
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalysis as an enabling tool for enantioselective ring-opening reactions of cyclopropanes
Efraim
Reyes
 ,
Uxue
Uria
,
Liher
Prieto
,
Luisa
Carrillo
and
Jose L.
Vicario
*
,
Uxue
Uria
,
Liher
Prieto
,
Luisa
Carrillo
and
Jose L.
Vicario
*
Department of Organic and Inorganic Chemistry, University of the Basque Country (UPV/EHU), P.O. Box 644, 48080 Bilbao, Spain. E-mail: joseluis.vicario@ehu.es
First published on 13th June 2024
Abstract
The rich reactivity profile of cyclopropanes has been extensively explored to trigger new organic transformations that enable unusual disconnective approaches to synthesize molecular motifs that are not easily reached through conventional reactions. In particular, the chemistry of cyclopropanes has received special attention in the last decade, with multiple new approaches that capitalize on the use of organocatalysis for the activation of the cyclopropane scaffold. This situation has also opened the possibility of developing enantioselective variants of many reactions that until now were only carried out in an enantiospecific or diastereoselective manner. Our group has been particularly active in this field, focusing more specifically on the use of aminocatalysis and Brønsted acid catalysis as major organocatalytic activation manifolds to trigger new unprecedented transformations involving cyclopropanes that add to the current toolbox of general methodologies available to organic chemists for the enantioselective synthesis of chiral compounds.
 Efraim Reyes | Efraim Reyes was born in Galdakao (Bizkaia, Spain) in 1978. He graduated from the University of the Basque Country (UPV/EHU) in 2001 and received his PhD in 2006, under the direction of Prof. Dr D. Badia and Prof. Jose L. Vicario. After postdoctoral studies in 2007-2008 at the Center for Catalysis of the University of Aarhus (Denmark) under the supervision of Prof. Dr K. A. Jørgensen, he returned to the Research Group of Asymmetric Synthesis (UPV/EHU), where he is currently Associate Professor. |
 Uxue Uria | Uxue Uria was born in Sopelana (Bizkaia) in 1982. She graduated from the University of the Basque Country, Basque Country, Spain in 2005 and received her PhD in 2009, working in the development of organocatalytic conjugate additions under the direction of Prof. Dr D. Badia and Prof. Jose L. Vicario. After postdoctoral studies in the group of Prof. Dr Magnus Rueping at RWTH Aachen (Germany) from 2010 to 2012 working in the use of chiral Brønsted acid catalysts, she returned to Bilbao, where she is currently Associate Professor. |
 Liher Prieto | Liher Prieto was born in Bilbao (Bizkaia) in 1989. He graduated from the University of the Basque Country in 2012 and received the PhD degree in 2017 under the direction of Prof. Dr Jose L. Vicario and Prof. E. Reyes. He carried out a predoctoral stay under the supervision of Prof. P. S. Baran at the Scripps Research Institute in the USA (2015) and postdoctoral studies in the group of Prof. Dr Mark Lautens at the University of Toronto (2018-2019) exploring transition metal catalyzed transformations, after which he was appointed Assistant Professor at the University of the Basque Country. |
 Luisa Carrillo | Luisa Carrillo was born in Bilbao in 1965. She graduated from the University of the Basque Country in 1988 and received her PhD in 1998, working in the Department of Organic Chemistry for the Faculty of Science at the same university. She then joined the Group of Asymmetric Synthesis under the direction of Prof. Dr D. Badía, where she was appointed associate professor in 2001 and full professor in 2020. |
 Jose L. Vicario | Jose L. Vicario was born in Elda (Alicante, Spain) in 1973. He graduated from the University of the Basque Country in 1996 and received his PhD in 2000 at the same university. After postdoctoral studies in 2002 at RWTH Aachen (Germany) under the supervision of Prof. Dieter Enders, he returned to the Department of Organic and Inorganic Chemistry of the University of the Basque Country to start his independent career, where he is currently Full Professor. Current research interests focus on the design of new methodologies in asymmetric synthesis, especially asymmetric organocatalysis, and stereocontrolled synthesis of pharmacologically active compounds. |
Introduction
Asymmetric organocatalysis has proved to be an extraordinarily useful and versatile methodology in organic synthesis and has contributed to significantly expanding the toolbox of available approaches that synthetic chemists can choose when facing the enantioselective preparation of a given chiral molecular target.1 The conceptual definition of organocatalysis is accepted to have been established in 2000,2 and, since then, the field has experienced exponential growth leading to the development and optimization of multiple organocatalytic methodologies and to the establishment of the foundations of different organocatalytic activation manifolds. The first conceptual advances and the mechanistic insights gathered thereof paved the way for the expansion of asymmetric organocatalysis into more complex systems, and, in fact, the exploration of less conventional and more elaborate substrates represents a natural progression in the evolution of this area, pushing the boundaries of the potential applicability of this concept as a general tool in synthesis.Among those unconventional substrates that have displayed a range of interesting reactivity patterns upon organocatalytic activation, cyclopropanes stand out as a particularly intriguing and challenging family of compounds and have contributed to expanding the range of reactions in which organocatalysis can be applied.3 The inherent ring strain associated with the presence of the cyclopropane scaffold provides a distinctive reactivity, typically leading to transformations driven by the release of strain associated with a ring-opening process in the presence of an external reagent.4 In particular, the combination of a cyclopropane-containing substrate with an organocatalyst has led in many cases to the observation of unconventional reactivity that has led to the development of very useful novel catalytic methodologies that enable the synthesis of molecular scaffolds that are not easily accessible through standard approaches.
Depending on the substitution pattern, cyclopropanes can be classified according to their reactivity (see Fig. 1) as electrophilic (those containing an electron-withdrawing substituent),5 nucleophilic (those with an electron-donating substituent)6 and donor–acceptor cyclopropanes7 (which incorporate an electron-donating and an electron-withdrawing substituent in adjacent positions).
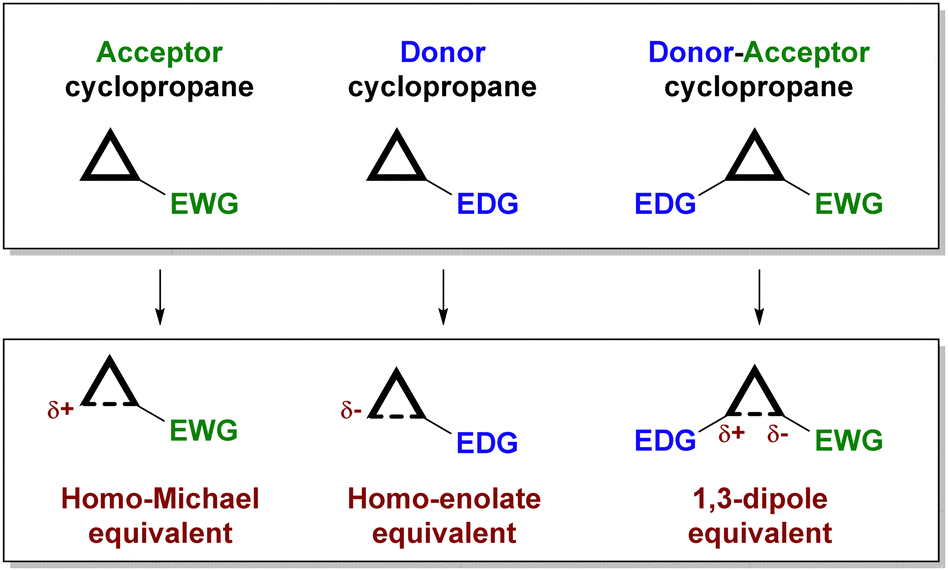 | ||
| Fig. 1 Classification of cyclopropanes according to their reactivity pattern. | ||
While electrophilic and nucleophilic cyclopropanes usually react directly with nucleophiles or electrophiles, respectively, in a concerted manner that involves a ring-opening event, donor–acceptor cyclopropanes typically engage in a ring-opening process to generate a zwitterionic 1,3-dipolar intermediate that subsequently undergoes reaction with an external reagent (a nucleophile and/or electrophile, or very often a dipolarophile, through formal cycloaddition chemistry). This particular behaviour of donor–acceptor cyclopropanes is a consequence of the high polarization of the cyclopropane C–C bond due to the synergistic contribution of the two substituents, which facilitates the ring-opening event from a kinetic point of view and contributes to the stabilization of the dipolar intermediate.
On the other hand, there are several ways for an organocatalyst to activate a cyclopropane-containing substrate, depending on the interaction between the two reagents (Fig. 2). On the one hand, covalent organocatalysis, such as aminocatalysis (enamine or iminium ion catalysis)8 or N-heterocyclic carbene9 catalysis, implies the condensation of the organocatalyst with the cyclopropane substrate, which must incorporate a formyl substituent for such a condensation to take place. In this way, an activated electron-donor site is generated in situ on the cyclopropane substrate upon enamine formation or after the Breslow-type intermediate is produced. Alternatively, the formation of an iminium ion leads to an activated electron-withdrawing moiety. The formation of all these covalently connected substrate/catalyst intermediates implies an increase in the polarization of cyclopropane C–C, which therefore results in activation towards the subsequent ring-opening process. On the other hand, a molecule operating through a noncovalent organocatalytic activation manifold, typically an H-bond donor10 or a stronger Brønsted acid11 catalyst, can interact with an electron-withdrawing substituent to increase its releasing ability, which also results in a more polarized cyclopropane C–C bond. In all these situations, the possibility of transferring stereochemical information from a chiral catalyst to the final product during the reaction arises.
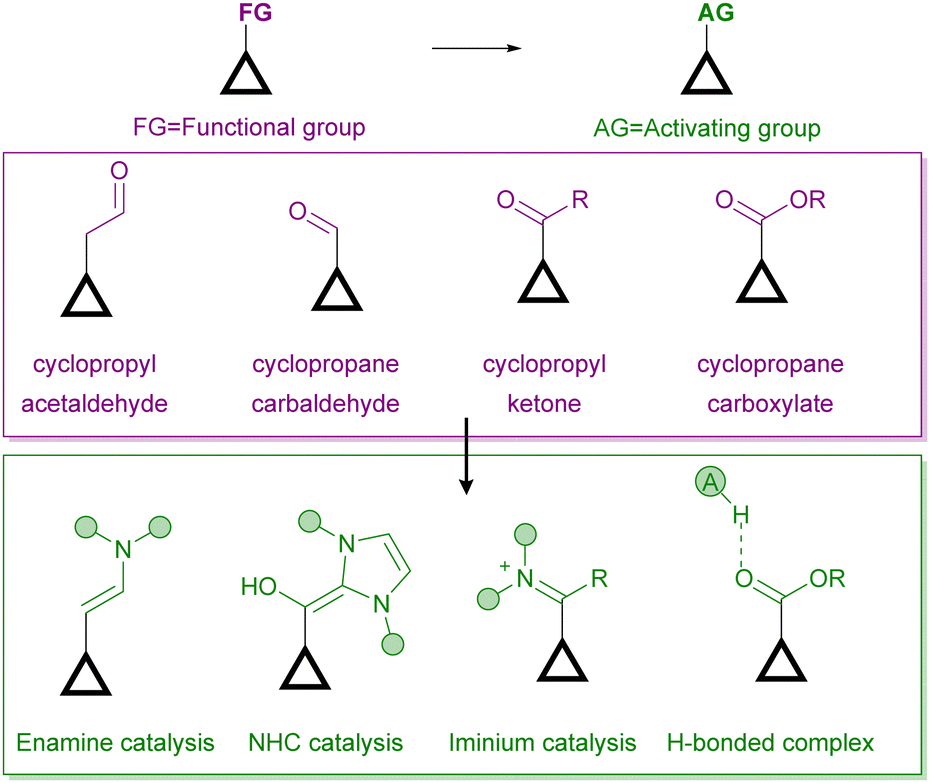 | ||
| Fig. 2 Organocatalytic activation manifolds typically employed for the activation of cyclopropanes in enantioselective ring-opening reactions. | ||
Some early precedents have already demonstrated the power of organocatalysis to trigger ring-opening processes in cyclopropane substrates (Scheme 1). For instance, with respect to covalent organocatalysis, in 2007, Wang and coworkers reported the use of a secondary amine catalyst to trigger the ring-opening of cyclopropanecarbaldehyde 1 containing strongly electron-withdrawing substituents at the two other positions of the cyclopropane core to quantitatively generate α-malonyl-β-aryl-substituted conjugated enal 2.12 The mechanistic proposal involved the participation of an activated iminium intermediate. On the contrary, the first use of N-heterocyclic carbene catalysis in this context was described one year earlier by Bode and coworkers, in a report in which 3-substituted 2-aroyl cyclopropanecarbaldehydes 3 were converted into acyclic δ-oxoesters 4 through reaction with methanol in the presence of imidazole-derived carbene catalyst C-2.13 The ability of Brønsted acids to promote ring-opening processes or rearrangements on acylcyclopropanes has been well documented in the literature since the beginning of the XX century.14 With the advent of the “organocatalysis era,” the first use of bifunctional thiourea/quinine C-3 engaging in H-bonding interactions to assist a deprotonation-induced desymmetrizative ring-opening of bicyclo[3.1.0]hexane 5 substrate was reported by Jørgensen in 2009.15
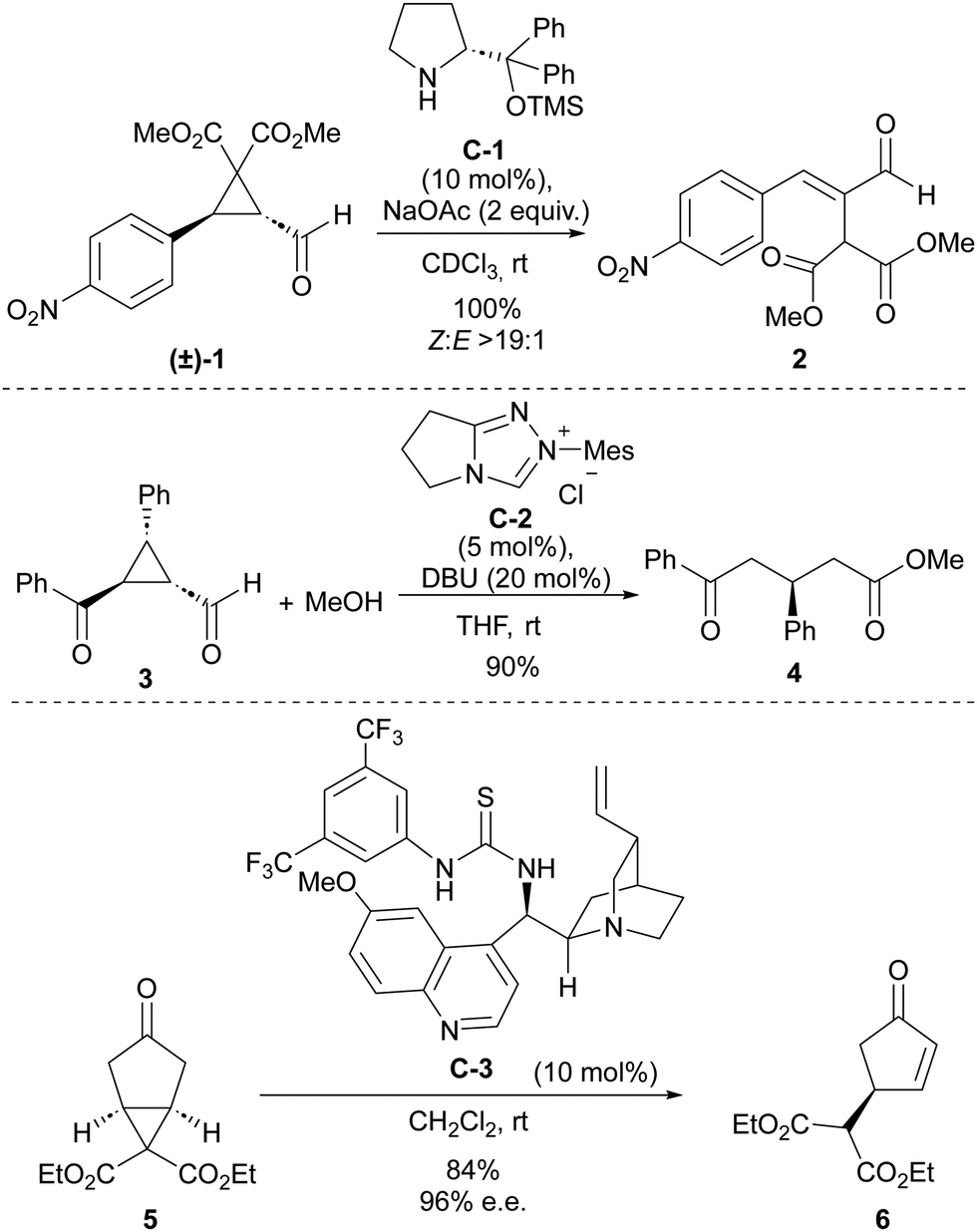 | ||
| Scheme 1 Pioneering examples of the use of organocatalysis for the activation of cyclopropanes. | ||
From these initial steps, various reactions have been reported, which demonstrate the enormous versatility offered by different organocatalytic activation manifolds to engage in the activation of cyclopropanes, provided that the correct functional group is placed within the structure of the starting material to enable the organocatalyst to play its role. The possibility of using this chemistry to develop enantioselective versions of such reactions is a highly appealing feature that increases the added value of these approaches. Our research group has been particularly active in developing this chemistry recently, and these contributions will be highlighted in the following text, providing the appropriate context with respect to other reports in the literature.
Iminium ion catalysis
The LUMO-lowering effect associated with the formation of an iminium ion compared to the corresponding cyclopropanecarbaldehyde starting material implies an increase in the polarization of the cyclopropane C–C bond, therefore enhancing its electrophilicity. This provides an activation effect towards the reaction with an external nucleophile, resulting in a formal homo-Michael-type addition process. One of the first attempts to develop such a ring-opening process under iminium ion catalysis involved the reaction between 2-aryl-substituted cyclopropanecarbaldehyde 7 and o-mercaptobenzaldehyde 8 catalyzed by L-proline C-4 (Scheme 2),12 providing benzothiepine 9 as final product resulting from an intramolecular aldol condensation reaction taking place after the ring-opening event. Interestingly, this reaction was found to be enantiospecific, providing enantioenriched materials in which the enantiomeric excess of the starting material had been mostly preserved, while racemic products were obtained when racemic cyclopropanes were used as starting materials. This is an indication that the C–C bond cleavage event operating during the ring-opening process takes place through an SN2-type process.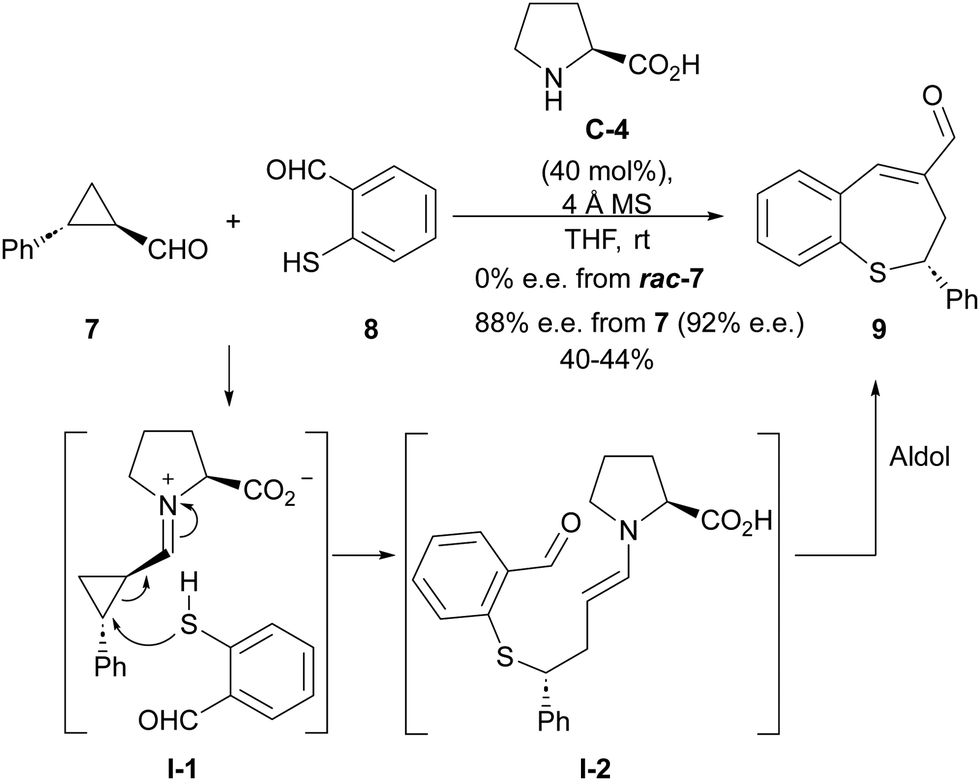 | ||
| Scheme 2 Iminium activation of cyclopropanecarbaldehydes in the reaction of o-mercaptobenzaldehydes. | ||
The fact that iminium activation implies enantiospecific SN2 ring-opening of cyclopropane led to the use of achiral meso-cyclopropanecarbaldehydes 10 as suitable substrates to implement an enantioselective version of this transformation. This was demonstrated for the first time by Gilmour and coworkers in the conversion of this type of cyclopropane-containing substrates into enantioenriched acyclic α,γ-dichloroaldehydes 13 through chloride-induced ring-opening followed by electrophilic α-chlorination with 11 and 12 (Scheme 3).16 In this transformation, the MacMillan-type imidazolidinone C-6 was identified as the best performing catalyst, which is involved in the stereodifferentiation between the two prostereogenic carbon atoms of cyclopropanecarbaledehyde iminium ion I-3 during the ring-opening event triggered by the nucleophilic addition of the chloride anion. After this, the generated nucleophilic enamine intermediate I-4 was subsequently α-chlorinated by hexachlorocyclohexa-2,4-diene-1-one 12, which was incorporated within the initial reaction mixture. This second reaction also took place under catalyst control, therefore leading to the preferential formation of one diastereoisomer with a very high enantiomeric excess.
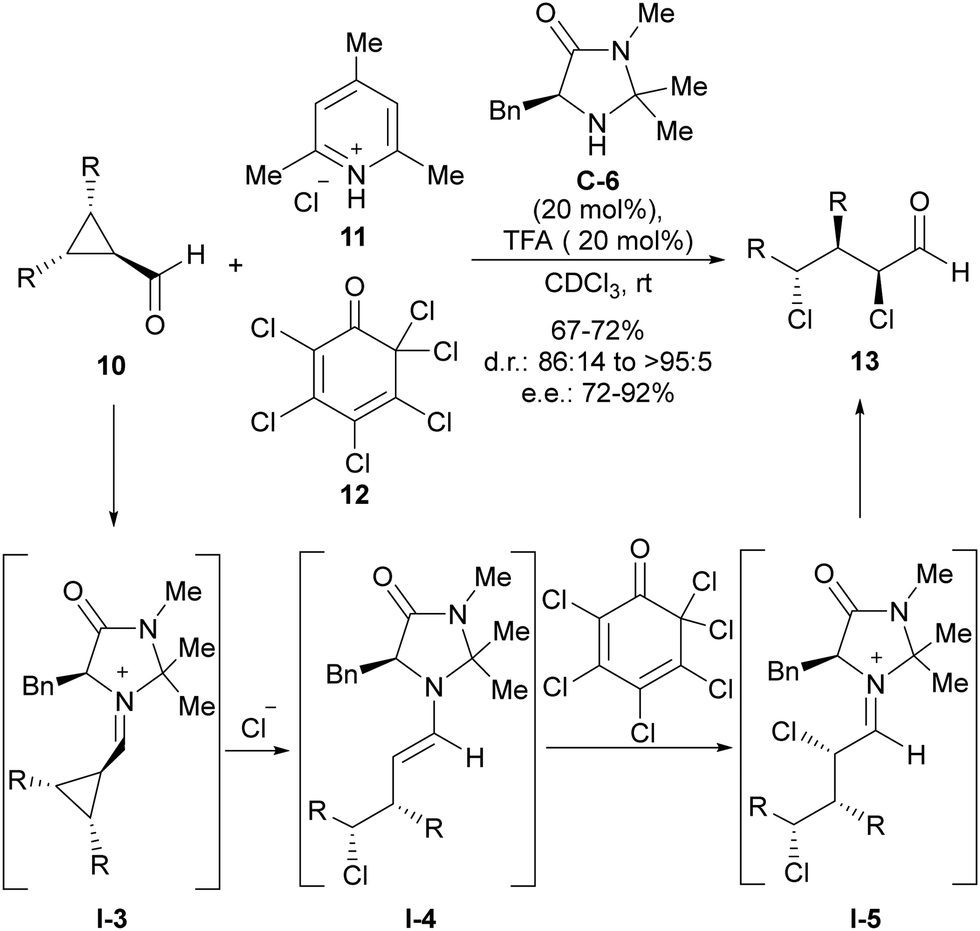 | ||
| Scheme 3 Chloride-addition-induced ring-opening of meso-cyclopropanecarbaldehydes under iminium ion activation. | ||
Following this reaction design, our group evaluated the possibility of using weak nucleophiles such as carboxylates to initiate the ring-opening process under this activation manifold without the need to incorporate any additional electrophilic reagent for the external trapping of the enamine intermediate (Scheme 4).17 In this case, a Jørgensen–Hayashi-type18O-methyldiphenylsilyl-substituted diarylprolinol C-7 catalyst was employed in a reaction that led to γ-acyloxy-substituted aldehydes 16 in good-to-excellent yields and high stereoselectivities in most cases. The reaction works perfectly well using various benzoic acids 15 as pronucleophiles containing either electron-withdrawing or electron-donating substituents at different positions of the aryl moiety, as well as heteroaromatics such as furfurilic acid. The ability of different meso-cyclopropanecarbaldehydes 14 was also evaluated, with the reaction performing well with bicyclic cyclopropanes or simpler 2,3-disubstituted cyclopropanecarbaldehydes with either two identical alkyl or aryl substituents. In all cases, a single diastereoisomer was formed in the reaction. Remarkably, aliphatic carboxylic acids could also be employed to trigger the ring-opening event, including α-aminoacids. In the latter case, the reaction was also observed to proceed under catalyst control, without any remarkable matched/mismatched scenario associated with the use of a chiral catalyst combined with a chiral pronucleophile.
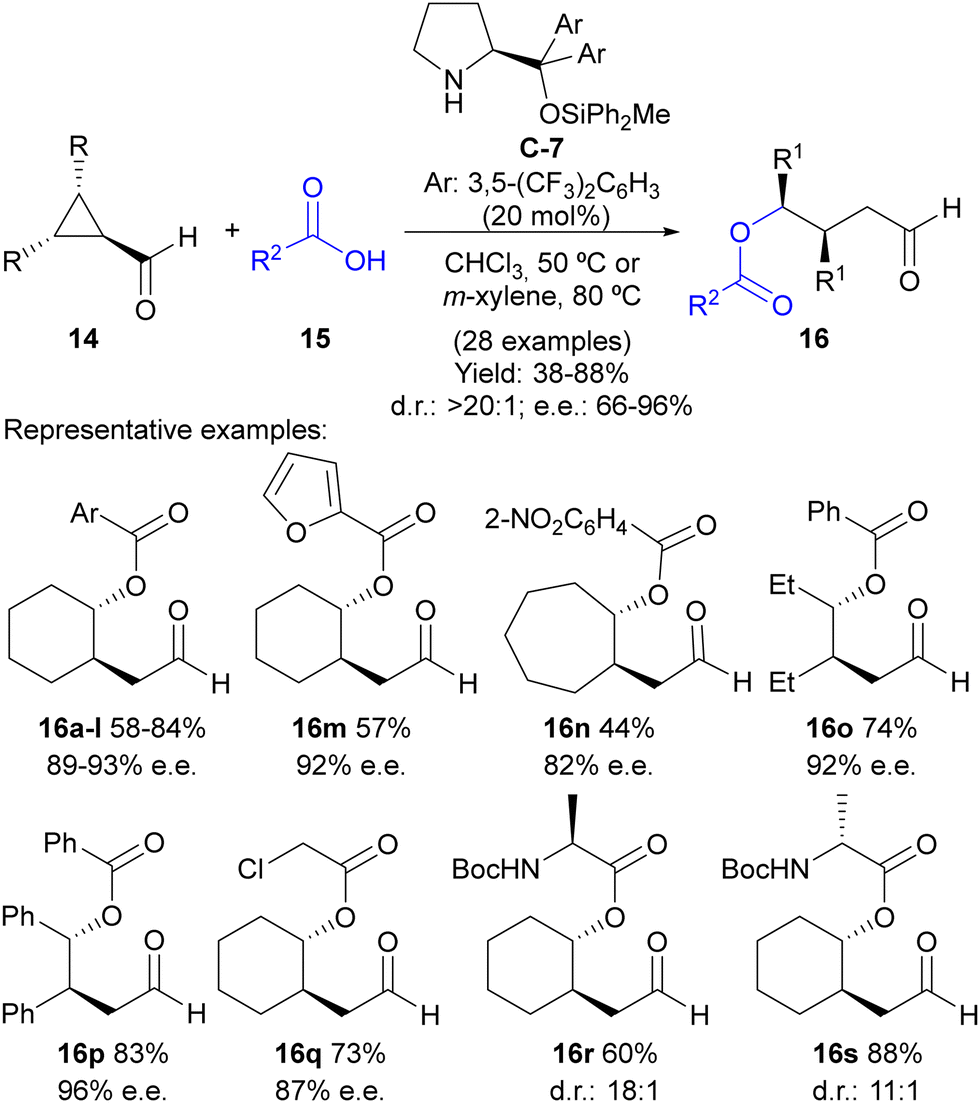 | ||
| Scheme 4 Desymmetrization of meso-cyclopropanecarbaldehydes through carboxylate-addition-initiated ring opening under iminium ion activation. | ||
Computational studies were carried out to fully understand the mechanism of the reaction. These confirmed the participation of the iminium ion intermediate that leads to a more polarized cyclopropane C–C bond and therefore to an activated electrophilic cyclopropane reagent. The carboxylic acid was also found to play a dual role in the reaction, initially activating the formyl group of starting cyclopropanecarbaldehyde 14a during the condensation with aminocatalyst C-1 and next acting as the nucleophile, in which it remains associated with the water molecule that had been released in the iminium ion formation step TS-1, stabilizing the iminium/carboxylate ion pair I-7. Remarkably, close inspection of TS-2, which corresponds to the ring-opening event, and its associated intrinsic reaction coordinate showed an important asynchronicity in the process. A more careful study of the progress of the reaction was therefore conducted and, in particular, we carried out a topological analysis of the electron localization function (ELF),19 which provides information about the evolution of electronic populations along the reaction coordinate from ion pair I-7 into I-8 through TS-2 for selected bonds and atoms (see Fig. 3). This ELF analysis indicated that when the cyclopropane C1–C3 bond is broken, N5 and the C3–C4 bond increase their electronic population, and at the same time, the electronic population of the C4–N5 bond decreases to values that are consistent with the presence of a single bond. This is consistent with an initial process that involves the beginning of the ring-opening of the cyclopropane ring and concomitant evolution of the iminium ion towards the enamine moiety. However, carboxylate oxygen maintains the same electron population throughout this process; it decreases only somewhat later as a consequence of the C–O bond formation, which is revealed by the higher electron density between those two atoms. The observed gap between the C–C bond cleavage event and C–O bond formation revealed the formal existence of a carbocation, which is not stable enough to be located as a minimum and also forms an intimate ion pair with the water-stabilized carboxylate anion.20 As a consequence, the mechanism of this ring-opening reaction should be regarded as an inverted SN1 process rather than a typical SN-type reaction. Since there are no intermediates, the overall process takes place in a single kinetic step but is regarded as a concerted asynchronous reaction since two events are identified along the reaction coordinate. The formation of such a contact ion pair21 explains the complete inversion of configuration observed and is also in good agreement with the differences in reactivity noted between cyclohexane-fused cyclopropanecarbaldehydes and related bicyclic substrates incorporating a fused cycloheptane or cyclopentane moiety. The lower reactivity experimentally observed for the latter is explained in terms of the reported lower stability of cyclopentyl and cycloheptyl carbocations with respect to cyclohexyl carbocations, as calculated through the solvolysis of corresponding 1-aryl-1-cycloalkanols.22 Finally, with respect to the high enantioselectivity observed during desymmetrization, calculations showed a clear preference (over 4.0 kcal mol−1) for the pathway leading to the experimentally observed enantiomer when comparing the energies of the TS associated with the reaction of the carboxylate nucleophile with each of the two carbon atoms of the cyclopropane moiety that can eventually lead to the two possible enantiomers.
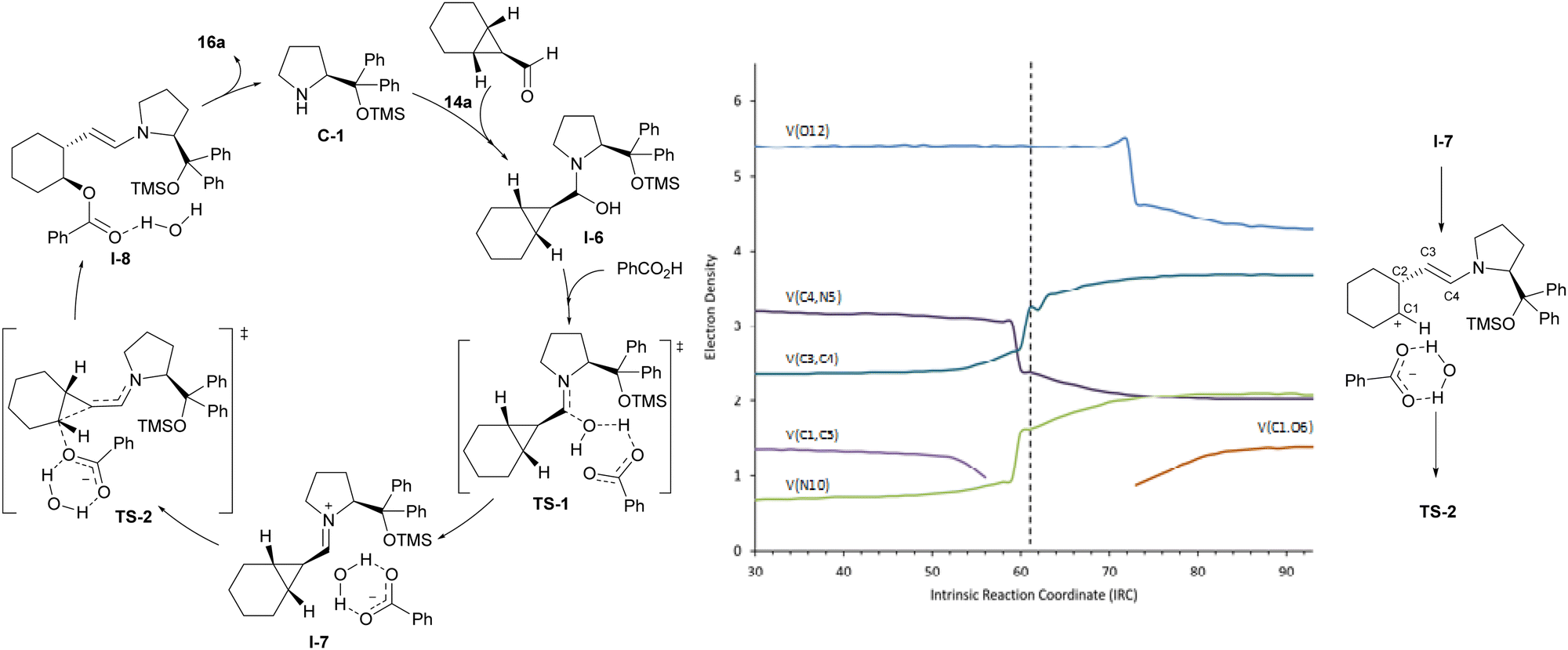 | ||
| Fig. 3 Mechanistic proposal and computational studies for the desymmetrizative ring-opening of meso-cyclopropanecarbaldehyde under iminium ion catalysis. | ||
The practical applicability of this reaction was highlighted by accomplishing the first total synthesis of the natural product speciosin H (Scheme 5), which is a metabolite isolated from Hexagonia speciosa, a basidiomycete fungus that grows in subtropical areas of China.23 The key step with respect to the installation of stereochemical information involved the desymmetrizative ring-opening of bicyclic cyclopropanecarbaldehyde 17 incorporating an internal alkene as the synthetic handle to introduce the additional secondary alcohol moiety present on the target. This key reaction proceeded with excellent yield and stereocontrol and on a multigram (5 gram) scale. Subsequent elaborations involved the protection of the formyl moiety in 19 and diastereoselective epoxidation of 20 followed by regioselective ring-opening with a hydride reagent. The final installation of the trisubstituted alkene was accomplished by deprotection of the formyl group to furnish 22, subsequent Wittig reaction and final ester cleavage. The complete synthetic route consisted of a linear six-step synthesis with an overall 31% yield.
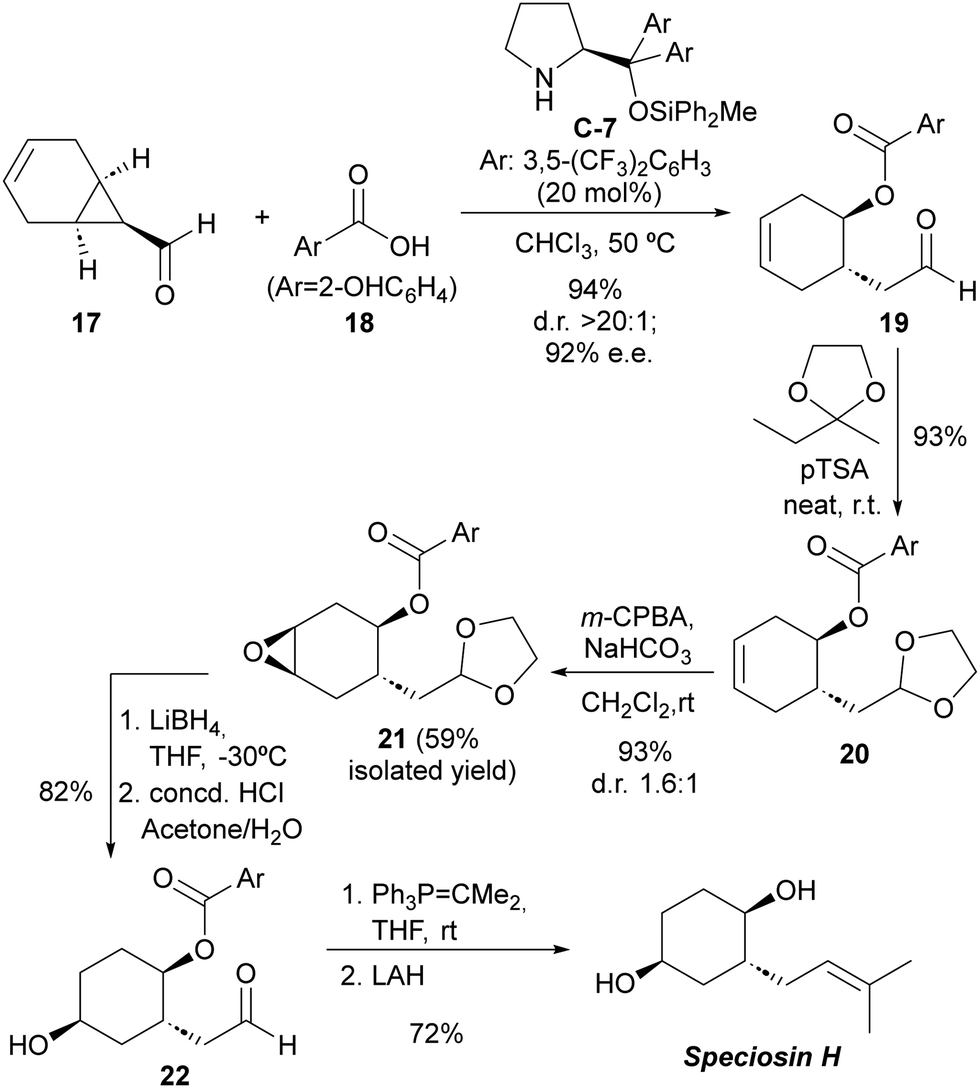 | ||
| Scheme 5 Total synthesis of speciosin H using the desymmetrization of meso-cyclopropanecarbaldehydes as the key step. | ||
Enamine catalysis
The HOMO-raising effect associated with the formation of an enamine moiety entails the formation of an electron-donating site being generated in a cyclopropaneacetaldehyde substrate that becomes activated towards the ring-opening event. This is especially evident in the case of cyclopropylacetaldehydes that incorporate one or more electron-withdrawing substituents at the adjacent positions in the cyclopropane scaffold, which leads to the formation of a highly polarized donor–acceptor cyclopropane. This idea was pioneered by Jørgensen, who demonstrated that cyclopropylacetaldehyde 23 containing two alkoxycarbonyl groups react with highly electrophilic Michael acceptor 24 in the presence of a chiral prolinol-derived secondary amine to provide complex cyclobutene adduct 25 in which the cyclopropane scaffold has undergone ring-opening (Scheme 6).24 The overall process consists of the initial generation of the donor–acceptor cyclopropane intermediate I-9 upon enamine formation, which, upon ring-opening, delivers an unsaturated iminium ion intermediate I-10 that is converted into its dienamine tautomer. This electron-rich dienamine is known to be reactive towards activated Michael acceptors in formal (2+2) cycloaddition through I-11, which is mechanistically understood as a Michael/Michael cascade reaction, generating cyclobutene scaffold I-12, and final product 25 is formed after the release of the catalyst through hydrolysis. Remarkably, racemic materials could be used as the cyclopropane starting material, as all stereochemical information is deleted in the ring-opening process and therefore, the chiral catalyst is directly involved in controlling four stereogenic centres that are generated in the cyclobutene formation step.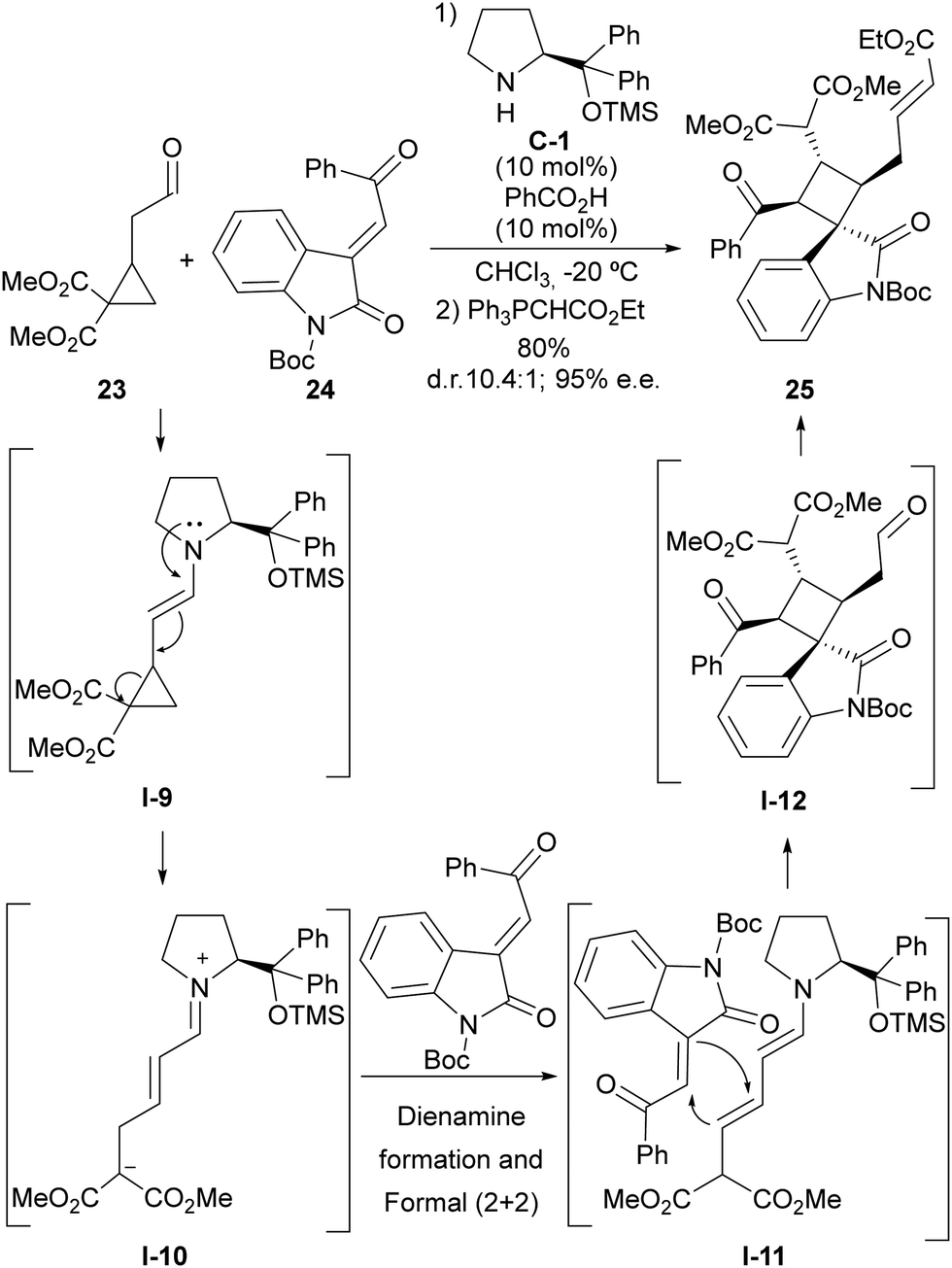 | ||
| Scheme 6 Enamine activation in the ring-opening reaction of cyclopropaneacetaldehydes followed by formal (2+2) cycloaddition. | ||
The authors showed through computational studies that the formation of enamine species I-9 also led to a significant increase in the C–C bond length with respect to starting cyclopropylacetaldehyde, an effect that was calculated to be especially enhanced on cyclopropane substrates that present the enamine moiety and two geminal adjacent ester moieties. Moreover, calculations indicated the presence of a predominant conformation in which the π-orbital of enamine and the σ*C–C orbital of the cyclopropane lay in a parallel arrangement, thus resulting in a more kinetically favourable C–C bond cleavage process.
Soon afterwards, our group also made use of this reaction design in the direct synthesis of dihydroquinolines 28 through the reaction of substituted cyclopropylacetaldehydes 26 with 2-aminobenzaldehydes 27 (Scheme 7).25 In this particular case, we succeeded in engaging conjugated iminium ion I-13 intermediate formed after the ring-opening event in the subsequent reaction with the external reagent, which was a distinctive feature with respect to the previous report by Jorgensen, which involved dienamine-type reactivity. Therefore, 2-aminobenzaldehyde reagent 27 underwent a cascade aza-Michael reaction followed by intramolecular aldol reaction, which, as shown in I-14, generated dihydroisoquinoline scaffold I-15 as a highly enantioenriched compound. In addition, these adducts were directly transformed into pyrroloquinolines 28 through a one-pot, three-step reaction sequence that capitalized on the innate reactivity of the two alkoxycarbonyl groups present at the pendant lateral chain and that originated from the starting cyclopropane, making full use of all reactive points of cyclopropylacetaldehyde starting material 26.
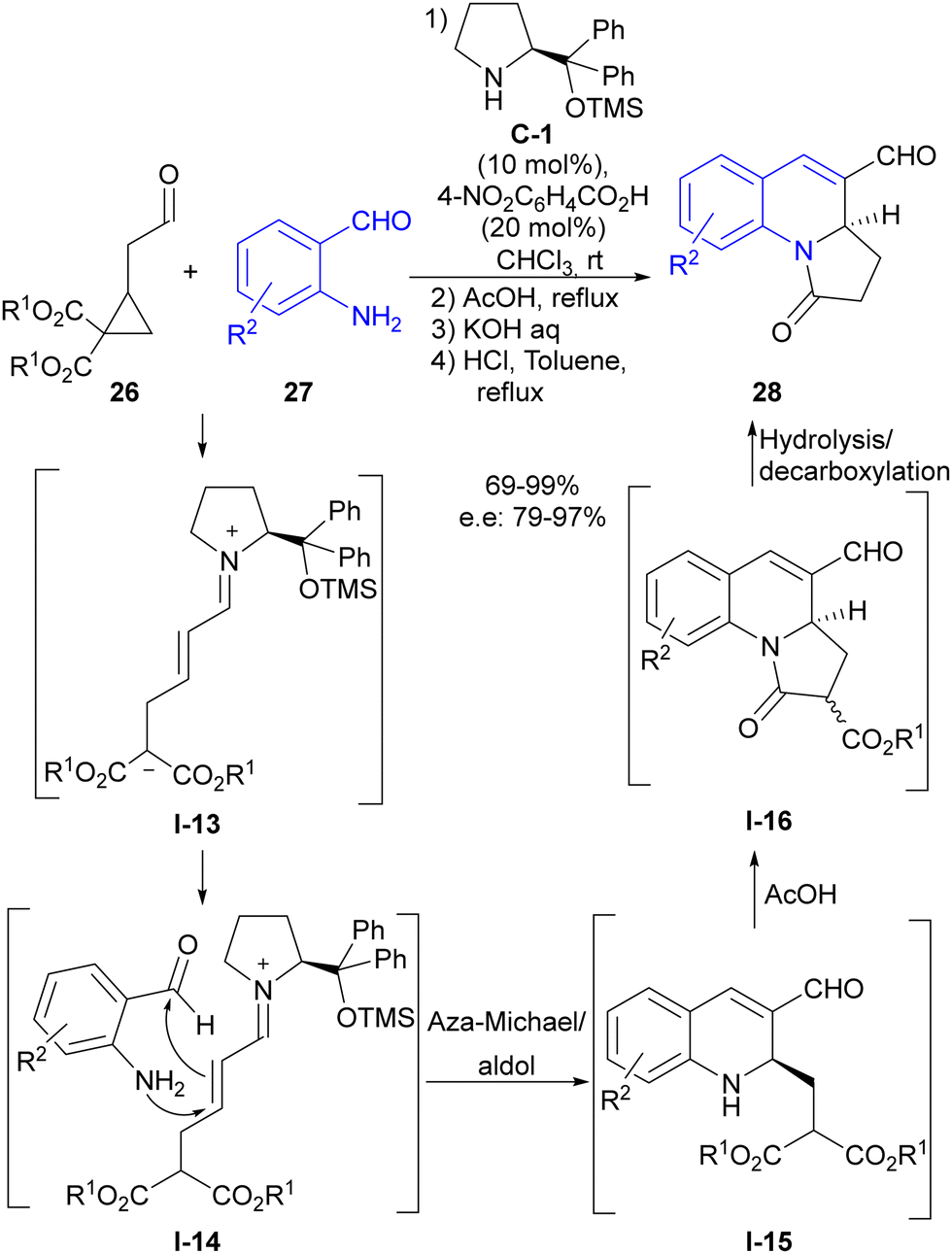 | ||
| Scheme 7 Enamine activation in the ring-opening of cyclopropaneacetaldehydes followed by aza-Michael/aldol cascade reaction. | ||
Using this activation manifold, we also reported a catalytic and enantioselective version of the vinylcyclopropane/cyclopentene (VCP/CP) rearrangement starting from cyclopropylacetaldehydes 29, which incorporate an unsaturation inserted between the cyclopropane moiety and electron-withdrawing substituent (Scheme 8).26 This type of substrates also generate donor–acceptor cyclopropane I-17 upon enamine formation with aminocatalyst C-8, and the subsequent ring-opening process leads to the formation of intermediate I-18 that simultaneously contains a dienolate nucleophilic site and an unsaturated iminium ion as a convenient Michael acceptor ready to undergo cyclization through intramolecular conjugate addition. This reaction provides a cyclopentene intermediate I-19 as a mixture of diastereoisomers in which alkene isomerization was carried out through the in situ addition of DBU to provide the more stable conjugated acylcyclopentene 30, which was isolated as a highly enantioenriched material. Importantly, all stereocentres present in the starting material were removed after ring-opening, but the configurational information was translated into conformational information due to the generation of a single iminium/dienolate conformer that underwent fast catalyst-controlled intramolecular Michael reaction, providing high facial enantioselectivity.
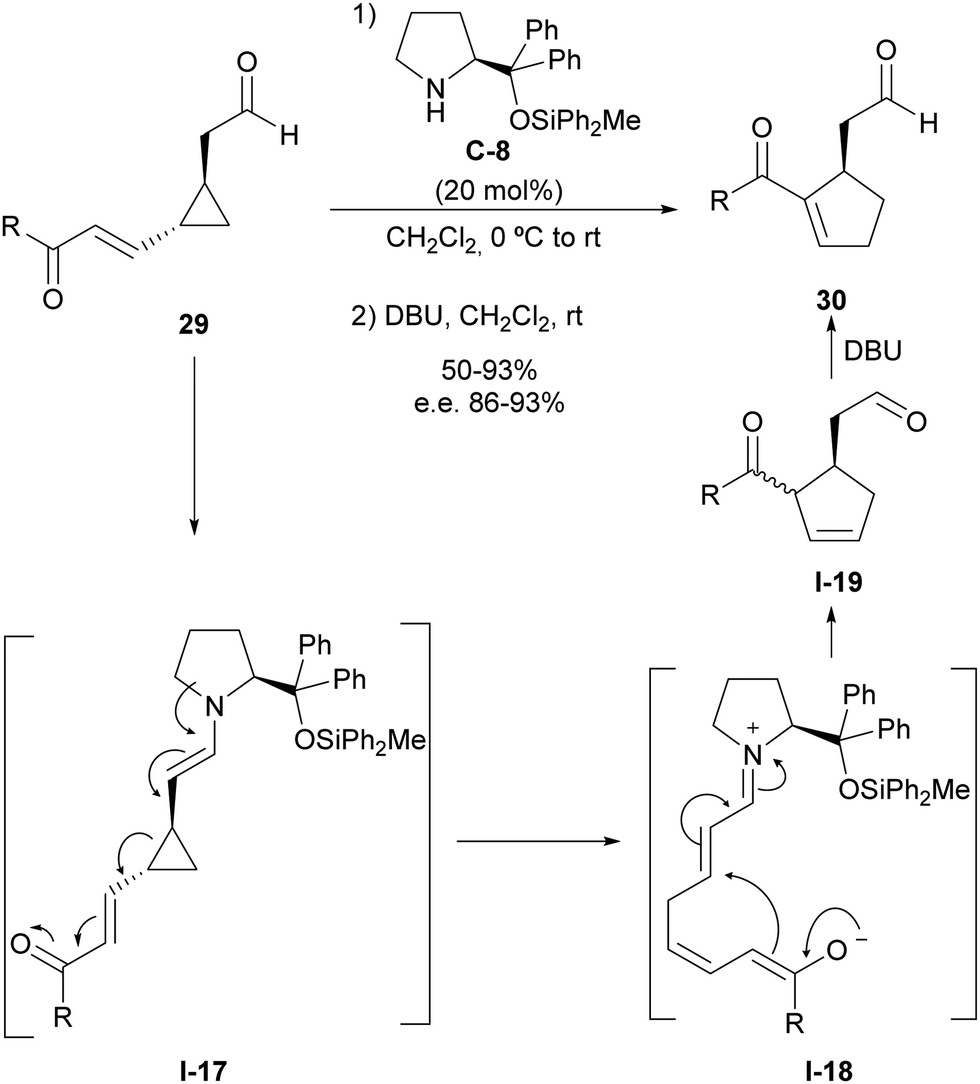 | ||
| Scheme 8 Enamine activation in the enantioselective vinylcyclopropane/cyclopentene rearrangement. | ||
The tendency of this type of conjugated substrates contrasts with the chemical behaviour shown by closely related cyclopropanes in which an additional unsaturation has been installed, this time in the chain that connects the formylmethyl substituent with cyclopropane.27 As can be seen in Scheme 9, this type of cyclopropanes such as 31 also undergoes activation through enamine formation as shown in I-20, in this case with a dienamine moiety playing the role of the electron-donating group. However, this intermediate was found to undergo Cope rearrangement rather than a ring-opening/ring-closing process, leading to cycloheptadienylacetaldehyde product 32 after catalyst turnover. The occurrence of such an enantiospecific Cope rearrangement was confirmed when enantioenriched cyclopentanes were employed as substrates and retained their enantiomeric excesses in the final products (chiral prolinol-type catalyst C-1 was only needed for providing high yields), while racemic starting materials always provided the racemic products. In addition, the fact that trans-substituted cyclopropane substrates were unreactive was consistent with this concerted Cope-type rearrangement.
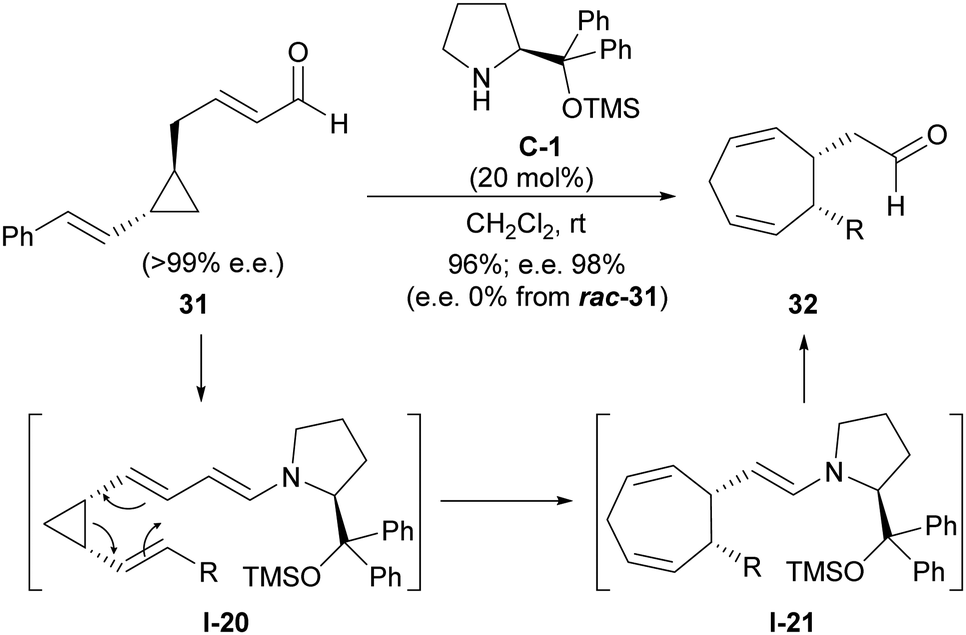 | ||
| Scheme 9 Divinylcyclopropane-cycloheptadiene rearrangement proceeding under enamine activation. | ||
N-heterocyclic carbene catalysis
As indicated in the introduction, the condensation between a formyl substituent with an N-heterocyclic carbene (NHC) leads to the formation of an enaminol intermediate (regarded as the Breslow intermediate), which can play the role of an electron-donating substituent if appropriately placed on a cyclopropane substrate (see Fig. 2). Our group has explored this possibility for the activation of cyclopropanecarbaldehydes with two alkoxycarbonyl substituents at the adjacent position, which lead to the catalytic generation of a donor–acceptor cyclopropane upon activation by the NHC.28 This intermediate also presents a highly polarized cyclopropane C–C bond that shows a pronounced tendency for a subsequent ring-opening process that forms an intermediate in which the stereocentres present at the cyclopropane starting material have been removed. This provided an opportunity to employ chiral triazolium salt C-9 as the precatalyst in the reaction between this type of cyclopropanecarbaldehydes 34 and β,γ-unsaturated α-oxoesters 33 for the enantioselective synthesis of pyranones 35 (Scheme 10). The mechanism of this reaction involves the initial catalytic generation of donor–acceptor cyclopropane I-22 upon activation by the NHC, which after ring-opening leads to an open-chain intermediate I-23 that contains an acyl-azolium-derived enol moiety and a malonate-derived enolate. A proton transfer event results in the formation of acyl azolium enolate I-24, which is known to have the ability to participate in a formal [4+2] cycloaddition with electron-poor dienes as shown in TS-3. Once this formal cycloaddition process has generated the heterocyclic scaffold I-25 under stereocontrol by chiral catalyst C-9, the good leaving-group ability of the triazolium unit enables regeneration of the carbene catalyst, leading to final product 35.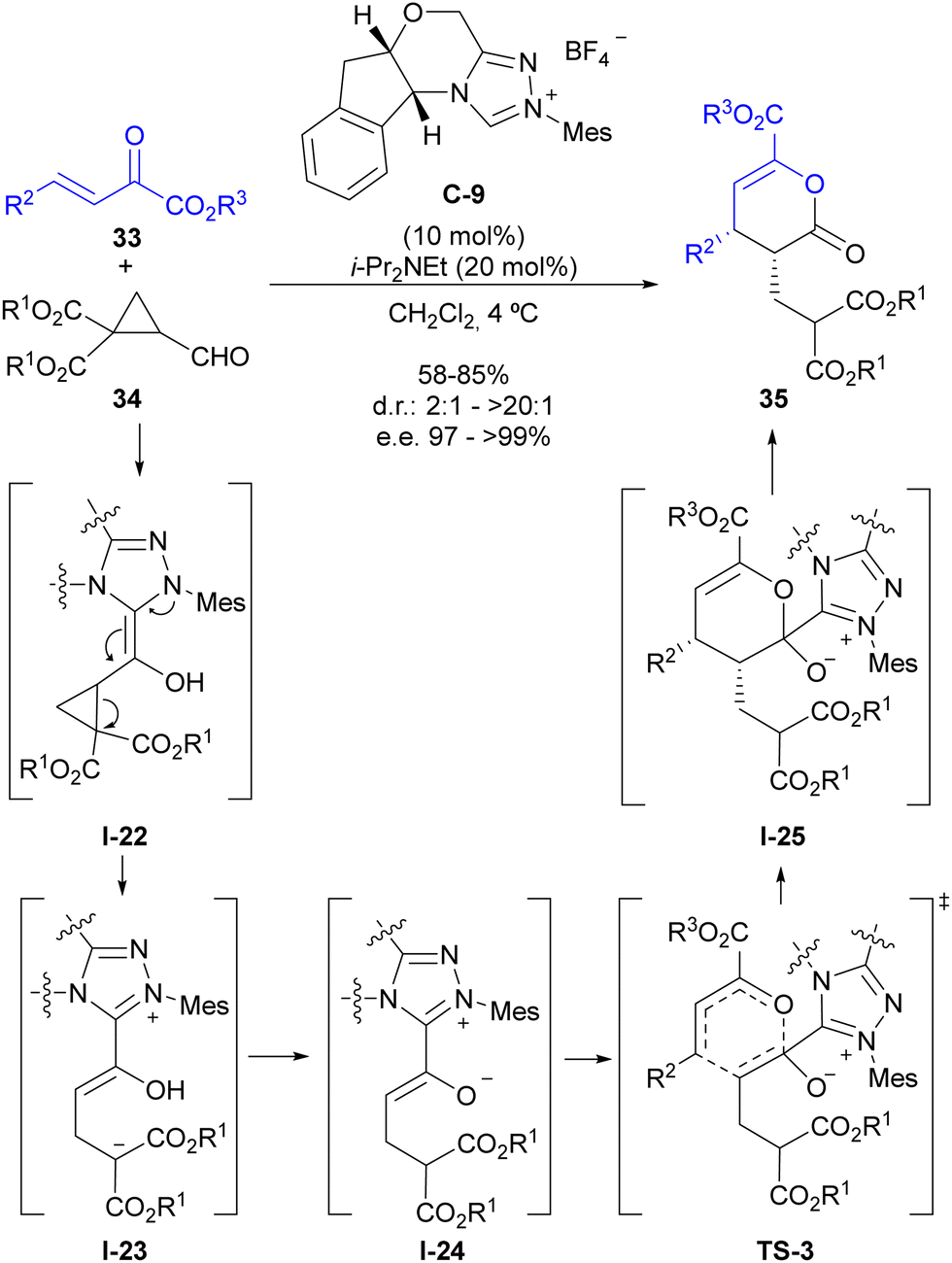 | ||
| Scheme 10 Enantioselective synthesis of dihydropyranones through NHC activation of cyclopropanecarbaldehydes. | ||
The same reaction manifold was employed for the enantioselective synthesis of complex pyranoindolones 37 by using alkylideneoxindoles 36 as the oxidiene counterpart in the formal [4+2] cycloaddition step (Scheme 11). In addition, a subsequent report studied the mechanism of this reaction in detail,29 confirming the initial proposal. Calculations also indicated that the increase in the polarization of the cyclopropane C–C bond associated with the formation of the Breslow intermediate is responsible for the cyclopropane ring-opening reaction, which alleviates the high electron density between two contiguous atoms at the catalytically generated donor–acceptor cyclopropane scaffold.
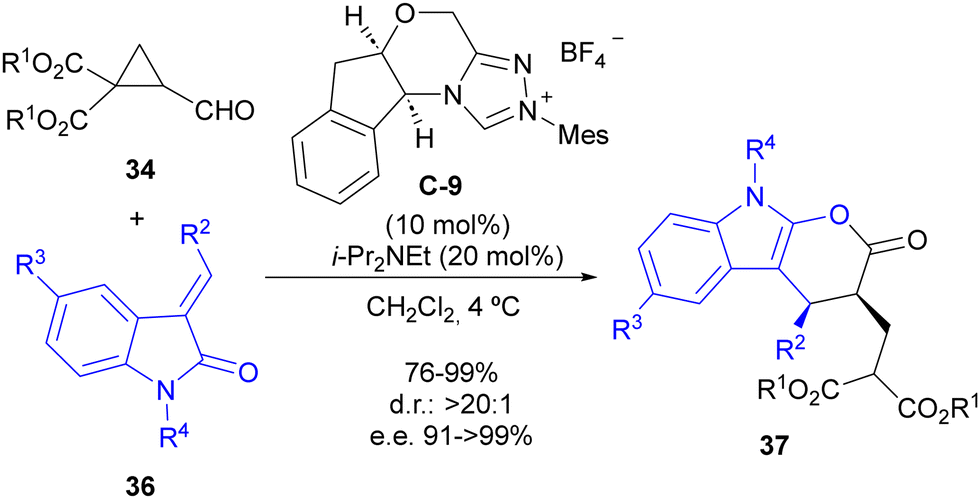 | ||
| Scheme 11 Enantioselective synthesis of pyranoindolones through cyclocondensation between cyclopropanecarbaldehydes and alkylideneoxindoles under NHC catalysis. | ||
Brønsted acid catalysis
The ability of a Brønsted acid to protonate a carbonyl group directly attached to the cyclopropane scaffold entails the activation of this substituent as a more effective electron-withdrawing group that can be used to trigger a ring-opening event. As mentioned, this reactivity was documented very early on,14 although it remains almost unexplored in the context of implementing enantioselective transformations on cyclopropanes. Quite recently, we developed a catalytic and enantioselective version of the Cloke–Wilson rearrangement under chiral Brønsted acid catalysis,30 which is a transformation that converts an acyl cyclopropane into a dihydrofuran in a single step. Literature examples dealing with the application of this reaction to the formation of enantioenriched chiral compounds have relied exclusively on enantiospecific cases in which the chiral information of an enantioenriched starting material was transferred into the final product. In our reaction design, we capitalized on the ability of strong Brønsted acids such as phosphoric acids to trigger a ring-opening event on suitably functionalized 2-arylcyclopropanecarbaldehyde 38 as an archetypical donor–acceptor cyclopropane (Scheme 12). The activation of this substrate by protonation of one of the two carbonyl moieties facilitates the ring-opening event through I-26 and leads to the formation of achiral carbocation/enol intermediate I-27, which remains in the close proximity to the Brønsted acid catalyst through the formation of an ion pair with its conjugated base. This enables transfer of the chiral information during the cyclization step to form final dihydrofuran products 39 as highly enantioenriched materials. Analysis of the origin of the enantioselectivity carried out via computational methods indicated that the key to enantiocontrol relied on the presence of stabilizing cation–π interactions between the carbocationic site and 9-phenanthryl lateral chain present at the chiral phosphoric acid catalyst in the transition state that led to the major enantiomer, while these interactions were absent in the alternative arrangement that could provide the opposite enantiomer, which is only observed in a very minor amount.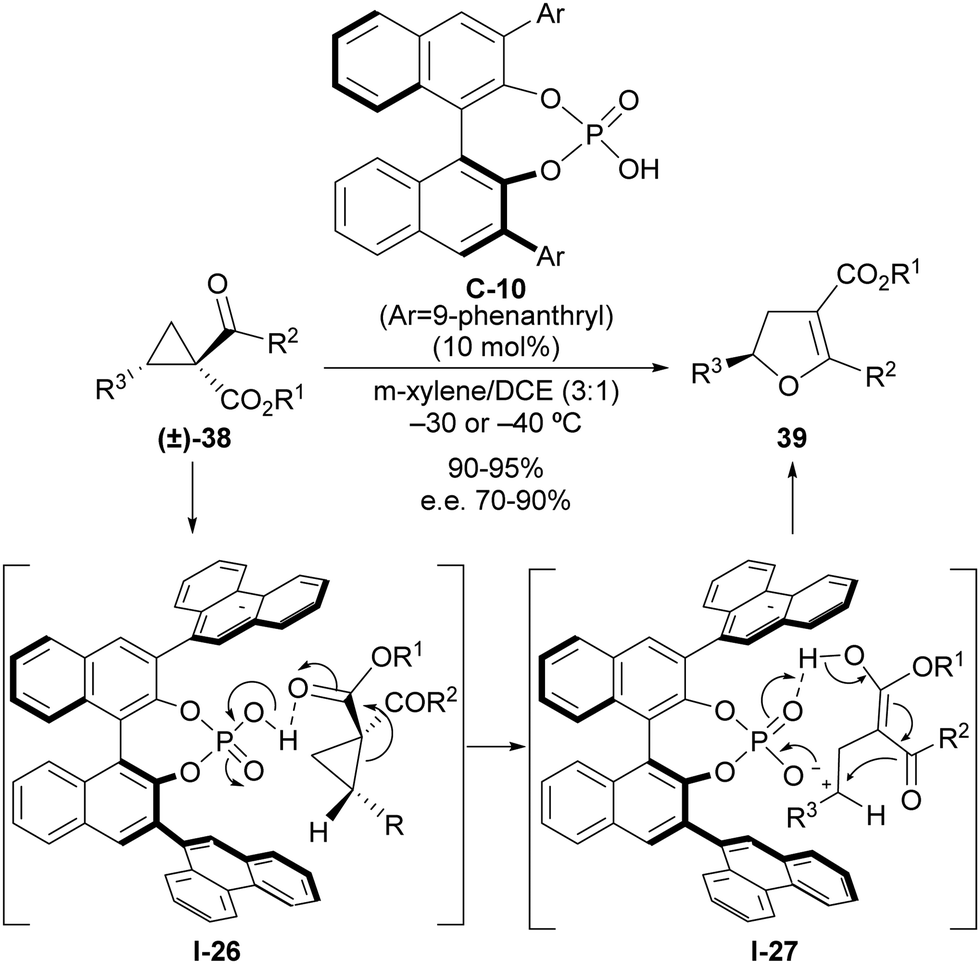 | ||
| Scheme 12 Catalytic enantioselective Cloke–Wilson rearrangement on acylcyclopropanes through chiral phosphoric acid catalysis. | ||
The same type of 2-aryl acylcyclopropanes 38 have also been employed by us under the same organocatalytic activation manifold for the synthesis of 8,9-dihydropyrido[1,2-a]indoles 41 through cyclocondensation with 3-substituted indoles 40 (Scheme 13).31 In this case, the presence of a highly nucleophilic external reagent such as the indole partner enabled trapping of the carbocationic intermediate, which reacted preferentially at the 2-position of the indole scaffold due to the fact that the 3-position is already substituted. Subsequent intramolecular 1,2-addition followed by elimination accounts for the formation of observed cyclocondensation products. The reaction required the use of N-trifluoromethanesulfonamide C-11 as a stronger Brønsted acid with respect to the parent phosphoric acid for best performance. However, in this case, when the implementation of an enantioselective version of this reaction was attempted, low enantiomeric excesses were observed under various tested conditions.
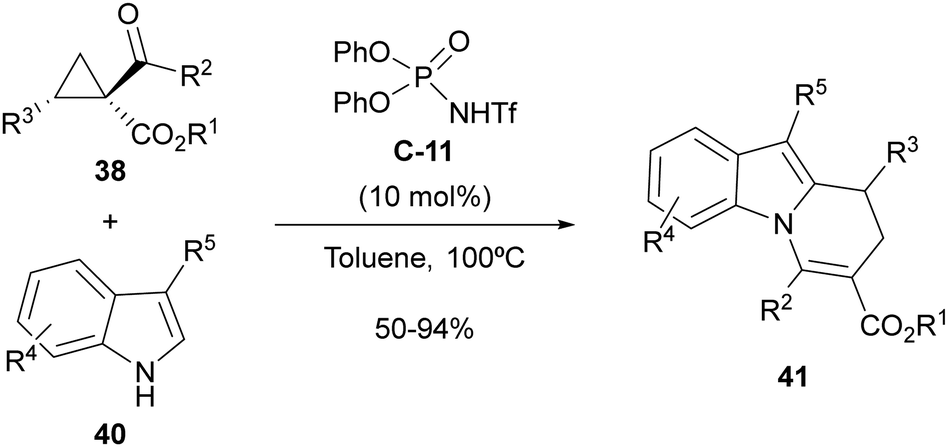 | ||
| Scheme 13 Brønsted-acid-catalyzed cyclocondensation between acylcyclopropanes and 3-substituted indoles. | ||
Conclusions
In this feature article, we have shown that the inherent reactivity of cyclopropanes to undergo ring-opening reactions driven by the release of the ring strain associated with the three-membered carbocyclic scaffold can be capitalized upon to trigger new reactions when combined with an appropriate organocatalytic activation manifold. This implies that the overall process can be controlled by the presence of the catalyst, and therefore, new opportunities appear in which the effective transfer of stereochemical information enables such reactions to be performed in an enantioselective fashion. In our particular case, we have made extensive use of two archetypical aminocatalytic activation manifolds (iminium and enamine catalysis) to trigger new, unprecedented reactions in which the activation of the cyclopropane-containing starting material (formylcyclopropane for iminium activation and cyclopropylacetaldehyde for enamine catalysis) takes place through the formation of covalently bound intermediates. As an alternative, Brønsted acid catalysts, and in particular, chiral phosphoric acids or derivatives, have been demonstrated to be highly proficient catalysts for performing a catalytic and enantioselective version of the Cloke–Wilson rearrangement that converts acylcyclopropanes into dihydrofuranes. This reaction involves the formation of a ring-opened carbocationic intermediate in which all the stereochemical information from the racemic starting material has been deleted, and therefore enables converting the racemic starting materials into enantiopure compounds. The same type of combination of acylcyclopropane/Brønsted acid catalysis has also been explored by us to capitalize on the reactivity of this carbocationic intermediate to trigger cyclocondensation reactions. This transformation is effective under phosphoric acid catalysis, but does not provide high enantiocontrol. As a general conclusion, we must state that despite important advances described in this area, especially in recent years, this is a type of reactivity that remains significantly underdeveloped. The number of reactions that employ asymmetric organocatalysis as the vehicle for enantiocontrol is still limited, and even the examples of the application of those already developed to challenging cases of target-directed synthesis is scarce. The same applies to the potential application of this type of reactivity to industrial production. In addition, new organocatalytic activation approaches using different intermediates or alternative mechanistic manifolds that are currently being expanded, such as many possibilities offered by photoredox catalysis or electrocatalysis, are still in their infancy in terms of their application to cyclopropane-containing substrates and exploitation of the strain-release concept together with enantioselective radical chemistry. As a consequence, important contributions are expected to be produced in following years, which will significantly expand the potential of functionalized cyclopropanes as useful reagents in synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank all past and present group members for their significant contributions and especially those directly involved in the chemistry that appears in corresponding cited references from our group. Financial support by the Spanish AEI (PID2020-118422GB-I00 and RED2022-134331-T funded by MICIU/AEI/10.13039/501100011033), the Basque Government (Grupos IT1558-22) and the University of the Basque Country (EHU-N23/48 and EHU-N23/30) is also gratefully acknowledged. We also wish to thank Prof. Dr P. Merino and Prof. Dr T. Tejero from the University of Zaragoza for the fruitful collaboration and continuous support directed toward a better understanding of the mechanisms of the new reactions developed by us through computational studies.Notes and references
- For general reviews on asymmetric organocatalysis, see: (a) B. Han, X.-H. He, Y.-Q. Liu, G. He, C. Peng and J.-L. Li, Chem. Soc. Rev., 2021, 50, 1522 RSC; (b) S.-H. Xiang and B. Tan, Nat. Commun., 2020, 11, 3786 CrossRef CAS PubMed; (c) J. M. Lassaletta, Nat. Commun., 2020, 11, 3787 CrossRef CAS PubMed; (d) M. P. van Der Helm, B. Klemm and R. Eelkema, Nat. Rev. Chem., 2019, 3, 491 CrossRef CAS; (e) M. Silvi and P. Melchiorre, Nature, 2018, 554, 41 CrossRef CAS PubMed; (f) Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications, ed. P. I. Dalko, Wiley-VCH, 2013 Search PubMed; (g) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS PubMed; (h) B. List, Chem. Rev., 2007, 107, 5413 CrossRef CAS.
- (a) B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS; (b) K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243 CrossRef CAS.
- E. Reyes, U. Uria, L. Prieto, L. Carrillo and J. L. Vicario, Tetrahedron Chem., 2023, 7, 100041 CrossRef.
- For selected reviews on cyclopropane reactivity, see: (a) V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227 CrossRef CAS PubMed; (b) V. Ganesh and S. Chandrasekaran, Synthesis, 2016, 4347 CAS; (c) J. R. Green and V. Snieckus, Synlett, 2014, 2258 CrossRef CAS; (d) P. Tang and Y. Qin, Synthesis, 2012, 2969 CAS; (e) F. De Simone and J. Waser, Synthesis, 2011, 589 CAS; (f) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051 RSC; (g) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (h) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213 CrossRef CAS PubMed; (i) H. N. C. Wong, M. Y. Hon, C. W. Tse, Y. C. Yip, J. Tanko and T. Hudlicky, Chem. Rev., 1989, 89, 165 CrossRef CAS; (j) C. Apel and M. Christmann, Tetrahedron, 2021, 82, 131760 CrossRef CAS.
- For reviews focused on electrophilic cyclopropanes, see: (a) R. Verhé and N. de Kimpe, In the Chemistry of the Cyclopropyl Group, ed. Z. Rappoport, Wiley, Great Britain, 1987, pp. 445–564 Search PubMed; (b) S. Danishefsky, Acc. Chem. Res., 1979, 12, 66 CrossRef CAS.
- For reviews focused on nucleophilic cyclopropanes, see: (a) D. Guijarro and M. Yus, Curr. Org. Chem., 2005, 9, 1713 CrossRef CAS; (b) O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597 CrossRef CAS PubMed; (c) J. Salaun, Chem. Rev., 1983, 83, 619 CrossRef CAS.
- For selected recent reviews on donor-acceptor cyclopropanes, see: (a) Y. Xia, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 9192 CrossRef CAS PubMed; (b) K. Ghosh and S. Das, Org. Biomol. Chem., 2021, 19, 965 RSC; (c) E. M. Budynina, K. L. Ivanov, I. D. Sorokin and M. Ya’Melnikov, Synthesis, 2017, 3035 CrossRef CAS; (d) H. K. Grover, M. R. Emmett and M. A. Kerr, Org. Biomol. Chem., 2015, 13, 655 RSC; (e) T. F. Schneider, K. Johannes and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef CAS PubMed; (f) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804 RSC; (g) F. De Nanteuil, F. De Simone, R. Frei, F. Benfatti, E. Serrano and J. Waser, Chem. Commun., 2014, 50, 10912 RSC; (h) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804 RSC.
- For selected reviews on aminocatalysis, see: (a) A. D. Vecchio, A. Sinibaldi, V. Nori, G. Giorgianni, G. D. Carmine and F. Pesciaioli, Chem. – Eur. J., 2022, 28, e202200818 CrossRef PubMed; (b) J. Lv, Q. Zhang, M. Cai, Y. Han and S. Luo, Chem. – Asian J., 2018, 13, 740 CrossRef CAS PubMed; (c) L. Albrecht, H. Jiang and K. A. Jorgensen, Chem. – Eur. J., 2014, 20, 358 CrossRef CAS PubMed; (d) I. D. Jurberg, I. Chatterjee, T. Tannert and P. Melchiorre, Chem. Commun., 2013, 49, 4869 RSC; (e) M. Nielsen, D. Worgull, T. Zweifel, B. Gschwend, S. Bertelsen and K. A. Jorgensen, Chem. Commun., 2011, 47, 632 RSC; (f) B. List, Angew. Chem., Int. Ed., 2010, 49, 1730 CrossRef CAS PubMed; (g) P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef CAS PubMed; (h) B. List, Chem. Commun., 2006, 819 RSC.
- For selected reviews on NHC catalysis, see: (a) C. D. Risi, A. Brandolese, G. D. Carmine, D. Ragno, A. Massi and O. Bortolini, Chem. – Eur. J., 2023, 29, e202202467 CrossRef PubMed; (b) K. Liu, M. Schwenzer and A. Studer, ACS Catal., 2022, 12, 11984 CrossRef CAS; (c) A. Ghosh and A. T. Biju, Angew. Chem., Int. Ed., 2021, 60, 13712 CrossRef CAS PubMed; (d) P. Bellotti, M. Koy, F. Glorius and M. N. Hopkinson, Nat. Rev. Chem., 2021, 5, 711 CrossRef CAS PubMed; (e) T. Ishii, K. Nagao and H. Ohmiya, Chem. Sci., 2020, 11, 5630 RSC; (f) K. J. R. Murauski, A. A. Jaworski and K. A. Scheidt, Chem. Soc. Rev., 2018, 47, 1773 RSC; (g) C. Zhang, J. F. Hooper and D. W. Lupton, ACS Catal., 2017, 7, 2583 CrossRef CAS; (h) E. Reyes, U. Uria, L. Carrillo and J. L. Vicario, Synthesis, 2017, 451 CAS; (i) M. H. Wang and K. A. Scheidt, Angew. Chem., Int. Ed., 2016, 55, 14912 CrossRef CAS PubMed; (j) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307 CrossRef CAS PubMed; (k) R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040 RSC; (l) J. Mahatthananchai, J. W. Bode, M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef PubMed.
- For selected reviews about H-bonding organocatalysis, see: (a) M. C. Gimeno and R. P. Herrera, Eur. J. Org. Chem., 2020, 1057 CrossRef CAS; (b) A. Rouf and C. Tanyeli, Curr. Org. Chem., 2016, 20, 2996 CrossRef CAS; (c) F. E. Held and S. B. Tsogoeva, Cat. Sci. Technol., 2016, 6, 645 RSC; (d) P. Chauhan, M. Pankaj, S. Mahajan, U. Suruchi, U. Kaya, D. Hack and D. Enders, Adv. Synth. Catal., 2015, 357, 253 CrossRef CAS; (e) T. J. Auvil, A. G. Schafer and A. E. Mattson, Eur. J. Org. Chem., 2014, 2633 CrossRef CAS; (f) M. Tsakos and C. G. Kokotos, Tetrahedron, 2013, 69, 10199 CrossRef CAS; (g) J. Alemán, A. Parra, H. Jiang and K. A. Jørgensen, Chem. – Eur. J., 2011, 17, 6890 CrossRef PubMed; (h) Y. Takemoto, Chem. Pharm. Bull., 2010, 58, 593 CrossRef CAS PubMed; (i) C. Palomo, M. Oiarbide and R. López, Chem. Soc. Rev., 2009, 38, 632 RSC; (j) S. J. Connon, Chem. Commun., 2008, 2499 RSC; (k) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed.
- For some selected reviews on chiral Brønsted acid catalysis see: (a) J. Merad, C. Lalli, G. Bernadat, J. Maury and G. Masson, Chem. – Eur. J., 2018, 24, 392 CrossRef PubMed; (b) R. Maji, S. C. Mallojjala and S. E. Wheeler, Chem. Soc. Rev., 2018, 47, 1142 RSC; (c) T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277 CrossRef CAS PubMed; (d) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed; (e) M. Terada, Synthesis, 2010, 1929 CrossRef CAS; (f) D. Kampen, C. M. Reisinger and B. List, Top. Curr. Chem., 2010, 291, 395 CrossRef CAS PubMed; (g) A. Zamfir, S. Schenker, M. Freund and S. B. Tsogoeva, Org. Biomol. Chem., 2010, 8, 5262 CAS; (h) M. Terada, Chem. Commun., 2008, 4097 RSC; (i) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS PubMed.
- L. Li, Z. Li and Q. Wang, Synlett, 2009, 1830 CAS.
- S. S. Sohn and J. W. Bode, Angew. Chem., Int. Ed., 2006, 45, 6021 CrossRef CAS PubMed.
- E. P. Kohler and J. B. Conant, J. Am. Chem. Soc., 1917, 39, 1404–1420 CrossRef CAS.
- (a) G. Dickmeiss, V. De Sio, J. Udmark, T. B. Poulsen, V. Marcos and K. A. Jørgensen, Angew. Chem., Int. Ed., 2009, 48, 6650 CrossRef CAS PubMed For a previous related example using stoichiometric amount of cinchona alkaloids, see: ; (b) P. Müller and D. Riegert, Tetrahedron, 2005, 61, 4373 CrossRef.
- (a) C. Sparr and R. Gilmour, Angew. Chem., Int. Ed., 2011, 50, 8391 CrossRef CAS PubMed See also ; (b) J. Wallbaum, L. K. B. Garve, P. Jones and D. B. Werz, Chem. – Eur. J., 2016, 22, 18756 CrossRef CAS PubMed.
- E. Diaz, E. Reyes, U. Uria, L. Carrillo, T. Tejero, P. Merino and J. L. Vicario, Chem. – Eur. J., 2018, 24, 8764 CrossRef CAS PubMed.
- Some reviews: (a) G. J. Reyes-Rodríguez, N. M. Rezayee, A. Vidal-Albalat and K. A. Jørgensen, Chem. Rev., 2019, 119, 4221 CrossRef PubMed; (b) D. McLeod, M. K. Thøgersen, N. I. Jessen, K. A. Jørgensen, C. S. Jamieson, X.-S. Xue, K. N. Houk, F. Liu and R. Hoffmann, Acc. Chem. Res., 2019, 52, 3488 CrossRef CAS PubMed; (c) B. S. Donslund, T. K. Johansen, P. H. Poulsen, K. S. Halskov and K. A. Jørgensen, Angew. Chem., Int. Ed., 2015, 54, 13860 CrossRef CAS PubMed; (d) S. Meninno and A. Lattanzi, Chem. Commun., 2013, 49, 3821 RSC; (e) K. L. Jensen, G. Dickmeiss, H. Jiang, L. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248 CrossRef CAS PubMed; (f) A. Mielgo and C. Palomo, Chem. – Asian J., 2008, 3, 922 CrossRef CAS PubMed.
- (a) B. Silvi and A. Savin, Nature, 1994, 371, 683 CrossRef CAS; (b) A. Savin, R. Nesper, S. Wengert and T. F. Fässler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808 CrossRef CAS.
- Stabilization of iminium ion through binding of the OH leaving group by a H-bond donor has been reported to happen with bifunctional pyrrolidine-containing organocatalysts: (a) Y. Gu, Y. Wang, T. Y. Yu, Y. M. Liang, Y. C. Luo and P. F. Xu, Angew. Chem., Int. Ed., 2014, 53, 14128 CrossRef CAS PubMed; (b) V. Juste-Navarro, L. Prieto, I. Delso, R. Manzano, T. Tejero, E. Reyes, J. L. Vicario and P. Merino, Adv. Synth. Catal., 2017, 359, 4122 CrossRef CAS.
- S. Winstein, E. Clippinger, A. H. Fainberg, R. Heck and G. C. Robinson, J. Am. Chem. Soc., 1956, 28, 328 CrossRef.
- H. C. Brown and M. Periasamy, J. Org. Chem., 1981, 46, 3161 CrossRef CAS.
- M.-Y. Jiang, L. Zhang, R. Liu, Z.-J. Dong and J.-K. Liu, J. Nat. Prod., 2009, 72, 1405 CrossRef CAS PubMed.
- K. S. Halskov, F. Kniep, V. H. Lauridsen, E. H. Iversen, B. S. Donslund and K. A. Jørgensen, J. Am. Chem. Soc., 2015, 137, 1685 CrossRef CAS PubMed.
- E. Sanchez-Diez, D. L. Vesga, E. Reyes, U. Uria, L. Carrillo and J. L. Vicario, Org. Lett., 2016, 18, 1270 CrossRef CAS PubMed.
- G. Garay, J. Hurtado, M. Pedron, L. Garcia, E. Reyes, E. Sanchez-Diez, T. Tejero, L. Carrillo, P. Merino and J. L. Vicario, Angew. Chem., Int. Ed., 2023, 62, e202302416 CrossRef CAS PubMed.
- C. Apel, S. S. Hartmann, D. Lentz and M. Christmann, Angew. Chem., Int. Ed., 2019, 58, 5075 CrossRef CAS PubMed.
- L. Prieto, E. Sanchez-Diez, U. Uria, E. Reyes, L. Carrillo and J. L. Vicario, Adv. Synth. Catal., 2017, 359, 1678 CrossRef CAS.
- Y. Wang, Y. Qiao, Y. Lan and D. Wei, Catal. Sci. Technol., 2021, 11, 332 RSC.
- A. Ortega, R. Manzano, U. Uria, L. Carrillo, E. Reyes, T. Tejero, P. Merino and J. L. Vicario, Angew. Chem., Int. Ed., 2018, 57, 8225 CrossRef CAS PubMed.
- A. Ortega, U. Uria, T. Tejero, L. Prieto, E. Reyes, P. Merino and J. L. Vicario, Org. Lett., 2021, 23, 2326 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |