
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLayered transition metal oxides (LTMO) for oxygen evolution reactions and aqueous Li-ion batteries
Yohan
Kim
 ,
Eunjin
Choi
,
Seunggu
Kim
and
Hye Ryung
Byon
*
,
Eunjin
Choi
,
Seunggu
Kim
and
Hye Ryung
Byon
*
Department of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), 291, Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea. E-mail: hrbyon@kaist.ac.kr
First published on 1st September 2023
Abstract
This perspective paper comprehensively explores recent electrochemical studies on layered transition metal oxides (LTMO) in aqueous media and specifically encompasses two topics: catalysis of the oxygen evolution reaction (OER) and cathodes of aqueous lithium-ion batteries (LiBs). They involve conflicting requirements; OER catalysts aim to facilitate water dissociation, while for cathodes in aqueous LiBs it is essential to suppress water dissociation. The interfacial reactions taking place at the LTMO in these two distinct systems are of particular significance. We show various strategies for designing LTMO materials for each desired aim based on an in-depth understanding of electrochemical interfacial reactions. This paper sheds light on how regulating the LTMO interface can contribute to efficient water splitting and economical energy storage, all with a single material.
1. Introduction
Electrochemical conversion and storage systems are currently receiving significant attention for utilizing green and sustainable energy sources. Carbon dioxide emissions from fossil fuels are the primary driver of the climate crisis. Thus, it is imperative for society to shift towards alternative and clean energy sources while also developing effective means of storing them as electrical energy.Among the promising energy sources, hydrogen (H2) stands out prominently.1,2 Extensive research efforts have been dedicated to exploring green H2 production through electrochemical water splitting. However, the water-splitting process faces a significant hurdle in the form of the sluggish oxygen evolution reaction (OER), which acts as the counter-reaction to hydrogen production in electrolyzers.3,4 Consequently, the development of efficient OER catalysts becomes a critical undertaking in realizing the vision of efficient and sustainable green H2 production.
Meanwhile, rechargeable batteries are quintessential energy storage systems, offering unparalleled capabilities in storing and releasing energy. At the heart of these batteries lies the lithium-ion (Li+).5,6 This charge carrier with light weight and high electrochemical reduction potential holds promise for achieving high energy density. In order to maximize energy density, it becomes critical to develop electrodes that can accommodate a greater influx of Li+ ions alongside the flow of electrons. Layered, spinel, and olivine structures have emerged as notable electrodes.7,8 Among them, layered oxides provide superior capacity and stable cyclability, thus being investigated considerably for achieving high energy density.9,10 In particular, lithium cobalt oxide LiCoO2 (referred to as LCO) and lithium nickel cobalt manganese oxide (LiNixCoyMnzO2, x + y + z = 1, denoted as NCM) as members of layered transition metal oxide (LTMO) are commercially successful cathodes of LiBs. Further, beyond their utilization in non-aqueous media, new research approaches have recently been explored in aqueous media. Aqueous lithium-ion batteries (LiBs) are cheaper and have a low fire risk, making them suitable for grid-scale energy storage systems (ESSs) linked to sustainable energy devices.11
These demands and scientific curiosity have spurred numerous material studies for OER catalysts and cathodes of aqueous LiBs. In particular, LTMO shows intriguing characteristics when applied for both purposes. The LTMO is vulnerable and prone to deformation in water. However, the resulting surface or bulk structural transformations can manifest OER activity. For example, after Li+ extraction from LCO, the oxidized transition metals facilitate the OER.12 Continuous Li+ removal further activates the oxide (consisting of O2− anions) of LCO by tuning the electronic structure, eventually providing multiple OER active sites. Importantly, these enriched parameters are attractive to understanding the origin of OER active sites and rendering better activity. As a cathode in aqueous LiBs, LTMO is seemingly undesired due to extreme water sensitivity and the corresponding structural distortion. In particular, protons (H+) are inserted into LTMO during the charging and discharging process which significantly exacerbates structural degradation and cell failure.13 Therefore, regulating electrochemical interfacial reactions for selective Li+ inflow while preventing H+ access is essential to protect LTMO structures in aqueous LiBs.
Interestingly, two vital applications require conflicting properties for LTMO, which presents a challenge.4,14–17 An efficient OER catalyst necessitates a high affinity and fast adsorption of OH− to facilitate oxidation to O2, while reversible Li+ storage requires minimizing the incorporation of water molecules to prevent irreversible H+ insertion. Addressing these contradicting demands calls for novel approaches that involve a profound understanding of the interfacial regions through a combination of in situ/ex situ electrochemical analyses and computational simulations. This approach enables a comprehensive examination of electrochemical reactions and facilitates the design of LTMO materials tailored to specific electrochemical conditions.18,19
In the following sections, we discuss these different purposes separately and the versatile tuning of LMTO according to the target aims. The crystal and electronic structure background of LTMO is introduced in Section 2. In Section 3, we present various strategies for designing OER catalysts through structural modifications of LTMO. Section 4 focuses on the origin of cathode degradation in aqueous LiBs. In addition, we introduce various in situ/ex situ observations and computational simulations to provide insight into the interfacial reactions where Li+ intercalation competes with H+ insertion.
2. Layered transition metal oxide (LTMO)
The development of LTMO reached significant milestones after demonstrating LCO with the reversible extraction and incorporation of Li+ ions. This breakthrough led to the successful implementation of LCO as the cathode in conjunction with a graphite anode, giving rise to the first commercially viable LiBs in 1991.20Structurally, LTMO consists of an alternating alkali metal ion layer and transition metal (M) oxide layer, denoted as MO2, and crystallizes in the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (no. 166) space group21 (Fig. 1(a)). The MO2 layers consist of edge-sharing MO6 octahedral units. Within the layered arrangement, alkali metal ions are coordinated with the oxygen lattice in the MO2 layer, adopting octahedral, tetrahedral, or prismatic configurations. The oxygen atoms can also occupy three possible sites on a hexagonal lattice. For example, LCO adopts an O3-type structure, indicating octahedral (O) oxygen coordination for Li+ and three (3) transition metal layers in the stacking unit.9,22–24
m (no. 166) space group21 (Fig. 1(a)). The MO2 layers consist of edge-sharing MO6 octahedral units. Within the layered arrangement, alkali metal ions are coordinated with the oxygen lattice in the MO2 layer, adopting octahedral, tetrahedral, or prismatic configurations. The oxygen atoms can also occupy three possible sites on a hexagonal lattice. For example, LCO adopts an O3-type structure, indicating octahedral (O) oxygen coordination for Li+ and three (3) transition metal layers in the stacking unit.9,22–24
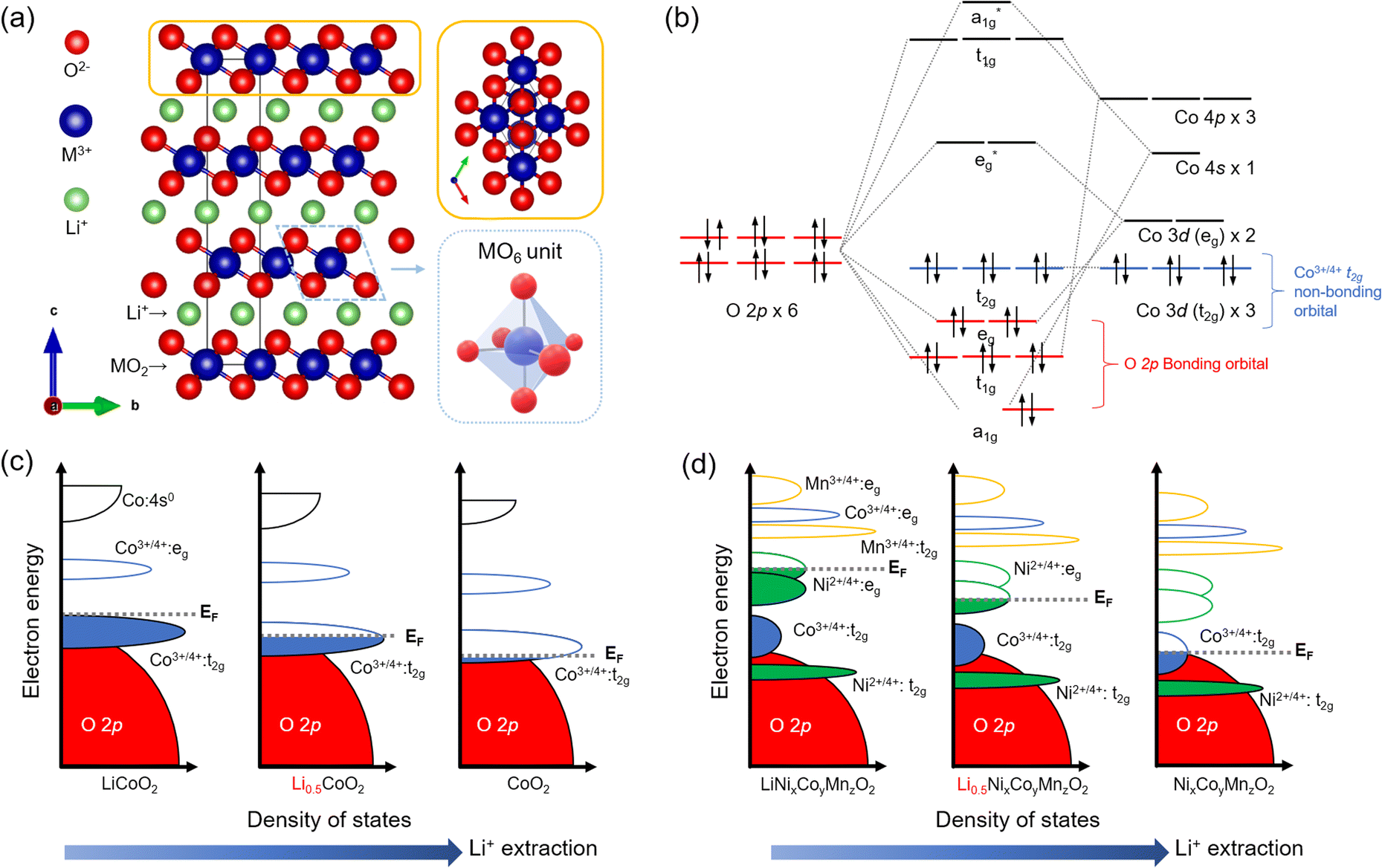 | ||
| Fig. 1 Crystal and electronic structures of LTMO. (a) Rm layered LTMO. (b) Molecular orbital hybridization of an MO6 octahedral unit in LiCoO2 (LCO). (c and d) Electronic band structures of (c) LCO and (d) LiNixCoyMnzO2 (x + y + z = 1, NCM) by Li+ extraction. | ||
For electrochemistry, an oxidation process (i.e., an anodic reaction) enables the extraction of Li+, which is indicated as delithiation, Li+ deintercalation, or Li+ deinsertion. This process increases the valence state (n+) of the Mn+ to balance the overall charge (eqn (1)).24 For instance, during the charging process, LCO undergoes delithiation, accompanied by the oxidation of Co3+ to Co4+ at approximately 4.0 V vs. Li/Li+ (equivalent to 0.96 V vs. SHE). The amount of extracted Li+ is restricted to 50% of the total Li+ quantity for LCO when the hexagonal O3 phase is transformed to the monoclinic O1 phase,12,25 and the Co ions exist in a mixed valence state, Co3+/Co4+. Further extensive delithiation (x > 0.5 in eqn (1)) triggers an irreversible phase transition, which is undesired.26,27 Conversely, the reverse reduction (i.e., cathodic) reaction occurs during discharge, and Li+ incorporation, indicated as Li+ intercalation, Li+ lithiation, or Li+ insertion, restores LCO to its original state. As another LTMO, NCMs involve redox events of Ni2+/Ni4+ along with Co3+/Co4+, contributing to higher capacity electrodes in LiBs.
| LiMn+O2 ⇄ Li1−xMn+1−xM(n+1)+xO2 + xLi+ + xe− | (1) |
Understanding the electronic structure of LTMO is pivotal for designing electrochemical and interfacial properties during the charging and discharging process. Considering the minimal unit of an MO6 octahedral structure, the O 2p orbitals are fully occupied by electrons in the O 2p–M nd hybridization in the molecular orbital diagram (where n is the principal quantum number of d-orbital, typically 3 for d-block elements), indicating a bonding character (Fig. 1(b)). On the other hand, because of the higher energy level of the M 3d orbital than O 2p, the occupancy of t2g and eg orbitals exclusively depends on the valence states of Mn+. The redox events occur primarily in these t2g and eg orbitals, underpinning the close association between Li+ deintercalation/intercalation and the nature of the M 3d orbitals.28
The molecular orbital concept extends to band theory when bulk LTMO crystals composed of arrays of edge-sharing MO6 units are considered. In the band diagram, the O 2p band, located at a low energy level, is fully filled, while the t2g and eg bands originating from the M character are positioned at higher energy levels (Fig. 1(c)). The crucial aspect is the comparison between the position of the Fermi level (EF) and the upper boundary level of the electron-filled state (i.e., valence band). During the anodic (delithiation) processes, the depletion of electrons from the t2g bands leads to a downward shift of EF. In the case of LCO, where 50% of the total Li+ is extracted, EF is lowered towards the t2g band.9 Continued oxidation drives EF to lower energy levels, eventually reaching the O 2p band. The EF lying in the O 2p band activates the oxygen lattice in LCO to participate in the redox process. However, severe activation compromises the structural stability of the material and exacerbates irreversible phase transitions.26,27
In the band diagram for NCM, both Ni2+ and Co3+ are involved in the redox reaction. Ni2+/Ni4+ undergoes a two-electron transfer process in the eg band, while Co3+/Co4+ participates in a one-electron transfer process in the t2g band (Fig. 1(d)). The Mn4+/Mn5+ band, located at a lower energy level, is fully occupied with electrons and remains inactive during the charging and discharging processes. Severe delithiation induces cation mixing, with Mn+ occupying the Li vacancy sites and causing deformation of the layered structure to spinel (space group Fdm (no. 227)) or rock-salt phases (space group Fmm (no. 225)).29 This structural transformation is particularly prominent at the surface and is more serious at higher Ni ion contents due to the instability of phases derived from Ni3+.30
The deformation of LTMO in LiBs has traditionally been considered undesirable due to irreversible and unstable electrochemical reactions. However, recent studies have explored structure designs of LTMO by deformation to gain insights into new interfacial reactions. In aqueous electrolyte solutions, harsh anodic reactions not only facilitate the extraction of the original alkali metal ions (e.g., Li+) but also allow the insertion of foreign cations, including H+, into the Li vacancy sites. Activating the oxide under stringent conditions leads to the participation of the oxygen lattice in the electrode reaction. These approaches have deepened our understanding of the characteristics of LTMO and broadened the scope of applications.
3. LTMO as an electrocatalyst for oxygen evolution
The OER from an aqueous electrolyte solution has been developed as the counterpart reaction of green-fuel processes such as hydrogen evolution (HER), carbon dioxide reduction, and nitrate reduction. The OER has gained wide utilization due to the availability of simple and low-cost anodic processes that can be coupled with green fuel in the aqueous electrolyte solution. However, the sluggish nature of the OER governs the overall kinetics in green-fuel electrochemistry. For instance, in the production of green hydrogen through water splitting, one H2 molecule is formed via a two-electron transfer reduction. In comparison, the OER process involves a four-electron transfer for the evolution of one O2 molecule (Fig. 2(a)). These multiple electron-transfer processes in the OER limit the overall efficiency of the process.31,32 Therefore, electrocatalysts are necessary to improve the OER kinetics. While RuOx and IrOx are known to be efficient OER catalysts, their high costs make them impractical for widespread use. Alternatively, various transition metal oxides, including LTMO (e.g., LixCoO2 and NaxCoO2 (0 < x < 1)),33–36 spinel, and perovskite structures, have been developed and utilized as OER catalysts. These cost-effective catalysts can be tailored through alkali metal ion extraction,12 element doping,37 and surface reconstruction.34 Although we focus on the LTMO catalyst for the OER in this perspective (Section 3.3), the fundamental mechanisms and descriptors of the OER are first introduced in Sections 3.1 and 3.2, and are applied for all transition metal oxide catalysts in the OER.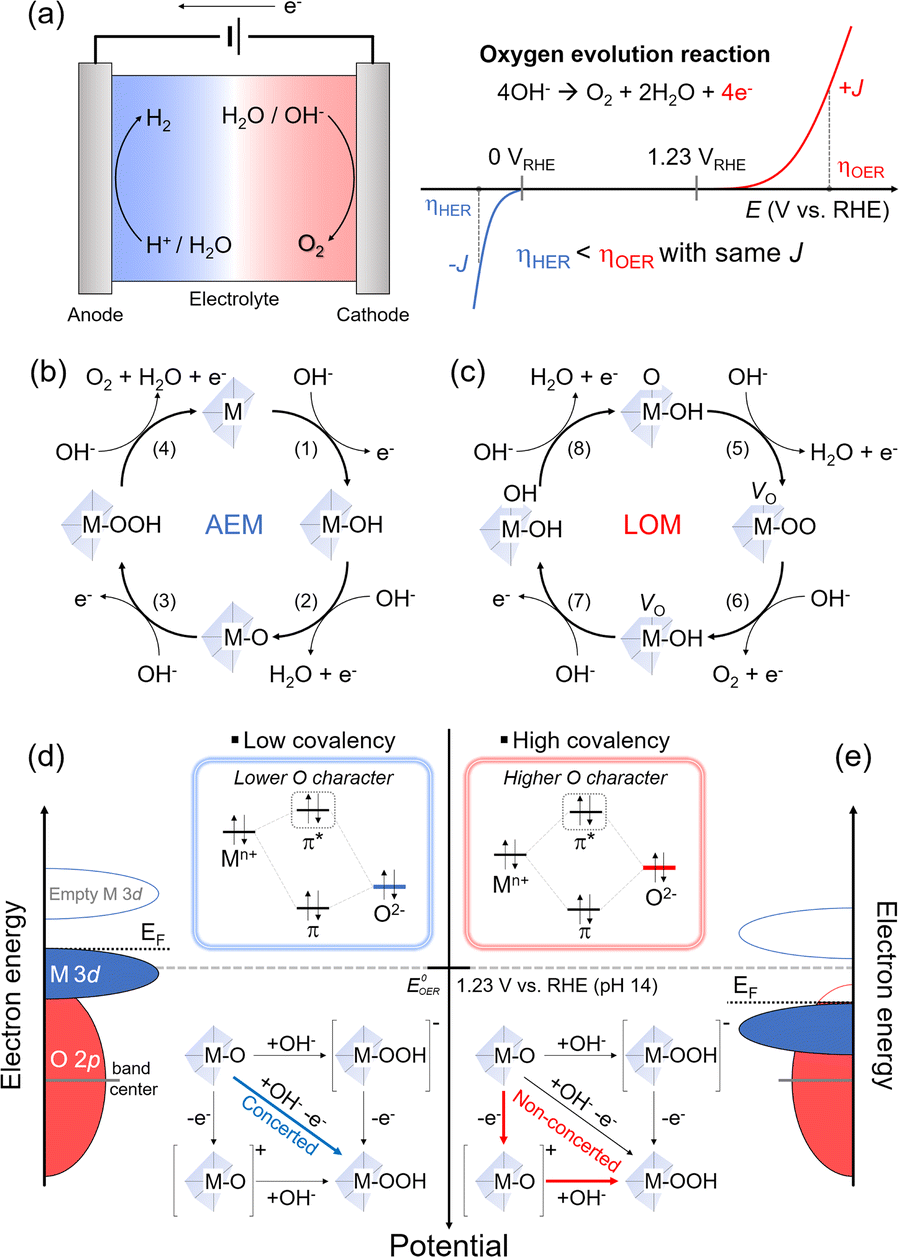 | ||
| Fig. 2 Two principal OER processes. (a) Schematic illustration of the water splitting cell and I–V curve for the HER and OER. Mechanism scheme of the 4-electron transferring oxygen evolution reaction by (b) the adsorbate evolution mechanism (AEM) and (c) lattice oxygen mediated (LOM) pathway. Schematic band diagram of (d) low covalent and (e) high covalent transition metal oxides. The concerted and non-concerted one-electron transfer processes are illustrated, respectively. | ||
3.1. OER processes: adsorbate evolution vs. lattice oxygen-mediated mechanisms
Alkaline electrolyte solutions, which are enriched in hydroxide ions (OH−), have been extensively utilized for the OER due to the instability of most transition metal oxide catalysts in acidic conditions and the slower electrochemical kinetics in neutral conditions. In alkaline environments, the OER occurs through four steps involving OH− adsorption and electron transfer processes, as depicted in eqn (2)–(5)3,38,39 (Fig. 2(b)). Provided that the oxide surface of transition metal oxide catalysts is very stable and inert during the OER, we only consider the adsorption of OH− (or H2O) above M of the catalysts as a key descriptor. Following electron transfer is faster than the adsorption as typically accompanied by the proton coupling process.3,38 Thus, the Mn+ acts as the active site, where OH− is bound via the one-electron transfer (eqn (2)). The subsequent OH− adsorption steps and proton-coupled electron transfer form M–O with H2O leaving, followed by formation of M–OOH as the intermediate (eqn (3) and (4)), and the last step evolves O2 gas with H2O leaving (eqn (5)). This process was indicated to follow the ‘adsorbate-evolution mechanism’ (AEM). The activity of OER catalysts is typically predicted from the free energy difference between M–OH and M–O intermediates (eqn (2) and (3)) in density functional theory (DFT) calculations and showed a scaling relationship with different catalytic materials and intermediates.38| M + OH−(aq) → M–OH + e− | (2) |
| M–OH + OH−(aq) → M–O + H2O(l) + e− | (3) |
| M–O + OH−(aq) → M–OOH + e− | (4) |
| M–OOH + OH−(aq) → M + O2(g) + H2O(l) + e− | (5) |
Unlike the assumption of the AEM for the ideal catalyst structures, practical transition metal oxide structures are imperfect and have many defects in the as-prepared states and during OER processes. These crystal defects in oxide40–43 play a significant role as the active sites and in surface reconstruction34,36,44 during the OER also change the activity. This suggests that the oxide is also involved in OER activity and has developed an alternative mechanism. As the transition metal oxide surface comprises O–M–OH in the alkaline solution, the first electron transfer and OH− adsorption form O–M–O and H2O. Concurrently, the lattice oxygen of the oxide diffuses to the deprotonated oxide to participate in O–O coupling, while leaving behind the oxygen vacancy, denoted as VO. Thus, the oxide surface becomes VO–M–OO (eqn (6)). The subsequent electron-transfer process produces O2 gas through the removal of the O–O lattice, called ‘lattice oxygen redox activation (O2−/O2)′. The catalyst surface retains HO–M–VO by adsorption of OH− (eqn (7)). The vacancy is then filled with another OH− and electron transfer, resulting in HO–M–OH (eqn (8)). The subsequent deprotonation with the fourth electron transfer recovers the catalyst to the original form (eqn (9)). A substantial difference from the AEM is the participation of lattice oxygen in the OER and the formation of a VO intermediate. This process is indicated to follow the ‘lattice oxygen-mediated mechanism’ (LOM), and the above processes are summarized in the following four consecutive equations.4,45,46 (Fig. 2(c))
| O–M–OH + OH−(aq) → VO–M–OO + H2O(l) + e− | (6) |
| VO–M–OO + OH−(aq) → HO–M–VO + O2(g) + e− | (7) |
| HO–M–VO + OH−(aq) → HO–M–OH + e− | (8) |
| HO–M–OH + OH−(aq) → O–M–OH + H2O(l) + e− | (9) |
For the LOM, the lattice oxygens (in other words, oxygen ligands for Mn+) should be activated, and this activation is determined from the EF position lying in the O 2p band. Namely, as the oxidation lowers EF, a part of the O 2p band below the EF initiates the ligand oxygen redox activation.46 This concept is applied for catalyst designs to modulate electronic structure. For example, the O 2p band upshifts close to EF by lattice distortion,41,47 or the transition metal d band and EF downshift using the high valence state of transition metals.48,49 These approaches cause a significant band overlap between the M 3d and the O 2p (without considerable extraction of alkali metal ions in the case of LTMO) and enhance the hybridization between Mn+ and the oxygen ligand, indicating strong ‘covalency’ of the Mn+–O bond.
The concept of covalency is developed to explain the enhanced OER kinetics based on the oxide surface. It refers to hybridized orbitals that mix the M 3d t2g and O 2p molecular orbitals. A large energy gap between M and O orbitals leads to shallow hybridization and causes low covalency (Fig. 2(d)). This indicates more ionic property in the orbital and low O character in π* orbitals in hybridization. A typical transition metal oxide catalyst shows low covalency and stronger ionic character. In addition, as the EF and the thermodynamic OER potential (EoOER) are close to the occupied M 3d t2g band, the Mn+ character determines the catalyst property. However, hybridized orbitals are altered when the M 3d t2g band moves downward and close to the O 2p band; when the M 3d t2g orbitals overlap with the O 2p deeply, the covalency becomes stronger (Fig. 2(e)). The π* orbitals have a strong O character in the hybridization, thus making the oxygen lattice participate in the redox reaction. The LOM is predominant as the EF lies in the O 2p band and is lower than EoOER while the electronic conductivity is high.
When the EF is located below EoOER, the surface charge is built in equilibrium with the electrolyte solution. The negative charges, e.g., electron (from the electrode) and OH− (from the electrolyte solution), are accumulated at the metal oxide catalyst surface. In the presence of the highly electronic conductive catalysts (e.g., semi-metal or metal), the OH− adsorption rate becomes the rate-determining step due to deprotonation or acid–base pre-equilibrium process4 (Fig. 2(e)). However, this OH− adsorption process becomes faster with increasing OH− concentrations at high pH.46,50 Thus, the LOM is highly sensitive to the solution pH; low OH− concentrations at lower pH limit the OER kinetics relative to the electron transfer in the electronically conductive catalyst.14
The above two mechanisms, AEM and LOM, rely on different key factors determining OER activity and guiding the design of OER catalysts. Conversely, the OER process of new catalysts can be addressed by the valence state (n+) of Mn+, pH-dependent OER activity, isotopic labeling of oxygen in catalysts or water,46 oxygen stoichiometry of catalysts,48,50 structural defects of catalysts,41,48 and so on.
3.2. Descriptors of OER activity
Identifying OER descriptors is pivotal to predicting the activity trend and guiding catalyst design principles. Here we introduce four descriptors of transition metal oxide catalysts.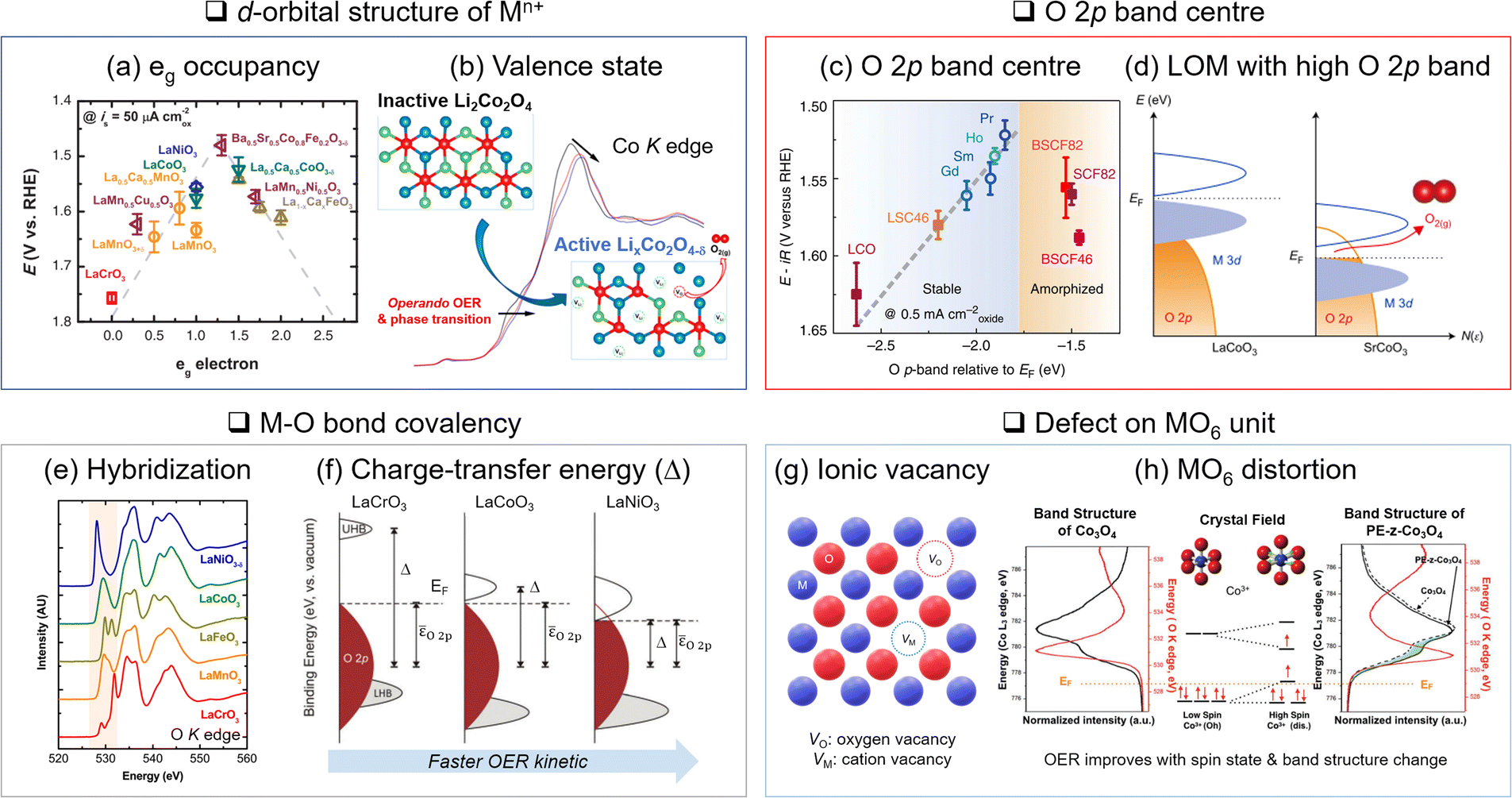 | ||
| Fig. 3 Descriptors of OER activity. (a) Relationship between eg occupancy and OER activity for perovskite oxide. Adapted with permission from ref. 31. Copyright 2011 The American Association for the Advancement of Science. (b) In situ monitoring of valence state change through X-ray absorption spectroscopy and active phase generation on Li2Co2O4. Adapted with permission from ref. 57. Copyright 2019 American Chemical Society. (c) Higher OER activity with a closer O 2p band centre position to the Fermi level (EF). Adapted with permission from ref. 72. Copyright 2013 Springer Nature. (d) Lattice oxygen mediated (LOM) OER activated by the EF position in the O 2p band. Adapted with permission from ref. 46. Copyright 2017 Springer Nature. (e) Estimation of metal–oxygen bond hybridization with O K-edge X-ray absorption. Adapted with permission from ref. 80. Copyright 2014 American Chemical Society. (f) Relationship between covalency and charger transfer energy enhancing OER activity. Adapted with permission from ref. 14. Copyright 2017 Royal Society of Chemistry. (g) Schematic illustration of ionic vacancy. (h) MO6 distortion and corresponding changes in molecular orbital and electronic band structure. Adapted with permission from ref. 87. Copyright 2018 John Wiley and Sons. | ||
The valence and spin states of Mn+ in the d orbital also determine M and oxygen ligand interactions. Another important aspect is the valence state (n+) of Mn+. The OER activity is typically better with higher valence states of M. The delithiated LCO (Li0.5CoO2), where Co3+ and Co4+ co-exist, improves electrophilicity, electrical conductivity, Co–O bond covalency, and OH− binding affinity compared to the pristine LCO where Co3+ only exists.12 This is attributed to the downshift of the d band and EF level with the increasing valence state of Co. The lowered d band can be overlapped with the O 2p band and enhances the Co–O covalency. In addition, the lowered EF activates the lattice oxygen.49 Thus, the higher valence state of Mn+ increases OER activity via the LOM.
The valence state can also be tuned by cation doping48 or anodic potential increase.49,55,56 The extraction of Li+ or Na+ from LTMO is a good example, where the valence state of M is increased.57–59 Zhang and coworkers reported that spontaneous delithiation on spinel Li2Co2O4 oxidized Co3+ to Co4+ below or at OER potentials and created an OER active surface57 (Fig. 3(b)).
To observe the alternation of the valence state of Mn+ during the OER, in situ/operando X-ray absorption near edge structure (XANES) spectroscopy is a suitable tool.60 In addition, extended X-ray absorption fine structure (EXAFS) spectroscopy also identifies the local structures of catalysts, including the neighbor atom distance and the coordination numbers, which are sensitively changed under the OER conditions. Thus, these X-ray absorption spectroscopies have been widely utilized to address the d-orbital structures.
XANES in O K-edge reveals the electron transition from the O 1s to the M 3d–O 2p hybridized band and addresses the M–O bonding character. Shao-Horn and coworkers identified the origin of the OER activities of metal oxide catalysts, attributing them to their different covalency, although they have the same eg occupancy based on XANES80 (Fig. 3(e)). The energy gap between the unoccupied M 3d–O 2p band centre and the occupied O 2p band centre is also estimated to be the charge transfer energy.14 The partial density of states for each M 3d–O 2p and O 2p band was obtained by O Kα X-ray emission and O K-edge X-ray absorption, respectively. The smaller charge transfer energy indicates stronger covalency and improves OER kinetics (Fig. 3(f)).
Distortion of the MO6 unit is also critical. Distortion defects are generally formed by lattice mismatches on grain boundaries,41 lattice expansion/compression,40,47,87,88 and A-site cation vacancies on perovskite.42 The distortion of the octahedral structure engenders the d-orbital splitting according to ligand field theory. It changes the spin state and band structure to expedite the charge transfer to the OER intermediates. For instance, the surface lattice expansion of Co3O4 induced a high spin state Co3+ (t2g4eg2) and increased eg occupancy, which optimized the binding strength of intermediates to the catalyst surface87 (Fig. 3(h)).
3.3. Applications of LTMO as an OER electrocatalyst
This section focuses on the LTMO structure and various LTMO designs to improve OER activity. The representative LTMO examples are LCO and NaCoO2. Shao-Horn and coworkers investigated the OER of LCO in different pH electrolyte solutions89 (Fig. 4). Using cyclic voltammetry (CV), the Li+ extraction (eqn (1)) was observed at 0.9 V vs. NHE in neutral KxH3−xPO4 solutions (pH 7) and at 0.7 V vs. NHE in alkaline KOH (pH 13) solution36,90,91 (Fig. 4(a and b)). After the delithiation, the OER engaged at 1.7 V (pH 7) and 1.5 V vs. RHE (pH 13). Unfortunately, OER activity was reduced during cycling due to surface deformation, from the layered structure to the non-active spinel structure at pH 7 (Fig. 4(c and d)) or amorphousness at pH 13 (Fig. 4(e)). Further, the OER activity and the catalytic stability of LTMO have been improved through doping foreign elements, nanostructuring catalysts, and in situ surface reconstruction during the OER. Table 1 shows representative examples of LTMO catalysts and summarizes their overpotential, Tafel slope, and stability.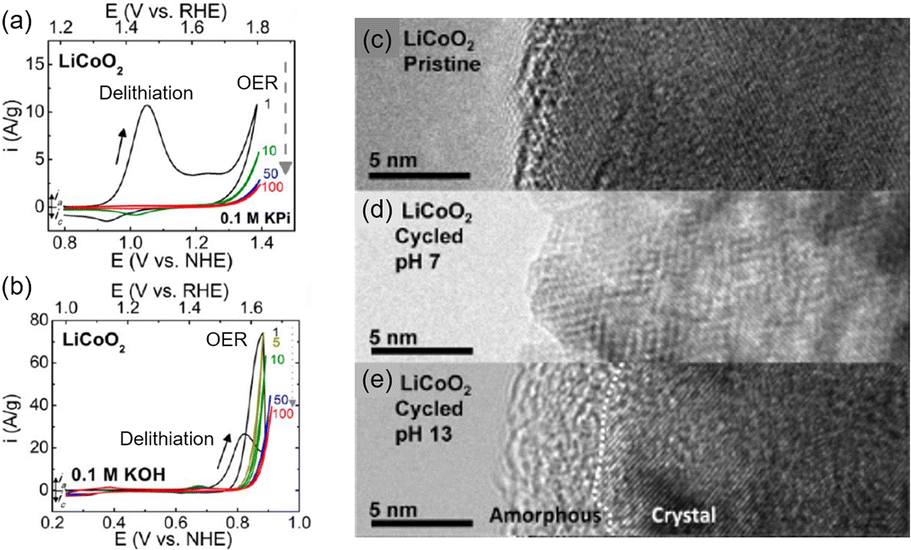 | ||
| Fig. 4 Cyclic voltammogram (CV) of LiCoO2 (LCO) in (a) neutral and (b) alkaline electrolytes. (c–e) TEM image of LCO (c) pristine, (d) after cycling at pH 7, and (e) after cycling at pH 13. Adapted with permission from ref. 89. Copyright 2012 American Chemical Society. | ||
| Electrocatalyst | Overpotential (mV)/Ja | Tafel slope (mV dec−1) | Stabilityb | Electrolyte | Substrate | Ref. |
|---|---|---|---|---|---|---|
| a J: current density at the overpotential. ‘geo’ = current normalization with the geometrical surface area of the substrate. ‘ox’ = current normalization with oxide surface area. b Stability tests were examined using CV cycling, chronopotentiometry (CP), or chronoamperometry (CA). | ||||||
| LiCoO2 | 440/10 mA cmgeo−2 | 98 | — | 0.1 M KOH | Glassy carbon | 47 |
| LiCoO2 | 430/10 mA cmgeo−2 | 89 | — | 0.1 M KOH | Glassy carbon | 37 |
| LiCoO2 | 360/0.1 mA cmgeo−2 | 48 | — | 0.1 M KOH | Carbon paper | 12 |
| De-LiCoO2 | 295/0.1 mA cmgeo−2 | 50 | 1000 cycles | 0.1 M KOH | Carbon paper | 12 |
| LiCo0.33Ni0.33Fe0.33O2 | 320/0.1 mA cmgeo−2 | 45 | — | 0.1 M KOH | Carbon paper | 12 |
| De-LiCo0.33Ni0.33Fe0.33O2 | 240/0.1 mA cmgeo−2 | 35 | 1000 cycles | 0.1 M KOH | Carbon paper | 12 |
| NaCoO2 | 388/10 mA cmgeo−2 | 51 | — | 1 M NaOH | Glassy carbon | 61 |
| NaCoO2 | 380/10 mA cmgeo−2 | 113.4 | — | 1 M KOH | Carbon paper | 62 |
| Na0.67CoO2 | 290/10 mA cmgeo−2 | 39 | 5.56 h @ 1.6 V vs. RHE | 0.1 M KOH | Glassy carbon | 33 |
| Na0.75CoO2 | 370/10 mA cmgeo−2 | 49 | 5000 cycles | 1 M NaOH | Glassy carbon | 61 |
| Na0.6CoO2 | 392/10 mA cmgeo−2 | 53 | — | 1 M NaOH | Glassy carbon | 63 |
| Mg-doped LCO-NS | 329/10 mA cmgeo−2 | 33.8 | — | 1 M KOH | Glassy carbon | 64 |
| LiCo0.8Fe0.2O2 | 350/10 mA cmgeo−2 | 50 | 5 h @ 10 mA cmgeo−2 | 0.1 M KOH | Glassy carbon | 37 |
| Na0.86Co0.95Fe0.05O2 | 450/10 mA cmgeo−2 | 60 | 3 h @ 5 mA cmgeo−2 | 0.1 M KOH | Glassy carbon | 65 |
| Na0.67Mn0.5Co0.3Fe0.2O2 | 390/10 mA cmgeo−2 | 67 | 2 h @ 5 mA cmgeo−2 | 0.1 M KOH | Glassy carbon | 66 |
| Ag-doped Na0.7CoO2 | 236/10 mA cmgeo−2 | 48 | 30 h @ 1.522 V vs. RHE | 1 M KOH | Carbon paper | 62 |
| 1% La-doped LCO | 330/10 mA cmgeo−2 | 48 | 10 h @ 10 mA cmgeo−2 | 0.1 M KOH | Glassy carbon | 47 |
| LCO-NS | 410/10 mA cmgeo−2 | 88 | 6 h @ 1.7 V vs. RHE | 0.1 M KOH | Glassy carbon | 67 |
| AD-LCO | 184/10 mA cmgeo−2 | 35.4 | 200 h @ 50 mA cmgeo−2 | 1 M KOH | Glassy carbon | 68 |
| LCO-NS/NS | 289/10 mA cmgeo−2 | 75.8 | 20 h @ 1.52 V vs. RHE | 1 M KOH | Carbon cloth | 69 |
| Pt-LCO-NS | 285/10 mA cmgeo−2 | 46.8 | 20 h @ 10 mA cmgeo−2 | 1 M KOH | Glassy carbon | 70 |
| Cs+-inserted LCO | 392/10 mA cmgeo−2 | 47.1 | 8 h @ 10 mA cmgeo−2 | 0.1 M CsOH | Glassy carbon | 36 |
| K+-inserted LCO | 416/10 mA cmgeo−2 | 60.0 | 2 h @ 10 mA cmgeo−2 | 0.1 M KOH | Glassy carbon | 36 |
| α-Li2IrO3 | 290/10 mA cmox−2 | 50 | 40 h @ 10 mA cmox−2 | 0.1 M KOH | Glassy carbon | 71 |
| LiCoO1.8Cl0.2 | 270/10 mA cmgeo−2 | 55.4 | 500 h @ 20 mA cmgeo−2 | 1 M KOH | Glassy carbon | 34 |
| Co3O4 | 460/10 mA cmgeo−2 | 76 | — | 1 M KOH | Glassy carbon | 33 |
| IrO2 | 408/10 mA cmgeo−2 | 109.3 | — | 1 M KOH | Glassy carbon | 68 |
| IrO2 | 450/10 mA cmgeo−2 | 83 | 5 h @ 10 mA cmgeo−2 | 0.1 M KOH | Glassy carbon | 37 |
| IrO2 | 310/10 mA cmgeo−2 | 57 | — | 1 M KOH | Glassy carbon | 33 |
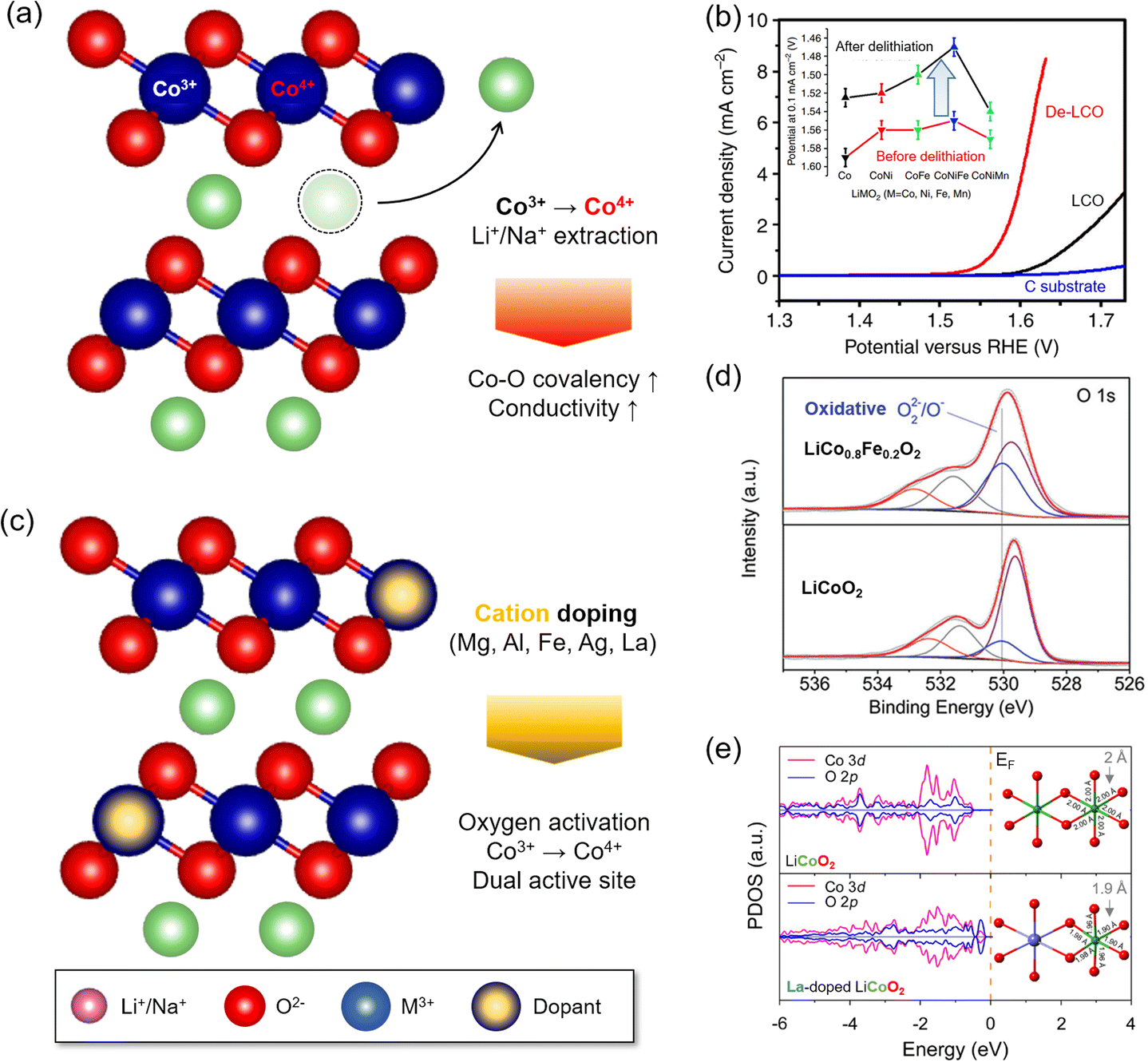 | ||
| Fig. 5 Local structure modulation for OER enhancement of LTMO. (a) Schematic illustration of alkali metal ion extraction. (b) Linear sweep voltammograms (LSVs) of delithiated LCO (De-LCOs) and pristine LCO. The inset indicates the potential value at 0.1 mA cm−2 for various LTMO compositions before and after delithiation. Adapted with permission from ref. 12. Copyright 2014 Springer Nature. (c) Schematic illustration of foreign cation doping. (d) O 1s spectra from XPS analysis indicating the formation of highly oxidative oxygen species by Fe doping. Adapted with permission from ref. 37. Copyright 2015 John Wiley and Sons. (e) DFT simulation of the projected density of states and local geometry for LCO with and without La doping. Adapted with permission from ref. 47. Copyright 2019 American Chemical Society. | ||
Similarly, the OER kinetics of NaCoO2 was enhanced by forming Co4+ through the Na+ deintercalation.33,61,63 Cheng and coworkers suggested the increased VO concentrations and improved electronic conductivity in Na+ deintercalation. In addition, the optimum design was suggested to be ∼40% Na+ deintercalation states (i.e., Na0.6CoO2).63 Ren and coworkers highlighted the role of Co–O covalency in NaxCoO2.61 The O 2p band upshifted toward EF with lower Na+ content, resulting in stronger Co–O hybridization and participation of the oxygen lattice in the OER. They are the central OER descriptors, as described in Section 3.2. Another important aspect was the short O–O distance in the CoO6 unit, observed from Na0.67CoO2.33 Due to the strong Co4+–O2− bond in the Rc space group, two O2− ligands had <2.4 Å distance and easily formed the peroxide ion (O22−) in leaving behind the VO, which was similar to the LOM path in eqn (6). This peroxide evolved O2 gas, and the OER overpotential was only 290 mV at 10 mA cm−2.
The impurity of Fe ions in the electrolyte solution often significantly improved the OER, and this behavior developed the idea of Fe ion doping to layered double hydroxide (LDH) or perovskite oxides.37,65,66 In the LDH electrocatalysts (e.g., nickel (oxy)hydroxide, NiOOH), the Fe dopant served as dynamic OER active sites as Fe ions were dissolved and deposited in LDH repeatedly during the OER.98–100 For the perovskite LaNiO3, the incorporation of Fe ions distorted the local lattice structures, and the occupied Fe 3d states beneath the EF accelerated charge transfer from M3+–O(OH*)− to M4+−OO*2−.93,101 Shao and coworkers showed that substituting 20% Co with Fe in LCO, thus forming LiCo0.8Fe0.2O2, reduced the overpotential to 350 mV at 10 mA cm−2.37 X-ray photoelectron spectroscopy (XPS) revealed partial oxidation of Co3+ to Co4+ and an increased amount of O22− or O−, which might be generated from partial oxidation of the O2− ligand near 530.1 eV (Fig. 5(d)). The electrophilicity of the oxygen ligand and VO generation at the surface caused enhancement of OER activity (Section 3.2.4).50,94,102–104 In addition, the Fe dopant increased the electronic conductivity, demonstrated by the reduced charge transfer resistance. Similar effects from the Fe dopant were also found in Na0.86Co0.95Fe0.05O2 (ref. 65) and Na0.67Mn0.5Co0.3Fe0.2O2,66 where both Co and Fe acted as OER active sites.105,106
Because the dopant size was often mismatched to the host Mn+ size of catalysts, the foreign ion doping imposed the strain on the surface lattice. La doping to LCO shortened the Co–O length (<2 Å) in the CoO6 octahedral unit and distorted the symmetry47 (Fig. 5(e)). This mechanical strain upshifted the O 2p band centre and induced stronger Co–O covalency. As a result, La doping in LCO resulted in a 330 mV overpotential at 10 mA cm−2, which was better than that of LCO (440 mV). A similar effect was also observed from Ag-doped Na0.7CoO2.62
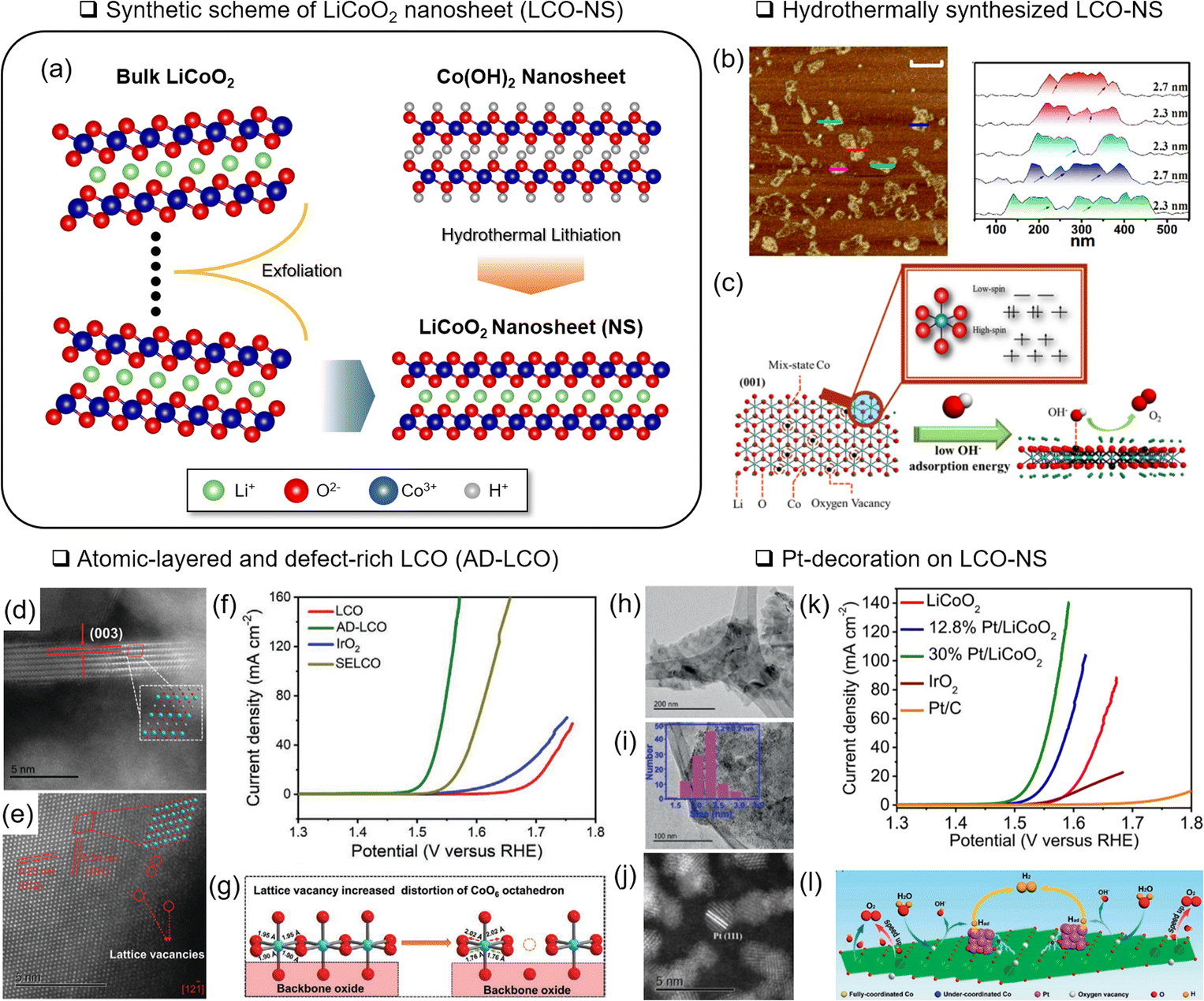 | ||
| Fig. 6 Nanostructuring of LCO. (a) Schematic illustration of an LCO nanosheet (LCO-NS) obtained by an exfoliation and hydrothermal lithiation method. (b) Atomic force microscopy analysis of LCO-NS. (c) Schematic illustration of LCO-NS with rich VO and multiple spin states of Co. Reproduced with permission from ref. 67. Copyright 2017 American Chemical Society. Scanning TEM image of atomic-layered and defect-rich LCO (AD-LCO) for (d) the edge step and (e) basal plane. (f) Corresponding LSV analysis with various reference materials. (g) Schematic illustration of the cation vacancy effect on AD-LCO for the OER. Reproduced with permission from ref. 68. Copyright 2022 John Wiley and Sons. (h–j) Structure analysis of Pt-decorated LCO-NS and TEM analysis of (h) LCO-NS and (i) anchored Pt nanoparticles. (j) Corresponding STEM image. (k) LSV analysis of Pt/LCO with various Pt loading. (l) Schematic illustration of the synergistic effect of Pt/LCO. Reproduced with permission from ref. 70. Copyright 2020 John Wiley and Sons. | ||
Sun and coworkers designed atomic-layered defect-rich LCO, denoted as AD-LCO, using a mechanical shear-assisted exfoliation method.68 The 3–5 nm thick AD-LCO contained cationic Co vacancies and distorted CoO6 (Fig. 6(d and e)). In addition, as the Co valence states became higher (4+), the Co–O covalency became stronger. Modulating the electronic structure decreased the OER overpotential to 280 mV at 10 mA cm−2 (Fig. 6(e and f)).
Along with the nanostructured OER catalysts, the effects of micro-structured substrates69 or decorated nanoparticles were investigated.70,107 Hierarchical growth of LCO-NS on carbon cloth formed microstructures and showed a 289 mV overpotential of the OER at 10 mA cm−2 after delithiation.69 Sun and coworkers attached Pt nanoparticles (around 2.2 nm diameter) to LCO-NS (10–25 nm thickness) and demonstrated a 285 mV OER overpotential at 10 mA cm−2 (ref. 70) (Fig. 6(h–k)). The charge transfer between Pt and Co3+ was presumed to form Pt2+, Co2+, and VO. This VO and under-coordinated Co in LCO-NS served as the active OH− adsorption site (Fig. 6(l)).
Byon and coworkers demonstrated the surface reconstruction of De-LCO by inserting foreign alkali metal ions such as Na+, K+, and Cs+ during the OER process and investigated the related mechanisms.36 Using NaOH or KOH electrolyte solutions, hydrated Na+ or K+ was inserted into De-LCO, creating the Li0.25Na0.33CoO2·(H2O)0.04 and Li0.33K0.17CoO2·(H2O)0.37 structures, respectively. The OER activity of Li0.33K0.17CoO2·(H2O)0.37 was better than that of Li0.25Na0.33CoO2·(H2O)0.04, because less K+ insertion induced higher Co4+ concentration (Fig. 7(a)). These bulk structural reconstructions enhanced the Co4+ concentration and the Co–O covalency, and the AEM governed the OER process. In comparison, little Cs+ was intercalated into De-LCO in the CsOH electrolyte solution, and the resulting Li0.55Cs0.03CoO2·(H2O)0.07 underwent negligible bulk phase transition. This shallow Cs+ insertion preserved the bulk LCO structure, causing better OER stability than the K+ and Na+ intercalations. In addition, Li0.55Cs0.03CoO2·(H2O)0.07 showed the best OER activity despite the small Co4+ concentrations (Fig. 7(b)). This was attributed to the surface strain caused by large-size Cs+, leading to the CoO2 slab edge bending (Fig. 7(c and d)). Its strong pH dependency indicated LOM as the main OER mechanism, distinguished from the above K+ and Na+ intercalated catalysts.
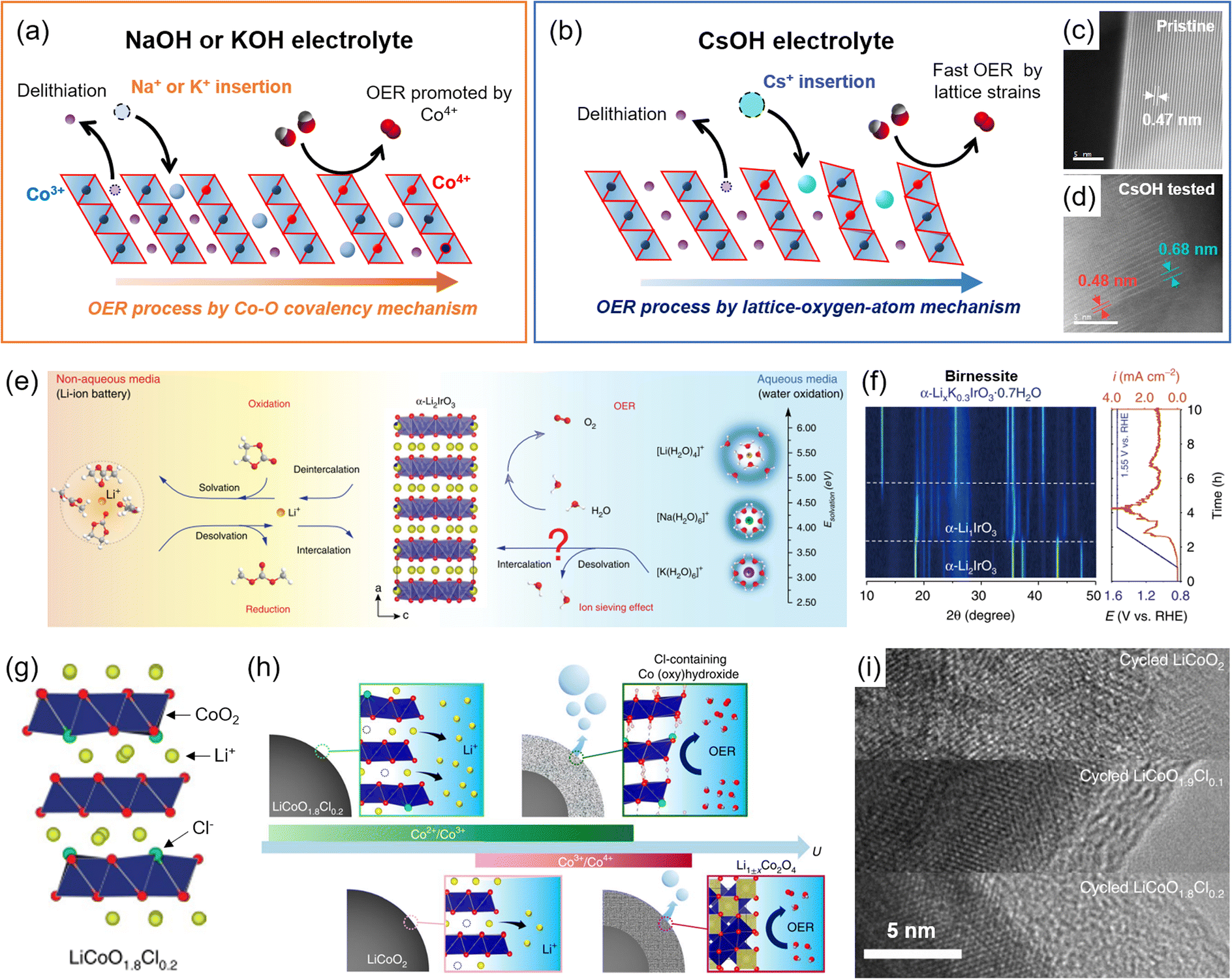 | ||
| Fig. 7 Surface reconstructions on LCO during the OER. (a and b) Schematic illustration of foreign alkali metal ion insertion on the Li+ vacant site with (a) small size ions (Na+ and K+) exhibiting OER enhancement from different Co4+ development and covalency, and (b) large size Cs+ following LOM. Comparison of the interlayer distances of (c) pristine and (d) OER-tested LCO after Cs+ insertion. Adapted with permission from ref. 36. Copyright 2022 Royal Society of Chemistry. (e) Schematic illustration of hydrated K+ intercalation on α-Li2IrO3. (f) Operando XRD analysis for birnessite phase formation during the OER of α-Li2IrO3. Reproduced from ref. 72. Copyright 2020 Springer Nature. (g) Crystal structure of Cl-doped LCO, LiCoO1.8Cl0.2. (h) Schematic illustrations of the surface reconstruction of LCO and LiCoO1.8Cl0.2 during the OER. (i) Corresponding TEM images after OER cycling. Reproduced with permission from ref. 34. Copyright 2021 Springer Nature. | ||
Grimaud and coworkers also reported similar phenomena for the layered α-Li2IrO3 (ref. 71) (Fig. 7(e)). The delithiated α-Li2IrO3 produced an α-Li1IrO3 structure, which had a high-valence state (5+) of Ir5+, and induced hydrated K+ insertion, promoting chemical OER. During this process, α-LixK0.3IrO3·0.7H2O was entirely converted to a birnessite structure and showed OER activity109 (Fig. 7(f)). The hydrated K+ continuously underwent reversible deintercalation and intercalation during the OER process, and this behavior was not observed in Li+ and Na+ electrolytes.
Lim and coworkers doped Cl− into LCO, forming LiCoO1.8Cl0.2, and induced structural reconstruction34 (Fig. 7(g)). The introduction of Cl− reduced Co3+ to Co2+ and caused the irreversible Co2+/Co3+ redox at <1.4 V vs. RHE through delithiation. As a result, a low redox potential developed a new pathway of surface reconstruction (Fig. 7(h)). The LiCoO1.8Cl0.2 formed an amorphous and Cl-doped cobalt (oxy)hydroxide surface during the OER, which prevented the Li+ extraction from the bulk structure and terminated the reconstruction process. This result contrasted with the continuous surface reconstruction of the LCO, becoming the spinel phase of Li1±xCo2O4 (Fig. 7(i)). Thanks to the highly OER-active Cl-doped cobalt (oxy)hydroxide and the increased conductivity of LiCoO1.8Cl0.2 in the bulk structure, the reconstructed catalysts exhibited a 270 mV overpotential at 10 mA cm−2.
4. LTMO as cathodes for aqueous LiBs
In this LiB section, we discuss the Li+ intercalation and deintercalation process in LTMO electrodes, which competes with H+ intercalation in an aqueous electrolyte solution.In non-aqueous media, LTMO serves as the stable cathode in LiBs. The representative LCO electrode provides a capacity of 140 mA h g−1 by extracting 50% of total Li+, which occurs at around 4.0 V vs. Li/Li+.124 Layered NCM further improves Li+ storage capacity by storage of two Li moieties through Ni2+/Ni4+ redox chemistry. Mn helps maintain the thermal stability of NCM, while Co provides high electronic conductivity.125 In addition, a small Co content in NCM compared to that in LCO diminishes the cost and toxicity. NCM811 (811 represents the ratio of transition metals Ni, Co, and Mn) has 200 mA h g−1 capacity and 3.88 V (vs. Li/Li+) charging voltage.126,127 Nonetheless, non-aqueous LiBs suffer from the risk of catching fire and the high cost of electrolyte solutions. They are particularly unsuitable for grid-scale ESSs.
An aqueous electrolyte solution has been introduced in LiBs to tackle this issue. Dahn and coworkers first reported rechargeable aqueous LiBs with 5 M LiNO3 electrolyte in 1994.128 However, the narrow electrochemical potential window of water, thermodynamically in the range of 2.62–3.85 V vs. Li/Li+ (converted from 0.0–1.23 V vs. RHE), restricts the use of graphite and metallic Li electrodes operating at 0–0.1 V vs. Li/Li+. It is also the reason for the significantly low energy density. For the cathode side, LTMO is unstable in water because Li+ intercalation competes with H+ intercalation, which originates from the dissociated water in neutral and weakly alkaline conditions. Thus, the interfacial reaction of LTMO with water provides significant challenges in aqueous LiBs. Here, we focus on the LTMO-based Li+/H+ insertion chemistry in aqueous cathodes.
4.1. H+ intercalation competing with Li+ intercalation in salt-in-water electrolyte solutions
The layered-structure LCO undergoes Li+ intercalation and deintercalation below the OER potential and forms Li1−xCoO2 where x is between 0.5 and 1. However, LiV3O8|LCO cells with a saturated LiNO3 electrolyte in water provided only 55 mA h g−1 capacity at an average charging/discharging potential of 4.38 V vs. Li/Li+, where LiV3O8 and LCO are the anode and cathode, respectively114,115 (Table 2). Water causes low capacity and rapid capacity fading for cycling. As another example, NCM electrodes had higher theoretical capacity than LCO and comparable charging/discharging voltages. However, an LiNi0.81Co0.19O2 or NCM111 electrode coupled with an LiV3O8 anode delivered only 45 mA h g−1 with 1–2 M Li2SO4 aqueous solution.117,118 Low NCM capacities relative to LCO were due to their extreme water sensitivity and severe Ni ion dissolution.126| Cell types | Cell configuration | Electrolytea | E range (V) | J or C-rateb | Capacity retention | Initial charge capacity (mA h g−1) | CE (%) | Cycle number | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Sat'd = saturated. b J stands for current density. c GPE: PVDF (poly(vinyl difluoride))–PMMA (poly(methyl methacrylate))–PVDF saturated in 1 M LiClO4. d LISICON: Li2O–Al2O3–SiO2–P2O5–TiO2–GeO2. e EMImTFSI: 1-ethyl-3-methylimidazolium TFSI. f TMSB: tris(trimethylsilyl) borate. | |||||||||
| Half cell | LiCoO2 (Ag/AgCl RE, Li0.5Mn2O4 CE) | 5 M LiNO3 | 3.59–4.19 (vs. Li+/Li) | 1C | — | 105 | 99.70 | 90 | 110 |
| LiCoO2 (Ag/AgCl RE, Li0.5Mn2O4 CE) | 5 M LiNO3 | 3.59–4.24 (vs. Li+/Li) | 1C | 82–86% | 135 | 82.00 | 200 | 111 | |
| LiNi0.6Mn0.2Co0.2O2 (Ag/AgCl RE, Pt foil CE) | 20 M LiTFSI | 3.25–4.45 | 20 mA g−1 | — | 152 | 81 | 3 | 112 | |
| Full cell | Activated carbon|LCO | 0.5 M Li2SO4 | 0–1.8 | 7C | — | 143 | 92.50 | 40 | 113 |
| LiV3O8|LiCoO2 | Sat'd LiNO3 | 0.5–1.5 | 1C | 65% | 55 | — | 40 | 114 | |
| LiV3O8|LiCoO2 | Sat'd LiNO3 | 0.5–1.5 | 0.2 mA cm−2 | — | 60 | — | 12 | 115 | |
| Li metal(GPEc + LISICONd)|LiCoO2 | 0.5 M Li2SO4 | 3.5–4.3 | 150 mA g−1 | — | 130 | — | 20 | 116 | |
| Li9/7−xNb2/7Mo3/7O2|LiCoO2 | 3 m Li2SO4 | 0–1.5 | 0.5C | 74% | 44 | 99.80 (20th) | 500 | 17 | |
| LiV3O8|LiNi0.81Co0.19O2 | 1 M Li2SO4 | 0.5–2 | 1 mA cm−2 | 40% | 45 | — | 100 | 117 | |
| LiV3O8|LiNi1/3Co1/3Mn1/3O2 | 2 M Li2SO4 | 0.5–1.5 | 0.2 mA cm−2 | 54.70% | 55.2 | — | 10 | 118 | |
| LiV2.9Ni0.05Mn0.05O8|LiNi1/3Co1/3Mn1/3O2 | 1 M LiNO3, 5 M LiNO3, Sat'd LiNO3 | 0.5–1.5 | 0.5C | 65% | 98.2 | 92 | 50 | 119 | |
| LiV3O8|10 wt% PPy–LiNi1/3Co1/3Mn1/3O2 | 5 M LiNO3 | 0–1.4 | 0.2 mA cm−2 | 71% | 70 | — | 50 | 120 | |
| Li4Ti5O12|Ni0.8Mn0.1Co0.1O2 | 40 m LiTFSI + 20 m EMImTFSIe | 0.8–2.75 | 1C | — | ∼67 | 99.4 | 300 | 121 | |
| Mo6S8|LiCoO2 | 21 m LiTFSI + 0.1 wt% TMSBf | 1–2.5 | 2.5C | — | ∼40 | — | 1000 | 122 | |
| Li4Ti5O12|LiCoO2 | Li(TFSI)0.7(BETI)0.3·2H2O | 1.6–2.6 | 10C | 75% | 55.3 | ∼100 | 200 | 123 | |
There were several demonstrations of adverse water effects on LTMO. Thin LCO electrodes suffered from H2O vapor in all-solid-state Li cells, resulting in 21 mA h g−1 capacity for the first cycle, which was only 20% of the capacity for the H2O vapor-free LCO capacity13 (Fig. 8(a and b)). The depth profile evaluated by resonant nuclear reaction analysis showed the presence of hydrogen in 20–30 nm depth of LCO (Fig. 8(c)). In addition, when NCM532 (without delithiation) was stored at high humidity over one month, its capacity was reduced;129 galvanostatic testing in the non-aqueous electrolyte solution exhibited a lower charging capacity of this NCM532 at 166 mA h g−1 (vs. 198 mA h g−1 for the fresh NCM532) and lower coulombic efficiency (CE, indicating reversibility during charging and discharging processes) at 86.9% (vs. 90.4% for the fresh one). This was attributed to Li+ exchange with H+ during the storage period. Li+ migrated outward of NCM and formed LiOH and Li2CO3 on the electrode surface, while H+ from humid air moved inside LCO. The resulting structural deformation destabilized NCM. However, the H+ insertion rate for non-delithiated NCM was slower than for the delithiated one.117,118 It should also be noted that H+ intercalation is preferred in the close-packed hexagonal stacking LTMO compared to spinel (e.g., LiMn2O4) and olivine (e.g., LiFPO4) structures.130
 | ||
| Fig. 8 Adverse water effect on LCO. (a) Schematic illustration of H2O-vapor treatment of the LCO electrode. (b) Discharge voltage profile of H2O-vapor exposed LCO (red) and pristine LCO (black) with the current of 2 and 200 μA cm−2 for solid and dashed lines, respectively. Li3PO4 solid electrolyte and metallic Li anode were used. (c) Hydrogen depth profile of 40 nm LCO film from resonant nuclear reaction analysis. Reproduced with permission from ref. 13. Copyright 2023 American Chemical Society. (d) Energies above hull calculated by PBE + U for the phase diagram (0 K) of the HyLixCoO2 (x + y ≤ 1) structure. (e) O3 structure of LCO. (f) Partial stacking displacement of Li0.75H0.125CoO2 with Co (blue), O (red), and Li (green). Adapted with permission from ref. 133. Copyright 2021 American Chemical Society. | ||
The first-principles density functional theory (DFT) calculations showed that when H+ is inserted between the CoO2 layers of the delithiated LCO, it is bound to the CoO2 lattice and forms an O–H covalent bond131,132 (Fig. 8(e and f)). H+ and Li+ are stabilized at different stacking sites; H+ is inserted into the prismatic sites, while Li+ is located in the octahedral sites of the delithiated LCO.133 Unfortunately, the O–H bond formation at the prismatic sites raises the energetic barrier of Li+ diffusion and restricts the Li+ diffusion pathway.132 Conversely, it is also predicted that the O–H bond prevents the oxidation of the oxygen lattice, suppressing OER activity related to LOM (see Section 3).131 The computational model reveals the phase transition from O3 to P3 when Li0.75H0.125CoO2 is formed. However, this transformation is incomplete because the octahedral site of Li+ is pronouncedly distorted, and a high concentration of vacancies appears (Fig. 8(d)). It turns out that the total concentrations of Li+ and H+ cannot become unity by forming vacancies.133
In aqueous LiBs, water (∼55 M) is an unlimited H+ source in an aqueous medium compared to a limited Li+ from the electrolyte (typically ∼1 M in a salt-in-water system). Because H+ has a smaller volume size and faster mobility than Li+, it damages the LTMO structure seriously. The fatal H+ effect was found in low pH solutions and even in neutral conditions.16 LCO underwent significant capacity loss in the initial cycles at pH < 7. In contrast, better charging and discharging cyclability in LCO were observed at pH > 9 with 1 m (mol kg−1) Li2SO4.17,132 However, because a strong alkaline solution engendered the OER and the LTMO served as OER catalysts in this condition, the pH of the aqueous electrolyte solutions was typically adjusted to a mildly alkaline condition (pH 9–11).16
To shed light on the H+ insertion contending with Li+, it is imperative to understand interfacial reactions at the aqueous electrolyte solutions/LTMO surface. Above the point of zero charge (PZC) or applied positive bias, water and anions sit on the topmost LTMO surface, called the inner Helmholtz plane (IHP), and form a few interfacial layers regime. The water molecules are the majority in the IHP and become the potential source of H+. By comparison, anions of electrolyte salt are minor in the typical salt-in-water electrolyte solution. Byon and coworkers recently revealed that anions protected the LTMO surface from the H+ insertion. To demonstrate the anionic electrolyte effects, sulfate (SO42−), nitrate (NO3−), perchlorate (ClO4−), and bistriflimide (TFSI−) were examined with 0.5–1 m concentrations.17 None of these anions either formed a cathode electrolyte interface (CEI) or significantly changed the solution pH. However, LCO cell performances were significantly different. Cyclability with 0.5 m Li2SO4 outperformed, revealing the constant capacity for 10 cycles (Fig. 9(a)). Electrochemical impedance spectroscopy (EIS) showed a single semicircle, indicating the Li+ charge-transfer resistance during the charging and discharging process (Fig. 9(c)). In contrast, TFSI− exhibited a pronounced capacity decay under the same condition (Fig. 9(b)). Interestingly, EIS demonstrated additional semicircles at the low-frequency region, which belonged to H+ insertion into LCO (Fig. 9(d)). NO3− and ClO4− also showed H+ charge-transfer resistances in EIS, which were, however, moderate compared to TFSI−. These H+ inserting resistances corresponded to capacity decays for 100 cycles, verifying that H+ was the central source for LCO degradation (Fig. 9(e)). The anion-dependent LCO stability is presumably explained by the Hofmeister series and kosmotropic traits. SO42− has a strong kosmotropic character,17 namely, the presence of SO42− in water preserves the hydrogen-bond strength and induces the ordered ice-like structure. Thus, the H+ dissociation is likely difficult due to the strong hydrogen-bonding water network. In sharp contrast, the weak kosmotropic TFSI− (i.e., the strongest chaotropic anion) attenuates the hydrogen-bond strength in water and induces disordered water structures, where H+ dissociation is possibly more favorable.
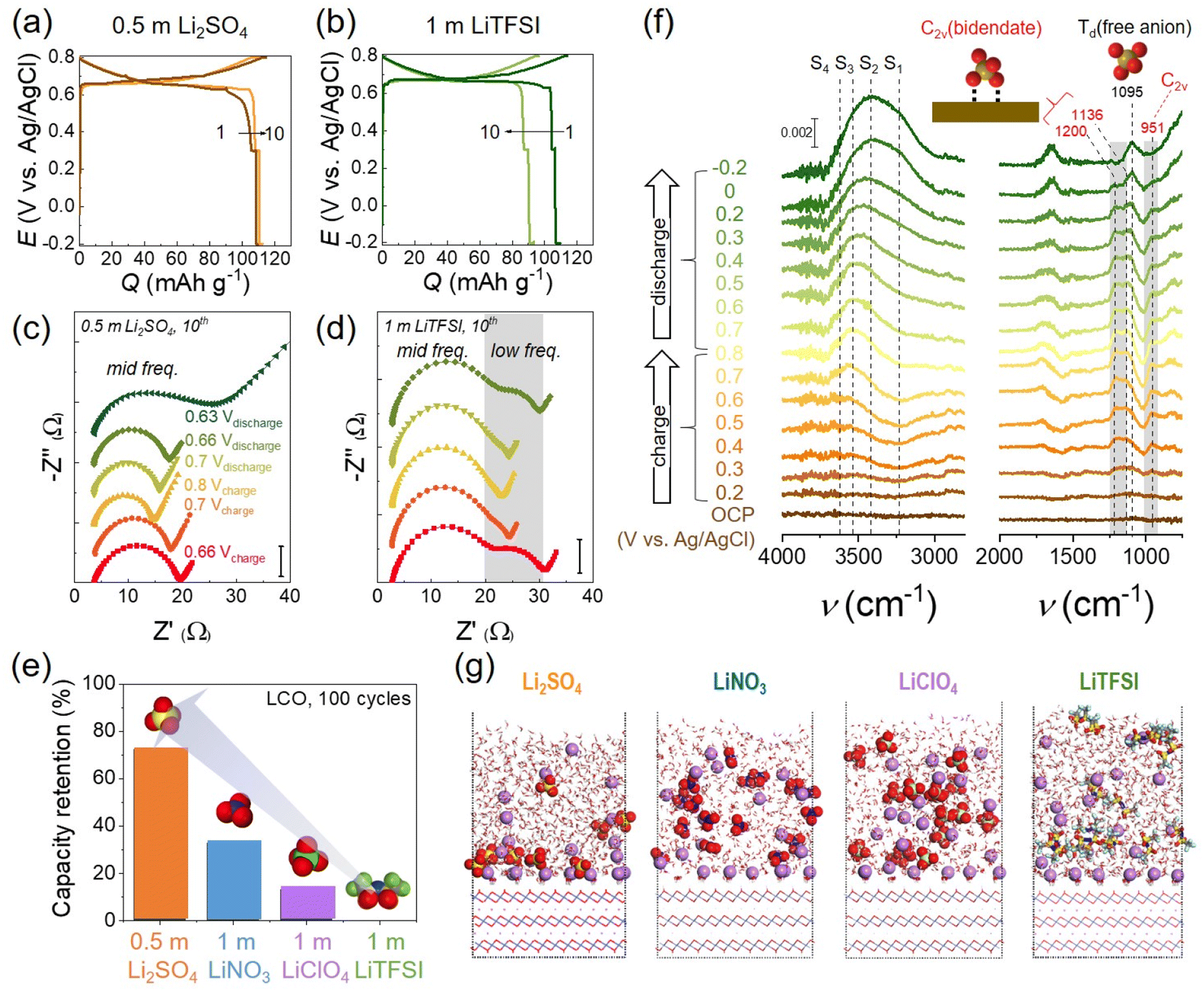 | ||
| Fig. 9 Anion effects on LCO in salt-in-water electrolytes. (a and b) The 1st and 10th galvanostatic cycles of the LCO half-cell at 0.5C in (a) 0.5 m Li2SO4 at pH 9.6 and (b) 1 m LiTFSI at pH 8.5. (c and d) Corresponding Nyquist plot obtained from EIS measurement during the 10th cycle in (c) 0.5 m Li2SO4 and (d) 1 m LiTFSI. EIS was measured during charging at 0.66 V, 0.7 V, and 0.8 V and subsequent discharging at 0.7 V, 0.66 V, and 0.63 V (Li2SO4 only). (e) The capacity retention of LCO for 100 cycles with different electrolytes. (f) In situ electrochemical SEIRAS spectra with 0.5 m Li2SO4. Ice-like water structures are designated as S1 and S2, liquid-like water structures as S3, and disordered free water molecules as S4, respectively. The peak at 1095 cm−1 is assigned to the Td point group of a free anion, SO42−, and the peaks at 951, 1136, and 1200 cm−1 are attributed to the bidentate coordinated C2v point group of SO42−. (g) QM/MM simulations of Li+ and anion adsorption on LCO having a negative surface charge (σ = −11.5 μC cm−2). Li+ (purple), O (red), S (yellow), C (gray), F (cyan), N (blue), and Cl (light green). Reproduced with permission from ref. 17. Copyright 2023 American Chemical Society. | ||
Further, in situ electrochemical surface-enhanced infrared absorption spectroscopy (SEIRAS) revealed the role of SO42− adsorption at the LCO surface and IHP. SO42− was coordinated with the LCO surface to form bidentate coordination (C2v point group), distinct from a typical tetrahedral (Td) free SO42− in the bulk solution (Fig. 9(f), C2v: 951, 1136, 1200 cm−1 and Td: 1095 cm−1). It demonstrated the complete SO42− adsorption on the LCO surface, where the water contacts and possible H+ access were prevented. By comparison, TFSI− adsorption was not evidenced in the IHP using electrochemical SEIRAS. Another concern was the H+ access at slightly below the PZC when SO42− was desorbed from LCO. This condition was often included before reaching the cut-off potential of galvanostatic tests and reasoned for severe capacity loss. Indeed, the SO42− vibration from the Td structure (1095 cm−1) was enhanced, while C2v-associated vibrations were attenuated below 0.2 V vs. Ag/AgCl during discharge in electrochemical SEIRAS (Fig. 9(f)). Mean-field quantum mechanics/molecular mechanics (QM/MM) simulation predicted that SO42− was more concentrated on the LCO surface than other anions (Fig. 9(g)). Below the PZC, Li+ is primarily distributed on LCO and hard Lewis base SO42− easily forms ion pairing with hard Lewis acid Li+ according to the hard and soft acids and bases (HSAB) concept. Thus, SO42− stays on the LCO surface and avoids H+ insertion. In sharp contrast, the soft Lewis base TFSI− is not closely associated with Li+, resulting in the exposure of delithiated LCO to water and H+.
From the above lesson, we can also understand the better performance of LTMO in aqueous LiBs with higher concentrations of electrolyte salts.110,134 The increased anion concentrations give rise to an anion-rich IHP and suppress the H+ insertion. A 3 m Li2SO4 solution extended the electrochemical potential window towards the positive potential compared to a 0.5 m Li2SO4. Further, a 3 m Li2SO4 had the widest potential window compared to 6 m LiNO3, 5 m LiClO4, and 6 m LiTFSI. This was attributed to the strong SO42− adsorption which led to forming bidentate coordination with the LCO surface compared to other anions.17 LCO with 3 m Li2SO4 electrolyte solution showed 87% capacity retention for 1500 cycles, which was better than the 66% retention with 7.5 m LiNO3.134 It was also reported that a 2–3 nm CoO layer was formed on LCO after 500 cycles with 3 m Li2SO4 solution, because of a chemical reaction between LCO and water. This surface layer delayed the LCO structural degradation. In comparison, LCO with 1 M LiNO3 electrolyte created a thicker (5–6 nm) and amorphous CoO layer.134
A similar approach was attempted at NCM. The electrochemical performance of NCM111 was examined with 1 M LiNO3 and saturated (7.5 m) LiNO3 in water.119 The anodic and cathodic peak separating potential (Ep,p) in CV was 0.356 and 0.25 V for 1 M and saturated LiNO3, respectively. It revealed more undesirable chemical reactions with lower electrolyte concentration. LiV2.9Ni0.05Mn0.05O8|NCM111 cells with the saturated LiNO3 solution delivered an initial capacity of 98.2 mA h g−1 at 0.5C and 62.8 mA h g−1 at 3C.
Apart from anions, artificial protective layers were also developed.16 The coating of the lithiated Nafion layer protected the LCO during the initial cycles. The hydrophobic domain of Nafion prevented water access to LCO, while the hydrophilic part, including the sulfonate group, played the role of the Li+ ion channel, which enhanced cyclability for the first 30 cycles with 1 m LiTFSI in water. However, the Nafion layer eventually underwent water swelling during long-term cyclability, causing inevitable LCO deformation. Polypyrrole (PPy) conducting polymer was utilized as a protective layer on NCM111.120 It showed an initial capacity of 70 mA h g−1 and 70% capacity retention for 50 cycles, compared to PPy-free NCM111 which exhibited 60 mA h g−1 initial capacity and ∼33% capacity retention for 40 cycles. The formed 2 nm thick spinel-Co3O4 layer on layered LCO also served as a protective layer and prevented Co ion dissolution.135 The Co3O4-LCO delivered a 1st cycle capacity of 83.6 mA h g−1 at 0.1 A g−1 and 84.5% capacity retention for 100 cycles with 1 M Li2SO4 in water. In comparison, Co3O4-free LCO exhibited a capacity of 84.8 mA h g−1 and 70.5% retention.
4.2. Diminishing water activity in water-in-salt electrolytes
Despite numerous attempts at electrode surface protection, LTMO underwent structural deformation and poor long-term cycling performance. An alternative and more fundamental strategy is to significantly diminish the source of H+, i.e., water. Dissolving extremely high electrolyte concentrations can reduce water volume remarkably, which causes decreased water activity (i.e., concentration). This concept was developed by discovering the dissolution of 21 m LiTFSI in water.136 It seemingly forms a water-in-salt electrolyte (WiSE), where the Li+ solvation structures are entirely changed, and Li+ and anion interactions are stronger.As water activity is significantly reduced, most water molecules solvate numerous Li+ ions, while the concentration of free water is low136 (Fig. 10(a)). In addition, Li+ is coordinated with a few water molecules instead of being shielded by primary and secondary water shells, which leads to strong Li+ and TFSI− attraction and the formation of aggregated ion pairs. Computational simulations and femtosecond IR spectroscopic observations demonstrated two separated domains, water channel and aggregated ion networks, in the bulk electrolyte137–139 (Fig. 10(b)). In this heterogeneous solvation structure, Li+ transport occurred in a bulk-like water molecule channel, explaining the higher ionic conductivity (∼9.5 mS cm−1 at 25 °C) than expected due to high viscosity.140 At the electrode surface, molecular dynamics (MD) simulations envisioned that the aggregated ion pairs are mostly occupied in the IHP while the water molecules were located away from the electrode surface141–143 (Fig. 10(c)). Experimentally, atomic force microscopy (AFM) force measurements detected two layers of thickness on gold electrodes, 4.3 and 6.4–6.7 Å at 0.3 V vs. Ag/Ag+. They were assigned to the TFSI−-rich layer and aggregated ion pair clusters ([Li(H2O)x]+([TFSI]−)y), respectively141 (Fig. 10(d)). These aggregated ion pairs had large sizes and were loosely bound to the surface. In comparison, the negatively charged surface was shielded by a hydrated Li+-rich layer.11 Both ionic layers push free water away from the electrode and extend the potential window to 3 V on stainless steel electrodes144 (Fig. 10(e)).
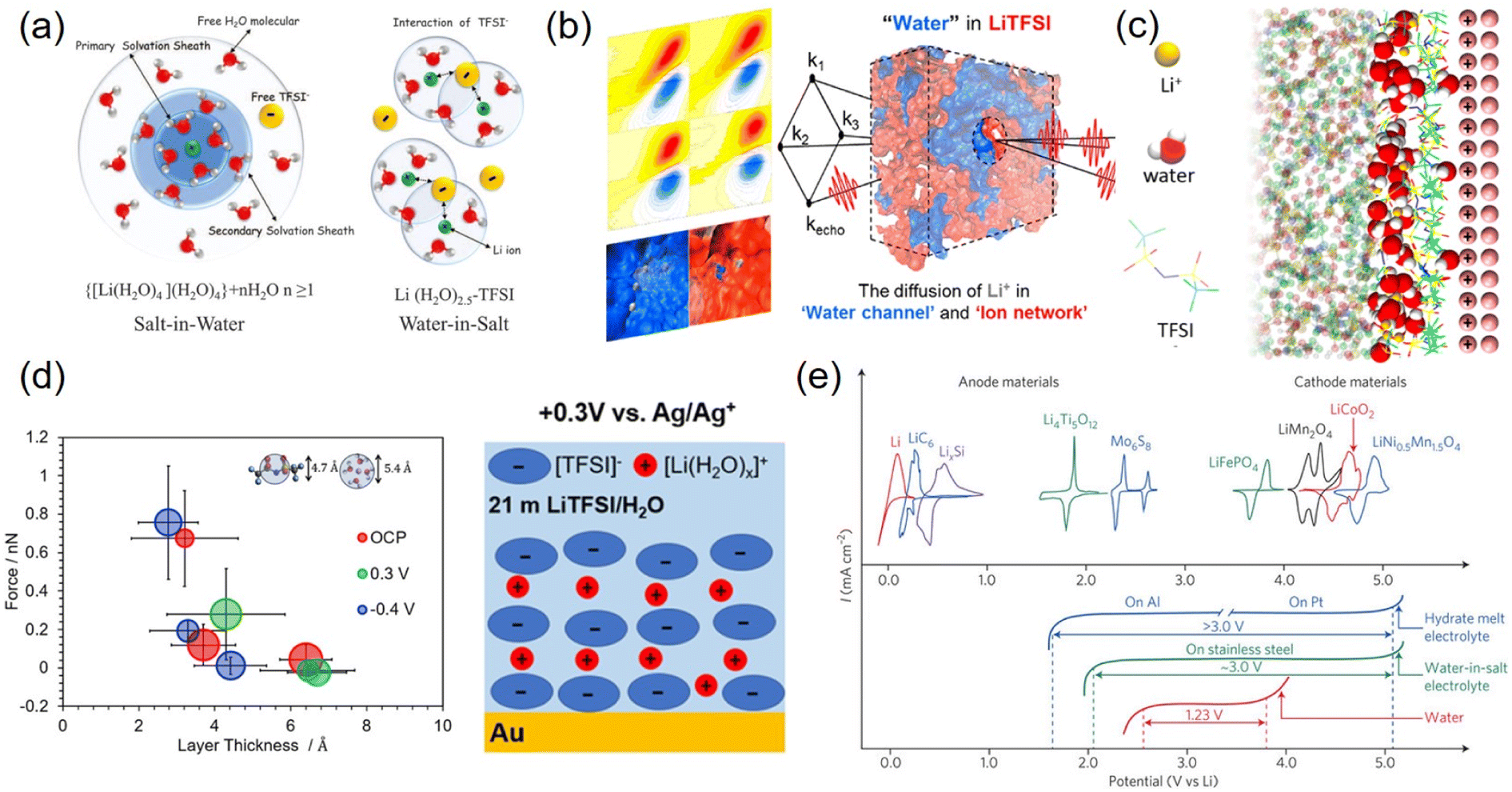 | ||
| Fig. 10 Water-in-salt electrolytes in bulk solution and at the interfacial region and electrochemical voltage. (a) Schematic illustration of the Li+ primary solvation sheath in salt-in-water (left) and water-in-salt electrolytes (right). Reproduced with permission from ref. 136. Copyright 2015 The American Association for the Advancement of Science. (b) Nano-heterogeneous domain of H-bond networks in water and ion networks obtained by 2D-IR measurements. Reproduced with permission from ref. 137. Copyright 2018 American Chemical Society. (c) Molecular dynamics (MD) simulation of 21 m LiTFSI with a positively charged (100) gold electrode. Reproduced with permission from ref. 142. Copyright 2018 American Chemical Society. (d) Diagram of layer thickness vs. force imposed by AFM for 21 m LiTFSI on (111) textured Au at OCP, 0.3 V, and −0.4 V (left). Schematic illustration of chemical species in the electrochemical double layer at 0.3 V (right). Reproduced with permission from ref. 141. Copyright 2020 American Chemical Society. (e) CV of various anode and cathode materials (top) and LSV of hydrate-melt electrolyte, water-in-salt electrolyte, and water (bottom). Reproduced with permission from ref. 144. Copyright 2016 Springer Nature. | ||
With a WiSE (20 m LiTFSI), NCM622 delivered 152 mA h g−1 capacity for the first cycle and maintained its capacity for the 3rd cycle. This result was compared to the same electrode with 9 M LiNO3, showing a 1st cycle capacity of 132 mA h g−1 and only ∼79% capacity retention for the subsequent three cycles.112 In addition, introducing additives to WiSE further stabilized LCO. Tris(trimethylsilyl) borate (TMSB) was sacrificially decomposed and formed a CEI layer.122 With 21 m LiTFSI and 0.1 wt% TMSB, a 2.5 V-class of Mo6S8|LCO cells provided a 1st cycle capacity of 40 mA h g−1 at 2.5C after electrochemical activation, and the average capacity fading rate was 0.013% per cycle for 1000 cycles.
To mitigate water activity further, bisalts,145 miscible non-aqueous solvents,146 or ionic liquid was utilized.121 LiTFSI was blended with LiBETI (BETI: N(SO2C2F5)2−, bis(pentafluoroethanesulfonyl)imide anion) to make Li(TFSI)0.7(BETI)0.3·2H2O. It was a room-temperature hydrate-melt electrolyte, where the eutectic LiTFSI and LiBETI composition greatly limited water content.123 A 2.4 V Li4Ti5O12|LCO cell with Li(TFSI)0.7(BETI)0.3·2H2O achieved 50 mA h g−1 capacity and 75% capacity retention after 200 cycles at 10C. A total 60 m electrolyte salt (40 m LiTFSI plus 20 m 1-ethyl-3-methylimidazolium TFSI (EMImTFSI)) attenuated Ni2+ dissolution and retarded capacity loss from NCM811, demonstrating better electrode stability than 21 m LiTFSI electrolyte.147 However, the significant cost associated with the large quantities of electrolyte is a considerable burden, as it is currently more expensive than the non-aqueous electrolyte solution.
Artificial solid-state protective layers were implemented along with WiSE to inhibit electrode deterioration and limit electrolyte concentrations. A gel polymer electrolyte consisting of WiSE and UV curable polymer extended the cathodic limit to 1.41 V and the anodic limit to 4.86 V, leading to a 3.86 V potential window.148 This approach was vital, in particular for the anode. Thus, graphite|LCO cells were first demonstrated using a gel electrolyte where 11 m LiTFSI in water and trimethylphosphate (TMP) was mixed with UV-curable monomers of poly(ethylene glycol) methyl ether acrylate (MPEGA), hydroxyethyl acrylate (HEA), and poly(ethylene glycol) diacrylate (PEGDA700), exhibiting a 3.8 V cell and 17 mA h capacity, which was 62% of the theoretical capacity.149 Developing advanced artificial protective layers has mitigated a rapid cell failure, which has addressed challenges in aqueous LiBs effectively in conjunction with electrolyte engineering.
5. Conclusions and perspectives
LTMO demonstrated tunable properties in an aqueous electrolyte solution and extended its applications for the OER. There are three controlling factors: alkali-metal-ion vacancies, transition metal states, and oxygen lattices. Moderate formation of alkali-metal-ion vacancies increased the valence state of the transition metal in the oxide layer. If OER-active transition metals constituted LTMO, they became active sites for the OER with suitable valence states. Besides, O 2p band engineering activated the oxygen lattice of the oxide layer. Notably, when the oxygen lattice of LTMO participated in the OER, the activity was enhanced to a greater extent than with the activation of transition metals alone. However, the number of oxygen vacancies should be optimized to prevent severe oxide degradation. Further, introducing dopants improved OER activity, and nanostructuring the LTMO catalyst increased the surface area and enhanced the current density.In addition to the above LTMO designs, we underlined that LTMO structures were often reconstructed during the OER due to the continuity of the deformed oxygen lattice and cation mixing. New crystalline or amorphous surfaces unexpectedly emerged and imposed strain and stress on LTMO. Various in situ X-ray and microscopy analytical tools were utilized to identify the reconstructed structures and address their OER activity linked with electrochemical evaluations. More importantly, even though the newly formed structure exhibited better OER activity, continuous structural transformation resulted in poor catalytic stability and reduced OER activity for long-term operation. For these reasons, the stability and consistency of the OER activity of LTMO have not yet become satisfactory for the practical level of water-splitting electrolyzers. Scrutinizing the time-dependent degradation mechanisms of LTMO and surface reconstruction trends associated with the above three controlling factors will help unveil the LTMO aging process. In this perspective, we showed several promising approaches to stabilize LTMO catalysts. The shallow insertion of Cs+ into delithiated LCO negligibly changed the bulk LCO structure, while surface strains improved OER activity.36 The cation electrolyte-mediated surface activation demonstrated improved catalytic stability compared to delithiated LCO. In another study, doping Cl− into LCO formed a new surface layer during the OER, which protected the bulk structure while performing OER activity.34 These studies will guide the design of practical OER catalysts for H2 production when considering electrochemical rebuilding processes.
On the other hand, investigations of LTMO cathodes for aqueous LiBs have been undertaken to enable cheap and grid-scale energy storage systems. However, vulnerability to water is a significant challenge for LTMO. Although the pH of the aqueous electrolyte solutions was selected at around 9 to avoid both the OER and H+ attacks, H+ permeation continued and led to electrode deformation and capacity fading in aqueous LiBs. To gain a better understanding of the interfacial reaction between LTMO and aqueous electrolyte solutions, various in situ and ex situ spectroscopic analyses have been conducted. These fundamental studies revealed that certain anions of the electrolyte, such as sulfate, chemically adsorbed onto LCO, acted as a barrier that prevented water and H+ from accessing the electrode surface. A water-in-salt electrolyte was developed to form protective aggregated ion pair layers on LTMO cathodes and diminish the water activity, which eliminated the source of H+. However, the insertion of water or H+ into LTMO was not reasonably suppressed, particularly during slow charging and discharging processes and long-term cyclability. Their performances were still inferior to non-aqueous LiBs, and the high cost of the massive amounts of electrolyte salts has not been resolved yet. Therefore, ground-breaking ideas are required for practical research approaches in aqueous LiBs.
We have exhibited the versatility of LTMO in two crucial applications in aqueous environments. We have gained a profound understanding of material properties through a wide range of approaches encompassing material designs, investigation of electrochemical processes, and evaluation of device performances. These efforts also highlighted key factors that need to be addressed to overcome the existing challenges. By leveraging the insights gained from these studies, we can drive forward the development of novel LTMO materials and technologies that will propel us toward a cleaner and more sustainable energy future.
Author contributions
Y. K., S. K., and E. C. wrote sessions 3, 2, and 4, respectively, under the direction of H. R. B., Y. K. and H. R. B. edited a whole manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2021R1A5A1030054) and the Korea Toray Science Foundation.References
- N. Dubouis and A. Grimaud, Chem. Sci., 2019, 10, 9165–9181 RSC
.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC
- J. Song, C. Wei, Z. F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196 RSC
- N. Zhang and Y. Chai, Energy Environ. Sci., 2021, 14, 4647–4671 RSC
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef
- J. W. Fergus, J. Power Sources, 2010, 195, 939–954 CrossRef CAS
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed
- C. M. Julien, A. Mauger, K. Zaghib and H. Groult, Inorganics, 2014, 2, 132–154 CrossRef CAS
-
Z. I. Radzi, B. Vengadaesvaran, S. Ramesh and N. A. Rahim, in Energy Materials, ed. S. J. Dhoble, N. T. Kalyani, B. Vengadaesvaran and A. Kariem Arof, Elsevier, 2021, pp. 285–312 Search PubMed
- C. Yang, J. Chen, T. Qing, X. Fan, W. Sun, A. Von Cresce, M. S. Ding, O. Borodin, J. Vatamanu, M. A. Schroeder, N. Eidson, C. Wang and K. Xu, Joule, 2017, 1, 122–132 CrossRef CAS
- Z. Lu, H. Wang, D. Kong, K. Yan, P.-C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 4345 CrossRef CAS PubMed
- S. Kobayashi, K. Nishio, M. Wilde, K. Fukutani, R. Shimizu and T. Hitosugi, J. Phys. Chem. C, 2023, 127, 4684–4688 CrossRef CAS
- W. T. Hong, K. A. Stoerzinger, Y.-L. Lee, L. Giordano, A. Grimaud, A. M. Johnson, J. Hwang, E. J. Crumlin, W. Yang and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 2190–2200 RSC
- Y. Li, X. Du, J. Huang, C. Wu, Y. Sun, G. Zou, C. Yang and J. Xiong, Small, 2019, 15, e1901980 CrossRef PubMed
- H. Oh, H. Yamagishi, T. Ohta and H. R. Byon, Mater. Chem. Front., 2021, 5, 3657–3663 RSC
- H. Oh, S.-J. Shin, E. Choi, H. Yamagishi, T. Ohta, N. Yabuuchi, H.-G. Jung, H. Kim and H. R. Byon, JACS Au, 2023, 3, 1392–1402 CrossRef CAS
- J. Wandt, A. T. S. Freiberg, A. Ogrodnik and H. A. Gasteiger, Mater. Today, 2018, 21, 825–833 CrossRef CAS
- J. Hwang, S. Myeong, W. Jin, H. Jang, G. Nam, M. Yoon, S. H. Kim, S. H. Joo, S. K. Kwak, M. G. Kim and J. Cho, Adv. Mater., 2020, 32, 2001944 CrossRef CAS
- T. Nagaura and K. Tozawa, Prog. Batteries Sol. Cells, 1990, 9, 209–217 CAS
- M. A. Marquardt, N. A. Ashmore and D. P. Cann, Thin Solid Films, 2006, 496, 146–156 CrossRef CAS
- C. Delmas, C. Fouassier and P. Hagenmuller, Physica B+C, 1980, 99, 81–85 CrossRef CAS
- J. N. Reimers and J. R. Dahn, J. Electrochem. Soc., 1992, 139, 2091 CrossRef CAS
- M. Okubo and A. Yamada, ACS Appl. Mater. Interfaces, 2017, 9, 36463–36472 CrossRef CAS
- T. Okumura, Y. Yamaguchi, M. Shikano and H. Kobayashi, J. Mater. Chem., 2012, 22, 17340–17348 RSC
- S.-K. Jung, H. Gwon, J. Hong, K.-Y. Park, D.-H. Seo, H. Kim, J. Hyun, W. Yang and K. Kang, Adv. Energy Mater., 2014, 4, 1300787 CrossRef
- P. Yan, L. Xiao, J. Zheng, Y. Zhou, Y. He, X. Zu, S. X. Mao, J. Xiao, F. Gao, J.-G. Zhang and C.-M. Wang, Chem. Mater., 2015, 27, 975–982 CrossRef CAS
- R. Hausbrand, G. Cherkashinin, H. Ehrenberg, M. Gröting, K. Albe, C. Hess and W. Jaegermann, Mater. Sci. Eng., B, 2015, 192, 3–25 CrossRef CAS
- W. Liu, P. Oh, X. Liu, M.-J. Lee, W. Cho, S. Chae, Y. Kim and J. Cho, Angew. Chem., Int. Ed. Engl., 2015, 54, 4440–4457 CrossRef CAS PubMed
- H. Sun and K. Zhao, J. Phys. Chem. C, 2017, 121, 6002–6010 CrossRef CAS
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef CAS PubMed
- H. Wang, J. Wu, A. Dolocan, Y. Li, X. Lu, N. Wu, K. Park, S. Xin, M. Lei, W. Yang and J. B. Goodenough, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 23473–23479 CrossRef CAS PubMed
- J. Wang, S.-J. Kim, J. Liu, Y. Gao, S. Choi, J. Han, H. Shin, S. Jo, J. Kim, F. Ciucci, H. Kim, Q. Li, W. Yang, X. Long, S. Yang, S.-P. Cho, K. H. Chae, M. G. Kim, H. Kim and J. Lim, Nat. Catal., 2021, 4, 212–222 CrossRef CAS
- S. Jiang, H. Suo, X. Zheng, T. Zhang, Y. Lei, Y. X. Wang, W. H. Lai and G. Wang, Adv. Energy Mater., 2022, 12, 2201934 CrossRef CAS
- Y. Kim, S. Kim, M. Shim, Y. Oh, K.-S. Lee, Y. Jung and H. R. Byon, J. Mater. Chem. A, 2022, 10, 10967–10978 RSC
- Y. Zhu, W. Zhou, Y. Chen, J. Yu, M. Liu and Z. Shao, Adv. Mater., 2015, 27, 7150–7155 CrossRef CAS PubMed
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS
- J. Bak, Y. Heo, T. G. Yun and S. Y. Chung, ACS Nano, 2020, 14, 14323–14354 CrossRef CAS
- J. R. Petrie, V. R. Cooper, J. W. Freeland, T. L. Meyer, Z. Zhang, D. A. Lutterman and H. N. Lee, J. Am. Chem. Soc., 2016, 138, 2488–2491 CrossRef CAS
- Y. Heo, S. Choi, J. Bak, H.-S. Kim, H. B. Bae and S.-Y. Chung, Adv. Energy Mater., 2018, 8, 1802481 CrossRef
- M. J. Choi, T. L. Kim, J. K. Kim, T. H. Lee, S. A. Lee, C. Kim, K. Hong, C. W. Bark, K. T. Ko and H. W. Jang, Nano Lett., 2020, 11, 8040–8045 CrossRef
- R. Zhang, L. Pan, B. Guo, Z. F. Huang, Z. Chen, L. Wang, X. Zhang, Z. Guo, W. Xu, K. P. Loh and J. J. Zou, J. Am. Chem. Soc., 2023, 145, 2271–2281 CrossRef CAS
- Y. Duan, S. Sun, Y. Sun, S. Xi, X. Chi, Q. Zhang, X. Ren, J. Wang, S. J. H. Ong, Y. Du, L. Gu, A. Grimaud and Z. J. Xu, Adv. Mater., 2019, 31, e1807898 CrossRef PubMed
- X. Rong, J. Parolin and A. M. Kolpak, ACS Catal., 2016, 6, 1153–1158 CrossRef CAS
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y. L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed
- Z. Zhang, C. Liu, C. Feng, P. Gao, Y. Liu, F. Ren, Y. Zhu, C. Cao, W. Yan, R. Si, S. Zhou and J. Zeng, Nano Lett., 2019, 19, 8774–8779 CrossRef CAS PubMed
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed
- N. Zhang, X. Feng, D. Rao, X. Deng, L. Cai, B. Qiu, R. Long, Y. Xiong, Y. Lu and Y. Chai, Nat. Commun., 2020, 11, 4066 CrossRef CAS PubMed
- Y. Pan, X. Xu, Y. Zhong, L. Ge, Y. Chen, J. M. Veder, D. Guan, R. O'Hayre, M. Li, G. Wang, H. Wang, W. Zhou and Z. Shao, Nat. Commun., 2020, 11, 2002 CrossRef CAS
- C. Wei, Z. Feng, G. G. Scherer, J. Barber, Y. Shao-Horn and Z. J. Xu, Adv. Mater., 2017, 29, 1606800 CrossRef
- H. Li, S. Sun, S. Xi, Y. Chen, T. Wang, Y. Du, M. Sherburne, J. W. Ager, A. C. Fisher and Z. J. Xu, Chem. Mater., 2018, 30, 6839–6848 CrossRef CAS
- J. Zhou, J. Li, L. Zhang, S. Song, Y. Wang, X. Lin, S. Gu, X. Wu, T.-C. Weng, J. Wang and S. Zhang, J. Phys. Chem. C, 2018, 122, 14447–14458 CrossRef CAS
- J. O. 'M. Bockris and T. Otagawa, J. Electrochem. Soc., 1984, 131, 290–302 CrossRef CAS
- Y. Liu, H. Wang, D. Lin, C. Liu, P.-C. Hsu, W. Liu, W. Chen and Y. Cui, Energy Environ. Sci., 2015, 8, 1719–1724 RSC
- H. Y. Wang, S. F. Hung, H. Y. Chen, T. S. Chan, H. M. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 36–39 CrossRef CAS
- S. Zhang, S. Gu, Y. Wang, C. Liang, Y. Yu, L. Han, S. Zheng, N. Zhang, X. Liu, J. Zhou and J. Li, ACS Catal., 2019, 9, 7389–7397 CrossRef CAS
- J. Gao, C. Q. Xu, S. F. Hung, W. Liu, W. Cai, Z. Zeng, C. Jia, H. M. Chen, H. Xiao, J. Li, Y. Huang and B. Liu, J. Am. Chem. Soc., 2019, 141, 3014–3023 CrossRef CAS
- J. Zhou, L. Zhang, Y. C. Huang, C. L. Dong, H. J. Lin, C. T. Chen, L. H. Tjeng and Z. Hu, Nat. Commun., 2020, 11, 1984 CrossRef CAS
- J. Timoshenko and B. Roldan Cuenya, Chem. Rev., 2021, 121, 882–961 CrossRef CAS PubMed
- S. Song, Y. Wang, R. C. Davis, Z. Ren, X. Xiao, G. Yang, D. Wang, J. Bao, Q. Zhang, S. Chen and Z. Ren, Chem. Mater., 2021, 33, 6299–6310 CrossRef CAS
- L. Sun, Z. Dai, L. Zhong, Y. Zhao, Y. Cheng, S. Chong, G. Chen, C. Yan, X. Zhang, H. Tan, L. Zhang, K. N. Dinh, S. Li, F. Ma and Q. Yan, Appl. Catal., B, 2021, 297, 120477 CrossRef CAS
- X. Li, W. Sun, C. Hao, Y. Bai, Z. Fu, Y. Lu, X. Wang and Z. Cheng, J. Phys. Chem. Lett., 2022, 13, 784–791 CrossRef CAS PubMed
- X. Zheng, Y. Chen, X. Zheng, G. Zhao, K. Rui, P. Li, X. Xu, Z. Cheng, S. X. Dou and W. Sun, Adv. Energy Mater., 2019, 9, 1803482 CrossRef
- J. Dai, Y. Zhu, Y. Chen, W. Zhou and Z. Shao, ACS Appl. Mater. Interfaces, 2017, 9, 21587–21592 CrossRef CAS PubMed
- S. Chu, H. Sun, G. Chen, Y. Chen, W. Zhou and Z. Shao, ACS Appl. Mater. Interfaces, 2019, 11, 25227–25235 CrossRef CAS
- J. Wang, L. Li, H. Tian, Y. Zhang, X. Che and G. Li, ACS Appl. Mater. Interfaces, 2017, 9, 7100–7107 CrossRef CAS PubMed
- X. Zheng, Y. Chen, W. Lai, P. Li, C. Ye, N. Liu, S. X. Dou, H. Pan and W. Sun, Adv. Funct. Mater., 2022, 32, 2200663 CrossRef CAS
- Z. Dai, Y. Liu, T. Liang, S. Lenus, Q. Liu, X. Zhang and P. Vijayakumar, Appl. Mater. Today, 2021, 25, 2200663 Search PubMed
- X. Zheng, P. Cui, Y. Qian, G. Zhao, X. Zheng, X. Xu, Z. Cheng, Y. Liu, S. X. Dou and W. Sun, Angew Chem. Int. Ed. Engl., 2020, 59, 14533–14540 CrossRef CAS PubMed
- C. Yang, G. Rousse, K. Louise Svane, P. E. Pearce, A. M. Abakumov, M. Deschamps, G. Cibin, A. V. Chadwick, D. A. Dalla Corte, H. Anton Hansen, T. Vegge, J. M. Tarascon and A. Grimaud, Nat. Commun., 2020, 11, 1378 CrossRef CAS PubMed
- A. Grimaud, K. J. May, C. E. Carlton, Y. L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 CrossRef
- T. Wang, Y. Sun, Y. Zhou, S. Sun, X. Hu, Y. Dai, S. Xi, Y. Du, Y. Yang and Z. J. Xu, ACS Catal., 2018, 8, 8568–8577 CrossRef CAS
- J. Li, Nano-Micro Lett., 2022, 14, 112 CrossRef CAS PubMed
- Y.-L. Lee, J. Kleis, J. Rossmeisl, Y. Shao-Horn and D. Morgan, Energy Environ. Sci., 2011, 4, 3966 RSC
- H. Ding, H. Liu, W. Chu, C. Wu and Y. Xie, Chem. Rev., 2021, 121, 13174–13212 CrossRef CAS PubMed
- K. J. May, C. E. Carlton, K. A. Stoerzinger, M. Risch, J. Suntivich, Y.-L. Lee, A. Grimaud and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 3264–3270 CrossRef CAS
- Y. Zhou, S. Sun, J. Song, S. Xi, B. Chen, Y. Du, A. C. Fisher, F. Cheng, X. Wang, H. Zhang and Z. J. Xu, Adv. Mater., 2018, 30, 1802912 CrossRef
- D. A. Kuznetsov, B. Han, Y. Yu, R. R. Rao, J. Hwang, Y. Román-Leshkov and Y. Shao-Horn, Joule, 2018, 2, 225–244 CrossRef CAS
- J. Suntivich, W. T. Hong, Y.-L. Lee, J. M. Rondinelli, W. Yang, J. B. Goodenough, B. Dabrowski, J. W. Freeland and Y. Shao-Horn, J. Phys. Chem. C, 2014, 118, 1856–1863 CrossRef CAS
- Z. Xiao, Y. C. Huang, C. L. Dong, C. Xie, Z. Liu, S. Du, W. Chen, D. Yan, L. Tao, Z. Shu, G. Zhang, H. Duan, Y. Wang, Y. Zou, R. Chen and S. Wang, J. Am. Chem. Soc., 2020, 142, 12087–12095 CrossRef CAS
- E. Marelli, J. Gazquez, E. Poghosyan, E. Muller, D. J. Gawryluk, E. Pomjakushina, D. Sheptyakov, C. Piamonteze, D. Aegerter, T. J. Schmidt, M. Medarde and E. Fabbri, Angew Chem. Int. Ed. Engl., 2021, 60, 14609–14619 CrossRef CAS
- R. Zhang, Y.-C. Zhang, L. Pan, G.-Q. Shen, N. Mahmood, Y.-H. Ma, Y. Shi, W. Jia, L. Wang, X. Zhang, W. Xu and J.-J. Zou, ACS Catal., 2018, 8, 3803–3811 CrossRef CAS
- J.-W. Zhao, H. Zhang, C.-F. Li, X. Zhou, J.-Q. Wu, F. Zeng, J. Zhang and G.-R. Li, Energy Environ. Sci., 2022, 15, 3912–3922 RSC
- B. Liu, Y. Wang, H. Q. Peng, R. Yang, Z. Jiang, X. Zhou, C. S. Lee, H. Zhao and W. Zhang, Adv. Mater., 2018, 30, 1803144 CrossRef PubMed
- X. Miao, L. Wu, Y. Lin, X. Yuan, J. Zhao, W. Yan, S. Zhou and L. Shi, Chem. Commun., 2019, 55, 1442–1445 RSC
- S.-H. Hsu, S.-F. Hung, H.-Y. Wang, F.-X. Xiao, L. Zhang, H. Yang, H. M. Chen, J.-M. Lee and B. Liu, Small Methods, 2018, 2, 1800001 CrossRef
- H. Liu, J. Qi, H. Xu, J. Li, F. Wang, Y. Zhang, M. Feng and W. Lü, ACS Catal., 2022, 12, 4119–4124 CrossRef CAS
- S. W. Lee, C. Carlton, M. Risch, Y. Surendranath, S. Chen, S. Furutsuki, A. Yamada, D. G. Nocera and Y. Shao-Horn, J. Am. Chem. Soc., 2012, 134, 16959–16962 CrossRef CAS
- B. Han, D. Qian, M. Risch, H. Chen, M. Chi, Y. S. Meng and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 1357–1362 CrossRef CAS PubMed
- N. Colligan, V. Augustyn and A. Manthiram, J. Phys. Chem. C, 2015, 119, 2335–2340 CrossRef CAS
- Z. Lu, G. Chen, Y. Li, H. Wang, J. Xie, L. Liao, C. Liu, Y. Liu, T. Wu, Y. Li, A. C. Luntz, M. Bajdich and Y. Cui, J. Am. Chem. Soc., 2017, 139, 6270–6276 CrossRef CAS
- T. G. Yun, Y. Heo, H. Bin Bae and S. Y. Chung, Nat. Commun., 2021, 12, 824 CrossRef CAS
- J. I. Jung, H. Y. Jeong, J. S. Lee, M. G. Kim and J. Cho, Angew Chem. Int. Ed. Engl., 2014, 53, 4582–4586 CrossRef CAS
- J. Bak, H. Bin Bae and S. Y. Chung, Nat. Commun., 2019, 10, 2713 CrossRef
- A. Gupta, W. D. Chemelewski, C. Buddie Mullins and J. B. Goodenough, Adv. Mater., 2015, 27, 6063–6067 CrossRef CAS
- X. Li, Q. Wang, H. Guo, N. Artrith and A. Urban, ACS Appl. Energy Mater., 2022, 5, 5730–5741 CrossRef CAS
- D. Y. Chung, P. P. Lopes, P. Farinazzo Bergamo Dias Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik, Y. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 222–230 CrossRef
- P. P. Lopes, D. Y. Chung, X. Rui, H. Zheng, H. He, P. Farinazzo Bergamo Dias Martins, D. Strmcnik, V. R. Stamenkovic, P. Zapol, J. F. Mitchell, R. F. Klie and N. M. Markovic, J. Am. Chem. Soc., 2021, 143, 2741–2750 CrossRef CAS
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS
- J. Bak, T. G. Yun, J.-S. An, H. B. Bae and S.-Y. Chung, Energy Environ. Sci., 2022, 15, 610–620 RSC
- Y. Wang, J. Ren, Y. Wang, F. Zhang, X. Liu, Y. Guo and G. Lu, J. Phys. Chem. C, 2008, 112, 15293–15298 CrossRef CAS
- F. Liang, Y. Yu, W. Zhou, X. Xu and Z. Zhu, J. Mater. Chem. A, 2015, 3, 634–640 RSC
- G. Cheng, T. Kou, J. Zhang, C. Si, H. Gao and Z. Zhang, Nano Energy, 2017, 38, 155–166 CrossRef CAS
- L. Bai, C. S. Hsu, D. T. L. Alexander, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2019, 141, 14190–14199 CrossRef CAS
- Y. Miyahara, T. Fukutsuka, T. Abe and K. Miyazaki, Chem. Mater., 2020, 32, 8195–8202 CrossRef CAS
- L. Gui, Y. Liu, J. Zhang, B. He, Q. Wang and L. Zhao, J. Mater. Chem. A, 2020, 8, 19946–19953 RSC
- T. Wu, S. Sun, J. Song, S. Xi, Y. Du, B. Chen, W. A. Sasangka, H. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. Wang, H. Li, A. Grimaud and Z. J. Xu, Nat. Catal., 2019, 2, 763–772 CrossRef CAS
- Q. Cheng, T. Yang, Y. Li, M. Li and C. K. Chan, J. Mater. Chem. A, 2016, 4, 6902–6910 RSC
- R. Ruffo, C. Wessells, R. A. Huggins and Y. Cui, Electrochem. Commun., 2009, 11, 247–249 CrossRef CAS
- R. Ruffo, F. La Mantia, C. Wessells, R. A. Huggins and Y. Cui, Solid State Ionics, 2011, 192, 289–292 CrossRef CAS
- M. Kunduraci, R. N. Mutlu and A. M. Gizir, Ionics, 2020, 26, 1663–1672 CrossRef CAS
- W. Tang, L. L. Liu, S. Tian, L. Li, Y. B. Yue, Y. P. Wu, S. Y. Guan and K. Zhu, Electrochem. Commun., 2010, 12, 1524–1526 CrossRef CAS
- G. Wang, L. Fu, N. Zhao, L. Yang, Y. Wu and H. Wu, Angew. Chem., Int. Ed. Engl., 2007, 46, 295–297 CrossRef CAS PubMed
- G. J. Wang, N. H. Zhao, L. C. Yang, Y. P. Wu, H. Q. Wu and R. Holze, Electrochim. Acta, 2007, 52, 4911–4915 CrossRef CAS
- X. Wang, Q. Qu, Y. Hou, F. Wang and Y. Wu, Chem. Commun., 2013, 49, 6179–6181 RSC
- J. Köhler, H. Makihara, H. Uegaito, H. Inoue and M. Toki, Electrochim. Acta, 2000, 46, 59–65 CrossRef
- G. J. Wang, L. J. Fu, B. Wang, N. H. Zhao, Y. P. Wu and R. Holze, J. Appl. Electrochem., 2008, 38, 579–581 CrossRef CAS
- L. Liu, F. Tian, X. Wang, Z. Yang, Q. Chen and X. Wang, J. Solid State Electrochem., 2012, 16, 491–497 CrossRef CAS
- R. B. Shivashankaraiah, H. Manjunatha, K. C. Mahesh, G. S. Suresh and T. V. Venkatesha, J. Solid State Electrochem., 2012, 16, 1279–1290 CrossRef CAS
- M. Becker, D. Rentsch, D. Reber, A. Aribia, C. Battaglia and R. S. Kühnel, Angew. Chem., Int. Ed. Engl., 2021, 60, 14100–14108 CrossRef CAS
- F. Wang, Y. Lin, L. Suo, X. Fan, T. Gao, C. Yang, F. Han, Y. Qi, K. Xu and C. Wang, Energy Environ. Sci., 2016, 9, 3666–3673 RSC
- Y. Yamada, K. Usui, K. Sodeyama, S. Ko, Y. Tateyama and A. Yamada, Nat. Energy, 2016, 1, 16129 CrossRef CAS
- J. Qian, L. Liu, J. Yang, S. Li, X. Wang, H. L. Zhuang and Y. Lu, Nat. Commun., 2018, 9, 4918 CrossRef PubMed
- L. Noerochim, S. Suwarno, N. H. Idris and H. K. Dipojono, Batteries, 2021, 7, 84 CrossRef CAS
- C. Lee, Y. Miyahara, T. Abe and K. Miyazaki, J. Power Sources, 2022, 524, 231081 CrossRef CAS
- S. S. Zhang, J. Chen and C. Wang, J. Electrochem. Soc., 2019, 166, A487–A492 CrossRef CAS
- W. Li, J. R. Dahn and D. S. Wainwright, Science, 1994, 264, 1115–1118 CrossRef CAS
- I. A. Shkrob, J. A. Gilbert, P. J. Phillips, R. Klie, R. T. Haasch, J. Bareño and D. P. Abraham, J. Electrochem. Soc., 2017, 164, A1489–A1498 CrossRef CAS
- R. Benedek, M. M. Thackeray and A. Van De Walle, Chem. Mater., 2008, 20, 5485–5490 CrossRef CAS
- J. Choi, E. Alvarez, T. A. Arunkumar and A. Manthiram, Electrochem. Solid-State Lett., 2006, 9, A241 CrossRef CAS
- X. Gu, J.-L. Liu, J.-H. Yang, H.-J. Xiang, X.-G. Gong and Y.-Y. Xia, J. Phys. Chem. C, 2011, 115, 12672–12676 CrossRef CAS
- S. Posada-Pérez, G.-M. Rignanese and G. Hautier, Chem. Mater., 2021, 33, 6942–6954 CrossRef
- A. Ramanujapuram, D. Gordon, A. Magasinski, B. Ward, N. Nitta, C. Huang and G. Yushin, Energy Environ. Sci., 2016, 9, 1841–1848 RSC
- H. J. Lee, J. H. Lee, I. H. Son, S. Han, P. Byeon, M.-S. Park, S.-Y. Chung and J. W. Choi, ACS Appl. Energy Mater., 2018, 1, 5726–5734 CAS
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed
- J. Lim, K. Park, H. Lee, J. Kim, K. Kwak and M. Cho, J. Am. Chem. Soc., 2018, 140, 15661–15667 CrossRef CAS PubMed
- O. Borodin, L. Suo, M. Gobet, X. Ren, F. Wang, A. Faraone, J. Peng, M. Olguin, M. Schroeder, M. S. Ding, E. Gobrogge, A. von Wald Cresce, S. Munoz, J. A. Dura, S. Greenbaum, C. Wang and K. Xu, ACS Nano, 2017, 11, 10462–10471 CrossRef CAS PubMed
- X. Liu, S.-C. Lee, S. Seifert, L. He, C. Do, R. E. Winans, G. Kwon, Y. Zhang and T. Li, Chem. Mater., 2023, 35, 2088–2094 CrossRef CAS
- J. Kim, B. Koo, J. Lim, J. Jeon, C. Lim, H. Lee, K. Kwak and M. Cho, ACS Energy Lett., 2022, 7, 189–196 CrossRef CAS
- R. Zhang, M. Han, K. Ta, K. E. Madsen, X. Chen, X. Zhang, R. M. Espinosa-Marzal and A. A. Gewirth, ACS Appl. Energy Mater., 2020, 3, 8086–8094 CrossRef CAS
- M. McEldrew, Z. A. H. Goodwin, A. A. Kornyshev and M. Z. Bazant, J. Phys. Chem. Lett., 2018, 9, 5840–5846 CrossRef CAS PubMed
- M. Han, R. Zhang, A. A. Gewirth and R. M. Espinosa-Marzal, Nano Lett., 2021, 21, 2304–2309 CrossRef CAS PubMed
- K. Xu and C. Wang, Nat. Energy, 2016, 1, 16161 CrossRef CAS
- L. Suo, O. Borodin, W. Sun, X. Fan, C. Yang, F. Wang, T. Gao, Z. Ma, M. Schroeder, A. Von Cresce, S. M. Russell, M. Armand, A. Angell, K. Xu and C. Wang, Angew. Chem., Int. Ed. Engl., 2016, 55, 7136–7141 CrossRef CAS PubMed
- Y. Huang, W. Sun, K. Xu, J. Zhang, H. Zhang, J. Li, L. He, L. Cai, F. Fu, J. Qin and H. Chen, Energy Storage Mater., 2022, 46, 577–582 CrossRef
- M. Becker, W. Zhao, F. Pagani, C. Schreiner, R. Figi, W. Dachraoui, R. Grissa, R.-S. Kühnel and C. Battaglia, ACS Appl. Energy Mater., 2022, 5, 11133–11141 CrossRef CAS
- J. Zhang, C. Cui, P.-F. Wang, Q. Li, L. Chen, F. Han, T. Jin, S. Liu, H. Choudhary, S. R. Raghavan, N. Eidson, A. Von
Cresce, L. Ma, J. Uddin, D. Addison, C. Yang and C. Wang, Energy Environ. Sci., 2020, 13, 2878–2887 RSC
- A. Cresce, N. Eidson, M. Schroeder, L. Ma, Y. Howarth, C. Yang, J. Ho, R. Dillon, M. Ding, A. Bassett, J. Stanzione, R. Tom, T. Soundappan, C. Wang and K. Xu, J. Power Sources, 2020, 469, 228378 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2023 |