
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomic-scale insight into the lattice volume plunge of LixCoO2 upon deep delithiation†
Yufang
He
a,
Li
Wang
 *a,
Bo
Zhang
a,
Hiep
Pham
b,
Hong
Xu
a,
Jonghyun
Park
*b and
Xiangming
He
*a
*a,
Bo
Zhang
a,
Hiep
Pham
b,
Hong
Xu
a,
Jonghyun
Park
*b and
Xiangming
He
*a
aInstitute of Nuclear and New Energy Technology, Tsinghua University, Beijing 100084, China. E-mail: wang-l@tsinghua.edu.cn; hexm@tsinghua.edu.cn
bDepartment of Mechanical Engineering and Aerospace Engineering, Missouri University of Science and Technology, Rolla, 65401, USA. E-mail: parkjonghy@mst.edu
First published on 2nd November 2022
Abstract
The practical capacity utilization of LiCoO2 is limited to 50–70% due to the dramatic volume shrinkage induced cracks and subsequent interface parasitic reactions at high voltage. However, the fundamental understanding of the dramatic lattice volume shrinkage remains unclear. In this work, we discover that the delithiation paths have an impact on the lattice volume turning point of LixCoO2, where the corresponding capacity utilization can be as low as 62.5% or as high as 75% consequently. In addition, the O ↔ O interlayer distance across the Li layer (O(d3)) mainly contributes to the volume increase before 62.5–75% delithiation, and the O ↔ O interlayer distance across the Co layer (O(d2)) is the dominant factor for the dramatic volume decrease after the volume turning point. The electron localization function (ELF) around O keeps increasing during delithiation and it increases significantly after more than 62.5% delithiation, indicating that the lattice oxygen participates in charge compensation during the whole delithiation process and it becomes the main contributor to the charge compensation at a high delithiated state compared with Co. This work unravels the fundamental reason for the dramatic volume shrinkage of LixCoO2 at high voltage. It claims that the antisite defects (CoLi) of LiCoO2 should be designed carefully owing to the weaker Co–O bond strength and further oxygen release, which accelerate the degradation of LiCoO2, although it can increase the voltage of LiCoO2. More importantly, LiCoO2 material design with elemental doping, oxygen defects (VO), Li defects (VLi), and Co defects (VCo) will contribute to the capacity utilization of LiCoO2.
Introduction
LiCoO2 was the first commercialized cathode material for lithium-ion batteries (LIBs) by the Sony Corporation in 1991 and it remains one of the most heavily used cathodes for consumer electronics owing to its high tap density, high initial coulombic efficiency, and high volumetric energy density.1–5 For a long time, only about half of the Li was utilized in the LiCoO2 cathode material. Researchers try to achieve more capacity while maintaining the stability of the LiCoO2 cathode material to fully utilize the energy density, such as doping and coating, which enables reversible Li-utilization up to 0.7 at high voltage,4 and little or no oxygen release from the bulk.6 However, deeper delithiation would lead to oxygen release or particle breakage, leading to capacity fading because of prominent parasitic reactions at the LixCoO2/electrolyte interface and material loss.4,7–9 These indicate that keeping the particle integrity during deep lithiation is important to achieve LiCoO2 with high capacity.Previous studies showed that the lattice volume of LiCoO2 experiences a dramatic decrease at high voltage and the volume turning points differ depending on different LiCoO2 samples.4,9–13 The existence of the sample difference results in the variation of capacity utilization of LiCoO2. The reported dramatic volume turning point of LixCoO2 ranges between 50% and 70% delithiation rates. Dahn et al. reported that the volume of LiCoO2 decreases around 4.2 V and the corresponding Li utilization is around 50%.12 Besides, the volume change of LiCoO2 is thought to be dominated by lattice c, and Tarascon et al. proposed that the lattice c parameter stably increased owing to the increasing electrostatic repulsion of O ↔ O interactions of adjacent CoO6 layers during delithiation. Then, the c parameter underwent a dramatic decrease when the delithiation ratio was 54%, stemming from a transformation from monoclinic distortion to a hexagonal structure.10,14 Van der Ven et al. also demonstrated that the lattice c parameter increases and then decreases dramatically when delithiation was over 60% Li.11 Liu et al. claimed that the reversible Li utilization of La- and Al-co-doped LiCoO2 was increased to 70% with the cut-off voltage increasing to 4.5 V.4 Barker et al. showed that the Li utilization in LiCoO2 reached up to 64% with 100% coulombic efficiency.13 It is well accepted that the dramatic volume change of LixCoO2 at high voltage is closely related to the O ↔ O interaction environments in LiCoO2, including O ↔ O interaction environments in the CoO6 layer and O ↔ O interaction environment across the Li layer. Recently, the oxygen redox reaction is regarded as the predominant factor for the electrochemical performance of transition metal oxide cathodes at high voltage,15–20 where O contributes much more to charge compensation than Co.6 The oxygen redox reaction at high voltage has an impact on the volume change of LixCoO2. However, there is no clear understanding of the variation of the dramatic volume turning point of LixCoO2 at high voltage, as well as the explicit perception regarding the change of O ↔ O interaction environment.
In this work, the fundamental reasons for the lattice volume shrinkage of LiCoO2 during delithiation are investigated via theoretical calculations, especially at high voltage. We found that there exists a relationship between delithiation paths and the lattice volume plunging point during charging, which determines the reversible capabilities of the LiCoO2 cathode. Four different random delithiation paths are considered. The O ↔ O distances with three different O ↔ O interaction environments in LixCoO2 along delithiation path 1 are further investigated, which are O ↔ O intralayer distance (O(d1)), O ↔ O interlayer distance across the Co layer (O(d2)), and O ↔ O interlayer distance across the Li layer (O(d3)). To further identify the electronic properties, the electron localization function (ELF) of LixCoO2 is calculated. There is peroxo-like O–O formation during delithiation. The analysis of the total density of states (TDOS) of LixCoO2 and the projected density of states (PDOS) of O and Co uncovers that the oxygen oxidation and the dramatic volume shrinkage point of LixCoO2 are correlated. In addition, the phase transition of LixCoO2 is examined for the reliability of these simulation results. This work provides theoretical guidance for reasonable materials design and synthesis to achieve a higher capacity utilization of LiCoO2 cathode.
Methods
The lattice volume plunge of LixCoO2 during delithiation is crucial for the energy density of LIBs. To study the fundamental reason for the lattice volume shrinkage of LixCoO2 during delithiation, density functional theory (DFT) calculations are performed by using the Vienna Ab initio Simulation Package (VASP) code.21 The Perdew–Burke–Ernzerhof (PBE) functional is employed to approximate the exchange–correlation function. The projector augmented wave (PAW) method22–24 is used for electron and core interactions. A Hubbard U parameter of 4.91 eV is applied to the delocalization correction of the Co d-orbital.25 The cutoff energy is set at 520 eV. The convergence criterion in the electronic self-consistent iteration is 10−4 eV and the convergence criterion for ionic relaxation is set at 0.03 eV Å−1. The layered LiCoO2 (space group: R3m) structure is used for calculations, which consists of 48 Li, 48 Co, and 96 O. The cell volume is allowed to change26 and the delithiated LixCoO2 structure is fully relaxed during structure optimization. The volume calculation of LixCoO2 is performed by considering four different delithiation paths. The electron localization function (ELF) of LixCoO2 at different delithiation states is examined27 to further study the valence states and bonding states of Co and O. The ELF and the phase transformation of LixCoO2 is visualized with the aid of VESTA software.28 The density of states (DOS)29 of LixCoO2 is examined via executing the ‘split_dos’ script from the VASP output. Besides, the Bader charge analysis30–33 is performed to calculate the charge states of Co and O in LixCoO2 during the delithiation process. In addition, the volume change of the LixCoO2 cathode during delithiation is characterized using in situ X-ray diffraction (XRD).34Results and discussion
To reveal the fundamental reason for the volume change of LixCoO2 during Li extraction, the lattice volumes of LixCoO2 at different delithiation states are first calculated. The layered LiCoO2 structure with the R3m space group used for density functional theory (DFT) calculations is shown in Fig. 1(a) and Fig. S1a (ESI†). Fig. 1(a) also demonstrates that the lattice volume of LiCoO2 is closely related to the intralayer O ↔ O distance O(d1), the O ↔ O interlayer distance across the Co layer O(d2), and the O ↔ O interlayer distance across the Li layer O(d3). As shown in Fig. 1(b), the volume of LixCoO2 with four different delithiation paths stably increases and then decreases dramatically when 62.5–75% Li is removed from the host, which has the same trend as the volume change of LixCoO2 measured with the in situ XRD experiment (Fig. S1b, ESI†). The simulation results are different from the experimental results because the simulation results are based on an ideal model, assuming that the LiCoO2 particles are uniform and pure single crystals. However, in reality, it is challenging to reach the theoretical value. In addition, the practical LiCoO2 particles in experiments possess different kinds of defects. There are four types of defects in LiCoO2, including antisite cation (CoLi), Li-ion vacancy (VLi), Co-ion vacancy (VCo), and O-ion vacancy (VO).35 The antisite defects and VO defects lead to an increase in intercalation potential. A higher intercalation potential of LiCoO2versus Li/Li+ indicates a higher discharge voltage.36 Meanwhile, the LiCoO2 with antisite defects (CoLi) contributes to weaker Co–O bond strength and further oxygen release,37,38 which might be responsible for the variation of the dramatic volume change point of LiCoO2. Whilst the VLi and VCo defects are responsible for the electron conductivity and charge compensation during delithiation. It is also reported that oxygen defects can ensure a high degree of oxygen redox.39 Furthermore, it is claimed that the phase transitions and volume variation are very sensitive to the impurity levels of LiCoO2 particles and it can be used as a quality check for the synthetic LiCoO2 material.13,40 Fig. S1c (ESI†) shows that the lattice c parameter of LixCoO2 increases with delithiation and then decreases after more than 62.5–75% delithiation, which shows the same trend as the volume change of LixCoO2. Meanwhile, the lattice a parameter of LixCoO2 decreases and then increases after more than 62.5–75% removal of Li (Fig. S1d, ESI†), which is consistent with reported experimental results.41 It also shows that the volume change of LixCoO2 is influenced by the lattice c parameter of LixCoO2, which is consistent with reported work.11 More importantly, it is reported that the ordering of lithium and vacancy has a profound effect on the electrochemical performance of Li-ion batteries.42 The lattice volume of LixCoO2 shrinks at different delithiation capacities by considering four different delithiation paths. Fig. 1(b) clearly shows that different delithiation paths in LixCoO2 produce different volume turning points, where it is between 62.5% and 75%, explaining why there exists a dramatic volume turning point difference between different experimental works. The difference in delithiation paths during the charging process may come from material defects and synthesis process of LiCoO2. To improve the delithiation performance of LiCoO2, the antisite defects of LiCoO2 should be designed carefully because although it contributes to the increase of the voltage of LiCoO2, it will cause a weaker Co–O bond strength and further oxygen release as well. More crucially, LiCoO2 material design with VO, VLi and VCo will contribute to the improvement of voltage, electron conductivity, and charge compensation during the delithiation process.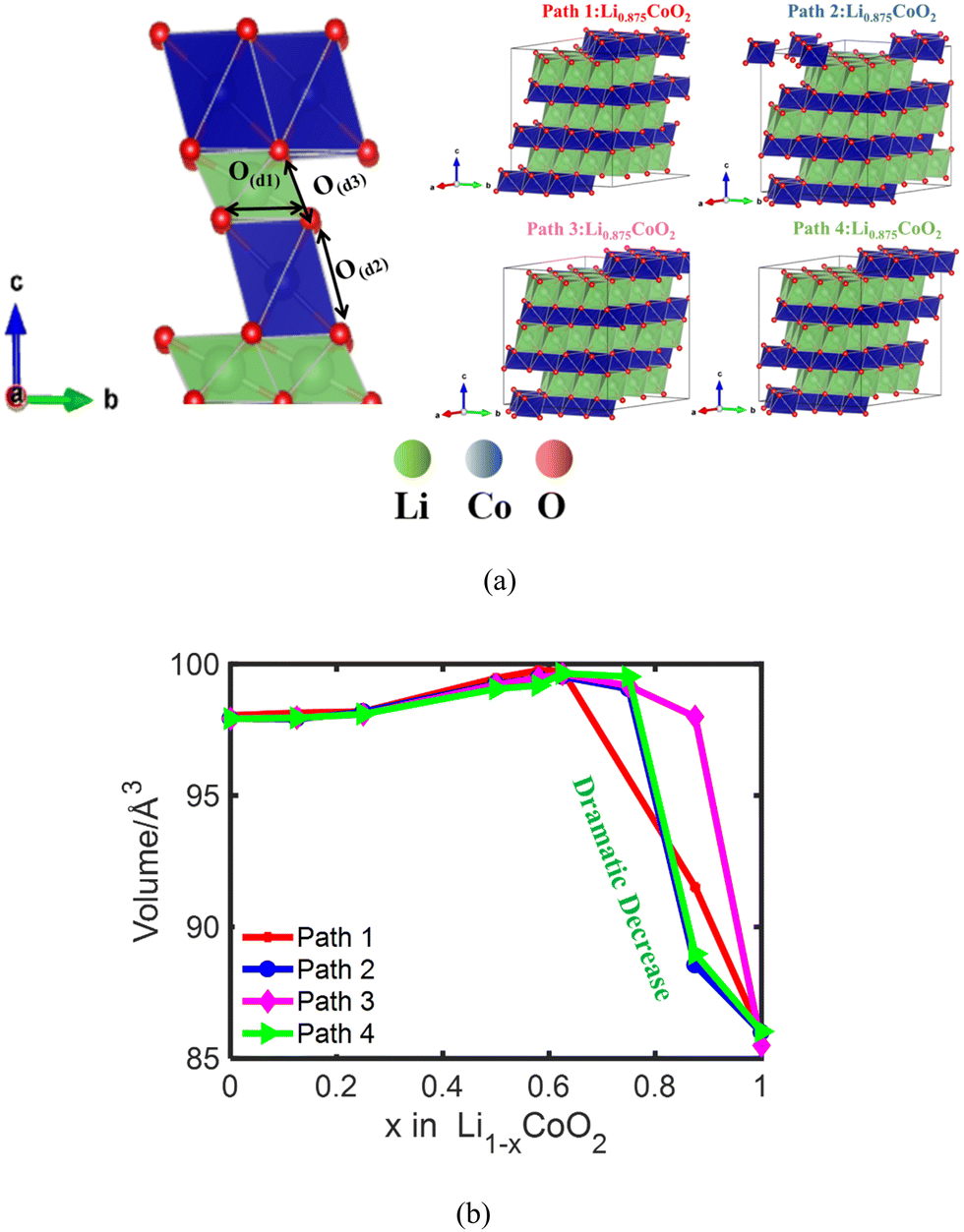 | ||
| Fig. 1 (a) The three different O ↔ O interactions including the O ↔ O distances O(d1), O(d2) and O(d3), and schematic of four different random delithiation paths in LixCoO2 considered during volume calculations using DFT; (b) the volume change of LixCoO2 along four different random delithiation paths. It demonstrates that the volume of LixCoO2 stably increases and then decreases dramatically when 62.5–75% Li is removed from the host, which has the same tendency as the volume change of LixCoO2 measured with the XRD experiment. | ||
To further identify the fundamental reason for the volume changes of LixCoO2, one of the four delithiation paths is chosen to analyze the change of O ↔ O interactions and the Co ↔ O interaction during delithiation. The initial delithiation structure of LixCoO2 along four different delithiation paths is shown in Fig. 2(a) and the Li vacancy formation energy of the four different delithiation paths is demonstrated in Fig. 2(b). The Li formation energy can measure how easily for Li to be extracted from the host, where a lower Li formation energy indicates the easier formation of Li vacancy. It shows that Li prefers to be extracted from the host during the whole delithiation owing to the negative value of Li vacancy formation energy. Besides, the Li vacancy formation energy of the four delithiation paths is almost the same when delithiation is less than 75%, except that there is a small difference when 12.5% Li is extracted. The Li vacancy formation energy of path 3 becomes the lowest and that of path 4 becomes the highest when delithiation is more than 75%. Based on the Li vacancy formation energy of the four delithiation paths, path 1 is selected for further analysis because the Li vacancy formation energy values of path 1 are located in the middle of the four paths during delithiation process. Fig. 2(c) shows that the Co ↔ O distance keeps decreasing from 1.93 Å to 1.88 Å, coinciding with the increasing charge state of Co attributed to the charge compensation delithiation. Besides, the O ↔ O intralayer distance O(d1) decreases slightly from 2.83 Å to 2.79 Å before delithiation reaching 62.5% and then increases rapidly to 2.86 Å between 62.5% and 100% delithiation (Fig. 2(c)). Meanwhile, the O ↔ O distance across the Co layer O(d2) is almost unchanged until 37.5% delithiation and the O ↔ O distance across the Co layer O(d2) decreases dramatically from 2.62 Å to 2.46 Å after the delithiation is more than 62.5%. In contrast, the O ↔ O interlayer distance across the Li layer O(d3) increases slightly and then increases dramatically after 62.5% Li is removed (Fig. 2(c)). The lowest O ↔ O distance is around 2.46 Å, which is almost the same as the reported results.6 The O ↔ O interlayer distance across the Co layer and Li layer dominates the volume change of LiCoO2 during delithiation. The O ↔ O interlayer distance across the Li layer O(d3) dominates the volume change of LixCoO2 when delithiation is less than 62.5% due to the electrostatic repulsion interaction between negatively charged O (Fig. 2(d)). More importantly, the O ↔ O interlayer distance across the Co layer O(d2) is mainly responsible for the dramatic volume turning point of LixCoO2 when delithiation is more than 62.5% owing to the oxygen oxidation at high voltage.
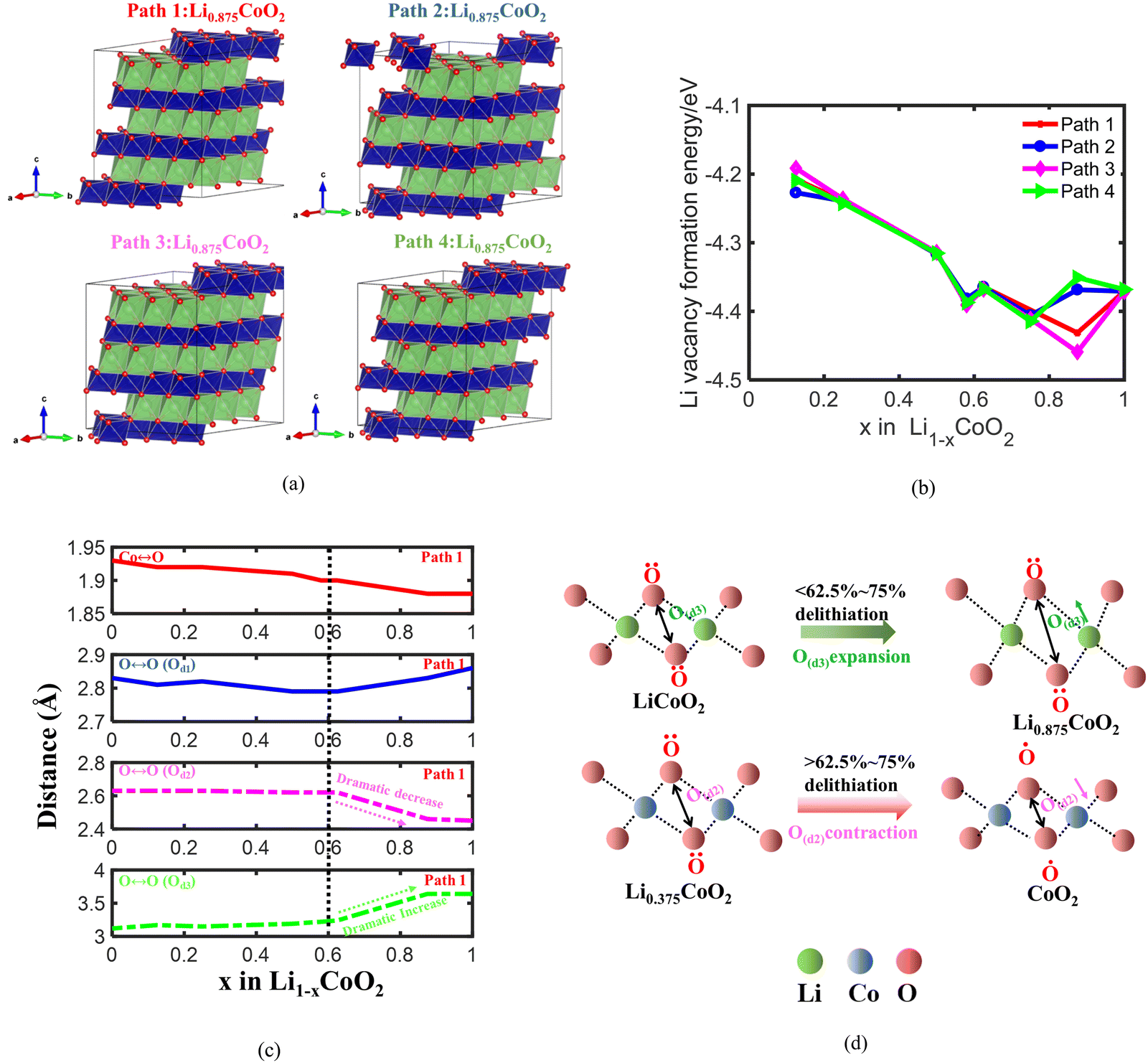 | ||
| Fig. 2 The LixCoO2 structure during initial delithiation with four different delithiation paths (a); the Li vacancy formation energy of LixCoO2 along four different delithiation paths (b); the Co ↔ O distance, the O ↔ O intralayer distance O(d1), the O ↔ O interlayer distance across the Co layer O(d2), and the O ↔ O interlayer distance across the Li layer O(d3) along delithiation path 1 (c); the schematic illustrating the mechanism of O ↔ O interaction induced volume change at different delithiation states. The O ↔ O interlayer distance across the Li layer O(d3) mainly contributes to the increasing volume of LixCoO2 when delithiation is less than 62.5% due to the electrostatic repulsion interaction between negatively charged O (d). | ||
To further investigate the electronic structure of the three O ↔ O interaction environments that induced the dramatic volume change of LixCoO2, the electron localization function (ELF) of LixCoO2 is calculated.43 The ELF results show the bonding type and lone pair electrons of LixCoO2, where the red color means fully localized and the blue color represents fully delocalized electrons. The fully localized electrons indicate the existence of a lone-pair electron, while the delocalized electrons imply the bonding state. Fig. 3(a) and Fig. S2a (ESI†) illustrate that the ELF around O keeps increasing during delithiation, implying that there exist more lone-pair electrons and the valence state of oxygen gradually changes from O2− to (O2)n− (1 ≤ n ≤ 3), which indicates that the oxygen contributes to the charge compensation and there is O–O bond formation during the delithiation process. But Yang et al. claimed that there is no O–O bond formation via COOP analysis.6 We believe the ELF calculation is more comprehensive and reasonable because the electron localization function is calculated by considering the entire LixCoO2, whereas the COOP analysis is conducted only checking a specific O–O bonding state in LixCoO2, which is not sufficient to represent the whole LixCoO2 structure during delithiation. The 2D electron localization function of LixCoO2 shown in Fig. 3(b) and Fig. S2b (ESI†) is sliced with half of lattice b and the Miller index is (010), and it clearly shows that the ELF around O increases significantly after more than 62.5% delithiation. Therefore, the lattice oxygen in LixCoO2 participates in charge compensation during the whole delithiation process and it becomes the main contributor to the charge compensation at a highly delithiated state compared with Co, where the oxygen redox reaction occurs.
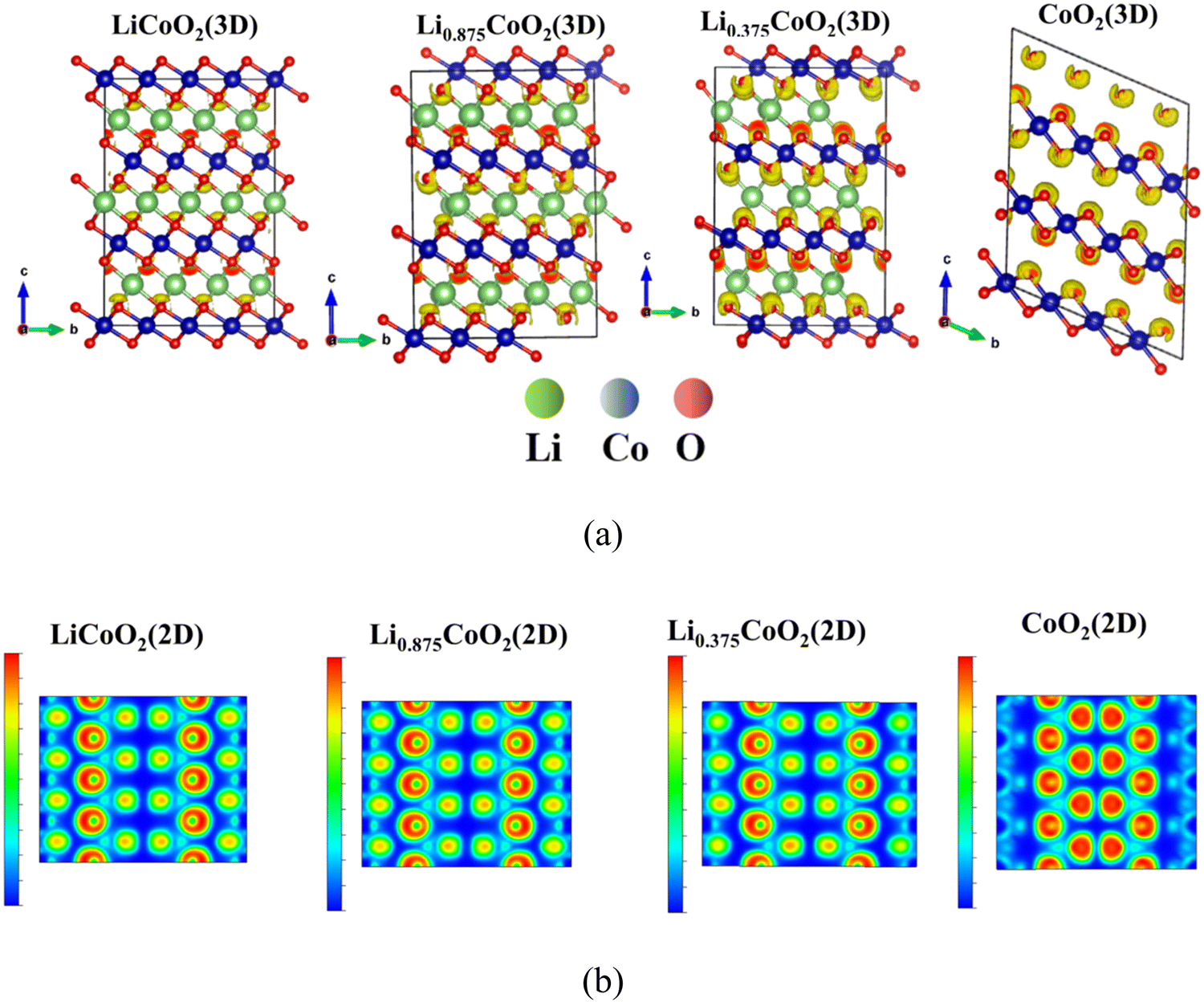 | ||
| Fig. 3 The 3D (a) and 2D (b) electron localization function of LixCoO2 during delithiation (the red color means electron localization, indicating the existence of lone pair electrons, while the blue color means electron delocalization showing that there is a bonding state). The electrons localized around O increase during delithiation and the ELF increases dramatically after delithiation is more than 62.5%, implying that O oxidation occurs and the lattice oxygen mainly contributes to the charge compensation at a high delithiation state. | ||
To further reveal the electronic properties of LixCoO2 during delithiation, the total density of states (TDOS) and the projected density of states (PDOS) of O and Co in LixCoO2 are calculated. Fig. 4(a) and Fig. S3a (ESI†) display that the highest energy peak of TDOS shifts to a lower energy level during delithiation. The lower energy state of TDOS suggests that the electrons in an atomic orbital are more difficult to escape. Therefore, it becomes harder for Li and electrons to escape from the LixCoO2 host during delithiation, which is reasonable due to the limited reversibility of LiCoO2 cathode materials. Then, the electronic properties of O in LixCoO2 are analyzed and the lattice oxygen atoms with and without surrounded Li extracted are considered. The Onumber![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30 and Onumber76 are in the oxygen sites with and without surrounded Li extracted respectively. The atomic environments of Onumber30 and Onumber76 are shown in Fig. S3b (ESI†). As illustrated in Fig. 4(b) and Fig. S3c (ESI†), the energy level of the PDOS peak of Onumber30 initially increases and then decreases when delithiation is around 62.5%, which indicates that oxygen oxidation occurs more easily when delithiation increases to 62.5%. However, after the dramatic volume change turning point, the energy level of the PDOS peak decreases, implying that it becomes more difficult for oxygen oxidation to occur when the dramatic volume change of LixCoO2 starts. It agrees well with the volume change and the O ↔ O distance calculation. It further provides the fundamental reason for the dramatic volume change occurring after more than 62.5% delithiation, corresponding to the reported experimental results that the reversible capacity of LixCoO2 is around 50–70%.12,13 Therefore, the oxygen oxidation and the dramatic volume change point of LixCoO2 are correlated. The energy level of the PDOS peak of Onumber76 also increases and then decreases, demonstrating that the oxygen oxidation capability is influenced by the whole delithiation state instead of the local atomic environment (Fig. 4(c) and Fig. S3d, ESI†). In addition, the PDOS of Co is checked and the atomic environments of Conumber20 and Conumber38 are demonstrated in Fig. S3b (ESI†). As shown in Fig. 4(d) and Fig. S3e (ESI†), the energy level of the PDOS peak of Conumber20 increases with delithiation and then decreases because the dramatic volume change of LixCoO2 starts. The energy level of the PDOS of Conumber38 shows the same trend as Conumber20 (Fig. 4(e) and Fig. S3f, ESI†), which suggests that the Co oxidation is also mainly related to the whole delithiation state of LixCoO2 instead of the local atomic environment.
30 and Onumber76 are in the oxygen sites with and without surrounded Li extracted respectively. The atomic environments of Onumber30 and Onumber76 are shown in Fig. S3b (ESI†). As illustrated in Fig. 4(b) and Fig. S3c (ESI†), the energy level of the PDOS peak of Onumber30 initially increases and then decreases when delithiation is around 62.5%, which indicates that oxygen oxidation occurs more easily when delithiation increases to 62.5%. However, after the dramatic volume change turning point, the energy level of the PDOS peak decreases, implying that it becomes more difficult for oxygen oxidation to occur when the dramatic volume change of LixCoO2 starts. It agrees well with the volume change and the O ↔ O distance calculation. It further provides the fundamental reason for the dramatic volume change occurring after more than 62.5% delithiation, corresponding to the reported experimental results that the reversible capacity of LixCoO2 is around 50–70%.12,13 Therefore, the oxygen oxidation and the dramatic volume change point of LixCoO2 are correlated. The energy level of the PDOS peak of Onumber76 also increases and then decreases, demonstrating that the oxygen oxidation capability is influenced by the whole delithiation state instead of the local atomic environment (Fig. 4(c) and Fig. S3d, ESI†). In addition, the PDOS of Co is checked and the atomic environments of Conumber20 and Conumber38 are demonstrated in Fig. S3b (ESI†). As shown in Fig. 4(d) and Fig. S3e (ESI†), the energy level of the PDOS peak of Conumber20 increases with delithiation and then decreases because the dramatic volume change of LixCoO2 starts. The energy level of the PDOS of Conumber38 shows the same trend as Conumber20 (Fig. 4(e) and Fig. S3f, ESI†), which suggests that the Co oxidation is also mainly related to the whole delithiation state of LixCoO2 instead of the local atomic environment.
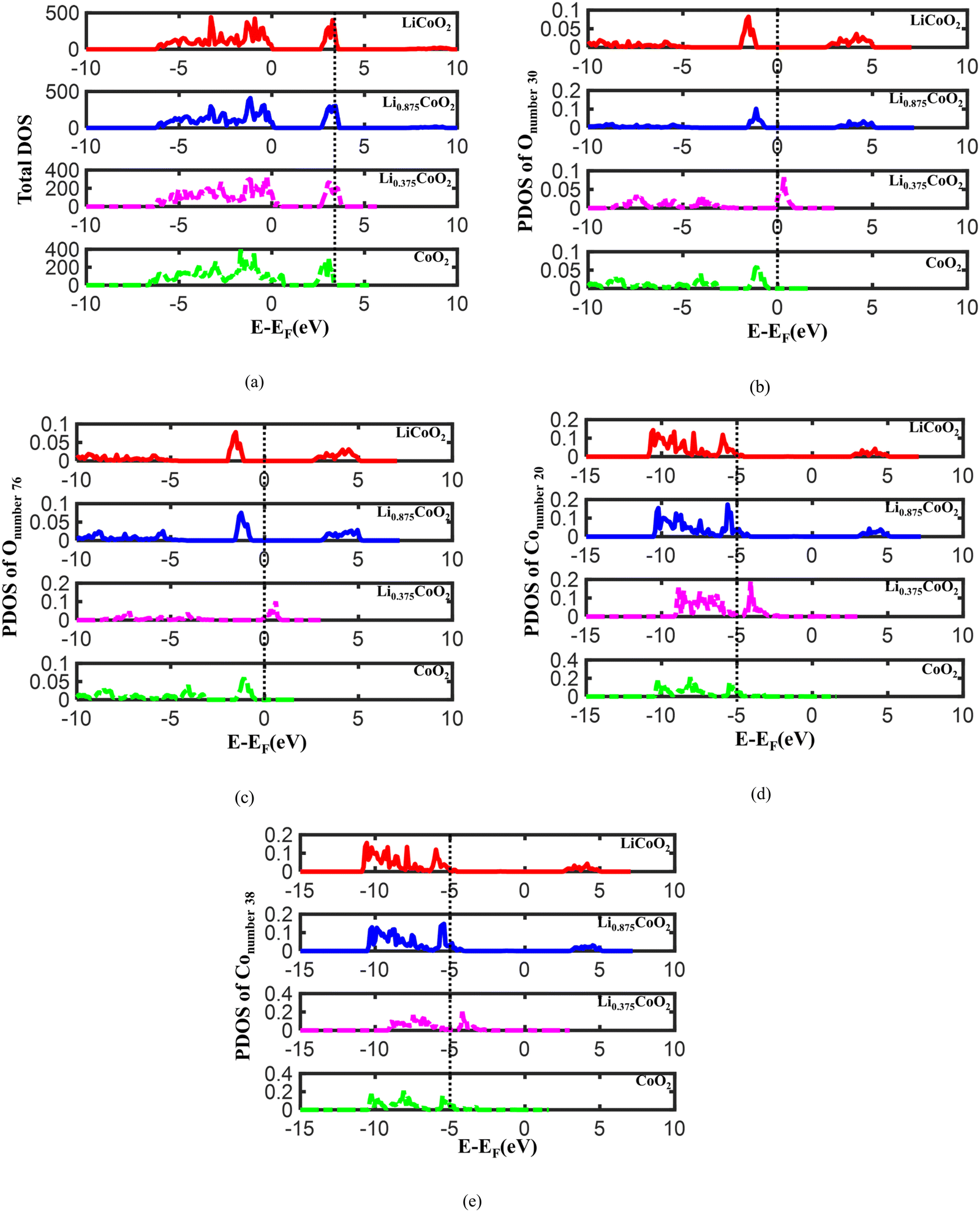 | ||
| Fig. 4 The TDOS of LixCoO2 during delithiation (a), the PDOS of Onumber30 with surrounded Li extracted (b), the PDOS of Onumber76 without surrounded Li extracted (c), and the PDOS of Conumber20 (d) and Conumber38 (e) during delithiation. The energy level of the PDOS peak of O keeps increasing until 62.5% Li is extracted from LixCoO2, implying that it becomes easier for O oxidation to occur with increasing delithiation, while the energy level of the PDOS peak shifts to a lower energy level when delithiation is more than 62.5%, indicating that there is a correlation between O oxidation and the dramatic volume change of LixCoO2. | ||
To uncover the effect of oxygen charge compensation for delithiation on the dramatic volume change of LixCoO2, the average charge states of all O and Co, and the charge states of Onumber30, Onumber76, Conumber20, and Conumber38 are calculated via Bader analysis.30–33 The average charge state of O increases from −1.05 to −0.67 (Fig. 5(a)) and the average charge state of Co keeps increasing from +1.25 to +1.33 (Fig. 5(b)), which is close to previously calculated results.6,11 This reveals that both Co and O participate in the charge compensation during the delithiation process but for different stages. The charge compensation for delithiation is predominated by Co via changing Co3+ to Co4+ when less than 62.5% Li is removed and the charge compensation for delithiation is dominated by O at a high delithiation state through changing from O2− to (O2)n−. Besides, the charge states of individual Onumber30, Onumber76, Conumber38, and Onumber76 in LixCoO2 are calculated (Fig. 5(a) and (b)), which show that there exists a slight difference between the average charge state and individual Co and O in LixCoO2. The charge states of Co and O are almost linear with the delithiation state, while the charge states of individual Co and O change dramatically around 62.5% delithiation. This reveals that the charge compensation of Co and O in LixCoO2 for delithiation is not uniform. A reasonable elemental doping, such as La and Al, can mitigate this non-uniformity and stabilize the crystalline structure of LxCoO2 at a high delithiation state.4,44–47
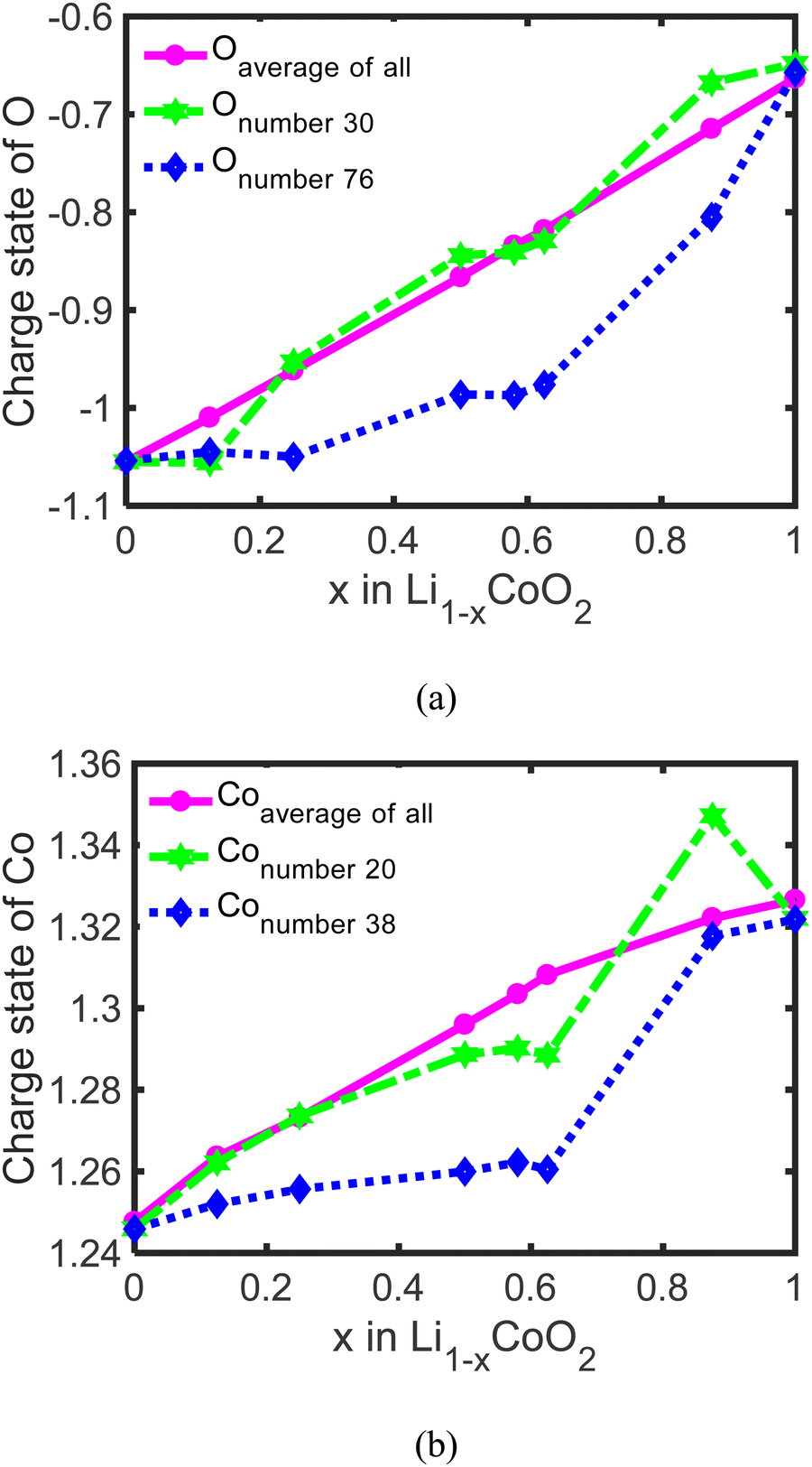 | ||
| Fig. 5 The average charge states of all O, individual Onumber30 with surrounded Li extracted, and individual Onumber76 without surrounded Li extracted in LixCoO2 during delithiation (a); the average charge states of all Co, individual Conumber20, and individual Conumber38 in LixCoO2 during delithiation (b). | ||
In addition, the change of O ↔ O interaction environments in the presence of CoO6 slab migration during delithiation, which is correlated with the phase transformation of the LixCoO2 structure. The phase transformation of LixCoO2 directly influences the volume change of LixCoO2. Fig. 6 demonstrates that the Co layer has a slight glide when 12.5% and 25% Li are extracted from LiCoO2. The Co layer undergoes a huge displacement when the delithiation is more than 62.5%. The CoO6 slab continues to glide, and the gradual angle, defined as the Co stacking in the LiCoO2 along the 001 direction, decreases with further delithiation, which is consistent with recently reported in situ XRD and ex situ STEM.48 This also indicates that the LixCoO2 evolves from the O3 phase to the O1 phase, which also agrees well with reported observation.48 More notably, this demonstrates that there is an accumulation of vacancy phenomenon, which is one of the main factors causing the phase transformation of LiCoO2. The accumulation of Li vacancies becomes more severe with delithiation, accompanied by the rich-vacancy phase formation owing to the lower energy state. Therefore, this theoretically confirms that the CoO6 slab glides continuously and is an intelligent dynamic response to the delithiation process, disclosing the existence of the accumulation of vacancy phenomenon in LixCoO2 cathode materials during delithiation.
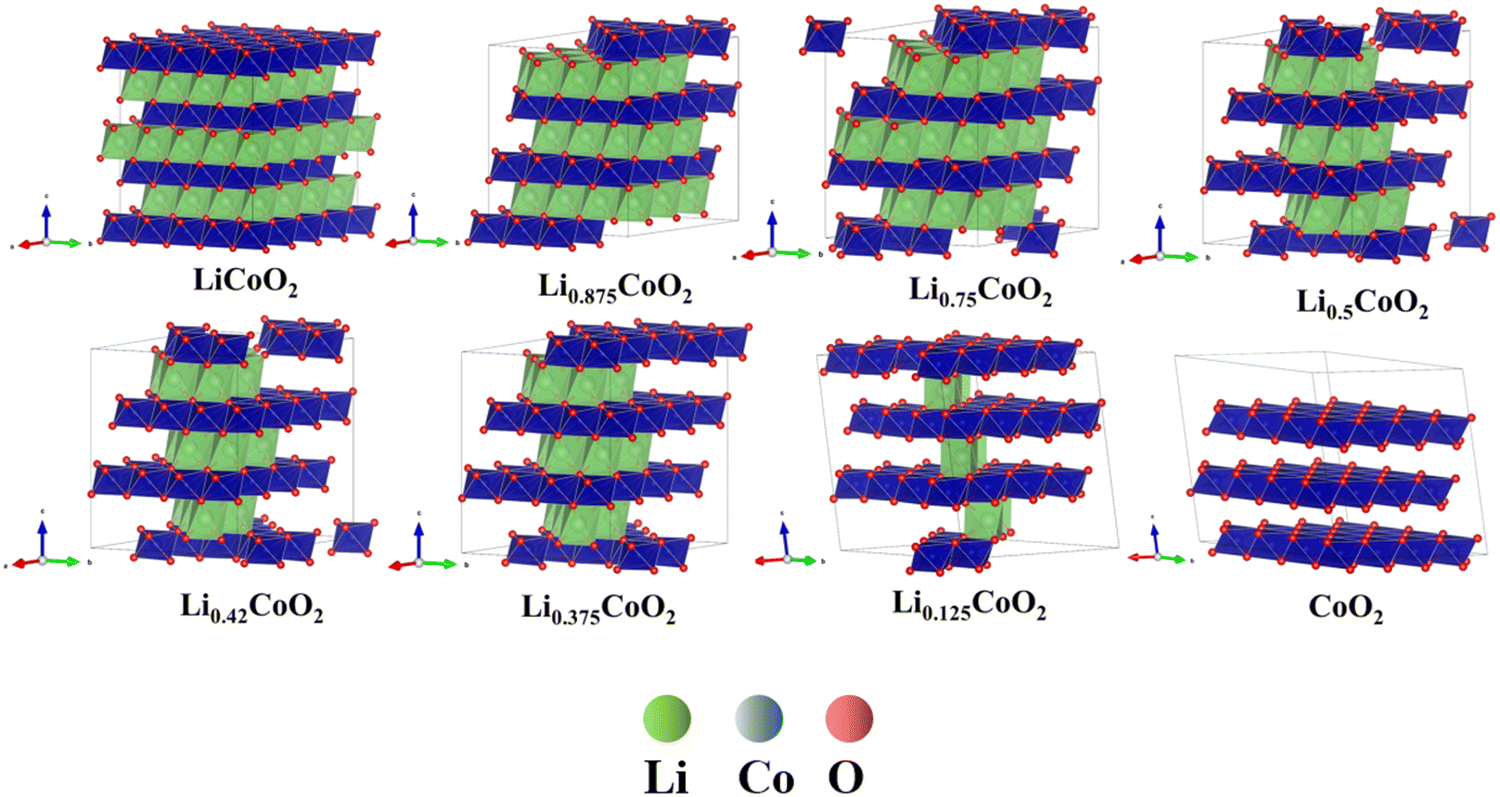 | ||
| Fig. 6 The phase transformation of LixCoO2 during delithiation: the LixCoO2 evolves from the O3 phase to the O1 phase and the gradual angle keeps decreasing during delithiation.48 | ||
Conclusion
In this work, it reveals that the different delithiation paths have an impact on the initiation of volume shrinkage of LixCoO2, which ranges between 62.5% and 75% delithiation. This means that the available capacity of the LiCoO2 cathode is tunable by manipulating Li extraction sites and LiCoO2 with poor reversible capacity can be improved via materials synthesis. In addition, the O ↔ O interlayer distance across the Li Layer (O(d3)) mainly contributes to the volume increase when less than 62.5–75% removal of Li and the O ↔ O interlayer distance across the Co layer (O(d2)) dominates the dramatic volume shrinkage of LixCoO2 when delithiation is more than 62.5%. The ELF calculation shows that there is peroxo-like O ↔ O formation during delithiation, which is more reasonable than the reported COOP analysis for only specific O–O bonding state. Furthermore, the oxygen oxidation and the dramatic volume change point of LixCoO2 are correlated. The delithiation state of LixCoO2 will influence oxygen oxidation, which will induce dramatic volume changes in LixCoO2. The volume shrinkage of LixCoO2 has an impact on the oxygen oxidation in return. Moreover, the volume shrinkage of LixCoO2 stems from the oxygen charge compensation, and the charge compensation of Co and O in LixCoO2 for delithiation is not uniform, which can be remitted by implementing a doping strategy. This provides new insight into the fundamental reason for the dramatic volume change of LixCoO2 from the electronic and atomic levels. We propose that the antisite defects of LiCoO2 should be designed carefully, which lead to weaker Co–O bond strength and further oxygen release, although they enable a higher intercalation potential of LiCoO2. More importantly, LiCoO2 material design with elemental doping, oxygen defects, Li defects, and Co defects contributes to the improvement of the reversible capacity of LiCoO2.Data availability
The LixCoO2 structures at different delithiation states are provided in the ESI.† All structures used for simulations and other data supporting the findings of this work are available upon request.Author contributions
Yufang He conducted the simulations and wrote the manuscript; Xiangming He and Li Wang provided guidance and suggestions for this work; Jonghyun Park and Hiep Pham provided suggestions and modified the manuscript; Bo Zhang and Hong Xu provided suggestions for the calculations and manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was sponsored by the National Natural Science Foundation of China (No. U21A20170), the National Science and Technology Major Project (No. 2019YFE0100200) and China Postdoctoral Science Foundation (No. 2022M711791 and 2021M701873). We also appreciate the Joint Work Plan for Research Projects under the Clean Vehicles Consortium at U.S. and China – Clean Energy Research Center (CERC-CVC) and the Tsinghua University – Zhangjiagang Joint Institute for Hydrogen Energy and Lithium-Ion Battery Technology.References
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenough, LixCoO2 (0 < x < −1): A new cathode material for batteries of high energy density, Mater. Res. Bull., 1980, 15(6), 783–789 CrossRef CAS.
- Y. Chen, Y. Kang, Y. Zhao, L. Wang, J. Liu, Y. Li, Z. Liang, X. He, X. Li, N. Tavajohi and B. Li, A review of lithium-ion battery safety concerns: The issues, strategies, and testing standards, J. Energy Chem., 2021, 59, 83–99 CrossRef CAS.
- L. Wang, J. Ma, C. Wang, X. Yu, R. Liu, F. Jiang, X. Sun, A. Du, X. Zhou and G. Cui, A novel bifunctional self-stabilized strategy enabling 4.6 V LiCoO2 with excellent long-term cyclability and high-rate capability, Adv. Sci., 2019, 6(12), 1900355 CrossRef.
- Q. Liu, X. Su, D. Lei, Y. Qin, J. Wen, F. Guo, Y. A. Wu, Y. Rong, R. Kou, X. Xiao, F. Aguesse, J. Bareño, Y. Ren, W. Lu and Y. Li, Approaching the capacity limit of lithium cobalt oxide in lithium ion batteries via lanthanum and aluminium doping, Nat. Energy, 2018, 3(11), 936–943 CrossRef CAS.
- P. Zhao, J. Yang, Y. Shang, L. Wang, M. Fang, J. Wang and X. He, Surface modification of polyolefin separators for lithium ion batteries to reduce thermal shrinkage without thickness increase, J. Energy Chem., 2015, 21(2), 138–144 CrossRef.
- E. Hu, Q. Li, X. Wang, F. Meng, J. Liu, J. Zhang, K. Page, W. Xu, L. Gu, R. Xiao, H. Li, X. Huang, L. Chen, W. Yang, X. Yu and X.-Q. Yang, Oxygen-redox reactions in LiCoO2 cathode without O–O bonding during charge-discharge, Joule, 2021, 5(3), 720–736 CrossRef CAS.
- J. L. Tebbe, A. M. Holder and C. B. Musgrave, Mechanisms of LiCoO2 Cathode Degradation by Reaction with HF and Protection by Thin Oxide Coatings, ACS Appl. Mater. Interfaces, 2015, 7(43), 24265–24278 CrossRef CAS.
- A. Yano, M. Shikano, A. Ueda, H. Sakaebe and Z. Ogumi, LiCoO2 degradation behavior in the high-voltage phase transition region and improved reversibility with surface coating, J. Electrochem. Soc., 2017, 164, A6116–A6122 CrossRef CAS.
- J. Qian, L. Liu, J. Yang, S. Li, X. Wang, H. L. Zhuang and Y. Lu, Electrochemical surface passivation of LiCoO2 particles at ultrahigh voltage and its applications in lithium-based batteries, Nat. Commun., 2018, 9(1), 4918 CrossRef.
- G. G. Amatucci, J. M. Tarascon and L. C. Klein, CoO2, the end member of the LixCoO2 solid solution, J. Electrochem. Soc., 1996, 143(3), 1114–1123 CrossRef CAS.
- A. Van der Ven, M. K. Aydinol, G. Ceder, G. Kresse and J. Hafner, First-principles investigation of phase stability in LixCoO2, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58(6), 2975–2987 CrossRef CAS.
- J. N. Reimers and J. R. Dahn, Electrochemical and in situ X-ray diffraction studies of lithium intercalation in LixCoO2, J. Electrochem. Soc., 1992, 139(8), 2091–2097 CrossRef CAS.
- J. Barker, R. Pynenburg, R. Koksbang and M. Y. Saidi, An electrochemical investigation into the lithium insertion properties of LixCoO2, Electrochim. Acta, 1996, 41(15), 2481–2488 CrossRef CAS.
- T. Ohzuku and A. Ueda, Why transition metal (di) oxides are the most attractive materials for batteries, Solid State Ionics, 1994, 69(3), 201–211 CrossRef CAS.
- E. McCalla, A. M. Abakumov, M. Saubanère, D. Foix, E. J. Berg, G. Rousse, M.-L. Doublet, D. Gonbeau, P. Novák, G. Van Tendeloo, R. Dominko and J.-M. Tarascon, Visualization of O–O peroxo-like dimers in high-capacity layered oxides for Li-ion batteries, Science, 2015, 350(6267), 1516–1521 CrossRef CAS.
- R. A. House, U. Maitra, M. A. Pérez-Osorio, J. G. Lozano, L. Jin, J. W. Somerville, L. C. Duda, A. Nag, A. Walters, K.-J. Zhou, M. R. Roberts and P. G. Bruce, Superstructure control of first-cycle voltage hysteresis in oxygen-redox cathodes, Nature, 2020, 577(7791), 502–508 CrossRef CAS PubMed.
- M. Sathiya, G. Rousse, K. Ramesha, C. P. Laisa, H. Vezin, M. T. Sougrati, M. L. Doublet, D. Foix, D. Gonbeau, W. Walker, A. S. Prakash, M. Ben Hassine, L. Dupont and J. M. Tarascon, Reversible anionic redox chemistry in high-capacity layered-oxide electrodes, Nat. Mater., 2013, 12(9), 827–835 CrossRef CAS PubMed.
- D.-H. Seo, J. Lee, A. Urban, R. Malik, S. Kang and G. Ceder, The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials, Nat. Chem., 2016, 8(7), 692–697 CrossRef CAS PubMed.
- S. Li, Z. Liu, L. Yang, X. Shen, Q. Liu, Z. Hu, Q. Kong, J. Ma, J. Li, H. J. Lin, C. T. Chen, X. Wang, R. Yu, Z. Wang and L. Chen, Anionic redox reaction and structural evolution of Ni-rich layered oxide cathode material, Nano Energy, 2022, 98, 107335 CrossRef CAS.
- H. Chen and M. S. Islam, Lithium extraction mechanism in Li-rich Li2MnO3 involving oxygen hole formation and dimerization, Chem. Mater., 2016, 28, 6656–6663 CrossRef CAS.
- J. Hafner and G. Kresse, The Vienna Ab-Initio Simulation Program VASP: An Efficient and Versatile Tool for Studying the Structural, Dynamic, and Electronic Properties of Materials, in Properties of Complex Inorganic Solids, ed. A. Gonis, A. Meike and P. E. A. Turchi, Springer, US, Boston, MA, 1997, pp.69–82 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- J. M. Lim, T. Hwang, D. Kim, M. S. Park, K. Cho and M. Cho, Intrinsic origins of crack generation in Ni-rich LiNi0.8Co0.1Mn0.1O2 layered oxide cathode material, Sci. Rep., 2017, 7(1), 39669 CrossRef CAS.
- B. Zhang, Y. He, H. Gao, X. Wang, J. Liu, H. Xu, L. Wang and X. He, Unraveling the doping mechanisms in lithium iron phosphate, Energy Mater., 2022, 2(2), 200013 CrossRef.
- B. Silvi and A. Savin, Classification of chemical bonds based on topological analysis of electron localization functions, Nature, 1994, 371(6499), 683–686 CrossRef CAS.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- Y. Kang, X. Guo, Z. Guo, J. Li, Y. Zhou, Z. Liang, C. Han, X. He, Y. Zhao, N. Tavajohi and B. Li, Phosphorus-doped lithium- and manganese-rich layered oxide cathode material for fast charging lithium-ion batteries, J. Energy Chem., 2021, 62, 538–545 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, A grid-based Bader analysis algorithm without lattice bias, J. Phys.: Condens. Matter, 2009, 21(8), 084204 CrossRef CAS.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, Improved grid-based algorithm for Bader charge allocation, J. Comput. Chem., 2007, 28(5), 899–908 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36(3), 354–360 CrossRef.
- M. Yu and D. R. Trinkle, Accurate and efficient algorithm for Bader charge integration, J. Chem. Phys., 2011, 134(6), 064111 CrossRef.
- M. A. Rodriguez, D. Ingersoll and D. H. Doughty, In situ X-ray diffraction studies on lithium-ion battery cathodes, MRS Online Proc. Libr., 1999, 575(1), 65–70 CrossRef.
- W. Hu, Y. Chen, H. Kou, Y. Wang, H. Wan and H. Li, First-principles study on the electronic structure of LiCoO2 with intrinsic defects, Ionics, 2022, 28(7), 3139–3143 CrossRef CAS.
- C. Liu, Z. G. Neale and G. Cao, Understanding electrochemical potentials of cathode materials in rechargeable batteries, Mater. Today, 2016, 19(2), 109–123 CrossRef CAS.
- E. Antolini, LiCoO2: Formation, structure, lithium and oxygen nonstoichiometry, electrochemical behaviour and transport properties, Solid State Ionics, 2004, 170(3), 159–171 CrossRef CAS.
- Y. Lyu, X. Wu, K. Wang, Z. Feng, T. Cheng, Y. Liu, M. Wang, R. Chen, L. Xu, J. Zhou, Y. Lu and B. Guo, An overview on the advances of LiCoO2 cathodes for lithium-ion batteries, Adv. Energy Mater., 2021, 11(2), 2000982 CrossRef CAS.
- C. Zheng, Z. Yang, J. Feng, J. Zhong, Z. Wei and J. Li, Bifunctional surface modification coupled with oxygen defect engineering enables high performance Li-rich cathodes, J. Mater. Chem. A, 2022, 10(30), 16046–16060 RSC.
- J. N. Reimers, J. R. Dahn and U. G. V. Sacken, Effects of impurities on the electrochemical properties of LiCoO2, J. Electrochem. Soc., 1993, 140, 2752–2754 CrossRef CAS.
- J. M. Tarascon, G. Vaughan, Y. Chabre, L. Seguin, M. Anne, P. Strobel and G. Amatucci, In situ structural and electrochemical study of Ni1−xCoxO2 metastable oxides prepared by soft chemistry, J. Solid State Chem., 1999, 147(1), 410–420 CrossRef CAS.
- Y. Shao-Horn, L. Croguennec, C. Delmas, E. C. Nelson and M. A. O'Keefe, Atomic resolution of lithium ions in LiCoO2, Nat. Mater., 2003, 2(7), 464–467 CrossRef.
- P. Fuentealba; E. Chamorro and J. C. Santos, Chapter 5 Understanding and using the electron localization function, in Theoretical and Computational Chemistry, ed. A. Toro-Labbé, Elsevier, 2007, pp.57–85, vol. 19 Search PubMed.
- J. N. Zhang, Q. Li, C. Ouyang, X. Yu, M. Ge, X. Huang, E. Hu, C. Ma, S. Li, R. Xiao, W. Yang, Y. Chu, Y. Liu, H. Yu, X. Q. Yang, X. Huang, L. Chen and H. Li, Trace doping of multiple elements enables stable battery cycling of LiCoO2 at 4.6 V, Nat. Energy, 2019, 4(7), 594–603 CrossRef CAS.
- S. T. Myung, N. Kumagai, S. Komaba and H. T. Chung, Effects of Al doping on the microstructure of LiCoO2 cathode materials, Solid State Ionics, 2001, 139(1), 47–56 CrossRef CAS.
- F. E. Er-Rami, M. Duffiet, S. Hinkle, J. Auvergniot, M. Blangero, P. E. Cabelguen, K. Song, F. Weill, C. Delmas and D. Carlier, Understanding the role of Al doping of LiCoO2 on the mechanisms upon cycling up to high voltages (≥4.6 V vs. Li+/Li), Chem. Mater., 2022, 34(10), 4384–4393 CrossRef CAS.
- S. Song, Y. Li, K. Yang, Z. Chen, J. Liu, R. Qi, Z. Li, C. Zuo, W. Zhao, N. Yang, M. Zhang and F. Pan, Interplay between multiple doping elements in high-voltage LiCoO2, J. Mater. Chem. A, 2021, 9(9), 5702–5710 RSC.
- S. Li, Y. Sun, A. Gao, Q. Zhang, X. Lu and X. Lu, Sustainable LiCoO2 by collective glide of CoO6 slabs upon charge/discharge, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(20), e2120060119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ya00278g |
| This journal is © The Royal Society of Chemistry 2023 |