
Intramolecular singlet fission and triplet exciton harvesting in tetracene oligomers for solar energy conversion
Tianying
Wang†
a,
Heyuan
Liu†
 *a,
Xianyuan
Wang
a,
Linglong
Tang
a,
Jun
Zhou
a,
Xiaojuan
Song
a,
Liping
Lv
a,
Wenmiao
Chen
*b,
Yanli
Chen
a and
Xiyou
Li
*a
*a,
Xianyuan
Wang
a,
Linglong
Tang
a,
Jun
Zhou
a,
Xiaojuan
Song
a,
Liping
Lv
a,
Wenmiao
Chen
*b,
Yanli
Chen
a and
Xiyou
Li
*a
aSchool of Materials Science and Engineering, China University of Petroleum (East China), Qingdao, Shandong 266580, China. E-mail: liuheyuan123@upc.edu.cn; Xiyouli@upc.edu.cn
bDepartment of Science, Texas A&M University at Qatar, Education City, P.O. Box 23874, Doha, Qatar. E-mail: cwm-tamu@tamu.edu
First published on 10th March 2023
Abstract
Singlet fission (SF), which can convert one singlet exciton into two triplet excitons, has the potential to enhance the solar energy conversion efficiency in devices such as photovoltaic cells and processes such as photocatalysis. Recently, intramolecular SF (iSF), which is observed in covalently linked molecular oligomers, has attracted significant research interest due to its unique advantages in revealing the SF mechanism and practical applications. In the case of exothermal chromophores (their singlet state energy (ES1) is more than twice their triplet state energy (ET1), such as pentacene), efficient iSF is easily achieved. However, efficient iSF is limited in endothermic/isothermal chromophores (ES1 ≤ 2ET1, such as tetracene). Compared to exothermal systems, the energy loss in the SF of endothermic/isothermal systems is much lower, which is more favorable for enhancing the solar energy conversion efficiency. Therefore, achieving efficient iSF in endothermic/isothermal systems is necessary. Unfortunately, the design strategy for efficient iSF based on endothermic/isothermal chromophores is not clear. Herein, we summarize the SF studies on covalently linked tetracene oligomers and analyze the key influencing factors on iSF in these systems. Additionally, we review the harvesting of the two formed triplets from the iSF of tetracene oligomers. It is hoped that the results of this review will provide some new insights into the design of highly efficient iSF materials based on endothermic/isothermic chromophores for practical applications.
 Tianying Wang | Tianying Wang received her Bachelor's Degree from China University of Petroleum (East China), Qingdao, China, in 2022. Presently, she is a Master's Degree candidate at China University of Petroleum (East China). Her research mainly focuses on the design and application of tetracene derivatives in photon down-conversion. |
 Heyuan Liu | Dr Heyuan Liu received his PhD from Shandong University, Jinan, China, in 2015. Presently, he is an Associate Professor at the School of Materials Science and Engineering, China University of Petroleum (East China). His research interests include the design, synthesis and mechanism study of novel photon up/down conversion materials, as well as their applications in solar energy conversion. |
 Xiyou Li | Prof. Xiyou Li received his PhD from Institute of Photographic Chemistry Chinese Academy of Sciences, Beijing, China, in 1998. Presently, he is a Full Professor at the School of Materials Science and Engineering, China University of Petroleum (East China). His research interests mainly include photoelectric conversion, photochemical conversion and photocatalytic hydrogen production. |
1 Introduction
Singlet fission (SF) is a spin-allowed multiple-exciton generation (MEG) process, which can split one high-energy singlet exciton into two low-energy triplet excitons. When SF is incorporated in the solar energy conversion process, it is expected to successfully achieve single-photon excitation to produce two electrons and enhance the efficiency of the related process.1–5 For example, the Shockley–Queisser (SQ) limit of the photoelectric conversion efficiency of single-junction solar cells was improved from 33% to 45% upon the incorporation of SF.6,7 Beside solar cells, SF has also been applied to enhance the quantum efficiencies of organic light-emitting diodes,8 organic photodetectors,9,10 photon multiplier devices11 and nonlinear optical responses.12,13 Recently, it has been reported that SF also shows potential application in quantum information and computing.14SF was first observed in anthracene crystals in 1965,15 and subsequently confirmed in tetracene in the 1970s.16–18 However, in the following decades, further advances in this field were negligible. It was only in 2006 that Hanna and Nozik found that SF has great potential for enhancing the efficiency of solar cells.1 Subsequently, SF has shown vigorous development, and great efforts have been devoted to developing new efficient SF materials and revealing the SF mechanism. Many chromophores have been found to meet the energy requirement of SF, where the singlet state energy (ES1) should be larger or equal to twice the triplet state energy (ET1) (ES1 ≤ 2ET1), such as acenes (anthracene,19 tetracene,20 pentacene,21–24 hexacene25–27 and their derivatives), polyene,28–34 1,3-diphenylisobenzofuran,35 diketopyrrolopyrrole,36–40 perylenediimide,41–46 boron dipyrromethene (BODIPY),47–50 and other chromophores.37,51–61 These molecules have been summarized in some comprehensive reviews on this field.7,53,62–65
Based on these chromophores, several intermolecular SF (xSF) materials, including crystal films, colloidal nanoparticles and concentrated solutions, have been developed. The study of the SF in these systems gave some insights into the SF mechanism and showed that the SF rate and efficiency are strongly dependent on the intermolecular packing, morphology and defects. However, the determination of the structures of xSF materials is challenging, and consequently it is difficult to establish their structure–property relationship. Therefore, intramolecular SF (iSF), which is observed in covalently linked molecular oligomers, has attracted significant research interest due to the well-defined structure and facile structural modification of these materials.24,66,67 These advantages are more feasible for the establishment of the relationship between the molecular structure and SF properties.7,32,68–71 More importantly, the inherent nature of iSF in these oligomers is not restricted by external conditions and iSF materials can be easily incorporated in solar energy conversion devices via solution processing techniques and still maintain their SF properties.72
To date, several iSF materials based on different chromophores have been prepared and their SF properties investigated.32,44,73–85 In the case of exothermic chromophores ES1 > 2ET1, such as pentacene, regardless of their configuration, efficient iSF can be easily achieved.86,87 However, efficient iSF is difficult to achieve in endothermic or isothermic systems (ES1 ≤ 2ET1) (such as tetracene, diketopyrrolopyrrole, and perylene diimide). Compared to exothermic systems, endothermic or isothermic systems exhibit lower energy loss in the SF process, which have greater potential to enhance the overall solar energy conversion efficiency. Thus, it is necessary to achieve efficient iSF in endothermic or isothermic systems. Presently, although there are some comprehensive reviews on the SF mechanism in the literature,7,26,62,88–93 strategies for the design of efficient iSF materials based on endothermic or isothermic chromophores remain unclear. The revelation of the key factors influencing the iSF of endothermic or isothermic systems is crucial for designing novel efficient iSF materials based on these chromophores. In this review, firstly, we summarize SF studies on covalently linked tetracene oligomers, and then list several key factors on the iSF in these systems. Additionally, we review the study of harvesting two triplets from iSF in tetracene oligomers. Taking tetracene as an example, it is hoped that this work will provide some insights for the design of efficient iSF materials based on endothermic or isothermic chromophores and the application of iSF in solar energy conversion processes.
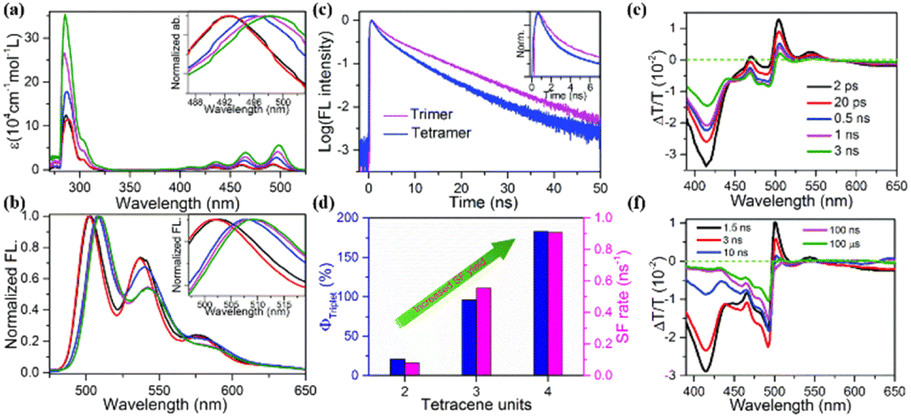
2 SF mechanism
Here, the generally accepted SF process is roughly depicted in Fig. 1.7,65 Upon photoexcitation, the singlet state (S1) is generated, which collides with another adjacent molecule in the ground state (S0) to produce the correlated triplet pair state (1(TT)) with an overall singlet spin character. This process can proceed via a direct or indirect mechanism. In the direct mechanism, the S1 state converts into the 1(TT) state without the involvement of any intermediate. Alternatively, the S1 state can transform into the 1(TT) state via an intermediate state that has a charge-transfer (CT) nature, which is the so-called indirect mechanism.7,63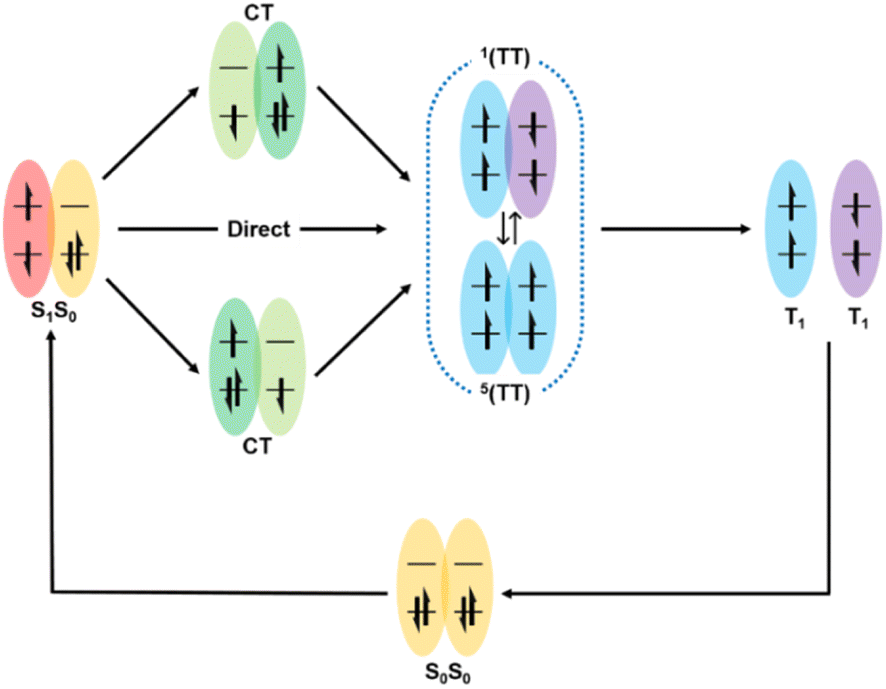 | ||
| Fig. 1 Description of SF mechanism. S0S0: ground state; S1S0: singlet state; and CT: charge transfer state, where this intermediate CT state may be virtual (in the form of a super-exchange mechanism) or real. 1(TT): triplet pair state and 5(TT): quintet state (spin evolution can be performed between the multiexciton 1(TT) and multiexciton 5(TT) state). T1: free triplet state. | ||
Additionally, a coherent superposition state of the lowest lying absorbing state, CT state, and multiexciton state is formed upon photoexcitation, which can drive the SF process. This is the so-called quantum coherent mechanism.88,89,94 Then, the two free triplets (T1) can be formed via the decoherence of either the 1(TT) state or the 5(TT) state. Generally, the 5(TT) state is formed via the spin evolution of the 1(TT) state.7,89,92 Initially, most of the research on SF only focused on the first step, namely, the formation of the 1(TT) state. Subsequently, the dissociation of the 1(TT) state into the T1 state has also received attention. However, due to the similar transient absorption spectral feature between the 1(TT) state and the T1 state as well as the possible intermediate state (3(TT) or 5(TT)), this dissociation process is difficult to be clarified by transient absorption spectra alone.95 Fortunately, the electron spin change in these states allows the use of spin resonance techniques to resolve this dissociation process.96,97
The above-mentioned rough SF mechanism is suitable for xSF and iSF. Many iSF molecules demonstrate a direct SF process,68,98–108 in which the iSF rate is dependent on the direct coupling strength between the S1 state and the 1(TT) state. The covalent linker plays an important role in iSF by controlling the coupling strength via through-bond or through-space interactions. Generally, conjugated linker-bridged oligomers exhibit much faster SF than bridged by nonconjugated linkers due to the stronger electronic coupling.106 However, a CT-assisted mechanism is also adopted in other iSF materials, as evident by the solvent-polarity-dependent SF rate and yield.24,42,45,74,77,82,109–118 The CT state couples strongly with the S1 and 1(TT) state via superexchange to serve as an intermediate state for the formation of 1(TT) whether directly populated or only virtually involved. With the help of this CT-assisted mechanism, highly efficient iSF can still occur in weakly coupled systems. Besides the above-mentioned two mechanisms, several other new models have been established for iSF. For example, the ‘vibronic coupling’ or ‘vibrationally coherent’ mechanism has been proven in several systems,73,119–121 corresponding to the case in which S1 and 1(TT) are nonadiabatically coupled through vibrational coordinates or form vibronically mixed states when their energy levels are resonant owing to excited vibrational quanta.73,122 This mechanism demonstrates an intimate link between the nuclear motions and SF, and can be approached from conical intersection, vibronically mixed S1–1(TT) manifolds or symmetry-breaking vibrations.73,119–121,123,124 Recently, a quantum interference-assisted mechanism was proposed for iSF.84 Constructive quantum interference can promote the formation of the 1(TT) state; however, destructive quantum interference will slow down the formation of 1(TT). Besides the CT intermediate state, the excimer state is also observed to act as an intermediate for the formation of 1(TT).44 However, this mechanism is still controversial. In some covalent link oligomers,23,100 the formation of the excimer state is considered to compete with SF. Thus, the real role of the excimer state in the SF process should be further investigated in the future. Additionally, a new spin exchange mechanism has been demonstrated to enable efficient SF via spin density localization,125 which is fundamentally different from the CT-mediated mechanism. In this mechanism, S1 already has partial 1(TT) characteristics and can quickly relax to 1(TT).
3 Covalently linked tetracene oligomers
Firstly, we discuss tetracene dimers, and then oligomers with more than two tetracene units. Dimers, as the smallest units capable of SF, can offer a better understanding of the SF mechanism. When the structural unit is increased, some extra factors including exciton delocalization and entropy increase should be considered.3.1 Tetracene dimers
In the past few years, several tetracene dimers with different conformations have been prepared. Depending on the connection position of tetracene, we classified these dimers into three groups, including 5,5′-linked dimers, 2,2′-linked dimers and other dimers linked via multiple-positions of tetracene (Fig. 2).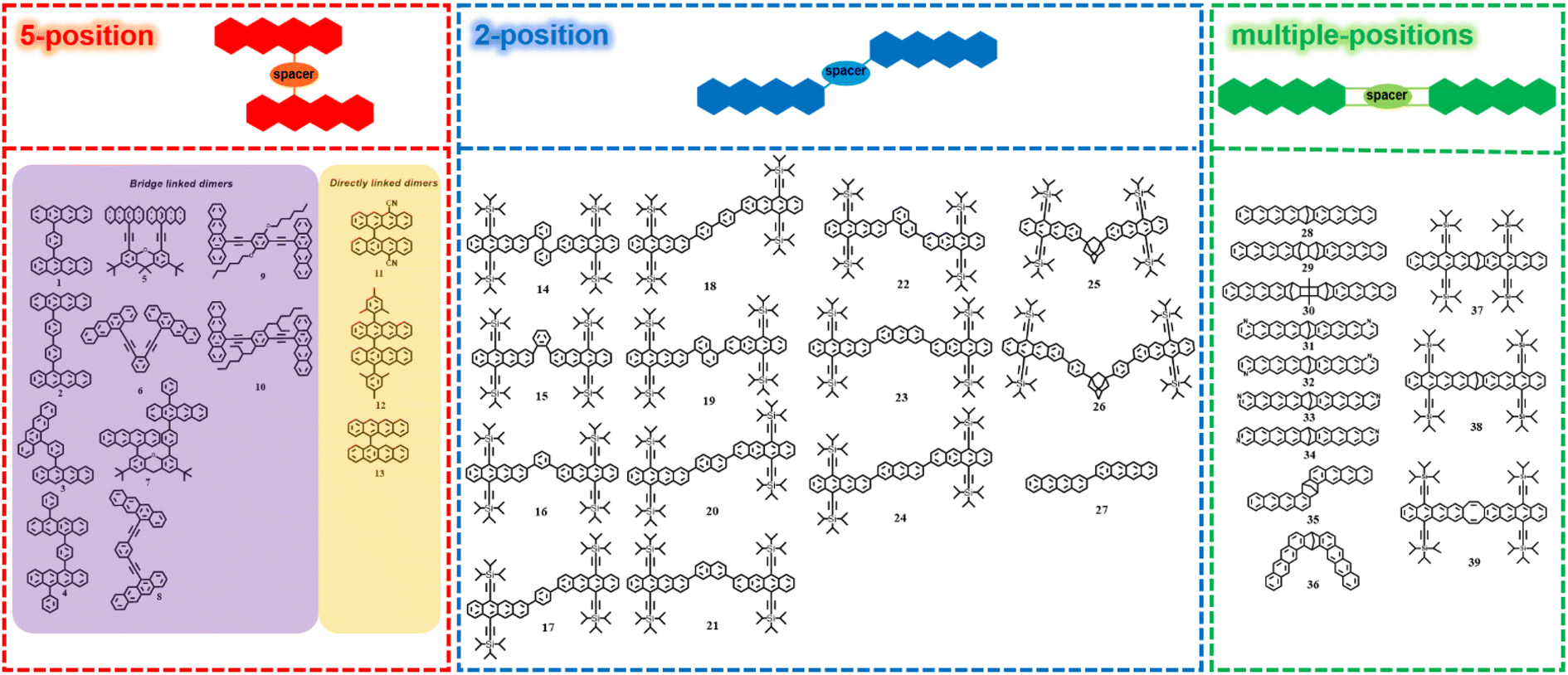 | ||
| Fig. 2 Molecular structures of tetracene dimers linked via 5-position of tetracene, 2-position of tetracene and multiple-positions of tetracene. | ||
3.1.1.1 Tetracene dimers bridged via 5-position of tetracene. In this part, initially we focus on tetracene dimers bridged through the 5-position of tetracene via a spacer (1–10, Fig. 2).
The pioneering work utilizing molecular dimers to study iSF was reported in 2006 by Bardeen's group, in which three tetracene dimers linked by para-phenylene (1 and 2) and meta-phenylene (3) groups were prepared.68,126 The electronic coupling in these three dimers is dominated by through-bond interactions, which is very weak, as revealed by the small redshift in their absorption spectra compared to that of monomeric tetracene (Fig. 3a).
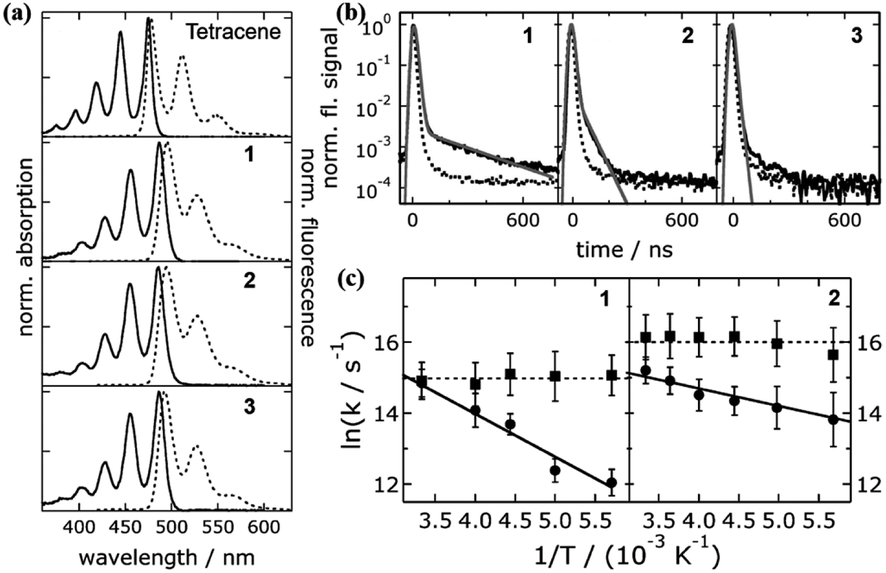 | ||
| Fig. 3 (a) Absorption and fluorescence spectra of dimers 1–3 and tetracene. (b) Fluorescence dynamics of dimers 1–3. (c) Temperature dependence of fission (circle) and fusion (square) rates of dimers 1 and 2. Reproduced with permission.68 Copyright 2007, the American Chemical Society. | ||
The presence of a delayed fluorescence signal in the fluorescence dynamics of dimers 1 and 2 indicates that these two dimers undergo SF (Fig. 3b). In contrast, SF is absent in dimer 3, which may be caused by its much smaller through-bond interactions than that in dimers 1 and 2. Notably, the SF efficiencies of dimers 1 and 2 are only ∼3%, which is much lower than that of tetracene crystal films.127–131 This should be ascribed to their weak intramolecular electronic coupling in comparison with the strong coupling in the solid. Additionally, this study revealed that the fission process (S1 → 1(TT)) is temperature-dependent in dimers 1 and 2, but the fusion process (1(TT) → S1) is independent of temperature (Fig. 3c). This is consistent with the fact that the fission process in tetracene derivatives is endothermic. Thus, this study provided some insights into the design of iSF molecules. Our group also synthesized a para-phenylene-linked tetracene dimer (4) but with two extra phenyl groups on the two tetracene units, which improved the solubility of the dimer. However, its SF efficiency was still very low, which should also be caused by the weak intramolecular electronic coupling.132 These results suggest that phenylene-bridged tetracene dimers are not favorable for SF due to their weak through-bond electronic coupling.
To strength the intramolecular electronic coupling, a “face-to-face” stacked tetracene dimer linked by a xanthane bridge was prepared by our group (5).133 Its absorption spectrum changed significantly compared to that of the corresponding monomer (Fig. 4a), suggesting the presence of strong electronic coupling between the two tetracene units. This strong coupling leads to the formation of an “excimer-like” state, as confirmed by its broad and structureless emission band.
 | ||
| Fig. 4 (a) Absorption and fluorescence spectra of dimers 5 and 6. (b) fs-TA spectra of dimer 5 in solution. (c) and (d) fs-TA spectra of dimer 6 in solution (c) and solid film (d). (e) Kinetic model for dimer 6 in solution (left) and solid film (right). Reproduced with permission.100 Copyright 2016, the American Chemical Society. | ||
However, the “excimer-like” state shows a short lifetime (∼0.23 ns), which is different from the traditional excimer-like state with a long lifetime.134–137 This phenomenon is similar to the observation in the excimer-mediated SF in a concentrated bis(triisopropylsilylethynyl)[TIPS]-tetracene solution,138 suggesting the presence of SF in this tetracene dimer.
Unfortunately, we did not find direct evidence for SF in this dimer due to the use of single detection technology. Subsequently, Thompson's group investigated its excited-state dynamics in detail using femtosecond transient absorption (fs-TA) spectra and found that this dimer undergoes structural relaxation to form an excimer-like structure that decays into the 1(TT) state within 200 fs (Fig. 4b).100 Nevertheless, the 1(TT) state cannot separate into the free triplet state due to its low energy and the strong coupling between the 1(TT) state and the S0 state, and decays directly into the S0 state quickly within 500 ps. It can be seen that the first step (S1 → 1(TT)) of SF can be conducted fast in this class of strongly coupled tetracene dimers, but the second step (1(TT) → T1 + T1) is impeded due to the strong exchange interaction between the two triplets of the 1(TT) state.139 Specifically, to achieve the whole SF process efficiently, the intramolecular electronic coupling should be decreased appropriately in molecular dimers.
Thus, a cofacial tetracene dimer (6) linked by ortho-phenylene with a twisted arrangement between tetracenes, exhibiting less overlap between the tetracene π-orbitals in comparison with dimer 5, was prepared by Thompson's group.100 The electronic coupling between the two tetracene units in dimer 6 is still strong, but weaker than that in dimer 5, as can be seen from the smaller degree of the inversion of the 0–0 and 0–1 peaks (Fig. 4a). Similar to dimer 5, the fluorescence of dimer 6 was almost quenched completely in solution (ΦF = 0.6%). However, the emission line shape retained the vibronic structure (Fig. 4a), suggesting that its emission does not originate from a deep-trap excimer state like that in dimer 5. The fs-TA experiment revealed that the 1(TT) state is formed quickly in dimer 6 upon photoexcitation in solution with a time constant of ∼2 ps (Fig. 4c). Similar to dimer 5, the 1(TT) state could not further dissociate into free triplets in solution and decayed directly to the ground state with a time constant of ∼500 ps (Fig. 4e). When dimer 6 was prepared as a solid film, the 1(TT) state was also formed quickly (τ = ∼0.8 ps) (Fig. 4d and e).
Notably, regardless of solution or solid film, the formation rate of the 1(TT) state was much faster than that in crystalline tetracene, suggesting that the arrangement of the two tetracenes in dimer 6 is more favorable for SF than that in crystalline tetracene. More importantly, the 1(TT) state can further separate into free triplets rapidly (τ = ∼0.2 ps) in the solid film via triplet energy transfer to the neighboring tetracene unit with a high triplet yield of 154%. This indicates that the triplet energy transfer is an essential second step required for the production of free triplets in endothermic SF systems. Additionally, to decrease the coupling strength, we synthesized a slip-stacked tetracene dimer composed of a tetracene unit and 5,12-diphenyltetracene (DPT) unit (7).140 In contrast to the “face-to-face” stacked structure of dimer 5, the slip-stacked structure of dimer 7 causes a large decrease in the degree of overlap between the two chromophores. Consequently, the electronic coupling between the two tetracene units in dimer 7 is almost negligible. The TA experiment revealed that ultrafast singlet energy transfer from the tetracene unit to the DPT unit proceeded with a time constant of ∼0.32 ps. This ultrafast singlet energy transfer successfully prevents the useless nonradioactive decays of the singlet state of tetracene and stores the excitation energy of tetracene as the singlet state of DPT. Unfortunately, no efficient SF was observed in this dimer, which may be caused by its weak intramolecular interaction.
Recently, Thompson's group prepared meta- and para-bis(ethynyl tetracenyl)benzene dimers (8–10), which are analogues to the above-mentioned tetracene dimer 6, to study the effect of through-bond versus through-space coupling on the overall SF process.99 For meta-bis(ethynyl tetracenyl)benzene dimer (8), the two tetracene units is cross-conjugated. The inter-chromophore interactions are dominated by through-bond coupling, but the interaction strength is very small, and thus it can be deduced from its nearly unchanged absorption spectrum compared to that of the corresponding monomer. Alternatively, ortho- and para-ethynylbenzene linkers enable conjugation between the two tetracene units. Thus, the through-bond coupling between the two tetracene units in ortho- (6) and para-ethynylbenzene-linked dimers (9 and 10) is strong.
In the case of dimer 6, through-space coupling is also present besides through-bond coupling. Analogously to dimer 6,100 dimers 9 and 10 are weakly emissive in solution (ΦF < 1%), suggesting the presence of a nonradiative decay pathway in them. Instead, dimer 8 is highly emissive with a quantum yield of 62%. TA spectra revealed that ultrafast SF proceeded in dimers 9 and 10 (τSF = 0.1 and 0.4 ps for dimers 9 and 10, respectively) similar to that in dimer 6 (τSF = 2 ps) (Fig. 5b–d). However, no obvious triplet signal was observed in the TA spectra of dimer 8 (Fig. 5a), indicating the absence of efficient SF. This suggests that the conjugation linked mode is more favorable for SF than the cross-conjugation linked mode in endothermic SF systems. In contrast, a series of pentacene dimers linked by the same linker (ortho-, meta- and para-ethynylbenzene), where the energetics are favorable for SF, displayed ultrafast SF.24
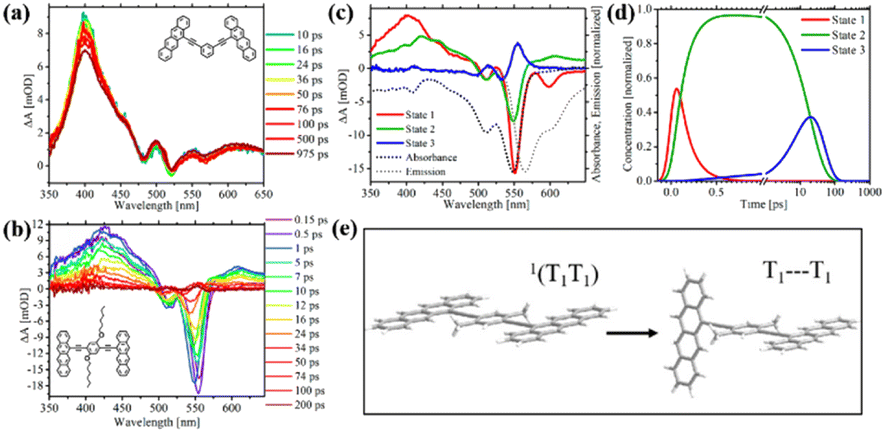 | ||
| Fig. 5 (a) and (b) TA spectra of dimer 8 (a) and 9 (b) in solution. (c) and (d) Species associated difference spectra (c) and concentration curves (d) from the target analysis of the TA data of dimer 9. (e) Proposed decoupling of the 1(TT) state in dimer 10 to give rise to two independent triplets. Reproduced with permission.99 Copyright 2018, the American Chemical Society. | ||
Additionally, the faster SF rate of dimers 9 and 10 compared to that of dimer 6 indicates that the through-bond coupling operating in the para dimers is stronger than the combined through-space and through-bond coupling operating in the ortho dimer. Compared to the slow SF rate in tetracene dimer 1, in which the two tetracene units are directly attached to the para-phenyl group, dimers 9 and 10, where the two tetracene units are linked to the para-phenyl group via an ethynyl group, exhibited a much faster SF rate. This may be caused by the presence of two ethynyl groups, which can perturb the S1 and T1 state energies in a favorable way and ensure stronger electronic coupling between the two tetracene units.
Notably, the formed 1(TT) state can further dissociate into two free triplets in the para dimers 9 and 10 instead of the ortho dimer 6, which is attributed to the rotational flexibility of the two tetracenes in dimers 9 and 10. This breaks the coupling between the two tetracenes and allows the triplets to separate from the 1(TT) state. The rotation of the tetracene units in dimer 6 is hindered because they are confined to a cofacial orientation. This provides another method to facilitate the separation of the 1(TT) state via the molecular rotation besides the above-mentioned triplet energy transfer. Although free triplets could be obtained in dimers 9 and 10via the molecular rotation, the yield is relatively low (ΦT = 70% and 94% for dimers 9 and 10, respectively), which is ascribed to the fast recombination rate of the triplets (20 and 76 ps for dimer 9 and 10) compared to the rate of their formation (19 and 48 ps for dimers 9 and 10), respectively. This may be a disadvantage of molecular dimers, where the formed two triplets are confined in one molecule, and thus recombine rapidly.
Moreover, dimer 9 linked by hexyloxy-bis(ethynyl)benzene (τSF = 0.1 ps) showed a faster SF rate compared to dimer 10 linked by 2-ethylhexylbis(ethynyl)benzene (τSF = 0.4 ps), which is because the hexyloxy-bis(ethynyl)benzene linker has a much closer molecular orbital energy to that of tetracene, inducing better mixing of the linker orbital with tetracene, and thus stronger coupling in dimer 9. This highlights the important role of the linker energetics in SF for endothermic SF systems. Nevertheless, it is important to note that different types of substituents may increase different degrees of steric hindrance when linking substituents on phenylene spacer, thus limiting the conformational flexibility and blocking SF.101
3.1.1.2 Directly linked tetracene dimers via 5-position of tetracene. Here, we discuss the directly linked dimer via the 5-position of tetracene (11–13, Fig. 2),118,141,142 in which the two tetracene units are linked directly through a C–C bond and interact via through-bond coupling.
Notably, the two tetracene units can rotate around this single bond, inducing the formation of the symmetry breaking charge transfer (SBCT) state, which shows a significant solvation effect, similar to that in single-bond-linked anthracene and pentacene dimers.114,115,144–146 The presence of the charge transfer (CT) state in this type of dimer enables the role of the CT state in the SF process to be determined.
In 2016, a stable directly linked tetracene dimer (11) was synthesized by our group by introducing an electron-withdrawing cyano group in the tetracene skeleton.141 Its singlet state is delocalized on the two tetracene units and mixed strongly with the CT state. Unfortunately, we failed to observe the occurrence of SF. This gives the impression that the orthogonal conformation linked by a single bond is not sufficient to support the occurrence of SF. However, in 2019, this vague understanding was broken by Musser's group.118 They found that activated iSF could occur in a single-bond-linked orthogonal tetracene dimer with two phenyl groups in the tetracene skeleton (12). As documented in anthracene and pentacene dimers with an orthogonal conformation,114,115,144–146 its photophysical properties are strongly dependent on the torsional angle (Φ) between the two tetracene units. The displacement along Φ leads to mixing between the localized excited state (LE) and the CT state (Fig. 6a), reaching the maximum mixing at the local minima of the LE state at 70°. This introduces a strong solvation effect on its fluorescence (Fig. 6b). In low-polar solvents, the emission is dominated by a well-defined vibronic progression, which is attributed to the relaxed LE state. With an increase in the solvent polarity, the emission becomes featureless and gradually shows a redshift, which indicates the participation of the CT state.115,144 The transient absorption spectra showed that activated iSF could proceed with the excitation energy above the threshold energy (2.8 eV). Notably, iSF is also strongly dependent on the solvent polarity (Fig. 6d). In nonpolar solvents, the 1(TT) yield is nearly constant above the threshold energy, but the 1(TT) yield shows an obvious gradual onset in polar solvents. Moreover, in intermediate-polarity solvents iSF has the largest 1(TT) yield and the fastest rate. This iSF behaviour was attributed to the large mixing of the CT state and the 1(TT) state in intermediate-polarity solvents, which makes SF coherent and accelerates the SF rate (Fig. 6c).143 In low-polarity and high-polarity solvents, the mixing is minimal and makes SF incoherent. This result deeply reveals the role of the CT state in the SF process, in which the hot iSF can be mediated by the CT state via the coherent or incoherent mechanism and these two mechanisms can be switched by the solvent polarity by controlling the mixing degree between the CT state and the 1(TT) state. Additionally, Shizu et al. predicted that the 1(TT) state was formed efficiently in a directly linked tetracene dimer (13) due to the small 1(S0S1) − 1(TT) energy difference and the large vibronic coupling at the 1(S0S1) geometry.142
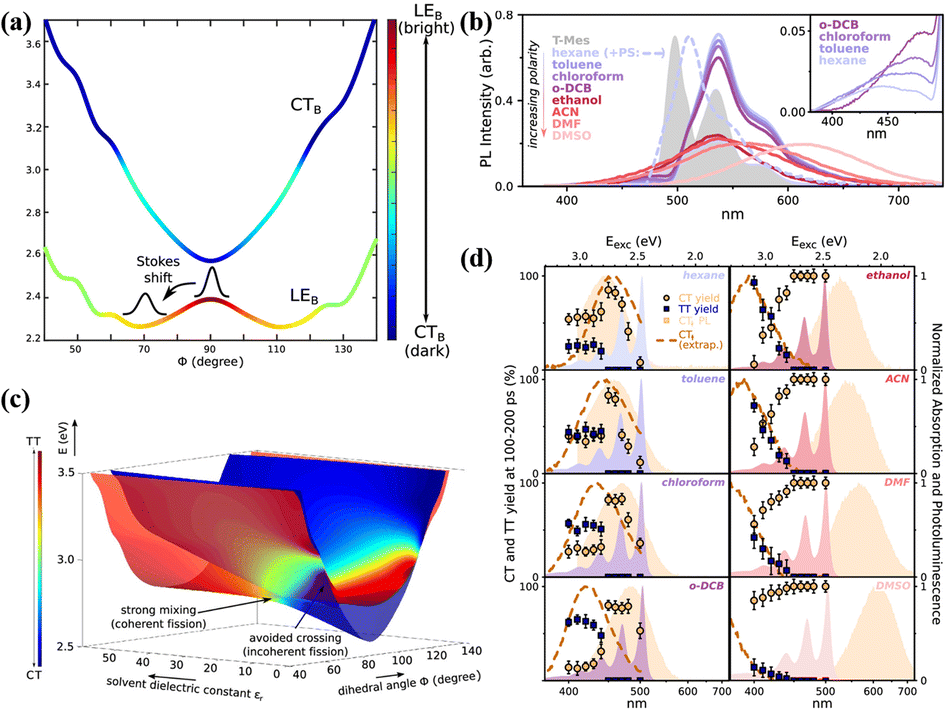 | ||
| Fig. 6 (a) Potential energy surfaces of the LEB and CTB states along the torsional coordinate (Φ) for dimer 12. (b) Steady-state photoluminescence of dimer 12 in a range of solvents (solid lines). (c) Adiabatic potential energy surfaces along the dihedral angle Φ and the solvent dielectric constant, showing their CT (blue) and TT (red) character. (d) Yield of long-lived CT (circles) and TT pairs (squares), extracted from excitation-dependent transient absorption measurements for solvents of increasing polarity (left to right). Reproduced with permission.143 Copyright 2019, the American Chemical Society. | ||
Recently, Mattos et al. further studied the role of the SBCT state in SF through a computational study of dimer 13.82 They quantified the SF mechanism proposed by Alvertis et al.,118 in which solvent-induced symmetry breaking leads to a high-energy CT state that interacts with the 1(TT) state, resulting in SF. An approximate assessment of the nonadiabatic interactions between the different electronic states highlighted that the CT state is essential in facilitating the transition from the bright excitonic state to the 1(TT) state, leading to SF. Additionally, several types of symmetry-breaking inter- and intra-fragment vibrations play a crucial role in a concerted mechanism with the solvent environment and with the symmetric interfragment torsion, which tune the admixture of excitonic and CT states. This work offers a new perspective on how solvent-induced SBCT can be understood and how it cooperates with the intramolecular mechanisms in SF. The result of the three latter works suggests that the orthogonal tetracene dimeric structure linked directly by the single-bond is advantageous for achieving efficient iSF, and the CT state here is generally considered to be an intermediate state for the occurrence of SF.
Although efficient iSF has been achieved in the above-mentioned tetracene dimers linked via the 5-position of tetracene, the lifetime of the obtained triplet is very short. Thus, the short-lived triplets are difficult to be harvested in solar energy conversion. Consequently, realizing the long-lived triplet state is necessary in tetracene derivatives for the application of SF. The 2,2′-biphenyl linker has been employed to construct pentacene dimers for achieving a high yield of long-lived triplets.149 Inspired by this, Nakamura et al. prepared a 2,2′-biphenyl-linked tetracene dimer (14, Fig. 2) with a bent-shaped structure.75 The steady-state absorption and fluorescence spectra demonstrated that the electronic coupling between the two tetracene units is relatively weak in this dimer.
The fs-TA spectra showed that the triplet signal appears rapidly with a time constant of ∼14 ps (Fig. 7A). Then, the triplet state decays with a biexponential function (Fig. 7B). The faster component originates from the mixture of the singlet and 1(TT) states because of the competitive reaction between iSF and triplet–triplet annihilation (TTA). The other much slower one is attributed to the free triplet state,150 which could be confirmed by its ultra-long lifetime (0.29 ms) (Fig. 7C). The quantum yield of the free triplet state is up to ∼175% in this dimer, which should be attributed to the weak interchromophore electronic coupling. However, the reason for the fast SF rate in this dimer with very weak electronic coupling is still unclear and should be further investigated in the future.
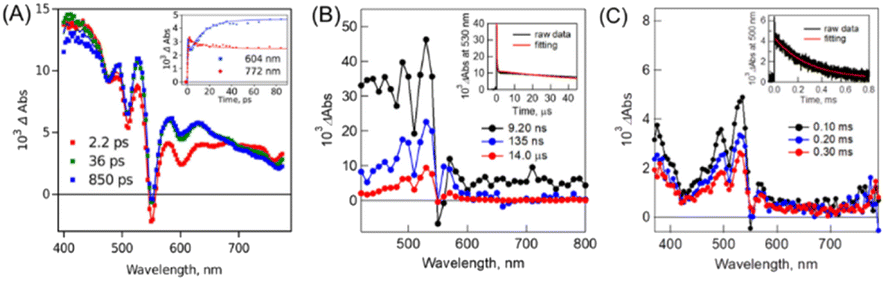 | ||
| Fig. 7 (A) fs-TA spectra of dimer 14 in PhCN (excited at 350 nm). The inset shows the time profiles at 604 nm (blue) and 772 nm (red). (B) ns-TA spectra of dimer 14 in PhCN (excited at 550 nm). The inset shows the time profile at 530 nm (black) and fitting curve at 530 nm (red). (C) μs-TA spectra of dimer 14 in PhCN (excited at 532 nm). The inset shows the time profile at 500 nm (black) and fitting curve at 500 nm (red). Reproduced with permission.75 Copyright 2019, the American Chemical Society. | ||
This is useful for the design of novel iSF materials with a high yield of long-lived triplet states. Besides electronic coupling, the conformational flexibility of the dimer structure may play an important role in SF.99 Thus, Nakamura et al. studied the synergistic role of conformational flexibility and electronic coupling in SF utilizing four tetracene dimers (15–18) linked by different phenylene-based linkers.98 The steady-state spectra and electrochemical measurements revealed that the intramolecular electronic coupling of dimer 16 linked by the meta-phenyl group is relatively weaker than the ortho- (dimer 15) and para- (dimer 17) phenyl-linked dimers. The electronic coupling of dimer 18 bridged by the 4,4′-biphenyl group should be somehow weaker than that of the other three dimers. The energies of the rotational barriers of these four dimers were calculated using DFT to discuss the conformational flexibility between the two tetracene units. The rotational barriers of dimers 18, 16 and 17 are 0.075, 0.13, and 0.081 eV, respectively. In contrast, the barrier of dimer 15 could not be determined because of the bulkiness of the TIPS acetylene unit. This suggests that dimers 17 and 18 possess larger conformational flexibilities than dimer 15 and 16. Thus, these four dimers are categorized into four different characters, i.e., dimer 15 with strong electronic coupling and low conformational flexibility, dimer 16 with weak electronic coupling and low conformational flexibility, dimer 17 with strong electronic coupling and high conformational flexibility, and dimer 18 with weak electronic coupling and low conformational flexibility. The TA experiment revealed that SF could proceed in all four dimers (Fig. 8a–h). However, the formation and separation rate of the 1(TT) state and the corresponding inverse process (TTA) are significantly dependent on the electronic coupling strength and the conformational flexibility degree. Dimers 15 and 17 with strong electronic coupling have a faster 1(TT) formation and TTA rate than dimer 16 and 18 with weak electronic coupling, suggesting that the larger electronic coupling will induce faster SF and TTA rate. Although the strong electronic coupling could enable a faster SF rate, it also accelerated the TTA process, which will decrease the overall triplet yield. Thus, the electronic coupling strength should be balanced to ensure a fast SF rate and slow TTA rate for achieving a high triplet yield. Notably, dimer 18 with the largest conformational flexibility has the fastest 1(TT) separation rate among the four dimers, suggesting that high conformational flexibility will accelerate the dissociation of the 1(TT) state. Additionally, the quintet (5(TT)) intermediate state for the dissociation of the 1(TT) state into the free triplet state was observed in the TREPR spectra of dimers 15, 17 and 18 (Fig. 8i). Different from dimers 15 and 17, the weakly coupled triplet state (T + T) also appeared in the TREPR spectra of dimer 18. This is caused by its flexible conformation, as confirmed by the calculated conformation of the 5(TT) and (T + T) states (Fig. 8j). The 5(TT) state possesses a nearly planer conformation with a strong spin–spin exchange interaction (J) induced by the large direct orbital overlap. However, an orthogonal conformation is adopted in the T1 state, and consequently the two triplets are no longer correlated.
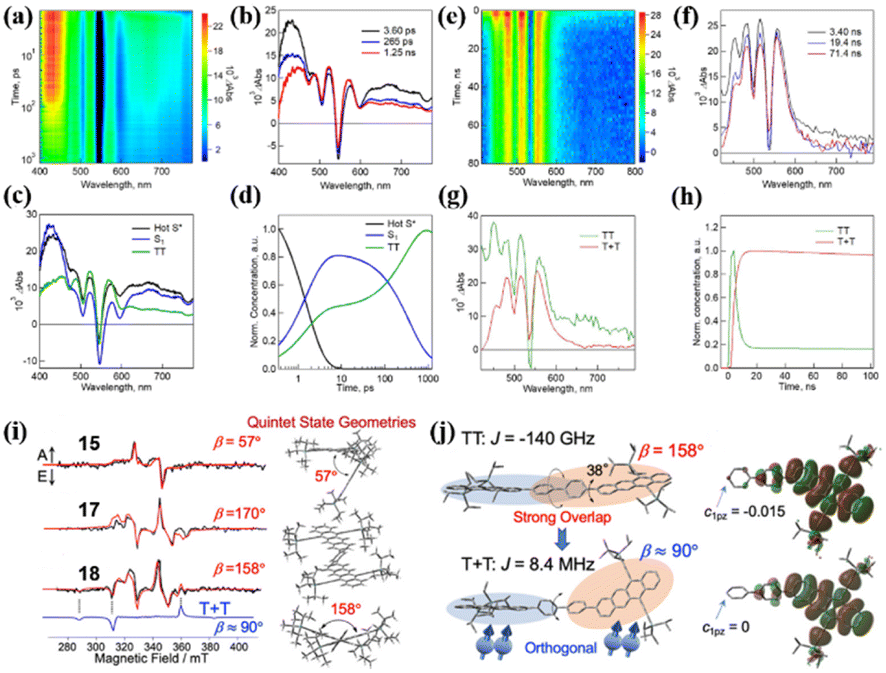 | ||
| Fig. 8 (a) and (b) fs-TA spectra of dimer 18 in THF (λex = 350 nm). (c) and (d) Species-associated TA spectra and corresponding decay profile of dimer 18. (e) and (f) ps-TA spectra of dimer 18 in THF (λex = 350 nm). (g) and (h) Species-associated TA spectra and corresponding decay profile of dimer 18. (i) TREPR spectra at 0.4 μs after the laser irradiation of dimers 15, 17 and 18 in MTHF at 77 K. Simulated spectra of the strongly coupled quintet multiexcitons are shown by the red lines with the dihedral angles (β) between the triplet excitons, as indicated by the right geometries. The spin-correlated triplet pair (SCTP) spectrum of the weakly coupled T + T state for dimer 18 was also computed by applying the electron spin polarization transfer (ESPT) model with J = 8.4 MHz, as shown by the blue line, and added to the quintet spectrum to obtain the red line. (j) Geometries of the 5(TT) and T + T multiexcitons in dimer 18 from the simulations of the TREPR spectra (left) and the corresponding electron density distribution (right). Dihedral angle change between the adjacent aromatic planes is indicated by the arrows at the centers, denoting that an almost orthogonal conformation leads to the decoupling in J. Reproduced with permission.98 Copyright 2021, the American Chemical Society. | ||
Specifically, intramolecular decoupling occurs in the 1(TT) state of dimer 18 by the conformation change, and thus leads to the highest free triplet yield (196%) among the dimers (67%, 125% and 117% for dimers 15, 16 and 17, respectively). This also highlights the important role of molecular conformation change for the separation of the 1(TT) state, similar to the report on dimers 9 and 10 by Thompson's group.99 This work provides a new perspective for the development of novel iSF materials with long-lived triplets.
Campos' group reported the preparation of a series of tetracene dimers (16, 17, and 19–24, Fig. 2) linked by a bridge with constructive (CQI) or destructive (DQI) quantum interference effect to reveal the effect of quantum interference (QI) on the formation dynamics of the 1(TT) state (Fig. 9a).84 The CQI bridges include 14Ph (17), 15N (19), 26N (20) and 26A (24), while 13Ph (16), 27N (21), 16N (22) and 27A (23) show the DQI effect (Fig. 9a). As revealed by the TA experiment, the formation rate of the 1(TT) state in the dimers with the DQI effect is always slower than that with the CQI effect across different bridge lengths and geometries (Fig. 9b), emphasizing the important role of CQI in SF.
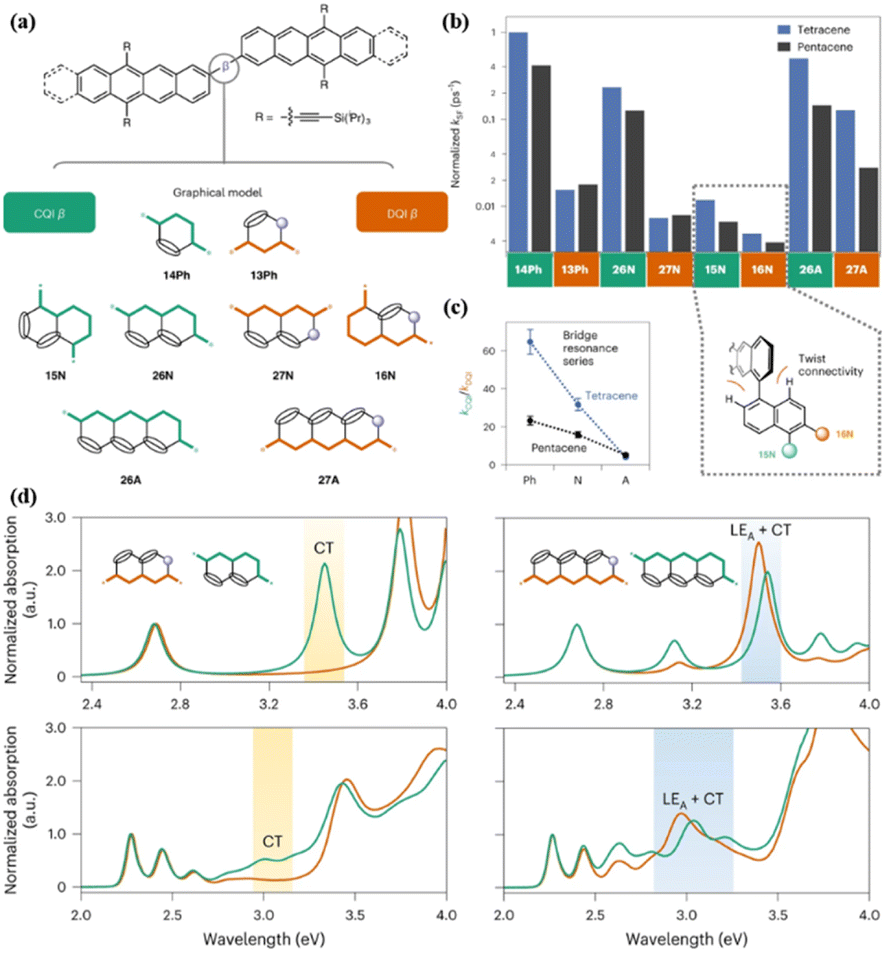 | ||
| Fig. 9 (a) Bridged tetracene and pentacene dimers (top), where β = 14Ph (17), 13Ph (16), 15N (19), 26N (20), 27N (21), 16N (22), 26A (23) and 27A (24). (b) Comparison of SF rates (normalized to the largest kSF) across all Ph, N and A bridges in this work. Inset: chromophores attached to the 1 and/or 5 positions and the 2, 6 or 7 positions of naphthalene. (c) Changes in the CQI and DQI SF rate constants as the bridge length increases. (d) Calculated (top) and experimental (bottom) normalized absorption spectra for naphthalene-bridged tetracene dimer (left) and anthracene-bridged tetracene dimer (right). Green traces correspond to a CQI bridge (26N or 26A) and orange traces correspond to a DQI bridge (27N or 27A). The calculations assume a planar conformation. The inclusion of intermonomer rotation decreases the intensity of the CT absorption, but the absorption energy is largely unaffected. Reproduced with permission.84 Copyright 2022, Nature. | ||
This phenomenon may be due to the more pronounced CT character in the dimers with the CQI effect compared to that with the DQI effect, as shown in the 26N- and 27N-bridged dimers (20 and 21), respectively (Fig. 9d). Notably, although the 26A- and 27A-bridged dimers show a similar CT strength, the 1(TT) formation rate of the 27A dimer is much slower than that of the 26A dimer (Fig. 9d), which is caused by the near-zero transition dipole moment between the ground state and the CT state in the 27A dimer. Thus, strong dipole coupling should also be considered for SF. As the bridge length increased, the difference between the 1(TT) formation rate constants of the CQI and DQI dimers became smaller (Fig. 9c). This is mainly due to the larger resonance interactions between tetracene and the longer bridge caused by the much closer molecular orbital energy of the bridge to tetracene, which can accelerate SF. This effect will compensate the DQI effect to some extent. Additionally, the chromophore attached to the 1 and/or 5 positions of naphthalene (dimer 19) has a larger chromophore distortion than the chromophore attached to the 2, 6 or 7 positions (dimer 22), which will reduce the orbital overlap of the chromophores and slow down the SF. However, the dimer attached to the 1 and/or 5 positions of naphthalene has a larger SF rate compared to that attached to the 2, 6 or 7 positions. This suggests that QI remains a substantial effect in the limit of both strong and weak interchromophore coupling. This work reveals critical considerations of the bridge topology and frontier molecular orbital energies in applying QI conductance principles to predict the rates of multiexciton generation.
The above-mentioned tetracene dimers are almost covalently bonded via conjugated or cross-conjugated linkages. Generally, these linkers result in a decrease in the S1 and T1 energies, which makes SF unfavorable thermodynamically.151 Thus, non-conjugated linkages including sp3-hybridized atoms may be a good choice for maintaining the intrinsic high energies of S1 and Tl and enable the moderate interactions needed to trigger iSF. Thus, Matsui et al. prepared two tetracene dimers (25 and 26) linked by an adamantane moiety.147 The two tetracene units in dimer 25 are bonded to the adamantane moiety directly, but in dimer 26, the two tetracene units are bonded to the adamantane moiety via the phenyl group. According to their steady-state spectra (Fig. 10a), their S1 and T1 state energies were estimated to be 2.27 and 1.02 eV, respectively, suggesting that the SF in these two dimers is exergonic. Notably, the T1 energy is stabilized a lot compared to the pure tetracene (1.2 eV), which is due to the presence of silylethynyl groups. The decreased T1 energy is contradictory to the value of the T1 energy (1.2 eV) for the same tetracene monomer with two substituted silylethynyl groups reported by Anthony et al.138 In anthracene derivatives, the introduction of two silylethynyl groups could indeed disturb the S1 and T1 energies and make SF more feasible.19,152 The role of the silylethynyl group in the decrease of the T1 energy should be further investigated. The presence of the delayed fluorescence (Fig. 10b) and the quintet state signal in time-resolved EPR spectra (Fig. 10c) of dimer 25 suggest that iSF occurs in dimer 25. In contrast, no SF signal was observed in dimer 26 due to the extremely weak electronic coupling induced by the large inter-chromophore distance.
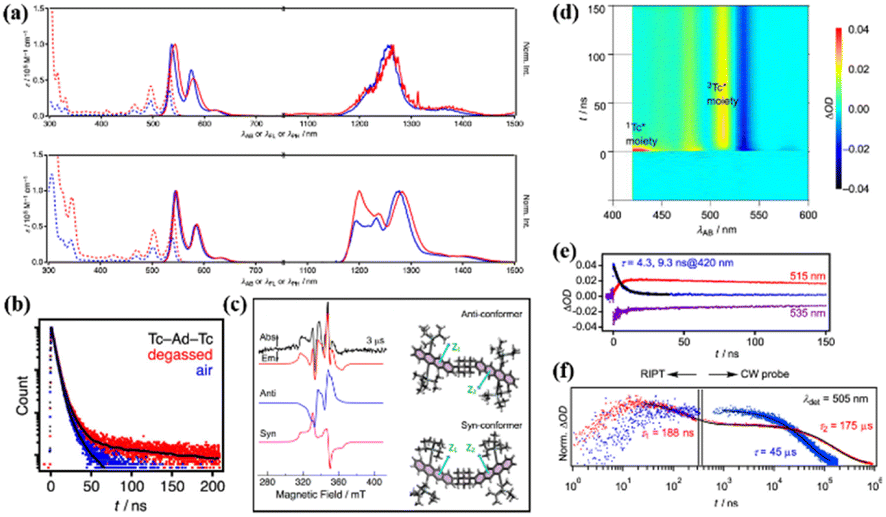 | ||
| Fig. 10 (a) UV-vis absorption (dotted), fluorescence, and phosphorescence (solid) spectra of dimers 25 and 26 (red) and the corresponding monomer (blue) in MCH. Phosphorescence spectra were measured using methylcyclohexane (MCH) matrices at 77 K. (b) Fluorescence decay profiles of dimer 25 in degassed (red) and aerated (blue) MCH detected at the respective λFL maxima. Black curves are fitted lines. (c) Time-resolved EPR spectra of dimer 25 in degassed MTHF at 110 K (left). (d) TA spectra of dimer 25 in degassed MCH. (e) Single-wavelength dynamics probed at 420, 515 and 535 nm. (f) Nano-to-millisecond decay profiles for (red) dimer 25 and (blue) corresponding monomer traced at 505 nm using RIPT and CW probe methods for the region shorter and longer than 200 ns, respectively. ΔOD values were normalized at 200 ns. Two components of the decay profile possess time constants of 188 ns and 175 μs for dimer 25. Reproduced with permission.147 Copyright 2019, the American Chemical Society. | ||
The presence of iSF in dimer 25 was also confirmed by the TA experiment (Fig. 10d–f). The triplet state decayed with a two-exponential function, i.e., 188 ns and 175 μs. The fast component is ascribed to the TTA process, and the long-lived component originated from the free triplet state. Notably, two different conformers were presented in the quintet state of dimer 25 (Fig. 10c), suggesting that the quintet state is populated by the spin relaxation of the 1(TT) state induced by the conformation change.153 The triplet yield of dimer 25 is still relatively high (92%), resulting from the slow TTA and the fast separation of the 1(TT) state, which is a consequence of the weak electronic coupling. As can be seen, the incorporation of rigid aliphatic linkages in dimers provides a way to control the 3D orientations of π-conjugated systems, while leaving the intrinsic S1 and T1 levels of the original monomers, causing exergonic iSF to take place and preventing the undesirable TTA.
Additionally, a directly linked tetracene dimer (27) via the 2-position of tetracene was reported by Müller et al. and the photophysical property of its solid state was investigated in detail.148 However, no SF signal was observed in its solid film, which is because the large decrease in the S1 state energy caused the energy requirement of SF to no longer be fulfilled.
This can be broken by vibration, and an effective conversion from S1 to 1(TT) can be achieved as long as there is moderate nonadiabatic coupling.123,156,157 Thus, the bridge geometry, including the length of the bridge and the orientation of the bridge, was used to control the SF thermodynamics and the electronic coupling between the chromophores in these dimers.154
Firstly, the effect of bridge length on the SF driving force and electronic coupling was investigated theoretically using three dimers (28–30).154 With the elongation of the bridge length from dimer 28 to 30, the interchromophore electronic coupling becomes weak gradually. However, the SF driving force does not change with the length of the bridge. Despite the sizeable electronic coupling and reasonable CT energy in these three dimers, the SF rate is expected to be nonoptimal due to the nonhorizontal CT matrix elements being zero as a result of the C2V symmetry. This was also confirmed by the experimental results, where dimer 28 showed slow iSF with a low triplet yield (∼6%).158 Thus, vibrational motions that break the long-axis reflection plane will be required.
Based on their vibronic coupling predictions,159 the norbornyl bridge was attached at the 1,2- instead of the 2,3-positions of tetracene to construct two isomers (trans-dimer (35) and cis-dimer (36)) and break the C2V symmetry.156 A more favorable SF driving force was achieved for dimers 35 and 36 compared to dimers 28–30, which can be understood as manifesting from the enhanced wave function delocalization inclusive of the bridge in dimers 35 and 36, serving to lower both the T1 and S1 energies. Notably, in these two dimers, it appears possible to increase the interchromophore electronic coupling (manifested by the larger Davydov splitting), while simultaneously increasing the SF driving force. Furthermore, the coupling strength of cis-dimer 36 is considered to be greater than that of the trans-one due to three reasons. Firstly, the transition dipole moments of the two tetracene units in the cis-dimer are very close, which enhances the Coulomb interaction. Secondly, the two cis-unit edges provide a larger steric contribution to the exchange interaction. Thirdly, given that the direct through-space orbital interaction occurs not only in the region of the bridge but also inside each tetracene arm, the cis-dimer through electron transfer and hole transfer electron coupling is stronger.154 Besides, the CT energy in the cis-dimer is significantly lower than that in the trans-dimer, and all these factors are favorable to improve the SF efficiency in the cis-dimer.156 In addition, heteroatom-substituted tetracene dimers (33 and 34) were also used to break the C2V symmetry.156 As expected, the electronic coupling is asymmetric. In these dimers, the “horizontal” coupling shows a common sign given that both chromophores in each dimer are juxtaposed directly across from each other via the norbornyl bridge, resulting in a fixed value. This means that SF will occur via the constructive interference of electron transfer and hole transfer pathways in the C2-symmetric species. This constructive interference increases the diabatic coupling significantly and leads to faster SF.
Additionally, the effect of the substituent position on iSF was investigated utilizing two isomers (37 and 38), where the acetylene substitution pattern on each chromophore in dimer 38 is moved outward by a ring relative to the bridge (Fig. 11a).123,155,157 The steady-state absorption and fluorescence spectra showed that there is nearly no difference between these two dimers (Fig. 11b), suggesting that their S1 state energy is almost identical. The TA experiment revealed that ultrafast iSF occurs in these two dimers (τSF = 10 ps and 5 ps for dimer 37 and 38) (Fig. 11c and e), respectively. The fast iSF rate is initially surprising given the absence of a reaction driving force and the unfavorable structural symmetry in these two dimers, which is expected to limit the diabatic coupling between the reactant and product.154,159 However, their analysis suggests that the few-parameter rate constant expression of the Marcus theory explains both individual and comparative findings in the set of systems, thus establishing benchmarks for the diabatic coupling and reorganization energy needed for efficient 1(TT) formation.159,161,162 This highlights the importance of diabatic coupling and reorganization energy for SF with the Marcus theory for the first time. Due to the close energy alignment of the S1 and 1(TT) state, fast equilibrium between them is established in these two dimers, as confirmed by the perfect match between the TA spectrum at 120 ps and TA spectrum obtained via the superposition of the S1 and T1 states (Fig. 11d and f). This is also consistent with their relatively high fluorescence quantum yields (∼72%). The equilibrium constant (K) was calculated to be 0.1 and 1 for dimers 38 and 38, respectively. According to the K value, the 1(TT) yields of dimers 37 and 38 were estimated to be ∼10% and ∼50%, respectively. These markedly different 1(TT) yields are caused by the different exchange interactions induced by the different placement of the acetylene side groups in these two dimers.97 In dimer 38, the relative placement of the acetylene side groups draws the triplet excitons further away from each another, thereby lowering the overall energy of the 1(TT) state and enabling its substantial participation (K ∼ 1) in equilibrium with the S1 singlet exciton state. This suggests that the magnitude of the exchange interaction in the correlated triplet manifold plays a critical role in dictating the 1(TT) yield in tetracene systems. However, no free triplet state has been found in these dimers.
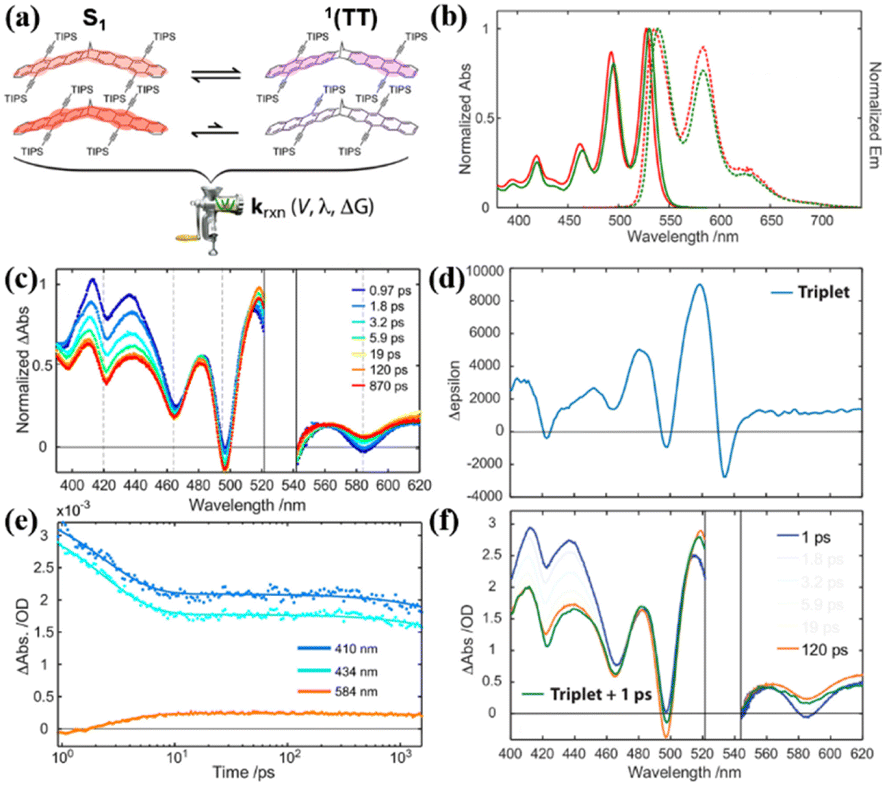 | ||
| Fig. 11 (a) Illustration of electronic coupling strength in the S1 and 1(TT) states for dimers 37 and 38. (b) Normalized steady-state electronic absorption (solid) and emission (dashed) spectra for dimers 37 (red) and 38 (green) in toluene. (c) TA spectra of dimer 38 in toluene following ultrafast excitation at 530 nm. The spectral region around the excitation wavelength is removed due to pump scatter. (d) Triplet Δε spectrum for dimer 38 from sensitization experiment in toluene. (e) Selected single wavelength kinetic traces (data points) taken from the full-spectrum data with applied model fits (lines) retrieved from global analysis. (f) Selected spectral slices for dimer 38 at 1 ps (blue) and 120 ps (red) along with a reconstructed TA spectrum (green), which is comprised of a superposition between the 1 ps TA spectrum and the sensitized triplet Δε spectrum from (d). Reproduced with permission.123 Copyright 2019, the American Chemical Society. | ||
Yamakado et al. also reported a dimer (39) linked via multiple-positions of tetracene in which the two tetracene units are connected by an 8π cyclooctatetraene (COT) ring. This linkage is different from the freedom rotation of a single bond73,103 and the fixed effect of the rigid spacer,106,157,158,160 and it is characterized by flapping motion (Fig. 12a).The electronic coupling between the two tetracene units of this dimer is relatively weak. This dimer exhibits C2V symmetry at its equilibrium geometries (Fig. 12d), whereas the electronic coupling between S1 and 1(TT) is zero.143,154,158,159 Thus, there should be no SF in this dimer. However, efficient iSF is observed in it, as revealed by TA spectra (Fig. 12c), which is attributed to the flexible COT structure. This structure can promote SF by inducing conformational flexibility to cause symmetry-breaking vibrational modes (Fig. 12d). Different from the COT-linked anthracene dimer, the conformational planar dynamics is absent in this dimer because the energy potential no longer gives a minimum in its planar conformation as a consequence of the decreased electronic contribution of the 8π COT ring with the extension of the acene unit.163 This work shows a new platform for the design of iSF materials utilizing the fused COT linkage with conformation flexibility, and again highlights the importance to break the molecular symmetry for activating SF.
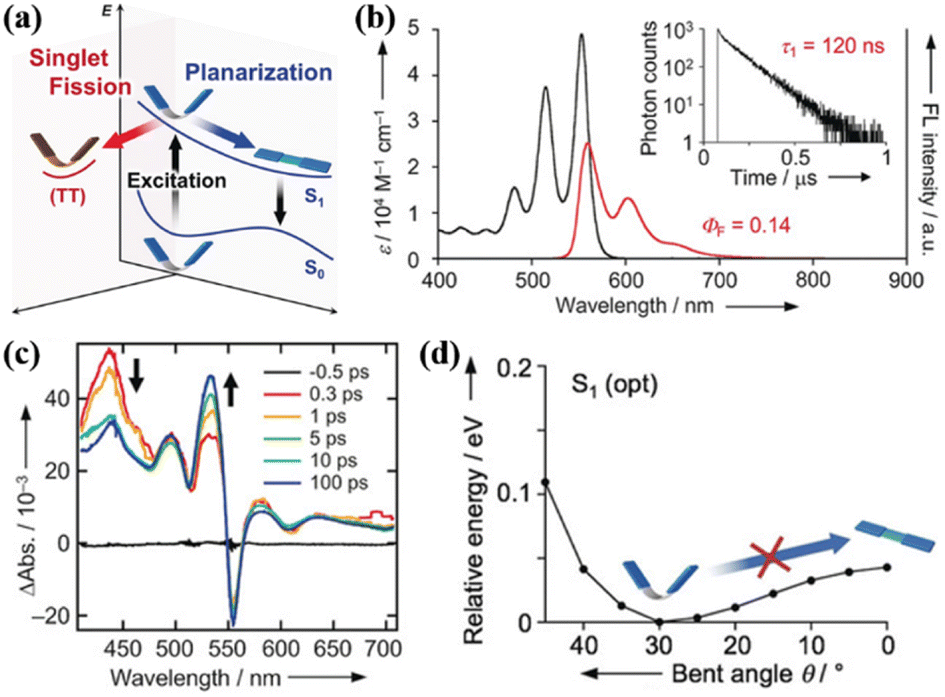 | ||
| Fig. 12 (a) Energy diagram of the COT-fused dimers. Two possible dynamics, conformational planarization in S1 and SF to a correlated triplet pair 1(TT), are indicated. (b) UV/vis absorption (black) and FL (red) spectra of dimer 39. Fluorescence quantum yield (ΦF) is considered in the area of the fluorescence spectra. The fluorescence decay curve of dimer 39 is shown in the inset of (b). (c) TA spectra of dimer 39 in CH2Cl2. (d) S1 energy profiles with respect to conformational planarization of dimer 39. Reproduced with permission.163 Copyright 2018, Wiley-VCH. | ||
Next, we summarize the SF parameters of these dimers in Table 1. Among the 5,5′-linked dimers, dimers 1–4 bridged by para-phenylene or meta-phenylene have weak electronic coupling between the two tetracene units and show a low SF efficiency. When a triple bond is inserted between the tetracene unit and para-phenylene or meta-phenylene (dimers 6, 9 and 10), the electronic coupling is enhanced significantly due to the free rotation of the two tetracene units along the triple bond, which accelerates the SF dramatically because the SF rate is proportional to the square of the effective electronic coupling.164 This indicates that relatively strong electronic coupling is necessary for efficient SF. However, relatively strong electronic coupling will hinder the dissociation of the 1(TT) state and result in fast decay of the 1(TT) state. Furthermore, too strong coupling will lead to the formation of an excimer state, which competes strongly with the occurrence of SF, similar to dimer 5. Compared to 5,5′-linked dimers, 2,2′-linked dimers are more conducive for the occurrence of SF and achieving a high yield of long-lived free triplet states, which can be attributed to the proper electronic coupling and the larger conformational flexibility in 2,2′-linked dimers.75,98,147 In these dimers, the electronic coupling is not only sufficient to ensure fast 1(TT) formation, but also weak enough to guarantee the 1(TT) dissociation into the free triplet state. More importantly, the strong conformational flexibility can also decrease the intertriplet spin–spin exchange interaction (J), and then decelerate the TTA process and accelerate the dissociation of the 1(TT) state into the free triplet state, resulting in a high yield of free triplet states. In comparison with 5,5′-linked and 2,2′-linked dimers, most of the reported multiple-positions linked dimers show no SF, which is due to their orbital symmetry, making the adiabatic coupling from S1 to 1(TT) is zero, and then blocking SF. Only when the symmetry is broken, can SF occur. In this case, the symmetry can be broken by vibration. For example, in dimer 39, the conformationally flexible cyclooctatetraene linkage could break the molecular symmetry to activate SF. This highlights the important role of the vibration on SF. Through a systematic comparison of these dimers, we conclude that 2,2′-linked dimers are more favourable for the occurrence of SF in the tetracene chromophore.
| Connection position | Dimer | τ SF/ps | Φ TT/% | τ TT/ns | Φ T⋯T/T1/% | τ T⋯T/T1/μs | Solvent | Ref. |
|---|---|---|---|---|---|---|---|---|
| 5-Position | 1 | 3.6 × 105 | 2–3 | — | — | — | Toluene | 68 |
| 2 | 2.5 × 105 | 2–3 | — | — | — | Toluene | 68 | |
| 3 | — | — | — | — | — | Toluene | 68 | |
| 4 | 1.23 × 104 | — | — | 21 | 10.9 | Toluene | 132 | |
| 5 | — | 0 | — | — | — | — | 100 and 133 | |
| 6 | 2 | — | 0.5 | — | — | THF | 165 | |
| 1 | — | 5 × 10−3 | 154 | 4 × 10−4 | Film | 100 and 139 | ||
| 7 | — | — | — | — | — | — | 140 | |
| 8 | 1.5 × 104 | 1 | — | — | — | CH2Cl2 | 99 | |
| 9 | 0.1 | — | 1.9 × 10−2 | 94% | 2 × 10−5 | CH2Cl2 | 99 | |
| 10 | 0.4 | — | 4.8 × 10−2 | 70% | 7.6 × 10−5 | CH2Cl2 | 99 | |
| 11 | — | — | — | — | — | — | 141 | |
| 12 | 3–40 | — | — | — | — | — | 118 | |
| 13 | — | ∼10 | — | — | — | — | 142 | |
| 2-Position | 14 | 14.1 | — | — | 175 ± 5 | 294 | PhCN | 75 |
| 15 | 14.3 | — | 33.3 | 67 ± 4 | 222 | THF | 98 | |
| 16 | 90.9 | — | 12.5 | 125 ± 8 | 277.8 | THF | 98 | |
| 453 | 192 | 129 | — | 145 | Toluene | 84 | ||
| 17 | 23.3 | — | 7.7 | 117 ± 8 | 294 | THF | 98 | |
| 7 | 199 | 121 | — | 141 | Toluene | 84 | ||
| 18 | 400 | — | 2.9 | 196 ± 12 | 333.3 | THF | 98 | |
| 19 | 30 | 199 | 140 | — | 136 | Toluene | 84 | |
| 20 | 952 | 185 | 129 | — | 145 | Toluene | 84 | |
| 21 | 591 | 190 | 141 | — | 170 | Toluene | 84 | |
| 22 | 1437 | 178 | 23 | — | 158 | Toluene | 84 | |
| 23 | 55 | 199 | 126 | — | 155 | Toluene | 84 | |
| 24 | 14 | 199 | 157 | — | 133 | Toluene | 84 | |
| 25 | 6.6 × 103 | 126 | 13.5 | 92 | 175.4 | Methylcyclohexane | 147 | |
| 26 | — | 0 | — | — | — | Methylcyclohexane | 147 | |
| 27 | — | — | — | — | — | — | 148 | |
| Multiple-positions | 28 | 7.1 × 104 | 6.3 | — | — | — | — | 158 |
| 29 | — | — | — | — | — | — | 154 | |
| 30 | — | — | — | — | — | — | 154 | |
| 31 | — | — | — | — | — | — | 156 | |
| 32 | — | — | — | — | — | — | 156 | |
| 33 | — | — | — | — | — | — | 156 | |
| 34 | — | — | — | — | — | — | 156 | |
| 35 | — | — | — | — | — | — | 154 and 156 | |
| 36 | — | — | — | — | — | — | 154 and 156 | |
| 37 | 9.1 | <0.1 | — | — | — | Toluene | 157 | |
| 38 | 5 ± 0.5 | 50 ± 8 | — | — | — | Toluene | 123 | |
| 39 | 3 | 180 | — | — | — | CH2Cl2 | 163 |
3.2 Tetracene oligomers with more than two tetracene units
In molecular dimers, the two triplets formed via SF are confined to the two subunits and easily annihilated by each other (TTA) to the ground state rapidly due to the strong electronic coupling.103,106,166 This, together with the strong spin-exchange interaction between the two correlated triplets, may slow down the spin dephasing of the correlated triplet state.167,168 Thus, the separation of the two correlated triplets into free triplets is difficult in molecular dimers. In endothermic SF systems, such as tetracene, the recombination of the two correlated triplets into the S1 state can also proceed. However, the free triplets with long lifetime are more convenient to be harvested in solar energy conversion process. Consequently, achieving the separation of the two correlated triplets to yield free triplets is urgent for the application of SF, especially in endothermic SF systems. In crystalline tetracene and pentacene films,92,169–171 the two correlated triplets can diffuse apart from each other via triplet energy transfer to the adjacent molecule at the S0 state, resulting in the generation of free triplets. Thus, applying this concept to iSF materials may also promote the dissociation of the two correlated triplets into two free triplets.To verify this hypothesis, our group synthesized a series of tetracene oligomers including trimer (40, Fig. 13) and tetramer (41, Fig. 13), with dimer 4 as a comparison.132,172,173 The electronic coupling in all these oligomers is relatively weak, as revealed by the small redshift in their absorption peak compared to that of the corresponding monomer. However, the degree of redshift from the dimer to tetramer gradually became large (Fig. 14a), suggesting that the corresponding electronic coupling gradually becomes stronger. This may be attributed to the increased planarity between/among the tetracene subunits in this series of molecules.132,174 Their fluorescence spectra showed a similar vibrational structure (Fig. 14b), but a gradually decreased fluorescence quantum yield and a gradually accelerated fluorescence decay rate were observed on going from the dimer to tetramer, indicating the presence of an accelerated nonradiative decay pathway in these oligomers (Fig. 14c).175 In the fs-TA spectra, the triplet state signal emerged at ∼490 nm with a decrease in the S1 absorption intensity in these oligomers (Fig. 14e), which originated from iSF. The triplet formation rate gradually became faster from dimer to tetramer, suggesting accelerated iSF (Fig. 14d). More importantly, the triplet state showed a long lifetime (>100 μs) (Fig. 14f), which suggests that the free triplet state is formed in these oligomers. A gradually enhanced free triplet yield was obtained in these oligomers (21%, 96% and 128% for dimer, trimer and tetramer, respectively) (Fig. 14d). This was the highest iSF yield with long-lived triplet state achieved in a tetracene compound in solution at that time, and it is also the only case with a triplet quantum yield exceeding 100%. The high long-lived triplet yield of tetramer 41 makes it a good candidate for the application of SF in solar energy conversion processes.
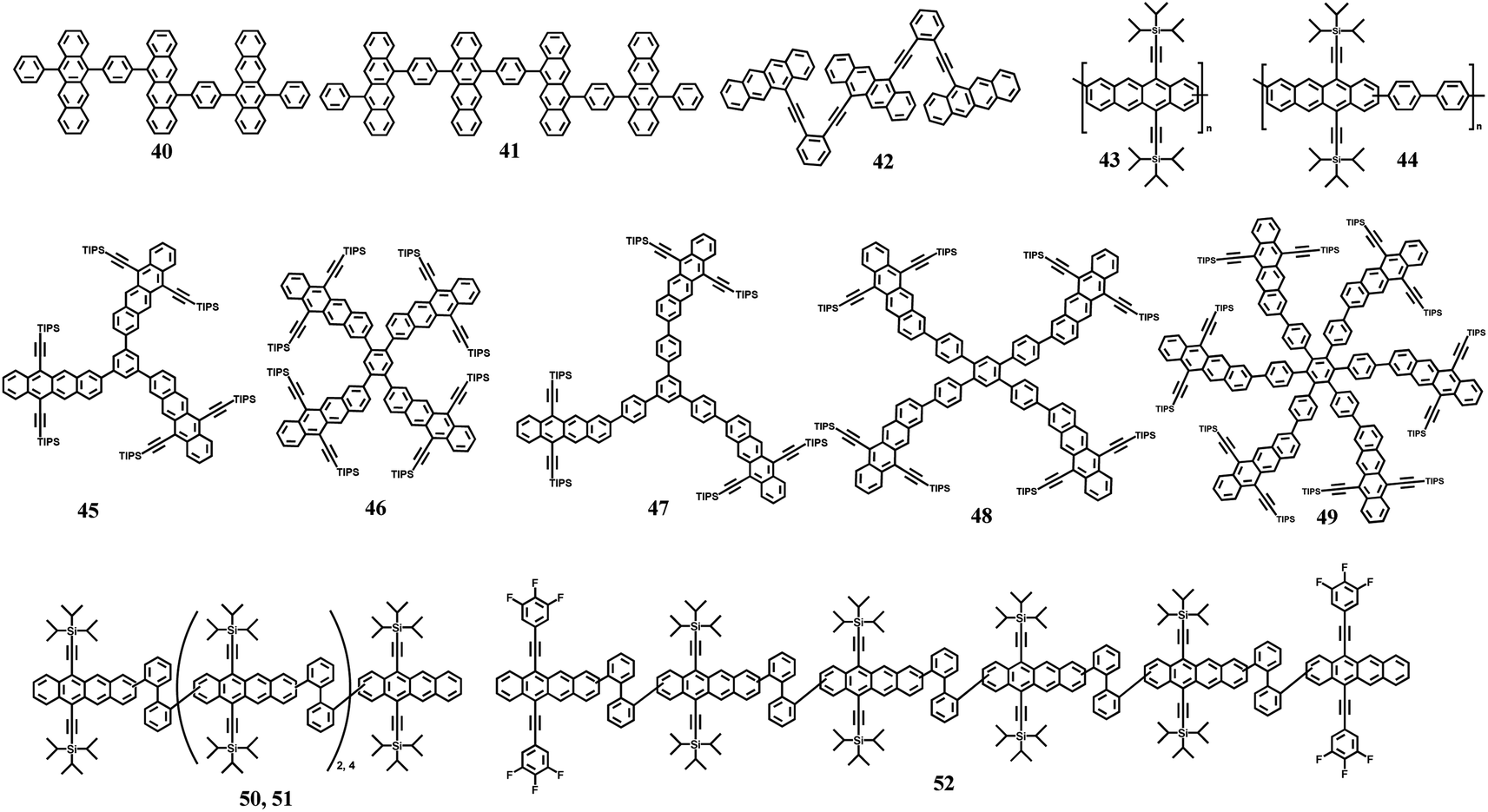 | ||
| Fig. 13 Molecular structures of tetracene oligomers. | ||
 | ||
| Fig. 14 (a) and (b) Absorption (a) and fluorescence (b) spectra of dimer 4, trimer 40 and tetramer 41 and their corresponding monomers. (c) Fluorescence dynamics of trimer 40 and tetramer 41. (d) Triplet yield and SF rate of dimer 4, trimer 40 and tetramer 41. (e) and (f) fs- (e) and ns- (f) TA of tetramer 41. Reproduced with permission.172 Copyright 2018, The Royal Society of Chemistry. | ||
Subsequently, in cooperation with Zhang's group, the iSF mechanism in these oligomers was illustrated clearly using magnetic-field-dependent time-resolved fluorescence and TA spectroscopic technology.173 In these oligomers, the 1(TT) state is formed first, and it then dissociates directly into free triplets in the dimer (Fig. 15a) or free triplets in the trimer and tetramer via a spatially separated 1(TT) state (denoted as 1(T⋯T)) (Fig. 15b). However, the dissociation of the 1(TT) state is very difficult in the dimer due to the limited diffusion space. The presence of the 1(TT) and 1(T⋯T) state in the trimer and tetramer was confirmed by the two-rise stage (∼10 ps and ∼400 ps) in their triplet dynamics (Fig. 15c–h). The fast and slow components are ascribed to the formation of the 1(TT) and 1(T⋯T) state, respectively. The assignment of the slow rise component to the 1(T⋯T) state in the trimer and tetramer was further proven by its suppression with an external magnetic field. That is because the dynamics of the 1(T⋯T) state with a weak exchange interaction is much more sensitive to the Zeeman interaction induced by the external magnetic field (Fig. 16b).168,176–178
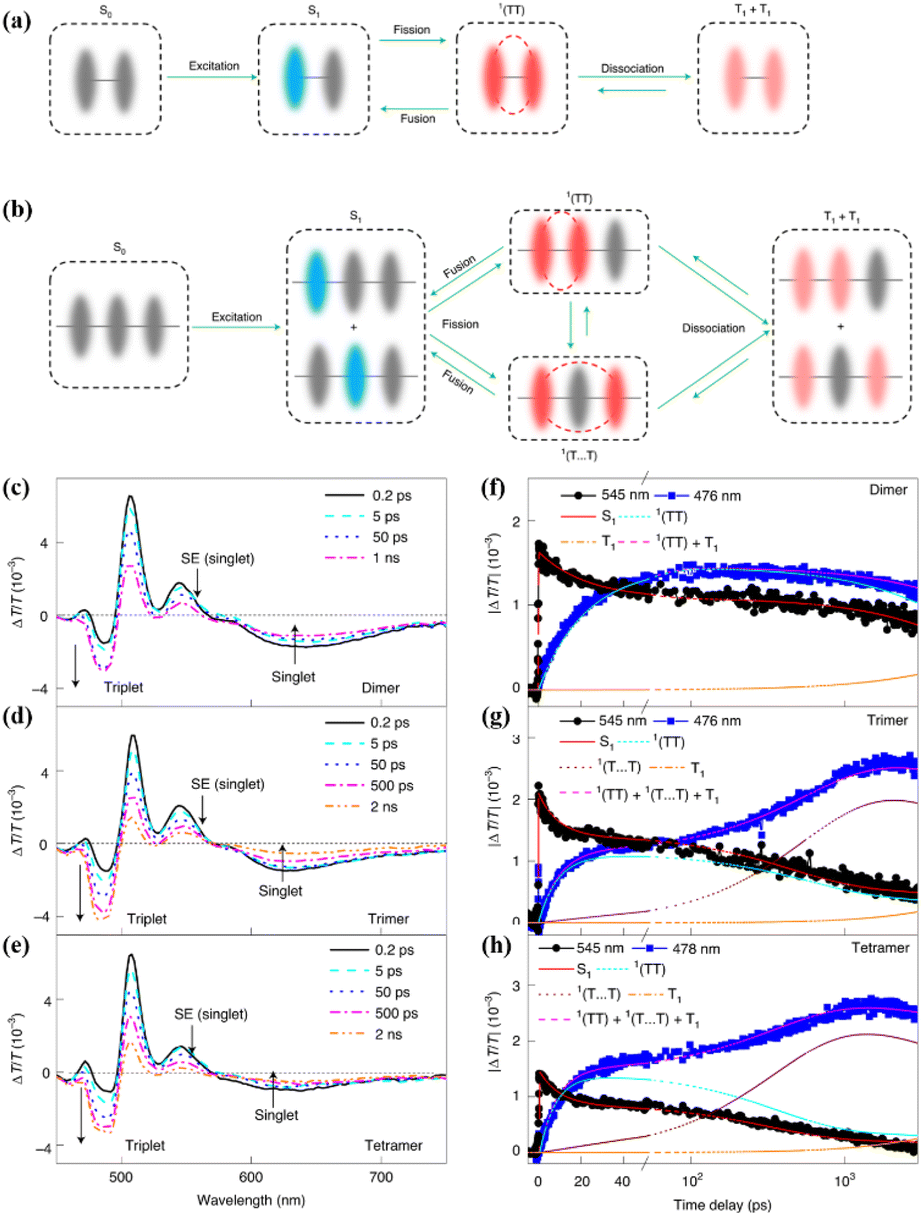 | ||
| Fig. 15 (a) and (b) Scheme of the iSF process in the dimer (a), trimer and larger array systems (b). (c)–(e) fs-TA spectra of dimer (c), trimer (d) and tetramer (e). (f)–(h) Kinetic curves probed at the characteristic wavelengths showing the dynamics of the singlet and triplet states in dimer (f), trimer (g) and tetramer (h). The lines represent the dynamics of the different species (S1, 1(TT), 1(T⋯T), T1 and the sum of the triplet populations) derived with the kinetic model (a) and (b) by a global fitting analysis. Reproduced with permission.173 Copyright 2021, Springer Nature. | ||
 | ||
| Fig. 16 (a) and (b) Dynamics of the singlet and triplet states of dimer (a) and trimer (b) with and without application of an external magnetic field of 0.8 T. (c) TA spectra of these oligomers recorded at a time delay of 100 ns, while applying a magnetic field of different magnitudes. (d) Fluorescence spectra of these oligomers with an applied magnetic field of different magnitudes. Reproduced with permission.173 Copyright 2021, Springer Nature. | ||
Due to the suppressed generation of the 1(T⋯T) state, the free triplet yields of the trimer and tetramer also decreased when applying an external magnetic field (Fig. 16c), confirming the important contribution of the 1(T⋯T) state to free triplet generation. Additionally, the fluorescence intensity of the trimer and tetramer was enhanced when an external magnetic field was applied (Fig. 16d), which is because the suppressed generation of the 1(T⋯T) state makes a certain portion of the singlet population that is originally converted into free triplets through the 1(T⋯T) state recombine through the radiative emission pathway with a longer fluorescence lifetime.
However, the fast component, representing the formation of the 1(TT) state, is nearly not independent of the magnetic field due to its strong intertriplet coupling (Fig. 16b).178 Thus, the triplet dynamics, triplet yield and fluorescence intensity of the dimer are not dependent on the magnetic field (Fig. 16a and d). Different from the report by Korovina et al.,99 rotational motions of the chromophores are important for the dissociation of the 1(TT) state. This result highlights the important role of the 1(T⋯T) state in the generation of the free triplet and provides an alternative route for highly efficient 1(TT) dissociation, representing an important step towards the practical applications of iSF materials because the free triplets can be harvested much more efficiently than the 1(TT) state through interfacial charge transfer.179
In addition, we also prepared a tetracene trimer with a face-to-face stacked structure (42), which showed strong interchromophore electronic coupling. Different from the corresponding dimer (6), the TA spectrum of the triplet obtained from iSF for trimer 42 more closely resembled that of the free triplet obtained from the sensitization experiment. Furthermore, the lifetime of the triplet of trimer 42 is much longer than that of the 1(TT) state obtained in dimer 6, but shorter than that of the free triplet. These results suggest that the 1(T⋯T) state may be formed in trimer 42. However, no long-lived free triplet is formed in this trimer, similar to the above-mentioned trimer 40 and tetramer 41, which may be due to the strong electronic coupling between the two adjacent tetracene units.
Campos' group reported two tetracene polymers linked via the 2-positon of tetracene, where one was bridged directly (43), while the other bridged via a diphenyl group (44).180 The TA experiment revealed that ultrafast iSF proceeded in these two polymers, which had a short lifetime (∼8 ns). No long-lived free triplet was generated in these two polymers. However, in tetracene dimer 18 reported by Hasobe's group,98 with a similar structure to polymer 44, a high yield of long-lived free triplet was achieved. This difference may because the conformational flexibility that can promote the 1(TT) separation is hampered in polymer 44 with long chain character.
Recently, Kim et al. reported a series of tetracene dendrimers (45–49) to mimic complicated SF dynamics in amorphous solids.107 These dendrimers can be classified into two categories, i.e., one type containing 45 and 46, showing relatively fast iSF, and the other including 47–49, which exhibit slow iSF. Notably, iSF proceeded with two time constants for dimer 46, which is attributed to the formation of two 1(TT) states of different boundness. The less-bound 1(TT) state at para-linked tetracene is formed via through-bond interactions with a fast rate, whereas the close bound 1(TT) state at ortho-linked tetracene is formed via through-space interactions with a slow rate. This work demonstrates the effect of structural heterogeneity on SF and highlights the importance of microscopic ordering between SF chromophores to maximize the efficiency of the exciton multiplication process for application in organic photovoltaic devices.
Under the guidance of dimer 14, Hasobe's group further synthesized a series of linear homo-oligomers (tetramer (Tc)4 (50), hexamer (Tc)6 (51)) and hetero-hexamer (TcF3-(Tc)4-TcF3, 52) with a 3,4,5-trifluoropheny substituent on the terminal position of the two tetracenes (Fig. 13).181 TcF3 acts as an energy acceptor, exciton marker and vibronic promotor (Fig. 17a). The systematic comparison of these homo-oligomers and hetero-oligomer enables the effect of oligomerization on the 1(TT) dissociation to be clarified. The TA spectra showed that iSF proceeded in all these compounds (Fig. 17c and d). The free triplet yield of homo-oligomers was enhanced gradually with an increase in the number of number of tetracene units (165%, 173% and 182% for (Tc)2, (Tc)4, and (Tc)6, respectively), which is attributed to the increased entropy change (ΔS) of 1(TT) dissociation with the increase in the number of tetracene units caused by the larger change (ΔΔW) in the number of T1 + T1 with respect to 1(TT), as revealed by the calculation of the thermodynamic parameters (Fig. 17e and f). More importantly, the trapped triplet excitons were observed (ΦTrT = 176%, τTrT = 0.30 ms) after the exciton migration in TcF3-(Tc)4-TcF3 due to the introduction of the energy acceptor (Fig. 17d). Furthermore, the ΔS value increased in TcF3-(Tc)4-TcF3 compared to (Tc)6, which is ascribed to the enhanced ΔΔW caused by the vibronic levels by the rotation of the trifluorophenyl groups at the terminal TcF3. This effect also contributed to the 1(TT) dissociation in TcF3-(Tc)4-TcF3 besides the increase in the number of tetracene units. This work presents a new design strategy for achieving efficient exciton transport processes in iSF oligomers by promoting 1(TT) dissociation and controlling the sequential exciton trapping process.
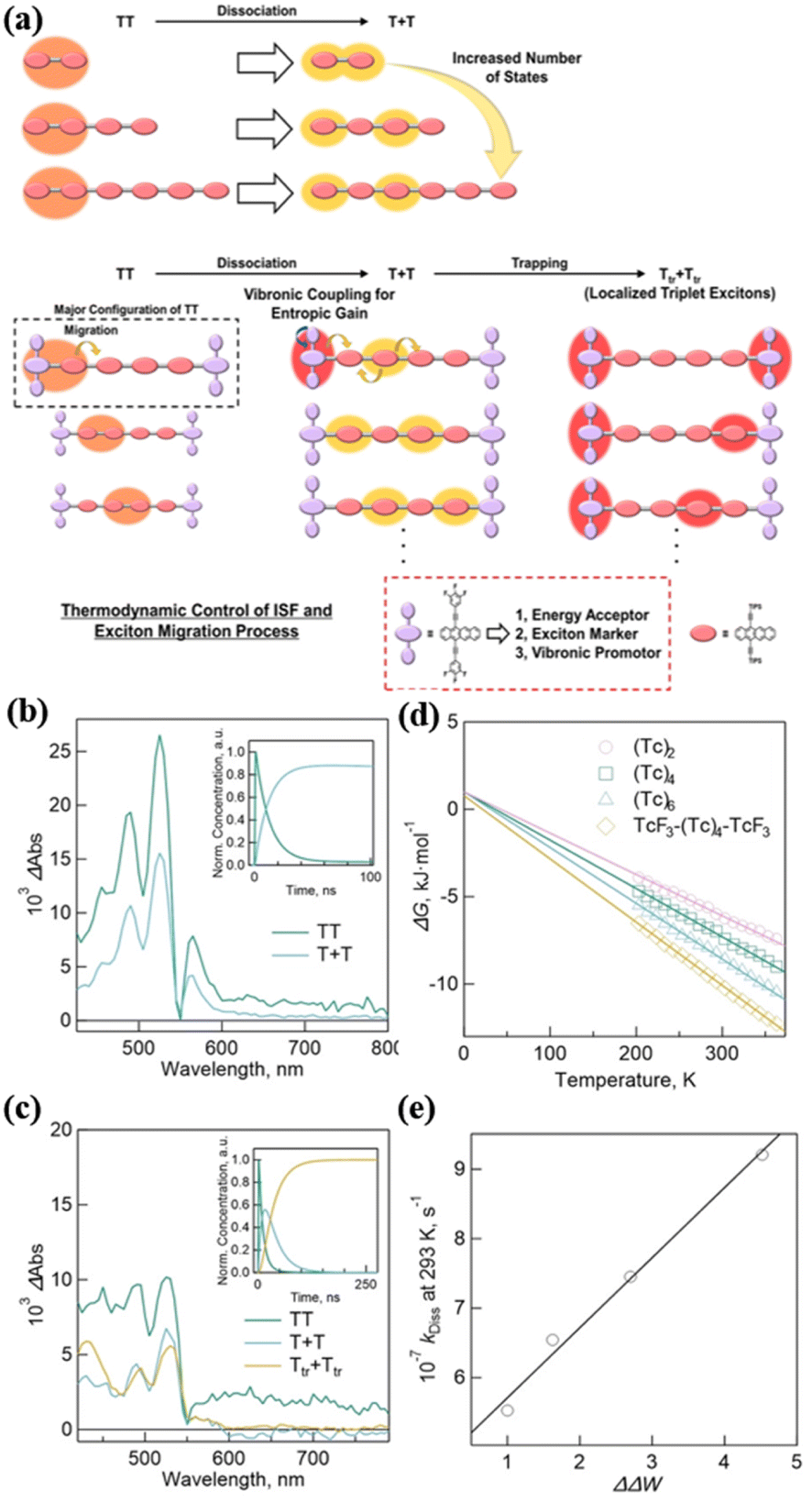 | ||
| Fig. 17 (a) Conceptual schemes for the triplet exciton migration including 1(TT) dissociation and exciton trapping processes in hetero-hexamer. (b) Species-associated spectra (SAS) of (Tc)6. The inset shows decay profiles of 1(TT) (green) and T + T (blue). (c) SAS of TcF3-(Tc)4-TcF3. The inset shows decay profiles of 1(TT) (green), T + T (blue) and Ttr + Ttr (orange). (d) The plot of ΔG vs. temperatures. (e) Plot of kDissvs. ΔΔW. Reproduced with permission.181 Copyright 2023, Wiley. | ||
4 Triplet harvesting in tetracene oligomers
Harvesting the two triplet states obtained from SF efficiently is a precondition for achieving its practical application. To achieve highly efficient extraction of the two triplets, the triplet state should have a long lifetime. Interestingly, dimer 14 shows a high yield (∼175%) of long-lived triplet (0.29 ms).75 With this dimer in hand, Nakamura et al. investigated the triplet electron transfer from dimer 14 to the electron acceptor chloranil.The TA experiment using a mixture of dimer 14 and chloranil demonstrated that the ESA signal of the tetracene cation and the chloranil anion emerges as the triplet signal vanishes (Fig. 18b and c), suggesting the presence of electron transfer from the triplet state of the dimer to chloranil. With an increase in the chloranil concentration, the electron transfer rate gradually became fast (Fig. 18d), confirming that the electron transfer is a diffusion-controlled process.182,183 More importantly, the quantum yield of the electron transfer reached ∼173%, which is comparable to the triplet yield (∼175%). This suggests that quantitative two-electron transfer proceeded from the two triplets of dimer 14 to chloranil. This provides a new perspective for the construction of future solar energy conversion systems with SF.
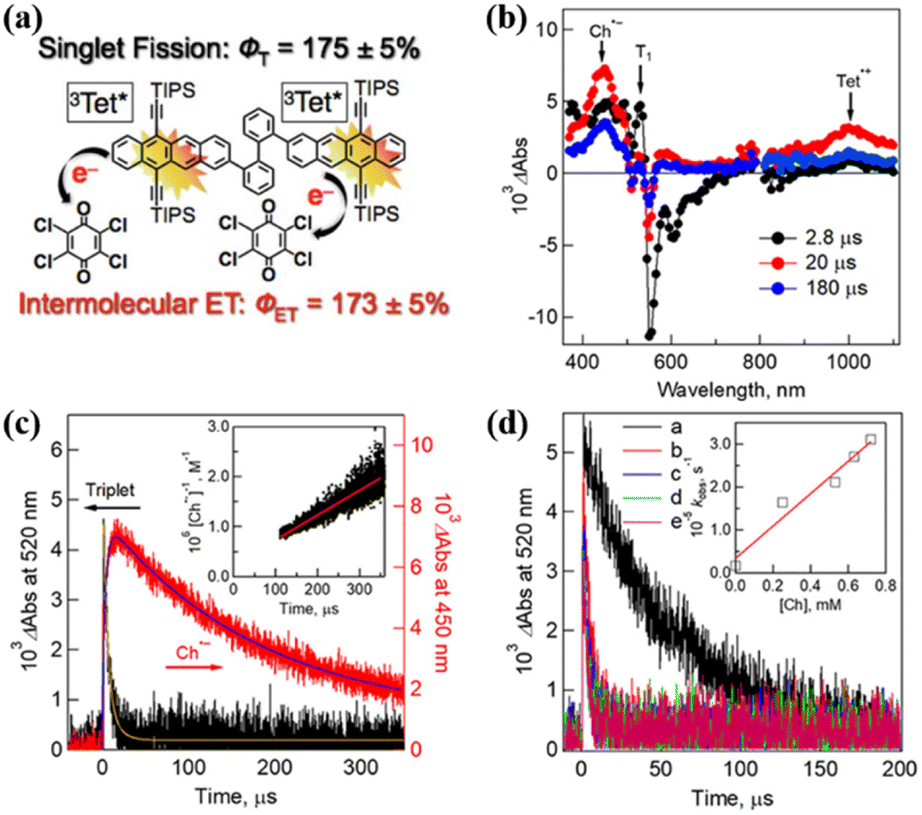 | ||
| Fig. 18 (a) Schematic diagram of intermolecular electron transfer from iSF of tetracene dimer 14 to chloranil. (b) μs-TA spectra of dimer 14 (50 μM) in the presence of 250 μM chloranil in Ar-saturated PhCN after excitation at 532 nm. (b) Corresponding time profiles at 450 and 520 nm. The inset shows the second-order plots. (c) Time profiles of absorption at 520 nm in the presence of different concentrations of chloranil: (a) 0 mM, (b) 0.25 mM, (c) 0.53 mM, (d) 0.63 mM, and (e) 0.72 mM. The inset shows the plot of the pseudo-first-order rate constant (kobs) vs. the concentration of chloranil. Reproduced with permission.75 Copyright 2019, the American Chemical Society. | ||
To harvest two triplet excitons of singlet fission (SF) via two-electron transfer efficiently, revelation of the key factors that influence the two-electron transfer process is necessary. Thus, our group investigated the effect of the intertriplet distance on the two-electron transfer process utilizing our previously prepared tetracene oligomers (dimer 4, trimer 40 and tetramer 41) (electron donor) and TCNQ (electron acceptor) for the first time (Fig. 19).184 In these compounds, the lifetimes of the triplet state are almost identical, but the separation distances between the two triplet states are different. This allowed us to reveal the effect of the separation distance between the two triplet excitons on the electron-transfer process independently. The TA spectra of the mixture of oligomers and TCNQ showed that the ESA signal of the TCNQ anion appears with the decay of the triplet state (Fig. 19c and d), suggesting that the electron transfer occurs from the triplet state of the oligomers to TCNQ. With an increase in the concentration of TCNQ, the electron transfer rate gradually became faster (Fig. 19e), indicating that the electron transfer is a diffusion-controlled process. More importantly, the electron transfer efficiency reached 100% at high TCNQ concentrations, suggesting that a two-electron transfer process occurred. Notably, at the same TCNQ concentration, the electron transfer efficiency from dimer 4 to trimer 40, and then tetramer 41 increased gradually, and the corresponding electron transfer rate increased. This suggests that a larger intertriplet distance in iSF molecules is more favorable for the occurrence of two-electron transfer. This is attributed to the more negative free energy change induced by the smaller Coulomb repulsion energy between the two positive charges generated after the two-electron transfer.185,186 A small intertriplet distance leads to a large Coulomb repulsion energy, which can cause a more positive free energy change and make the second-electron transfer less efficient or even be hindered. This work demonstrates the important role of the Coulomb repulsion energy in the two-electron transfer from the two triplets of iSF molecules for the first time and provides useful guidelines to design iSF molecules that can exhibit efficient two-triplet-electron transfer.
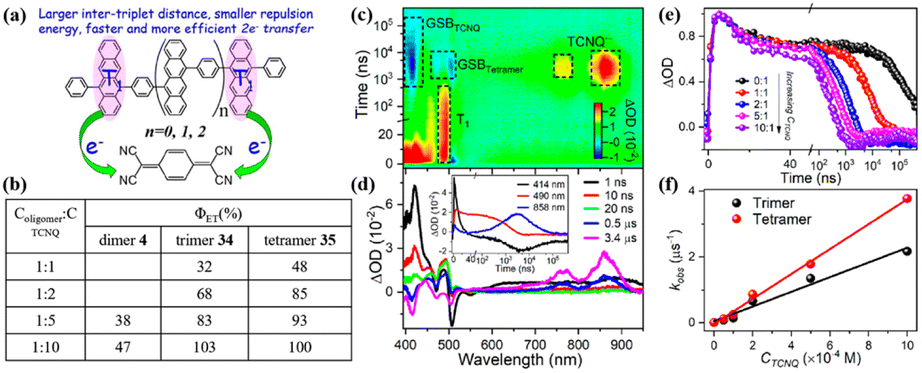 | ||
| Fig. 19 (a) Schematic diagram of intermolecular electron transfer from iSF of dimer 4, trimer 40 and tetramer 41 to TCNQ. (b) Electron transfer efficiency (Φ) from dimer 4, trimer 40 and tetramer 41 to TCNQ. (c) ns-TA absorption spectra of tetramer 41 in the presence of TCNQ in degassed PhCN. (d) Single-wavelength dynamics for a mixture of the tetramer 41 and TCNQ monitored at different wavelengths. (e) Triplet dynamics of the tetramer probed at 490 nm in the presence of different concentrations of TCNQ in degassed PhCN. (f) Plots of the pseudo-first-order rate constant (kobs) vs. concentration of TCNQ. Reproduced with permission.184 Copyright 2022, the American Chemical Society. | ||
Additionally, Compos; group fabricated active layers composed of polymer 43 and a series of acceptor molecules (PC60BM, C60 and PDIF-CN2, Fig. 20a) into organic photovoltaic (OPV) devices.180 OPV devices that primarily harvest triplets from an SF material should show an increased photocurrent at low magnetic fields, followed by a decrease and eventual saturation with an increase in the magnetic field.187,188 The reason for this feature is that the number of triplet–triplet pairs with singlet character increases from three at a low magnetic field. Instead, the number of triplet–triplet pairs with singlet character decreases to two at a high magnetic field. This change increases, and then decreases the SF rate, leading to a positive, and then negative change in photocurrent if the device operates via majority triplet transport as a result of SF.
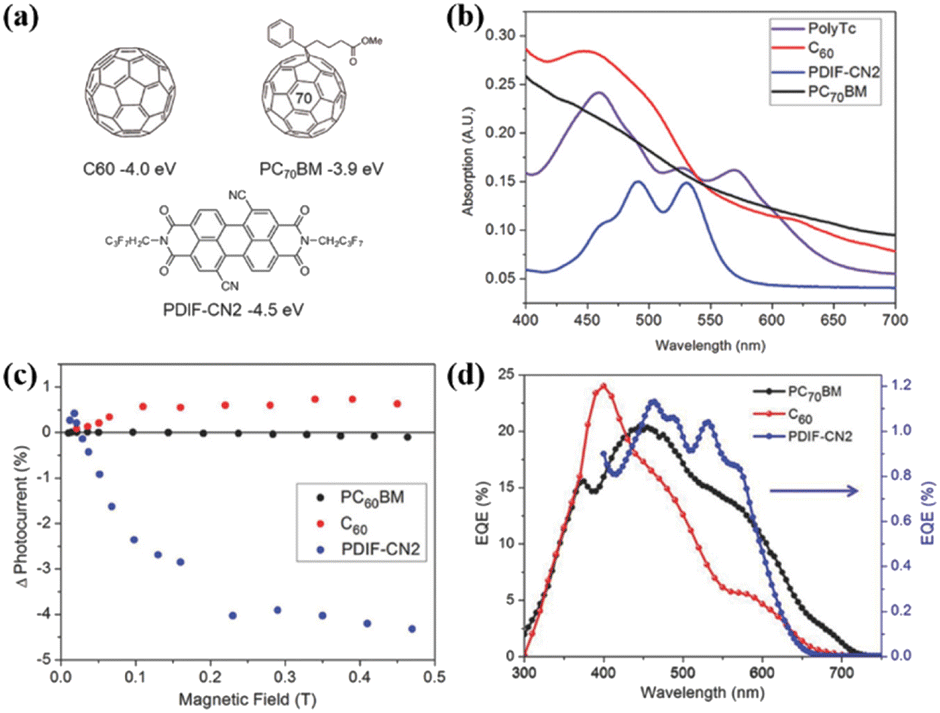 | ||
| Fig. 20 (a) Structures of the acceptors used in this work, with their LUMO levels relative to vacuum. (b) Steady-state absorption profiles of the individual layers of acceptors and polymer 43. (c) Change in photocurrent with an applied magnetic field on polymer OPVs incorporating PC60BM (black) C60 (red) or PDIF-CN2 (blue) as the electron acceptor. (d) EQE characteristics of polymer OPVs incorporating PC70BM (black) C60 (red) or PDIF-CN2 (blue) as the electron acceptor. EQE values for the PDIF-CN2-containing device were scaled by 20 for clarity. Reproduced with permission.180 Copyright 2017, Wiley-VCH. | ||
This characteristic effect of photocurrent enhancement at low magnetic field, and reduction at high magnetic field is observed for the acceptor PDIF-CN2, but not for C60 or PC60BM. Furthermore, the external quantum efficiency (EQE) is enhanced, benefitting from the contribution from polymer 43 (Fig. 20d). This, together with the characteristic behaviour of the photocurrent as function of magnetic field, indicate the extraction of multiple charge carriers generated from iSF in polymer 43 when PDIF-CN2 is used as the acceptor. This is because the much lower LUMO of PDIF-CN2 in comparison to that of fullerenes provides a suitable driving force for the dissociation of the triplets generated.189 This work represents the first example of iSF-generated triplet extraction in devices, exhibiting the potential of iSF materials for use in next-generation devices.
Recently, Guldi's group prepared a photoelectrode composed of carboxylate-anchored pentacene dimer and ZnO/indium-zinc oxide (IZO), and successfully injected the triplet excitons of the pentacene dimer generated via iSF into ZnO/IZO.190 This photoelectrode achieved an electron injection rate of 4.4 × 1011 s−1 and carrier multiplication rate of 130%. Unfortunately, the overall device efficiencies of the above-mentioned two examples are rather moderate. Thus, further optimization of iSF molecules and the device structure is needed for achieving high device efficiencies.
5 Influence factors on endothermic SF systems
A high yield of long-lived triplet states from SF is more favorable for its application in solar energy conversion. Next, we summarize the factors influencing the overall SF dynamics (including the formation and separation of the 1(TT) state) of tetracene oligomers. This will give significant insights for achieving high yield of long-lived free triplet states in endothermic SF systems.5.1 Electronic coupling
Electronic coupling plays a crucial role in the overall SF dynamics of tetracene oligomers. A relatively strong interchromophore electronic coupling is needed for the fast and efficient formation of the 1(TT) state in tetracene derivatives. Weakly coupled tetracene oligomers show slow and inefficient 1(TT) formation. This is consistent with the fact that the SF rate is proportion to the strength of the electronic coupling predicted by the Fermi Golden Rule.191 However, in exothermic SF systems, such as pentacene, efficient formation of the 1(TT) state can be still realized although weak coupling is adopted.86 These studies indicate that this effect is particularly pronounced in endothermic and isothermal systems compared to that in exothermal systems. However, the strong electronic coupling will accelerate the direct deactivation process of the 1(TT) state to the ground state (1(TT) → S0 + S0) and the reverse reaction of SF (TTA), and thus prevent the dissociation of the 1(TT) state into the free triplet state with long lifetime.81,139 Furthermore, too strong electronic coupling will decrease the S1 energy seriously. This will make SF more endothermic, and thus slow down the formation rate of the 1(TT) state. Thus, a balanced electronic coupling strength is necessary for achieving a high yield of the overall SF process. As is known, electronic coupling is dependent on the intermolecular orientation and distance, which can be controlled by various linkers. Notably, it is difficult to achieve efficient overall SF in tetracene oligomers via simple electronic coupling regulation. Thus, other factors should be considered to achieve high efficiency of long-lived free triplet states.5.2 Conformational flexibility
As discussed by Hasobe's and Thompson's group,98,99 conformational flexibility plays an important role in the dissociation of the 1(TT) state to generate free triplet states in tetracene derivatives. The conformational change at the 1(TT) state can decrease the intertriplet spin–spin exchange interaction (J) by reducing the orbital overlap, and then decelerate the TTA process and accelerate the dissociation of the 1(TT) state to the free triplet state, resulting in a high yield of free triplet states. For example, the two tetracene units of compound 18 at the 1(TT) state are nearly planer, which shows strong orbital overlap. However, the conformation at the T1 state is changed to orthogonal with a negligible orbital overlap.48 The reduced orbital overlap induces electronic decoupling between the two triplets, and then leads to the generation of free triplets. This pivotal role has also been highlighted in some other oligomers composed of pentacene and perylene.58,101,105,111,192,193 In perylene oligomers, the torsional disorder results in the formation of free triplet states from the separation of the 1(TT) state.58 Thus, a too rigid structure of tetracene oligomer is not a preferred conformation to achieve a highly efficient overall SF process. Based on the premise of the appropriate intermolecular electronic coupling strength, a more flexible conformation that is determined by the linker may result in a high free triplet yield. Besides, for oligomers with a highly symmetrical structure, their conformation change may break the molecular symmetry to activate SF.43,1635.3 Covalent linker energy-level alignment
The effect of energy-level alignment of the covalent linker on SF dynamics has been illustrated by several groups.68,84,87,99 The closer energy of the frontier molecular orbital of the linker with that of the chromophore, the better mixing of the linker orbitals with that of the chromophores, and thus the stronger resonance coupling between the chromophores. Stronger resonance coupling will result in the faster formation of the 1(TT) state. To promote the dissociation of the 1(TT) state into the T1 state, a larger interchromophore distance will be more favorable. However, a decreased SF rate will be induced simultaneously. Fortunately, the resonance effect can overcome this negative effect. For example, compound 23 showed faster SF than of 16, despite having a longer linker.84 Similarly, this phenomenon was also observed in a series of pentacene dimers.84 Specifically, the selection of a covalent linker with an appropriate energy level and length will be more favorable for achieving a high yield of long-lived triplet states. Thus, the energy-level alignment, length and flexibility of the covalent linker should be considered when choosing the linker to connect the chromophores for achieving efficient SF.5.4 Oligomer size
As discovered by us and Zhang's group,132,172,173 with an increase in the number of repeating unit, the formation rate and yield of the free triplet state increase significantly. This is because the increased number of structural units can provide sufficient statistical space to promote the formation of the spatially separated triplet pair state (1(T⋯T)), which mediates the generation of the free triplet state. This effect is more important for endothermic SF systems because the formation of the 1(T⋯T) state can effectively avoid the recombination of the 1(TT) state to the S1 state, which is a competing process with its separation into free triplet states. This provides an alternative route for the highly efficient dissociation of the 1(TT) state into free triplet states. This effect also contributes to SF in a series of perylene oligomers.58 Additionally, an increase in the number of repeat units in oligomers can result in an increase in entropy, which is beneficial for the formation and separation of the 1(TT) state.58,191,1945.5 Aggregation state behaviour
The ultimate goal for SF is its integration in photovoltaic devices. Considering that the active layer of solar cells is in the solid state, the effect of the aggregation behaviour of SF molecules in the solid state on SF dynamics should be addressed. Many crystal films and nanoparticles of tetracene derivatives with different arrangements show SF. However, the SF rate and efficiency are strongly dependent on the molecular packing. In this case, herringbone packing may be the preferred conformation for SF in the solid state. For example, unsubstituted tetracene, rubrene and 5,12-bis(phenylethynyl) tetracene films, with herringbone accumulation, generally show efficient SF.195–198 Alternatively, a 5,12-diphenyl tetracene film with a coplanar arrangement demonstrated relatively inefficient SF.20 This is consistent with the computation result that SF can occur efficiently in acene when the molecule adopts a herringbone arrangement.199,200 However, very little work has been reported to reveal the effect of the aggregating behavior of iSF molecules on SF in the solid state, especially for tetracene.100,180,200–202 When an iSF molecule is prepared in the solid state, its intermolecular interactions are dominated by through-space coupling besides intramolecular interactions. Thus, iSF and xSF will be both accessed possibly, resulting in their competition. This depends on the relative strength of the intramolecular and intermolecular interactions. For example, Korovina et al. observed ultrafast iSF in amorphous films of dimer 6 with strong intramolecular coupling.100 More importantly, 1(TT) dissociation was also observed due to the entropic gains, which is often an issue in covalent dimers in solution. However, when the intramolecular coupling was weak, the through-space intermolecular coupling led to xSF instead of iSF. This was proven by the study of the terrylene-3,4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11,12-bis(dicarboximide) dimer in the solid state, which exhibited strong xSF.201 This provides an approach to achieve high yield of long-lived free triplet states by combining strong intramolecular coupling and weak intermolecular coupling. Xia's group demonstrated that the behaviour of changing the aggregation state of the iSF material from the solution phase to the film could effectively cut off the transportation channel of the triplet excitons and obtain long-lived triplet excitons without affecting the rapid generation of triplet states.200
:11,12-bis(dicarboximide) dimer in the solid state, which exhibited strong xSF.201 This provides an approach to achieve high yield of long-lived free triplet states by combining strong intramolecular coupling and weak intermolecular coupling. Xia's group demonstrated that the behaviour of changing the aggregation state of the iSF material from the solution phase to the film could effectively cut off the transportation channel of the triplet excitons and obtain long-lived triplet excitons without affecting the rapid generation of triplet states.200
As can be seen, there are many factors that affect the overall SF process including the formation and separation of the 1(TT) state. It is easy to understand that among the factors listed above, electronic coupling plays a decisive role in the SF process, which directly determines whether the material can undergo SF, especially for endothermic and isothermal systems. Furthermore, the electronic coupling also influences the dissociation of the 1(TT) state. A moderate electronic coupling strength can ensure that both the formation and separation of the 1(TT) state proceed simultaneously. The aggregation behaviour of iSF molecules in the solid state also plays an important role in the overall SF process by controlling the intramolecular and intermolecular interactions. In the case of covalent linker energy-level alignment, it will accelerate the formation of the 1(TT) state by increasing the resonance interaction with the chromophore. Instead, the conformational flexibility and oligomer size mainly influence the separation process of the 1(TT) state. Increasing the conformational flexibility and oligomer size can promote the dissociation of the 1(TT) state by suppressing the TTA process and the reverse process of SF. Sometimes, different factors show the opposite effect. Consequently, it is difficult to make an absolute definition on how to get a high yield of long-lived free triplet excitons. However, through a systematic comparison of the above-mentioned tetracene oligomers, we can get some insights into the design of efficient SF materials based on endothermic chromophores. A structure with an appropriate electronic coupling strength and large conformational flexibility is more favorable for the generation of free triplet excitons. Additionally, an increase in the oligomer size based on the premise of proper electronic coupling can also promote the dissociation of the 1(TT) state into free triplet states.
6 Conclusions and outlook
In this review, initially, we described the progress in iSF of tetracene oligomers, especially the relationship between their molecular structures and SF properties. Then, the factors that affect the overall SF process in tetracene oligomers were summarized, including electronic coupling, conformational flexibility, covalent linker energy, oligomer size and aggregation behaviour. The results showed that a molecular structure with appropriate electronic coupling strength and large conformational flexibility may be a good choice for achieving a high yield of long-lived free triplet states to construct iSF materials based on endothermic SF chromophores. Additionally, based on the premise of an appropriate electronic coupling strength, molecular oligomers with three or more structural units will promote the dissociation of the 1(TT) state into free triplet states.Many breakthroughs have been made in SF field in recent years. Several new series of SF materials have been developed, and their related SF mechanism has been established. However, several challenges still exist, which should be addressed in the future application of SF in solar conversion processes. Firstly, the number of SF materials, with fast rate, high triplet state energy (1.2–1.3 eV), high yield (>100%) of long-lived free triplet states and good light/heat stability, is still small. This will limit the application of SF to some extent. A fast SF rate can avoid extra competing processes, such as energy and charge transfer occurring from the singlet state at molecule/semiconductor interfaces in the device. The high triplet state energy is more feasible for tandem with solar cells (such as Si). The long-lived triplet state guarantees the efficient harvesting of two triplet states in solar conversion processes. Notably, a high yield of long-lived triplet states is difficult to be achieved. This is because it needs highly efficient 1(TT) formation and separation simultaneously. Generally, efficient 1(TT) formation needs strong electronic coupling, especially for endothermic and isothermic systems, but strong electronic coupling suppresses the dissociation of the 1(TT) state into long-lived free triplet states. Therefore, achieving a high yield of long-lived triplet states by controlling the electronic coupling strength and other factors listed above is the main difficulty before the application of SF. Secondly, how to efficiently harvest the two triplet states of SF is another difficulty. Triplet electron transfer and triplet energy transfer have been demonstrated to be useful methods to harvest the two triplet states with quantum dots and metal oxides as well as organic molecules as energy or electron acceptors,4,75,189,190,203–212 but there are few examples with high efficiency. Furthermore, the real extraction mechanism of the electron transfer or the energy transfer is controversial. Additionally, some photovoltaic devices incorporating SF showed that several competitive routes exist in the extraction of the two triplets, such as energy/electron transfer from the singlet state, TTA and the direct decay of the 1(TT) state into the ground state.93,187,213–215 More importantly, the exact working mechanism of SF-based devices remains ambiguous. Thus, the resolution of all the above-mentioned difficulties is urgent to achieve the practical application of SF.
Endothermic iSF systems have some advantages for application in solar energy conversion process due to the lower energy losses in these SF systems. Although highly efficient iSF has been achieved in tetracene oligomers, tetracene derivatives are less stable toward to light and oxygen, which limits their practical application. Therefore, it is urgent to achieve highly efficient iSF in some molecular oligomers constructed by stable endothermic chromophores. Of course, improving the stability of tetracene is also a good choice. In this case, several methods can be used to improve the stability of the tetracene. Firstly, the introduction of some specific functional groups in tetracene has a positive effect on its oxidative stability. For example, the ethynyl group can improve the oxidation stability of tetracene.216 It also avoids the thermal decomposition of the resulting endoperoxides (EPOs). Moreover, the free radical stabilization of the intermediate produced by the photooxidation of tetracene is conducive to C–O cleavage, and thus it can promote the re-conversion of EPOs to the parent tetracene. Secondly, the introduction of heteroatoms is another method to enhance the stability of tetracene by affecting its electronic and optical properties.217–221 For example, the introduction of nitrogen and fluorine (or chlorine) atoms as electron-withdrawing groups can reduce the energy of the frontier molecular orbital, which is expected to improve the stability of tetracene. With an increase in the number of heteroatoms, this effect is more obvious. Thirdly, the introduction of heterocycles (such as thiophene and pyridine) in the tetracene framework can also increase its stability.218,222 The stability of the oligomers prepared with the modified tetracene will be enhanced significantly. Additionally, to maximize the efficiency of solar cells, it is necessary to broaden the absorption of tetracene in the red or NIR region. However, although the polymerization of tetracenes into polymers is a choice, their solubility and processability will decrease remarkably due to the increase in their molecular rigidity. In this case, the introduction of solubilizing groups (such as long branched aliphatic chains) in the tetracene skeleton can improve the polymer solubility efficiently. This method has been widely applied in the design of polymer photovoltaic materials.223,224 Furthermore, the introduction of conjugated groups (such as alkynyl group and cyano group) in the tetracene parent can redshift its absorption significantly. More importantly, these groups can also increase the solubility of tetracene to some extent. Thus, polymerizing tetracenes with redshifted absorption into polymers may be another effective method.
Author contributions
T. Wang and H. Liu wrote the manuscript. X. Wang, L. Tang, J. Zhou, X. Song and L. Lv, helped search related literature. W. Chen helped revise the manuscript. H. Liu and X. Li conceived the idea, designed the structure of the review, revised the draft of the manuscript and supervised the work. X. Li and Y. Chen provided funding support.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Financial support from Key Program of National Natural Science Foundation of China (22133006), the National Natural Science Foundation of China (21703287), the Natural Science Foundation of Shandong Province (ZR2022MB065, ZR2017MB006 and ZR2017BB027), and the Fundamental Research Funds for the Central Universities (19CX05002A) are acknowledged. X. L. thanks Taishan Scholar program of Shandong Province (ts201712019) and Yankuang Group 2019 Science and Technology Program (YKKJ2019AJ05JG-R60) for financial support.Notes and references
- M. C. Hanna and A. J. Nozik, J. Appl. Phys., 2006, 100, 074510 CrossRef.
- M. Einzinger, T. Wu, J. F. Kompalla, H. L. Smith, C. F. Perkinson, L. Nienhaus, S. Wieghold, D. N. Congreve, A. Kahn, M. G. Bawendi and M. A. Baldo, Nature, 2019, 571, 90–94 CrossRef CAS PubMed.
- A. R. Bowman, S. D. Stranks and B. Monserrat, Chem. Mater., 2022, 34, 4865–4875 CrossRef CAS PubMed.
- T. Wang, B.-Y. Zhang and H.-L. Zhang, Macromol. Rapid Commun., 2022, 43, 2200326 CrossRef CAS PubMed.
- W. Ni, Y. Wu, L. Sun and G. G. Gurzadyan, ACS Appl. Nano Mater., 2022, 5, 801–809 CrossRef CAS.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- R. Nagata, H. Nakanotani, W. J. Potscavage Jr and C. Adachi, Adv. Mater., 2018, 30, 1801484 CrossRef PubMed.
- X. Qiao and D. Ma, Mater. Sci. Eng., R, 2020, 139, 100519 CrossRef.
- J. Lee, P. Jadhav and M. A. Baldo, Appl. Phys. Lett., 2009, 95, 033301 CrossRef.
- D. T. W. Toolan, M. P. Weir, S. Dowland, J. F. Winkel, J. R. Willmott, Z. Zhang, V. Gray, J. Xiao, A. J. Petty, J. E. Anthony, N. C. Greenham, R. H. Friend, A. Rao, R. A. L. Jones and A. J. Ryan, J. Mater. Chem. C, 2022, 10, 11192–11198 RSC.
- Y. Liu, C. Zhang, R. Wang, B. Zhang, Z. Tan, X. Wang and M. Xiao, Angew. Chem., Int. Ed., 2015, 54, 6222–6226 CrossRef CAS PubMed.
- M. Zhao, K. Liu, Y.-D. Zhang, Q. Wang, Z.-G. Li, Y.-L. Song and H.-L. Zhang, Mater. Horiz., 2015, 2, 619–624 RSC.
- K. E. Smyser and J. D. Eaves, Sci. Rep., 2020, 10, 18480 CrossRef CAS PubMed.
- S. Singh, W. Jones, W. Siebrand, B. Stoicheff and W. Schneider, J. Chem. Phys., 1965, 42, 330–342 CrossRef CAS.
- N. Geacintov, M. Pope and F. Vogel, Phys. Rev. Lett., 1969, 22, 593–596 CrossRef CAS.
- R. E. Merrifield, P. Avakian and R. P. Groff, Chem. Phys. Lett., 1969, 3, 386–388 CrossRef CAS.
- C. E. Swenberg and W. T. Stacy, Chem. Phys. Lett., 1968, 2, 327–328 CrossRef CAS.
- Y. J. Bae, G. Kang, C. D. Malliakas, J. N. Nelson, J. Zhou, R. M. Young, Y.-L. Wu, R. P. Van Duyne, G. C. Schatz and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 15140–15144 CrossRef CAS PubMed.
- S. T. Roberts, R. E. McAnally, J. N. Mastron, D. H. Webber, M. T. Whited, R. L. Brutchey, M. E. Thompson and S. E. Bradforth, J. Am. Chem. Soc., 2012, 134, 6388–6400 CrossRef CAS PubMed.
- G. Mayonado, K. T. Vogt, J. D. B. Van Schenck, L. Zhu, G. Fregoso, J. Anthony, O. Ostroverkhova and M. W. Graham, J. Phys. Chem. C, 2022, 126, 4433–4445 CrossRef CAS.
- P. J. Budden, L. R. Weiss, M. Müller, N. A. Panjwani, S. Dowland, J. R. Allardice, M. Ganschow, J. Freudenberg, J. Behrends, U. H. F. Bunz and R. H. Friend, Nat. Commun., 2021, 12, 1527 CrossRef CAS PubMed.
- S. Paul and V. Karunakaran, J. Phys. Chem. B, 2022, 126, 1054–1062 CrossRef CAS PubMed.
- J. Zirzlmeier, D. Lehnherr, P. B. Coto, E. T. Chernick, R. Casillas, B. S. Basel, M. Thoss, R. R. Tykwinski and D. M. Guldi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5325–5330 CrossRef CAS PubMed.
- E. Busby, T. C. Berkelbach, B. Kumar, A. Chernikov, Y. Zhong, H. Hlaing, X. Y. Zhu, T. F. Heinz, M. S. Hybertsen, M. Y. Sfeir, D. R. Reichman, C. Nuckolls and O. Yaffe, J. Am. Chem. Soc., 2014, 136, 10654–10660 CrossRef CAS PubMed.
- G. B. Piland, J. J. Burdett, R. J. Dillon and C. J. Bardeen, J. Phys. Chem. Lett., 2014, 5, 2312–2319 CrossRef CAS PubMed.
- Y. Qian, Z.-C. Huang-Fu, T. Zhang, X. Li, A. R. Harutyunyan, G. Chen, H. Chen and Y. Rao, J. Phys. Chem. C, 2022, 126, 8377–8383 CrossRef CAS.
- M. J. Llansola-Portoles, K. Redeckas, S. Streckaite, C. Ilioaia, A. A. Pascal, A. Telfer, M. Vengris, L. Valkunas and B. Robert, Phys. Chem. Chem. Phys., 2018, 20, 8640–8646 RSC.
- Y. Huang, I. A. Buyanova, C. Phansa, M. E. Sandoval-Salinas, D. Casanova, W. K. Myers, N. C. Greenham, A. Rao, W. M. Chen and Y. Puttisong, Cell Rep. Phys. Sci., 2021, 2, 100339 CrossRef CAS.
- R. Pandya, Q. Gu, A. Cheminal, R. Y. S. Chen, E. P. Booker, R. Soucek, M. Schott, L. Legrand, F. Mathevet, N. C. Greenham, T. Barisien, A. J. Musser, A. W. Chin and A. Rao, Chem, 2020, 6, 2826–2851 CAS.
- K. J. Fallon, N. Sawhney, D. T. W. Toolan, A. Sharma, W. Zeng, S. Montanaro, A. Leventis, S. Dowland, O. Millington, D. Congrave, A. Bond, R. Friend, A. Rao and H. Bronstein, J. Am. Chem. Soc., 2022, 144, 23516–23521 CrossRef CAS PubMed.
- O. Millington, S. Montanaro, A. Leventis, A. Sharma, S. A. Dowland, N. Sawhney, K. J. Fallon, W. Zeng, D. G. Congrave, A. J. Musser, A. Rao and H. Bronstein, J. Am. Chem. Soc., 2023, 145, 2499–2510 CrossRef CAS PubMed.
- D. Manawadu, T. N. Georges and W. Barford, J. Phys. Chem. A, 2023, 127, 1342–1352 CrossRef CAS PubMed.
- Y. Sonoda, R. Katoh, N. Tohnai, T. Yago and M. Wakasa, J. Phys. Chem. C, 2022, 126, 8742–8751 CrossRef CAS.
- E. A. Buchanan and J. Michl, Photochem. Photobiol. Sci., 2019, 18, 2112–2124 CrossRef CAS PubMed.
- C. M. Mauck, P. E. Hartnett, E. A. Margulies, L. Ma, C. E. Miller, G. C. Schatz, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2016, 138, 11749–11761 CrossRef CAS PubMed.
- L. Wang, T.-S. Zhang, L. Fu, S. Xie, Y. Wu, G. Cui, W.-H. Fang, J. Yao and H. Fu, J. Am. Chem. Soc., 2021, 143, 5691–5697 CrossRef CAS PubMed.
- L. Wang, W. Cai, J. Sun, Y. Wu, B. Zhang, X. Tian, S. Guo, W. Liang, H. Fu and J. Yao, J. Phys. Chem. Lett., 2021, 12, 12276–12282 CrossRef CAS PubMed.
- N. Maity, W. Kim, N. A. Panjwani, A. Kundu, K. Majumder, P. Kasetty, D. Mishra, R. Bittl, J. Nagesh, J. Dasgupta, A. J. Musser and S. Patil, Nat. Commun., 2022, 13, 5244 CrossRef CAS PubMed.
- L. Wang, W. Jiang, S. Guo, S. Wang, M. Zhang, Z. Liu, G. Wang, Y. Miao, L. Yan, J.-Y. Shao, Y.-W. Zhong, Z. Liu, D. Zhang, H. Fu and J. Yao, Chem. Sci., 2022, 13, 13907–13913 RSC.
- J. Zhou, H. Liu, W. Wang, T. Li, Z. Li, Z. Liu, Y. Chen, Y. Dong and X. Li, J. Photochem. Photobiol., A, 2023, 438, 114473 CrossRef CAS.
- I. Papadopoulos, D. Gutiérrez-Moreno, Y. Bo, R. Casillas, P. M. Greißel, T. Clark, F. Fernández-Lázaro and D. M. Guldi, Nanoscale, 2022, 14, 5194–5203 RSC.
- B. Carlotti, I. K. Madu, H. Kim, Z. Cai, H. Jiang, A. K. Muthike, L. Yu, P. M. Zimmerman and T. Goodson, Chem. Sci., 2020, 11, 8757–8770 RSC.
- Y. Hong, M. Rudolf, M. Kim, J. Kim, T. Schembri, A.-M. Krause, K. Shoyama, D. Bialas, M. I. S. Röhr, T. Joo, H. Kim, D. Kim and F. Würthner, Nat. Commun., 2022, 13, 4488 CrossRef CAS PubMed.
- Y. Hong, J. Kim, W. Kim, C. Kaufmann, H. Kim, F. Würthner and D. Kim, J. Am. Chem. Soc., 2020, 142, 7845–7857 CrossRef CAS PubMed.
- F. S. Conrad-Burton, T. Liu, F. Geyer, R. Costantini, A. P. Schlaus, M. S. Spencer, J. Wang, R. H. Sánchez, B. Zhang, Q. Xu, M. L. Steigerwald, S. Xiao, H. Li, C. P. Nuckolls and X. Zhu, J. Am. Chem. Soc., 2019, 141, 13143–13147 CrossRef CAS PubMed.
- L. Xue, X. Song, Y. Feng, S. Cheng, G. Lu and Y. Bu, J. Am. Chem. Soc., 2020, 142, 17469–17479 CrossRef CAS PubMed.
- I. García-Moreno, V. Postils, E. Rebollar, M. J. Ortiz, A. R. Agarrabeitia and D. Casanova, Phys. Chem. Chem. Phys., 2022, 24, 5929–5938 RSC.
- L. Mencaroni, B. Carlotti, F. Elisei, A. Marrocchi and A. Spalletti, Chem. Sci., 2022, 13, 2071–2078 RSC.
- Y. Zhou, W. Ni, L. Ma, L. Sun, J. Zhao and G. G. Gurzadyan, J. Phys. Chem. C, 2022, 126, 17212–17222 CrossRef CAS.
- W. Zeng, O. El Bakouri, D. W. Szczepanik, H. Bronstein and H. Ottosson, Chem. Sci., 2021, 12, 6159–6171 RSC.
- X. Fei, S. Zhang, D. Zhai, Z. Wang, J.-L. Lin, Q. Xiao, C.-L. Sun, W. Deng, C. Zhang, W. Hu and H.-L. Zhang, Chem. Sci., 2022, 13, 9914–9920 RSC.
- K. J. Fallon and H. Bronstein, Acc. Chem. Res., 2021, 54, 182–193 CrossRef CAS PubMed.
- Q. Sun, Y. Wu, Y. Cui, C. Gao, Q. Ou, D. Hu, L. Wang, Y. Wang, H. Dong, J. Zhao, C. Zhang, Z. Shuai, H. Fu and Q. Peng, Mater. Horiz., 2022, 9, 2518–2523 RSC.
- L. Wang, L. Lin, J. Yang, Y. Wu, H. Wang, J. Zhu, J. Yao and H. Fu, J. Am. Chem. Soc., 2020, 142, 10235–10239 CrossRef CAS PubMed.
- S. Sun, F. S. Conrad-Burton, Y. Liu, F. Ng, M. Steigerwald, X. Zhu and C. Nuckolls, J. Phys. Chem. A, 2022, 126, 7559–7565 CrossRef CAS PubMed.
- W. Ni, L. Sun and G. G. Gurzadyan, Sci. Rep., 2021, 11, 5220 CrossRef CAS PubMed.
- N. V. Korovina, C. H. Chang and J. C. Johnson, Nat. Chem., 2020, 12, 391–398 CrossRef CAS PubMed.
- L. Wang, S. Bai, Y. Wu, Y. Liu, J. Yao and H. Fu, Angew. Chem., Int. Ed., 2020, 59, 2003–2007 CrossRef CAS PubMed.
- Y. Wu, Y. Wang, J. Chen, G. Zhang, J. Yao, D. Zhang and H. Fu, Angew. Chem., Int. Ed., 2017, 56, 9400–9404 CrossRef CAS PubMed.
- J. Choi, S. Kim, M. Ahn, J. Kim, D. W. Cho, D. Kim, S. Eom, D. Im, Y. Kim, S. H. Kim, K.-R. Wee and H. Ihee, Commun. Chem., 2023, 6, 16 CrossRef PubMed.
- K. C. Krishnapriya, A. J. Musser and S. Patil, ACS Energy Lett., 2019, 4, 192–202 CrossRef CAS.
- M. B. Smith and J. Michl, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS PubMed.
- T. Ullrich, D. Munz and D. M. Guldi, Chem. Soc. Rev., 2021, 50, 3485–3518 RSC.
- A. J. Carrod, V. Gray and K. Börjesson, Energy Environ. Sci., 2022, 15, 4982–5016 RSC.
- B. S. Basel, J. Zirzlmeier, C. Hetzer, S. R. Reddy, B. T. Phelan, M. D. Krzyaniak, M. K. Volland, P. B. Coto, R. M. Young, T. Clark, M. Thoss, R. R. Tykwinski, M. R. Wasielewski and D. M. Gulditla, Chem, 2018, 4, 1092–1111 CAS.
- H. Sakai, R. Inaya, H. Nagashima, S. Nakamura, Y. Kobori, N. V. Tkachenko and T. Hasobe, J. Phys. Chem. Lett., 2018, 9, 3354–3360 CrossRef CAS PubMed.
- A. M. Mueller, Y. S. Avlasevich, W. W. Schoeller, K. Muellen and C. J. Bardeen, J. Am. Chem. Soc., 2007, 129, 14240–14250 CrossRef CAS PubMed.
- E. A. Margulies, C. E. Miller, Y. Wu, L. Ma, G. C. Schatz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2016, 8, 1120–1125 CrossRef CAS PubMed.
- E. Busby, J. Xia, Q. Wu, J. Z. Low, R. Song, J. R. Miller, X. Y. Zhu, L. M. Campos and M. Y. Sfeir, Nat. Mater., 2015, 14, 426–433 CrossRef CAS PubMed.
- J. Xia, S. N. Sanders, W. Cheng, J. Z. Low, J. Liu, L. M. Campos and T. Sun, Adv. Mater., 2017, 29, 1601652 CrossRef PubMed.
- J. Z. Low, S. N. Sanders and L. M. Campos, Chem. Mater., 2015, 27, 5453–5463 CrossRef CAS.
- E. G. Fuemmeler, S. N. Sanders, A. B. Pun, E. Kumarasamy, T. Zeng, K. Miyata, M. L. Steigerwald, X. Y. Zhu, M. Y. Sfeir, L. M. Campos and N. Ananth, ACS Cent. Sci., 2016, 2, 316–324 CrossRef CAS PubMed.
- J. Hu, K. Xu, L. Shen, Q. Wu, G. He, J.-Y. Wang, J. Pei, J. Xia and M. Y. Sfeir, Nat. Commun., 2018, 9, 2999 CrossRef PubMed.
- S. Nakamura, H. Sakai, H. Nagashima, Y. Kobori, N. V. Tkachenko and T. Hasobe, ACS Energy Lett., 2019, 4, 26–31 CrossRef CAS.
- R. Montero, V. Martínez-Martínez, A. Longarte, N. Epelde-Elezcano, E. Palao, I. Lamas, H. Manzano, A. R. Agarrabeitia, I. López Arbeloa, M. J. Ortiz and I. Garcia-Moreno, J. Phys. Chem. Lett., 2018, 9, 641–646 CrossRef CAS PubMed.
- D. Bansal, A. Kundu, V. P. Singh, A. K. Pal, A. Datta, J. Dasgupta and P. Mukhopadhyay, Chem. Sci., 2022, 13, 11506–11512 RSC.
- Y. Duan, G. Zhang, X. Liu, F. Shi, T. Wang, H. Yan, H. Xu and L. Zhang, J. Org. Chem., 2022, 87, 8841–8848 CrossRef CAS PubMed.
- Y. J. Bae, D. Shimizu, J. D. Schultz, G. Kang, J. Zhou, G. C. Schatz, A. Osuka and M. R. Wasielewski, J. Phys. Chem. A, 2020, 124, 8478–8487 CrossRef CAS PubMed.
- N. V. Korovina, N. F. Pompetti and J. C. Johnson, J. Chem. Phys., 2020, 152, 040904 CrossRef CAS PubMed.
- T. Hasobe, S. Nakamura, N. V. Tkachenko and Y. Kobori, ACS Energy Lett., 2022, 7, 390–400 CrossRef CAS.
- R. S. Mattos, I. Burghardt, A. J. A. Aquino, T. M. Cardozo and H. Lischka, J. Am. Chem. Soc., 2022, 144, 23492–23504 CrossRef CAS PubMed.
- L. M. Yablon, S. N. Sanders, K. Miyazaki, E. Kumarasamy, G. He, B. Choi, N. Ananth, M. Y. Sfeir and L. M. Campos, Mater. Horiz., 2022, 9, 462–470 RSC.
- K. R. Parenti, R. Chesler, G. He, P. Bhattacharyya, B. Xiao, H. Huang, D. Malinowski, J. Zhang, X. Yin, A. Shukla, S. Mazumdar, M. Y. Sfeir and L. M. Campos, Nat. Chem., 2022, 15, 339–346 CrossRef PubMed.
- R. Casillas, I. Papadopoulos, T. Ullrich, D. Thiel, A. Kunzmann and D. M. Guldi, Energy Environ. Sci., 2020, 13, 2741–2804 RSC.
- C. Hetzer, D. M. Guldi and R. R. Tykwinski, Chem. Eur. J., 2018, 24, 8245 CrossRef CAS PubMed.
- K. R. Parenti, G. He, S. N. Sanders, A. B. Pun, E. Kumarasamy, M. Y. Sfeir and L. M. Campos, J. Phys. Chem. A, 2020, 124, 9392–9399 CrossRef CAS PubMed.
- N. Monahan and X.-Y. Zhu, Annu. Rev. Phys. Chem., 2015, 66, 601–618 CrossRef CAS PubMed.
- W.-L. Chan, T. C. Berkelbach, M. R. Provorse, N. R. Monahan, J. R. Tritsch, M. S. Hybertsen, D. R. Reichman, J. Gao and X. Y. Zhu, Acc. Chem. Res., 2013, 46, 1321–1329 CrossRef CAS PubMed.
- P. M. Zimmerman, C. B. Musgrave and M. Head-Gordon, Acc. Chem. Res., 2013, 46, 1339–1347 CrossRef CAS PubMed.
- M. W. B. Wilson, A. Rao, B. Ehrler and R. H. Friend, Acc. Chem. Res., 2013, 46, 1330–1338 CrossRef CAS PubMed.
- K. Miyata, F. S. Conrad-Burton, F. L. Geyer and X. Y. Zhu, Chem. Rev., 2019, 119, 4261–4292 CrossRef CAS PubMed.
- A. Rao and R. H. Friend, Nat. Rev. Mater., 2017, 2, 17063 CrossRef CAS.
- P. E. Hartnett, E. A. Margulies, C. M. Mauck, S. A. Miller, Y. Wu, Y.-L. Wu, T. J. Marks and M. R. Wasielewski, J. Phys. Chem. B, 2016, 120, 1357–1366 CrossRef CAS PubMed.
- M. J. Y. Tayebjee, S. N. Sanders, E. Kumarasamy, L. M. Campos, M. Y. Sfeir and D. R. McCamey, Nat. Phys., 2017, 13, 182–188 Search PubMed.
- S. L. Bayliss, A. D. Chepelianskii, A. Sepe, B. J. Walker, B. Ehrler, M. J. Bruzek, J. E. Anthony and N. C. Greenham, Phys. Rev. Lett., 2014, 112, 238701 CrossRef PubMed.
- L. R. Weiss, S. L. Bayliss, F. Kraffert, K. J. Thorley, J. E. Anthony, R. Bittl, R. H. Friend, A. Rao, N. C. Greenham and J. Behrends, Nat. Phys., 2017, 13, 176–181 Search PubMed.
- S. Nakamura, H. Sakai, H. Nagashima, M. Fuki, K. Onishi, R. Khan, Y. Kobori, N. V. Tkachenko and T. Hasobe, J. Phys. Chem. C, 2021, 125, 18287–18296 CrossRef CAS.
- N. V. Korovina, J. Joy, X. Feng, C. Feltenberger, A. I. Krylov, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2018, 140, 10179–10190 CrossRef CAS PubMed.
- N. V. Korovina, S. Das, Z. Nett, X. Feng, J. Joy, R. Haiges, A. I. Krylov, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2016, 138, 617–627 CrossRef CAS PubMed.
- R. Ringström, F. Edhborg, Z. W. Schroeder, L. Chen, M. J. Ferguson, R. R. Tykwinski and B. Albinsson, Chem. Sci., 2022, 13, 4944–4954 RSC.
- L. M. Yablon, S. N. Sanders, H. Li, K. R. Parenti, E. Kumarasamy, K. J. Fallon, M. J. A. Hore, A. Cacciuto, M. Y. Sfeir and L. M. Campos, J. Am. Chem. Soc., 2019, 141, 9564–9569 CrossRef CAS PubMed.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. T. Trinh, B. Choi, J. Xia, E. J. Taffet, J. Z. Low, J. R. Miller, X. Roy, X. Y. Zhu, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, J. Am. Chem. Soc., 2015, 137, 8965–8972 CrossRef CAS PubMed.
- H. M. Bergman, G. R. Kiel, R. J. Witzke, D. P. Nenon, A. M. Schwartzberg, Y. Liu and T. D. Tilley, J. Am. Chem. Soc., 2020, 142, 19850–19855 CrossRef CAS PubMed.
- A. Aster, F. Zinna, C. Rumble, J. Lacour and E. Vauthey, J. Am. Chem. Soc., 2021, 143, 2361–2371 CrossRef CAS PubMed.
- E. Kumarasamy, S. N. Sanders, M. J. Y. Tayebjee, A. Asadpoordarvish, T. J. H. Hele, E. G. Fuemmeler, A. B. Pun, L. M. Yablon, J. Z. Low, D. W. Paley, J. C. Dean, B. Choi, G. D. Scholes, M. L. Steigerwald, N. Ananth, D. R. McCamey, M. Y. Sfeir and L. M. Campos, J. Am. Chem. Soc., 2017, 139, 12488–12494 CrossRef CAS PubMed.
- J. Kim, H. T. Teo, Y. Hong, J. Oh, H. Kim, C. Chi and D. Kim, Angew. Chem., Int. Ed., 2020, 59, 20956–20964 CrossRef CAS PubMed.
- A. B. Pun, A. Asadpoordarvish, E. Kumarasamy, M. J. Y. Tayebjee, D. Niesner, D. R. McCamey, S. N. Sanders, L. M. Campos and M. Y. Sfeir, Nat. Chem., 2019, 11, 821–828 CrossRef CAS PubMed.
- C. Hetzer, B. S. Basel, S. M. Kopp, F. Hampel, F. J. White, T. Clark, D. M. Guldi and R. R. Tykwinski, Angew. Chem., Int. Ed., 2019, 58, 15263–15267 CrossRef CAS PubMed.
- B. S. Basel, J. Zirzlmeier, C. Hetzer, S. R. Reddy, B. T. Phelan, M. D. Krzyaniak, M. K. Volland, P. B. Coto, R. M. Young, T. Clark, M. Thoss, R. R. Tykwinski, M. R. Wasielewski and D. M. Guldi, Chem, 2018, 4, 1092–1111 CAS.
- I. Papadopoulos, S. R. Reddy, P. B. Coto, D. Lehnherr, D. Thiel, M. Thoss, R. R. Tykwinski and D. M. Guldi, J. Phys. Chem. Lett., 2022, 13, 5094–5100 CrossRef CAS PubMed.
- J. Zirzlmeier, D. Lehnherr, P. B. Coto, E. T. Chernick, R. Casillas, B. S. Basel, M. Thoss, R. R. Tykwinski and D. M. Guldi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5325–5330 CrossRef CAS PubMed.
- J. Zirzlmeier, R. Casillas, S. R. Reddy, P. B. Coto, D. Lehnherr, E. T. Chernick, I. Papadopoulos, M. Thoss, R. R. Tykwinski and D. M. Guldi, Nanoscale, 2016, 8, 10113–10123 RSC.
- S. Lukman, A. J. Musser, K. Chen, S. Athanasopoulos, C. K. Yong, Z. Zeng, Q. Ye, C. Chi, J. M. Hodgkiss, J. Wu, R. H. Friend and N. C. Greenham, Adv. Funct. Mater., 2015, 25, 5452–5461 CrossRef CAS.
- S. Lukman, K. Chen, J. M. Hodgkiss, D. H. P. Turban, N. D. M. Hine, S. Dong, J. Wu, N. C. Greenham and A. J. Musser, Nat. Commun., 2016, 7, 13622 CrossRef CAS PubMed.
- I. Papadopoulos, J. Zirzlmeier, C. Hetzer, Y. J. Bae, M. D. Krzyaniak, M. R. Wasielewski, T. Clark, R. R. Tykwinski and D. M. Guldi, J. Am. Chem. Soc., 2019, 141, 6191–6203 CrossRef CAS PubMed.
- J. P. Mora-Fuentes, I. Papadopoulos, D. Thiel, R. Álvarez-Boto, D. Cortizo-Lacalle, T. Clark, M. Melle-Franco, D. M. Guldi and A. Mateo-Alonso, Angew. Chem., Int. Ed., 2020, 59, 1113–1117 CrossRef CAS PubMed.
- A. M. Alvertis, S. Lukman, T. J. H. Hele, E. G. Fuemmeler, J. Feng, J. Wu, N. C. Greenham, A. W. Chin and A. J. Musser, J. Am. Chem. Soc., 2019, 141, 17558–17570 CrossRef CAS PubMed.
- J. D. Schultz, J. Y. Shin, M. Chen, J. P. O'Connor, R. M. Young, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2021, 143, 2049–2058 CrossRef CAS PubMed.
- A. J. Musser, M. Liebel, C. Schnedermann, T. Wende, T. B. Kehoe, A. Rao and P. Kukura, Nat. Phys., 2015, 11, 352–357 Search PubMed.
- L. Yu, J. F. Yang, B. Y. Guan, Y. Lu and X. W. Lou, Angew. Chem., Int. Ed., 2018, 57, 1 CrossRef CAS.
- W. Kim and A. J. Musser, Adv. Phys.: X, 2021, 6, 1918022 Search PubMed.
- A. T. Gilligan, E. G. Miller, T. Sammakia and N. H. Damrauer, J. Am. Chem. Soc., 2019, 141, 5961–5971 CrossRef CAS PubMed.
- H. L. Stern, A. Cheminal, S. R. Yost, K. Broch, S. L. Bayliss, K. Chen, M. Tabachnyk, K. Thorley, N. Greenham, J. M. Hodgkiss, J. Anthony, M. Head-Gordon, A. J. Musser, A. Rao and R. H. Friend, Nat. Chem., 2017, 9, 1205–1212 CrossRef CAS PubMed.
- K. C. Krishnapriya, P. Roy, B. Puttaraju, U. Salzner, A. J. Musser, M. Jain, J. Dasgupta and S. Patil, Nat. Commun., 2019, 10, 33 CrossRef CAS PubMed.
- A. M. Müller, Y. S. Avlasevich, K. Müllen and C. J. Bardeen, Chem. Phys. Lett., 2006, 421, 518–522 CrossRef.
- S. Arnold, R. R. Alfano, M. Pope, W. Yu, P. Ho, R. Selsby, J. Tharrats and C. E. Swenberg, J. Chem. Phys., 1976, 64, 5104–5114 CrossRef CAS.
- S. Arnold and W. B. Whitten, J. Chem. Phys., 1981, 75, 1166–1169 CrossRef CAS.
- G. Klein, Chem. Phys. Lett., 1978, 57, 202–206 CrossRef CAS.
- R. P. Groff, P. Avakian and R. E. Merrifield, Phys. Rev. B, 1970, 1, 815–817 CrossRef.
- G. Fleming, D. Millar, G. Morris, J. Morris and G. Robinson, Aust. J. Chem., 1977, 30, 2353–2359 CrossRef CAS.
- H. Liu, R. Wang, L. Shen, Y. Xu, M. Xiao, C. Zhang and X. Li, Org. Lett., 2017, 19, 580–583 CrossRef CAS PubMed.
- H. Liu, V. M. Nichols, L. Shen, S. Jahansouz, Y. Chen, K. M. Hanson, C. J. Bardeen and X. Li, Phys. Chem. Chem. Phys., 2015, 17, 6523–6531 RSC.
- H. Liu, L. Shen, Z. Cao and X. Li, Phys. Chem. Chem. Phys., 2014, 16, 16399–16406 RSC.
- R. J. Lindquist, K. M. Lefler, K. E. Brown, S. M. Dyar, E. A. Margulies, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2014, 136, 14912–14923 CrossRef CAS PubMed.
- E. A. Margulies, L. E. Shoer, S. W. Eaton and M. R. Wasielewski, Phys. Chem. Chem. Phys., 2014, 16, 23735–23742 RSC.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- H. L. Stern, A. J. Musser, S. Gelinas, P. Parkinson, L. M. Herz, M. J. Bruzek, J. Anthony, R. H. Friend and B. J. Walker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7656–7661 CrossRef CAS PubMed.
- X. Feng and A. I. Krylov, Phys. Chem. Chem. Phys., 2016, 18, 7751–7761 RSC.
- H. Liu, X. Wang, L. Pan, L. Shen, X. Wang, Q. Chen and X. Li, J. Photochem. Photobiol., A, 2017, 340, 21–28 CrossRef CAS.
- T. Sun, L. Shen, H. Liu, X. Sun and X. Li, J. Mol. Struct., 2016, 1116, 200–206 CrossRef CAS.
- K. Shizu, C. Adachi and H. Kaji, ACS Omega, 2021, 6, 2638–2643 CrossRef CAS PubMed.
- K. Miyata, Y. Kurashige, K. Watanabe, T. Sugimoto, S. Takahashi, S. Tanaka, J. Takeya, T. Yanai and Y. Matsumoto, Nat. Chem., 2017, 9, 983–989 CrossRef CAS PubMed.
- J. J. Piet, W. Schuddeboom, B. R. Wegewijs, F. C. Grozema and J. M. Warman, J. Am. Chem. Soc., 2001, 123, 5337–5347 CrossRef CAS PubMed.
- N. Mataga, H. Yao, T. Okada and W. Rettig, J. Phys. Chem., 1989, 93, 3383–3386 CrossRef CAS.
- K. Elich, M. Kitazawa, T. Okada and R. Wortmann, J. Phys. Chem. A, 1997, 101, 2010–2015 CrossRef CAS.
- Y. Matsui, S. Kawaoka, H. Nagashima, T. Nakagawa, N. Okamura, T. Ogaki, E. Ohta, S. Akimoto, A. Sato-Tomita, S. Yagi, Y. Kobori and H. Ikeda, J. Phys. Chem. C, 2019, 123, 18813–18823 CrossRef CAS.
- U. Müller, L. Roos, M. Frank, M. Deutsch, S. Hammer, M. Krumrein, A. Friedrich, T. B. Marder, B. Engels, A. Krueger and J. Pflaum, J. Phys. Chem. C, 2020, 124, 19435–19446 CrossRef.
- T. Sakuma, H. Sakai, Y. Araki, T. Mori, T. Wada, N. V. Tkachenko and T. Hasobe, J. Phys. Chem. A, 2016, 120, 1867–1875 CrossRef CAS PubMed.
- H. Sakai, R. Inaya, H. Nagashima, S. Nakamura, Y. Kobori, N. V. Tkachenko and T. Hasobe, J. Phys. Chem. Lett., 2018, 9, 3354–3360 CrossRef CAS PubMed.
- A. K. Le, J. A. Bender, D. H. Arias, D. E. Cotton, J. C. Johnson and S. T. Roberts, J. Am. Chem. Soc., 2018, 140, 814–826 CrossRef CAS PubMed.
- B. Manna, A. Nandi and R. Ghosh, J. Phys. Chem. C, 2018, 122, 21047–21055 CrossRef CAS.
- S. Yamauchi, A. Takahashi, Y. Iwasaki, M. Unno, Y. Ohba, J. Higuchi, A. Blank and H. Levanon, J. Phys. Chem. A, 2003, 107, 1478–1485 CrossRef CAS.
- P. J. Vallett, J. L. Snyder and N. H. Damrauer, J. Phys. Chem. A, 2013, 117, 10824–10838 CrossRef CAS PubMed.
- T. J. Carey, J. L. Snyder, E. G. Miller, T. Sammakia and N. H. Damrauer, J. Org. Chem., 2017, 82, 4866–4874 CrossRef CAS PubMed.
- N. H. Damrauer and J. L. Snyder, J. Phys. Chem. Lett., 2015, 6, 4456–4462 CrossRef CAS PubMed.
- J. D. Cook, T. J. Carey, D. H. Arias, J. C. Johnson and N. H. Damrauer, J. Phys. Chem. A, 2017, 121, 9229–9242 CrossRef CAS PubMed.
- J. D. Cook, T. J. Carey and N. H. Damrauer, J. Phys. Chem. A, 2016, 120, 4473–4481 CrossRef CAS PubMed.
- E. C. Alguire, J. E. Subotnik and N. H. Damrauer, J. Phys. Chem. A, 2015, 119, 299–311 CrossRef CAS PubMed.
- T. J. Carey, E. G. Miller, A. T. Gilligan, T. Sammakia and N. H. Damrauer, Org. Lett., 2018, 20, 457–460 CrossRef CAS PubMed.
- S. R. Yost, J. Lee, M. W. B. Wilson, T. Wu, D. P. McMahon, R. R. Parkhurst, N. J. Thompson, D. N. Congreve, A. Rao, K. Johnson, M. Y. Sfeir, M. G. Bawendi, T. M. Swager, R. H. Friend, M. A. Baldo and T. Van Voorhis, Nat. Chem., 2014, 6, 492–497 CrossRef CAS PubMed.
- J. J. Burdett, G. B. Piland and C. J. Bardeen, Chem. Phys. Lett., 2013, 585, 1–10 CrossRef CAS.
- T. Yamakado, S. Takahashi, K. Watanabe, Y. Matsumoto, A. Osuka and S. Saito, Angew. Chem., Int. Ed., 2018, 57, 5438–5443 CrossRef CAS PubMed.
- S. D. Jadhav, D. Sasikumar and M. Hariharan, Phys. Chem. Chem. Phys., 2022, 24, 16193–16199 RSC.
- X. Wang, R. Wang, L. Shen, Z. Tang, C. Wen, B. Dong, H. Liu, C. Zhang and X. Li, Phys. Chem. Chem. Phys., 2018, 20, 6330–6336 RSC.
- Y. Wan, Z. Guo, T. Zhu, S. Yan, J. Johnson and L. Huang, Nat. Chem., 2015, 7, 785–792 CrossRef CAS PubMed.
- B. S. Basel, J. Zirzlmeier, C. Hetzer, B. T. Phelan, M. D. Krzyaniak, S. R. Reddy, P. B. Coto, N. E. Horwitz, R. M. Young, F. J. White, F. Hampel, T. Clark, M. Thoss, R. R. Tykwinski, M. R. Wasielewski and D. M. Guldi, Nat. Commun., 2017, 8, 15171 CrossRef CAS PubMed.
- M. Wakasa, M. Kaise, T. Yago, R. Katoh, Y. Wakikawa and T. Ikoma, J. Phys. Chem. C, 2015, 119, 25840–25844 CrossRef CAS.
- W.-L. Chan, M. Ligges, A. Jailaubekov, L. Kaake, L. Miaja-Avila and X.-Y. Zhu, Science, 2011, 334, 1541–1545 CrossRef CAS PubMed.
- R. D. Pensack, E. E. Ostroumov, A. J. Tilley, S. Mazza, C. Grieco, K. J. Thorley, J. B. Asbury, D. S. Seferos, J. E. Anthony and G. D. Scholes, J. Phys. Chem. Lett., 2016, 7, 2370–2375 CrossRef CAS PubMed.
- I. Breen, R. Tempelaar, L. A. Bizimana, B. Kloss, D. R. Reichman and D. B. Turner, J. Am. Chem. Soc., 2017, 139, 11745–11751 CrossRef CAS PubMed.
- H. Liu, Z. Wang, X. Wang, L. Shen, C. Zhang, M. Xiao and X. Li, J. Mater. Chem. C, 2018, 6, 3245–3253 RSC.
- Z. Wang, H. Liu, X. Xie, C. Zhang, R. Wang, L. Chen, Y. Xu, H. Ma, W. Fang, Y. Yao, H. Sang, X. Wang, X. Li and M. Xiao, Nat. Chem., 2021, 13, 559–567 CrossRef CAS PubMed.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, Chem, 2016, 1, 505–511 CAS.
- E. A. Margulies, C. E. Miller, Y. Wu, L. Ma, G. C. Schatz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2016, 8, 1120–1125 CrossRef CAS PubMed.
- S. L. Bayliss, L. R. Weiss, A. Rao, R. H. Friend, A. D. Chepelianskii and N. C. Greenham, Phys. Rev. B, 2016, 94, 045204 CrossRef.
- S. L. Bayliss, L. R. Weiss, A. Mitioglu, K. Galkowski, Z. Yang, K. Yunusova, A. Surrente, K. J. Thorley, J. Behrends, R. Bittl, J. E. Anthony, A. Rao, R. H. Friend, P. Plochocka, P. C. M. Christianen, N. C. Greenham and A. D. Chepelianskii, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5077–5082 CrossRef CAS PubMed.
- R. Xu, C. Zhang and M. Xiao, Trends Chem., 2022, 4, 528–539 CrossRef CAS.
- M. T. Trinh, A. Pinkard, A. B. Pun, S. N. Sanders, E. Kumarasamy, M. Y. Sfeir, L. M. Campos, X. Roy and X.-Y. Zhu, Sci. Adv., 2017, 3, e1700241 CrossRef PubMed.
- A. B. Pun, S. N. Sanders, E. Kumarasamy, M. Y. Sfeir, D. N. Congreve and L. M. Campos, Adv. Mater., 2017, 29, 1701416 CrossRef PubMed.
- S. Nakamura, H. Sakai, M. Fuki, R. Ooie, F. Ishiwari, A. Saeki, N. V. Tkachenko, Y. Kobori and T. Hasobe, Angew. Chem., Int. Ed., 2023, 62, e202217704 CrossRef CAS PubMed.
- C. M. Davis, Y. Kawashima, K. Ohkubo, J. M. Lim, D. Kim, S. Fukuzumi and J. L. Sessler, J. Phys. Chem. C, 2014, 118, 13503–13513 CrossRef CAS.
- F. D'Souza and O. Ito, Coord. Chem. Rev., 2005, 249, 1410–1422 CrossRef.
- H. Liu, X. Wang, L. Ma, W. Wang, S. Liu, J. Zhou, P. Su, Z. Liu, Z. Li, X. Lin, Y. Chen and X. Li, J. Am. Chem. Soc., 2022, 144, 15509–15518 CrossRef CAS PubMed.
- D. Gosztola, M. P. Niemczyk and M. R. Wasielewski, J. Am. Chem. Soc., 1998, 120, 5118–5119 CrossRef CAS.
- M. P. Debreczeny, W. A. Svec and M. R. Wasielewski, Science, 1996, 274, 584–587 CrossRef CAS.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334–337 CrossRef CAS PubMed.
- P. J. Jadhav, A. Mohanty, J. Sussman, J. Lee and M. A. Baldo, Nano Lett., 2011, 11, 1495–1498 CrossRef CAS PubMed.
- P. J. Jadhav, P. R. Brown, N. Thompson, B. Wunsch, A. Mohanty, S. R. Yost, E. Hontz, T. Van Voorhis, M. G. Bawendi, V. Bulović and M. A. Baldo, Adv. Mater., 2012, 24, 6169–6174 CrossRef CAS PubMed.
- A. Kunzmann, M. Gruber, R. Casillas, J. Zirzlmeier, M. Stanzel, W. Peukert, R. R. Tykwinski and D. M. Guldi, Angew. Chem., Int. Ed., 2018, 57, 10742–10747 CrossRef CAS PubMed.
- A. B. Kolomeisky, X. Feng and A. I. Krylov, J. Phys. Chem. C, 2014, 118, 5188–5195 CrossRef CAS.
- Y. Kobori, M. Fuki, S. Nakamura and T. Hasobe, J. Phys. Chem. B, 2020, 124, 9411–9419 CrossRef CAS PubMed.
- S. Matsuda, S. Oyama and Y. Kobori, Chem. Sci., 2020, 11, 2934–2942 RSC.
- W.-L. Chan, M. Ligges and X. Y. Zhu, Nat. Chem., 2012, 4, 840–845 CrossRef CAS PubMed.
- S. M. Ryno, C. Risko and J.-L. Brédas, ACS Appl. Mater. Interfaces, 2016, 8, 14053–14062 CrossRef CAS PubMed.
- R. Tom, S. Gao, Y. Yang, K. Zhao, I. Bier, E. A. Buchanan, A. Zaykov, Z. Havlas, J. Michl and N. Marom, Chem. Mater., 2023, 35, 1373–1386 CrossRef CAS.
- A. Nandi, B. Manna and R. Ghosh, J. Phys. Chem. C, 2021, 125, 2583–2591 CrossRef CAS.
- Y. Lei, Y. Sun, Y. Zhang, H. Zhang, H. Zhang, Z. Meng, W.-Y. Wong, J. Yao and H. Fu, Nat. Commun., 2018, 9, 4358 CrossRef PubMed.
- S. E. Strong and J. D. Eaves, J. Phys. Chem. Lett., 2015, 6, 1209–1215 CrossRef CAS PubMed.
- H. Huang, G. He, K. Xu, Q. Wu, D. Wu, M. Y. Sfeir and J. Xia, Chem, 2019, 5, 2405–2417 CAS.
- M. Chen, A. F. Coleman, R. M. Young and M. R. Wasielewski, J. Phys. Chem. C, 2021, 125, 6999–7009 CrossRef CAS.
- N. A. Pace, W. Zhang, D. H. Arias, I. McCulloch, G. Rumbles and J. C. Johnson, J. Phys. Chem. Lett., 2017, 8, 6086–6091 CrossRef CAS PubMed.
- B. Daiber, K. van den Hoven, M. H. Futscher and B. Ehrler, ACS Energy Lett., 2021, 6, 2800–2808 CrossRef CAS PubMed.
- E. Sundin, R. Ringström, F. Johansson, B. Küçüköz, A. Ekebergh, V. Gray, B. Albinsson, J. Mårtensson and M. Abrahamsson, J. Phys. Chem. C, 2020, 124, 20794–20805 CrossRef CAS.
- N. A. Pace, N. V. Korovina, T. T. Clikeman, S. Holliday, D. B. Granger, G. M. Carroll, S. U. Nanayakkara, J. E. Anthony, I. McCulloch, S. H. Strauss, O. V. Boltalina, J. C. Johnson, G. Rumbles and O. G. Reid, Nat. Chem., 2020, 12, 63–70 CrossRef CAS PubMed.
- W. Zukun, R. Wu, Z. Chen, L. Ye, H. Li and H. Zhu, J. Phys. Chem. A, 2020, 124, 4185–4192 CrossRef CAS PubMed.
- N. A. Pace, D. H. Arias, D. B. Granger, S. Christensen, J. E. Anthony and J. C. Johnson, Chem. Sci., 2018, 9, 3004–3013 RSC.
- N. J. Thompson, M. W. B. Wilson, D. N. Congreve, P. R. Brown, J. M. Scherer, T. S. Bischof, M. Wu, N. Geva, M. Welborn, T. V. Voorhis, V. Bulović, M. G. Bawendi and M. A. Baldo, Nat. Mater., 2014, 13, 1039–1043 CrossRef CAS PubMed.
- M. Tabachnyk, B. Ehrler, S. Gélinas, M. L. Böhm, B. J. Walker, K. P. Musselman, N. C. Greenham, R. H. Friend and A. Rao, Nat. Mater., 2014, 13, 1033–1038 CrossRef CAS PubMed.
- H. Kim, B. Keller, R. Ho-Wu, N. Abeyasinghe, R. J. Vázquez, T. Goodson III and P. M. Zimmerman, J. Am. Chem. Soc., 2018, 140, 7760–7763 CrossRef CAS PubMed.
- J. R. Allardice, A. Thampi, S. Dowland, J. Xiao, V. Gray, Z. Zhang, P. Budden, A. J. Petty II, N. J. L. K. Davis, N. C. Greenham, J. E. Anthony and A. Rao, J. Am. Chem. Soc., 2019, 141, 12907–12915 CrossRef CAS PubMed.
- G. He, K. R. Parenti, L. M. Campos and M. Y. Sfeir, Adv. Mater., 2022, 34, 2203974 CrossRef CAS PubMed.
- L. M. Pazos-Outón, J. M. Lee, M. H. Futscher, A. Kirch, M. Tabachnyk, R. H. Friend and B. Ehrler, ACS Energy Lett., 2017, 2, 476–480 CrossRef PubMed.
- R. W. MacQueen, M. Liebhaber, J. Niederhausen, M. Mews, C. Gersmann, S. Jäckle, K. Jäger, M. J. Y. Tayebjee, T. W. Schmidt, B. Rech and K. Lips, Mater. Horiz., 2018, 5, 1065–1075 RSC.
- B. Ehrler, M. W. B. Wilson, A. Rao, R. H. Friend and N. C. Greenham, Nano Lett., 2012, 12, 1053–1057 CrossRef CAS PubMed.
- W. Fudickar and T. Linker, J. Am. Chem. Soc., 2012, 134, 15071–15082 CrossRef CAS PubMed.
- Y. Chen, L. Shen and X. Li, J. Phys. Chem. A, 2014, 118, 5700–5708 CrossRef CAS PubMed.
- Y.-D. Zhang, Y. Wu, Y. Xu, Q. Wang, K. Liu, J.-W. Chen, J.-J. Cao, C. Zhang, H. Fu and H.-L. Zhang, J. Am. Chem. Soc., 2016, 138, 6739–6745 CrossRef CAS PubMed.
- J. Bosson, G. M. Labrador, S. Pascal, F.-A. Miannay, O. Yushchenko, H. Li, L. Bouffier, N. Sojic, R. C. Tovar, G. Muller, D. Jacquemin, A. D. Laurent, B. LeGuennic, E. Vauthey and J. Lacour, Chem. Eur. J., 2016, 22, 18394–18403 CrossRef CAS PubMed.
- S. Lou, J. Yu, Y. Jiang, G. Guo and Q. Zhang, Chin. Sci. Bull., 2008, 53, 2940–2945 CAS.
- K. Liu, C.-L. Song, Y.-C. Zhou, X.-Y. Zhou, X.-J. Pan, L.-Y. Cao, C. Zhang, Y. Liu, X. Gong and H.-L. Zhang, J. Mater. Chem. C, 2015, 3, 4188–4196 RSC.
- R. Xiong, A.-B. Bornhof, A. I. Arkhypchuk, A. Orthaber and K. E. Borbas, Chem. Eur. J., 2017, 23, 4089–4095 CrossRef CAS PubMed.
- C. An and J. Hou, Acc. Mater. Res., 2022, 3, 540–551 CrossRef CAS.
- C. An, Z. Zheng and J. Hou, Chem. Commun., 2020, 56, 4750–4760 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |