strong band edge of TiO2 is reduced when TiO2 is amalgamated with another material.57,58 Also, doping non-metals into wide band gap metal oxide photocatalysts may effectively improve their visible light absorption but at the same time reduces UV absorption.59,60
This last observation is also consistent with the current observation: the absorption for all NCs tails into the visible region beyond 700 nm, with the tailing in the order of C500KT < C400KT < C600KT < C300KT. This clearly shows that all NCs can absorb some visible light; this is in strong contrast to P-TiO2, where the absorption is largely confined to the UV region.
The band gaps of P-TiO2 and the NCs were determined using Tauc analysis as illustrated in Fig. 2c and a summary of the band gap for all samples are presented in Table 1. For P-TiO2, a wide band gap of 3.08 eV is obtained while band gaps of 3.06, 2.99, 3.05, and 2.88 eV were obtained for C300KT, C400KT, C500KT, and C600KT, respectively. The slightly lower band gap energies of the NCs compared to P-TiO2 are likely due to impurities or defects within these materials, thereby shifting the absorption window slightly into the visible range.61–63 The slight reduction in the band gap of the band gaps in the NCs suggests that the introduction of C into the TiO2 lattice likely modifies the TiO2 band structure by creating impurity midgaps and thus tuning its bandgap.15,16,59
| Materials | Degradation efficiency (%) at 120 min | TOC mineralization (%) | Band gap (eV) | Kinetic parameter | |||
|---|---|---|---|---|---|---|---|
| UV | Sunlight | UV | Sunlight | k (min−1) | t 1/2 (min) | ||
| C300KT | 82 | 80 | 53 | 44 | 3.06 | 0.012 | 57.8 |
| C400KT | 88 | 84 | 55 | 46 | 2.99 | 0.022 | 30.1 |
| C500KT | 90 | 84 | 50 | 50 | 3.05 | 0.028 | 24.8 |
| C600KT | 92 | 89 | 60 | 50 | 2.88 | 0.029 | 23.9 |
| P-TiO2 | 97 | 91 | 58 | 39 | 3.08 | — | — |
| TET (photolysis in solution) | 20 | 18 | — | — | — | 0.0014 | 495 |
Fig. 2d shows the PL spectra of the composite materials. PL analysis has been widely used to understand the behavior of photo-induced charge carriers and the recombination process.64 With an excitation wavelength (λex) of 385 nm, broad emissions peaks in the visible region (420–800 nm) with very low intensities are observed for P-TiO2 and the NCs. Generally, a low emission intensity indicates a decreased recombination rate of the photogenerated electron/hole pairs and thus a higher photocatalytic activity.15,65,66
Alternatively, it is also possible that the carbonaceous species generate defects in the TiO2 crystal structure that either deepen existing trap states or act as trap states themselves.67,68 However, if those traps are too deep, they can act as recombination centers if located within the bulk or innermost part of the nanoparticles thereby reducing degradation activity.68,69 This could explain, why the degradation efficiency of C300KT is the lowest (see below for details).
In this study, the spectra of all the NCs exhibit a similar curve shape, and the peak intensities are nearly the same. However, the position the bands in the spectra of C300KT and C600KT is more red-shifted. This suggests that these NCs are active emitters in the visible region of the electromagnetic spectrum.16,70 Contrary to our data, other data suggest that the hybrid clay components do not necessarily contribute to the photoactive nature of the nanocomposites but merely acted as substrates for the development of the nanocomposites.71,72 The reason for this difference is currently unknown.
XPS analysis was performed to investigate the chemical state of the elements present in P-TiO2 and C600KT. Fig. 3a shows the survey spectra of P-TiO2 and C600KT from 0 to 1100 eV, which show fairly strong O 1s, Ti 2p, and C 1s peaks for both samples, while Si 2p and Al 2p signals are only weakly visible in C600KT due to the small amount of kaolinite combined with P-TiO2 (Fig. S2†). Particularly, compared to the data of P-TiO2, the survey spectrum of C600KT revealed the highly pronounced C 1s peak due to the introduction of carbon species from the biochar during the pyrolysis.
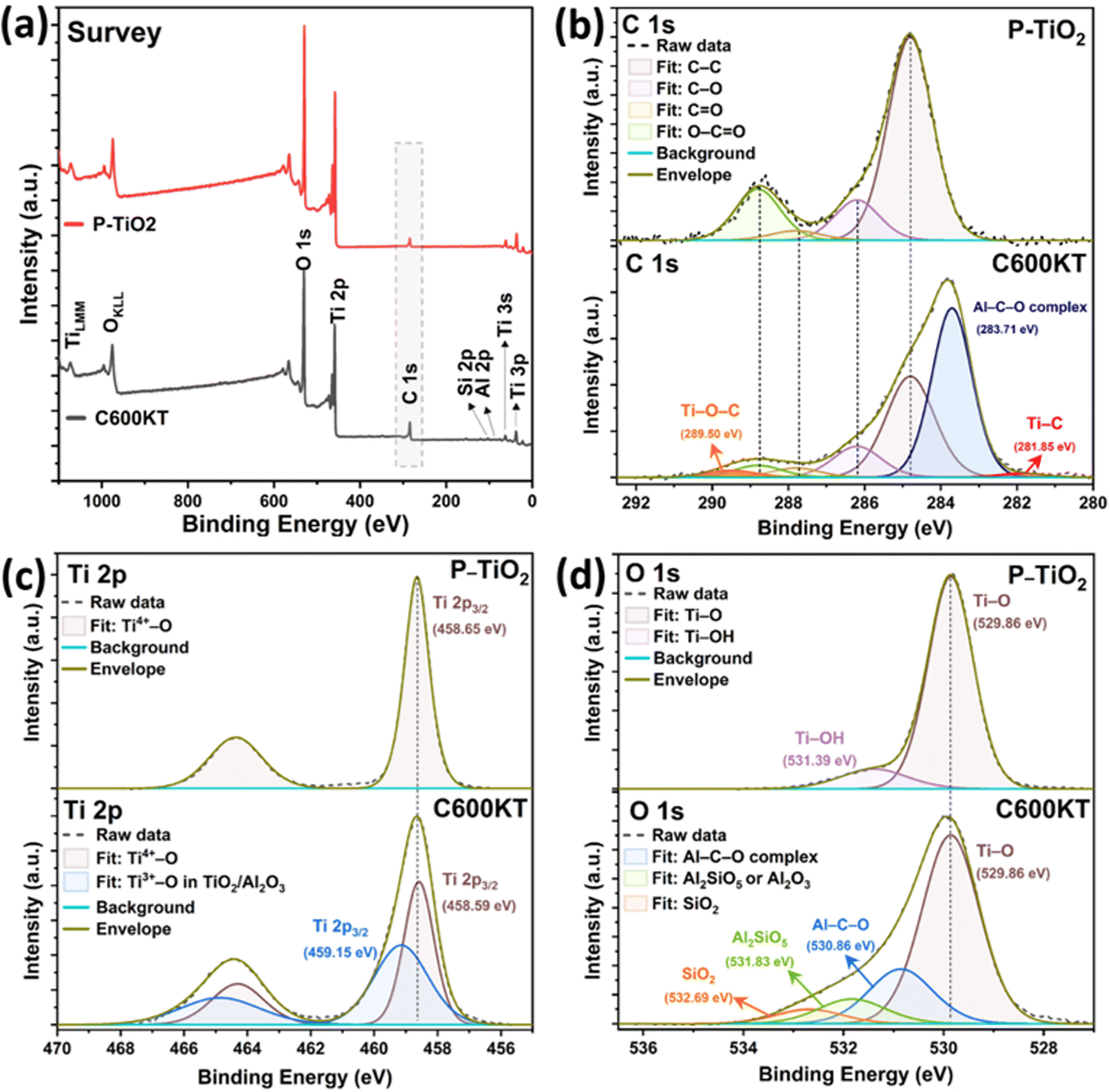 | ||
| Fig. 3 High-resolution XPS spectra of survey (a), C 1s (b), O 1s (c), and Ti 2p (d) of P-TiO2 and C600KT, respectively. | ||
In Fig. 3b, the binding energies of C–C bonds in both samples were charge-corrected by setting it as equal to 284.8 eV, and the oxygen-bound species of C 1s were fitted as C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, O–CO bonds at 286.2 eV, 287.8 eV, and 288.8 eV equally in both samples, respectively.73–75 Different from the XPS peak fitting of P-TiO2, the C 1s of C600KT revealed three more peaks at 281.85 eV, 283.07 eV, and 289.5 eV, where the most intense peak at 283.07 eV could be designated as binding energy of Al–C–O organometallic complex,76–78 which suggests that various alcohol and carbonyl groups in orange peel, including those from cellulose, lignin, and hemicellulose, may lead to aluminum-mediated organometallic compounds with the chemical bonds (e.g., Al–O–Al or Al–O–Si) in kaolinite during the pyrolysis reaction (Fig. S2a†).44,79
O, O–CO bonds at 286.2 eV, 287.8 eV, and 288.8 eV equally in both samples, respectively.73–75 Different from the XPS peak fitting of P-TiO2, the C 1s of C600KT revealed three more peaks at 281.85 eV, 283.07 eV, and 289.5 eV, where the most intense peak at 283.07 eV could be designated as binding energy of Al–C–O organometallic complex,76–78 which suggests that various alcohol and carbonyl groups in orange peel, including those from cellulose, lignin, and hemicellulose, may lead to aluminum-mediated organometallic compounds with the chemical bonds (e.g., Al–O–Al or Al–O–Si) in kaolinite during the pyrolysis reaction (Fig. S2a†).44,79
In addition, C600KT showed two more peaks at 281.85 eV and 289.5 eV. Basically, in the binding energy shift of XPS, the more chemical bonds with electronegative atoms are formed in the chemical structure, the stronger the positive XPS chemical shift is observed.80,81 Therefore, when considering the pyrolysis reaction of orange peel and kaolinite with P-TiO2, the higher binding energy at 289.5 eV can be assigned to Ti–O–C bonds because Ti atoms can be partially replaced by the more electronegative C atoms,53 while the lower binding energy at 281.85 eV can be ascribed to Ti–C bonds due to the low electronegativity of C atom relative to O atom.53 Thus, it can be inferred that some amount of carbon from the biochar is incorporated inside the TiO2 lattice of the composite materials.
Fig. 3c shows photoelectron signals from Ti 2p in two samples, which consist of doublet peaks with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in intensity due to their degeneracies. In the case of P-TiO2, the binding energy of Ti 2p3/2 and Ti 2p1/2 for P-TiO2 were determined at 458.65 eV and 464.37 eV, respectively, with the doublet separation as 5.72 eV between them, which mostly indicates a Ti4+ valency state,82 matched with a form of anatase TiO2.22,23,83 Contrarily, the Ti 2p of C600KT requires two components to be fitted, where the second component at 459.15 eV can be attributed to the Ti3+–O bonds originated from TiO/Al2O3.84–87 Also, the Ti4+–O (Ti 2p3/2) peak of C600KT was slightly shifted toward lower binding energy from 458.65 eV to 458.59 eV as compared to that of P-TiO2, which might be attributed to the strong interaction between the anatase TiO2 with newly formed Ti3+–O bonds and the kaolinite substrate.53,88
:1 ratio in intensity due to their degeneracies. In the case of P-TiO2, the binding energy of Ti 2p3/2 and Ti 2p1/2 for P-TiO2 were determined at 458.65 eV and 464.37 eV, respectively, with the doublet separation as 5.72 eV between them, which mostly indicates a Ti4+ valency state,82 matched with a form of anatase TiO2.22,23,83 Contrarily, the Ti 2p of C600KT requires two components to be fitted, where the second component at 459.15 eV can be attributed to the Ti3+–O bonds originated from TiO/Al2O3.84–87 Also, the Ti4+–O (Ti 2p3/2) peak of C600KT was slightly shifted toward lower binding energy from 458.65 eV to 458.59 eV as compared to that of P-TiO2, which might be attributed to the strong interaction between the anatase TiO2 with newly formed Ti3+–O bonds and the kaolinite substrate.53,88
Similar to this trend of the chemical interaction between P-TiO2 kaolin clay, and biochar, in Fig. 3d, the O 1s spectrum of C600KT also revealed three more peaks centered at 530.86 eV, 531.83 eV, and 532.69 eV besides Ti–O bonds (529.86 eV), where the binding energy at 530.86 eV can be attributed to Al–C–O organometallic bonds.76–78 This data may indirectly support the existence of an Al–C–O organometallic complex detected in the C 1s scan. Also, the highly shifted binding energy, peaking at 531.83 eV and 532.69 eV, can be assigned to the Al2SiO5 or Al2O3, and SiO2, respectively.89–94
3.2 Degradation study
Fig. 4a shows the photocatalytic degradation of TET under UVB irradiation (using the photoreactor described in the Experimental section) vs. irradiation time. With no catalyst present in the mixture, only a slight decrease (19%) in the TET concentration is observed after 120 min of irradiation. This indicates that TET is not easily degraded by UVB irradiation only.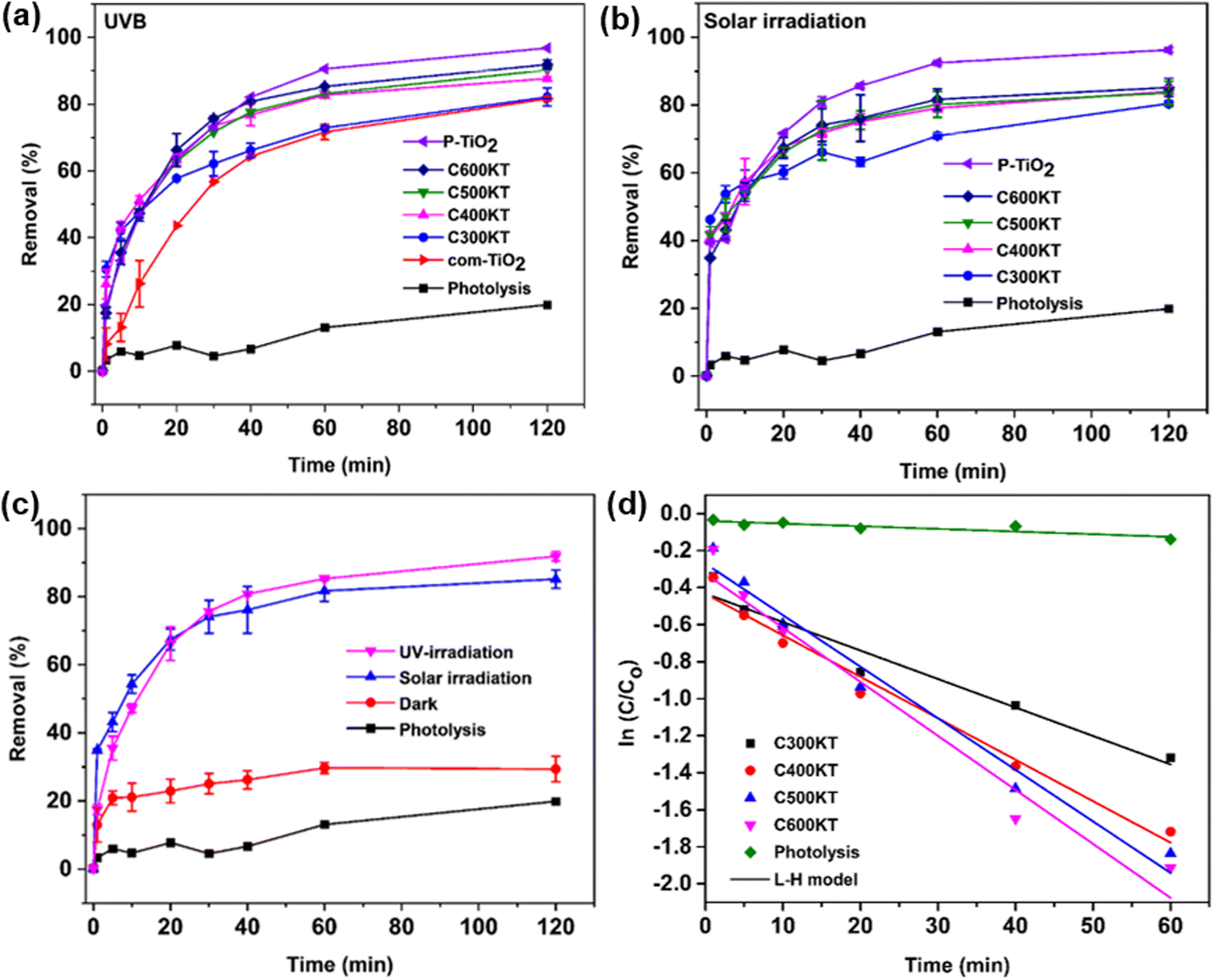 | ||
| Fig. 4 Degradation of TET under (a) UVB and (b) solar irradiation using com-TiO2, P-TiO2, C300KT, C400KT, C500KT, and C600KT along with the control photolysis experiment (no catalyst present), (c) comparison of the efficiency of C600KT with the dark experiment, and (d) Langmuir–Hinshelwood kinetic fits for all measurements. Some of the error bars are small and directly on the plot symbols, the same figure only showing the error bars can be found in the Fig. S3.† | ||
In the presence of all photocatalytic NCs, however, the degradation is effective and between 82 and 93% of TET is degraded within 120 minutes of irradiation (Table 1). As a result, we conclude that all composites show a rather high photocatalytic activity and are highly suited for the removal and degradation of TET under UVB irradiation. Among the four NCs studied, C600KT has the best performance with about 85% TET removal/degradation after 60 min and 93% after 120 min of irradiation.
It must be noted, however, that P-TiO2 (the control material not containing biochar and clay) prepared in this study has a slightly higher catalytic activity than even the best of the NCs, C600KT, resulting in a final degradation of 96% of TET after 120 minutes. However, P-TiO2 is much more difficult to separate from the liquid phase than any of the composites, Fig. S8, ESI.† As a result, the NC photocatalyst C600KT has a clear advantage over com-TiO2 and P-TiO2 in that the performance is comparable to P-TiO2, and separation and recycling is much more straightforward.
Fig. 4b shows the photocatalytic degradation of TET under solar irradiation (as opposed to UVB lamps, Fig. 4a). All NCs again exhibit a good performance in TET degradation and their degradation efficiencies (>80%) are essentially identical to the data obtained via UVB irradiation (Table 1). Overall, Fig. 4 therefore demonstrates that the NCs are highly effective, regardless of whether UVB or natural light is used. The visible-light absorption ability of the NCs for TET degradation can be attributed to:
(1) their slightly lower band gap (Fig. 2b and c) compared to com-TiO2 and P-TiO2 allowing for more photons to be harnessed for the photocatalytic degradation,
(2) the presence of carbonaceous species, providing improved surface areas and also acting as a carbon source both of which promote the narrowing of the band gap, and enhancing charge-carrier separation (electron sink),95–97
(3) their relatively small crystalline size (about 60 nm obtained from the Scherrer equation),44 and
(4) the larger specific surface area (70–163 m2 g−1)44 which offers a larger contact area between the TET molecules and the catalyst surface, thus reducing the charge recombination rate on the catalyst surface.
From the Tauc plot, C600KT has the lowest band gap (2.88 eV, Table 1) suggesting that the carbonaceous species could introduce lattice or surface defects in the titania that effectively lower the band gap, similar to previous reports by Oseghe et al.,15 Alfred et al.,16 and Li et al.41 and improve the visible light catalytic performance of TiO2 catalysts. The presence of carbon in the catalytic composites can lead to the formation of new active sites, which may be responsible for the more pronounced absorption tail into the visible region of the spectrum.63 These concepts should also apply to the current materials because elemental analysis and nitrogen sorption results indicate that the photocatalytic composites contain some amount of carbon and that the composites have a larger specific surface area than the unmodified titania, respectively.44
Furthermore, to differentiate the adsorption from photocatalytic properties of C600KT in the degradation of TET, a dark experiment was carried out (Fig. 4c). About 29% of TET is removed in the dark compared to the reaction in the presence of light, where the removal (or degradation) is about >89%. This confirms that C600KT is photoactive and that the most significant contribution to TET removal is via photocatalysis.
Fig. 4d shows that TET degradation follows pseudo-first-order kinetics (eqn (5)). The corresponding kinetic parameters (rate constant k and half-life t1/2) are presented in Table 1. Composites with the highest k and lowest t1/2 are considered the best materials.15 Among the materials studied, C600KT has the highest k and the lowest t1/2, consistent with the highest efficiency found earlier, Fig. 4a and b. In contrast, photolytic degradation (that is, irradiation only, with no catalyst present) has the lowest rate constant and largest t1/2. These data also illustrate why generally TET degradation in natural systems is rather slow in the absence of a catalyst.
Table S1† compares the photo-efficiency of the photocatalysts to other materials and processes that have been reported for the removal of TET molecules in water. Given the lower weight of the composite utilized in this study, the efficiency of C600KT is better than some previously reported in terms of kinetics, percentage of TET removal, and TOC removal efficiency.
3.3 Effect of operating variables
As C600KT is the most efficient material among those described here, it has been studied in detail in the following sections. The degradation efficiencies (% removal) vs. various operating variables for C600KT are shown in Fig. 5.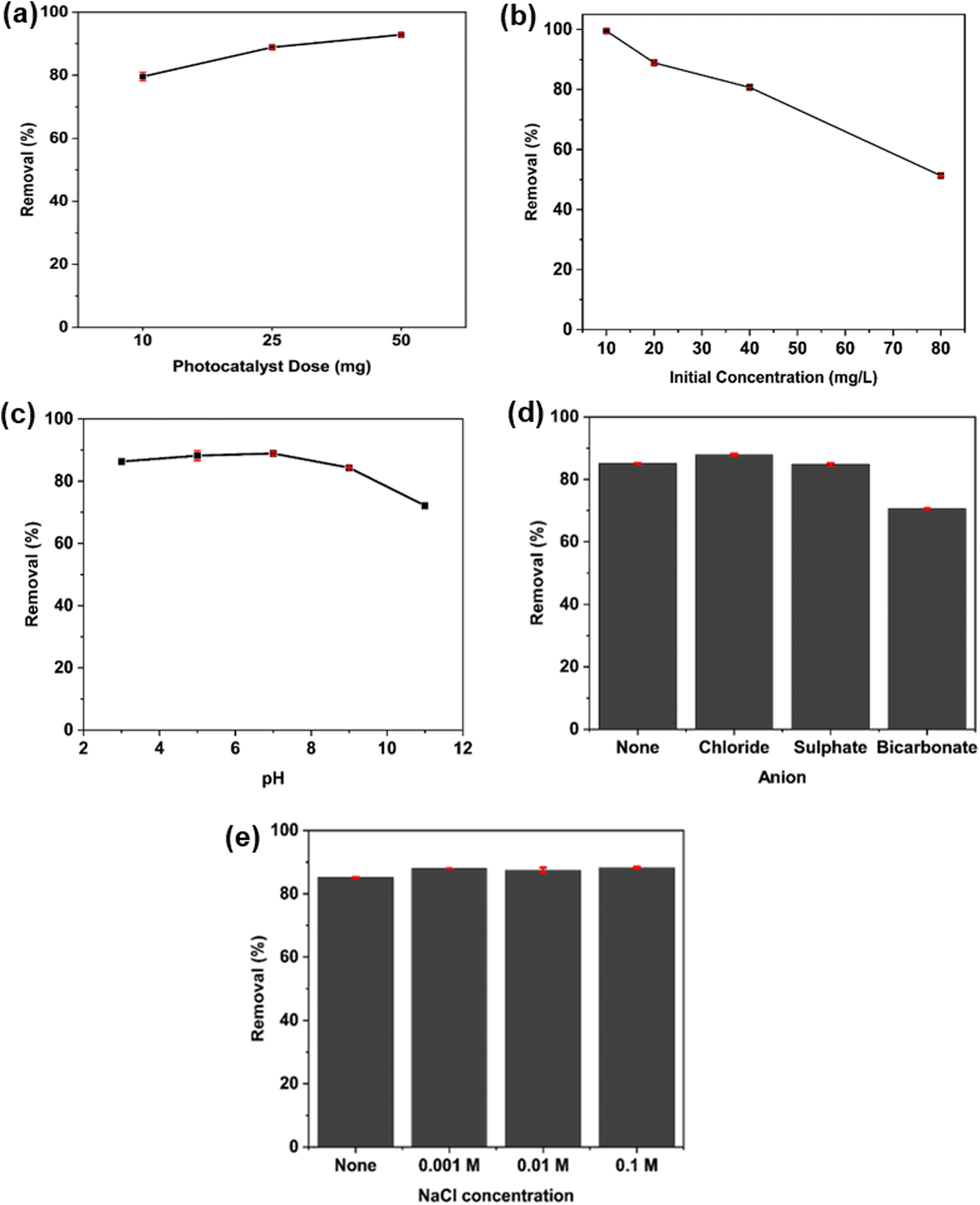 | ||
| Fig. 5 Effect of (a) photocatalyst dose, (b) initial concentration of TET at fixed catalyst dose, (c) initial TET solution pH, (d) anion species, and (e) ionic strength on the TET degradation efficiency of C600KT. Some of the error bars are small and directly on the plot symbols, the same figure only showing the error bars can be found in the Fig. S4.† | ||
The effect of the mass of the photocatalyst composites on degradation was studied in order to prevent excess catalyst usage and to determine the optimum mass needed for maximum TET degradation. Fig. 5a shows the effect of the catalyst dose on the removal rate at a fixed TET concentration of 20 mg L−1. TET degradation increases from 80% to 90% as the composite dose increases from 10 to 50 mg. This increase in degradation efficiency is attributed to the higher number of available active (photocatalytic) sites on the composite surface. This enables a more effective formation of reactive oxygen species (ROS) upon irradiation. The resulting higher number of ROS can then accelerate TET degradation.98
Fig. 5b shows that the amount of TET degraded by the composite decreases with increasing TET concentration (10–80 mg L−1). This is likely because as concentration increases, more TET molecules and degradation intermediates are present in the solution. These will then absorb a higher fraction of the incoming light and, as a result, lower the number of photons reaching the photocatalyst surface. This results in a decrease in the removal efficiency.99–102 Moreover, the photolysis experiment (Fig. 4a) demonstrates that light absorption by TET molecules does not significantly contribute to TET photodegradation since TET is not easily degraded by UVB irradiation alone.
In addition, when the initial TET concentration is high, a greater number of intermediate products are released into the solution during degradation. This might result in direct competition between the TET molecule itself and the intermediate products for the active sites, thereby slowing down TET degradation.103
Fig. 5c shows the influence of the solution pH on the degradation efficiency of C600KT. The TET photodegradation efficiency slightly increases from ca. 80 to ca. 85% from pH 3 to 7. Above pH 7, the efficiency of the photocatalyst drastically decreases and reaches ca. 70% at pH 11. From our previous study,44 pHpzc of C600KT is 6.23 while TET exists as a cationic species at pH < 3.32, as a neutral species at pH 3.32–7.68, and as an anionic species at pH > 7.68. Taking this information into account it can expected that at pH < pHpzc the composite is positively charged. As a result, the lower the pH the more positively charged the photocatalyst surface is. In contrast, if pH > pHpzc, the surface is negatively charged; this effect is more pronounced at higher pH.104 Similarly, TET is more negatively charged at higher pH values so that at high pH, a rather weak interaction can be expected. Due to negative–negative coulombic repulsion, TET will not interact as effectively with C600KT than when the pH is lower; this in turn reduces the system's overall effectiveness.
As a result, proper choice of the solution pH can improve the interaction between the photocatalyst and TET. In the current case, a pH around 7 works best. This suggests that electrostatics may not be the major driving force for interaction between photocatalysts and TET. We currently speculate that π–π and van der Waals forces along with hydrogen bonding may be important for TET adsorption onto the catalyst and subsequent photocatalytic degradation.
To explain the different photocatalytic activities at low and high pH, there are different aspects to consider. While the photocatalytic efficiency is only slightly lower at lower pH (that is, at higher H+ concentrations) and significantly lower at higher pH (pH < 9, higher OH− concentrations), this could be attributed to repulsion between the surface charge of the C600KT and the charged TET species inhibiting the adsorption of TET on C600KT, as stated above. On the other hand, the concentration of OH˙ influences the photodegradation and at a high solution pH (>9.7) more OH˙ can be generated,105 while TET is also present as H(TET)2−. This negatively charged TET species could attract the electrophilic OH˙, due to the high electrical density on its ring system, leading to enhanced photocatalytic activities.105,106 Apparently, based on the adsorption mechanism and the concentration of OH˙, there may be an optimum condition for TET photocatalytic degradation. In this study, only a slightly alkaline pH (pH = 9) was suitable for photocatalytic degradation of TET. However, the maximum degradation efficiency of C600KT is at pH 7 (≈89%), similar to ref. 100.
Fig. 5d shows the influence of foreign anions present in the system. Cl− and SO42− show no inhibitory effect on the photocatalytic activity of C600KT. Interestingly, Cl− improved catalytic performance slightly. Previous studies have noted that Cl− can facilitate the degradation of certain organic substance containing electron-rich groups, although the authors did of this study not provide a detailed explanation of this observation.107,108 In contrast, HCO3− reduces the activity of C600KT by 10–15% compared to the control reaction. This may be a result of this anion competing with organic molecules such as TET for active sites on the composite surface.109 Furthermore, HCO3− is a free radical scavenger, which consumes some active free radicals.110 This would then lead to a reduction in the photocatalytic activity due to two competing reactions and a concurrent, reduced degradation of TET.
Finally, Fig. 5e shows the effect of added salt on TET degradation. Typically, in water, the presence of Na+ and Cl− (and related ions) is inevitable. In this study, Na+ and Cl− have no significant effect on the efficiency of C600KT in the photodegradation of TET up to a concentration of 0.1 M. The efficiency of the composite is maintained at ≈87% irrespective of the ionic strength.
3.4 Reusability study
One major advantage of the C600KT over P-TiO2 and com-TiO2 is its ability to be reused over several cycles without losing its photocatalytic activity. In part this is due to the fact that titania NPs are difficult to separate post-treatment, which complicate the recovery process. The incorporation of the clay component into the composites facilitate a fast and uncomplicated recovery of the photocatalyst NCs (see Fig. S5†), allowing for a much easier reuse than P-TiO2 and com-TiO2. This easy separation of composites can be assigned to much larger particle sizes in the composites than in the P-TiO2; this advantage is caused by the large clay particles. This is consistent with our previous study that has shown that the addition of clay to the composites greatly enhances the recyclability.44The photocatalyst was reused for four cycles to degrade TET (20 mg L−1) under the same conditions as described previously. Fig. 6a shows that the degradation efficiency observed in these four cycles is ca. 85%, 79%, 68% and 71%, respectively. The slight decrease in efficiency is likely caused by the loss of catalyst particles and the gradual buildup of TET molecules and their intermediates on the active sites during each cycle, which may hinder the generation of reactive species.111,112 Overall the data show that the degradation process and the recycling of the nanocomposites are viable and are candidates for effective low cost photocatalytic water treatment.
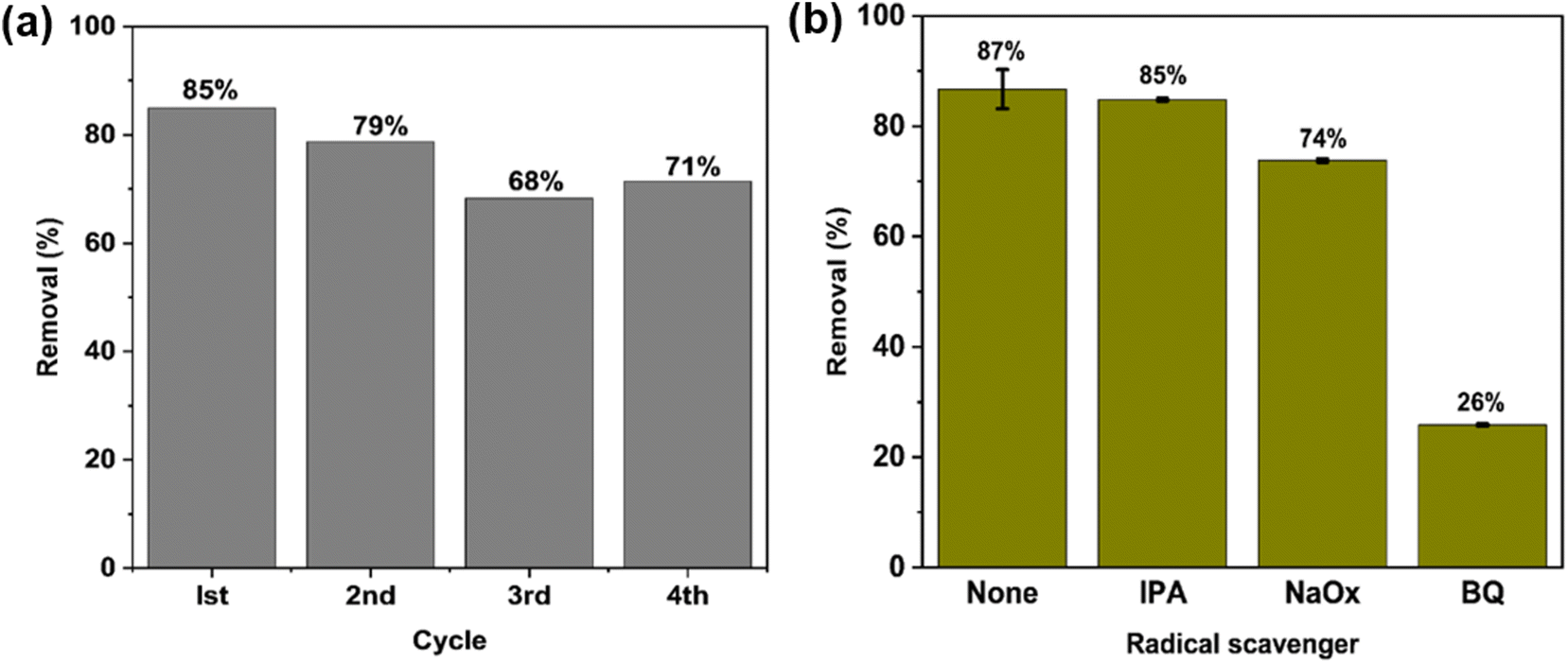 | ||
| Fig. 6 (a) Percentage of TET removal upon C600KT recycling and (b) effect of radical scavengers on TET removal from aqueous solution with C600KT after 60 min. Fig. 6a does not have error bars because each bar represents a single experiment. The error bars in Fig. 6b are small showing a high reproducibility. | ||
3.5 Identification of main reactive species
To understand the photocatalytic mechanism and to identify the reactive species involved in the photocatalytic TET degradation with C600KT, three different radical scavengers – isopropyl alcohol (IPA), sodium oxalate (NaOx), and benzoquinone (BQ) – were used for quenching hydroxyl radicals (˙OH), holes (h+), and superoxide radicals (˙O2−), respectively. Fig. 6b shows that the addition of IPA hardly affects the TET degradation over 60 min reaction time. NaOx leads to a visible decrease and only 74% of the TET is degraded. The addition of BQ leads to a significant decrease in TET degradation down to only 26% TET degradation. This implies that the composite releases more ˙O2− than h+ and ˙OH radicals in solution when exposed to irradiation. Superoxide radicals ˙O2− appear to be the major active species involved in TET degradation via the C600KT photocatalyst. Despite this, h+ and ˙OH also play a pivotal role in the reaction system because these two ROS contribute to the TET degradation.3.6 Mineralization efficiency
Fig. 7a and b shows the efficiency of the different photocatalyst composites for TET mineralization as evaluated via measurements of the total organic carbon (TOC)113 of the effluent after UVB and solar irradiations in selected conditions (see Experimental section for details). Upon irradiation, C600KT mineralizes approximately 60% (UVB irradiation) and 50% (solar irradiation) of TET, respectively. This is higher than the values obtained for the other composites investigated in this study. Under visible light irradiation, C600KT also shows a higher TET mineralization efficiency than P-TiO2.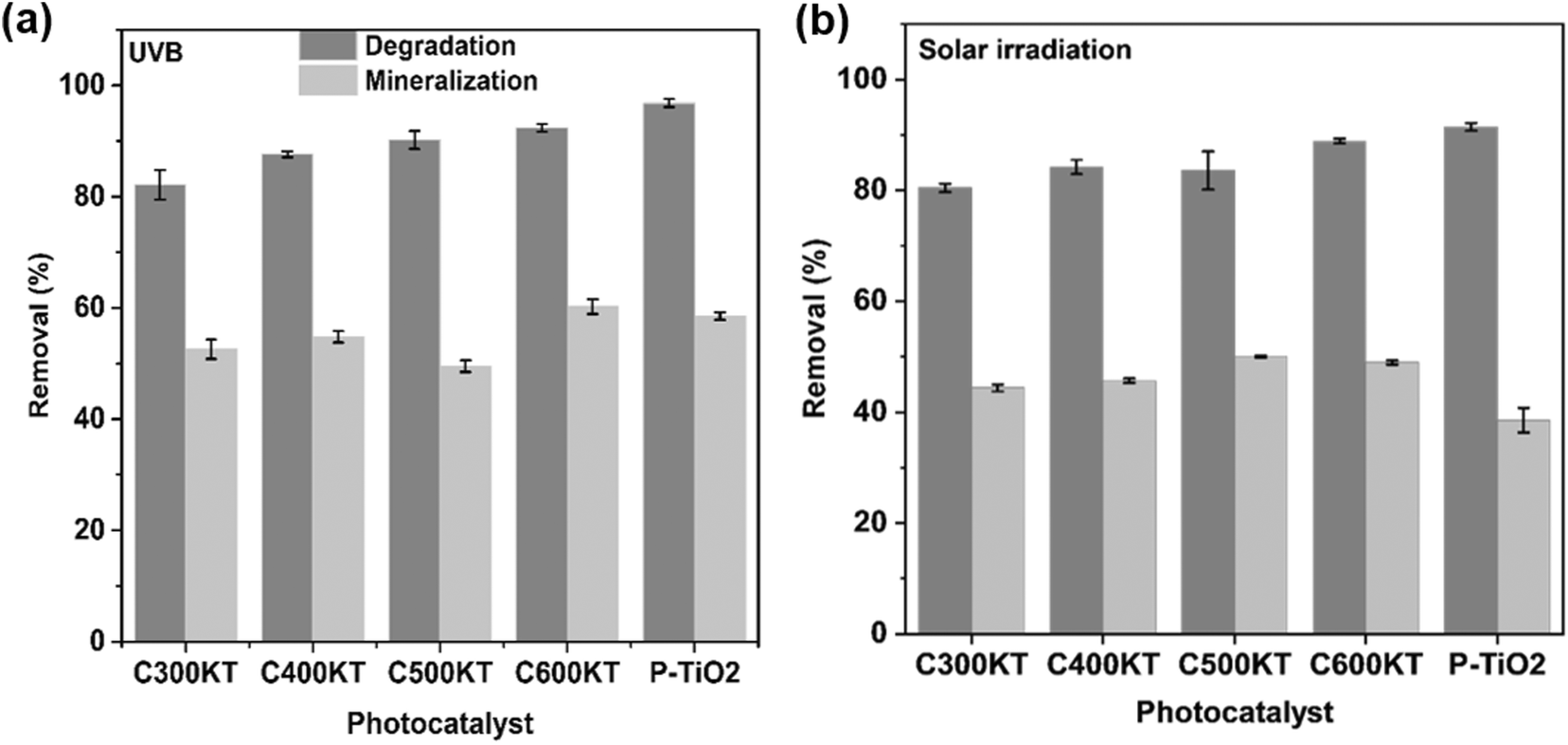 | ||
| Fig. 7 Mineralization and degradation of TET in aqueous solution by P-TiO2 and all composites under (a) UVB and (b) solar irradiation for 120 min. Dark grey is degradation, light grey indicates mineralization in both panels. | ||
The intermediate products obtained during the TET photodegradation over C600KT were detected by liquid chromatography-mass spectrometry using electrospray ionization (ESI-LC-MS). Fig. S3† shows the information on the extracted ion chromatograms of the TET solution before and after degradation. The primary characteristic peak with a retention time (rt) of 2.31 min and mass-to-charge ratio (m/z) of 445 corresponds to the deprotonated TET molecule ion. After photodegradation, three main intermediates with m/z of 198 (rt = 2.71 min), 170 (rt = 1.39 min), and 156 (rt = 0.91 min) were detected.
According to Fig. S6–S9 (ESI†), two of the intermediates exhibit shorter retention times than TET, indicating that they are more hydrophilic than TET. This is consistent with the TOC data, which shows that complete mineralization is not achieved within the 120 min degradation time proving that prolonged irradiation duration is required for complete mineralization of the residual fragments. The results suggest that the generation of these intermediates results from a loss of functional groups and various reactions such as ring-opening, bond cleavage, demethylation, dehydration, dehydroxylation, deamination, and oxidation.114–116
Moreover, we have shown above (Fig. 6b) that superoxide radicals are key players in TET degradation with C600KT. As a result, based on the data shown in Fig. 7 and the mass spectrometry data, a number of intermediates (Fig. 8) can be postulated. These can form upon irradiation and are present in the liquid phase after 120 minutes of irradiation. Upon longer irradiation, they can be transformed further or ultimately be fully mineralized to CO2 and H2O.
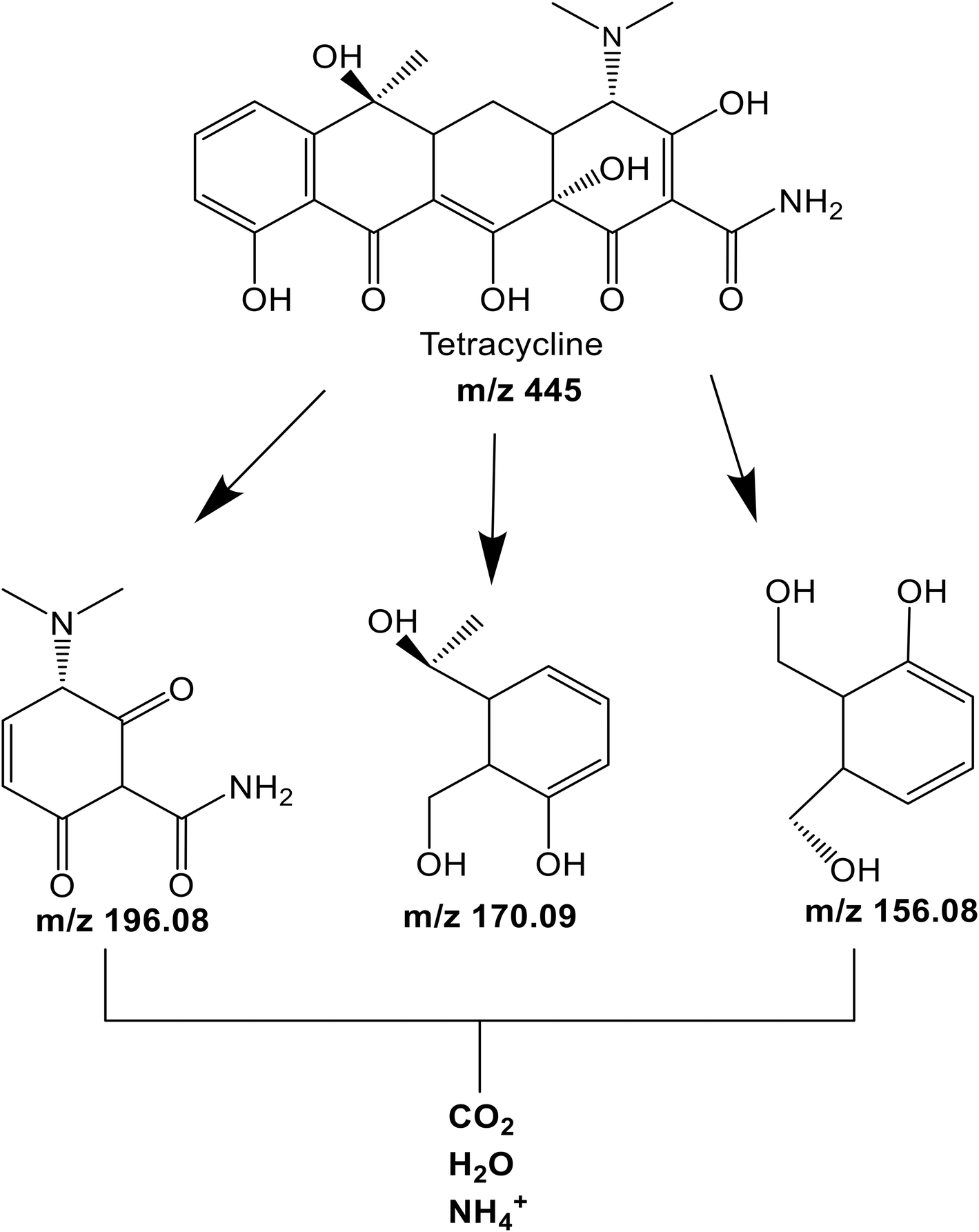 | ||
| Fig. 8 Proposed structure of degradation intermediates identified from mass spectrometry. | ||
3.7 Toxicity assessment
One of the key setbacks of the advanced oxidation process is the possibility of producing a photodegradation product with higher toxicity to the environment and humans due to incomplete mineralization. As a result, the toxicity of C600KT-treated aqueous solutions against E. coli and S. xylosus bacteria was investigated. Effluents collected after 15–120 min treatment with C600KT show no toxicity to both bacteria. The starting material (t = 0 min; 20 mg L−1 TET) shows a clear growth inhibition around the paper disk. With increasing incubation time of the water sample treated with C600KT the size of the inhibition zone becomes smaller. Already after 15 min, the inhibition zone is nearly the size of the paper disk. This means that at this time point around 1.25 mg L−1 TET is present in the water sample.If TET is used undiluted, almost no infection of the growth can be seen after 15 minutes. This means that the TET concentration must be less than 10 mg L−1 (Table 2, see reference plate). If the sample is concentrated by a factor of 10, this results in a radius of inhibition of ≈1.5 mm, which corresponds to a concentration of 15 mg L−1 of the 10-fold concentrated sample or less than 1.5 mg L−1 of the undiluted sample. This corresponds to a reduction in TET of over 90%. These data also show that the rate of TET degradation is fast and the intermediate products generated are less toxic than the TET molecules, at least to the two bacterial strains investigated here (Fig. 9).
| Plate | Sample | Radius of inhibition area [mm] (radius additional to paper disc) | |||
|---|---|---|---|---|---|
| E. coli | S. xylosus | ||||
| a The factor due to concentrating the samples (10×) was taken into account. The radius of the inhibition area of the water sample was compared to the dilution series. | |||||
| Reference plate | 20 mg L−1 | 2 | 3.6–3.8 | ||
| 15 mg L−1 | 1.5 | 3.6 | |||
| 12.5 mg L−1 | 1 | 2.5 | |||
| 10 mg L−1 | — | 1.8 | |||
| 7.5 mg L−1 | — | 1.4 | |||
| 5 mg L−1 | — | 1.1 | |||
| 1 mg L−1 | — | — | |||
| Negative control | — | — | |||
| TET concentration in water samplea | |||||
| E. coli | S. xylosus | ||||
| Plate with 10× concentrated process samples | Blank | 6–7.5 | 9 | 20 mg L−1 | >2 mg L−1 |
| 5 min | 4.5 | 6.5 | >20 mg L−1 | >2 mg L−1 | |
| 15 min | 1.5 | 2.7–3.1 | ∼1.25 mg L−1 | ∼1.25 mg L−1 | |
| 30 min | — | — | n.d. | n.d. | |
| 60 min | — | — | n.d. | n.d. | |
| 120 min | — | — | n.d. | n.d. | |
| Negative control | — | ||||
 | ||
| Fig. 9 Toxicity test for untreated (t = 0) and treated water collected at different time intervals against Escherichia coli SCS1 and Staphylococcus xylosus. | ||
4. Conclusion
Contamination of water bodies with antibiotics is a severe issue and low cost yet effective solutions to treat contaminated water are highly sought after. The current study demonstrates that the combination of a high surface area biochar and clay composite, which is rendered photoactive by the addition of titania nanoparticles is an effective means for removing tetracycline from an aqueous solution. While the composite also works in the dark, the combination of multiple components providing high surface areas, good recyclability, and photoactivity shows its full potential when irradiated with UV lamps or natural sunlight. Under appropriate pH, tetracycline concentrations, photocatalyst dose, and appropriate light sources, up to ca. 90% of tetracycline can be degraded using these new low cost photocatalysts. The comparison of the chemical characterization of the reaction products and degradation time with a biological readout shows very good comparability and also demonstrates that no toxic products are generated during the photodegradation process.Author contributions
Morenike O. Adesina: writing – original draft, investigation, formal analysis. Moses O. Alfred: methodology, review & editing. Harald Seitz: investigation, resources, review & editing, Katlen Brennenstuhl: analysis, resources. Harshadrai M. Rawel: formal analysis, visualization, writing – review & editing, Pablo Wessig: resources. Jiyong Kim: investigation, resources, writing – review & editing. Armin Wedel: investigation and resources. Wouter Koopman: investigation, resources, review & editing. Christina Günter: investigation, and review & editing. Emmanuel I. Unuabonah: conceptualization, supervision, writing – review & editing. Andreas Taubert: conceptualization, supervision, visualization, resources, writing – review & editing and funding acquisition.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank A. Nitschke and S. Mies for support with chemicals and supplies, P. Bíró and Prof. S. Oswald for access to the TOC analyzer, and J. Reinicke for technical assistance. We also thank the German Academic Exchange Service (DAAD, Grant No. 57552338, to M. O. A.) and the University of Potsdam (Grant No. 53170000, to A. T.) for financial support.References
- L. Xu, H. Zhang, P. Xiong, Q. Zhu, C. Liao and G. Jiang, Occurrence, fate, and risk assessment of typical tetracycline antibiotics in the aquatic environment: A review, Sci. Total Environ., 2021, 753, 141975 CrossRef CAS PubMed.
- R. Daghrir and P. Drogui, Tetracycline antibiotics in the environment: a review, Environ. Chem. Lett., 2013, 11, 209–227 CrossRef CAS.
- K. A. Azalok, A. A. Oladipo and M. Gazi, UV-light-induced photocatalytic performance of reusable MnFe-LDO–biochar for tetracycline removal in water, J. Photochem. Photobiol., A, 2021, 405, 112976 CrossRef CAS.
- I. M. Al-Riyami, M. Ahmed, A. Al-Busaidi and B. Choudri, Antibiotics in wastewaters: a review with focus on Oman, Appl. Water Sci., 2018, 8, 1–10 CrossRef CAS.
- A. A. Borghi and M. S. A. Palma, Tetracycline: production, waste treatment and environmental impact assessment, Braz. J. Pharm. Sci., 2014, 50, 25–40 CrossRef.
- A. Faleye, A. Adegoke, K. Ramluckan, F. Bux and T. Stenström, Antibiotic residue in the aquatic environment: status in Africa, Open Chem., 2018, 16, 890–903 CAS.
- S. M. Zainab, M. Junaid, N. Xu and R. N. Malik, Antibiotics and antibiotic resistant genes (ARGs) in groundwater: A global review on dissemination, sources, interactions, environmental and human health risks, 2020, 187, p. 116455.
- M. Bilal, S. Mehmood, T. Rasheed, H. M. Iqbal and Health, Antibiotics traces in the aquatic environment: persistence and adverse environmental impact, Current opinion in environmental science & health, 2020, 13, 68–74 Search PubMed.
- M. Bilal, S. Mehmood, T. Rasheed and H. M. Iqbal, Antibiotics traces in the aquatic environment: persistence and adverse environmental impact, Current Opinion in Environmental Science & Health, 2020, 13, 68–74 Search PubMed.
- D. Wang, Y. Zheng, Q. Deng and X. Liu, Water-Soluble Synthetic Polymers: Their Environmental Emission Relevant Usage, Transport and Transformation, Persistence, and Toxicity, Environ. Sci. Technol., 2023, 57, 6387–6402 CrossRef CAS PubMed.
- P. Kokkinos, D. Mantzavinos and D. Venieri, Current trends in the application of nanomaterials for the removal of emerging micropollutants and pathogens from water, Molecules, 2020, 25, 2016 CrossRef CAS PubMed.
- S. Mathew, P. Ganguly, S. Rhatigan, V. Kumaravel, C. Byrne, S. J. Hinder, J. Bartlett, M. Nolan and S. C. Pillai, Cu-doped TiO2: visible light assisted photocatalytic antimicrobial activity, Appl. Sci., 2018, 8, 2067 CrossRef CAS.
- A. A. Oladipo and F. S. Mustafa, Bismuth-based nanostructured photocatalysts for the remediation of antibiotics and organic dyes, Beilstein J. Nanotechnol., 2023, 14, 291–321 CrossRef CAS PubMed.
- M. A. Henderson, A surface science perspective on TiO2 photocatalysis, Surf. Sci. Rep., 2011, 66, 185–297 CrossRef CAS.
- E. O. Oseghe and A. E. Ofomaja, Facile microwave synthesis of pine cone derived C-doped TiO2 for the photodegradation of tetracycline hydrochloride under visible-LED light, J. Environ. Manage., 2018, 223, 860–867 CrossRef CAS PubMed.
- M. O. Alfred, M. O. Omorogie, O. Bodede, R. Moodley, A. Ogunlaja, O. G. Adeyemi, C. Günter, A. Taubert, I. Iermak and H. Eckert, Solar-active clay-TiO2 nanocomposites prepared via biomass assisted synthesis: Efficient removal of ampicillin, sulfamethoxazole and artemether from water, Chem. Eng. J., 2020, 398, 125544 CrossRef.
- R. Hickman, E. Walker and S. Chowdhury, TiO2-PDMS composite sponge for adsorption and solar mediated photodegradation of dye pollutants, J. Water Proc. Engineering, 2018, 24, 74–82 CrossRef.
- X. Gao, X. Liu, Z. Zhu, Y. Gao, Q. Wang, F. Zhu and Z. Xie, Enhanced visible light photocatalytic performance of CdS sensitized TiO2 nanorod arrays decorated with Au nanoparticles as electron sinks, Sci. Rep., 2017, 7, 973 CrossRef PubMed.
- S. Liu, N. Zhang, Z.-R. Tang and Y.-J. Xu, Synthesis of one-dimensional CdS@ TiO2 core–shell nanocomposites photocatalyst for selective redox: the dual role of TiO2 shell, ACS Appl. Mater. Interfaces, 2012, 4, 6378–6385 CrossRef CAS PubMed.
- M. Milani, M. Mazzanti, S. Caramori, G. Di Carmine, G. Magnacca and A. Molinari, Composite CdS/TiO2 Powders for the Selective Reduction of 4-Nitrobenzaldehyde by Visible Light: Relation between Preparation, Morphology and Photocatalytic Activity, Catalysts, 2022, 13, 74 CrossRef.
- M. Nasirian, Y. Lin, C. Bustillo-Lecompte and M. Mehrvar, Enhancement of photocatalytic activity of titanium dioxide using non-metal doping methods under visible light: a review, Int. J. Environ. Sci. Technol., 2018, 15, 2009–2032 CrossRef CAS.
- E. K. Tetteh, S. Rathilal, D. Asante-Sackey and M. N. Chollom, Prospects of synthesized magnetic TiO2-based membranes for wastewater treatment: A review, Materials, 2021, 14, 3524 CrossRef CAS PubMed.
- K. P. Gopinath, N. V. Madhav, A. Krishnan, R. Malolan and G. Rangarajan, Present applications of titanium dioxide for the photocatalytic removal of pollutants from water: A review, J. Environ. Manage., 2020, 270, 110906 CrossRef CAS PubMed.
- B. Srikanth, R. Goutham, R. B. Narayan, A. Ramprasath, K. Gopinath and A. Sankaranarayanan, Recent advancements in supporting materials for immobilised photocatalytic applications in waste water treatment, J. Environ. Manage., 2017, 200, 60–78 CrossRef CAS PubMed.
- H. M. Ali, F. A. Roghabadi and V. Ahmadi, Solid-supported photocatalysts for wastewater treatment: Supports contribution in the photocatalysis process, J. Sol. Energy, 2023, 255, 99–125 CrossRef CAS.
- A. J. Jafari, M. Moslemzadeh, A. Esrafili and R. R. Kalantary, Synthesis of new composite based on TiO2 immobilized in glass fibers for photo-catalytic degradation of chlorobenzene in aqueous solutions, Environ. Res., 2022, 204, 112018 CrossRef CAS PubMed.
- M. Mazzanti, S. Caramori, M. Fogagnolo, V. Cristino and A. Molinari, Turning waste into useful products by photocatalysis with nanocrystalline TiO2 thin films: Reductive cleavage of azo bond in the presence of aqueous formate, Nanomaterials, 2020, 10, 2147 CrossRef CAS PubMed.
- R. Kumar, R. Choudhary, S. Kolay, O. Pandey, K. Singh and P. Bhargava, Carbon coated titanium dioxide (CC-TiO 2) as an efficient material for photocatalytic degradation, Energy Adv., 2022, 1, 926–934 RSC.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, P25-graphene composite as a high performance photocatalyst, ACS Nano, 2010, 4, 380–386 CrossRef CAS PubMed.
- X. Zhang, D. Han, M. Dai, K. Chen, Z. Han, Y. Fan, Y. He, D. Han and L. Niu, Enhanced photocatalytic degradation of tetracycline by constructing a controllable Cu 2 O–TiO 2 heterojunction with specific crystal facets, Catal. Sci. Technol., 2021, 11, 6248–6256 RSC.
- S. Dong, J. Feng, M. Fan, Y. Pi, L. Hu, X. Han, M. Liu, J. Sun and J. Sun, Recent developments in heterogeneous photocatalytic water treatment using visible light-responsive photocatalysts: a review, RSC Adv., 2015, 5, 14610–14630 RSC.
- M. Jain, A. Mudhoo, D. L. Ramasamy, M. Najafi, M. Usman, R. Zhu, G. Kumar, S. Shobana, V. K. Garg and M. Sillanpää, Adsorption, degradation, and mineralization of emerging pollutants (pharmaceuticals and agrochemicals) by nanostructures: a comprehensive review, Environ. Sci. Pollut. Res., 2020, 27, 34862–34905 CrossRef CAS PubMed.
- W. Kong, F. Zhao, H. Guan, Y. Zhao, H. Zhang and B. Zhang, Highly adsorptive mesoporous carbon from biomass using molten-salt route, J. Mater. Sci., 2016, 51, 6793–6800 CrossRef CAS.
- S. D. Perera, R. G. Mariano, K. Vu, N. Nour, O. Seitz, Y. Chabal and K. J. Balkus Jr, Hydrothermal synthesis of graphene-TiO2 nanotube composites with enhanced photocatalytic activity, ACS Catal., 2012, 2, 949–956 CrossRef CAS.
- H. Gao, X. Li, J. Lv and G. Liu, Interfacial charge transfer and enhanced photocatalytic mechanisms for the hybrid graphene/anatase TiO2 (001) nanocomposites, J. Phys. Chem. C, 2013, 117, 16022–16027 CrossRef CAS.
- W.-J. Ong, L.-L. Tan, S.-P. Chai, S.-T. Yong and A. R. Mohamed, Self-assembly of nitrogen-doped TiO 2 with exposed {001} facets on a graphene scaffold as photo-active hybrid nanostructures for reduction of carbon dioxide to methane, Nano Res., 2014, 7, 1528–1547 CrossRef CAS.
- C. Thambiliyagodage, Efficient photocatalysis of carbon coupled TiO2 to degrade pollutants in wastewater–A review, Environ. Nanotechnol., Monit. Manage., 2022, 100737 CAS.
- N. R. Reddy, P. M. Reddy, N. Jyothi, A. S. Kumar, J. H. Jung and S. W. Joo, Versatile TiO2 bandgap modification with metal, non-metal, noble metal, carbon material, and semiconductor for the photoelectrochemical water splitting and photocatalytic dye degradation performance, J. Alloys Compd., 2023, 935, 167713 CrossRef CAS.
- S. Ullah, E. P. Ferreira-Neto, A. A. Khan, I. P. Medeiros and H. Wender, Supported nanostructured photocatalysts: the role of support-photocatalyst interactions, Photochem. Photobiol. Sci., 2023, 22, 219–240 CrossRef CAS PubMed.
- Y. Tan, W. Zhao, L. Sun, R. Zhang, J. Hou, S. Fu, W. Xu and R. Zhang, Photocatalytic abatement of formaldehyde under visible light irradiation via bamboo carbon/TiO2 modified by plasma at low temperature, J. Cleaner Prod., 2022, 369, 133280 CrossRef CAS.
- C. Li, N. Zhu, X. Dong, X. Zhang, T. Chen, S. Zheng and Z. Sun, Tuning and controlling photocatalytic performance of TiO2/kaolinite composite towards ciprofloxacin: Role of 0D/2D structural assembly, Adv. Powder Technol., 2020, 31, 1241–1252 CrossRef CAS.
- G. Cheng, F. Xu, J. Xiong, F. Tian, J. Ding, F. J. Stadler and R. Chen, Enhanced adsorption and photocatalysis capability of generally synthesized TiO2-carbon materials hybrids, Adv. Powder Technol., 2016, 27, 1949–1962 CrossRef CAS.
- L. Lu, R. Shan, Y. Shi, S. Wang and H. Yuan, A novel TiO2/biochar composite catalysts for photocatalytic degradation of methyl orange, Chemosphere, 2019, 222, 391–398 CrossRef CAS PubMed.
- M. O. Adesina, I. Block, C. Günter, E. I. Unuabonah and A. Taubert, Efficient Removal of Tetracycline and Bisphenol A from Water with a New Hybrid Clay/TiO2 Composite, ACS Omega, 2023, 8(24), 21594–21604 CrossRef CAS PubMed.
- L. Luo, Y. Yang, M. Xiao, L. Bian, B. Yuan, Y. Liu, F. Jiang and X. Pan, A novel biotemplated synthesis of TiO2/wood charcoal composites for synergistic removal of bisphenol A by adsorption and photocatalytic degradation, Chem. Eng. J., 2015, 262, 1275–1283 CrossRef CAS.
- Z. Mengting, T. A. Kurniawan, R. Avtar, M. H. D. Othman, T. Ouyang, H. Yujia, Z. Xueting, T. Setiadi and I. Iswanto, Applicability of TiO2 (B) nanosheets@ hydrochar composites for adsorption of tetracycline (TC) from contaminated water, J. Hazard. Mater., 2021, 405, 123999 CrossRef CAS PubMed.
- U. Suzuki, T. Kameda, S. Kumagai, Y. Saito and T. Yoshioka, Adsorption of bisphenol A by TiO2-based organic–inorganic hybrid materials, J. Water Proc. Engineering, 2022, 49, 103081 CrossRef.
- J. Leichtweis, Y. Vieira, N. Welter, S. Silvestri, G. L. Dotto and E. Carissimi, A review of the occurrence, disposal, determination, toxicity and remediation technologies of the tetracycline antibiotic, Process Saf. Environ. Prot., 2022, 160, 25–40 CrossRef CAS.
- P.-P. Sun, D. R. Kripalani, M. Hao, W. Chi, W. Li and K. Zhou, Emissive nature and molecular behavior of zero-dimensional organic–inorganic metal halides Bmpip2MX4, J. Phys. Chem. Lett., 2020, 11, 5234–5240 CrossRef CAS PubMed.
- S. Landi Jr, I. R. Segundo, C. Afonso, O. Lima Jr, M. F. Costa, E. Freitas and J. Carneiro, Evaluation of band gap energy of TiO2 precipitated from titanium sulphate, Phys. B, 2022, 639, 414008 CrossRef.
- C. Wagner, L. Davis, M. Zeller, J. Taylor, R. Raymond and L. Gale, Empirical atomic sensitivity factors for quantitative analysis by electron spectroscopy for chemical analysis, Surf. Interface Anal., 1981, 3, 211–225 CrossRef CAS.
- A. O. Oluwole and O. S. Olatunji, Photocatalytic degradation of tetracycline in aqueous systems under visible light irridiation using needle-like SnO 2 nanoparticles anchored on exfoliated gC 3 N 4, Environ. Sci. Eur., 2022, 34, 1–14 CrossRef.
- C. Li, X. Dong, N. Zhu, X. Zhang, S. Yang, Z. Sun, Y. Liu, S. Zheng and D. D. Dionysiou, Rational design of efficient visible-light driven photocatalyst through 0D/2D structural assembly: Natural kaolinite supported monodispersed TiO2 with carbon regulation, Chem. Eng. J., 2020, 396, 125311 CrossRef CAS.
- S. G. Ullattil, J. Zavašnik, K. Maver, M. Finšgar, N. Novak Tušar and A. Pintar, Defective Grey TiO2 with Minuscule Anatase–Rutile Heterophase Junctions for Hydroxyl Radicals Formation in a Visible Light-Triggered Photocatalysis, Catalysts, 2021, 11, 1500 CrossRef CAS.
- S. M. Piontek, Characterizing Heterogeneously Charged Mineral Oxide Surfaces Using Nonlinear Spectroscopy, PhD, Temple University, 2019 Search PubMed.
- N. Fajrina and M. Tahir, 2D-montmorillonite-dispersed g-C3N4/TiO2 2D/0Dnanocomposite for enhanced photo-induced H2 evolution from glycerol-water mixture, Appl. Surf. Sci., 2019, 471, 1053–1064 CrossRef CAS.
- R. Jaiswal, N. Patel, D. Kothari and A. Miotello, Improved visible light photocatalytic activity of TiO2 co-doped with Vanadium and Nitrogen, Appl. Catal., B, 2012, 126, 47–54 CrossRef CAS.
- N. U. M. Nor and N. A. S. Amin, Glucose precursor carbon-doped TiO2 heterojunctions for enhanced efficiency in photocatalytic reduction of carbon dioxide to methanol, J. CO2 Util., 2019, 33, 372–383 CrossRef CAS.
- A. S. Alshammari, L. Chi, X. Chen, A. Bagabas, D. Kramer, A. Alromaeh and Z. Jiang, Visible-light photocatalysis on C-doped ZnO derived from polymer-assisted pyrolysis, RSC Adv., 2015, 5, 27690–27698 RSC.
- O. Haibo, H. J. Feng, L. Cuiyan, C. Liyun and F. Jie, Synthesis of carbon doped ZnO with a porous structure and its solar-light photocatalytic properties, Mater. Lett., 2013, 111, 217–220 CrossRef.
- K. Palanivelu, J.-S. Im and Y.-S. Lee, Carbon doping of TiO 2 for visible light photo catalysis-a review, Carbon Lett., 2007, 8, 214–224 CrossRef.
- R. Kavitha and L. G. Devi, Synergistic effect between carbon dopant in titania lattice and surface carbonaceous species for enhancing the visible light photocatalysis, J. Environ. Chem. Eng., 2014, 2, 857–867 CrossRef CAS.
- H. Xu, S. Ouyang, L. Liu, P. Reunchan, N. Umezawa and J. Ye, Recent advances in TiO 2-based photocatalysis, J. Mater. Chem. A, 2014, 2, 12642–12661 RSC.
- S. Lettieri, M. Pavone, A. Fioravanti, L. Santamaria Amato and P. Maddalena, Charge carrier processes and optical properties in TiO2 and TiO2-based heterojunction photocatalysts: A review, Materials, 2021, 14, 1645 CrossRef CAS PubMed.
- M. Das, D. Das, S. Sil and P. P. Ray, Development of hierarchical copper sulfide–carbon nanotube (CuS–CNT) composites and utilization of their superior carrier mobility in efficient charge transport towards photodegradation of Rhodamine B under visible light, Nanoscale Adv., 2023, 5, 3655–3663 RSC.
- X. Xing, H. Zhu, M. Zhang, L. Hou, Q. Li and J. Yang, Interfacial oxygen vacancy layer of a Z-scheme BCN–TiO 2 heterostructure accelerating charge carrier transfer for visible light photocatalytic H 2 evolution, Catal. Sci. Technol., 2018, 8, 3629–3637 RSC.
- S. G. Kumar and L. G. Devi, Review on modified TiO2 photocatalysis under UV/visible light: selected results and related mechanisms on interfacial charge carrier transfer dynamics, J. Phys. Chem. A, 2011, 115, 13211–13241 CrossRef CAS PubMed.
- A. Kubacka, M. Fernandez-Garcia and G. Colon, Advanced nanoarchitectures for solar photocatalytic applications, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed.
- M. J. Elser, E. Neige, T. Berger, M. Chiesa, E. Giamello, K. McKenna, T. Risse and O. Diwald, On the Importance of Nanoparticle Necks and Carbon Impurities for Charge Trapping in TiO2, J. Phys. Chem. C, 2023, 127, 8778–8787 CrossRef CAS PubMed.
- S. Patil, S. Deshmukh, K. More, V. Shevale, S. Mullani, A. Dhodamani and S. Delekar, Sulfated TiO2/WO3 nanocomposite: An efficient photocatalyst for degradation of Congo red and methyl red dyes under visible light irradiation, Mater. Chem. Phys., 2019, 225, 247–255 CrossRef CAS.
- C. G. Ugwuja, O. O. Adelowo, A. Ogunlaja, M. O. Omorogie, O. D. Olukanni, O. O. Ikhimiukor, I. Iermak, G. A. Kolawole, C. Guenter and A. Taubert, Visible-light-mediated photodynamic water disinfection@ bimetallic-doped hybrid clay nanocomposites, ACS Appl. Mater. Interfaces, 2019, 11, 25483–25494 CrossRef CAS PubMed.
- B. P. D. Oliveira, K. C. Blanco and V. S. Bagnato, Contamination control in a portable-materials with photochemical process, Int. J. Chem., 2019, 11, 86–94 CrossRef.
- P. Swift, Adventitious carbon—the panacea for energy referencing?, Surf. Interface Anal., 1982, 4, 47–51 CrossRef CAS.
- T. L. Barr and S. Seal, Nature of the use of adventitious carbon as a binding energy standard, J. Vac. Sci. Technol., A, 1995, 13, 1239–1246 CrossRef CAS.
- M. C. Biesinger, Accessing the robustness of adventitious carbon for charge referencing (correction) purposes in XPS analysis: Insights from a multi-user facility data review, Appl. Surf. Sci., 2022, 597, 153681 CrossRef CAS.
- S. Akhter, X.-L. Zhou and J. White, XPS study of polymer/organometallic interaction: Trimethyl aluminum on polyvinyl alcohol polymer, Appl. Surf. Sci., 1989, 37, 201–216 CrossRef CAS.
- J. M. Burkstrand, Metal-polymer interfaces: adhesion and x-ray photoemission studies, J. Appl. Phys., 1981, 52, 4795–4800 CrossRef CAS.
- P. Stoyanov, S. Akhter and J. White, XPS study of metal/polymer interaction: evaporated aluminum on polyvinyl alcohol polymer, Surf. Interface Anal., 1990, 15, 509–515 CrossRef CAS.
- M. Nielsen, A. Hafreager, R. Y. Brogaard, K. De Wispelaere, H. Falsig, P. Beato, V. Van Speybroeck and S. Svelle, Collective action of water molecules in zeolite dealumination, Catal. Sci. Technol., 2019, 9, 3721–3725 RSC.
- Z. Xu, S. Gu, Z. Sun, D. Zhang, Y. Zhou, Y. Gao, R. Qi and W. Chen, Synthesis of char-based adsorbents from cotton textile waste assisted by iron salts at low pyrolysis temperature for Cr (VI) removal, Environ. Sci. Pollut. Res., 2020, 27, 11012–11025 CrossRef CAS PubMed.
- J. F. Watts and J. Wolstenholme, An introduction to surface analysis by XPS and AES, John Wiley & Sons, 2019 Search PubMed.
- Z. Hua, Z. Dai, X. Bai, Z. Ye, H. Gu and X. Huang, A facile one-step electrochemical strategy of doping iron, nitrogen, and fluorine into titania nanotube arrays with enhanced visible light photoactivity, J. Hazard. Mater., 2015, 293, 112–121 CrossRef CAS PubMed.
- J. G. Fierro, L. Arrua, J. L. Nieto and G. Kremenic, Surface properties of Co-precipitated VTiO catalysts and their relation to the selectiveoxidation of isobutene, Appl. Catal., 1988, 37, 323–338 CrossRef CAS.
- S. Watanabe, X. Ma and C. Song, Characterization of structural and surface properties of nanocrystalline TiO2− CeO2 mixed oxides by XRD, XPS, TPR, and TPD, J. Phys. Chem. C, 2009, 113, 14249–14257 CrossRef CAS.
- L. Yang, Y. Peng, Y. Yang, J. Liu, Z. Li, Y. Ma, Z. Zhang, Y. Wei, S. Li and Z. Huang, Green and sensitive flexible semiconductor SERS substrates: Hydrogenated black TiO2 nanowires, ACS Appl. Nano Mater., 2018, 1, 4516–4527 CrossRef CAS.
- R. P. Netterfield, P. Martin, C. Pacey, W. Sainty, D. McKenzie and G. Auchterlonie, Ion-assisted deposition of mixed TiO2-SiO2 films, J. Appl. Phys., 1989, 66, 1805–1809 CrossRef CAS.
- M. A. Stranick, M. Houalla and D. M. Hercules, Spectroscopic characterization of TiO2Al2O3 and CoAl203·TiO2 catalysts, J. Catal., 1987, 106, 362–368 CrossRef CAS.
- X. Li, K. Peng, H. Chen and Z. Wang, TiO2 nanoparticles assembled on kaolinites with different morphologies for efficient photocatalytic performance, Sci. Rep., 2018, 8, 11663 CrossRef PubMed.
- R. Bertoncello, A. Glisenti, G. Granozzi, G. Battaglin, F. Caccavale, E. Cattaruzza and P. Mazzoldi, Chemical interactions in titanium-and tungsten-implanted fused silica, J. Non-Cryst. Solids, 1993, 162, 205–216 CrossRef CAS.
- M. Sundararajan, R. Rejith, R. Renjith, A. P. Mohamed, G. Gayathri, A. Resmi, K. Jinesh and V. Loveson, Raman-XPS spectroscopic investigation of heavy mineral sands along Indian coast, Geo-Mar. Lett., 2021, 41, 22 CrossRef CAS.
- M. Kuznetsov, J. F. Zhuravlev, V. Zhilyaev and V. Gubanov, XPS study of the nitrides, oxides and oxynitrides of titanium, J. Electron Spectrosc. Relat. Phenom., 1992, 58, 1–9 CrossRef CAS.
- A. Hughes, M. Hedges and B. Sexton, Reactions at the Al/SiO 2/SiC layered interface, J. Mater. Sci., 1990, 25, 4856–4865 CrossRef CAS.
- P. Anderson and W. Swartz, X-ray photoelectron spectroscopy of some aluminosilicates, Inorg. Chem., 1974, 13, 2293–2294 CrossRef CAS.
- F. Rueda, J. Mendialdua, A. Rodriguez, R. Casanova, Y. Barbaux, L. Gengembre and L. Jalowiecki, Characterization of Venezuelan laterites by X-ray photoelectron spectroscopy, J. Electron Spectrosc. Relat. Phenom., 1996, 82, 135–143 CrossRef CAS.
- S. Patnaik, S. Martha, S. Acharya and K. Parida, An overview of the modification of gC 3 N 4 with high carbon containing materials for photocatalytic applications, Inorg. Chem. Front., 2016, 3, 336–347 RSC.
- R. Leary and A. Westwood, Carbonaceous nanomaterials for the enhancement of TiO2 photocatalysis, Carbon, 2011, 49, 741–772 CrossRef CAS.
- A. Meng, L. Zhang, B. Cheng and J. Yu, Dual cocatalysts in TiO2 photocatalysis, Adv. Mater., 2019, 31, 1807660 CrossRef PubMed.
- Y. M. Hunge, A. Yadav, S.-W. Kang and H. Kim, Photocatalytic degradation of tetracycline antibiotics using hydrothermally synthesized two-dimensional molybdenum disulfide/titanium dioxide composites, J. Colloid Interface Sci., 2022, 606, 454–463 CrossRef CAS PubMed.
- H. Asadzadeh Patehkhor, M. Fattahi and M. Khosravi-Nikou, Synthesis and characterization of ternary chitosan–TiO2–ZnO over graphene for photocatalytic degradation of tetracycline from pharmaceutical wastewater, Sci. Rep., 2021, 11, 24177 CrossRef CAS PubMed.
- M. Ahmadi, H. R. Motlagh, N. Jaafarzadeh, A. Mostoufi, R. Saeedi, G. Barzegar and S. Jorfi, Enhanced photocatalytic degradation of tetracycline and real pharmaceutical wastewater using MWCNT/TiO2 nano-composite, J. Environ. Manage., 2017, 186, 55–63 CrossRef CAS PubMed.
- R. Mohammadi, B. Massoumi and M. Rabani, Photocatalytic decomposition of amoxicillin trihydrate antibiotic in aqueous solutions under UV irradiation using Sn/TiO 2 nanoparticles, Int. J. Photoenergy, 2012, 2012, 514856 CrossRef.
- G. Safari, M. Hoseini, M. Seyedsalehi, H. Kamani, J. Jaafari and A. Mahvi, Photocatalytic degradation of tetracycline using nanosized titanium dioxide in aqueous solution, Int. J. Environ. Sci. Technol., 2015, 12, 603–616 CrossRef CAS.
- T. Zhang, Y. Liu, Y. Rao, X. Li, D. Yuan, S. Tang and Q. Zhao, Enhanced photocatalytic activity of TiO2 with acetylene black and persulfate for degradation of tetracycline hydrochloride under visible light, Chem. Eng. J., 2020, 384, 123350 CrossRef CAS.
- Z. Wang, Y. Muhammad, R. Tang, C. Lu, S. Yu, R. Song, Z. Tong, B. Han and H. Zhang, Dually organic modified bentonite with enhanced adsorption and desorption of tetracycline and ciprofloxacine, Sep. Purif. Technol., 2021, 274, 119059 CrossRef CAS.
- P. Wang, P.-S. Yap and T.-T. Lim, C–N–S tridoped TiO2 for photocatalytic degradation of tetracycline under visible-light irradiation, Appl. Catal., A, 2011, 399, 252–261 CrossRef CAS.
- S. Jiao, S. Zheng, D. Yin, L. Wang and L. Chen, Aqueous photolysis of tetracycline and toxicity of photolytic products to luminescent bacteria, Chemosphere, 2008, 73, 377–382 CrossRef CAS PubMed.
- Y. Feng, P.-H. Lee, D. Wu and K. Shih, Rapid selective circumneutral degradation of phenolic pollutants using peroxymonosulfate–iodide metal-free oxidation: role of iodine atoms, Environ. Sci. Technol., 2017, 51, 2312–2320 CrossRef CAS PubMed.
- W. Ma, N. Wang, Y. Fan, T. Tong, X. Han and Y. Du, Non-radical-dominated catalytic degradation of bisphenol A by ZIF-67 derived nitrogen-doped carbon nanotubes frameworks in the presence of peroxymonosulfate, Chem. Eng. J., 2018, 336, 721–731 CrossRef CAS.
- A. A. Bayode, E. M. Vieira, R. Moodley, S. Akpotu, A. S. de Camargo, D. Fatta-Kassinos and E. I. Unuabonah, Tuning ZnO/GO pn heterostructure with carbon interlayer supported on clay for visible-light catalysis: Removal of steroid estrogens from water, Chem. Eng. J., 2021, 420, 127668 CrossRef CAS.
- F. Su, P. Li, J. Huang, M. Gu, Z. Liu and Y. Xu, Photocatalytic degradation of organic dye and tetracycline by ternary Ag2O/AgBr–CeO2 photocatalyst under visible-light irradiation, Sci. Rep., 2021, 11, 85 CrossRef CAS PubMed.
- M. Samy, M. G. Ibrahim, M. Fujii, K. E. Diab, M. ElKady and M. G. Alalm, CNTs/MOF-808 painted plates for extended treatment of pharmaceutical and agrochemical wastewaters in a novel photocatalytic reactor, Chem. Eng. J., 2021, 406, 127152 CrossRef CAS.
- A. Sharara, M. Samy, M. Mossad and M. Gar Alalm, Photodegradation of polyethylene debris in water by sulfur-doped TiO2: system optimization, degradation mechanism, and reusability, Environ. Sci. Pollut. Res., 2023, 1–13 Search PubMed.
- J. Rashid, M. Barakat, R. Mohamed and I. Ibrahim, Enhancement of photocatalytic activity of zinc/cobalt spinel oxides by doping with ZrO2 for visible light photocatalytic degradation of 2-chlorophenol in wastewater, J. Photochem. Photobiol., A, 2014, 284, 1–7 CrossRef CAS.
- Y. Chen, X. Yuan, L. Jiang, Y. Zhao, H. Chen, Z. Shangguan, C. Qin and H. Wang, Insights into periodate oxidation of antibiotics mediated by visible-light-induced polymeric carbon nitride: Performance and mechanism, Chem. Eng. J., 2023, 457, 141147 CrossRef CAS.
- Z. Ma, C. Zeng, L. Hu, Q. Zhao, Q. Yang, J. Niu, B. Yao and Y. He, A high-performance photocatalyst of ZnTCPP sensitized porous graphitic carbon nitride for antibiotic degradation under visible light irradiation, Appl. Surf. Sci., 2019, 484, 489–500 CrossRef CAS.
- H. Kaur, S. Singh and B. Pal, Effect of plasmonic metal (Cu, Ag, and Au) loading over the physicochemical and photocatalytic properties of Mg-Al LDH towards degradation of tetracycline under LED light, Appl. Surf. Sci., 2023, 609, 155455 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ew00037d |
| This journal is © The Royal Society of Chemistry 2024 |