
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactions of hypobromous acid with dimethyl selenide, dimethyl diselenide and other organic selenium compounds: kinetics and product formation†
Emanuel
Müller
 ab,
Urs
von Gunten
acb,
Julie
Tolu
ab,
Sylvain
Bouchet
ab and
Lenny H. E.
Winkel
*ab
ab,
Urs
von Gunten
acb,
Julie
Tolu
ab,
Sylvain
Bouchet
ab and
Lenny H. E.
Winkel
*ab
aSwiss Federal Institute of Aquatic Science and Technology, Department of Water Resources and Drinking Water (W+T), Eawag, Ueberlandstrasse 133, CH-8600 Duebendorf, Switzerland. E-mail: lenny.winkel@eawag.ch; Tel: +41 58 765 5601
bSwiss Federal Institute of Technology, Institute of Biogeochemistry and Pollutant Dynamics (IBP), Department of Environment Systems (D-USYS), ETH Zurich, Universitätsstrasse 16, 8092 Zürich, Switzerland
cSchool of Architecture, Civil and Environmental Engineering (ENAC), École Polytechnique Fédérale de Lausanne (EPFL), 1015, Lausanne, Switzerland
First published on 8th January 2024
Abstract
Selenium (Se) is an essential micronutrient for many living organisms particularly due to its unique redox properties. We recently found that the sulfur (S) analog for dimethyl selenide (DMSe), i.e. dimethyl sulfide (DMS), reacts fast with the marine oxidant hypobromous acid (HOBr) which likely serves as a sink of marine DMS. Here we investigated the reactivity of HOBr with dimethyl selenide and dimethyl diselenide (DMDSe), which are the main volatile Se compounds biogenically produced in marine waters. In addition, the reactivity of HOBr with further organic Se compounds was tested, i.e., SeMet (as N-acetylated-SeMet), and selenocystine (SeCys2 as N-acetylated-SeCys2), as well as the phenyl-analogs of DMSe and DMDSe, respectively, diphenyl selenide (DPSe) and diphenyl diselenide (DPDSe). Apparent second-order rate constants at pH 8 for the reactions of HOBr with the studied Se compounds were (7.1 ± 0.7) × 107 M−1 s−1 for DMSe, (4.3 ± 0.4) × 107 M−1 s−1 for DMDSe, (2.8 ± 0.3) × 108 M−1 s−1 for SeMet, (3.8 ± 0.2) × 107 M−1 s−1 for SeCys2, (3.5 ± 0.1) × 107 M−1 s−1 for DPSe, and (8.0 ± 0.4) × 106 M−1 s−1 for DPDSe, indicating a very high reactivity of all selected Se compounds with HOBr. The reactivity between HOBr and DMSe is lower than for DMS and therefore this reaction is likely not relevant for marine DMSe abatement. However, the high reactivity of SeMet with HOBr suggests that SeMet may act as a relevant quencher of HOBr.
Water impactSelenium is an essential element in low doses but toxic at higher levels. It can be found in natural and engineered aquatic systems in organic forms and potentially react with the important oxidant hypobromous acid (HOBr). Our study, focusing on seawater, shows that selected organic selenium compounds indeed react fast with HOBr and may thus act as relevant quenchers for reactive bromine in seawater and other aquatic systems. |
Introduction
Selenium (Se) is an essential micronutrient for many living organisms, including mammals, bacteria and archaea, and some species of microalgae, while the essentiality of Se for plants is still controversial.1–4 Multiple biological functions and beneficial effects on organisms' health are attributed to Se, based on the redox activity of Se compounds. Undersupply of Se has been associated with various human health issues, including cardio-myopathy (Keshan disease), cancer, immune and endocrine disfunction, muscle and bone disorder, neurodegenerative diseases, decreased thyroid function and male infertility.5,6 Where in many areas bedrock is low in Se, the atmosphere is an important source of Se to terrestrial food systems, mainly in the form of wet deposition.7–10 Marine environments play an important role in the atmospheric Se budget via biogenic emissions of the volatile organic compounds dimethyl selenide (DMSe) and dimethyl diselenide (DMDSe).9,11 In marine waters, DMSe was detected in concentrations ranging from 0.06 to 4.73 pM11,12 and its production was reported for macroalgae,13 marine bacteria14,15 and has been suggested for marine microalgae such as coccolithophorids (Table S1, ESI†).11,12,16–18 For DMDSe, which has been reported in concentrations between 0.02–0.26 pM Se in marine waters,11,12 the production pathways are largely unknown, although, various biotic/abiotic pathways have been suggested as well as different precursors (selenols, selenocysteine (SeCys), selenocystine (SeCys2) and selenomethionine (SeMet)).19–22Recently, we reported a fast reaction (second-order rate constant: k = 1.6 × 109 M−1 s−1) between the sulfur analog of DMSe, i.e., dimethyl sulfide (DMS) and the marine oxidant hypobromous acid (HOBr), produced via the oxidation of bromide (Br−) by H2O2, catalyzed by the enzyme vanadium-bromoperoxidase (V-BrPO).23 Therefore, we concluded that this reaction is likely an important sink for marine DMS.24,25 Compared to organic sulfur, redox reactions for the analogous organic Se compounds generally have been reported to be faster, with 1–2 orders of magnitude higher second-order rate constants than for their analogous S compounds.26 Therefore, organic Se compounds may be effective oxidant scavengers, although, previous studies reported diselenides to be less efficient in scavenging hydrogen peroxide (H2O2) and singlet oxygen (1O2) than selenols and selenides.27 Furthermore, the Se amino acids SeCys (also referred as the 21st amino acid) and SeMet, are quickly oxidized to the diselenide “selenocystine (SeCys2)” and the selenoxide “Selenomethionine-oxide”, respectively, but can also be rapidly reduced back to SeCys and SeMet.26,28–32 In contrast to the higher reactivity of organic Se than organic S, it has been demonstrated that the inorganic Se compound selenite has an almost 6 order of magnitude lower second-order rate constant for the reaction with HOBr than sulfite, the inorganic S analog.33 The reason for this difference is currently unknown.
The main objective of this study was to determine apparent second-order rate constants for the reactions between HOBr and DMSe, and HOBr and DMDSe. Furthermore, the reactivities of HOBr with SeMet (as N-acetylated-SeMet), and SeCys2 (as N-acetylated-SeCys2) were investigated. These compounds are likely precursors of volatile organic Se in marine waters and organisms. We also investigated the kinetics of the reactions of HOBr with the aryl-analogs of DMSe and DMDSe, i.e., diphenyl selenide (DPSe) and diphenyl diselenide (DPDSe), which are industrially produced organic Se compounds used in chemical synthesis and as green catalysts for medical and pharmacological purposes.34–36 These aromatic analogs of the aliphatic compounds DMSe and DMDSe, respectively, were selected to obtain further insights into the substitution effects of organo-Se compounds on the reactivity with HOBr. Furthermore, Se-containing oxidation products from the above reactions for different molar HOBr![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Se compound ratios were identified and if possible quantified using high-resolution mass spectrometry (HR-MS) and liquid chromatography coupled to inductively coupled plasma mass spectrometry (LC-ICP-MS/MS). Finally, the reactivity of HOBr with the selected organic Se compounds was compared to the analogous S compounds.
:Se compound ratios were identified and if possible quantified using high-resolution mass spectrometry (HR-MS) and liquid chromatography coupled to inductively coupled plasma mass spectrometry (LC-ICP-MS/MS). Finally, the reactivity of HOBr with the selected organic Se compounds was compared to the analogous S compounds.
Materials and methods
Chemicals and reagents
For treatment of glassware and handling of chemicals see Text S2, ESI.† A list with information of all used chemicals is provided in Table S2, ESI.†Se compounds. Stock solutions of dimethyl selenide (DMSe), dimethyl diselenide (DMDSe) and diphenyl selenide (DPSe) were produced in gastight 10 mL headspace amber crimp vials (ND20, 46 × 22.5 mm, BGB Analytics, Boeckten, Switzerland) by diluting the pure compound in methanol (0.25%; corresponding to ca. 25 mM of the Se compound concentration). Stock solutions of diphenyl diselenide (DPDSe), seleno-DL-methionine (SeMet) and seleno-L-cystine (SeCys2) were produced by weighing the required quantity of the solid material in a 10 mL headspace amber crimp vial and dissolving it either in ethanol or water (6 mM DPDSe in ethanol, 6 mM SeMet in water and 0.6 mM SeCys2 in water). Solutions of the volatile substances DMSe and DMDSe were produced daily, whereas the other Se stock solutions were produced monthly.
Acetylation of SeMet and SeCys2 amino groups. Di-tert-butyl dicarbonate (BOC2O) and sodium hydrogen carbonate (NaHCO3) were used to acetylate the amino groups of SeMet and SeCys2 to block them against reactions with HOBr (method described in Text S3, ESI†). The stability of the BOC2O-protection group for N-acetylated-SeMet and N-acetylated-SeCys2 was tested by chloramine formation during chlorination (see Table S2 and Text S4, ESI†).
HOBr. Stock solutions of HOBr were produced by mixing sodium hypochlorite (NaOCl) and potassium bromide (KBr), as described previously.24
Competitors for competition kinetics. Resorcinol and 1,3,5-trimethoxybenzene (TMB) were used as competitors for competition kinetics. A 250 mM resorcinol stock solution was produced in ultrapure water and further diluted to working concentrations of 2.5 mM and 250 μM. A saturated solution of TMB was produced in ultrapure water and its effective concentration was determined by a methanolic stock solution of known concentration (dissolved TMB concentration: 2.5 mM in water). Both resorcinol and TMB stock solutions were stored in 10 mL headspace amber crimp vials at 4 °C.
Potassium perchlorate (KClO4) was used to maintain constant ionic strength of the reaction solution. Perchlorate concentrations of 100 and 50 mM were used depending on the phosphate buffer concentration and solution pH.
Standard solutions of Se compounds and competitors were prepared freshly every day for the following calibration ranges: DMSe = 0–800 nM, DMDSe = 0–125 nM, DPSe, Res, TMB = 1.67–16.7 μM, DPDSe = 1–10 μM.
Kinetic experiments and determination of second-order rate constants
:1 ratio, targeting doses of 0, 2.5, 5, 7.5, 10, 12.5, 15 μM HOBr for experiments with DMSe, DPSe and N-acetylated-SeMet; 0, 5, 10, 15, 20, 25 μM HOBr for experiments with DMDSe and N-acetylated-SeCys2; and 0, 2.5, 3.75, 5, 6.25, 7.5 μM HOBr for experiments with DPDSe. To guarantee fast and complete HOBr consumption, i.e., within 1 min under magnetic stirring, all HOBr doses were lower than the sum of the concentrations of target compound and the competitor (see also below, determination of second-order rate constants). For the experiments with diselenides, the higher HOBr doses exceeded the concentration of the Se-compound. This is required because 3 moles of HOBr are consumed per mole of diselenide (see section: “Pathways for reactions between HOBr and the studied diselenides”). After 1 min reaction time, reaction solutions were diluted 1:100 to 10 mL amber crimp vials for DMSe and DMDSe quantification and transferred to HPLC vials for the quantification of DPSe, DPDSe, N-acetylated-SeMet, N-acetylated-SeCys2, resorcinol and TMB. DMSe, DMDSe, DPSe and DPDSe were immediately measured after experiments due to their volatility (DMSe and DMDSe) or observed instability in diluted aqueous solutions (DPSe and DPDSe). N-Acetylated-SeMet, N-acetylated-SeCys2 and resorcinol from experiments with DMSe and DMDSe were measured within one week and stored beforehand at 4 °C. The pH of reaction solutions was immediately measured after each experiment and did not deviate more than 0.05 units from the initial pH.
| Compound name | Compound structure | Molecular mass | k app,pH 8,HOBr+Se [M−1 s−1] | Reactivity ratio (kapp,pH 8,HOBr+Se:kapp,pH 8,HOBr+competitor) |
k app,pH 8,HOBr+S [M−1 s−1] |
|---|---|---|---|---|---|
| Experiments in buffered ultrapure water | |||||
| Dimethyl selenide (DMSe) |
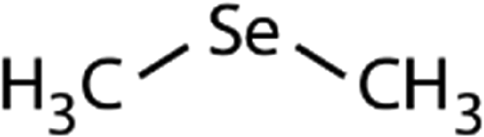
|
109 | (7.1 ± 0.7) × 107 | 3.9 | (1.1 ± 0.2) × 109a |
| Dimethyl-diselenide (DMDSe) |
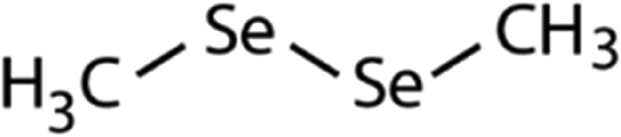
|
188 | (4.3 ± 0.4) × 107 | 2.3 | Not known |
| Diphenyl selenide (DPSe) |
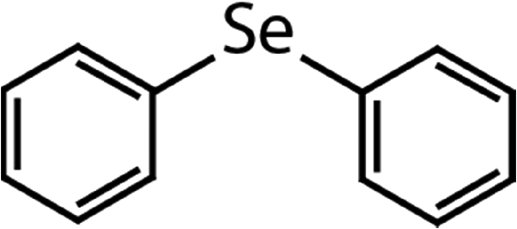
|
233 | (3.5 ± 0.1) × 107 | 1.9 | Not known |
| Diphenyl diselenide (DPDSe) |
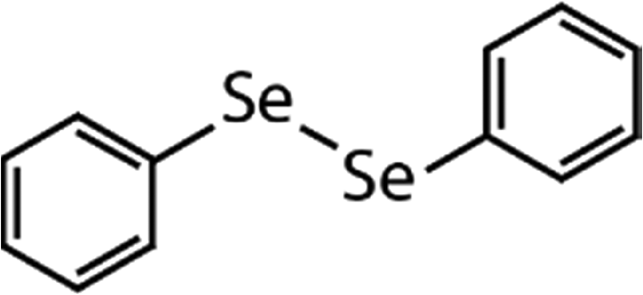
|
313 | (8.0 ± 0.4) × 106 | 2.8 | Not known |
| N-Acetylated-selenomethionine (N-acetylated-SeMet) |
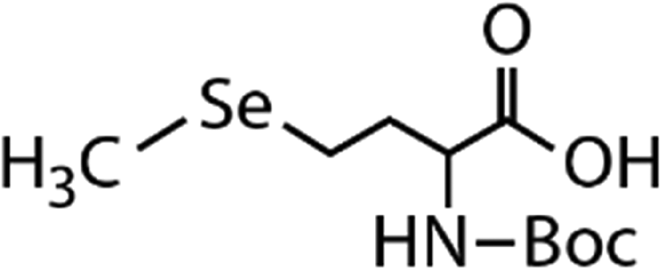
|
413.4 | (2.8 ± 0.3) × 108c | 15.3c | (3.6 ± 0.3) × 106b |
| N-Acetylated-selenocystine (N-acetylated-SeCys2) |
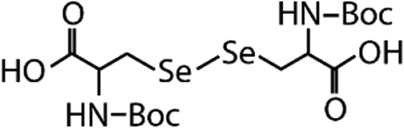
|
768.7 | (3.8 ± 0.2) × 107 | 2.1 | (3.4 ± 0.8) × 105b |
| Seawater experiments | |||||
| Dimethyl selenide (DMSe) |
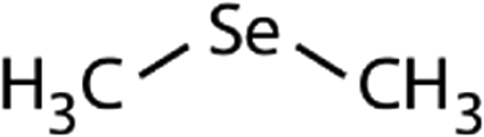
|
109 | (4.7 ± 0.5) × 108c | 26.0 | (1.2 ± 0.2) × 109a |
| Dimethyl-diselenide (DMDSe) |
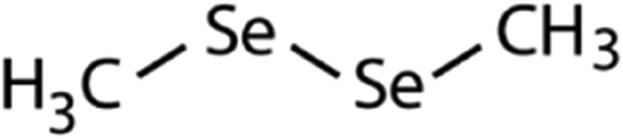
|
188 | (4.5 ± 0.2) × 107 | 2.5 | Not known |
| Second-order rate constants for the competitors | |||||
|---|---|---|---|---|---|
| Compound name | Compound structure | Molecular mass | k HOBr+X [M−1 s−1] | Reactivity ratio (kapp,pH 8,HOBr+Se:kapp,pH 8,HOBr+competitor) |
k app,pH 8,HOBr+X [M−1 s−1] |
| a Ref. 24. b Ref. 55, pH 7.2–7.5, T = 22 °C. c Since the reactivity ratio between kapp,pH 8,HOBr+N-acetylated-SeMet and kapp,pH 8,HOBr+resorcinol is 15, the reaction conditions are suboptimal and the determined second-order rate constant of the reaction between HOBr and N-acetylated-SeMet is associated with some uncertainty. Likewise, the determined rate constant for the reaction between DMSe and HOBr in artificial seawater medium is subject to uncertainty since the reactivity ratio between kapp,pH 8,HOBr+DMSe and kapp,pH 8,HOBr+resorcinol is 26. d Ref. 59. e See Table S4.† | |||||
| 1,3,5-Trimethoxy-benzene (TMB) |
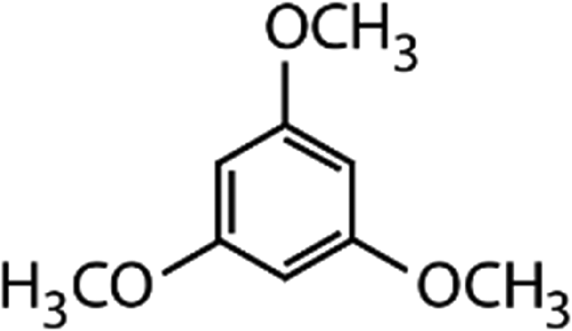
|
168.2 | 3.4 × 106d | — | 2.9 × 106 |
| Resorcinol |
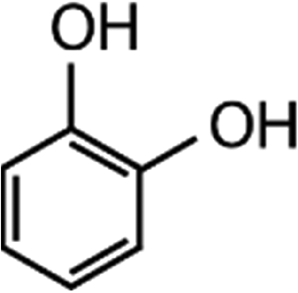
|
110.1 | 6.56 × 106e | — | 1.8 × 107 |
Identification and (semi)quantification of Se-containing oxidation products
To investigate the products of the reactions of HOBr with selected organic Se compounds, batch experiments were performed in 10 mL amber gastight headspace vials using a 1 mM NaHCO3-buffer solution (pH 8) and variable molar HOBr:Se compound ratios (i.e., 0:1 [Se compound blank], 1:1, 3:1 and 10:1). The initial concentration of the Se compounds was 6.25 μM. After 1 min of reaction time under magnetic stirring (t1/2 ≈ 0.01 seconds), the solutions were mixed with methanol containing 0.1% formic acid (volumetric 1:1 ratio) and analyzed by high resolution mass spectrometry (HR-MS) within the next 10 hours. The HR-MS system (QExactive HF Orbitrap™, Thermo Scientific, Switzerland) was equipped with an electrospray ionization source (ESI) and was calibrated before analyses (Pierce™ ESI solutions). All analyses were performed as direct infusion in the positive ionization mode. Selenium compounds in the recorded mass spectra were detected based on the specific Se isotopic pattern. The oxidation products were then identified using the software “Freestyle” (Thermo Scientific) and the Eawag tool “enviPath”.38 Once identified, a semi-quantification of the Se-containing oxidation products was carried out by recording single ion mode (SIM) spectra at their nominal mass for each condition, and the intensity was retrieved using the Freestyle software. Further details about HR-MS analyses are provided in the Text S9, ESI.† To quantify potential formation of inorganic Se (SeO32− [IV] and SeO42− [VI]), reaction products were additionally analyzed by liquid chromatography coupled to inductively coupled plasma tandem mass spectrometry (LC-ICP-MS/MS). The system consisted of an Agilent LC 1200 series coupled to an Agilent 8800 ICP-MS/MS. The chromatographic separation was adapted from a previous study,39i.e., using an anion exchange column (Hamilton PRP-X100, 100 × 4.1 mm, 10 μm, with the appropriate guard column) and a gradient elution of ammonium citrate (4 to 10 mM; pH 5.2). The mobile phase was delivered at 1.25 mL min−1 and the injection volume was 25 μL.
Results and discussion
Reactivities of the selected organic selenium compounds with HOBr in buffered ultrapure water and artificial seawater
The apparent second-order rate constants (pH 8) for the reactions between the selected organic Se compounds and HOBr are provided in Table 1 and are in the range of (8.0 ± 0.4) × 106 and (2.8 ± 0.3) × 108 M−1 s−1, indicating a very high reactivity of all selected Se compounds. Monoselenides had significantly higher reactivities than the analogous diselenides with apparent second-order rate constants for DMSe, DPSe and N-acetylated-SeMet, respectively, 1.7, 4.4 and 7.4 times higher than for their diselenide analogs, i.e., DMDSe, DPDSe, and N-acetylated-SeCys2 (Table 1). The relatively higher reactivity of monoselenides compared to diselenides can be explained by the lower oxidation state of Se in monoselenides (oxidation state of −II) as compared to diselenides (oxidation state of −I). Therefore, the electron density in Se in monoselenides is higher than for the corresponding diselenides, which makes monoselenides better nucleophiles. Apart from the effect of the Se oxidation state, we observed that Se compounds with alkyl substituents had a higher reactivity than those with aryl substituents. A reactivity enhancement by a factor of 2.1 and 5.3 was observed, respectively, for DMSe versus DPSe and DMDSe versus DPDSe (Table 1). Alkyl substituents have a positive inductive effect (i.e., electron donating effect, +I effect), while aryl substituents have a negative resonance and a negative inductive effect (i.e., electron withdrawing effect, −I effect).40 Therefore, alkyl substituents make the Se-atom more nucleophilic than aryl substituents, which is in line with the higher reactivities of alkylated compared to arylated Se species with the electrophilic HOBr.A significantly higher reactivity (almost 1 order of magnitude) was observed for the DMSe–HOBr reaction in buffered artificial seawater (kDMSe+HOBr,seaw. = (4.7 ± 0.5) × 108 M−1 s−1) than in ultrapure water (kDMSe+HOBr = (7.1 ± 0.7) × 107 M−1 s−1). It was tested if the ionic strength of seawater could explain the higher reactivity. Experiments were carried out in a perchlorate medium with the same ionic strength as the artificial seawater (but in absence of bromide (Br−) and chloride, to avoid alteration of the bromine speciation) (Table S6 and Fig. S5†).41 Based on these experiments it can be concluded that ionic strength effects as well as a higher Br− concentration contribute to the higher reactivity (Text S10, ESI†). In contrast, no change in reactivity was observed for the reaction between DMDSe and HOBr when comparing ultrapure water with artificial seawater (Table 1). We currently do not have an explanation for the different behavior between DMSe and DMDSe in these tests. However, given the suboptimal conditions for the measurement of second-order rate constant for the reaction of HOBr with DMSe (large difference between target compound and competitor), this value should be considered with caution.
Reactivity of SeMet with HOBr compared to other studies and oxidants
For the alkylated monoselenide compound N-acetylated-SeMet, an apparent second-order rate constant for the reaction with HOBr on the order of 3 × 108 M−1 s−1 was obtained, which is higher than for DMSe. Because both compounds are selenoethers with two methyl groups at the Se-atom, this indicates that the Se atom in SeMet is the preferred site of HOBr attack at pH 8 and that the amino group only plays a minor role (kapp,pH 8,R-NH2+HOBr ≈ 106 M−1 s−1).41–43 The apparent second-order rate constant determined here for the reaction between SeMet and HOBr is in the same order of magnitude as the values reported for the reactions between SeMet and SeCys with hypochlorous acid (HOCl) at pH 7.4, i.e., (3.2 × 108 M−1 s−1 and 8.5 ± 0.4 × 108 M−1 s−1, respectively),27,44 however, it is 20 times higher than a previous value (1.4 × 107 M−1 s−1) determined for the reaction between SeMet and HOBr.44 That value is also derived by competition kinetics (at a pH of 7.4), using N-acetyl-tyrosin (N-ac-Tyr) as a competitor. However, N-ac-Tyr has a much lower HOBr reactivity (kHOBr+N-ac-Tyr = 2.6 × 105 M−1 s−1) than SeMet, which is not ideal in experiments using competition kinetics (the ratio of the second-order rate constants is 50). In this study, resorcinol was applied, for which the reactivity is closer to HOBr–N-acetyl-SeMet reaction at the studied pH value (kHOBr+resorcinol = 1.8 × 107 M−1 s−1; pH 8, ratio of second-order rate constants is 15), and therefore the second-order rate constant determined in the current study is most likely closer to the expected value. Nevertheless, further experiments would be necessary to elucidate this discrepancy. To exclude a contribution of BOC2O to the observed reactivity (note that BOC2O was added in a 10-times molar excess relative to Se), we performed additional tests (Text S11, ESI†), indicating that the apparent second-order rate constant for the reaction of HOBr with BOC2O is about 10 M−1 s−1. Based on this observation, a contribution of the derivatization agent to the overall kinetics can be excluded. Further studies have investigated the reactions of organic selenium species with other oxidants than HOBr,27i.e., with hypothiocyanous acid,44 chloramine,45 peroxynitrous acid46 and singlet oxygen.27 From these studied oxidants, the reactivity between HOBr and SeMet determined in the current study was the highest.Se-containing products from the reactions between organic selenium compounds and HOBr
:DMSe ratio of 1:1 (data not shown). The HR-MS signal intensity for DMSeO indicated that its formation is complete at a molar HOBr:DMSe ratio of 1:1 and that even for HOBr:DMSe ratios up to 10:1, DMSeO is not further oxidized (Fig. 1A). Also, LC-ICP-MS/MS and LC-HRMS analyses indicated a single Se species for the experiment with a molar HOBr:DMSe ratio of 10:1 (Fig. S8–S10, ESI†) and neither selenite (Se[+IV]) nor selenate (Se[+VI]) were detected. Our results are consistent with previous studies using other oxidants (hydrogen peroxide [H2O2], ozone [O3] or hypochlorous acid [HOCl]), reporting DMSeO as main oxidation product.35,47,48 Higher oxidized products (i.e., selenones such as dimethyl selenone [DMSeO2]) were only reported in organic solvents with oxidants such as HOCl, permanganate (MnO4−), and others.47,49
| Se-containing target compound | Se-containing oxidation products | ||
|---|---|---|---|
| Name | Chemical formula | Chemical structure | |
| DMSe | Dimethyl selenoxide (DMSeO) | (CH3)2SeO |
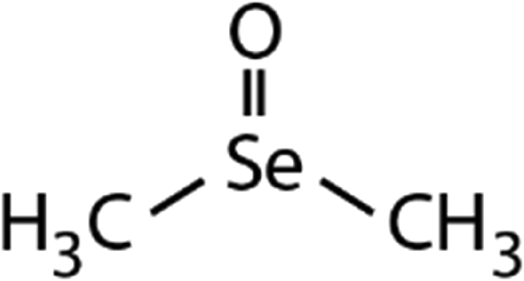
|
| DPSe | Diphenyl selenoxide (DPSeO) | (C6H5)2SeO |
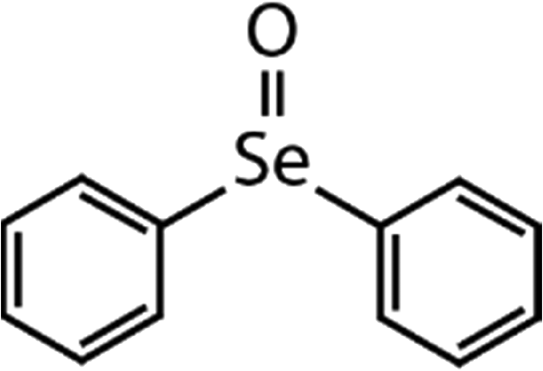
|
| N-Acetylated-SeMet | N-Acetylated-selenomethionine-oxide (N-acetylated-SeMetO) | C10H19O4NSeO |
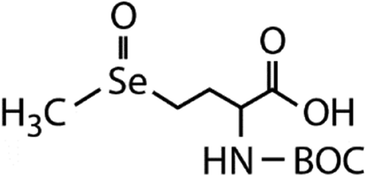
|
| DMDSe | Methane seleninic acid (MSeIA) | CH3SeO2H |
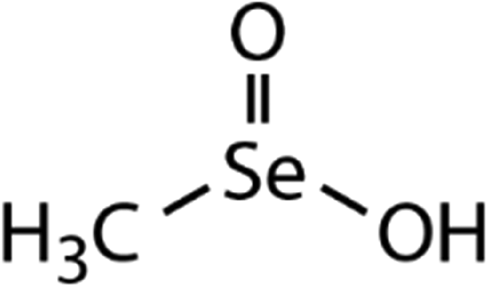
|
| Methane selenonic acid (MSeA) | CH3SeO3H |
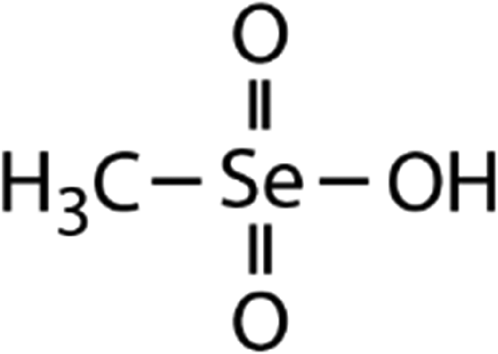
|
|
| DPDSe | Phenyl seleninic acid (PhSeIA) | (C6H5)SeO2H |
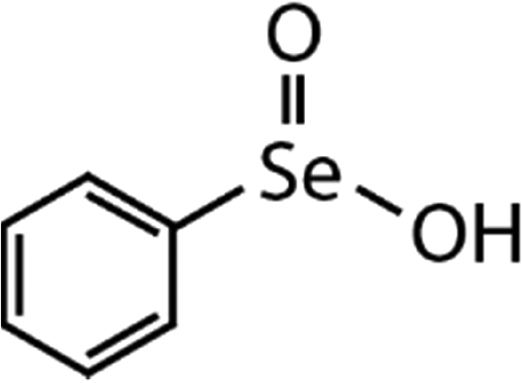
|
| Methyl phenyl selenone (MPSeO2) | C7H8SeO2 |
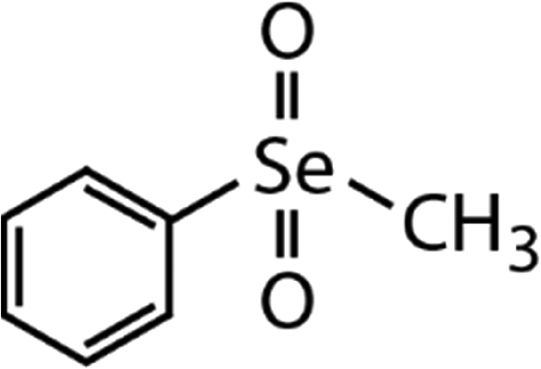
|
|
| N-Acetylated-SeCys2 | N-Acetylated-selenocysteine seleninic acid (N-acetylated-SeCysO2H) | C8H14O4NSeO2H |
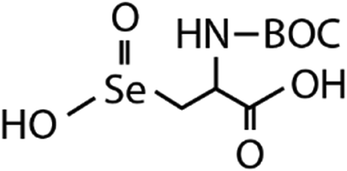
|
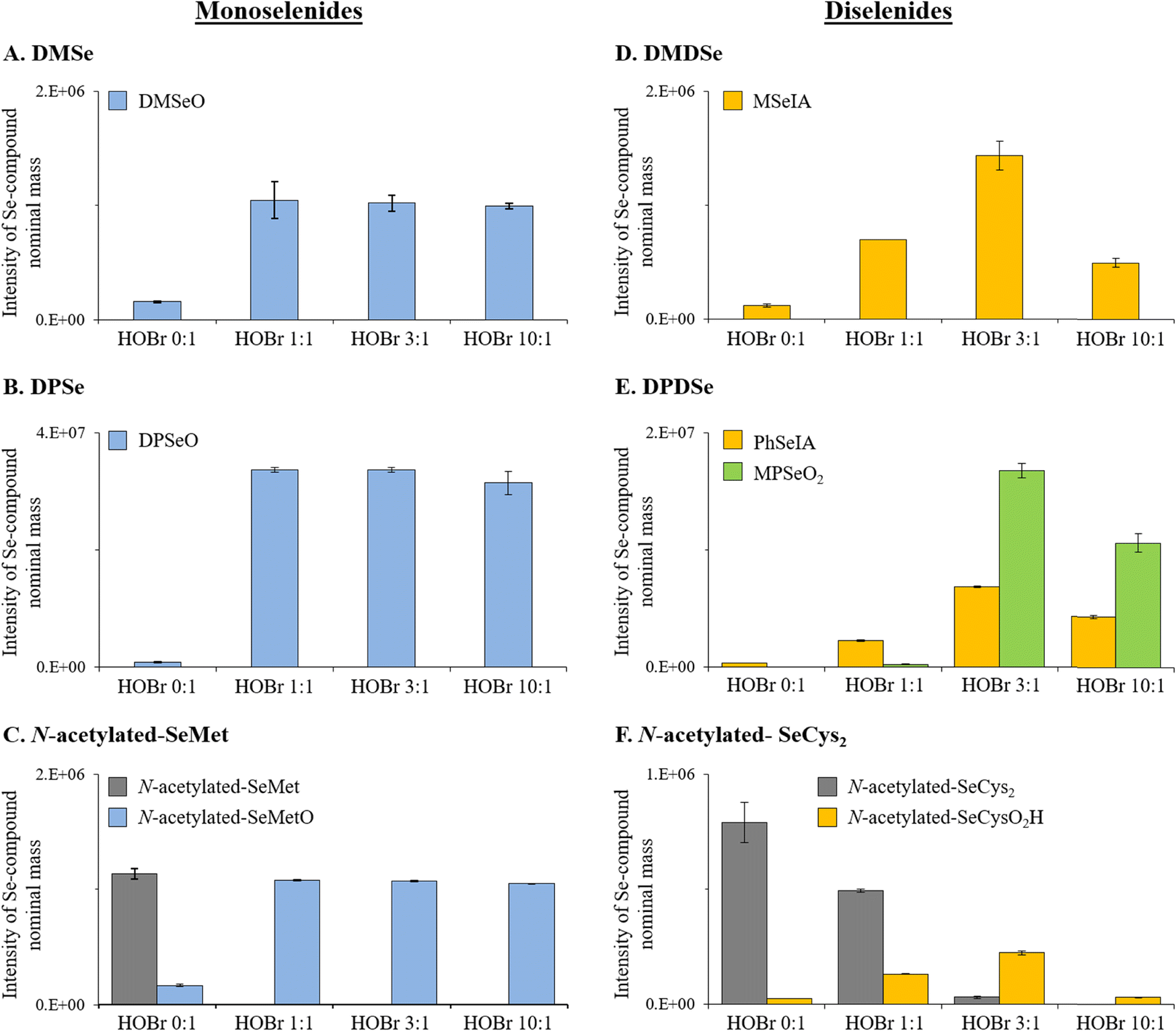 | ||
| Fig. 1 Semi-quantitative HR-MS data for the identified Se-containing products and the initial Se compounds when detected from the reactions between HOBr and (A) DMSe, (B) DPSe, (C) N-acetylated-SeMet, (D) DMDSe, (E) DPDSe and (F) N-acetylated-SeCys2. All reactions were performed with variable molar HOBr:Se compound ratios (i.e., 0:1, 1:1, 3:1 and 10:1) at pH 8. Initial concentrations of organic Se compounds were 6.25 μM. The signal intensities are those of the Se compounds detected with a Na+ adduct. Data in all panels give the average value for triplicate analyses. For all data indicated, error bars present standard deviations. | ||
Similarly, for the other organic monoselenide compounds, i.e., DPSe or N-acetylated-SeMet, only one oxidation product was detected, which were identified by HR-MS to be DPSeO and N-acetylated-SeMetO, respectively (Table 2, Fig. 1B and C, and S11 and S12, ESI†). HPLC/UV analyses confirmed that at a molar HOBr:Se compound ratio of 1:1, DPSe and N-acetylated-SeMet were completely consumed (data not shown). Also, the HR-MS signal intensities indicated that their formation was complete at a molar HOBr:Se compound ratio of 1:1 (Fig. 1B and C). Overall, these results indicate that the oxidation of monoselenides by HOBr proceeds via an oxygen transfer reaction with a 1:1 stoichiometry between HOBr and the target compounds as is known for organo sulfur halogenation, however, this was not specifically investigated in our study.
:DMDSe ratio was increased from 1:1 to 3:1, and then its intensity decreased for a ratio of 10:1 (Fig. 1D and S14 and S15, ESI†). The decrease of MSeIA when increasing the molar HOBr:DMDSe ratio from 3:1 to 10:1 indicates a further reaction of MSeIA with HOBr. Stoichiometric experiments indicate the consumption of 3 moles HOBr per mole of DMDSe (Text S12, Fig. S22, ESI†). While a new Se peak could be observed by LC-ICP-MS/MS at a molar HOBr:DMDSe ratio of 10:1 (which did neither correspond to Se[+IV] nor Se[+VI]; Fig. S13, ESI†), no other Se-containing compound was detected by HR-MS, apart from DMSeO2 (Fig. S12, ESI†), which is derived from a DMSe impurity in the DMDSe standard (Fig. S23, ESI†).
DPDSe and N-acetylated-SeCys2 were oxidized by HOBr to phenyl seleninic acid (PhSeIA) and N-acetylated-selenocysteine-seleninic-acid (N-acetylated-SeCysO2H), respectively (Table 2, Fig. 1E and F and S19–S23, ESI†). For DPDSe, an additional product was detected, i.e., methyl phenyl selenone (MPSeO2; Table 2 and Fig. S16, ESI†). PhSeIA, MPSeO2 and N-acetylated-SeCysO2H were already detected in blank samples of DPDSe and N-acetylated-SeCys2 (without HOBr addition) potentially due to their formation in the electrospray ionization. The concentrations of these compounds increased at molar HOBr:DPDSe or HOBr:N-acetylated-SeCys2 ratios of 1:1 and 3:1 (Fig. 1E and F), in line with the total consumption of DPDSe or N-acetylated-SeCys2 at a molar HOBr:Se compound ratio of 3:1 (HPLC/UV analyses, not shown). It is unclear why MPSeO2 is observed, but impurities of methylphenyldiselenide might be precursors. However, further investigations are needed to support this hypothesis. At molar HOBr:DPDSe or HOBr:N-acetylated-SeCys2 ratios of 10:1, PhSeIA, MPSeO2 and N-acetylated-SeCysO2, respectively, were present in lower concentrations than for the 3:1 ratios (Fig. 1E and F). A similar trend was also observed for MSeIA (Fig. 1D). This indicates that further products are formed, which could not be detected by HR-MS. Furthermore, neither Se(+IV) nor Se(+VI) were detected by LC-ICP-MS/MS for all investigated molar HOBr:DPDSe ratios. Only for the 3:1 and 10:1 molar HOBr:N-acetylated-SeCys2 ratios, a small proportion of Se(+IV) (i.e., <10%) was tentatively detected (see Fig. S18, ESI†).
Pathways for reactions between HOBr and the selected diselenides
Seleninic acids were the end products of reactions between diselenides and HOBr (Table 2, Fig. 1D and E). Our results indicate that 3 moles of HOBr per mole of diselenide were required for a complete abatement of these compounds (i.e., DMDSe, DPDSe and N-acetylated-SeCys2) (Fig. S22, ESI†). Overall, the diselenide–HOBr reaction proceeds by the transfer of 6 electrons (eqn (1)–(3)). Thereby, 3 moles of HOBr (Br[+I]) are reduced to Br− (Br[−I]) to oxidize 1 mole of diselenide (2× Se[−I]) to two moles of seleninic acid (Se[+II]), according to eqn (1)–(3).| Reduction reaction: 3HOBr + 6e− + 3H+ → 3Br− + 3H2O | (1) |
| Oxidation reaction: R–Se–Se–R + 4H2O → 2R–SeO–OH + 6e− + 6H+ | (2) |
| Redox: 3HOBr + R–Se–Se–R + H2O → 3Br− + 2R–SeO–OH + 3H+ | (3) |
The formation of seleninic acid involves the rupture of the diselenide bond likely due to the weaker Se–Se than Se–C bond.50 Seleninic acid was also identified as the main product in other studies that investigated the oxidation of diselenides by H2O2 (ref. 35, 51 and 52) and O3.48 Two out of these four studies35,48 also reported a 1:3 stoichiometric ratio with a slower initial reaction step followed by faster reactions of the intermediates.
Comparison of the bromine reactivities of selected Se compounds and the analogous S compounds
The apparent second-order rate constants for the reactions of organic Se compounds with HOBr can be compared to the analogous S compounds, i.e., DMS, N-acetylated-Met and N-acetylated-Cys2. Previously, similar comparisons were made for the reaction between 1O2 and SeMet vs. Met, indicating around 10- to 4-fold27,53 higher reactivity of SeMet. The reactivity of SeMet with HOCl was about 10-fold higher than for Met.54 The apparent second-order rate constants for the reactions of N-acetylated-SeMet (2.8 ± 0.3 × 108 M−1 s−1) and N-acetylated-SeCys2 (3.8 ± 0.2 × 107 M−1 s−1) with HOBr obtained in the current study are around 2 orders of magnitude higher than the corresponding values of the analogous S-containing compounds N-acetylated-Met (3.6 × 106 M−1 s−1)55 and N-acetylated-Cys2 (3.4 × 105 M−1 s−1).55 The higher reactivity of organic Se compounds compared to the analog organic S compounds is often attributed to the higher nucleophilicity and polarizability of the Se atom compared to the S atom.26,29,30,56 In a computational study on H2O2 oxidation, investigating the influence of different hydrogen, alkyl and aryl substituents and type of chalcogens, the lower activation energy for diselenides than for disulfides was explained by the type of chalcogen (which acts as a nucleophile) having a larger effect on reactivity than the hydrogen, alkyl and aryl substituent.57 Also, our results for HOBr reactivity (Table 1), demonstrate a larger difference between Se- and S-species (about 2 orders of magnitude) than between Se compounds with alkyl- and aryl-substituents (about a factor of 2 to 5).However, when comparing DMS and DMSe an opposite trend was observed. The apparent second-order rate constant of 1.2 × 109 M−1 s−1 (ref. 24) for the reaction between DMS and HOBr exceeds the value for the reaction between DMSe and HOBr (k = 7.1 ± 0.7 × 107 M−1 s−1) by more than one order of magnitude (Table 1). For the experiments carried out in a simplified seawater matrix, the reactivity difference is smaller but still about a factor of 3 (Table 1).
Environmental implications
To assess if HOBr could play a role in the fate of DMSe in marine waters, in analogy to DMS,24 first-order rate constants for the DMSe–HOBr reaction were calculated and compared to the kinetics of photochemical oxidation (currently considered as the main DMSe sink). With an apparent second-order rate constant of the DMSe–HOBr reaction of (7.1 ± 0.7) × 107 M−1 s−1 (4.7 ± 0.5 × 108 M−1 s−1 for seawater) at pH 8 (Table 1) and an estimated HOBr steady-state concentration of ≈3 × 10−14 M24 the calculated first-order rate constant of the DMSe-HOBr reaction is ≈2.1 × 10−6 s−1 (1.4 × 10−5 s−1 in synthetic seawater). These values are 1–2 orders of magnitude lower than the reported first-order rate constants for DMSe photooxidation (surface degradation) of 2.1 × 10−4 s−1 for the open ocean.58 Therefore, in contrast to DMS, it seems that HOBr for the boundary conditions in this estimation does not play an important role in the fate of marine DMSe.For the other studied species, which may occur naturally in seawater (i.e., DMDSe, SeMet and SeCys2), the relevance of their reaction with HOBr depends on the factors driving the concentrations of these species as well as the presence of further oxidants, e.g., 1O2 and H2O2 (ref. 27) and other processes such as UV-mediated degradation, which are to the best of our knowledge still not quantified.
Furthermore, it is unlikely that SeMet, SeCys2, DMSe and DMDSe affect HOBr concentrations in marine waters, since HOBr is controlled by iodide, DOM and DMS.24,25
Conclusions
The determined apparent second-order rate constants (pH 8) for the reactions between HOBr and the organic Se compounds DMSe, DMDSe, SeMet (as N-acetylated-SeMet), and SeCys2 (as N-acetylated-SeCys2) as well as the industrially produced aryl compounds DPSe and DPDSe ranged between (8.0 ± 0.4) × 106 and (2.8 ± 0.3) × 108 M−1 s−1. Generally, monoselenides had a significantly higher reactivity than the analogous diselenides, explained by the lower oxidation state of Se of −II in monoselenides than in diselenides (−I). Furthermore, it was demonstrated that monoselenides reacted in a 1:1 stoichiometry (HOBr:Se) to selenoxides, while diselenides reacted in a 3:1 stoichiometry (HOBr:Se) to seleninic acids. For the reaction between DMSe and HOBr, dimethyl selenoxide was the only oxidation product observed, which is in agreement with studies that investigated the reaction of DMSe with other oxidants. Also, for the other organic monoselenide compounds, i.e., DPSe or N-acetylated-SeMet, only one oxidation product was detected, i.e., DPSeO and N-acetylated-SeMetO, respectively. For the diselenides DMDSe, DPDSe and N-acetylated-SeCys2 respectively, MSeIA, phenyl seleninic acid (PhSeIA) and N-acetylated-selenocysteine-seleninic-acid (N-acetylated-SeCysO2H) were detected as (main) oxidation products. HOBr is likely not a generally relevant sink for DMSe, in contrast to DMS. Due to a lack of data on concentrations of the other studied natural Se compounds in seawater and their further sinks, the relevance of HOBr for their fate can currently not be assessed.
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
We acknowledge funding by the SNF (project number 200021_179104), as well as internal funds from Eawag and ETH Zurich. We thank Caroline Stengel for assistance in the lab, Ursula Schönenberger, Jakov Bolotin and Elisabeth Muck for their support with HPLC/UV, GC/MS, and quench-flow measurements, respectively. Furthermore, we thank Joanna Houska for providing HPLC instrumental methods and for measuring selected organic Se compounds and competitors and we thank Pauline Béziat for her assistance with marine biological pathways of Se. Finally, we thank Karim-Alexandre Essaied for providing information on treatment of SeMet and SeCys2 with BOC2O.References
- M. Schiavon, A. Ertani, S. Parrasia and F. Dalla Vecchia, Selenium accumulation and metabolism in algae, Aquat. Toxicol., 2017, 189, 1–8 CrossRef CAS PubMed.
- H. Zeng, Selenium as an essential micronutrient: roles in cell cycle and apoptosis, Molecules, 2009, 14(3), 1263–1278 CrossRef CAS PubMed.
- M. Gupta and S. Gupta, An overview of selenium uptake, metabolism, and toxicity in plants, Front. Plant Sci., 2017, 7, 2074 Search PubMed.
- E. Mangiapane, A. Pessione and E. Pessione, Selenium and selenoproteins: an overview on different biological systems, Curr. Protein Pept. Sci., 2014, 15(6), 598–607 CrossRef CAS PubMed.
- L. V. Papp, A. Holmgren and K. K. Khanna, Selenium and selenoproteins in health and disease, Antioxid. Redox Signaling, 2010, 12(7), 793–795 CrossRef CAS PubMed.
- E. Zoidis, I. Seremelis, N. Kontopoulos and G. P. Danezis, Selenium-dependent antioxidant enzymes: Actions and properties of selenoproteins, Antioxidants, 2018, 7(5), 66 CrossRef PubMed.
- T. Blazina, A. Läderach, G. D. Jones, H. Sodemann, H. Wernli, J. W. Kirchner and L. H. Winkel, Marine primary productivity as a potential indirect source of selenium and other trace elements in atmospheric deposition, Environ. Sci. Technol., 2017, 51(1), 108–118 CrossRef CAS PubMed.
- T. Blazina, Y. Sun, A. Voegelin, M. Lenz, M. Berg and L. H. Winkel, Terrestrial selenium distribution in China is potentially linked to monsoonal climate, Nat. Commun., 2014, 5(1), 1–7 Search PubMed.
- H. Wen and J. Carignan, Reviews on atmospheric selenium: emissions, speciation and fate, Atmos. Environ., 2007, 41(34), 7151–7165 CrossRef CAS.
- A. Feinberg, A. Stenke, T. Peter and L. H. Winkel, Constraining atmospheric selenium emissions using observations, global modelling, and Bayesian inference, Environ. Sci. Technol., 2020, 54(12), 7146–7155 CrossRef CAS PubMed.
- D. Amouroux, P. S. Liss, E. Tessier, M. Hamren-Larsson and O. F. Donard, Role of oceans as biogenic sources of selenium, Earth Planet. Sci. Lett., 2001, 189(3–4), 277–283 CrossRef CAS.
- D. Amouroux and O. F. Donard, Maritime emission of selenium to the atmosphere in eastern Mediterranean seas, Geophys. Res. Lett., 1996, 23(14), 1777–1780 CrossRef CAS.
- M. P. De Souza, I. J. Pickering, M. Walla and N. Terry, Selenium assimilation and volatilization from selenocyanate-treated Indian mustard and muskgrass, Plant Physiol., 2002, 128(2), 625–633 CrossRef CAS PubMed.
- J. H. Ansede and D. C. Yoch, Comparison of selenium and sulfur volatilization by dimethylsulfoniopropionate lyase (DMSP) in two marine bacteria and estuarine sediments, FEMS Microbiol. Ecol., 1997, 23(4), 315–324 CrossRef CAS.
- J. S. Dickschat, C. Zell and N. L. Brock, Pathways and substrate specificity of DMSP catabolism in marine bacteria of the Roseobacter clade, ChemBioChem, 2010, 11(3), 417–425 CrossRef CAS PubMed.
- K. E. Luxem, B. Vriens, R. Behra and L. H. Winkel, Studying selenium and sulfur volatilisation by marine algae Emiliania huxleyi and Thalassiosira oceanica in culture, Environ. Chem., 2017, 14(4), 199–206 CrossRef CAS.
- H. Araie and Y. Shiraiwa, Selenium utilization strategy by microalgae, Molecules, 2009, 14(12), 4880–4891 CrossRef CAS PubMed.
- H. Araie, K. Sakamoto, I. Suzuki and Y. Shiraiwa, Characterization of the selenite uptake mechanism in the coccolithophore Emiliania huxleyi (Haptophyta), Plant Cell Physiol., 2011, 52(7), 1204–1210 CrossRef CAS PubMed.
- N. Terry, A. Zayed, M. De Souza and A. Tarun, Selenium in higher plants, Annu. Rev. Plant Biol., 2000, 51(1), 401–432 CrossRef CAS PubMed.
- T. G. Sors, D. R. Ellis and D. E. Salt, Selenium uptake, translocation, assimilation and metabolic fate in plants, Photosynth. Res., 2005, 86(3), 373–389 CrossRef CAS PubMed.
- C. Gabel-Jensen, K. Lunøe and B. Gammelgaard, Formation of methylselenol, dimethylselenide and dimethyldiselenide in in vitro metabolism models determined by headspace GC-MS, Metallomics, 2010, 2(2), 167–173 CrossRef CAS PubMed.
- D. Amouroux, C. Pécheyran and O. F. Donard, Formation of volatile selenium species in synthetic seawater under light and dark experimental conditions, Appl. Organomet. Chem., 2000, 14(5), 236–244 CrossRef CAS.
- R. Wever and M. A. van der Horst, The role of vanadium haloperoxidases in the formation of volatile brominated compounds and their impact on the environment, Dalton Trans., 2013, 42(33), 11778–11786 RSC.
- E. Müller, U. von Gunten, S. Bouchet, B. Droz and L. H. Winkel, Hypobromous acid as an unaccounted sink for marine dimethyl sulfide?, Environ. Sci. Technol., 2019, 53(22), 13146–13157 CrossRef PubMed.
- E. Müller, U. von Gunten, S. Bouchet, B. Droz and L. H. Winkel, Reaction of DMS and HOBr as a Sink for Marine DMS and an Inhibitor of Bromoform Formation, Environ. Sci. Technol., 2021, 55(8), 5547–5558 CrossRef PubMed.
- D. Steinmann, T. Nauser and W. H. Koppenol, Selenium and sulfur in exchange reactions: a comparative study, J. Org. Chem., 2010, 75(19), 6696–6699 CrossRef CAS PubMed.
- L. Carroll, K. Gardiner, M. Ignasiak, J. Holmehave, S. Shimodaira, T. Breitenbach, M. Iwaoka, P. R. Ogilby, D. I. Pattison and M. J. Davies, Interaction kinetics of selenium-containing compounds with oxidants, Free Radical Biol. Med., 2020, 155, 58–68 CrossRef CAS PubMed.
- R. Huber and R. Criddle, Comparison of the chemical properties of selenocysteine and selenocystine with their sulfur analogs, Arch. Biochem. Biophys., 1967, 122(1), 164–173 CrossRef CAS PubMed.
- A. S. Rahmanto and M. J. Davies, Selenium-containing amino acids as direct and indirect antioxidants, IUBMB Life, 2012, 64(11), 863–871 CrossRef CAS PubMed.
- R. J. Hondal, S. M. Marino and V. N. Gladyshev, Selenocysteine in thiol/disulfide-like exchange reactions, Antioxid. Redox Signaling, 2013, 18(13), 1675–1689 CrossRef CAS PubMed.
- J. C. Pleasants, W. Guo and D. L. Rabenstein, A comparative study of the kinetics of selenol/diselenide and thiol/disulfide exchange reactions, J. Am. Chem. Soc., 1989, 111(17), 6553–6558 CrossRef CAS.
- B. Cardey and M. Enescu, Selenocysteine versus cysteine reactivity: a theoretical study of their oxidation by hydrogen peroxide, J. Phys. Chem. A, 2007, 111(4), 673–678 CrossRef CAS PubMed.
- S. Liu, E. Salhi, W. Huang, K. Diao and U. von Gunten, Kinetic and mechanistic aspects of selenite oxidation by chlorine, bromine, monochloramine, ozone, permanganate, and hydrogen peroxide, Water Res., 2019, 164, 114876 CrossRef CAS PubMed.
- R. M. Rosa, R. Roesler, A. L. Braga, J. Saffi and J. A. P. Henriques, Pharmacology and toxicology of diphenyl diselenide in several biological models, Braz. J. Med. Biol. Res., 2007, 40(10), 1287–1304 CrossRef CAS PubMed.
- G. Ribaudo, M. Bellanda, I. Menegazzo, L. P. Wolters, M. Bortoli, G. Ferrer-Sueta, G. Zagotto and L. Orian, Mechanistic insight into the oxidation of organic phenylselenides by H2O2, Chem. – Eur. J., 2017, 23(10), 2405–2422 CrossRef CAS PubMed.
- Sigma-Aldrich, Diphenyl selenide. 2021.
- B. Vriens, M. Mathis, L. H. Winkel and M. Berg, Quantification of volatile-alkylated selenium and sulfur in complex aqueous media using solid-phase microextraction, J. Chromatogr. A, 2015, 1407, 11–20 CrossRef CAS PubMed.
- M. Loos, C. Gerber, F. Corona, J. Hollender and H. Singer, Accelerated isotope fine structure calculation using pruned transition trees, Anal. Chem., 2015, 87(11), 5738–5744 CrossRef CAS PubMed.
- J. Tolu, I. Le Hécho, M. Bueno, Y. Thiry and M. Potin-Gautier, Selenium speciation analysis at trace level in soils, Anal. Chim. Acta, 2011, 684(1–2), 126–133 CrossRef CAS PubMed.
- R. P. Schwarzenbach, P. M. Gschwend and D. M. Imboden, Environmental Organic Chemistry, Amazon, USA, 2nd edn, 2003 Search PubMed.
- M. B. Heeb, J. Criquet, S. G. Zimmermann-Steffens and U. von Gunten, Oxidative treatment of bromide-containing waters: formation of bromine and its reactions with inorganic and organic compounds--a critical review, Water Res., 2014, 48, 15–42 CrossRef CAS PubMed.
- J. E. Wajon and J. C. Morris, Rates of formation of N-bromo amines in aqueous solution, Inorg. Chem., 1982, 21(12), 4258–4263 CrossRef CAS.
- M. B. Heeb, I. Kristiana, D. Trogolo, J. S. Arey and U. Von Gunten, Formation and reactivity of inorganic and organic chloramines and bromamines during oxidative water treatment, Water Res., 2017, 110, 91–101 CrossRef CAS PubMed.
- C. Storkey, D. I. Pattison, J. M. White, C. H. Schiesser and M. J. Davies, Preventing protein oxidation with sugars: scavenging of hypohalous acids by 5-selenopyranose and 4-selenofuranose derivatives, Chem. Res. Toxicol., 2012, 25(11), 2589–2599 Search PubMed.
- L. Carroll, D. I. Pattison, S. Fu, C. H. Schiesser, M. J. Davies and C. L. Hawkins, Reactivity of selenium-containing compounds with myeloperoxidase-derived chlorinating oxidants: second-order rate constants and implications for biological damage, Free Radical Biol. Med., 2015, 84, 279–288 CrossRef CAS.
- S. Padmaja, G. Squadrito, J.-N. Lemercier, R. Cueto and W. A. Pryor, Rapid oxidation of DL-selenomethionine by peroxynitrite, Free Radical Biol. Med., 1996, 21(3), 317–322 CrossRef CAS PubMed.
- A. Krief, W. Dumont, J.-N. Denis, G. Evrard and B. Norberg, Synthesis of selenones: a comparative study, J. Chem. Soc., Chem. Commun., 1985, 569–570 RSC.
- G. Ayrey, D. Barnard and D. Woodbridge, 401. The oxidation of organoselenium compounds by ozone, J. Chem. Soc., 1962, 2089–2099 RSC.
- J. M. Khurana, B. M. Kandpal and Y. K. Chauhan, Rapid oxidation of selenides, selenoxides, tellurides, and telluroxides with aqueous sodium hypochlorite, Phosphorus, Sulfur Silicon Relat. Elem., 2003, 178(6), 1369–1375 CrossRef CAS.
- F. Zaccaria, L. P. Wolters, C. Fonseca Guerra and L. Orian, Insights on selenium and tellurium diaryldichalcogenides: A benchmark DFT study, J. Comput. Chem., 2016, 37(18), 1672–1680 CrossRef CAS PubMed.
- K. Satheeshkumar, S. Raju, H. B. Singh and R. J. Butcher, Reactivity of Selenocystine and Tellurocystine: Structure and Antioxidant Activity of the Derivatives, Chem. – Eur. J., 2018, 24(66), 17513–17522 CrossRef CAS PubMed.
- M. Farina, N. Barbosa, C. Nogueira, V. Folmer, G. Zeni, L. Andrade, A. Braga and J. Rocha, Reaction of diphenyl diselenide with hydrogen peroxide and inhibition of delta-aminolevulinate dehydratase from rat liver and cucumber leaves, Braz. J. Med. Biol. Res., 2002, 35(6), 623–631 CrossRef CAS PubMed.
- R. Scurlock, M. Rougee, R. Bensasson, M. Evers and N. Dereu, Deactivation of singlet molecular oxygen by organo-selenium compounds exhibiting glutathione peroxidase activity and by sulfur-containing homologs, Photochem. Photobiol., 1991, 54(5), 733–736 CrossRef CAS PubMed.
- A. M. Casaril, M. T. Ignasiak, C. Y. Chuang, B. Vieira, N. B. Padilha, L. Carroll, E. J. Lenardão, L. Savegnago and M. J. Davies, Selenium-containing indolyl compounds: kinetics of reaction with inflammation-associated oxidants and protective effect against oxidation of extracellular matrix proteins, Free Radical Biol. Med., 2017, 113, 395–405 CrossRef CAS PubMed.
- D. I. Pattison and M. J. Davies, Kinetic analysis of the reactions of hypobromous acid with protein components: implications for cellular damage and use of 3-bromotyrosine as a marker of oxidative stress, Biochemistry, 2004, 43(16), 4799–4809 CrossRef CAS PubMed.
- H. Sies and G. E. Arteel, Strategies for controlling oxidative stress: Protection against peroxynitrite and hydroperoxides by selenoproteins and selenoorganic compounds. in Critical Reviews of Oxidative Stress and Aging: Advances in Basic Science, Diagnostics and Intervention, World Scientific, 2003, vol. 2, pp. 1313–1323 Search PubMed.
- M. Bortoli, F. Zaccaria, M. Dalla Tiezza, M. Bruschi, C. F. Guerra, F. M. Bickelhaupt and L. Orian, Oxidation of organic diselenides and ditellurides by H 2 O 2 for bioinspired catalyst design, Phys. Chem. Chem. Phys., 2018, 20(32), 20874–20885 RSC.
- R. P. Mason, A. L. Soerensen, B. P. DiMento and P. H. Balcom, The global marine selenium cycle: Insights from measurements and modeling, Global Biogeochem. Cycles, 2018, 32(12), 1720–1737 CrossRef CAS.
- S. S. Lau, R. P. Dias, K. R. Martin-Culet, N. A. Race, M. H. Schammel, K. P. Reber, A. L. Roberts and J. D. Sivey, 1, 3, 5-Trimethoxybenzene (TMB) as a new quencher for preserving redox-labile disinfection byproducts and for quantifying free chlorine and free bromine, Environ. Sci.: Water Res. Technol., 2018, 4(7), 926–941 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ew00787a |
| This journal is © The Royal Society of Chemistry 2024 |