
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pre-equilibrium reactions involving pendent relays improve CO2 reduction mediated by molecular Cr-based electrocatalysts†
Megan E.
Moberg
 ,
Amelia G.
Reid
,
Diane A.
Dickie
and
Charles W.
Machan
*
,
Amelia G.
Reid
,
Diane A.
Dickie
and
Charles W.
Machan
*
Department of Chemistry, University of Virginia, PO Box 400319, Charlottesville, VA 22904-4319, USA. E-mail: machan@virginia.edu
First published on 27th August 2024
Abstract
Homogeneous earth abundant transition-metal electrocatalysts capable of carbon dioxide (CO2) reduction to generate value-added chemical products are a possible strategy to minimize rising anthropogenic CO2 emissions. Previously, it was determined that Cr-centered bipyridine-based N2O2 complexes for CO2 reduction are kinetically limited by a proton-transfer step during C–OH bond cleavage. Therefore, it was hypothesized that the inclusion of pendent relay groups in the secondary coordination sphere of these molecular catalysts could increase their catalytic activity. Here, it is shown that the introduction of a pendent methoxy group favorably impacts a pre-equilibrium protonation prior to the catalytic resting state, resulting in a significant increase in catalytic activity without a loss of product selectivity for generating carbon monoxide (CO) from CO2. Interestingly, combining the pendent methoxy group with a cationic acid causes a positive shift of the catalytic reduction potential of the system, while maintaining increased activity and quantitative selectivity. This work suggests that tuning the secondary coordination sphere with respect to cationic proton sources can result in activity improvements by modifying the kinetic and thermodynamic aspects of proton transfer in the catalytic cycle.
Introduction
The industrial revolution marked the start of a dramatic increase in the amount of accumulated atmospheric carbon dioxide (CO2), a leading contributor to the global climate crisis.1 As of 2021 the amount of atmospheric CO2 is approximately 1.4 trillion tons, compared to approximately 45 billion tons in 1900.2 Therefore, there is significant interest in mitigating the impact of increased solar energy retention caused by elevated levels of atmospheric CO2.1 Electrochemically reducing CO2 to value-added chemical products through the use of renewable energy sources, such as wind or solar, is one way to lessen the concentration of CO2 in the atmosphere.3 The reduction of CO2 can be accomplished through proton-coupled multi-electron pathways, but the majority of molecular electrocatalysts reduce CO2via a two-electron, two-proton (2e−/2H+) mechanism, producing either carbon monoxide (CO) with water (H2O) as a co-product or formic acid (HCOOH).4 Both carbon-containing products are relevant to industrial applications: the Fischer–Tropsch process uses CO and H2 to produce fuels and commodity chemicals5 while formic acid is an energy dense carrier for fuel cells, as well as a useful chemical synthon.6Although the CO2 reduction reaction (CO2RR) can be mediated by homogeneous complexes based on expensive late transition metals (Pd,7 Ru,8 and Re![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9), a more sustainable approach would be to use earth-abundant transition metals to convert CO2 into these useful products for chemical feedstocks or fuels.10 In 1984, a [NiII(cyclam)]2+ catalyst for the selective reduction of CO2 to CO on a mercury cathode was reported by Beley et al.,11 following up on early work from Fisher and Eisenberg in 1980 on less-selective reaction conditions.12 Since then, there have been other examples of Earth-abundant transition metal catalysts for the CO2RR, most notably, manganese (Mn) carbonyl bipyridyls13 and iron (Fe) porphyrins.14 The most active molecular catalyst reported to date is the iron porphyrin, Fe-o-TMA (iron(III) 5,10,15,20-tetra-(o-N,N,N-trimethylanilium)-porphyrin), reported by Azcarate et al., which has a turnover frequency (TOF) of 106 s−1.15,16
9), a more sustainable approach would be to use earth-abundant transition metals to convert CO2 into these useful products for chemical feedstocks or fuels.10 In 1984, a [NiII(cyclam)]2+ catalyst for the selective reduction of CO2 to CO on a mercury cathode was reported by Beley et al.,11 following up on early work from Fisher and Eisenberg in 1980 on less-selective reaction conditions.12 Since then, there have been other examples of Earth-abundant transition metal catalysts for the CO2RR, most notably, manganese (Mn) carbonyl bipyridyls13 and iron (Fe) porphyrins.14 The most active molecular catalyst reported to date is the iron porphyrin, Fe-o-TMA (iron(III) 5,10,15,20-tetra-(o-N,N,N-trimethylanilium)-porphyrin), reported by Azcarate et al., which has a turnover frequency (TOF) of 106 s−1.15,16
Although for decades chromium (Cr) was thought to be nonprivileged for the CO2RR,17,18 more recently four examples of molecular Cr catalysts for the selective reduction of CO2 to CO have been reported.10,19–22 The first selective homogeneous Cr-based catalyst was Cr(tbudhbpy)Cl(H2O), where the ligand precursor, (tbudhbpy(H)2) is 6,6′-di(3,5-di-tert-butyl-2-hydroxybenzene)-2,2′-bipyridine.10,19,20 The catalytic cycle for the CO2 reduction reaction mediated by Cr(tbudhbpy)Cl(H2O) proposed by Hooe et al. under protic conditions is depicted in Fig. 1.19,23 After an initial overall two-electron reduction of the Cr complex with chloride loss to form the four-coordinate monoanionic Cr(II)(bpy˙−) active species, i, CO2 binds to the Cr center forming [Cr−CO2]−, ii. The CO2 adduct is subsequently protonated to form a neutral hydroxycarbonyl, iii. A second reduction with a standard potential more positive than 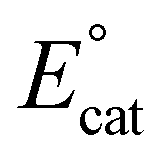 generates a monoanionic hydroxycarbonyl species [Cr−CO2H]−, iv. Another proton transfer reaction cleaves the C–OH bond and releases water, leaving CO bound to the Cr center, v. The release of CO from this neutral species is expected to be rapid and generate a neutral four-coordinate species, vi, from which one-electron reduction at
generates a monoanionic hydroxycarbonyl species [Cr−CO2H]−, iv. Another proton transfer reaction cleaves the C–OH bond and releases water, leaving CO bound to the Cr center, v. The release of CO from this neutral species is expected to be rapid and generate a neutral four-coordinate species, vi, from which one-electron reduction at 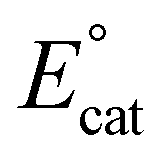 closes the catalytic cycle. Density Functional Theory (DFT) calculations suggest that the cleavage of the C–OH bond to release water is the rate-determining step (RDS),23 consistent with the experimental mechanistic data. During electrolysis, this Cr-based complex produced CO with a FE of 96 ± 8% from CO2 in the presence of 0.6 M phenol (PhOH).19
closes the catalytic cycle. Density Functional Theory (DFT) calculations suggest that the cleavage of the C–OH bond to release water is the rate-determining step (RDS),23 consistent with the experimental mechanistic data. During electrolysis, this Cr-based complex produced CO with a FE of 96 ± 8% from CO2 in the presence of 0.6 M phenol (PhOH).19
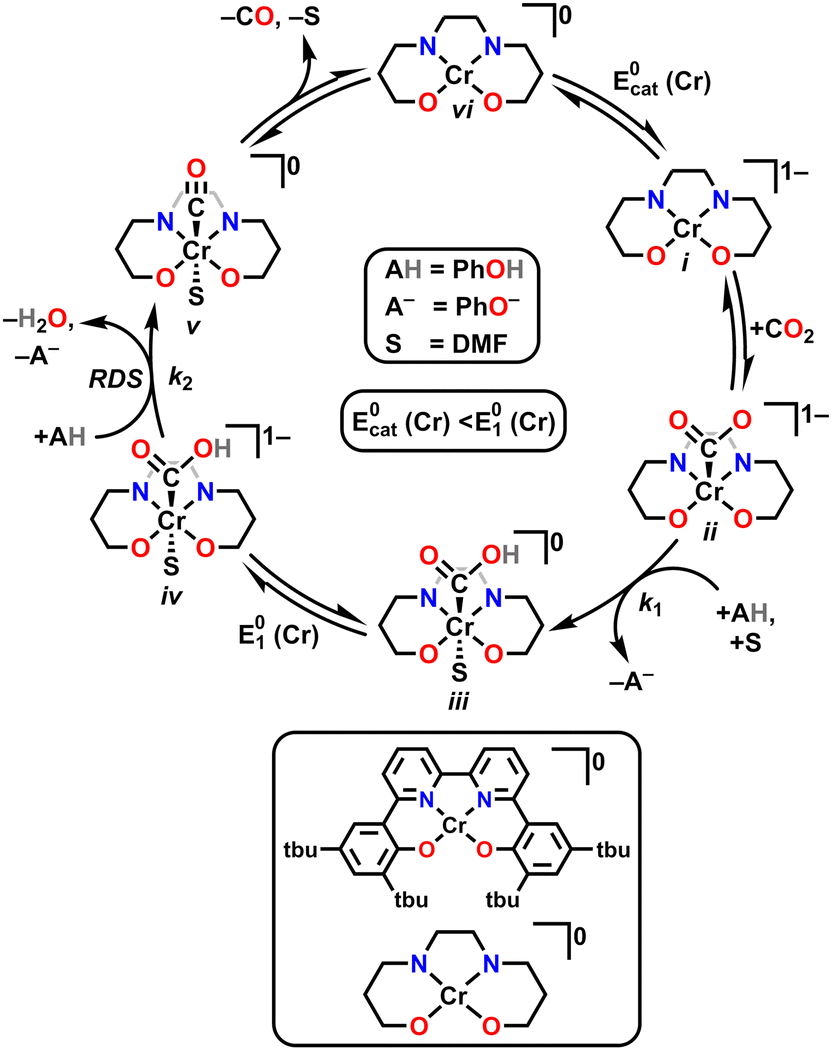 | ||
| Fig. 1 Proposed catalytic mechanism for electrocatalytic CO2 reduction mediated by Cr(tbudhbpy)Cl(H2O). | ||
Pendent proton (or proton donor) relays are commonly used to facilitate proton transfer by means of hydrogen bonding and proximity to the active site during electrochemical reactions.24 Pendent relays24–27 incorporated into the secondary coordination sphere have been studied extensively for reductive small molecule conversion in the context of the oxygen reduction reaction,28–33 the hydrogen evolution reaction,34–44 and the CO2RR.45–58 Since the kinetic enhancement originates from the overall balance of the equilibria involving proton transfer from solution to the active site via the relay,59 it is advantageous to have acid–base pairs with fast exchange reactions (e.g., O or N containing).24 For instance, in 2017, Ngo et al. described how the introduction of methoxy (–OMe) groups into a 2,2′-bipyridine-based ligand appended to a Mn(CO)3Br core led to an increase in CO2RR catalysis.58 For this Mn-based system, the pendent Lewis base is able to form noncovalent hydrogen-bonding interactions with the C–OH intermediate, resulting in an overall stabilization of the C–OH bond cleavage step by acting as a proton donor relay.58 Along with its increased catalytic activity, the system operated at lower overpotentials, as compared to the parent non-substituted catalyst, and controlled potential electrolysis (CPE) studies of the complex confirmed high product selectivity for CO in the presence of nonaqueous Brønsted acid proton sources.58 In the context of the CO2RR, we have similarly examined a molecular Fe-based CO2RR catalyst with methoxy-based pendent relays incorporated into an analogous coordination environment to the Cr-based compound described above.45 In this Fe system, the addition of the –OMe group increases the availability of the proton donor through non-covalent interactions, resulting in an increase in catalytic activity and improved selectivity for formate (HCO2−).45
Based on the proposed rate-determining C–OH bond cleavage step for Cr-based complexes during electrocatalytic CO2 reduction, it was hypothesized that the addition of pendent relays to the ligand framework would have a beneficial effect on the mechanism of proton transfer, without resulting in significant changes to the electronic structure of the Cr active site (changes in 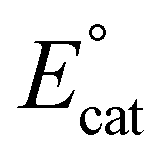 ). Here, the synthesis, characterization, and electrochemical behavior of two new Cr-based complexes with a series of proton sources under unbuffered (acid only) and buffered conditions (acid and conjugate base added) under electrocatalytic conditions for the CO2RR is reported. Notably, significant differences in activity and operating potential are observed when comparing a control complex, Cr(p-tbudhbpy)Cl(H2O) (1) (Fig. 2) with a compound containing pendent –OMe groups, Cr(nPrdhbpy)Cl(H2O) (2) (Fig. 2). Using cyclic voltammetry (CV) and CPE, the effects of the pendent relay on catalysis are elucidated, specifically the shift to a pre-equilibrium catalytic response that causes both a kinetic effect (higher TOF in CPE studies) and a thermodynamic effect (positive shift in catalyst operating potential) on CO2 reduction, without loss of quantitative product selectivity for CO. The pre-equilibrium approximation for catalysis applies when there is a fast initial step under equilibrium control prior to the rate-determining step, while the steady state approximation can be used to describe catalytic responses where the rate of intermediate formation is matched by the rate of intermediate consumption.
). Here, the synthesis, characterization, and electrochemical behavior of two new Cr-based complexes with a series of proton sources under unbuffered (acid only) and buffered conditions (acid and conjugate base added) under electrocatalytic conditions for the CO2RR is reported. Notably, significant differences in activity and operating potential are observed when comparing a control complex, Cr(p-tbudhbpy)Cl(H2O) (1) (Fig. 2) with a compound containing pendent –OMe groups, Cr(nPrdhbpy)Cl(H2O) (2) (Fig. 2). Using cyclic voltammetry (CV) and CPE, the effects of the pendent relay on catalysis are elucidated, specifically the shift to a pre-equilibrium catalytic response that causes both a kinetic effect (higher TOF in CPE studies) and a thermodynamic effect (positive shift in catalyst operating potential) on CO2 reduction, without loss of quantitative product selectivity for CO. The pre-equilibrium approximation for catalysis applies when there is a fast initial step under equilibrium control prior to the rate-determining step, while the steady state approximation can be used to describe catalytic responses where the rate of intermediate formation is matched by the rate of intermediate consumption.
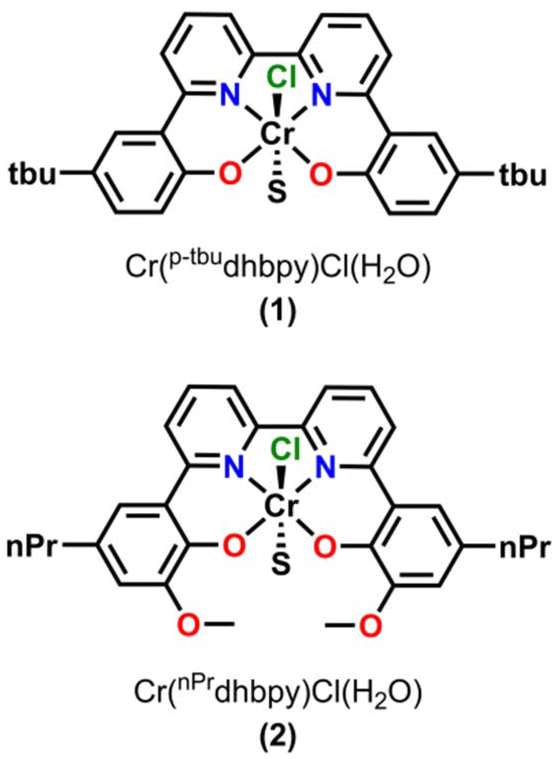 | ||
| Fig. 2 Structures of Cr(p-tbudhbpy)Cl(H2O) (1), and Cr(nPrdhbpy)Cl(H2O) (2), where S is a solvent adduct of water or DMF. | ||
Results
Synthesis and characterization
The ligand precursor complex 6,6′-di(5-tert-butyl-2-hydroxybenzene)-2,2′-bipyridine, (p-tbudhbpy(H)2) was synthesized as previously reported.60 The synthesis of the 6,6′-di(3-methoxy-5-n-propyl-2-hydroxybenzene)-2,2′-bipyridine, (nPrdhbpy(H)2) ligand was completed via a modification of our previously reported procedure (see ESI† for details).28 Metalation of (p-tbudhbpy(H)2) and (nPrdhbpy(H)2) to generate Cr(p-tbudhbpy)Cl(H2O) (1) and Cr(nPrdhbpy)Cl(H2O) (2), respectively, was achieved by refluxing a solution of ligand and Cr(II) chloride metal salt under inert atmosphere for 24 hours, analogous to previously reported procedures (see the ESI†).19 Purified products were characterized by UV-vis (Fig. S2–S4†), NMR (Tables S2 and S3†), and microanalysis. Crystals of 1 suitable for XRD studies were grown from a concentrated solution of 1 in N,N-dimethylformamide (DMF) at cold temperatures (Fig. S1†).Electrochemistry
To probe the electrochemical behavior of these complexes, cyclic voltammetry (CV) was performed on 1 and 2 in DMF with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte. Under argon (Ar) and CO2 saturation conditions, three redox features were observed for 1 at E1/2 = –1.59, Ep = –1.75, and E1/2 = –1.95 V versus ferrocenium/ferrocene (Fc+/Fc) (Fig. 3). The first two redox features of 1 coalesce upon the addition of excess chloride (Fig. S5 and S6†), consistent with the features being chemically related to each other by chloride loss. It is proposed that the more positive redox feature at E1/2 = –1.59 V vs. Fc+/Fc represents a solvento species generated by an equilibrium chloride displacement reaction, while the more negative at Ep = –1.75 V vs. Fc+/Fc corresponds to the neutral chloro adduct based on precedent.19,20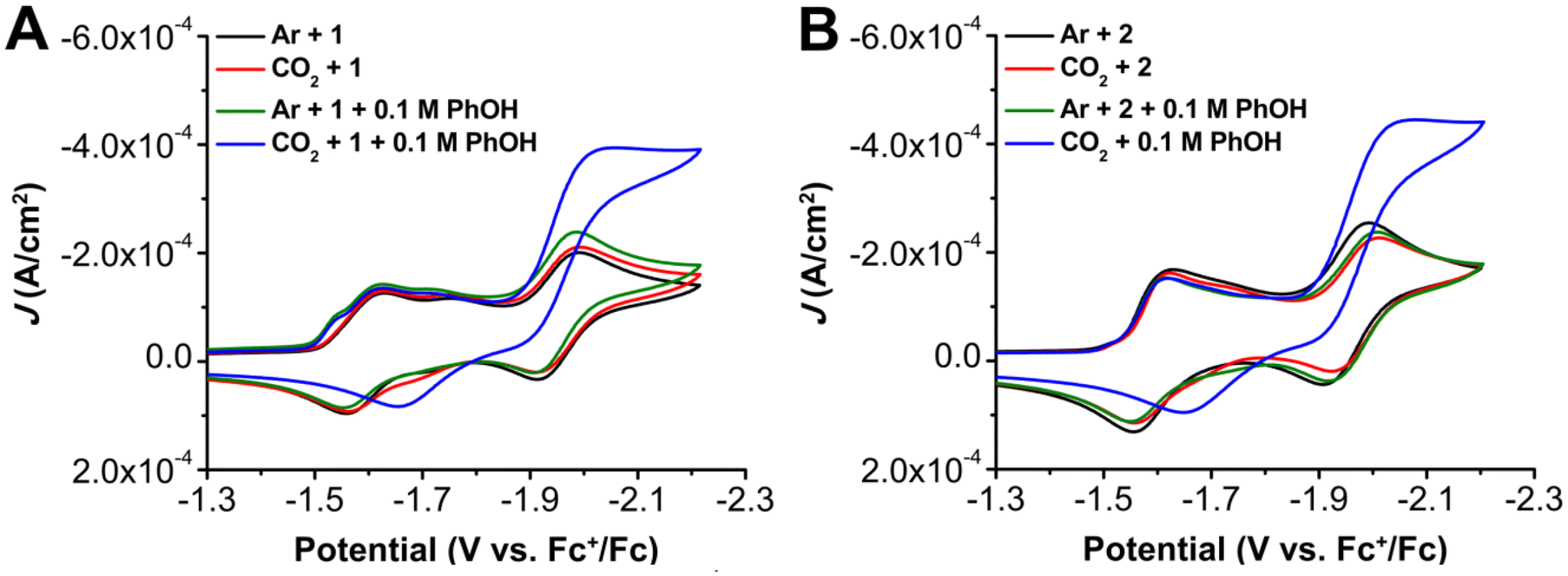 | ||
| Fig. 3 (A) Comparison CVs of Cr(p-tbudhbpy)Cl(H2O) 1 under Ar and CO2 saturation conditions with and without 0.1 M PhOH. (B) Comparison CVs of Cr(nPrdhbpy)Cl(H2O) 2 under Ar and CO2 saturation conditions with and without 0.1 M PhOH. Conditions: 1.0 mM catalyst, 0.1 M TBAPF6/DMF; glassy carbon disc working electrode, glassy carbon rod counter electrode, Ag/AgCl pseudoreference electrode; referenced to Fc+/Fc internal standard; 100 mV s−1 scan rate. | ||
For 1, under Ar and CO2 saturation conditions, there are minimal changes observed in the CV response (Fig. 3). However, under both Ar and CO2 saturation, the addition of PhOH results in the appearance of a feature at a potential positive of the first reduction feature in the forward trace with Ep = –1.51 V vs. Fc+/Fc (Fig. 3). To probe the nature of this prewave feature observed for 1, CV responses with an excess of tetrabutylammonium chloride (TBACl) were conducted. In the presence of an excess of TBACl under Ar and CO2 saturation, a shift in the first reduction feature from Ep = −1.59 to −1.63 V vs. Fc+/Fc was observed, along with the disappearance of the second redox feature at Ep = −1.75 V vs. Fc+/Fc (Fig. S6 and S7†). The combination of excess TBACl and PhOH under Ar and CO2 saturation conditions suppressed the appearance of the prewave observed in the presence of PhOH only (Fig. S8†), although it still could be observed at high scan rates (Fig. S6 and S7†). Similar to the addition of TBACl by itself, a shift to more negative reduction potentials for the original first reduction feature and disappearance of the original second redox feature was observed with the combination of TBACl and PhOH (Fig. S8†). Overall, these results agree with previously reported results19,23 and are consistent with the proposal that the two reduction features at E1/2 = −1.59 and Ep = −1.75 V vs. Fc+/Fc are related to an equilibrium chloride displacement reaction for complex 1. The pre-wave feature at Ep = −1.51 V vs. Fc+/Fc can be ascribed to reversible and weak adsorption to the electrode, based on its shape and the rinse tests conducted under electrolysis conditions described below. The enhancement of this pre-wave with added phenol suggests that the proton donor could assist with chloride loss and therefore alter electrode adsorption.
Variable scan rate studies under both Ar (Fig. S6†) and CO2 (Fig. S7†) were subsequently examined and the diffusion-controlled saturation response at the reversible third reduction feature (E1/2 = –1.95 V vs. Fc+/Fc) is representative of a homogeneous electrochemical response under both conditions (Fig. S6 and S7†). In the presence of PhOH (pKa = 18.8 in DMF)61 as a proton source under CO2 saturation, a current increase indicative of a catalytic response for the CO2RR was observed to originate from the redox wave at E1/2 = –1.95 V vs. Fc+/Fc (Fig. 3). Mechanistic analysis was attempted via variable concentration studies via a log–log plots in regions where catalytic current was sensitive to substrate concentration.62 These studies suggest a mixed-order concentration dependence of 1 (Fig. S9†), first-order dependence of PhOH (Fig. S10†), and mixed-order dependence of CO2 (Fig. S11†) on the observed electrocatalytic current. Variable scan rate studies establish that 1 is in the appropriate kinetically limited regime (Fig. S13†), suggesting that pre-equilibrium processes are being observed in the concentration dependence of the electrocatalytic response.
Analogous CV experiments for 2 under Ar and CO2 saturation similarly show three reduction features at E1/2 = –1.59, Ep = –1.72, and E1/2 = –1.95 V vs. Fc+/Fc (Fig. 3). The first two redox features of 2 coalesce with the addition of chloride (Fig. S33 and S34†), consistent with the features being chemically related to each other; as previously assigned, the more positive redox feature represents a solvento species generated by an equilibrium chloride displacement reaction while the more negative corresponds to the neutral chloro adduct.19,20 Unlike complex 1, no adsorption pre-feature is observed either at high-scan rates or upon the addition of added proton donor to complex 2 (Fig. 3B), implying an improved solubility. As was the case with 1, the addition of PhOH under CO2 saturation results in a large increase in current and loss of reversibility at the most negative redox feature (E1/2 = −1.95 V vs. Fc+/Fc). Variable concentration studies show that the current corresponding to electrochemical CO2 reduction mediated by complex 2 has a first-order concentration dependence with respect to complex 2 (Fig. S36†) and mixed-order dependences on CO2 (Fig. S37†) and PhOH (Fig. S38†). The relative current densities achieved for 1 and 2 at 0.6 M PhOH under CO2 saturation was approximately the same, but less than for the previously reported bpy-based catalyst under comparable conditions.10,19–21 Unlike complex 1, 2 also exhibited a 10 mV shift to more positive Ecat/2 as PhOH concentration increased (Fig. S38†). In the absence of CO2, this shift was not observed, indicating that this shift relates to a modification of either pre-equilibrium CO2 binding or protonation of a CO2-containing intermediate. A comparable shift was not observed for 1.
Next, a weaker proton source, trifluoroethanol (TFE) (estimated pKa = 35.4 in MeCN,63 scaled to ∼24.0 in DMF60) was tested to assess the role of the pendent relay. Similar to PhOH, a large increase in current density and loss of reversibility is observed at the third reduction of 1 and 2 with 0.6 M TFE. For 1 there was again no shift in Ecat/2 with TFE, however, a shift to more positive potentials by 30 mV was observed for the Ecat/2 of 2. Variable concentration studies with 1 and 2 show that the electrochemical reaction under these conditions has a first-order dependence on catalyst and CO2 concentration, as well as TFE for both complexes (Fig. S14–16 and S41–43†). Since these results do not appear to scale with the reported pKa when compared to PhOH, it can be speculated that solvation and homoconjugation effects for TFE impose slightly different constraints on the reaction environment. Finally, the more acidic and cationic acid triethylammonium hexafluorophosphate (TEAHPF6) (pKa = 9.25 in DMF)64 was examined. As was the case with PhOH, a large increase in current density accompanied by a loss of reversibility is observed at the third reduction feature of 1 under CO2 saturation and in the presence of 20 mM TEAHPF6 (Fig. 4). Variable concentration studies show that the electrochemical reaction under these conditions with 1 present has a first-order concentration dependence with respect to catalyst (Fig. S19†), however, mixed-order kinetics are again observed for TEAHPF6 (Fig. S20†), and the catalytic waveform with CO2 shows evidence of an overlapping heterogeneous interaction between the proton donor and the electrode at low CO2 concentrations (Fig. S21†). It is worth emphasizing that increasing to 0.1 M concentrations of TEAHPF6 also results in significant changes to the waveform, suggestive of a heterogeneous interaction between the proton donor and the electrode (Fig. 4 and S26†).65 Consistent with this, comparing the icat/ip ratio to the inverse square root of scan rate showed a linear response that deviated from intercept, indicating competing reactivity with the homogeneous process (Fig. S24†). Complex 1 was found to still exhibit an irreversible redox response consistent with the CO2RR under buffered conditions (equivalent molar addition of conjugate base triethylamine (TEA) to solutions containing TEAH+), although the observed current density diminished significantly (Fig. S22†).
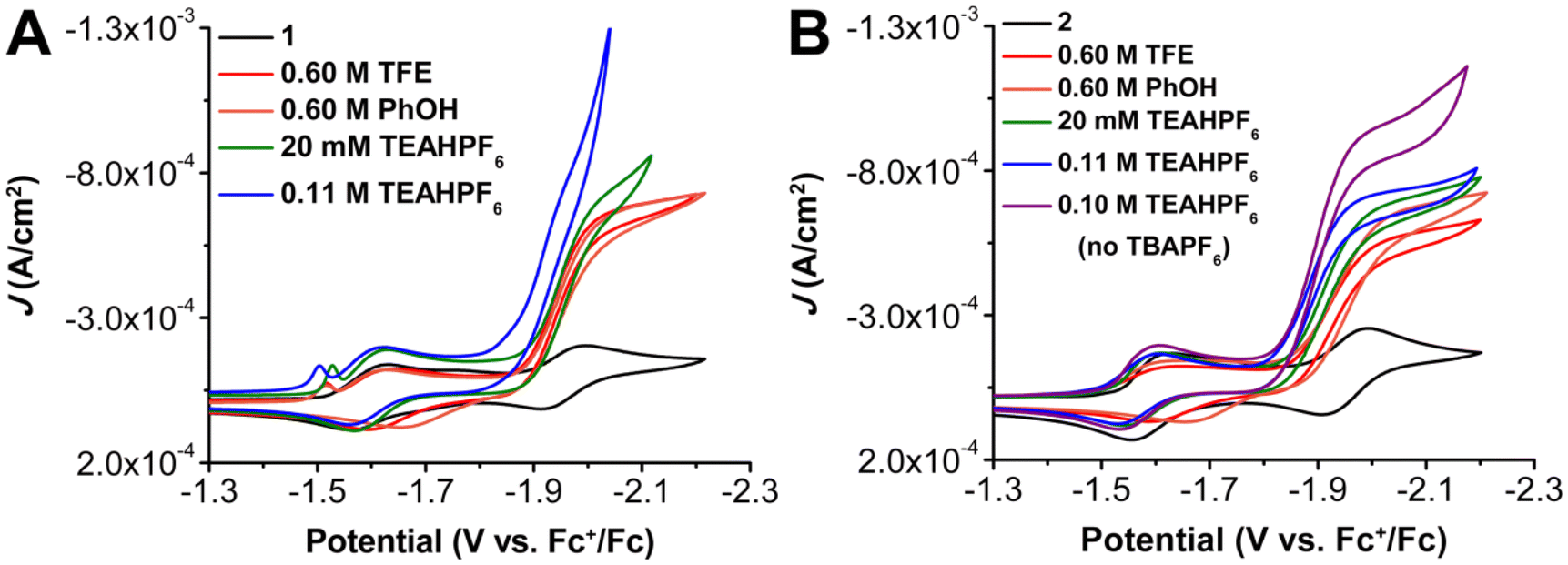 | ||
| Fig. 4 Comparison CVs of (A) Cr(p-tbudhbpy)Cl(H2O) 1 and (B) Cr(nPrdhbpy)Cl(H2O) 2 under CO2 saturation conditions with different proton sources. Conditions: 1.0 mM catalyst, 0.1 M TBAPF6/DMF unless otherwise noted; glassy carbon disc working electrode, glassy carbon rod counter electrode, Ag/AgCl pseudoreference electrode; referenced to Fc+/Fc internal standard; 100 mV s−1 scan rate. | ||
Under unbuffered conditions with 20 mM TEAHPF6 and complex 2, there is a slight increase in catalytic current density, with no waveform changes associated with an interaction between the proton donor and the electrode. Further, complex 2 experiences a 30 mV shift to more positive potentials, Ecat/2 = −1.92 V vs. Fc+/Fc (Fig. 4). It is proposed that the increased shift to more positive potentials for 2 in comparison to 1 is likely the result of the pendent methoxy groups interacting with the ammonium-based proton donor. Variable concentration studies show that the electrochemical reaction shows a mixed-order concentration dependence with respect to catalyst (Fig. S46†), TEAHPF6 (Fig. S47†), and CO2 (Fig. S48†); the concentration of the TEAHPF6 proton donor is fixed at 20 mM when the other reaction components are varied. Unlike 1, at 0.1 M TEAHPF6 concentrations complex 2 remained catalytic for the CO2RR (Ecat/2 = −1.89 V vs. Fc+/Fc) without being overwhelmed by a heterogeneous electrode response. Interestingly, variable concentration studies conducted in the presence of the higher fixed concentration of 0.1 M TEAHPF6 exhibited first-order dependence with respect to catalyst (Fig. S49†) but the reaction order dependence on CO2 still showed a mixed-order response (Fig. S50†). Like 1, complex 2 was found to experience a slight decrease in catalytic activity when under buffered conditions (Fig. S51†). For complex 2 under unbuffered conditions, increasing the concentration of TEAHPF6 resulted again in a corresponding shift of Ecat/2 to more positive potentials; similar titration studies under these conditions did not show a shift of Ecat/2 for 1. Interestingly, measurements under buffered conditions did not reflect as significant a suppression of catalysis for 2 as was observed for 1.
The acid TEAHPF6 is unique in that it has a relatively low saturation concentration in these CV systems (20 mM compared to 0.6 M PhOH or 1.1 M TFE with 2 in DMF) and has the potential to act as both an electrolyte and as an acid. CV studies were conducted on 1 in DMF with 0.1 M TEAHPF6 serving the dual roles of proton donor and supporting electrolyte. Under Ar saturation conditions, 1 has a redox feature that appears at E1/2 = –1.59 V vs. Fc+/Fc followed by a sharp increase in current that suggests the hydrogen evolution reaction (HER) is occurring, possibly originating from the electrode surface (Fig. S25†).65,66 Cross-tracing is also observed, implying either adsorption of homogeneous intermediates to the electrode surface or a slow chemical step leading to the accumulation of reactive species in the reaction-diffusion layer.66 Under CO2 saturation conditions, two redox features appear at E1/2 = –1.59 V and Ep = –1.88 V vs. Fc+/Fc for 1, but the second feature is very slight and is quickly overcome by a large increase consistent with an HER response (Fig. S26†).65 Analogous CV experiments for 2 in DMF with 0.1 M TEAHPF6 as both proton donor and supporting electrolyte under Ar exhibited two redox features at E1/2 = –1.57 V and Ep = –1.95 V vs. Fc+/Fc (Fig. S26†). Under CO2 saturation a large increase in current with a loss of reversibility is observed at the more negative redox feature, with Ecat/2 = –1.90 V vs. Fc+/Fc. The relative current density of this system with 2 and 0.1 M TEAHPF6 as electrolyte and proton donor is also decreased compared to the system which also includes TBAPF6 electrolyte.
Finally, CPE was performed to elucidate differences in product selectivity and assess TOF under preparative conditions for the different acids. CPE experiments for 1 with PhOH present under CO2 saturation at an applied potential of −2.10 V vs. Fc+/Fc showed CO production with a Faradaic efficiency (FECO) of 85 ± 5% accompanied by minor H2 production (Table 1), with a turnover frequency (TOF) of 0.29 s−1. CPE experiments for 2 under comparable conditions demonstrated quantitative efficiency for CO (99 ± 3%) and a slightly improved TOF of 0.93 s−1.10,21 CPE experiments with 1 where TFE was the proton donor produced CO with an efficiency of 103 ± 3% with a TOF of 1.03 s−1. Complex 2 also showed quantitative efficiency of 105 ± 12% and a TOF of 1.50 s−1 with TFE as the proton source. CPE experiments in the presence of TEAHPF6 showed a CO efficiency of 83 ± 1% with a TOF of 0.87 s−1 for complex 1, whereas complex 2 demonstrates quantitative FECO (104 ± 4%) with TEAHPF6, and an increased TOF of 13.70 s−1. Finally, a CPE experiment was conducted with 0.1 M TEAHPF6 serving the dual role of electrolyte and proton source for 2 demonstrated quantitative FECO and a TOF of 5.47 s−1. When TEAHPF6 is used as electrolyte and proton source, more positive potentials are required to suppress background HER, suggesting that the TBA+ component of the electrolyte coulombically screens the strength of the interaction between the electrode and TEAH+, improving the molecular electrocatalytic response.
| Conditions | Potential (V vs. Fc+/Fc) | FECO (%) | FEH2 (%) | TOFCPE (s−1) | η (V) | Turnovers of CO w.r.t [1] or [2] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a 0.75 mM catalyst and 1.5 M acid. b 0.5 mM catalyst and 1.0 M acid. c 0.5 mM catalyst and 20 mM acid. d 0.4 mM catalyst and 16 mM acid. e 0.5 mM catalyst and 0.10 M acid. f 0.1 M TBAPF6 present as electrolyte. g No electrolyte present. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 + PhOHa,f | −2.10 | 85 ± 5 | 5 ± 0 | 0.29 | 0.09 | 2.14 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 + TFEb,f | −2.10 | 103 ± 3 | n/a | 1.03 | — | 3.09 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 + TEAHPF6c,f | −2.10 | 83 ± 1 | 17 ± 0 | 0.87 | 0.70 | 3.50 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 + PhOHb,f | −2.10 | 99 ± 3 | n/a | 0.93 | 0.06 | 3.08 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 + TFEb,f | −2.10 | 105 ± 12 | n/a | 1.50 | — | 2.56 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 + TEAHPF6d,f | −2.10 | 104 ± 4 | n/a | 13.70 | 0.66 | 4.61 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 + TEAHPF6e,g | −2.05 | 103 ± 5 | n/a | 5.47 | 0.66 | 5.05 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Computational results
Given the observed activity difference for complexes 1 and 2, several of the key reaction steps were evaluated to see if this correlated to an intrinsic difference, Table 2. Using a DFT methodology developed in previous comprehensive computational studies,23 a minimal difference is predicted for the thermodynamics and kinetics of CO2 binding, the thermodynamics and kinetics for the rate-limiting protonation of the [Cr−CO2H]− intermediates by TEAH+, as well as for the thermodynamics of CO release. Calculation of the redox potential for the generation of the catalytically active species for complex 1, [Cr(p-tbudhbpy)]0/− = −1.96 V vs. Fc+/Fc, showed good agreement with experimental value of −1.95 V. Likewise, the calculated redox potential at which the catalytically relevant species is generated for complex 2, [Cr(nPrdhbpy)]0/− = –1.97 V vs. Fc+/Fc, was similarly aligned with the experimental value of −1.95 V.| Reaction | ΔG‡ (kcal mol−1) | ΔG (kcal mol−1) |
|---|---|---|
| [Cr(p-tbudhbpy)]− + CO2 ⇌ [Cr(p-tbudhbpy)(CO2)]− | +11.1 | +6.6 |
| [Cr(nPrdhbpy)]− + CO2 ⇌ [Cr(nPrdhbpy)(CO2)]− | +10.9 | +6.5 |
| [Cr(p-tbudhbpy)(CO2H)]− + TEAH+ → [Cr(p-tbudhbpy)(CO)]0 + H2O + TEA | +10.5 | –19.4 |
| [Cr(nPrdhbpy)(CO2H)]− + TEAH+ → [Cr(nPrdhbpy)(CO)]0 + H2O + TEA | +11.9 | –19.0 |
| [Cr(p-tbudhbpy)(CO)]0 ⇌ [Cr(p-tbudhbpy)]0 + CO | –7.4 | |
| [Cr(nPrdhbpy)(CO)]0 ⇌ [Cr(nPrdhbpy)]0 + CO | –7.8 |
Discussion
Using the mechanistic data described above, it is possible for us to propose a role for the pendent relay in modifying the intrinsic catalytic mechanism (Fig. 1 and eqn (1)–(6)). It is helpful here to first summarize the catalytic cycle of the base catalyst family as the following equilibrium and irreversible reaction steps (note for simplicity a neutral acid, HA, is considered, solvento species are not identified, and that the ligand, which remains tetradentate in all cases, is omitted):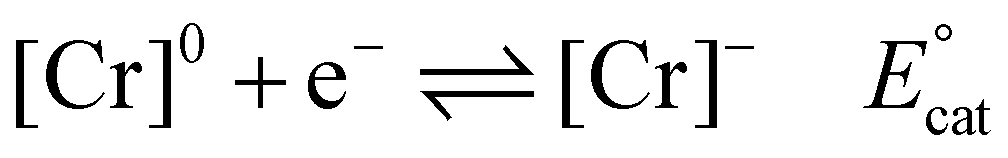 | (1) |
| [Cr]− + CO2 ⇌ [Cr(CO2)]−KCO2 | (2) |
| [Cr(CO2)]− + HA → [Cr(CO2H)]0 + A−\quadk1 | (3) |
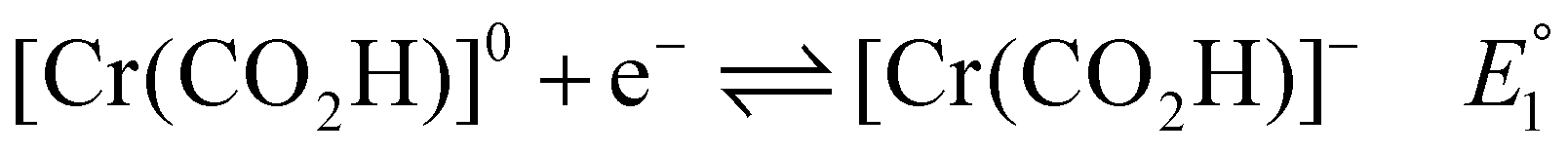 | (4) |
| [Cr(CO2H)]− + HA → [Cr(CO)]0 + H2O + A−\quadk2 | (5) |
| [Cr(CO)]0 ⇌ [Cr]0 + CO KCO | (6) |
Previous mechanistic and computational studies have established that 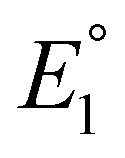 is more positive than
is more positive than 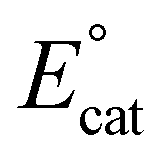 , that KCO2 is an endergonic process (<1), that CO release is facile (no CO adducts observed, KCO ≫ 1), and that k1 ≫ k2. Overall, this allows the electrocatalytic behavior of the original catalyst to be condensed to an ECEC′ framework, considering the two proton transfers to be the relevant chemical steps. Consistent behavior is observed here for complexes 1 and 2, where no CO2 binding is again observed under aprotic conditions (Fig. 3). However, several notable changes occur in conditions with CO2 and a proton donor present. In many cases, mixed-order kinetics are observed for the substrates and the metal complex. Further, in the case of complex 2, a shift to more positive voltages is observed for the catalytic potential with two of the three proton donors. Since the catalytically relevant reduction potentials of 1 and 2 are nearly identical, it is proposed that several pre-equilibrium reactions are contributing to the deviation of both 1 and 2 from the previously reported catalytic behavior (eqn (7) and (11)), as summarized by the modified reaction sequence below:
, that KCO2 is an endergonic process (<1), that CO release is facile (no CO adducts observed, KCO ≫ 1), and that k1 ≫ k2. Overall, this allows the electrocatalytic behavior of the original catalyst to be condensed to an ECEC′ framework, considering the two proton transfers to be the relevant chemical steps. Consistent behavior is observed here for complexes 1 and 2, where no CO2 binding is again observed under aprotic conditions (Fig. 3). However, several notable changes occur in conditions with CO2 and a proton donor present. In many cases, mixed-order kinetics are observed for the substrates and the metal complex. Further, in the case of complex 2, a shift to more positive voltages is observed for the catalytic potential with two of the three proton donors. Since the catalytically relevant reduction potentials of 1 and 2 are nearly identical, it is proposed that several pre-equilibrium reactions are contributing to the deviation of both 1 and 2 from the previously reported catalytic behavior (eqn (7) and (11)), as summarized by the modified reaction sequence below:
 | (1) |
| [Cr]− + HA ⇌ Cr(HA)]−\quadKHA1 | (7) |
| [Cr(HA)]− + CO2 ⇌ [Cr(CO2)(HA)]−\quadKHA\_CO2 | (8) |
| [Cr(CO2)(HA)]− → [Cr(CO2H)]0 + A−\quadk1 | (9) |
 | (10) |
| [Cr(CO2H)]− + HA ⇌ [Cr(CO2H)(HA)]−\quadKHA2 | (11) |
| [Cr(CO2H)(HA)]− → [Cr(CO)]0 + H2O + A−\quadk2 | (12) |
| [Cr(CO)]0 ⇌ [Cr]0 + CO KCO | (6) |
The data suggest that for both proton transfer steps (k1 and k2), a pre-equilibrium involving the association of the acid to reduced form of complex 2, KHA1 > 1, becomes kinetically relevant in the presence of certain acids. It is possible to extract an equilibrium binding constant for the equilibrium interaction between 2 and TEAH+ under Ar saturation of KHA1 = 380 ± 50 (Fig. S64†) at the reduction feature where catalysis is initiated under CO2 saturation. A comparable shift in reduction potential under Ar saturation is not observed for 2 with the weaker and uncharged PhOH and TFEOH proton donors or complex 1 with any of the three proton donors.
The shift observed for the Ecat/2 of complex 2 suggests that these pre-equilibria are catalytically relevant when the methoxy group is present. In a reductive ECEC′ framework, where the second reversible electron transfer occurs at potentials more positive than the first, shifts in catalytic potential can be described according to the following equation:66
 | (13) |
In this framework, positive shifts in potential will be observed as the second-order rate constant k1 increases relative to k2. For the complexes described here, 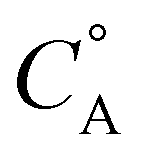 is taken as the concentration of the proton donor and the TOF derived from preparative electrolysis is assigned as
is taken as the concentration of the proton donor and the TOF derived from preparative electrolysis is assigned as 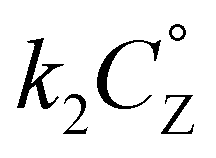 , given that k2 is the rate-limiting step of the reaction and that CVs of the catalytic waves under these conditions have the appropriate S-shape for a kinetically limited response and experience minimal change in current as proton donor concentration is varied. Using the relationship of Ecat/2 and acid concentration, a k1 of 11 ± 2 M−1 s−1 was determined with TFE as a proton donor by averaging values across a concentration range of 0.1 to 1.1 M TFE (Fig. S63†).66 However, comparable analysis of the shifts in Ecat/2 of 2 in relation to acid concentration under electrocatalytic conditions showed that the same k1 value steadily increased from 7.1 × 103 M−1 s−1 at 20 mM TEAH+ to 2.7 × 104 M−1 s−1 at a final concentration of 150 mM (Fig. S65†).67 It is reasoned that the cationic charge introduced in the secondary coordination sphere upon the association of TEAH+ to the anionic Cr complex contributes favorable kinetic and thermodynamic effects to the conversion of reduced CO2 species in a manner analogous to that previously observed for other complexes.15,16,68–74 It is important to acknowledge, however, that it is difficult to completely disentangle this effect from the increased proton donor activity of TEAH+ relative to TFE. As mentioned above, there was no shift in Ecat/2 observed for PhOH with complex 2 and no shift in Ecat/2 for complex 1 with any of the three proton donors. Consistent with the proposed relevance of pre-equilibrium steps when TEAH+ is used as the acid, there is an absence of any significant changes in the energetics and barriers of the key reaction steps for complexes 1 and 2 by DFT, Table 2.
, given that k2 is the rate-limiting step of the reaction and that CVs of the catalytic waves under these conditions have the appropriate S-shape for a kinetically limited response and experience minimal change in current as proton donor concentration is varied. Using the relationship of Ecat/2 and acid concentration, a k1 of 11 ± 2 M−1 s−1 was determined with TFE as a proton donor by averaging values across a concentration range of 0.1 to 1.1 M TFE (Fig. S63†).66 However, comparable analysis of the shifts in Ecat/2 of 2 in relation to acid concentration under electrocatalytic conditions showed that the same k1 value steadily increased from 7.1 × 103 M−1 s−1 at 20 mM TEAH+ to 2.7 × 104 M−1 s−1 at a final concentration of 150 mM (Fig. S65†).67 It is reasoned that the cationic charge introduced in the secondary coordination sphere upon the association of TEAH+ to the anionic Cr complex contributes favorable kinetic and thermodynamic effects to the conversion of reduced CO2 species in a manner analogous to that previously observed for other complexes.15,16,68–74 It is important to acknowledge, however, that it is difficult to completely disentangle this effect from the increased proton donor activity of TEAH+ relative to TFE. As mentioned above, there was no shift in Ecat/2 observed for PhOH with complex 2 and no shift in Ecat/2 for complex 1 with any of the three proton donors. Consistent with the proposed relevance of pre-equilibrium steps when TEAH+ is used as the acid, there is an absence of any significant changes in the energetics and barriers of the key reaction steps for complexes 1 and 2 by DFT, Table 2.
A pre-equilibrium condition can also be used to rationalize the observed increase in catalytic activity observed for complex 2 relative to complex 1 in preparative electrolysis, as outlined recently by Pattanayak and Berben.59 The experimental observations can also be contrasted with the Fe porphyrin derivative reported by Savéant and co-workers, who showed that KCO2 was impacted by the inclusion of pendent proton donors, thanks to intramolecular hydrogen-bonding interactions.47,56 For the Fe porphyrin derivative with pendent proton donors reported by Savéant and co-workers, hydrogen bonding stabilized the Fe–CO2 intermediate, shifting the corresponding redox feature to more positive potentials, however, the subsequent reduction that initiates the rate-determining proton-coupled electron transfer was not shifted by a similar amount, resulting in the appearance of a pre-wave to the catalytic feature. It is reasoned that the difference in the impact of the pendent relay site on reaction parameters reported here has to do with the position of the pendent relay relative to the site of CO2 binding.
A plot of log10(TOF s−1) versus Ecat/2 for the original family of Cr-based molecular catalysts,10,19,21 with PhOH, for CO2 to CO shows a normal Tafel relationship (black squares, Fig. 5), where more negative potentials show an increased TOF. Comparing this trend with the data collected for 1 and 2 with PhOH (blue and red square, respectively, Fig. 5), the more positive Ecat/2 of both species corresponds to a decreased TOF, consistent with a Tafel scaling relationship (more negative catalytic potentials result in greater activity for the same catalyst family).75 However, the response of complex 1 in this series is significantly below the linear trend established by the original complexes and 2. This deviation suggests that the propensity to reversible electrode adsorption limits the activity of 1. Although estimated to be slightly a weaker acid,63,76 TFE shows an increase in activity for both 1 and 2 relative to PhOH, with a slight positive shift in Ecat/2 observed for the latter. These data lead to the assumption that there are additional contributions to the effective proton activity of TFE, or that there are limitations to the estimated pKa. It is possible that the difference may have to do with a combination of solvation effects, homoconjugation, and binding of CO2 by the conjugate base of TFE, since the pKa of TFE is higher than PhOH in known solvents.63,77
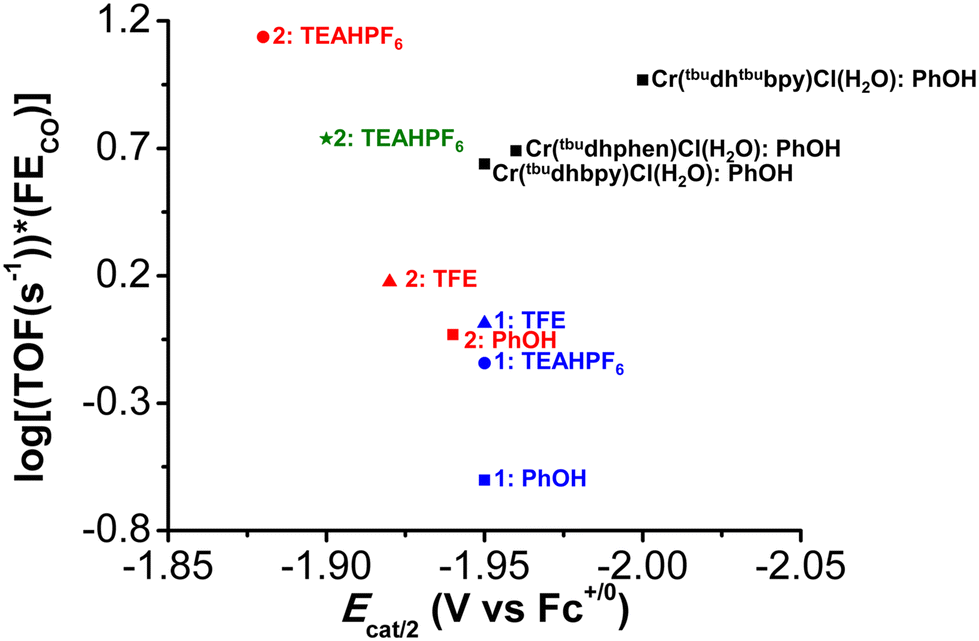 | ||
| Fig. 5 Comparison of activity and standard reduction potential in the Cr catalyst family. Squares are data taken with PhOH and added supporting electrolyte. Triangles are data taken with TFE and added supporting electrolyte. Circles are data taken with triethylammonium as the acid and added supporting electrolyte, while the star indicates data taken with triethylammonium in the absence of supporting electrolyte. | ||
In the presence of the stronger acid, TEAHPF6 (blue and red circles), a significant separation in Ecat/2 is observed between 1 and 2 (60 mV), with the more positive potential of complex 2 showing significantly higher activity. Since an equilibrium binding constant can be established for complex 2 but not 1 under Ar saturation with this acid (Fig. S64†), this effect must be correlated to the involvement of the methoxy group in proton transfer steps following CO2 binding at Cr. Further, the sensitivity of Ecat/2 to the concentration of the ammonium-based acid can be used to demonstrate that the second-order rate constant of the underlying proton transfer in question, k1, has increased significantly from TFE (Fig. S63 and S65†). Thus, it appears that the proton donor can render the pre-equilibrium steps to the formation of intermediate iii kinetically relevant with suitable functional groups in the secondary coordination sphere (Fig. 1).
The potential tuning effect resulting from modifying this pre-equilibrium between complexes 1 and 2 is also noteworthy, especially in light of their activity differences. While TEAH+ associates to the anionic complex i derived from 2, hydrogen-bonding interactions with the pendent methoxy groups would not stabilize CO2 binding based on relative positioning. However, for coulombic reasons, association of the ammonium proton donor to anionic states is overall favorable and observed to improve proton transfer to the electronegative O atoms of bound CO2 reduction intermediates, as negative charge develops there during C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond breaking. Indeed, Martin and Mayer demonstrated that it is the amount of positive charge, rather than the position of the charge, which matters in activity enhancements.16 However, the utility of using cationic acids to achieve this is tempered by the significant increase in overpotential required. Going forward, these data imply that in order to harness this improved operating potential, the pre-equilibrium proton donor association needs to be enhanced when weaker acids are used. This suggests that optimizing the pendent relay ligand design, rather than proton donor choice, will ultimately lead to the most significant performance increases in the long-term.
O bond breaking. Indeed, Martin and Mayer demonstrated that it is the amount of positive charge, rather than the position of the charge, which matters in activity enhancements.16 However, the utility of using cationic acids to achieve this is tempered by the significant increase in overpotential required. Going forward, these data imply that in order to harness this improved operating potential, the pre-equilibrium proton donor association needs to be enhanced when weaker acids are used. This suggests that optimizing the pendent relay ligand design, rather than proton donor choice, will ultimately lead to the most significant performance increases in the long-term.
While pendent functional groups increasing activity at a fixed reduction potential are known48,78 and the pre-equilibrium effect has been previously observed to impact speciation during electrocatalysis,59 future efforts could focus on the combination of these effects to achieve inverse potential scaling for homogeneous electrocatalytic activity.79 It is worth noting that alternative strategies for accessing inverse potential scaling based on the ligand and reaction system design also exist and offer complementary opportunities to tune performance.10,80 Since there has been general interest in the introduction of charge to impact the kinetics and operating potential of molecular CO2 reduction catalysts,15,16,80 in addition to the interest in pendent relays described above, it would be advantageous to leverage both effects.79
Conclusion
These experimental results demonstrate that the combination of a pre-equilibrium effect directed by a pendent functional group and the use of a cationic proton donor can achieve improved activity at lower operating potentials during homogeneous CO2 reduction. Mechanistic studies comparing neutral proton donors to the cationic one show minimal changes in activity or operating potentials unless the cationic acid is present, with a particularly strong interaction occurring in the presence of the pendent relay. Therefore, it is reasoned that the use of cationic acids can generally be regarded as beneficial when the catalytic reduction cycle involves several anionic intermediate species in organic solvents. Based on these conclusions, further studies exploring cationic acids and optimizing the ligand framework to interact with the metal complex following CO2 binding and activation are currently underway. Importantly, this strategy should be applicable to other proton-coupled electron transfer steps in small molecule reduction reactions, offering a new design strategy for optimizing activity and selectivity.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Catalysis Science Program, under Award DE-SC0022219. Single crystal X-ray diffraction experiments were performed on a diffractometer at the University of Virginia funded by the NSF-MRI program (CHE-2018870).References
- Carbon dioxide now more than 50% higher than pre-industrial levels. U.S. Department of Commerce, 2022https://www.noaa.gov/news-release/carbon-dioxide-now-more-than-50-higher-than-pre-industrial-levels.
- Cumulative CO2 Emissions by World Region. Our World in Data: https://ourworldindata.org/grapher/cumulative-co2-emissions-region?stackMode=absolute, 2023.
- A. Adamu, F. Russo-Abegão and K. Boodhoo, Process intensification technologies for CO2 capture and conversion – a review, BMC Chem. Eng., 2020, 2(1), 2, DOI:10.1186/s42480-019-0026-4.
- H. Ishida, Electrochemical/Photochemical CO2 Reduction Catalyzed by Transition Metal Complexes, in Carbon Dioxide Chemistry, Capture and Oil Recovery, ed. J. S. Iyad Karame and H. Srour, InTech Open, 2018 Search PubMed.
- M. Marchese, G. Buffo, M. Santarelli and A. Lanzini, CO2 from direct air capture as carbon feedstock for Fischer-Tropsch chemicals and fuels: Energy and economic analysis, J. CO2 Util., 2021, 46, 101487, DOI:10.1016/j.jcou.2021.101487.
- Z. Ma, U. Legrand, E. Pahija, J. R. Tavares and D. C. Boffito, From CO2 to Formic Acid Fuel Cells, Ind. Eng. Chem. Res., 2021, 60(2), 803–815, DOI:10.1021/acs.iecr.0c04711.
- J. W. Raebiger, J. W. Turner, B. C. Noll, C. J. Curtis, A. Miedaner, B. Cox and D. L. DuBois, Electrochemical Reduction of CO2 to CO Catalyzed by a Bimetallic Palladium Complex, Organometallics, 2006, 25(14), 3345–3351, DOI:10.1021/om060228g.
- Z. Chen, C. Chen, D. R. Weinberg, P. Kang, J. J. Concepcion, D. P. Harrison, M. S. Brookhart and T. J. Meyer, Electrocatalytic reduction of CO2 to CO by polypyridyl ruthenium complexes, Chem. Commun., 2011, 47(47), 12607–12609, 10.1039/C1CC15071E.
- K.-Y. Wong, W.-H. Chung and C.-P. Lau, The effect of weak Brönsted acids on the electrocatalytic reduction of carbon dioxide by a rhenium tricarbonyl bipyridyl complex, J. Electroanal. Chem., 1998, 453(1), 161–170, DOI:10.1016/S0022-0728(98)00116-8.
- A. G. Reid, J. J. Moreno, S. H. Hooe, K. R. Baugh, I. H. Thomas, D. A. Dickie and C. W. Machan, Inverse Potential Scaling in Co-Electrocatalytic Activity for CO2 Reduction Through Redox Mediator Tuning and Catalyst Design, Chem. Sci., 2022, 13, 9595–9606 RSC.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, Nickel(II)-cyclam: an extremely selective electrocatalyst for reduction of CO2 in water, J. Chem. Soc., Chem. Commun., 1984, 19, 1315–1316, 10.1039/C39840001315.
- B. J. Fisher and R. Eisenberg, Electrocatalytic reduction of carbon dioxide by using macrocycles of nickel and cobalt, J. Am. Chem. Soc., 1980, 102(24), 7361–7363, DOI:10.1021/ja00544a035.
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, [Mn(bipyridyl)(CO)3Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction, Angew. Chem., Int. Ed., 2011, 50(42), 9903–9906, DOI:10.1002/anie.201103616.
- C. Costentin, M. Robert and J.-M. Savéant, Current Issues in Molecular Catalysis Illustrated by Iron Porphyrins as Catalysts of the CO2-to-CO Electrochemical Conversion, Acc. Chem. Res., 2015, 48(12), 2996–3006, DOI:10.1021/acs.accounts.5b00262.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, Through-Space Charge Interaction Substituent Effects in Molecular Catalysis Leading to the Design of the Most Efficient Catalyst of CO2-to-CO Electrochemical Conversion, J. Am. Chem. Soc., 2016, 138(51), 16639–16644, DOI:10.1021/jacs.6b07014.
- D. J. Martin and J. M. Mayer, Oriented Electrostatic Effects on O2 and CO2 Reduction by a Polycationic Iron Porphyrin, J. Am. Chem. Soc., 2021, 143(30), 11423–11434, DOI:10.1021/jacs.1c03132.
- C. Jiang, A. W. Nichols and C. W. Machan, A look at periodic trends in d-block molecular electrocatalysts for CO2 reduction, Dalton Trans., 2019, 48(26), 9454–9468, 10.1039/C9DT00491B.
- R. Francke, B. Schille and M. Roemelt, Homogeneously Catalyzed Electroreduction of Carbon Dioxide—Methods, Mechanisms, and Catalysts, Chem. Rev., 2018, 118(9), 4631–4701, DOI:10.1021/acs.chemrev.7b00459.
- S. L. Hooe, J. M. Dressel, D. A. Dickie and C. W. Machan, Highly Efficient Electrocatalytic Reduction of CO2 to CO by a Molecular Chromium Complex, ACS Catal., 2020, 10(2), 1146–1151, DOI:10.1021/acscatal.9b04687.
- A. G. Reid, S. L. Hooe, J. J. Moreno, D. A. Dickie and C. W. Machan, Homogeneous Electrocatalytic Reduction of CO2 by a CrN3O Complex: Electronic Coupling with a Redox-Active Terpyridine Fragment Favors Selectivity for CO, Inorg. Chem., 2022, 61(43), 16963–16970, DOI:10.1021/acs.inorgchem.2c02013.
- A. G. Reid, M. E. Moberg, C. A. Koellner, J. J. Moreno, S. L. Hooe, K. R. Baugh, D. A. Dickie and C. W. Machan, Comparisons of bpy and phen Ligand Backbones in Cr-Mediated (Co-)Electrocatalytic CO2 Reduction, Organometallics, 2023, 42(11), 1139–1148, DOI:10.1021/acs.organomet.2c00600.
- M. E. Moberg and C. W. Machan, Design of Cr-Based Molecular Electrocatalyst Systems for the CO2 Reduction Reaction, Acc. Chem. Res., 2024, 57, 2326–2335, DOI:10.1021/acs.accounts.4c00283.
- J. J. Moreno, S. L. Hooe and C. W. Machan, DFT Study on the Electrocatalytic Reduction of CO2 to CO by a Molecular Chromium Complex, Inorg. Chem., 2021, 60(6), 3635–3650, DOI:10.1021/acs.inorgchem.0c03136.
- J.-M. Savéant, Proton Relays in Molecular Catalysis of Electrochemical Reactions: Origin and Limitations of the Boosting Effect, Angew. Chem., Int. Ed., 2019, 58(7), 2125–2128, DOI:10.1002/anie.201812375.
- B. Reuillard, C. Costentin and V. Artero, Deciphering Reversible Homogeneous Catalysis of the Electrochemical H2 Evolution and Oxidation: Role of Proton Relays and Local Concentration Effects, Angew. Chem., Int. Ed., 2023, 62(36), e202302779, DOI:10.1002/anie.202302779.
- M. R. Dubois and D. L. Dubois, Development of Molecular Electrocatalysts for CO2 Reduction and H2 Production/Oxidation, Acc. Chem. Res., 2009, 42(12), 1974–1982, DOI:10.1021/ar900110c.
- A. W. Nichols and C. W. Machan, Secondary-Sphere Effects in Molecular Electrocatalytic CO2 Reduction, Front. Chem., 2019, 7, 397, DOI:10.3389/fchem.2019.00397.
- A. W. Nichols, E. N. Cook, Y. J. Gan, P. R. Miedaner, J. M. Dressel, D. A. Dickie, H. S. Shafaat and C. W. Machan, Pendent Relay Enhances H2O2 Selectivity during Dioxygen Reduction Mediated by Bipyridine-Based Co–N2O2 Complexes, J. Am. Chem. Soc., 2021, 143(33), 13065–13073, DOI:10.1021/jacs.1c03381.
- S. Sinha, M. Ghosh and J. J. Warren, Changing the Selectivity of O2 Reduction Catalysis with One Ligand Heteroatom, ACS Catal., 2019, 9(3), 2685–2691, DOI:10.1021/acscatal.8b04757.
- L. Wang, M. Gennari, F. G. Cantú Reinhard, J. Gutiérrez, A. Morozan, C. Philouze, S. Demeshko, V. Artero, F. Meyer, S. P. de Visser and C. Duboc, A Non-Heme Diiron Complex for (Electro)catalytic Reduction of Dioxygen: Tuning the Selectivity through Electron Delivery, J. Am. Chem. Soc., 2019, 141(20), 8244–8253, DOI:10.1021/jacs.9b02011.
- J. Y. Yang, R. M. Bullock, W. G. Dougherty, W. S. Kassel, B. Twamley, D. L. DuBois and M. R. DuBois, Reduction of oxygen catalyzed by nickel diphosphine complexes with positioned pendant amines, Dalton Trans., 2010, 39(12), 3001–3010, 10.1039/B921245K.
- H. Kotani, T. Yagi, T. Ishizuka and T. Kojima, Enhancement of 4-electron O2 reduction by a Cu(II)–pyridylamine complex via protonation of a pendant pyridine in the second coordination sphere in water, Chem. Commun., 2015, 51(69), 13385–13388, 10.1039/C5CC03012A.
- R. L. Shook, S. M. Peterson, J. Greaves, C. Moore, A. L. Rheingold and A. S. Borovik, Catalytic Reduction of Dioxygen to Water with a Monomeric Manganese Complex at Room Temperature, J. Am. Chem. Soc., 2011, 133(15), 5810–5817, DOI:10.1021/ja106564a.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. R. DuBois and D. L. DuBois, Hydrogen Oxidation and Production Using Nickel-Based Molecular Catalysts with Positioned Proton Relays, J. Am. Chem. Soc., 2006, 128(1), 358–366, DOI:10.1021/ja056442y.
- R. M. Henry, R. K. Shoemaker, D. L. DuBois and M. R. DuBois, Pendant Bases as Proton Relays in Iron Hydride and Dihydrogen Complexes, J. Am. Chem. Soc., 2006, 128(9), 3002–3010, DOI:10.1021/ja057242p.
- A. D. Wilson, R. K. Shoemaker, A. Miedaner, J. T. Muckerman, D. L. DuBois and M. R. DuBois, Nature of hydrogen interactions with Ni(II) complexes containing cyclic phosphine ligands with pendant nitrogen bases, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(17), 6951–6956, DOI:10.1073/pnas.0608928104.
- M. R. DuBois and D. L. DuBois, The roles of the first and second coordination spheres in the design of molecular catalysts for H2 production and oxidation, Chem. Sci. Rev., 2009, 38(1), 62–72, 10.1039/B801197B.
- B. E. Barton and T. B. Rauchfuss, Hydride-Containing Models for the Active Site of the Nickel−Iron Hydrogenases, J. Am. Chem. Soc., 2010, 132(42), 14877–14885, DOI:10.1021/ja105312p.
- C. H. Lee, D. K. Dogutan and D. G. Nocera, Hydrogen Generation by Hangman Metalloporphyrins, J. Am. Chem. Soc., 2011, 133(23), 8775–8777, DOI:10.1021/ja202136y.
- M. M. Roubelakis, D. K. Bediako, D. K. Dogutan and D. G. Nocera, Proton-coupled electron transfer kinetics for the hydrogen evolution reaction of hangman porphyrins, Energy Environ. Sci., 2012, 5(7), 7737–7740, 10.1039/C2EE21123H.
- D. K. Bediako, B. H. Solis, D. K. Dogutan, M. M. Roubelakis, A. G. Maher, C. H. Lee, M. B. Chambers, S. Hammes-Schiffer and D. G. Nocera, Role of pendant proton relays and proton-coupled electron transfer on the hydrogen evolution reaction by nickel hangman porphyrins, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(42), 15001–15006, DOI:10.1073/pnas.1414908111.
- D. Dolui, S. Khandelwal, A. Shaik, D. Gaat, V. Thiruvenkatam and A. Dutta, Enzyme-Inspired Synthetic Proton Relays Generate Fast and Acid-Stable Cobalt-Based H2 Production Electrocatalysts, ACS Catal., 2019, 9(11), 10115–10125, DOI:10.1021/acscatal.9b02953.
- N. Queyriaux, D. Sun, J. Fize, J. Pécaut, M. J. Field, M. Chavarot-Kerlidou and V. Artero, Electrocatalytic Hydrogen Evolution with a Cobalt Complex Bearing Pendant Proton Relays: Acid Strength and Applied Potential Govern Mechanism and Stability, J. Am. Chem. Soc., 2020, 142(1), 274–282, DOI:10.1021/jacs.9b10407.
- S. Lin, S. Banerjee, M. T. Fortunato, C. Xue, J. Huang, A. Y. Sokolov and C. Turro, Electrochemical Strategy for Proton Relay Installation Enhances the Activity of a Hydrogen Evolution Electrocatalyst, J. Am. Chem. Soc., 2022, 144(44), 20267–20277, DOI:10.1021/jacs.2c06011.
- A. W. Nichols, S. L. Hooe, J. S. Kuehner, D. A. Dickie and C. W. Machan, Electrocatalytic CO2 Reduction to Formate with Molecular Fe(III) Complexes Containing Pendent Proton Relays, Inorg. Chem., 2020, 59(9), 5854–5864, DOI:10.1021/acs.inorgchem.9b03341.
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, Electrocatalytic reduction of carbon dioxide by nickel cyclam2+ in water: study of the factors affecting the efficiency and the selectivity of the process, J. Am. Chem. Soc., 1986, 108(24), 7461–7467, DOI:10.1021/ja00284a003.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst, Science, 2012, 338(6103), 90–94, DOI:10.1126/science.1224581.
- E. M. Nichols, J. S. Derrick, S. K. Nistanaki, P. T. Smith and C. J. Chang, Positional effects of second-sphere amide pendants on electrochemical CO2 reduction catalyzed by iron porphyrins, Chem. Sci., 2018, 9(11), 2952–2960, 10.1039/C7SC04682K.
- A. Chapovetsky, T. H. Do, R. Haiges, M. K. Takase and S. C. Marinescu, Proton-Assisted Reduction of CO2 by Cobalt Aminopyridine Macrocycles, J. Am. Chem. Soc., 2016, 138(18), 5765–5768, DOI:10.1021/jacs.6b01980 , From NLM.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille and P. J. A. Kenis, et al., Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation, Chem. Rev., 2013, 113(8), 6621–6658, DOI:10.1021/cr300463y.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, Molecular Cobalt Complexes with Pendant Amines for Selective Electrocatalytic Reduction of Carbon Dioxide to Formic Acid, J. Am. Chem. Soc., 2017, 139(10), 3685–3696, DOI:10.1021/jacs.6b11474.
- C. Costentin, M. Robert, J.-M. Savéant and A. Tatin, Efficient and selective molecular catalyst for the CO2-to-CO electrochemical conversion in water, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(22), 6882–6886, DOI:10.1073/pnas.1507063112.
- J. T. Bays, N. Priyadarshani, M. S. Jeletic, E. B. Hulley, D. L. Miller, J. C. Linehan and W. J. Shaw, The Influence of the Second and Outer Coordination Spheres on Rh(diphosphine)2 CO2 Hydrogenation Catalysts, ACS Catal., 2014, 4(10), 3663–3670, DOI:10.1021/cs5009199.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, Ultraefficient homogeneous catalyst for the CO2-to-CO electrochemical conversion, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(42), 14990–14994, DOI:10.1073/pnas.1416697111.
- A. Chapovetsky, M. Welborn, J. M. Luna, R. Haiges, T. F. Miller and S. C. Marinescu III, Pendant Hydrogen-Bond Donors in Cobalt Catalysts Independently Enhance CO2 Reduction, ACS Cent. Sci., 2018, 4(3), 397–404, DOI:10.1021/acscentsci.7b00607.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, Pendant Acid–Base Groups in Molecular Catalysts: H-Bond Promoters or Proton Relays? Mechanisms of the Conversion of CO2 to CO by Electrogenerated Iron(0)Porphyrins Bearing Prepositioned Phenol Functionalities, J. Am. Chem. Soc., 2014, 136(33), 11821–11829, DOI:10.1021/ja506193v.
- A. Sonea, K. L. Branch and J. J. Warren, The Pattern of Hydroxyphenyl-Substitution Influences CO2 Reduction More Strongly than the Number of Hydroxyphenyl Groups in Iron-Porphyrin Electrocatalysts, ACS Catal., 2023, 13(6), 3902–3912, DOI:10.1021/acscatal.2c06275.
- K. T. Ngo, M. McKinnon, B. Mahanti, R. Narayanan, D. C. Grills, M. Z. Ertem and J. Rochford, Turning on the Protonation-First Pathway for Electrocatalytic CO2 Reduction by Manganese Bipyridyl Tricarbonyl Complexes, J. Am. Chem. Soc., 2017, 139(7), 2604–2618, DOI:10.1021/jacs.6b08776.
- S. Pattanayak and L. A. Berben, Pre-Equilibrium Reaction Mechanism as a Strategy to Enhance Rate and Lower Overpotential in Electrocatalysis, J. Am. Chem. Soc., 2023, 145(6), 3419–3426, DOI:10.1021/jacs.2c10942.
- J. M. Dressel, E. N. Cook, S. L. Hooe, J. J. Moreno, D. A. Dickie and C. W. Machan, Electrocatalytic hydrogen evolution reaction by a Ni(N2O2) complex based on 2,2′-bipyridine, Inorg. Chem. Front., 2023, 10(3), 972–978, 10.1039/D2QI01928K.
- M. L. Pegis, J. A. S. Roberts, D. J. Wasylenko, E. A. Mader, A. M. Appel and J. M. Mayer, Standard Reduction Potentials for Oxygen and Carbon Dioxide Couples in Acetonitrile and N,N-Dimethylformamide, Inorg. Chem., 2015, 54(24), 11883–11888, DOI:10.1021/acs.inorgchem.5b02136.
- A. J. Sathrum and C. P. Kubiak, Kinetics and Limiting Current Densities of Homogeneous and Heterogeneous Electrocatalysts, J. Phys. Chem. Lett., 2011, 2(18), 2372–2379, DOI:10.1021/jz2008227.
- Y. C. Lam, R. J. Nielsen, H. B. Gray and W. A. Goddard III, A Mn Bipyrimidine Catalyst Predicted To Reduce CO2 at Lower Overpotential, ACS Catal., 2015, 5(4), 2521–2528, DOI:10.1021/cs501963v.
- K. Izutsu, Electrochemistry in Nonaqueous Solutions, Wiley-VCH, 2009 Search PubMed.
- B. D. McCarthy, D. J. Martin, E. S. Rountree, A. C. Ullman and J. L. Dempsey, Electrochemical Reduction of Brønsted Acids by Glassy Carbon in Acetonitrile—Implications for Electrocatalytic Hydrogen Evolution, Inorg. Chem., 2014, 53(16), 8350–8361, DOI:10.1021/ic500770k.
- C. Costentin and J.-M. Savéant, Elements of Molecular and Biomolecular Electrochemistry: An Electrochemical Approach to Electron Transfer Chemistry; John Wiley & Sons Inc., 2019. DOI:10.1002/9781119292364.
- N. Elgrishi, D. A. Kurtz and J. L. Dempsey, Reaction Parameters Influencing Cobalt Hydride Formation Kinetics: Implications for Benchmarking H2-Evolution Catalysts, J. Am. Chem. Soc., 2017, 139(1), 239–244, DOI:10.1021/jacs.6b10148.
- Y. Mao, M. Loipersberger, K. J. Kron, J. S. Derrick, C. J. Chang, S. M. Sharada and M. Head-Gordon, Consistent inclusion of continuum solvation in energy decomposition analysis: theory and application to molecular CO2 reduction catalysts, Chem. Sci., 2021, 12(4), 1398–1414, 10.1039/D0SC05327A.
- D. C. Grills, Y. Matsubara, Y. Kuwahara, S. R. Golisz, D. A. Kurtz and B. A. Mello, Electrocatalytic CO2 Reduction with a Homogeneous Catalyst in Ionic Liquid: High Catalytic Activity at Low Overpotential, J. Phys. Chem. Lett., 2014, 5(11), 2033–2038, DOI:10.1021/jz500759x.
- X. Li and J. A. Panetier, Computational Study for CO2-to-CO Conversion over Proton Reduction Using [Re[bpyMe(Im-R)](CO)3Cl]+ (R = Me, Me2, and Me4) Electrocatalysts and Comparison with Manganese Analogues, ACS Catal., 2021, 11(21), 12989–13000, DOI:10.1021/acscatal.1c02899.
- E. Vichou, Y. Li, M. Gomez-Mingot, M. Fontecave and C. M. Sánchez-Sánchez, Imidazolium- and Pyrrolidinium-Based Ionic Liquids as Cocatalysts for CO2 Electroreduction in Model Molecular Electrocatalysis, J. Phys. Chem. C, 2020, 124(43), 23764–23772, DOI:10.1021/acs.jpcc.0c07556.
- S. Sung, X. Li, L. M. Wolf, J. R. Meeder, N. S. Bhuvanesh, K. A. Grice, J. A. Panetier and M. Nippe, Synergistic Effects of Imidazolium-Functionalization on fac-Mn(CO)3 Bipyridine Catalyst Platforms for Electrocatalytic Carbon Dioxide Reduction, J. Am. Chem. Soc., 2019, 141(16), 6569–6582, DOI:10.1021/jacs.8b13657.
- S. Sahu, P. L. Cheung, C. W. Machan, S. A. Chabolla, C. P. Kubiak and N. C. Gianneschi, Charged Macromolecular Rhenium Bipyridine Catalysts with Tunable CO2 Reduction Potentials, Chem. – Eur. J., 2017, 23(36), 8619–8622, DOI:10.1002/chem.201701901.
- S. Sung, D. Kumar, M. Gil-Sepulcre and M. Nippe, Electrocatalytic CO2 Reduction by Imidazolium-Functionalized Molecular Catalysts, J. Am. Chem. Soc., 2017, 139(40), 13993–13996, DOI:10.1021/jacs.7b07709.
- C. Costentin and J.-M. Savéant, Homogeneous Molecular Catalysis of Electrochemical Reactions: Manipulating Intrinsic and Operational Factors for Catalyst Improvement, J. Am. Chem. Soc., 2018, 140(48), 16669–16675, DOI:10.1021/jacs.8b09154.
- S. L. Hooe, A. L. Rheingold and C. W. Machan, Electrocatalytic Reduction of Dioxygen to Hydrogen Peroxide by a Molecular Manganese Complex with a Bipyridine-Containing Schiff Base Ligand, J. Am. Chem. Soc., 2018, 140(9), 3232–3241, DOI:10.1021/jacs.7b09027.
- P. Ballinger and F. A. Long, Acid Ionization Constants of Alcohols. I. Trifluoroethanol in the Solvents H2O and D2O1, J. Am. Chem. Soc., 1959, 81(5), 1050–1053, DOI:10.1021/ja01514a010.
- K. Teindl, B. O. Patrick and E. M. Nichols, Linear Free Energy Relationships and Transition State Analysis of CO2 Reduction Catalysts Bearing Second Coordination Spheres with Tunable Acidity, J. Am. Chem. Soc., 2023, 145(31), 17176–17186, DOI:10.1021/jacs.3c03919.
- S. Bhunia, A. Ghatak, A. Rana and A. Dey, Amine Groups in the Second Sphere of Iron Porphyrins Allow for Higher and Selective 4e−/4H+ Oxygen Reduction Rates at Lower Overpotentials, J. Am. Chem. Soc., 2023, 145(6), 3812–3825, DOI:10.1021/jacs.2c13552.
- W. Nie, D. E. Tarnopol and C. C. L. McCrory, Enhancing a Molecular Electrocatalyst's Activity for CO2 Reduction by Simultaneously Modulating Three Substituent Effects, J. Am. Chem. Soc., 2021, 143(10), 3764–3778, DOI:10.1021/jacs.0c09357.
Footnote |
| † Electronic supplementary information (ESI) available: Additional material and methods, CV, UV-vis, NMR, and electrochemical data. CCDC 2330314 and 2330315. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01981d |
| This journal is © The Royal Society of Chemistry 2024 |