
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the evolution of catalytically active multi-metal sites in a bifunctional high-entropy alloy electrocatalyst for zinc–air battery application†
Chetna
Madan
 a,
Saumya R.
Jha
b,
Nirmal Kumar
Katiyar
cd,
Arkaj
Singh
a,
Rahul
Mitra
b,
Chandra Sekhar
Tiwary
*c,
Krishanu
Biswas
*b and
Aditi
Halder
*a
a,
Saumya R.
Jha
b,
Nirmal Kumar
Katiyar
cd,
Arkaj
Singh
a,
Rahul
Mitra
b,
Chandra Sekhar
Tiwary
*c,
Krishanu
Biswas
*b and
Aditi
Halder
*a
aSchool of Chemical Sciences, Indian Institute of Technology, Mandi, Himachal Pradesh 175005, India. E-mail: aditi@iitmandi.ac.in
bDepartment of Material Science and Engineering, Indian Institute of Technology, Kanpur, Uttar Pradesh 208016, India. E-mail: kbiswas@iitk.ac.in
cDepartment of Metallurgical and Materials Engineering, Indian Institute of Technology, Kharagpur, West Bengal 721302, India. E-mail: Chandra.tiwary@metal.iitkgp.ac.in
dAmity Institute of Applied Sciences, Amity University Noida, Sector 125, 201303 Uttar Pradesh, India
First published on 23rd October 2023
Abstract
Zinc–air batteries are known for high theoretical energy density and environmental friendliness. The successful commercial utilization of rechargeable zinc–air batteries is limited by unstable electrochemical interfaces and sluggish kinetics with poor round-trip efficiency. In this study, we report a nanocrystalline high entropy alloy (HEA) comprising Cu–Co–Mn–Ni–Fe (CCMNF) prepared by casting-cum-cryomilling method. This multi-component HEA embodies multiple catalytically active sites with diverse functionalities, thus enhancing the electrochemical redox reactions, e.g., oxygen reduction (ORR) and oxygen evolution reaction (OER). The bifunctional electrocatalytic performance of this HEA is comparable to that of standard catalysts, RuO2 and Pt/C, as evidenced by low overpotential requirements towards OER and ORR. The HEA was tested for use in the air electrode catalyst in the zinc–air battery, where it performed stable oxygen electrocatalysis that was durable over 1045 charging–discharging cycles for ∼90 hours of continuous operation. The microstructural analysis of HEA at different time scales (0, 24, 87 h) during the zinc–air battery operation suggested a dynamic participation of multiple metal active sites on the catalyst surface. Detailed studies revealed that despite leaching in harsh alkaline operation conditions, the synergistic electronic interactions between the component metal sites sustained good electrocatalytic performance and promoted oxygen electrocatalysis through the modification of electronic and chemical properties.
1. Introduction
Nanomaterials are an integral part of the advanced technological systems in every sector, such as electronics, energy, nanomedicines, computing, catalysis, etc.1 The domain of electrocatalysis has switched to nanocrystalline materials that prominently increase the efficiency and stability of the reactions. For various energy-related applications involved in electrochemical energy conversion or storage devices, there is still a dependence on electrocatalysts that belong to the class of noble metals. Precious metals, such as Pt, Ru, Au, etc., exhibit suitable electronic properties that make them ideal catalyst materials for multiple catalytic reactions, especially energy conversion reactions. However, their high cost and scarcity impede the commercial practicability of devices, such as fuel cells, water electrolyzers, and metal–air batteries. Society's primary concerns are environmental safety and lower energy costs which can be achieved by utilizing environmentally benign, low-cost, and abundant materials, such as 3d-transition metal compounds. A study by Olivetti et al.2 encompasses a plethora of metrics, such as cost, regional availability, and recyclability of various transition metals, which are utilized in fabricating high entropy alloys. Their results have confirmed the overall superiority of 3d-transition metals. The availability of vacant 3d orbitals allows the adoption of various oxidation states. The tunable electronic bands of the 3d transition metals enable modifying their electronic and chemical behavior leading to remarkable enhancement in the desired catalytic material.3–8 The 3d transition metals (Mn, Fe, Co, Ni, Cu) are relatively more abundant in comparison to the other transition metals (4d and 5d).9 They also have a lower environmental impact in terms of processing, recycling, and disposal.10 Hence, it is preferred to employ 3d-transition metal-based materials to fabricate high-entropy alloys that yield desirable catalytic properties without compromising environment, safety, and economy.In the past decade, the development of high-entropy alloys (HEAs) has attracted researchers’ attention due to their chemical homogeneity despite having multi-metallic elements.11–14 In an HEA, the number of participating metals is usually five or higher, which are present in nearly equiatomic compositions.15,16 This class of alloys is termed multi-composition or multi-principal metal alloys17 and is generally referred to as high-entropy alloys (HEAs) because entropy is the stabilization force.18 These HEAs exhibit interesting hybrids of structural and mechanical19 properties, such as better tensile strength, flexibility, ductility, corrosion resistance, etc. as well as electrical and electrochemical properties.20 These arise from four “core effects”,21 namely the high-entropy effect, the lattice distortion due to the interplay of different atomic sizes of the combining elements, the slow diffusion effect, and the cocktail effect.
The term “high entropy” signifies an increase in the configurational entropy of mixing five or more metals with different atomic sizes and electronegativity. The contribution of increased configurational entropy with a higher number of constituent metals to the total free energy outsteps the enthalpy change associated with the compound formation, thus forming a stable solid solution and preventing the formation of detrimental intermetallic systems.22 Therefore, the high entropy of mixing of different metals could lower the immiscibility gap and result in a more uniform concentration of constituents. The massive variety of possible configurations of atomic and surface arrangements in such multi-component systems allow optimized adsorption sites. The exceptional catalytic activity of HEA is revealed to be derived from plentiful active atomic sites on the surface23 that could be tuned for local electronic arrangement,24 defect sites,25 and tuned intermediate adsorption/desorption energies.26 An HEA has multiple principal metals arranged in infinitely possible combinations and microstructures at its surface, offering a large number of active sites. The lattice distortion present in an HEA causes its potential energy to rise, which reduces the energy barrier for the adsorption of catalytic intermediates.27 Various recent studies advertise the utility of HEAs in heterogeneous catalysis involving electrochemical energy-related applications.20,28,29 Sharma et al. reported low-cost equiatomic high-entropy alloys (Co–Fe–Ga–Ni–Zn) for efficient and stable OER (oxygen evolution reaction) reactions.30 The two well-known core effects of high-entropy alloys directly impact the electrochemical reaction. As a result of the cocktail effect, arising from atomic-level homogeneity, a high-entropy alloy exhibits a re-oriented electronic structure. According to the cocktail effect, where all the components of the alloy dandle over the surface (five or more than five metallic elements), the alloy becomes susceptible to the chemisorption of analytes. Another predominant effect is ‘lattice distortion’, which directly alters localized atomic spacing, both in the form of tensile and compressive stress due to differences in atomic radii.
The involved electrochemical redox reactions at the electrodes of a metal–air battery require the electrocatalyst to catalyze the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) at the air electrode efficiently. The center of d-bands in catalytic materials plays a significant role in adsorption energy and activation barrier. Electrochemical ORR takes place at an optimum d-orbital occupancy of the catalytically active sites near the Fermi level.31 A larger value of atomic spacing forces the oxygen molecules to dissociate before the adsorption, and therefore, a smaller value than the optimum reduces the repulsive force and forces analytes for dual site absorption,32 enhancing the catalytic mechanism for ORR. The tensile strain upshifts the d-band center, and the compressive strain downshifts the d-band center.32,33 In the case of high-entropy alloys, Katiyar et al.12 have reported in the Ag–Au–Cu–Pd–Pt catalyst for Formic acid oxidation, whereby all the metallic elements in the alloy system shifted their d-band center toward the Fermi energy or upshifted as compared to the pure metallic entity. Therefore, plentiful active atomic sites on the surface in HEAs34 could be tuned for a local electronic arrangement,35 defect sites,36 and modified intermediate adsorption/desorption energies37 by altering the elemental ratio and elemental composition. The HEA offers a huge variety of surface-active sites with various plausible combinations of atomic environments and local electronic arrangements, along with associated intermediate adsorption energies, thus proving to be a potential multifunctional electrocatalyst. For instance, a high-entropy alloy oxide synthesized by Yu et al. was composed of Cr, Fe, Co, and Ni and showed excellent OER performance owing to the formation of Cr2O3 microdomains that leached out of the cobalt ions; amorphization of interface between the different existing phases also took place.38
To date, platinum-based catalysts are the most sought-after for the ORR reaction, while ruthenium or iridium-based catalysts are most widely accepted for OER. HEAs offer a huge variety of surface-active sites with various plausible combinations of atomic environments, local electronic arrangements, and the associated intermediate adsorption energies, making them potential multifunctional electrocatalysts.
In the present study, an equiatomic high-entropy alloy of Cu–Co–Mn–Ni–Fe (CCMNF) has been synthesized via the casting-cum-cryomilling method and investigated for its electrochemical performance behavior in a three-electrode system for bifunctional oxygen electrocatalysis. The excellent performance of CCMNF prompted the study of its application in zinc–air battery (ZAB) energy storage devices. This study discusses ongoing modifications occurring at the surface of CCMNF at different time scales during zinc–air battery operation that impact the chemical structure and electronic properties of the catalysts over its performance. Thus, it establishes an understanding of the modification of behavior and stability of an HEA electrocatalyst during electrocatalytic energy storage applications.
2. Experimental section
2.1. Chemicals
All the metals were purchased from Alfa Aesar® with 99.99% purity in the form of metal chips and buttons. All the chemicals used in this study were of analytical grade. Potassium hydroxide flakes, isopropyl alcohol, and zinc acetate powder were purchased from Merck. Nafion D-520 (5% w/w in water and 1-propanol) was purchased from Alfa Aesar® and used as it is. Standard catalyst 20% platinum carbon and ruthenium oxide powder were also bought from Alfa Aesar. Zinc foil (thickness 0.25 mm and purity 99.9%) was purchased from Merck®. Conducting carbon paper was purchased from Vritra Technologies (hydrophilic, thickness 0.3 mm, resistance 3–5 ohms per sq).2.2. Synthesis procedure
The all-metallic elements, Cu, Co, Mn, Ni, and Fe, were melted together in a vacuum arc melting furnace. The concentration of all elements was selected in equiatomic proportion. To ensure chemical homogeneity, the casted ingot was melted three to four times and solidified. For homogenization, the casted HEA ingot was vacuum sealed inside a quartz tube, homogenized at 1000 °C for 10 hours, and then quenched in cold water to freeze the high-temperature phase. After ensuring single-phase formation using X-ray diffraction, the ingot was divided into smaller pieces and then cryomilled for 7 hours using custom-built cryomill to form high-entropy alloy nanoparticles.39,40 Liquid nitrogen (LN2) was used to reduce the milling temperature to below −150 °C in such a way that the LN2 and milling powder were never mixed; Ar gas (inert) was purged continuously in the milling chamber to protect nanoparticles from oxidation.2.3. Physicochemical characterization
X-ray diffraction (XRD) patterns of the as-synthesized HEA were recorded using a Rigaku smart lab diffractometer using Cu Kα radiation (λ = 1.54056 Å). X-ray photoelectron spectroscopy (XPS) was performed using ThermoFisher Scientific NEXSA surface analyzer with a micro-focused (400 μm, 72 W, 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 V) XPS source monochromatic Al-Kα source (hν = 1486.6 eV). The scanning electron microscopic (SEM) images of the electrodes were obtained using FEI Nova NanoSEM 450. Energy dispersive spectroscopy (EDS) was used to estimate the nanoparticles’ size and composition with transmission electron microscopy (TEM) using FEI TECNAI G2 T20. The nanoparticles were dispersed in ultrapure methanol by ultrasonication and drop cast over 400 mesh TEM Cu-grid. The sample was dried under a vacuum overnight before the TEM analysis. Inductively coupled plasma-mass spectrometry (ICP-MS) was performed using Agilent ICP-MS Model 7850 with Mass Hunter software.
000 V) XPS source monochromatic Al-Kα source (hν = 1486.6 eV). The scanning electron microscopic (SEM) images of the electrodes were obtained using FEI Nova NanoSEM 450. Energy dispersive spectroscopy (EDS) was used to estimate the nanoparticles’ size and composition with transmission electron microscopy (TEM) using FEI TECNAI G2 T20. The nanoparticles were dispersed in ultrapure methanol by ultrasonication and drop cast over 400 mesh TEM Cu-grid. The sample was dried under a vacuum overnight before the TEM analysis. Inductively coupled plasma-mass spectrometry (ICP-MS) was performed using Agilent ICP-MS Model 7850 with Mass Hunter software.
2.4. Electrochemical characterization
The working electrode was prepared by making an ink of the catalyst in a specific manner and loading it uniformly onto a 5 mm-diameter glassy carbon rotating disk electrode (RDE). To prepare the catalyst ink, 10 mg of HEA was added to 500 μL of isopropyl alcohol (IPA) with 10 μL of 5% Nafion solution, which was sonicated for 1 hour. Then, 10 μL of this suspension was drop cast on a clean glassy carbon RDE (0.196 cm2) and allowed to dry completely under ambient conditions. The inks for the standard catalyst, RuO2, and Pt/C standard were prepared using the same composition. A constant catalyst loading of 1000 μg cm−2 on the working electrode was maintained for all the catalysts. All the electrochemical measurements were conducted using PARSTAT multi-channel potentiostat PMC from AMETEK Scientific Instruments with an RDE (5 mm diameter) from Pine Research Instruments. Cyclic voltammetry (CV) sweeps were performed to pre-condition the working electrode surface at a 20 mV s−1 scan rate in a three-electrode configuration which consisted of a Pt electrode as the counter electrode, saturated in 3 M KCl silver–silver chloride (Ag/AgCl) electrode as the reference electrode, and a catalyst of coated glassy carbon RDE as described above for the working electrode. The potential of the reference electrode was recalculated against the reversible hydrogen electrode (RHE) using the equation:| ERHE = EAg/AgCl + 0.059 × pH + 0.197 |
Linear sweep voltammetry (LSV) scans were recorded at a 10 mV s−1 scan rate in the anodic scan direction (0 to 1 V) for evaluating the OER and in the cathodic scan direction (0 to −1 V) for evaluating the ORR. The alkaline electrolyte used for all these measurements was 1 M KOH which was de-aerated by nitrogen gas purging. Electrochemical impedance spectroscopy (EIS) was performed in the frequency range of 10 kHz to 0.1 Hz at an amplitude of 10 mV at the onset potential. Chronoamperometric tests were also performed for the OER and the ORR for 24 hours at specified potentials. To evaluate the intrinsic activity, electrochemically active surface area was calculated by measuring double-layer capacitance, whereas cyclic voltammetry was carried out in a non-faradaic region (0.85–1.15 V vs. RHE) at varying scan rates (5–30 mV s−1). Accelerated durability test (ADT) was performed by scanning 1000 cyclic voltammetry curves between 0.75–1.5 V vs. RHE at a scan rate of 20 mV s−1.
2.5. Zinc–air battery performance
Testing of aqueous zinc–air battery performance was conducted in a homemade setup using a zinc foil (2 × 1 cm2 long) as the anode, a catalyst-coated carbon paper (loading of 1 mg cm−2) as the air electrode working as the current collector and the gas diffusion layer, and 6 M KOH with 0.2 M zinc acetate solution as the electrolyte. The galvanostatic charge–discharge cycles were performed at a current density of 5 mA cm−2 with 5 minutes of charge and discharge time each for approximately 88 hours, leading to ∼1045 cycles. The same current density was provided for the galvanostatic discharge experiment. Standard catalyst ink (Pt/C–RuO2) was also coated on the carbon paper working electrode to compare with the bifunctional activity of CCMNF in an aqueous zinc–air battery device.3. Results and discussion
3.1. Structural characterizations
The high-entropy alloy with equiatomic composition Cu–Co–Mn–Ni–Fe (CCMNF) was prepared in nanocrystalline form using casting-cum-cryomilling process as the schematic shown in Fig. 1(a). In the nanocrystalline HEA, CCMNF, the crystalline phase was found to be face-centered cubic (FCC), which was revealed by an X-ray diffraction pattern shown in Fig. 1(b). The approximate average particle size was found to be 14 ± 5 nm, estimated using a bright field TEM micrograph as shown in Fig. 1(c). The ring diffraction pattern of the selected area corroborates the result of the X-ray diffraction pattern to be FCC as shown in Fig. 1(d). Compositional analysis was performed by energy dispersive spectroscopy (EDS) and X-ray photoelectron spectroscopy (XPS), and the composition estimation has been compared in Fig. 1(e–g). The EDS showed higher Cu content due to the Cu-TEM grid used for mounting the powder sample. The XPS showed high C and O content because the XPS is a surface-sensitive technique, and the powder is exposed to the environment during mounting over the carbon tape. However, cryomilling provides high-purity metallic nanoparticles.39,41 The chemical homogeneity of high-entropy alloys is its virtue and has been tested by EDS elemental mapping. In the nanoparticles, all elements are homogeneously distributed at the atomic level as shown in Fig. 1(h).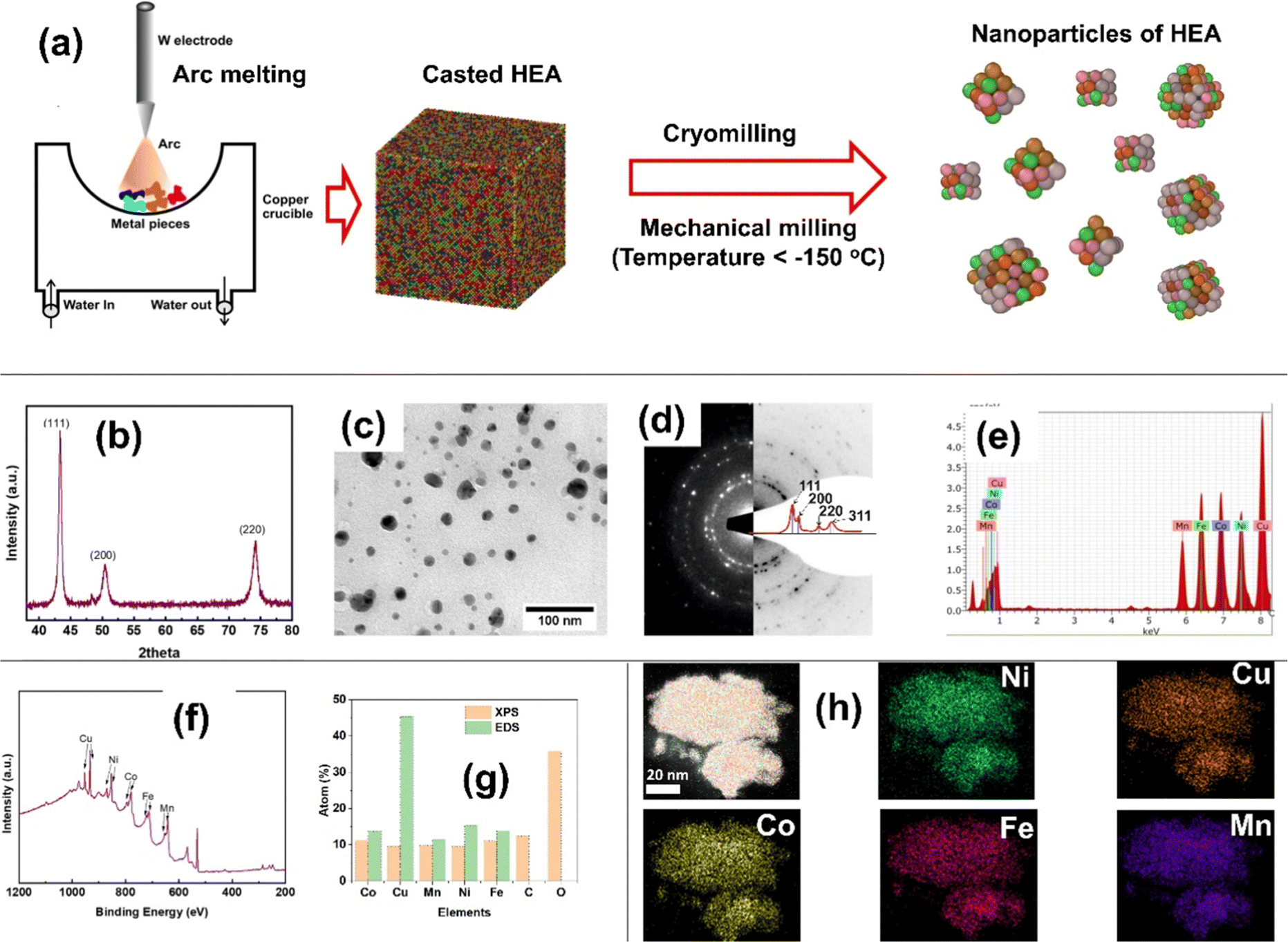 | ||
| Fig. 1 (a) Schematic of nanocrystalline HEA preparation by casting-cum-cryomilling, (b) X-ray diffraction peaks of the nanocrystalline CCMNF alloy, (c) bright-field TEM micrograph of CCMNF nanocrystalline powder, (d) ring diffraction pattern of a selected area of the nanoparticles, (e) EDS spectrum produced through TEM associated EDAX, (f) survey spectrum of CCMNF nanoparticles by XPS, (g) composition comparison between XPS and EDS, and (h) EDS elemental mapping by EDAX associated in TEM. | ||
It is desirable to have a single-phased FCC lattice structure HEA with homogeneously distributed active elements for superior oxygen electrocatalysis.42,43 The atomic radii of the selected 3d-transition metals (Fe, Co, Ni, Cu, and Mn) are similar enough to favor the stabilization of HEA into an FCC structure.42,44 High-entropy alloy systems stabilized with high configurational entropy have stressed lattice structures thus manifesting plenty of defect sites and coordinatively unsaturated sites that are randomly distributed across the multi-component surface.45 Such multi-metal sites have reorganized electronic arrangements leading to synergistic charge transfer and enhanced intrinsic activity46 that could serve as the epicenter of catalytic activity. Thus, motivated by the unique structural features that promise good catalytic activity, electrochemical studies were performed for the high-entropy CCMNF.
3.2. Electrochemical performance
The working electrode preparation is discussed in the Experimental section; it was subjected to cyclic voltammetry preconditioning in which the catalyst was scanned for multiple cycles at a high scan rate until constant overlapping curves were obtained. This step ensured that the cleaned surface of the catalyst was loaded on the working electrode. Linear sweep voltammetry scans were run for CCMNF in the anodic direction to analyze its performance in the oxidative environment as shown in Fig. 2(a). It was observed that CCMNF could initiate the catalysis of the oxygen evolution reaction at an onset potential of 1.57 V vs. RHE, while the standard catalyst RuO2, which is known as the state-of-the-art OER catalyst, initiates oxidation at the onset potential of 1.51 V vs. RHE. CCMNF requires only ∼60 mV more overpotential than the superior standard RuO2 thus proving it to be an excellent competitor catalyst. Also, CCMNF could obtain a current density of 10 mA cm−2 at an overpotential of 380 mV, while for RuO2, 320 mV overpotential was needed. Fig. 2(b) shows the Tafel slope of the catalyst in comparison to RuO2, where a small Tafel slope of 89.7 mV dec−1 indicates fast charge transfer kinetics in CCMNF. This indicates faster kinetics towards OER for CCMNF as compared to RuO2. The homogeneous distribution of surface-active sites in CCMNF reconfigured the electronic states and optimized adsorption energies for the various intermediates that were involved during the multiple steps of OER, leading to enhanced catalytic activity.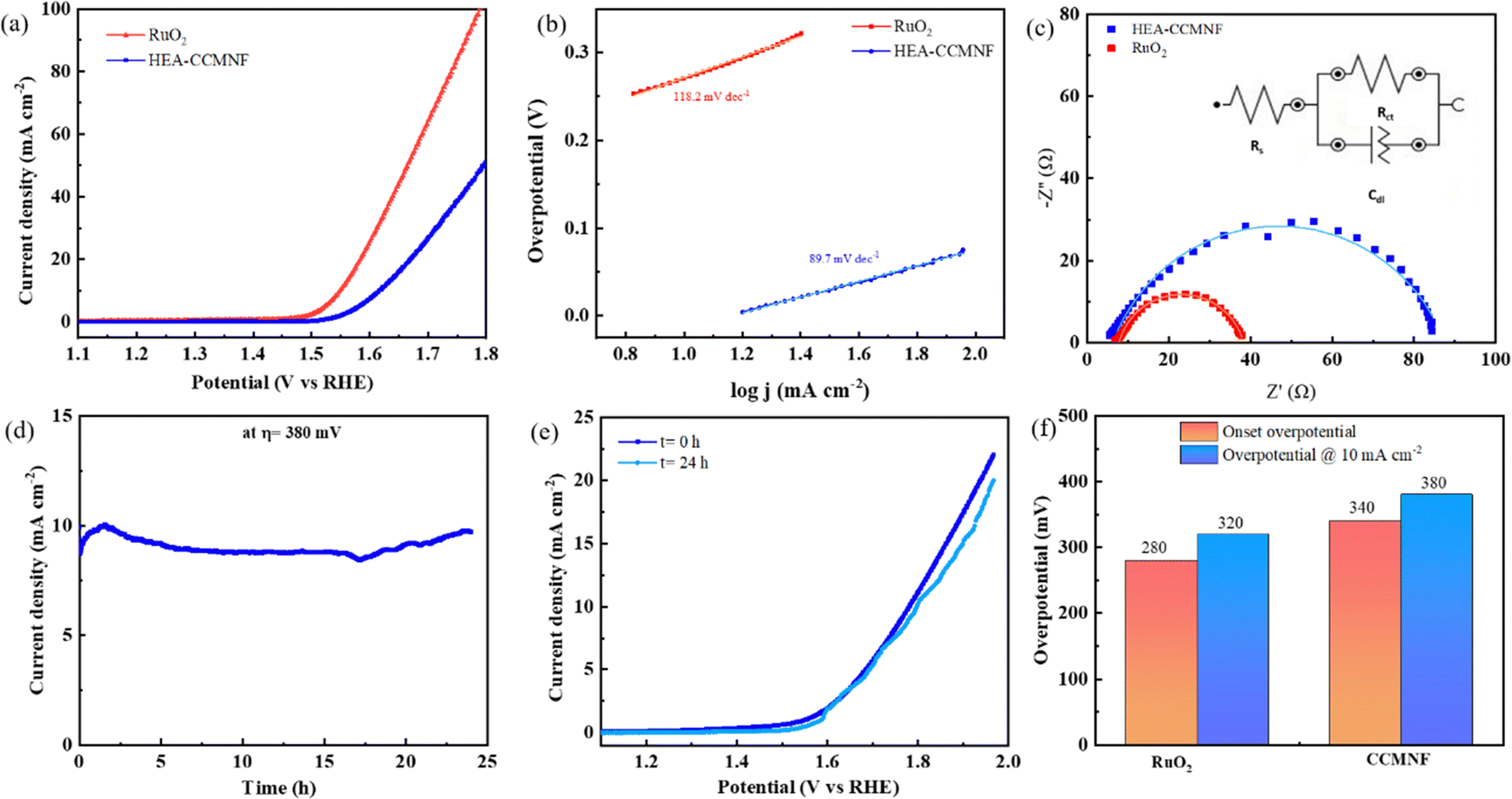 | ||
| Fig. 2 (a) Anodic linear sweep voltammetry (LSV) scans of CCMNF in comparison with the state-of-the-art catalysts (b) Tafel plots showing the Tafel slope, (c) Nyquist plots obtained from EIS giving information about interface resistance, (d) Long-term chronoamperometric tests of CCMNF run for 24 hours at a potential generating 10 mA cm−2 current density. (e) A comparison of undiminished OER activity in the LSV curves from before and after the chronoamperometric test, (f) a comparison of the overall OER performance. | ||
The EIS results in the form of Nyquist plots were fitted into the Randles circuit as shown in Fig. 2(c), where the charge transfer resistance was measured by the diameter of the semicircle. The smaller diameter of the RuO2 semicircle corresponds to a charge transfer resistance (Rct) of 34 Ω, which is much smaller than the Rct of CCMNF that was measured to be 80 Ω. This indicates a slightly higher resistance at the electrode–electrolyte interface of the CCMNF catalyst compared to the standard RuO2. Also, the long-term stability of the CCMNF catalyst in a highly oxidative anodic potential of 1.61 V (at 10 mA cm−2 current density) was tested in a chronoamperometric test for 24 hours as shown in Fig. 2(d). The catalyst retained a constant current density of approximately 10 mA cm−2 throughout the test, thus confirming its excellent operational stability during the OER. The small disturbances in the current density-time curve depict the fluctuations in the current arising from the release of oxygen bubbles evolving from the surface of the catalyst during the measurement. The LSV curve recorded after the chronoamperometric test shows a slightly more fluctuating current, due to more vigorous oxygen bubble evolution, which is possibly an effect of the modifications in the surface of the CCMNF catalyst after 24 hours of continuous operation. However, the current density and the OER onset potential are consistent with the performance before undergoing the chronoamperometric test as shown in Fig. 2(e). The long-term stability test of RuO2 was also conducted by measuring its chronoamperometric response at a potential of 1.55 V to obtain a current density of 10 mA cm−2, depicted in Fig. S1(a) (ESI†). The vigorous oxygen bubble evolution from the RuO2 catalyst surface led to fluctuations in the current density and was also responsible for the detachment of the catalyst from the electrode surface leading to a decline in the performance. The LSV curves shown in Fig. S1(b) (ESI†) were used to compare the OER performance after the long-term stability test, whereby RuO2 lost the catalytic activity significantly due to poor stability in anodic conditions. An overall OER performance comparison between CCMNF and the state-of-the-art OER catalyst, RuO2, is summarized in Fig. 2(f).
This confirms the good operational stability of CCMNF in the continuous oxygen evolution operation. The composition of CCMNF has nickel, iron, and cobalt sites that have been identified by many research studies47 as the sites of coherent electronic interaction required for superior oxygen evolution activity. Most importantly, the synergistic electronic interaction between nickel and iron promotes optimal binding energies for OER intermediates at β-NiOOH,48 which makes it a favorable catalytic site.
The surface structural features of CCMNF, possessing multiple transition metals (including Fe and Co) provide plentiful active sites and favorable reactant and intermediate adsorption energies. The synergistic electronic interactions between the component metal sites propose good oxygen reduction electrocatalytic activity, especially for HEA. To evaluate the catalytic behavior of CCMNF toward the oxygen reduction reaction (ORR), linear sweep voltammetry was performed in the cathodic region as shown in Fig. 3(a). The ORR performance of CCMNF was compared with the best-performing standard catalyst Pt/C; it was observed that CCMNF requires 120 mV more overpotential than Pt/C. However, an ORR onset potential of 0.78 V vs. RHE is significantly superior to many existing bifunctional catalysts. A comparison of the ORR performance of CCMNF with the various bifunctional oxygen electrocatalysts in the literature is summarized in Table S1 (ESI†).
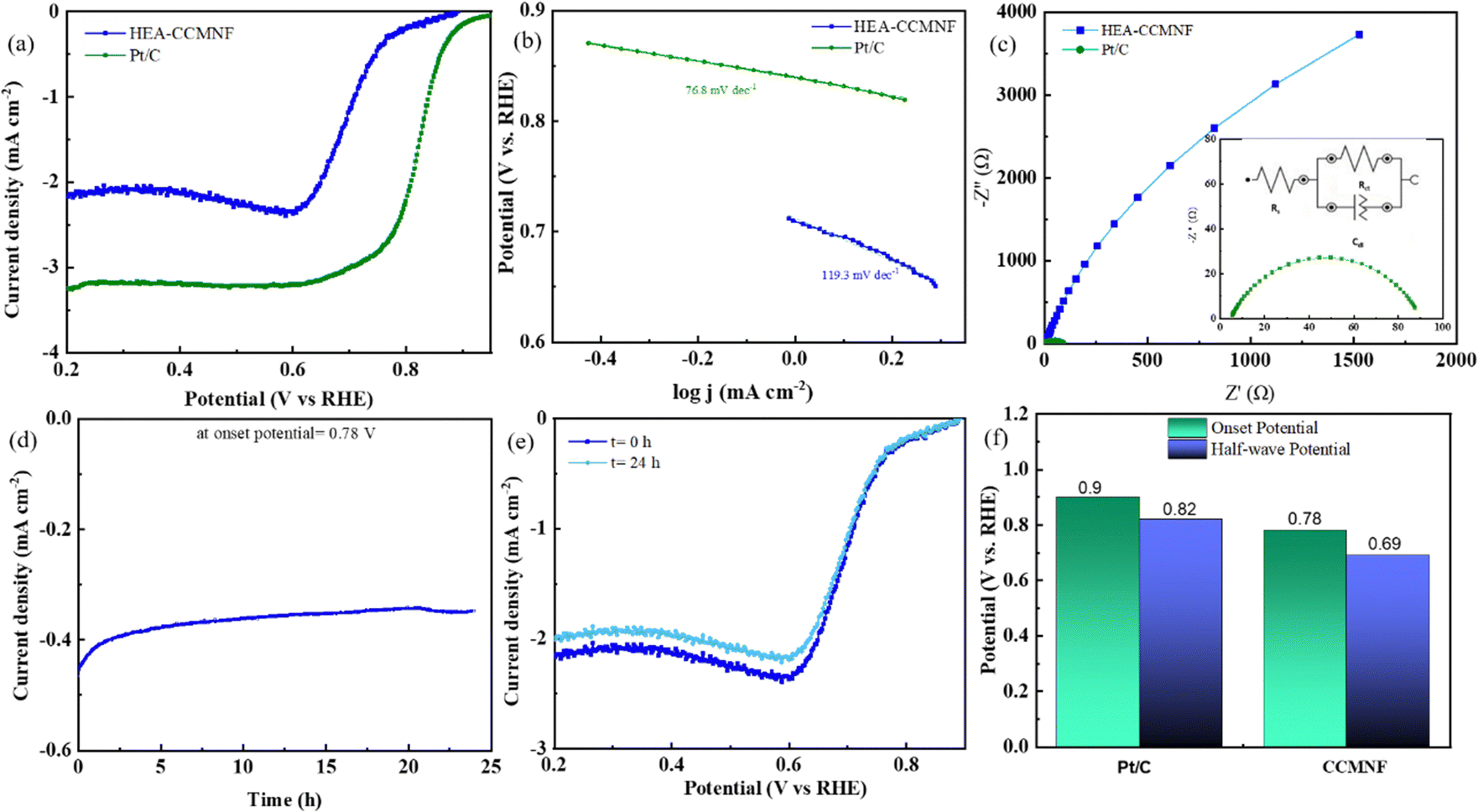 | ||
| Fig. 3 (a) A comparison of the linear sweep voltammetry (LSV) scans of CCMNF in the ORR region with the state-of-the-art catalyst, (b) Tafel plots indicating Tafel slope, (c) Nyquist plots obtained from EIS giving information about interface resistance, (d) long-term chronoamperometric tests of CCMNF run for 24 hours operation at the onset potential, (e) the comparison of undiminished ORR activity in the LSV curves from before and after the chronoamperometric test, (f) comparison of the overall ORR performance. | ||
The Tafel plots shown in Fig. 3(b) also suggest that CCMNF has relatively slow kinetics towards ORR as compared to Pt/C. The Nyquist plots shown in Fig. 3(c) also confirm the activity trend with a larger Rct value for CCMNF and a small Rct for Pt/C. The long-term stability of CCMNF was tested chronoamperometrically under the operational conditions of the ORR at the onset potential given in Fig. 3(d). The catalyst preserved a stable current response over 24 hours of continuous operation. Fig. 3(e) compares the ORR LSV curves before and after the chronoamperometric test, and the results complied with the chronoamperometry data, reflecting only a minor decrease in the current density over the long-term operation but no significant change in the onset potential was observed. To demonstrate the ORR stability of the synthesized catalyst CCMNF, its chronoamperometric performance was compared with Pt/C. Fig. S1(c) (ESI†) depicts the long-term chronoamperometric tests of the standard catalyst Pt/C at the ORR onset potential that was run for 24 hours to estimate its stability. Pt/C retained a fairly stable performance without fluctuations in the current density, similar to the long-term stability performance of CCMNF illustrated in Fig. 3(d). CCMNF retained excellent performance stability of the ORR, which was comparable to Pt/C. The LSV plots of Pt/C exhibited similar ORR performance after the 24 hour continuous operation test as shown in Fig. S1(d) (ESI†). A comparison of ORR performance between CCMNF and Pt/C is summarized in Fig. 3(f).
A progressive increase in the rotation rates of the working electrode led to an increase in the ORR current densities as well as depicted in the LSV plots in Fig. 4(a); the inset presents the Koutecky–Levich plot (K–L plot) at different potentials whereby the linearity between the parallel indicates the first-order kinetics of the reaction.49 Another important parameter to assess the intrinsic catalytic performance of an electrocatalyst is its electrochemically active surface area (ECSA). ECSA denotes the area on the surface of the catalysts that participate in interacting with the electrolyte for charge-transfer reaction. A higher ECSA implies more participation of the catalyst surface in the redox reactions, thus availing better performance. To gain insights into the electrocatalytic activity at the catalyst's surface, its ECSA was evaluated by the double-layer capacitance method50 since ECSA is directly related to double-layer capacitance (Cdl). Cyclic voltammetry scans were run in non-faradaic regions at different scan rates as shown in Fig. 4(b) inset. Fig. 4(b) shows the linear correlation between the difference between cathodic and anodic current density and the applied scan rate, where the slope of the linear plot represents the double-layer capacitance (Cdl) value which is representative of the ECSA. The Cdl value of 2.89 mF cm−2 for CCMNF indicates a high ECSA, thus more participation of the catalyst surface is evident in its electrocatalytic performance.
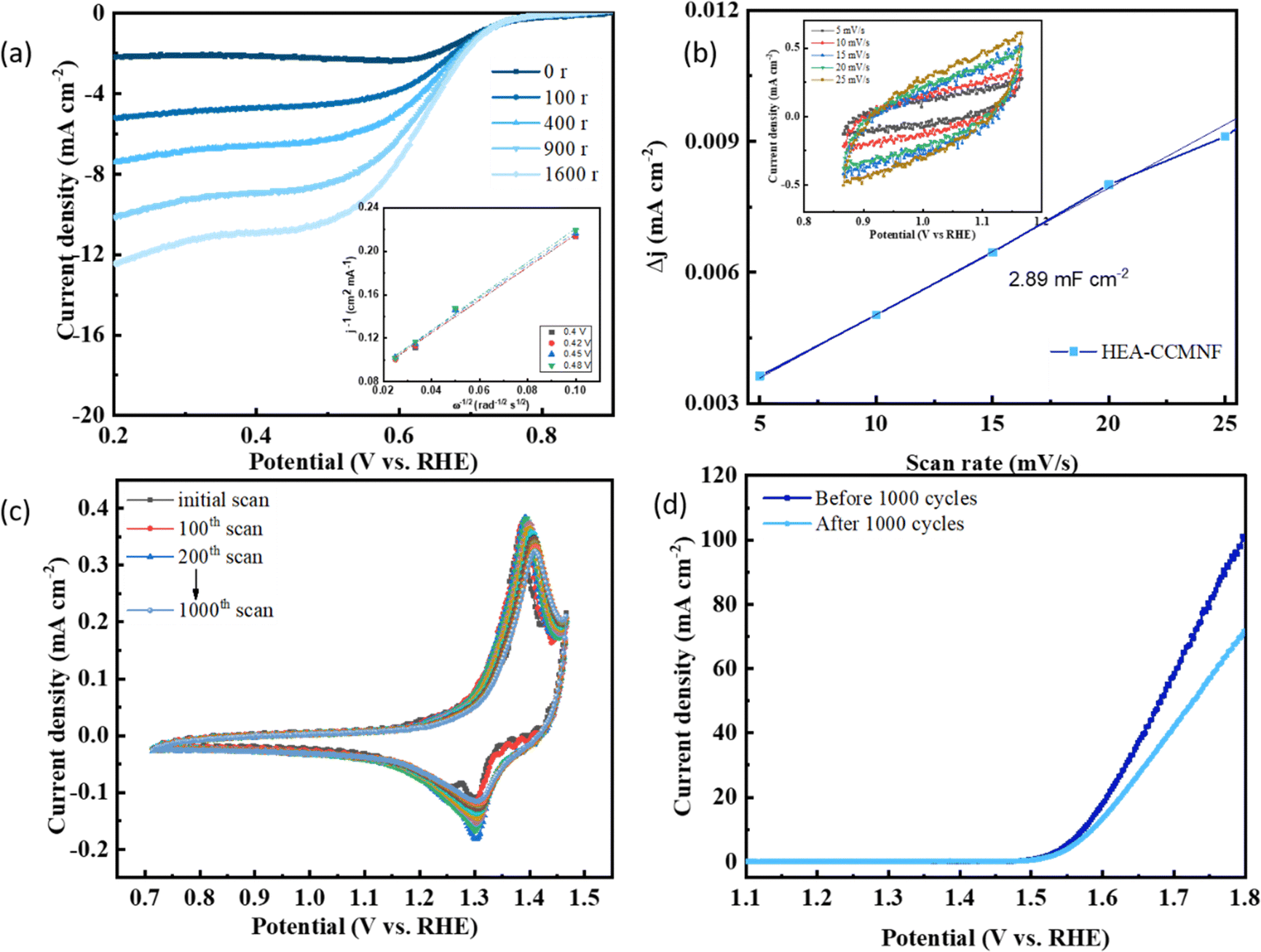 | ||
| Fig. 4 (a) ORR polarization curves at different rotation rates (inset: Koutecky–Levich plots at various potentials), (b) capacitive current density plot where the slope represents the Cdl value (inset: CV plots at a progressive scan rate in the non-faradaic region to calculate double layer capacitance (Cdl) which corresponds to the ECSA). (c) Accelerated durability test of CCMNF for 1000 CV scans at 20 mV s−1 scan rate. (d) LSV curves comparing the OER performance before and after the ADT test of 1000 scans. | ||
An accelerated durability test (ADT) of CCMNF was also performed to evaluate its stability during longer operations, as demonstrated in Fig. 4(c). The cyclic voltammetry curves remain unchanged in the potential values, with only a minor decrease in the current density, thus confirming the excellent stability of CCMNF in extreme operation durations. Fig. 4(d) depicts the OER performance of CCMNF from the initial scan of the ADT and the OER performance after 1000 scans, where the onset potential remained unshifted; only a small decrease in the current density was observed due to the loss in the number of active sites that leached into the electrolyte.
From the above experimental analysis, it is evident that CCMNF can catalyze both OER and ORR with not only excellent bifunctional oxygen electrocatalytic activity but also exceptional stability during both reactions. The outstanding electrocatalytic activity of CCMNF is attributed to the lattice effect and cocktail effect arising at the surface of the 3d transition metals.51Fig. 5(a and b) represents the bifunctional activity descriptor ΔE values calculated at the difference of potential required to reach 10 mA cm−2 current density in OER and the potential at the half-wave of the ORR curve. A catalyst that exhibits the minimum ΔE value requires the least overpotential to catalyze the redox couple reactions of oxygen evolution and oxygen reduction, thus being a superior bifunctional catalyst. CCMNF shows a ΔE value of 0.92 V, which is slightly more than Pt/C but lower than RuO2 standard catalysts. The promising bifunctional performance motivated us to use it as an oxygen/air electrode for an aqueous zinc–air battery.
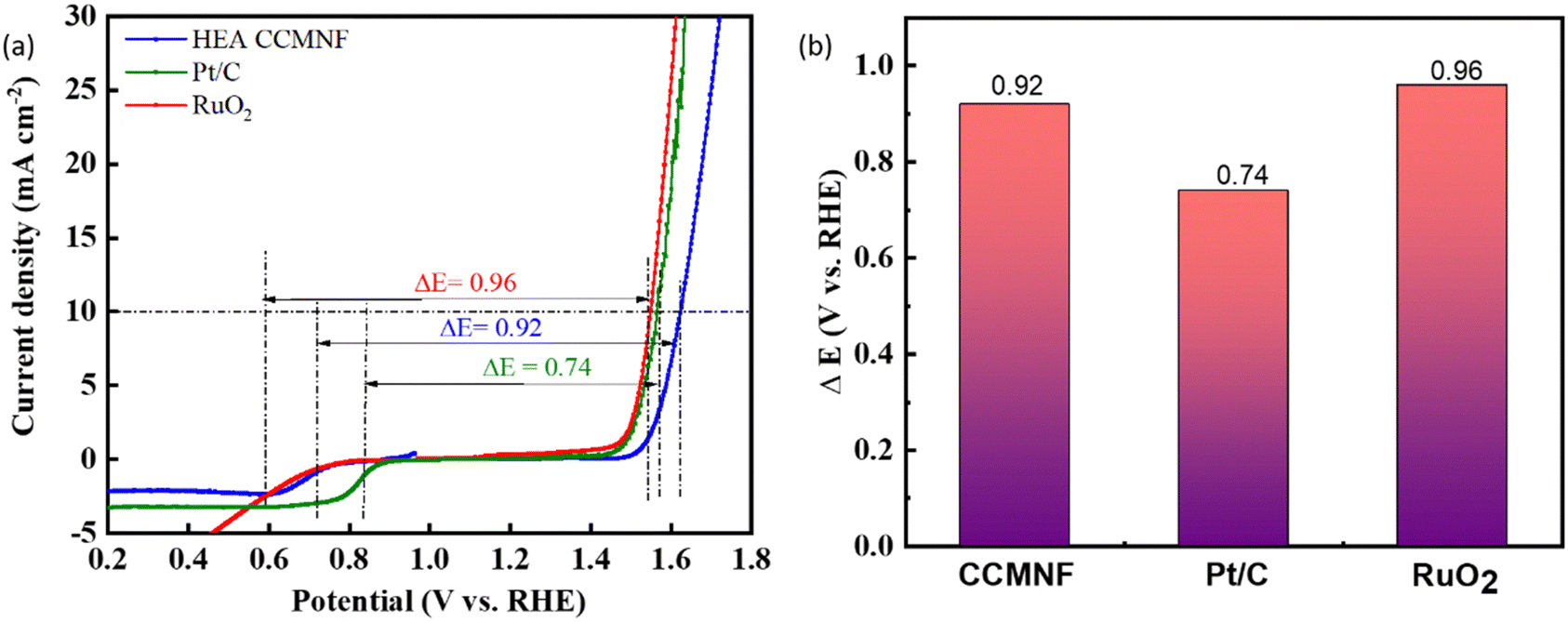 | ||
| Fig. 5 (a) LSV plots depicting the calculation of ΔE as the difference between the potential required to reach 10 mA cm−2 current density in the OER (Ej=10) and the potential at the half-wave of the ORR curve (E1/2). (b) The obtained ΔE values for different catalysts describing their bifunctional activity. | ||
3.3. Zinc–air battery performance
Fig. 6(a) depicts the typical set-up of an aqueous zinc–air battery cell, where the anode is a plate made of zinc foil while the cathode has a current collector and carbon paper as a gas diffusion layer. The active electrocatalyst material was coated as ink/paste on the carbon paper. The electrolyte is typically a highly concentrated alkali to achieve excellent conductivity, with a salt of zinc added to maintain uninterrupted redeposition of zinc50 from the electrolyte to the anode. The discharging polarization curves of the CCMNF catalyst, as shown in Fig. 6(b) reach a maximum current density of 70 mA cm−2 while having an open-circuit voltage of 1.52 V. The peak power density as obtained from the CCMNF battery in Fig. 6(c) was 16.5 mW cm−2. A typical galvanostatic charge–discharge curve for the battery assembled with CCMNF is shown in Fig. 6(d). At a current density of 5 mA cm−2, the charge–discharge cycles maintained a voltage gap of approximately 1.1 V and were fairly stable till 24 hours of continuous cycling. The energy density calculated for the zinc–air battery made from the CCMNF catalyst was found to be 481.5 W kg−1, while the specific capacity for this aqueous battery was calculated to be 688.6 mA h g−1 as represented in Fig. 6(e and f). These results confirm the excellent performance of CCMNF as a bifunctional electrocatalyst for the air-electrode of a zinc–air battery. The zinc–air battery assembled from the standard catalysts (Pt/C–RuO2) at the air electrode performed significantly poorly than CCMNF. Fig. S2(a) (ESI†) shows the lower power density of 12.2 mW cm−2 achieved from the zinc–air battery of Pt/C–RuO2; Fig. S2(b) (ESI†) represents the galvanostatic charge–discharge with 5 minutes of charging and discharging each at 5 mA cm−2 current density with a much wider, in fact, almost double, voltage gap (ΔE = 2.1 V) between charging–discharging than the one obtained for CCMNF zinc–air battery. The battery from the standard catalyst also could not last more than 33 hours of continuous cyclic operation indicating low durability of operation and unsatisfactory performance as the air electrode of the zinc–air battery.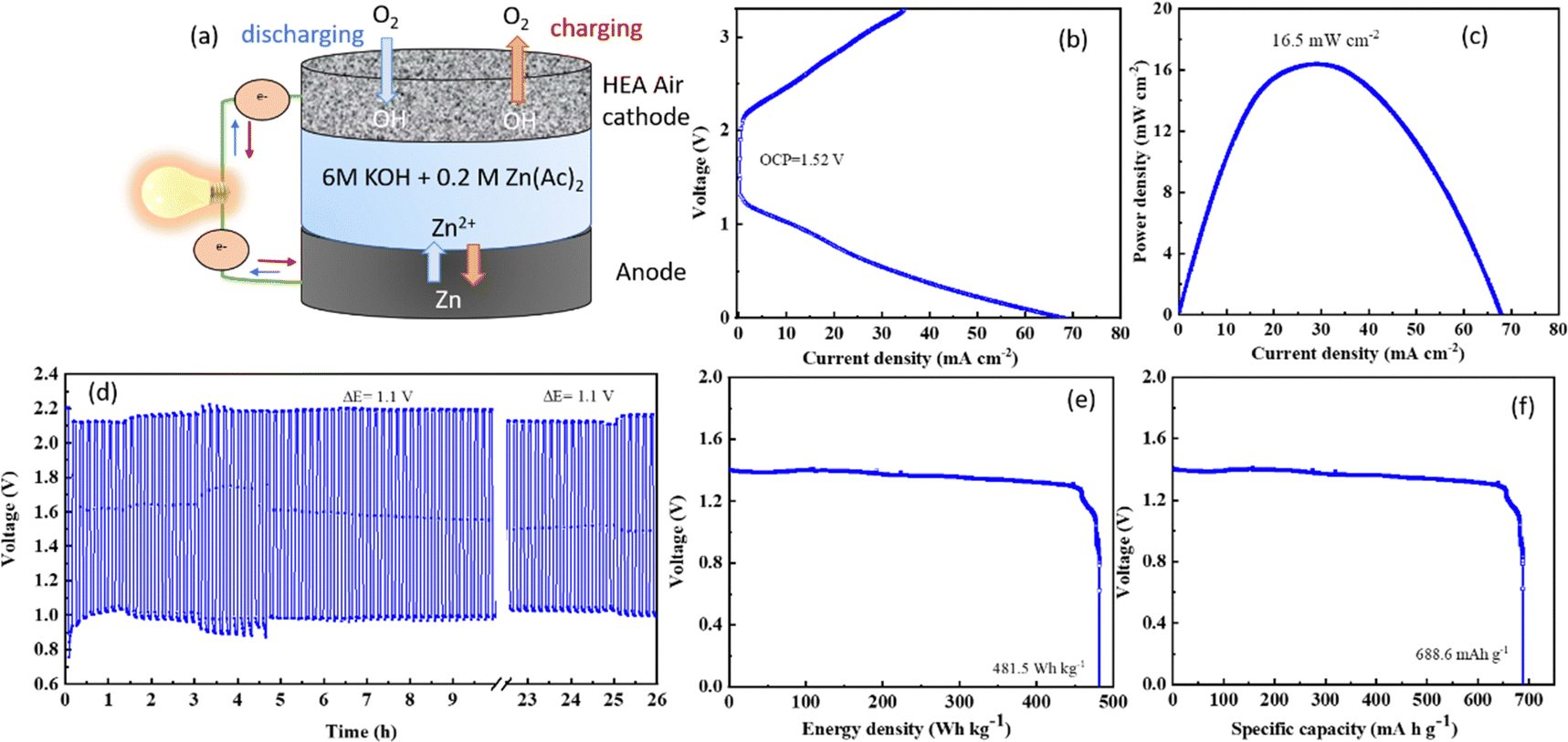 | ||
| Fig. 6 (a) A schematic representing the aqueous zinc–air battery set-up, (b) i–v polarization curve of CCMNF zinc–air battery depicting its open-circuit voltage. Various evaluation parameters to assess the performance of the zinc–air battery: (c) power density measurements, (d) galvanostatic charging and discharging at 5 mA cm−2 current density, (e) energy density, and (f) specific capacity measurements are presented. | ||
Given the extraordinary stability of the air electrode made by using CCMNF electrocatalyst, the charge–discharge cycles were carried out for an approximately 87 hour period as shown in Fig. 7(a). All the parameters for charge–discharge cycles were maintained as in the case of the previous study. It was noticed that even after 12 hours of continuous charge–discharge operation shown in Fig. 7(c) the voltage gap (ΔE) of 1.05 V was the same as what was present initially during the first two hours of cycling in Fig. 7(b). This indicates an outstanding interaction of the CCMNF-coated carbon paper with the electrolyte that does not need any long stabilization periods to deliver a stable activity. The prevention of loss of any catalytic activity of CCMNF after 12 hours of operation proves its durability. When the continuous cycling of the charge–discharge approaches 24 hours of operation, an increase in the voltage gap is evident which raises the ΔE to 1.3 V as indicated by Fig. 7(d). This suggests a loss in the activity of the CCMNF electrode owing to a drastic change in the structural or chemical properties that initiate the degradation of the catalyst. After 48 hours of charge–discharge cycles, shown in Fig. 7(e), the CCMNF catalyst had undergone further degradation and a decline in its catalytic activity as compared to its initial activity that could be inferred by an increase in the voltage gap to 1.7 V. After the catalyst had performed more than 84 hours of continuous charging and discharging, the zinc plate at the anode was cracked due to its poor stripping-plating preference originating from dendrite formation as illustrated by the SEM morphological in Fig. S3 (ESI†). This stopped the battery experiments. During every discharge cycle, the zinc from the anode enters the electrolyte as soluble zincate ions, thereby causing mass loss and morphology change at the anode. The subsequent charging cycles are incapable of homogeneously redistributing the deposited zinc as a result of the unevenness in the morphology; therefore, under the influence of the tip effect, a dendritic regrowth of the zinc occurs on the anode. In a highly alkaline environment, the oxidation of zinc dendrites into the insoluble zinc oxide is accelerated, which passivates the surface of the zinc anode from forming the Zn@ZnO core–shell interface. This electrically non-conducting ZnO severely limits the activity of zinc anode making it inaccessible for the electrochemical activity. The gradual growth of the Zn@ZnO dendritic morphology after numerous charge–discharge cycles makes the electrode brittle and pulverized, which is detrimental to the battery performance.52 The SEM image illustrated in Fig. S3 (ESI†) displays the dendritic morphology emerging on the zinc anode as a result of the tip effect during the constant charging and discharging cycles of the zinc–air battery. In the case of a solid-state zinc–air battery, these dendrites are found to pierce through the separator and electrolyte membrane and come in contact with the cathode thereby short-circuiting the battery.
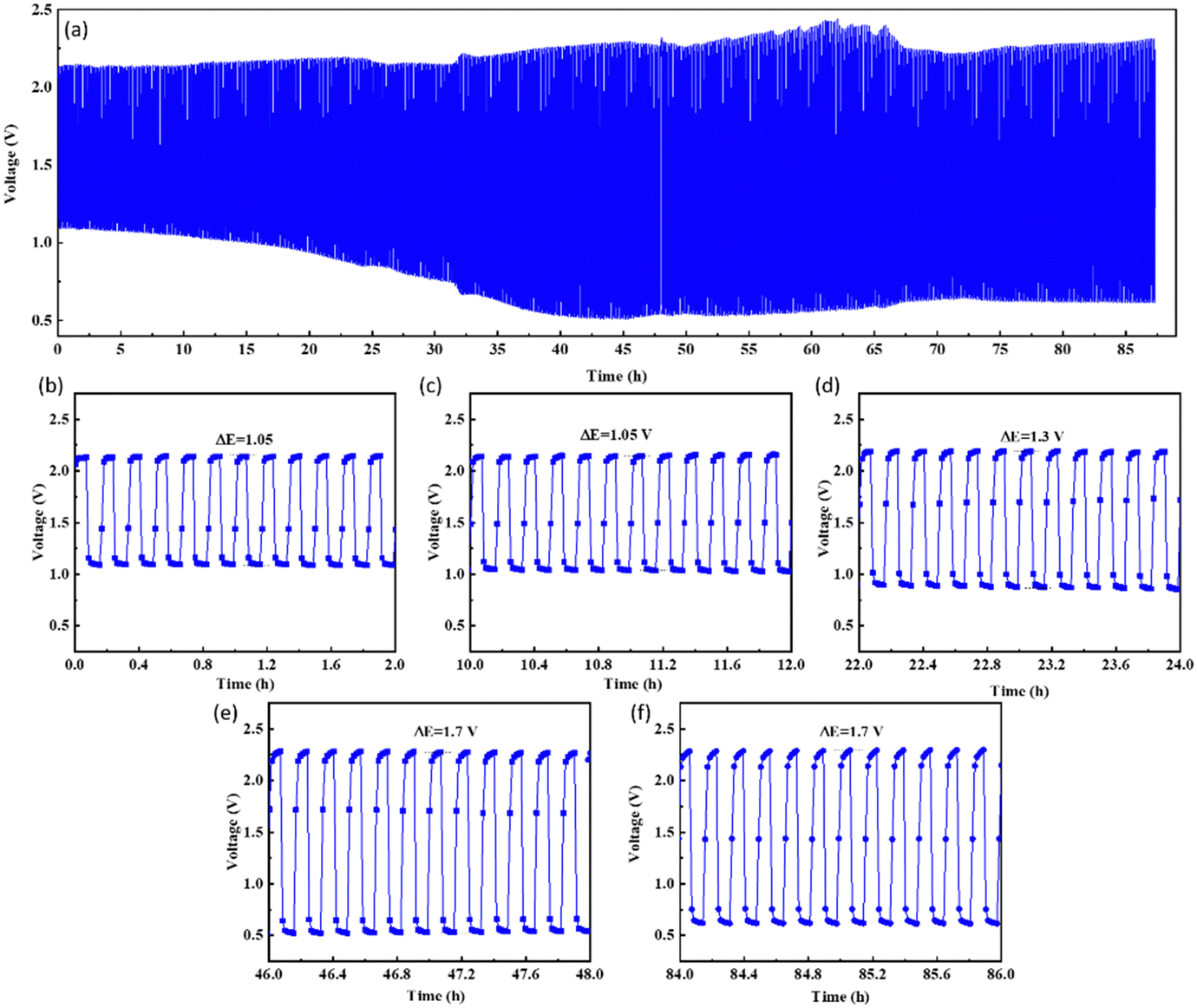 | ||
| Fig. 7 (a) Long-term charge–discharge cycles of CCMNF supporting its excellent performance as an oxygen electrocatalyst in the zinc–air battery, (b)–(f) the changing ΔE values over the continuous charging–discharging operation. | ||
Even after performing for >87 hours of continuous charge–discharge cycle as evident from Fig. 7(f), the voltage gap of the CCMNF catalyst between charging and discharging was still 1.7 V. The constant voltage gap suggests excellent stability and durability of the catalyst. Fig. S4 (ESI†) demonstrates subsequent TEM micrographs of the CCMNF catalyst after scrapping it from the battery electrode at the end of its operation. Fig. S4(a) (ESI†) presents a brightfield TEM micrograph, depicting agglomerated materials. This agglomeration was a result of nanoparticles being embedded within the binder, as evidenced by the corresponding ring diffraction patterns shown in Fig. S4(b) (ESI†). The diffraction rings, while clear, exhibit broadness due to the presence of an amorphous binder. The distinct ring pattern serves as compelling evidence of the nanocrystalline nature of the materials, with no significant signs of coarsening observed. Furthermore, the diffraction pattern continues to demonstrate the presence of the FCC crystal structure. Consequently, it can be concluded that the catalytic materials maintain their stability as single-phase high-entropy alloys.
3.4. Post zinc–air battery testing analysis
A high-entropy catalyst like CCMNF confining multiple quasi-equiatomic metals likely exhibits a synergistic electronic interaction at the atomic scale that offers considerable catalytic properties specifically towards the surface.53 The experimental characterizations that can identify the ongoing modifications at the catalyst surface during operation are the most reliable for understanding the behavior of the catalyst. In this study, the modifications happening in the CCMNF catalyst were monitored using X-ray photoelectron spectroscopy (XPS) as it is the best physical characterization technique to interpret the chemical changes occurring on the surface of the catalyst during electrocatalysis as these changes are interfacial phenomena arising majorly at the interface between the electrocatalyst and the electrolyte. The post-catalysis XPS and ICPMS studies are very useful approaches to understanding the mechanistic pathway and microstructural changes the catalysts endured during the operational condition while performing oxygen electrocatalysis in the zinc–air battery operation.XPS data were recorded for the CCMNF catalyst before testing its battery performance (t = 0 h) and after 24 hours (t = 24 h) and 87 hours (t = 87 h) of the battery operation. Fig. S5 (ESI†) depicts the XPS survey spectrum of the time-dependent stages of the device operation. All the metallic elements present in the catalyst remained after the test operation of the zinc–air battery, indicating good stability of the catalyst under operational conditions, however, a change in the intensity of peaks and the positions of binding energy could be observed. In the survey spectrum of t = 24 h and t = 87 h samples, the peak arising at ∼284.8 eV represents carbon from the carbon paper substrate used to load the CCMNF catalyst for the battery cycling tests. Also, in their survey spectrum, the presence of zinc on the CCMNF catalyst was detected. High-resolution XPS spectra of zinc 2p that were evident in CCMNF for t = 24 h and t = 87 h sample is shown in Fig. S6 (ESI†). The availability of zinc ions in the electrolyte solution or the stripping/plating of zinc at the anode occurring in tandem with the discharging and charging of the battery could be responsible for the appearance of zinc ions at the CCMNF electrocatalyst. It could be anticipated that the decline in the activity of CCMNF as suggested by the increasing voltage gap between the charge–discharge cycles is a consequence of the active catalytic sites being blocked by the involvement of zinc ions. The presence of zinc at the CCMNF catalyst alters the chemistry of electronic interactions occurring at the interface of the electrolyte and the catalyst surface. The surface atomic composition obtained from the XPS, Table S1 (ESI†), verified an increased percentage of zinc from 0.62 atom% to 0.80 atom% on the surface of the catalyst at 24 hours and 87 hours respectively. Also, there was a decline in the relative percentage of other metallic species present in the catalyst, suggesting the leaching of catalytically active metallic sites into the electrolyte over time, thus accelerating a decline in the electrocatalytic activity. As can be discerned from Fig. S7 (ESI†), the EDX data is no longer quasi-equimolar for the different metallic components as was at the beginning of the battery testing, i.e., at t = 0 h. As the catalyst performs charging–discharging cycles of the battery, the loss in the activity arises from the loss of catalytically active metallic sites. The loss of manganese and nickel is the most predominant, and the corresponding loss in the electrocatalytic activity of oxygen suggests them to be a major contributing active site. Table S2 (ESI†) compares the relative compositions of the metallic species present near the surface and throughout as evaluated from the XPS survey and EDX composition analysis. The detection of leached metallic species from the catalyst electrode into the electrolyte was confirmed by the ICP-MS analysis. The electrolyte was filtered thoroughly to avoid any undesirable solid particles and diluted with de-ionized water to prevent any ionic contamination before the ICP-MS measurements. Table S3 (ESI†) displays the increasing concentration of the leached metallic ions detected in the electrolyte, collected after 24 and 87 hours of battery cycling experiments (parts per billion scale).
From these results, it was confirmed that the metallic components from the HEA catalyst were leaching out in the harsh oxidative alkaline conditions of the electrolyte. However, there was no significant decline in the activity after 48 hours of operation, and remained almost the same till 87 hours. This indicates that there was no further leaching or degradation of the catalyst after some time; thus, it retained the activity thereafter. To understand the mechanism of catalyst evolution during battery operation, an in situ study would be very helpful, however, here we could not perform it. However, a study of changes in the local electronic structure affecting the activity was assessed using XPS data. The high-resolution XPS spectra of the metallic components are shown in Fig. 8(a–e).
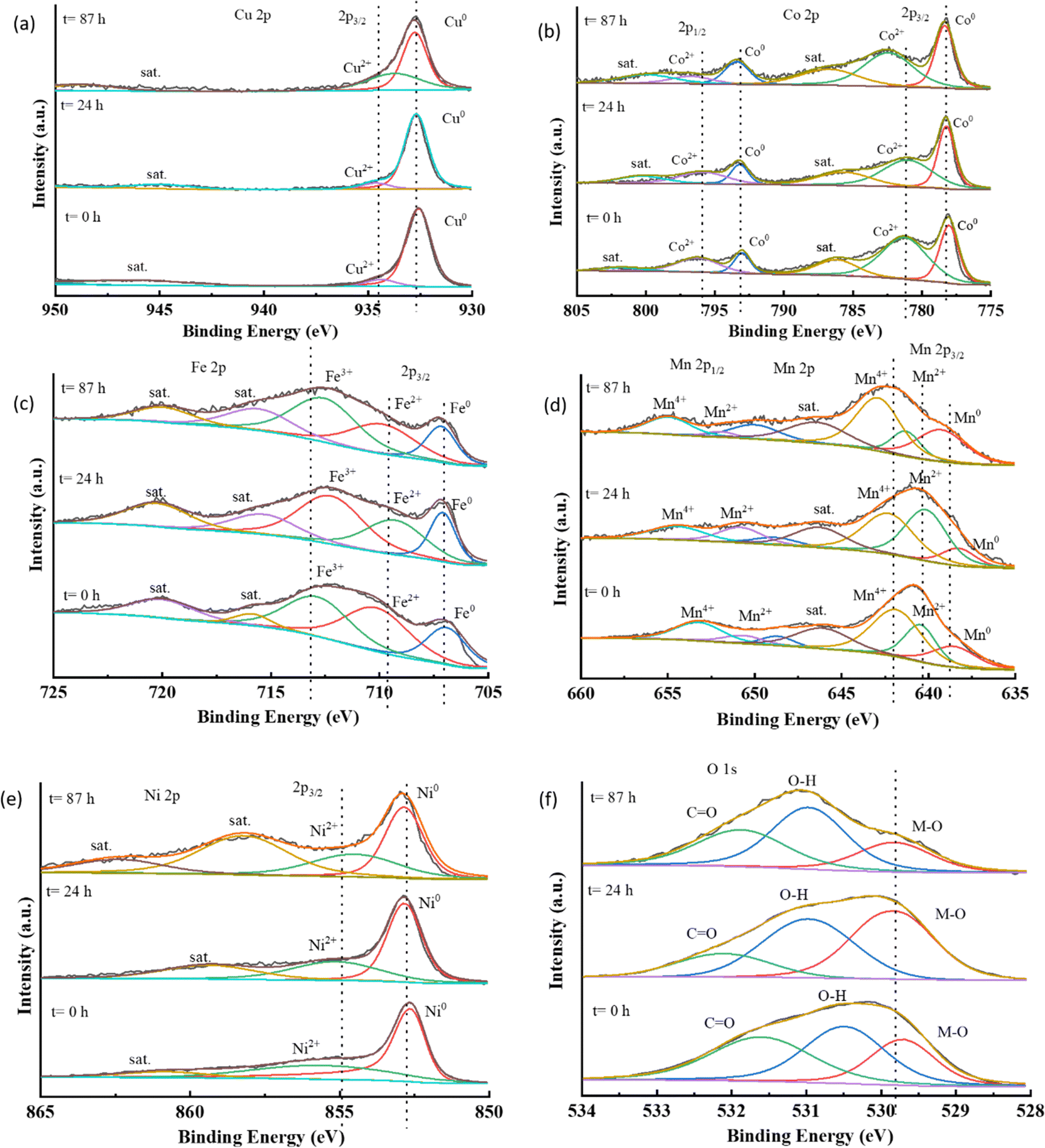 | ||
| Fig. 8 High-resolution XPS plots for the composition metals (a)–(e) in CCMNF and the modifications in their binding energies over the duration of charge–discharge operation of the zinc–air battery, (f) the deconvoluted O 1s spectra peaks of oxygen at the surface of CCMNF for each time set. | ||
The presence of metallic peaks M0 at their specific binding energies was consistent for all the composite metals of the HEA in all the stages of operation. However, various oxidation peaks corresponding to M2+ were found to have undergone changes in the binding energy during different stages of operation duration. The +2 state of copper moved towards a lower binding energy over the course of the operation. This could indicate an increased electron density at the copper sites, favoring the oxygen reduction reaction. For cobalt, the binding energy corresponding to the Co2+ peak increased after 87 hours of operation, suggesting the progress towards oxidation of Co2+ to Co3+ which is a critical step for catalysts supporting oxygen evolution reaction.7 In the case of iron, the binding energy corresponding to Fe0 and Fe2+ remained approximately the same, but for Fe3+, after 24 hours, a decrease was observed, while after 87 hours the binding energy increased again to nearly the same value as it was before the battery testing. This could have occurred due to iron's active participation in the oxygen reduction reaction initially, but the loss of activity over a long-term operation possibility occurs with the irreversible oxidation of the surface of iron to the highly active oxyhydroxide species responsible for the oxygen evolution reaction. It is a well-known fact from the literature that metal oxides undergo surface reconstruction to convert into oxy-hydroxide species via electrooxidation which behaves as the site for oxygen evolution reaction.54,55 The high-entropy catalyst, CCMNF, is also expected to have undergone surface and electronic reconstruction to form (oxy)hydroxide responsible for its superior OER activity. From this XPS data, the observance of negative shifts in the binding energy of high oxidation metal states after 87 hours of operation suggests the loss of catalytically active oxy-hydroxide species as a consequence of changing electronic interactions induced by reduction. Like the XPS binding energy trend of cobalt, manganese exhibits an almost unchanged metallic peak, while the Mn2+ and Mn4+ shift to a higher binding energy, thus indicating an active electron transfer from manganese sites. The Ni2+ binding energy shifts to a lower value after a continuous operation of 87 hours thus losing its activity as an efficient oxygen evolution reaction site. A positive shift occurred in the binding energy of Mn2+, Mn4+, and Co2+, while a negative shift occurred in the binding energy of Ni2+ and Cu2+, whereas the binding energy of Fe3+ remained the same essentially, which suggested an inconsistent stoichiometry, dynamic charge transfer, and lattice distortion occurring at the metallic sites, while sustaining ORR and OER catalytic activity during continuous operation. For a critical examination of the ongoing dynamics, operando techniques, such as SECM (scanning electrochemical microscopy), in situ XPS, and XAS (X-ray absorption spectroscopy) would be highly insightful in studying the surface microstructural changes as well as electronic changes. The continuous modification of oxidation states of the composite metals to compensate for the activity loss arising from cation vacancies created by the neighboring leaching metal sites proves the synergistic electronic interactions in the high-entropy alloy. This interplay of charge transfer also explains how CCMNF retains its electrocatalytic activity despite continuous leaching in harsh alkaline conditions. The synergistic electron transfer makes it an efficient electrocatalyst for two fundamentally opposite reactions of oxygen electrocatalysis. Loss in the performance of the zinc–air battery over 87 hours of operation is due to the leaching out of the catalyst from the gas diffusion layer, thus decreasing the availability of active sites, as well as leaving the electronic states irreversibly modified. This eventually leads to a decline in the zinc–air battery performance of the CCMNF as compared to its initial activity. From the O 1s spectra, in Fig. 8(f), the deconvoluted peaks indicate the presence of a metal-oxygen bond, adsorbed moisture, or the hydroxide bond as well as the carbonyl bond arising from the inevitable carbon contamination.
Another observation with the catalyst-coated electrode that caused a decline in the performance of the CCMNF catalyst was the incorporation of zinc on the sites of catalysis. The presence of zinc ions in the electrolyte as an additive facilitates the replenishment of zinc at the anode during charging by maintaining a reserve of zincate ions.50,56 However, as confirmed by the XPS survey spectra and high-resolution zinc 2p spectra, zinc was found to be incorporated at the CCMNF catalyst either by substituting for the leached metal sites or by replacing them. In either case, the electrocatalytic activity of CCMNF was diminished by the structural and electronic defects brought by unrequited zinc incorporation.57 Such constitutional changes in CCMNF electrocatalyst over the operation could be avoided by using a solid electrolyte membrane which would selectively allow the passage of hydroxide ions and inhibit the intrusion of zinc ions from the solution at the oxygen electrode, thus prolonging its excellent catalytic activity.
Initially, a parallel between the time-dependent declining performance of the CCMNF zinc–air battery and the gradual leaching of manganese and nickel catalytic sites could be recognized. Even though the leaching of all the catalytically active metal sites from CCMNF was observed from the EDX and ICP-MS results, the concentration loss was maximum for Mn and Ni. Synergistic electronic interaction between Mn and Ni has been recognized in the literature as responsible for bifunctional oxygen electrocatalytic activity.58–60 Hence, it led to the proposal of the predominant catalytic activity arising from Mn and Ni sites. The high-resolution XPS results of each metal indicated a positive shift in the binding energy of Mn2+, Mn4+, and Co2+, a negative shift in the binding energy of Ni2+ and Cu2+, and essentially constant binding energy of Fe3+, which implied an inconsistency in the stoichiometry, dynamic charge transfer, and lattice distortion occurring at these metallic sites during continuous catalytic operation. This observation propounded an in-tandem interaction between all metallic sites rather than being limited to only Mn and Ni.
4. Conclusion
High-entropy alloys offer the most suitable choice of catalytic materials owing to the virtually infinite number of combinations of surface microstructures and atomic interactions. The various features have several advantages for catalysis, such as lattice distortion exhibiting a favorable intermediate adsorption/desorption kinetics, thus enabling their utility for oxygen electrocatalysis. The bifunctional activity results and aqueous zinc–air battery test approve the deployment of CCMNF as the oxygen electrocatalyst. The long-term charging–discharging of the battery showed stable performance for a certain duration and then an increase in voltage gap. This trend followed as long as the zinc anode was functional. The loss of metallic sites due to leaching and the incorporation of zinc sites at the CCMNF could be the cause of this fall in the activity. The synergistic changes in electronic states might help circumvent the decline in the electrocatalytic activity associated with the loss of metal species through leaching. The XPS results corroborate our hypothesis that shifts in the binding energies of different constituent metals of the HEA catalyst occur because of the coordinated electron transfer among the metallic sites of the alloy. Thus, the reorganized oxidation states of the metallic sites sustain electrocatalytic performance till a certain degree before undergoing degradation. This suggests excellent synergism between the active sites of the catalyst CCMNF for extended activity and stability.Data availability
The raw/processed data required to reproduce these findings can be shared on request.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We are grateful to the Advanced Materials Research Centre (AMRC) at IIT, Mandi, for the research facility. We thank the Ministry of Human Resource Development (MHRD), India. AH extends thanks to the SERB core grant, DST (CRG/2019/005276) for funding. We also thank DST/FIST (SR/FST/CS-I/2017/20(C) for facility development.References
- J. Lu, Z. Chen, Z. Ma, F. Pan, L. A. Curtiss and K. Amine, The role of nanotechnology in the development of battery materials for electric vehicles, Nat. Nanotechnol., 2016, 11, 1031–1038 CrossRef CAS PubMed.
- X. Fu, C. A. Schuh and E. A. Olivetti, Materials selection considerations for high entropy alloys, Scr. Mater., 2017, 138, 145–150 CrossRef CAS.
- K. D. Vogiatzis, M. V. Polynski, J. K. Kirkland, J. Townsend, A. Hashemi, C. Liu and E. A. Pidko, Computational Approach to Molecular Catalysis by 3d Transition Metals: Challenges and Opportunities, Chem. Rev., 2019, 119, 2453–2523 CrossRef CAS PubMed.
- F. Dionigi, J. Zhu, Z. Zeng, T. Merzdorf, H. Sarodnik, M. Gliech, L. Pan, W. Li, J. Greeley and P. Strasser, Intrinsic Electrocatalytic Activity for Oxygen Evolution of Crystalline 3d-Transition Metal Layered Double Hydroxides, Angew. Chem., 2021, 133, 14567–14578 CrossRef.
- S. Anantharaj and V. Aravindan, Developments and Perspectives in 3d Transition-Metal-Based Electrocatalysts for Neutral and Near-Neutral Water Electrolysis, Adv. Energy Mater., 2020, 10, 1902666, DOI:10.1002/aenm.201902666.
- C. Maouche, Y. Zhou, Y. Wang and J. Yang, Recent advances of the key parameters of 3d block transition metal single and dual atoms catalysts: from their synthesis to their practical applications, Mater. Today Sustainable, 2023, 21, 100288 CrossRef.
- K. Wang, X. Wang, Z. Li, B. Yang, M. Ling, X. Gao, J. Lu, Q. Shi, L. Lei, G. Wu and Y. Hou, Designing 3d dual transition metal electrocatalysts for oxygen evolution reaction in alkaline electrolyte: Beyond oxides, Nano Energy, 2020, 77, 105162 CrossRef CAS.
- N. Kaplaneris and L. Ackermann, Earth-abundant 3d transition metals on the rise in catalysis, Beilstein J. Org. Chem., 2022, 18, 86–88 CrossRef CAS PubMed.
- Y. Feng, S. Long, X. Tang, Y. Sun, R. Luque, X. Zeng and L. Lin, Earth-abundant 3d-transition-metal catalysts for lignocellulosic biomass conversion, Chem. Soc. Rev., 2021, 50, 6042–6093 RSC.
- S. M. Jowitt, G. M. Mudd and J. F. H. Thompson, Future availability of non-renewable metal resources and the influence of environmental, social, and governance conflicts on metal production, Commun. Earth Environ., 2020, 1, 13 CrossRef.
- N. K. Katiyar, S. Dhakar, A. Parui, P. Gakhad, A. K. Singh, K. Biswas, C. S. Tiwary and S. Sharma, Electrooxidation of Hydrazine Utilizing High-Entropy Alloys: Assisting the Oxygen Evolution Reaction at the Thermodynamic Voltage, ACS Catal., 2021, 11, 14000–14007 CrossRef CAS.
- N. K. Katiyar, S. Nellaiappan, R. Kumar, K. D. Malviya, K. G. Pradeep, A. K. Singh, S. Sharma, C. S. Tiwary and K. Biswas, Formic acid and methanol electro-oxidation and counter hydrogen production using nano high entropy catalyst, Mater. Today Energy, 2020, 16, 100393 CrossRef.
- N. Kumar Katiyar, K. Biswas, J.-W. Yeh, S. Sharma and C. Sekhar Tiwary, A perspective on the catalysis using the high entropy alloys, Nano Energy, 2021, 88, 106261 CrossRef CAS.
- S. Nellaiappan, N. K. Katiyar, R. Kumar, A. Parui, K. D. Malviya, K. G. Pradeep, A. K. Singh, S. Sharma, C. S. Tiwary and K. Biswas, High-Entropy Alloys as Catalysts for the CO 2 and CO Reduction Reactions: Experimental Realization, ACS Catal., 2020, 10, 3658–3663 CrossRef CAS.
- D. B. Miracle, High entropy alloys as a bold step forward in alloy development, Nat. Commun., 2019, 10, 1–3 CrossRef CAS PubMed.
- C. Wen, Y. Zhang, C. Wang, D. Xue, Y. Bai, S. Antonov, L. Dai, T. Lookman and Y. Su, Machine learning assisted design of high entropy alloys with desired property, Acta Mater., 2019, 170, 109–117 CrossRef CAS.
- J. Li, B. Xie, L. Li, B. Liu, Y. Liu, D. Shaysultanov, Q. Fang, N. Stepanov and P. K. Liaw, Performance-oriented multistage design for multi-principal element alloys with low cost yet high efficiency, Mater. Horizons, 2022, 9, 1518–1525 RSC.
- X. Yan and Y. Zhang, Functional properties and promising applications of high entropy alloys, Scr. Mater., 2020, 187, 188–193 CrossRef CAS.
- J. Kumar, S. Jha, A. Raturi, A. Bajpai, R. Sonkusare, N. P. Gurao and K. Biswas, Novel Alloy Design Concepts Enabling Enhanced Mechanical Properties of High Entropy Alloys, Front. Mater., 2022, 9, 1–13 Search PubMed.
- T. A. A. Batchelor, J. K. Pedersen, S. H. Winther, I. E. Castelli, K. W. Jacobsen and J. Rossmeisl, High-Entropy Alloys as a Discovery Platform for Electrocatalysis, Joule, 2019, 3, 834–845 CrossRef CAS.
- M. Fu, X. Ma, K. Zhao, X. Li and D. Su, High-entropy materials for energy-related applications, iScience, 2021, 24, 102177, DOI:10.1016/j.isci.2021.102177.
- E. P. George, D. Raabe and R. O. Ritchie, High-entropy alloys, Nat. Rev. Mater., 2019, 4, 515–534 CrossRef CAS.
- H. Li, J. Lai, Z. Li and L. Wang, Multi-Sites Electrocatalysis in High-Entropy Alloys, Adv. Funct. Mater., 2021, 31, 2106715 CrossRef CAS.
- H. Li, Y. Han, H. Zhao, W. Qi, D. Zhang, Y. Yu, W. Cai, S. Li, J. Lai, B. Huang and L. Wang, Fast site-to-site electron transfer of high-entropy alloy nanocatalyst driving redox electrocatalysis, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- S. Zhao, Role of chemical disorder and local ordering on defect evolution in high-entropy alloys, Phys. Rev. Mater., 2021, 5, 1–11 Search PubMed.
- W. A. Saidi, Emergence of local scaling relations in adsorption energies on high-entropy alloys, npj Comput. Mater., 2022, 8, 86, DOI:10.1038/s41524-022-00766-y.
- N. Kumar Katiyar, K. Biswas, J.-W. Yeh, S. Sharma and C. Sekhar Tiwary, A perspective on the catalysis using the high entropy alloys, Nano Energy, 2021, 88, 106261 CrossRef CAS.
- Y. Zhang, D. Wang and S. Wang, Small, 2022, 18 Search PubMed.
- X. Cui, B. Zhang, C. Zeng and S. Guo, Electrocatalytic activity of high-entropy alloys toward oxygen evolution reaction, MRS Commun., 2018, 8, 1230–1235 CrossRef CAS.
- L. Sharma, N. K. Katiyar, A. Parui, R. Das, R. Kumar, C. S. Tiwary, A. K. Singh, A. Halder and K. Biswas, Low-cost high entropy alloy (HEA) for high-efficiency oxygen evolution reaction (OER), Nano Res., 2022, 15, 4799–4806 CrossRef CAS.
- N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, Activity Descriptor Identification for Oxygen Reduction on Nonprecious Electrocatalysts: Linking Surface Science to Coordination Chemistry, J. Am. Chem. Soc., 2013, 135, 15443–15449 CrossRef CAS PubMed.
- M. Luo and S. Guo, Strain-controlled electrocatalysis on multimetallic nanomaterials, Nat. Rev. Mater., 2017, 2, 17059 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Electronic factors determining the reactivity of metal surfaces, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- H. Li, J. Lai, Z. Li and L. Wang, Multi-Sites Electrocatalysis in High-Entropy Alloys, Adv. Funct. Mater., 2021, 31, 2106715 CrossRef CAS.
- H. Li, Y. Han, H. Zhao, W. Qi, D. Zhang, Y. Yu, W. Cai, S. Li, J. Lai, B. Huang and L. Wang, Fast site-to-site electron transfer of high-entropy alloy nanocatalyst driving redox electrocatalysis, Nat. Commun., 2020, 11, 5437 CrossRef CAS PubMed.
- S. Zhao, Role of chemical disorder and local ordering on defect evolution in high-entropy alloys, Phys. Rev. Mater., 2021, 5, 103604 CrossRef CAS.
- W. A. Saidi, Emergence of local scaling relations in adsorption energies on high-entropy alloys, npj Comput. Mater., 2022, 8, 86 CrossRef CAS.
- Z. Chen, T. Zhang, X. Gao, Y. Huang, X. Qin, Y. Wang, K. Zhao, X. Peng, C. Zhang, L. Liu, M. Zeng and H. Yu, Engineering Microdomains of Oxides in High-Entropy Alloy Electrodes toward Efficient Oxygen Evolution, Adv. Mater., 2021, 33, 2101845 CrossRef CAS PubMed.
- N. Kumar and K. Biswas, Fabrication of novel cryomill for synthesis of high purity metallic nanoparticles, Rev. Sci. Instrum., 2015, 86, 083903 CrossRef PubMed.
- N. K. Katiyar, K. Biswas and C. S. Tiwary, Cryomilling as environmentally friendly synthesis route to prepare nanomaterials, Int. Mater. Rev., 2021, 66, 493–532 CrossRef CAS.
- N. K. Katiyar, K. Biswas, C. S. Tiwary, L. D. Machado and R. K. Gupta, Stabilization of a Highly Concentrated Colloidal Suspension of Pristine Metallic Nanoparticles, Langmuir, 2019, 35, 2668–2673 CrossRef CAS PubMed.
- K. Huang, B. Zhang, J. Wu, T. Zhang, D. Peng, X. Cao, Z. Zhang, Z. Li and Y. Huang, Exploring the impact of atomic lattice deformation on oxygen evolution reactions based on a sub-5 nm pure face-centred cubic high-entropy alloy electrocatalyst, J. Mater. Chem. A, 2020, 8, 11938–11947 RSC.
- A. Sivanantham, H. Lee, S. W. Hwang, B. Ahn and I. S. Cho, Preparation, electrical and electrochemical characterizations of CuCoNiFeMn high-entropy-alloy for overall water splitting at neutral-pH, J. Mater. Chem. A, 2021, 9, 16841–16851 RSC.
- S. A. Kube, S. Sohn, D. Uhl, A. Datye, A. Mehta and J. Schroers, Phase selection motifs in High Entropy Alloys revealed through combinatorial methods: Large atomic size difference favors BCC over FCC, Acta Mater., 2019, 166, 677–686 CrossRef CAS.
- K. Li and W. Chen, Recent progress in high-entropy alloys for catalysts: synthesis, applications, and prospects, Mater. Today, Energy, 2021, 20, 100638 CAS.
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, I. Gueye, O. Seo, J. Kim, S. Hiroi, O. Sakata, S. Kawaguchi, Y. Kubota and H. Kitagawa, On the electronic structure and hydrogen evolution reaction activity of platinum group metal-based high-entropy-alloy nanoparticles, Chem. Sci., 2020, 11, 12731–12736 RSC.
- M. B. Stevens, L. J. Enman, E. H. Korkus, J. Zaffran, C. D. M. Trang, J. Asbury, M. G. Kast, M. C. Toroker and S. W. Boettcher, Ternary Ni-Co-Fe oxyhydroxide oxygen evolution catalysts: Intrinsic activity trends, electrical conductivity, and electronic band structure, Nano Res., 2019, 12, 2288–2295 CrossRef CAS.
- Y. Zhou and N. López, The Role of Fe Species on NiOOH in Oxygen Evolution Reactions, ACS Catal., 2020, 10, 6254–6261 CrossRef CAS.
- Q. Bai, F. Shen, S. Li, J. Liu, L. Dong, Z. Wang and Y. Lan, Cobalt@Nitrogen-Doped Porous Carbon Fiber Derived from the Electrospun Fiber of Bimetal–Organic Framework for Highly Active Oxygen Reduction, Small Methods, 2018, 2, 1800049 CrossRef.
- C. Madan, A. Mathur and A. Halder, Facile Generation of a Stable Bi-Functional Mixed Phase Fe 3 O 4/Fe-N 4 Electrocatalyst for Rechargeable Zinc-Air Battery, J. Electrochem. Soc., 2022, 169, 020516 CrossRef CAS.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, Transition Metal Oxides as Electrocatalysts for the Oxygen Evolution Reaction in Alkaline Solutions: An Application-Inspired Renaissance, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- D. Deckenbach and J. J. Schneider, A Long-Overlooked Pitfall in Rechargeable Zinc–Air Batteries: Proper Electrode Balancing, Adv. Mater. Interfaces, 2023, 10(15), 2202494, DOI:10.1002/admi.202202494.
- F. McKay, Y. Fang, O. Kizilkaya, P. Singh, D. D. Johnson, A. Roy, D. P. Young, P. T. Sprunger, J. C. Flake, W. A. Shelton and Y. Xu, CoCrFeNi High-Entropy Alloy as an Enhanced Hydrogen Evolution Catalyst in an Acidic Solution, J. Phys. Chem. C, 2021, 125, 17008–17018 CrossRef CAS PubMed.
- M. Kuang, J. Zhang, D. Liu, H. Tan, K. N. Dinh, L. Yang, H. Ren, W. Huang, W. Fang, J. Yao, X. Hao, J. Xu, C. Liu, L. Song, B. Liu and Q. Yan, Amorphous/Crystalline Heterostructured Cobalt-Vanadium-Iron (Oxy)hydroxides for Highly Efficient Oxygen Evolution Reaction, Adv. Energy Mater., 2020, 10, 2002215 CrossRef CAS.
- Q. Chen, X. Sheng, Y. Sun, K. Yu, Y. Cheng, H. Geng, J. Shen, Q. Zhang and J. Liu, Low-cost Trimetallic Ni-Fe-Mn Oxides/(Oxy)hydroxides Nanosheets Array for Efficient Oxygen Evolution Reaction, Eur. J. Inorg. Chem., 2022, 2022(21), e202200230, DOI:10.1002/ejic.202200230.
- Y. Li, M. Gong, Y. Liang, J. Feng, J.-E. Kim, H. Wang, G. Hong, B. Zhang and H. Dai, Advanced zinc-air batteries based on high-performance hybrid electrocatalysts, Nat. Commun., 2013, 4, 1805 CrossRef PubMed.
- W. Liu, J. Han, I. Yamada and S. Yagi, Effects of zinc ions at tetrahedral sites in spinel oxides on catalytic activity for oxygen evolution reaction, J. Catal., 2021, 394, 50–57 CrossRef CAS.
- G. Fu, X. Jiang, Y. Chen, L. Xu, D. Sun, J.-M. Lee and Y. Tang, Robust bifunctional oxygen electrocatalyst with a “rigid and flexible” structure for air-cathodes, NPG Asia Mater., 2018, 10, 618–629 CrossRef CAS.
- K.-L. Yan, X. Shang, W.-K. Gao, B. Dong, X. Li, J.-Q. Chi, Y.-R. Liu, Y.-M. Chai and C.-G. Liu, Ternary MnO 2/NiCo 2 O 4/NF with hierarchical structure and synergistic interaction as efficient electrocatalysts for oxygen evolution reaction, J. Alloys Compd., 2017, 719, 314–321 CrossRef CAS.
- T. Matthews, T. H. Dolla, S. S. Gwebu, T. A. Mashola, L. T. Dlamini, E. Carleschi, P. Ndungu and N. W. Maxakato, Mn–Ni–Co–O Spinel Oxides towards Oxygen Reduction Reaction in Alkaline Medium: Mn0.5Ni0.5Co2O4/C Synergism and Cooperation, Catalysts, 2021, 11, 1059 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: A supplementary file with the associated data from SEM, TEM, XPS, EDX, and ICP-MS used in this work is provided as ESI. See DOI: https://doi.org/10.1039/d3ya00356f |
| This journal is © The Royal Society of Chemistry 2023 |