
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA host potassiophilicity strategy for unprecedentedly stable and safe K metal batteries†
Zhibin
Li‡
a,
Liang
Ma‡
ab,
Kai
Han
ad,
Yingying
Ji
a,
Junpeng
Xie
a,
Likun
Pan
 c,
Jinliang
Li
*a and
Wenjie
Mai
*ad
c,
Jinliang
Li
*a and
Wenjie
Mai
*ad
aSiyuan Laboratory, Guangdong Provincial Engineering Technology Research Center of Vacuum Coating Technologies and New Energy Materials, Guangdong Provincial Key Laboratory of Nanophotonic Manipulation, Department of Physics, Jinan University, Guangzhou 510632, China. E-mail: lijinliang@email.jnu.edu.cn; wenjiemai@email.jnu.edu.cn
bSchool of Chemistry, Guangzhou Key Laboratory of Materials for Energy Conversion and Storage, South China Normal University, Guangzhou, 510006, China
cShanghai Key Laboratory of Magnetic Resonance, School of Physics and Electronic Science, East China Normal University, Shanghai 200241, China
dCAS Center for Excellence in Nanoscience, Beijing Key Laboratory of Micro–Nano Energy and Sensor, Beijing Institute of Nanoenergy and Nanosystems, Chinese Academy of Sciences, Beijing, 100083, China
First published on 6th August 2023
Abstract
Creating high-performance host materials for potassium (K) metal anodes remains a significant challenge due to the complex preparation process and poor K reversibility. In our work, we developed a potassiophilicity strategy using an oxygen-modified carbon cloth (O-CC) network as a host for K metal anodes. The O-CC network exhibited superior potassiophilic ability, and this improvement was also observed in other carbon hosts using the same process. The oxygen-induced epoxy group in the carbon network regulates interface electrons and enables strong binding of K adatoms through orbital hybridization, resulting in fewer side reactions with the electrolyte and promoting K-ion desolvation and uniform deposition. These factors result in unprecedented stability of the carbon network host, with a long lifespan of over 5500 hours at 0.5 mA cm−2/0.5 mA h cm−2 and 3500 h at 1 mA cm−2/0.5 mA h cm−2 in symmetric cells for K metal anodes, surpassing the cycle life of all previously reported K metal anodes. Furthermore, a high average coulombic efficiency of over 99.3% is demonstrated in O-CC//K cells during 210 cycles. The O-CC also exhibited a stable electrochemical performance, with a capacity retention of 73.3% in full cells coupled with a perylene-3,4,9,10-tetracarboxylic dianhydride cathode. We believe that this new strategy holds great promise for metal anodes in battery applications.
Introduction
The shortage of Li sources has caused a rapid increase in the cost, which has restricted the further spread of lithium-ion batteries (LIBs).1,2 Exploring a new high-efficiency energy storage system with low cost has become necessary. Among the new rechargeable battery systems, potassium-ion batteries (KIBs) have attracted great attention due to the low cost of K resources, high operating voltage, and fast diffusion kinetics.3–5 To achieve high energy density KIBs, developing the anode with a low charge plateau is a reliable approach.6,7 However, most anodes, including carbon and sulfide, still present high charge plateaus, resulting in the decrease of the KIB's open-circuit voltage.8,9 To achieve high output voltage, the K metal anode has gained wide attention due to its extremely low potential of −2.93 V vs. the standard hydrogen electrode (SHE) and vitally high theoretical capacity of 687 mA h g−1.10,11 However, the adverse effect of the high reactivity and the vigorous dendrite growth of the K metal anode during the plating/stripping process has caused serious safety hazards for KIBs, which seriously limits the practical application of K metal anodes.12,13To realize the practical applications of the K metal anode, scientists have made a lot of attempts to address critical issues, including dendrite growth and dead K.14,15 Among all of the attempts, designing the K metal host is undoubtedly one of the most reliable strategies.16,17 The introduction of the host in the K metal anode can reduce the side reaction caused by the direct contact between the electrolyte and K metal anode.18–20 The host frame buffers the large volume expansion during the plating/stripping process.21,22 Due to these properties, the K metal host has acquired a great deal of investigation. Mitlin's group developed a 3D copper current collector with reduced graphene oxide as a host for the K metal anode and this composite host exhibited a significantly enhanced adsorption capacity for K metal, which achieved stable cycling for 167 h at 0.5 mA cm−2.15 Xu's group reported a 3D SnO2 coated conductive porous carbon nanofiber framework as a host and this framework host presented ultra-long cycling stability over 1700 h at 1 mA cm−2 in a symmetric cell.23 Despite a relatively stable circulation being achieved in the above studies, the complex preparation methods of these hosts are still difficult to obtain at a large scale for K metal batteries (KMBs), resulting in the limitation of their practical application. So far, some researchers have attempted to explore the simplified preparation path to optimize the K metal host performance. Tu's group utilized Ag nanoparticles to decorate carbon cloth (Ag-CC) as a K metal host and the Ag-CC host maintained stable cycling for more than 700 cycles at 0.5 mA cm−2/0.5 mA h cm−2 in a symmetric cell without abnormality.24 Xu's group adopted biomass carbon as the K metal host and found that the carbon derived from the bacterial cellulose host presented stable cycling for 1400 h at 0.5 mA cm−2 with a low overpotential of 45 mV.16 Although these studies involving K metal hosts have made a series of advancements, the cycle life is still nowhere near enough for the batteries. Further development of the simple approaches to obtaining a K metal host with stable cycling remains to be explored.
In this work, we developed a one-step air heat treatment to obtain oxygen-modified carbon cloth (O-CC) as a host for the K metal anode. It is found that numerous epoxy groups were introduced onto the surface of O-CC after the air modification. According to our experiments and calculations, this introduction can decrease the surrounding electron density and hinder the transfer of electrons to the lowest unoccupied molecular orbital (LUMO) of the electrolyte. A lower Fermi level on the surface of the O-CC material can be obtained after introduction of epoxy groups, which causes fewer side reactions of the electrolyte in batteries. Additionally, due to orbital hybridization, the epoxy groups on the O-CC surface present strong forces, allowing easy desolvation of K atoms from the electrolyte which then deposited on the surface of the host. Benefiting from this behavior, the O-CC@K electrode in a symmetric cell delivers unprecedented electrochemical performance with an ultra-long lifespan of over 5500 h at 0.5 mA cm−2/0.5 mA h cm−2 and 3500 h at 1 mA cm−2/0.5 mA h cm−2, which outperforms the cycle life of all previously reported K metal anodes. The O-CC@K electrode coupled with a modified perylene-3,4,9,10-tetracarboxylic dianhydride (PTCDA) cathode is used to construct K metal full batteries, which present a stable capacity over 300 cycles. We suggest that our optimization path will provide a guide to designing a high-efficiency host for current K metal anodes.
Results and discussion
Fig. 1a shows the schematic of O-CC's fabrication process, which is obtained by heat treatment in the air to achieve the surface modification of oxidation. For comparison, we denote the pristine carbon cloth without heat treatment as P-CC. Fig. S1† shows the photographs of P-CC and O-CC. It is almost impossible to distinguish these two electrodes macroscopically. From the scanning electron microscope (SEM) images, we found that there is virtually no change in the macroscopic region between P-CC (Fig. 1b) and O-CC (Fig. 1e). In the enlarged SEM image (Fig. 1c), the relatively smooth surface of a carbon fiber in P-CC can be detected. After heat treatment, the surface of O-CC becomes rough (Fig. 1f). To identify the changed elements, the element mapping of P-CC (Fig. 1d and S2a†) and O-CC (Fig. 1g and S2b†) is provided. It is observed that the O content in O-CC is significantly increased and evenly distributed, indicating the introduction of oxygen into the carbon fiber surface after heat treatments in air. Since H won't be introduced into carbon at high temperatures, we suggest that the newly introduced O should be bonded with C to form the epoxy groups.25,26 To further determine the surface elemental composition, X-ray photoelectron spectroscopy (XPS) of P-CC and O-CC was conducted, and the results are shown in Fig. S3† and 1h and i. Four peaks of C 1s XPS spectra can be deconvoluted, corresponding to the conjugated C, non-conjugated C, epoxy group (–C(O)C–) and hydroxy group (–C–OH) bonds, respectively. The increased peak intensity at 286.8 eV indicates the increase of epoxy groups on the surface of O-CC after heat treatment in air.27,28 From the O 1s XPS spectra, two peaks corresponding to the epoxy group and hydroxy group are deconvoluted. It is observed that the peak intensity at 532.3 eV increases, also illustrating the reinforcement of the epoxy group. Due to the detection depth of XPS being less than 10 nm, except for the O inside carbon fibers, the increased O content should be derived from the interface's epoxy group. The Raman spectra of P-CC and O-CC were recorded, and are shown in Fig. 1j. Two typical carbon characteristic peaks of the D band and G band are observed. The D band arises from the vibration of amorphous carbon and the G band involves the vibration of graphitic carbon.29 It is found that the G-band intensity of O-CC increases, indicating that the heat treatment of carbon fibers in the air tends to the graphitization state.30 To further confirm the presence of epoxy groups, we also compared the FTIR spectra (Fig. S4†) of P-CC and O-CC. We found that at the position corresponding to the epoxy group (1077 cm−1), O-CC showed a significantly enhanced vibration peak, further demonstrating that air thermal treatment can introduce epoxy groups into the carbon network. The molten K metal cannot be adsorbed by P-CC even after 30 s contact, indicating P-CC presents potassiophobic properties (Fig. S5a and Video S1†). Therefore, the P-CC@K electrode is obtained by physical squeezing. Compared with P-CC, O-CC exhibits a superior potassiophilic ability with rapid molten K metal adsorption in a short time (less than 1 s, Fig. S5b and Video S2†). Fig. S6† shows the photographs of P-CC and O-CC obtained using an optical microscope and the SEM images. A very poor combination between P-CC and K metal can be found by the naked eye and at smaller scales. For O-CC, uniform and strong infiltration of K metal can be observed, with K metal plating uniformly on the surface of O-CC and combining tightly with each carbon fiber, indicating that O-CC can be a superior host for the K metal anode during the plating/stripping process. To demonstrate the universality, carbon felt and carbon paper were also used with the same procedure and demonstrate a similarly enhanced potassiophilicity. In Fig. S7 (Video S3†) and Fig. S8 (Video S4†), both show a rapid molten K metal adsorption.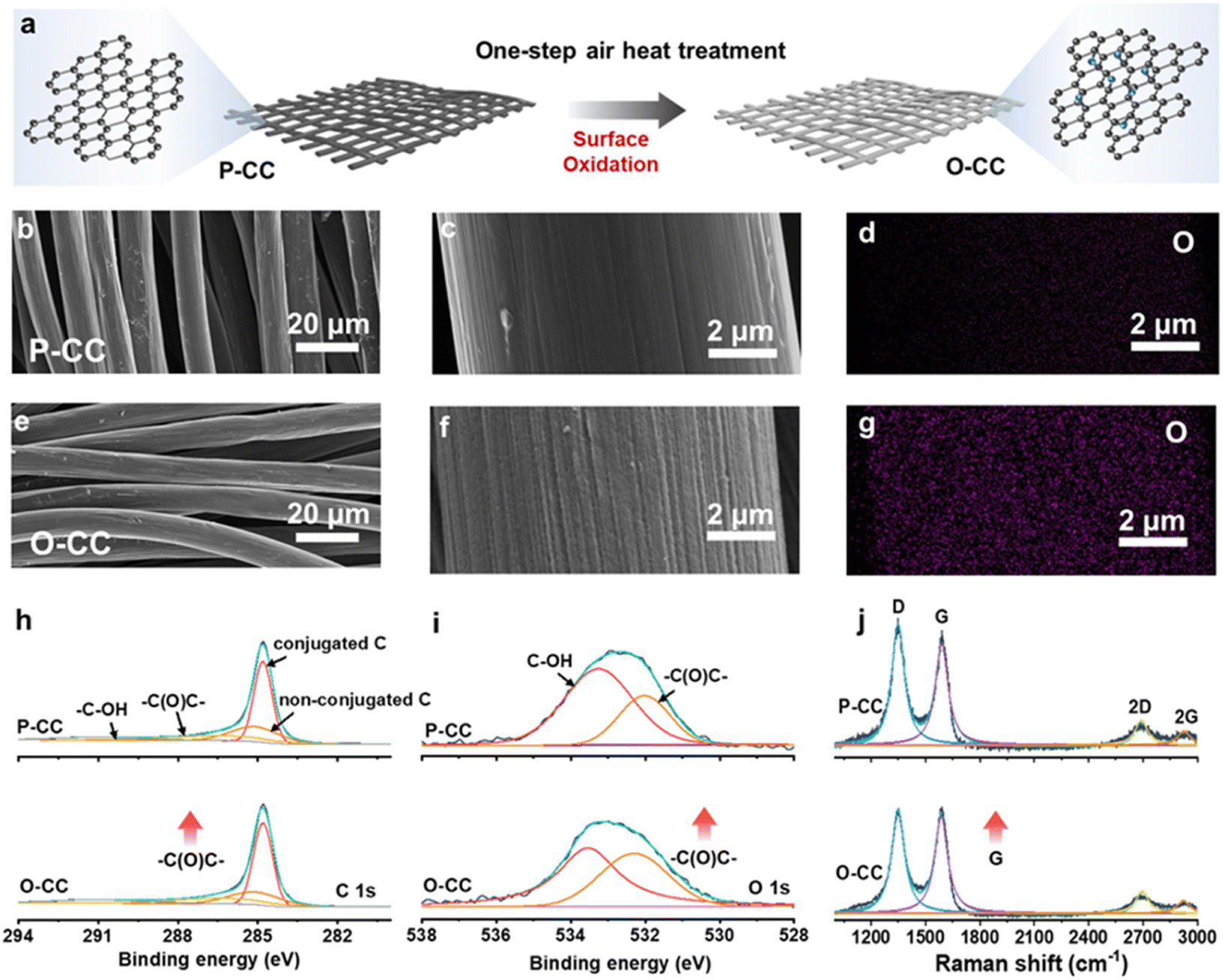 | ||
| Fig. 1 Schematic illustration and characterization. (a) Schematic illustration for the preparation of the O-CC. SEM images of (b and c) P-CC and (e and f) O-CC and the corresponding O elemental mapping images of (d) P-CC and (g) O-CC. (h) C 1s and (i) O 1s XPS spectra, and (j) Raman spectra of P-CC and O-CC. | ||
To evaluate the K plating/stripping efficiency of the host, a series of electrochemical tests were conducted. Fig. 2a and b show the reversibility of Cu@K, P-CC@K, and O-CC@K in symmetric cells during K plating/stripping. As shown in Fig. 2a, Cu@K and P-CC@K electrodes suffer a fast failure at 117 h and 330 h, respectively. After the introduction of the epoxy group, the O-CC@K electrode presents an ultra-long cycle life of over 5500 h at 0.5 mA cm−2/0.5 mA h cm−2 and an ultralow voltage hysteresis of less than 30 mV, which are much better than those of Cu@K and P-CC@K electrodes in symmetric cells. A slight periodic voltage hysteresis fluctuation can be observed in Fig. 2a, which is caused by the temperature difference between day and night. Fig. S9† also presents the reversibility of Cu@K, P-CC@K, and O-CC@K in symmetric cells during K plating/stripping in the first 500 h, confirming the ultralow voltage hysteresis of the O-CC@K electrode for the K metal anode. We also noticed that the voltage hysteresis of the O-CC@K electrode gradually decreases with the K plating/stripping process. Therefore, the electrochemical impedance spectroscopy (EIS) of O-CC@K electrodes at different K plating/stripping states and the EIS of bare K and P-CC@K before cycling are also provided, as shown in Fig. S10.† The decreased impedance of the O-CC@K electrode can be detected, indicating continuous optimization of the electrode interface with the proceeding of K plating/stripping.31 The O-CC@K electrode also undergoes stable cycling for 3500 h even at 1 mA cm−2/0.5 mA h cm−2, as shown in Fig. 2b. These long cycle life and low polarization voltage indicate that the O-CC can significantly inhibit dendrite growth. To better evaluate the electrochemical performance of our O-CC, all the previous performances of K metal hosts are provided for comparison, as shown in Fig. 2c and Table S1.† The results demonstrate that our O-CC presents unprecedented stability for K plating/stripping. Fig. 2d and S11† exhibit the cycling performance of the P-CC@K and O-CC@K electrodes in symmetric cells at different current densities. The P-CC@K electrode presents a short circuit at 1 mA cm−2 and the O-CC@K electrode still maintains stable cycling performance even at 3 mA cm−2. To further assess the electrochemical performance, we quantified the coulombic efficiencies (CEs) of the battery by O-CC//K and P-CC//K asymmetric cells with an areal capacity of 1 mA h cm−2, as shown in Fig. 2e and S12a and b.† P-CC//K and O-CC//K cells present the initial CE of 22.5% and 31.6% at 0.5 mA cm−2, respectively. The higher initial CE indicated less side reaction with the electrolyte during SEI formation. The O-CC//K cell presents excellent stability with a high average CE of 99.6% for 250 cycles at 0.5 mA cm−2 and average CE of 99.3% for 210 cycles at 1 mA cm−2. However, P-CC//K suffers from a break only after 35 cycles at 0.5 mA cm−2 (Fig. S12b and c†). The low initial CE of O-CC//K at 1 mA cm−2 indicates that a large amount of K is absorbed by the epoxy group in the surface of O-CC during SEI formation, while the high average CE indicates that the O-CC can inhibit the dendrite growth well for a long time. Besides, the CEs of Cu//P-CC@K and Cu//O-CC@K asymmetric cells are also provided with an areal capacity of 0.5 mA h cm−2, as shown in Fig. S12d.† Only an average CE of 82% in 40 cycles can be achieved for the Cu//P-CC@K cell, while the Cu//O-CC@K cell holds a high average CE of 91.2% for 50 cycles. Fig. S12e and f† show the galvanostatic charge–discharge (GCD) curves of Cu//P-CC@K and Cu//O-CC@K cells, respectively. Both initial GCD curves present less than 300 mV bias voltage gaps between charge and discharge plateaus, implying side reactions with the electrolyte.32 Compared with the Cu//P-CC@K cell, the Cu//O-CC@K cell shows an overlapping flat pattern with little fluctuation, indicating the stability of the O-CC electrode for KMBs. The K nucleation process in the different hosts is studied in Fig. 2f and the O-CC presents a lower nucleation overpotential (258 mV) than that of P-CC (386 mV), indicating that the O-CC exhibits a rapid nucleation process during K plating.33 The rapid nucleation process of O-CC demonstrates the formation of slight K accumulation, which appears as uniform K deposition macroscopically.
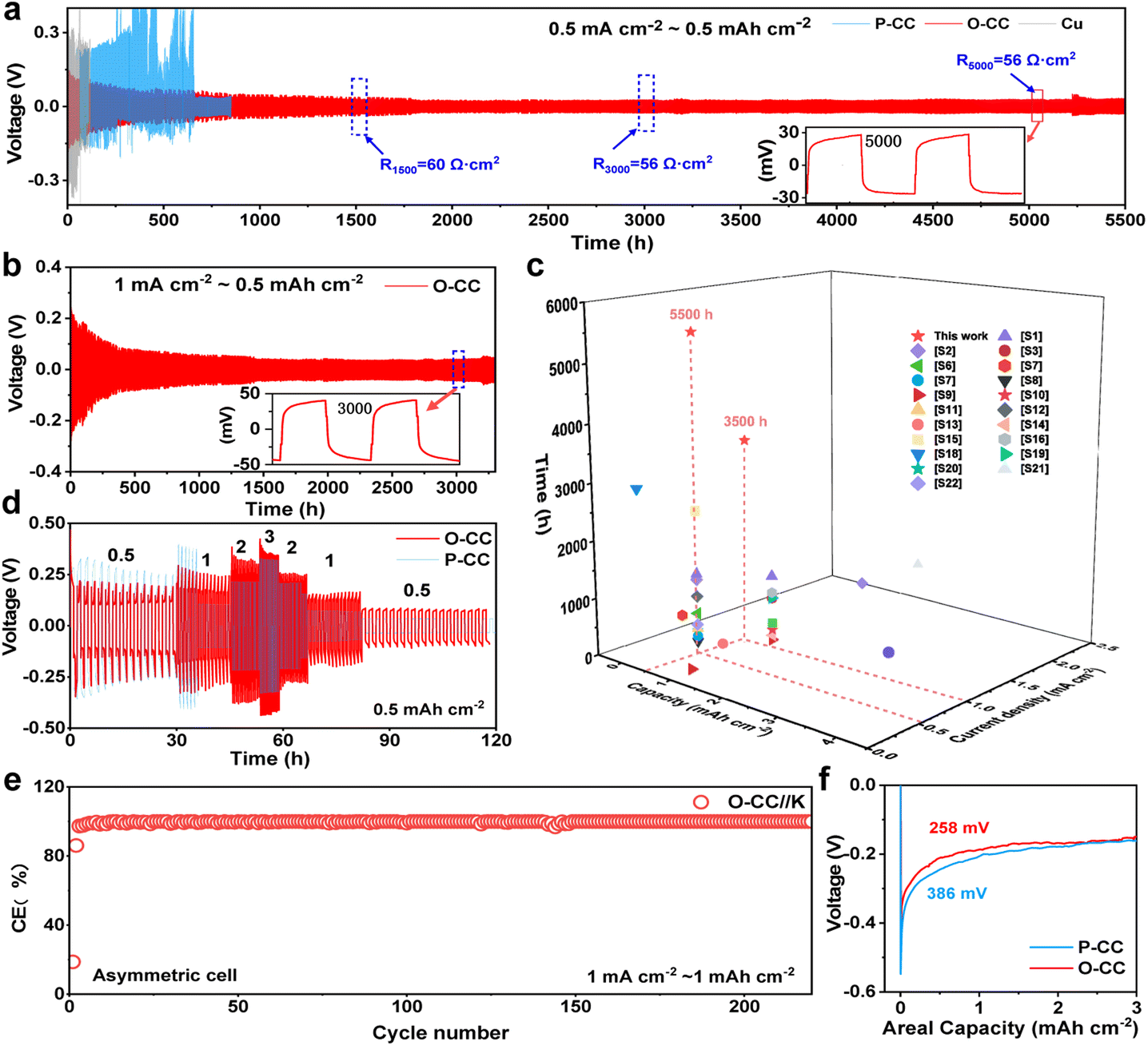 | ||
| Fig. 2 Electrochemical performance. Galvanostatic cycling performance of P-CC and O-CC in symmetric cells at (a) 0.5 mA cm−2 and (b) 1 mA cm−2; (c) comparison of all the host materials' performance for the K metal anode in symmetric cells; (d) cycling performance of P-CC and O-CC in symmetric cells at different current densities with the areal capacity of 0.5 mA h cm−2; (e) CE curves for O-CC//K asymmetric cells at 1 mA cm−2/1 mA h cm−2; (f) nucleation overpotential of P-CC and O-CC at 0.5 mA cm−2. | ||
We investigated the charge distribution, Fermi levels, surface binding energies, and the corresponding partial density of states (pDOS) to explore the electrochemical enhancement mechanism of the epoxy group by density functional theory (DFT) calculations. From Fig. 3a, the P-CC model presents high electron density for all atoms, and the O-CC model exhibits a decrease in charge distribution after the introduction of the epoxy group at this level. This behavior indicates that O-CC exerts strong electron binding to passivate the interface due to electron regulation by the epoxy group. Automatically, the surface of O-CC shows a Fermi level of −2.84 eV, which is lower than that of P-CC (−2.31 eV), as shown in Fig. 3b. Compared with P-CC, a larger energy difference between O-CC and the LUMO level of dimethoxyethane (DME) electrolyte indicates that it is difficult for the electron to jump from the Fermi level of O-CC to the LUMO level of DME. This behavior illustrates the suppression of electron transfer in O-CC, resulting in the reduction of the side reaction between the electrode and DME solvent during the stripping/plating process.34,35 From the charge distribution in Fig. 3c, a relatively high electron density around the epoxy group can be observed in the O-CC model, indicating that the introduction of the epoxy group causes interface electron regulation. Relative to the P-CC model, this interface electron regulation will create a region of very strong localized electrons for the adsorption of K atoms. Furthermore, we calculated the density of states (DOS) of the K atom on P-CC and O-CC for comparison, as shown in Fig. 3d. Compared with the K atom on P-CC, a DOS broadening of the K atom on O-CC can be observed, indicating stronger orbital hybridization of the K atom and the epoxy group. Fig. 3e shows the calculation model of P-CC and O-CC for K atom adsorption. It is found that the binding energy between K atoms and P-CC is −1.21 eV. For O-CC, the binding energy greatly decreases to −3.15 eV, indicating that K will preferentially adsorb on O-CC. To verify this behavior, the Raman spectra of the primary electrolyte and the electrolyte filtered by P-CC and O-CC are provided in Fig. S13.† According to fitting results, the Raman peaks of the electrolyte can be deconvoluted into three peaks, which correspond to the solvent-separated ion pair (SSIP), contact ion pair (CIP), and aggregate (AGG) coordination.36 It is observed that the solvation structures of aggregate (AGG) coordination in the electrolyte filtered by O-CC decrease and show almost no change in the electrolyte filtered by P-CC, indicating the strong K adsorption ability of O-CC after epoxy group introduction. In all, we suggest that the K adsorption ability influences the solvated structures of electrolyte and changes the constituent of the solid electrolyte interphase (SEI) layer, which contributes to the improved electrochemical behavior of the battery.37 To further confirm this behavior, we calculated the adsorption energy (Ea) of P-CC and O-CC with DME electrolyte (Fig. S14†). O-CC presents an adsorption energy of −0.12 eV with DME molecules, more negative than that of P-CC (0.06 eV). Therefore, for the solvated structure of K ions, the more negative adsorption energy indicates the strong adsorption of K ions by O-CC, which facilitates the desolvation process and the interfacial transfer process of K ions.
 | ||
| Fig. 3 DFT calculations and the corresponding experimental characterization of P-CC and O-CC. (a) Charge density on the surface; (b) plots of the surface Fermi levels of the host to the electrolyte; (c) 3D charge density and cross profile of O-CC's charge density; (d) pDOS results, (e) surface binding energies to K, (f) TEM images of the SEI layers after 1 cycle; (g) XPS spectra of SEI layers after 1 cycle. | ||
The contact angles of P-CC and O-CC with electrolyte (Fig. S15†) were compared to verify the calculation results and it was found that O-CC presents an angle of 11° at 0.07 s after the electrolyte drops, lower than that of P-CC (20°), demonstrating an improvement of electrolyte wettability of the O-CC electrode. Due to this reason and good potassiophilic ability, O-CC has a larger active area of K metal with electrolyte while K covers the surface of P-CC like a two-dimensional plane by physical squeezing. Then a larger amount of K-ions can translate faster in the host and avoid localized deposition. We disassembled the symmetric cells after cycling to observe the change in electrode structure. The TEM images in Fig. 3f indicate that the SEI layer in the O-CC@K//O-CC@K symmetric cell is about 8 nm, thinner than in the P-CC@K//P-CC@K cell (25 nm). To further explore the SEI composition, the XPS of P-CC and O-CC is also performed, as shown in Fig. S16, 3g, and S17.† Considering that the detection depth of XPS is less than 10 nm, we believe that the detection results based on XPS after cycling are from the SEI layer of the electrode.38 From the F 1s XPS, the K–F bond (682.7 eV and 684.6 eV) and S–F bond (687.1 eV) can be deconvoluted.39 It is found that the SEI layer of the O-CC electrode contains more K–F ingredients compared with the P-CC electrode. The high K–F content of the SEI layer contributes to the enhancement of the mechanical strength of the electrode during the K plating/stripping process, resulting in better stability of the batteries.40,41
Fig. 4a and b show the in situ optical microscope photographs of P-CC@K and O-CC@K electrodes at a plating areal current density of 0.1 mA cm−2 for 60 min, respectively. The corresponding photograph of the in situ optical microscope observation system is shown in Fig. S18.† Both electrodes present a smooth interface in the initial stage. However, clear K dendrites arise quickly in the P-CC@K electrode after plating for 30 min (Fig. 4a and Video S5†), which is ascribed to the uneven deposition caused by the high adsorption energy between K and P-CC. With the increase of the plating process to 60 min, the growth of K dendrites is more pronounced. In contrast, the O-CC@K electrode still has a smooth interface from 0 min to 60 min (Fig. 4b and Video S6†), indicating that the K dendrite can be inhibited significantly with the introduction of the epoxy group. Fig. 4c and d show the SEM images of P-CC@K and O-CC@K which transfer under an Ar atmosphere after 100 cycles at a current density of 0.5 mA cm−2 with a capacity of 0.5 mA h cm−2. The P-CC still has plenty of carbon fiber that can't be wrapped by K, indicating that P-CC easily accumulates K non-uniformly, which causes the growth of K dendrites. In contrast, the carbon fiber of O-CC is evenly and densely wrapped by K without dendrites. This micro-scale feature ensures the absence of dendrites in the macro-scale. After that, we also attempted to model the growth of K dendrites of P-CC and O-CC by finite element analysis (FEA). Given the different adsorption abilities of K with P-CC and O-CC electrodes, we set the P-CC and O-CC electrodes with the initial conditions of inhomogeneous K-ions and homogeneous K-ions for deposition, respectively (Fig. 4e and f). From the FEA results, some inhomogeneous deposition of the P-CC electrode begins to appear at 300 s and intensifies at 600 s. As a comparison, the O-CC electrode delivers uniform K deposition and seems almost dendrite-free at 600 s.
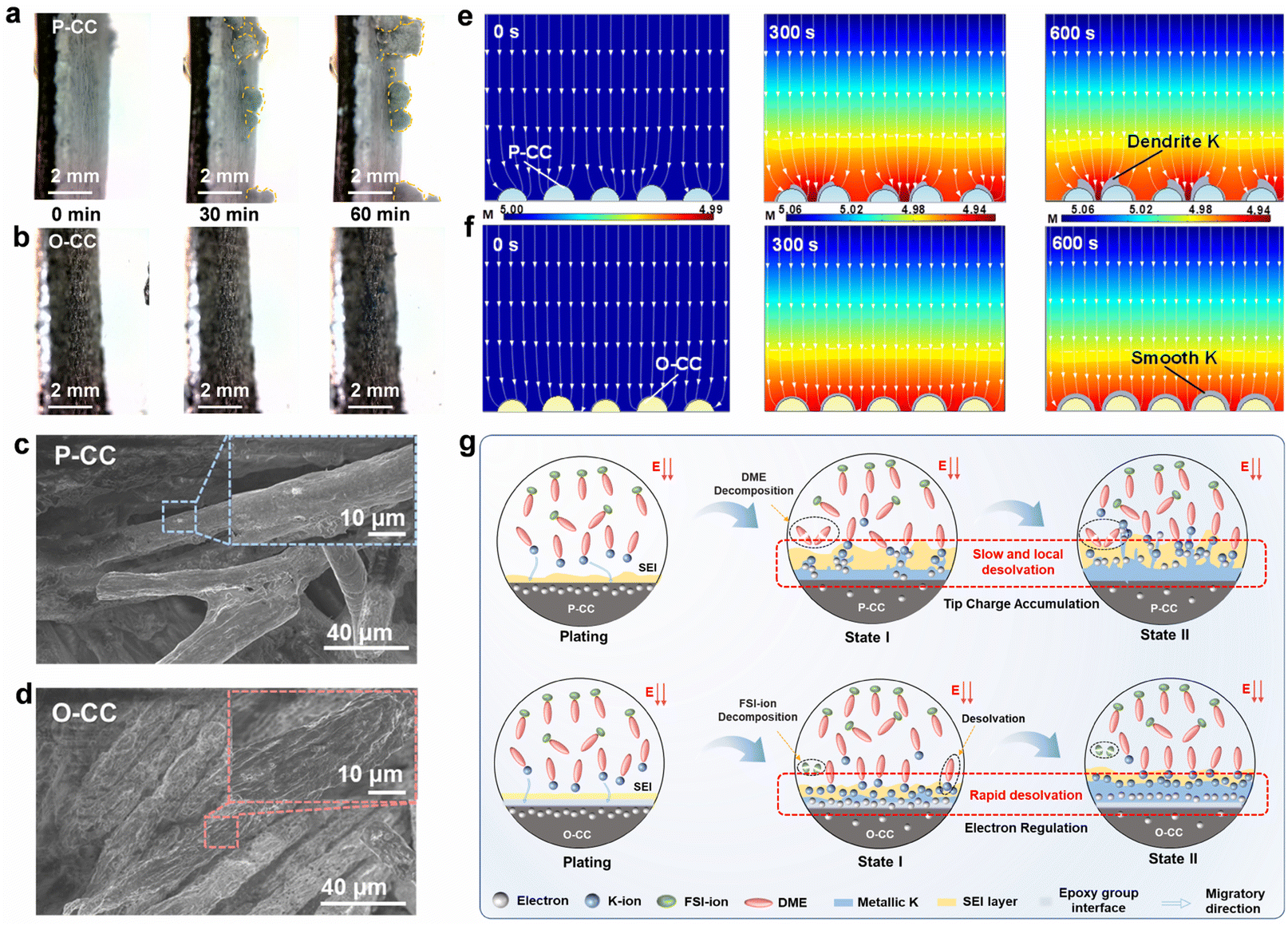 | ||
| Fig. 4 Process and mechanism of the K metal plating–stripping process. In situ optical microscope photographs for (a) P-CC@K and (b) O-CC@K at a plating areal current density of 0.7 mA cm−2. SEM images of (c) P-CC@K and (d) O-CC@K after 100 cycles at 0.5 mA cm−2–0.5 mA h cm−2; surface morphology simulations of K metal deposition for (e) P-CC@K and (f) O-CC@K, the color represents K-ion concentration; (g) schematic of the P-CC and O-CC multifunctional host for the K-ion plating process. | ||
Based on the aforementioned discussions, we propose the electrochemical enhancement schematic of the O-CC multifunctional host for KIBs, as shown in Fig. 4g. Generally, the electron distribution in the interface of the host should be homogeneous and the number of electrons exceeds the number of K-ions in the initial state in both P-CC and O-CC electrodes. Therefore, the K-ion desolvation rate is critical for the uniform deposition of K metal. For P-CC, due to the slow K-ion desolvation, the electrons in the interface of the host tend to accumulate at the local region, triggering the enhancement of K-ion desolvation in the local region and causing the rapid growth of K dendrites. In addition, the high Fermi level of P-CC also reinforces the electron transfer from the electrode to DME, resulting in the decomposition of DME solvent. After the introduction of the epoxy group, the strong electron binding of the epoxy group means that the O-CC electrode has a strong interaction with K-ions. Therefore, it exhibits a rapid K-ion desolvation during the K plating state. In this process, the uniformly distributed electrons can quickly react with the desolvated K-ions, giving rise to the uniform K metal deposition. This homogeneous K metal deposition results in dendrite-free growth on the O-CC electrode. Besides, the O-CC electrode also exhibits a weak interaction with DME, indicating the electrode is inclined to react with the anion, which contributes to forming a robust KF-rich SEI layer.
We assembled an O-CC@K//PTCDA full battery to further evaluate practical applicability. The corresponding full battery schematic is shown in Fig. S19.† The full cells delivered a reversible specific capacity of 91 mA h g−1 at a current density of 20 mA h g−1 after 50 cycles (Fig. 5a) (Fig. S20a†). The corresponding GCD curves show a high degree of reversibility and almost overlapping GCD curves in different potassiation–depotassiation processes (Fig. S21†). Fig. 5b presents the rate performance of the O-CC@K//PTCDA full cell, which delivers the stable reversible discharge capacities of 89, 80, 63, 54, and 37 mA h g−1 at current densities of 20, 40, 80, 100, and 200 mA g−1, respectively. Fig. 5c presents the long-term cyclic performance of O-CC@K//PTCDA and the full cell has a high reversible specific capacity of 44 mA h g−1 at a current density of 100 mA h g−1 after 300 cycles with a capacity retention of 73.3%. The full cell presents a super high average CE at 300 cycles (Fig. S20b†) and a stable charge–discharge plateau (Fig. S22†). To demonstrate its excellent discharge performance, a high-power LED bulb (1 W) can be lit up by the CC@K//PTCDA cell (Fig. S23†), indicating a high-power discharge capacity of the CC@K//PTCDA cell. The schematic in Fig. 5d presents a prospect for the potential application of KMBs charged by a triboelectric nanogenerator (TENG).42 As a validation example, we used our O-CC@K//PTCDA full cell as an energy storage system, which is charged by a TENG and discharged at a galvanostatic current density of 100 mA g−1 for 3 cycles (Fig. 5e). This result indicates the great potential of the O-CC@K//PTCDA cell for constructing a self-charging power pack.
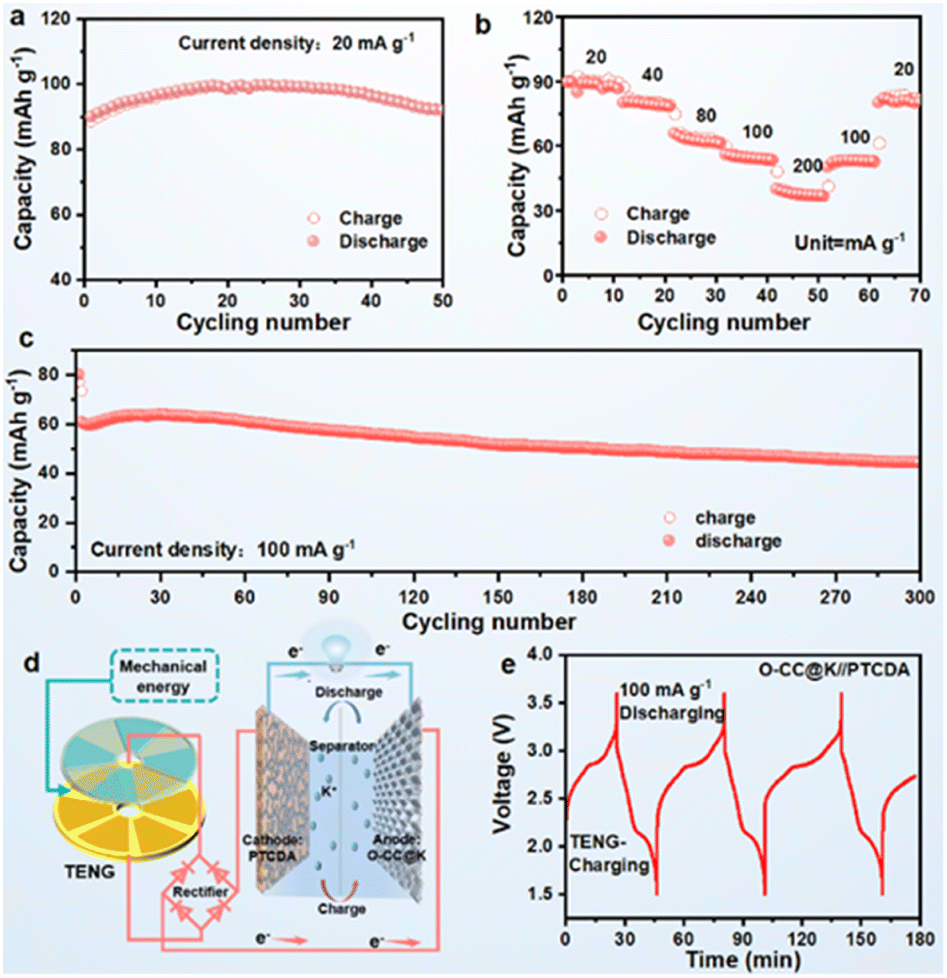 | ||
| Fig. 5 Electrochemical performance of PTCDA//O-CC@K full cells. (a) Cyclic performance at 20 mA g−1 and (c) rate performance at different current densities; (c) long-term cyclic performance at 100 mA g−1; (d) schematic of TENG charge and galvanostatic discharge of the self-powered system; (e) TENG charge and galvanostatic discharge performance of the self-powered system by O-CC@K//PTCDA batteries. All cells were tested after a 20-cycle activation process at 20 mA g−1. | ||
Conclusions
In summary, we constructed an O-CC as a multifunctional host for a K metal anode. The epoxy groups in O-CC can suppress electron transfer and form a surface with lower Fermi levels, resulting in fewer side reactions with the electrolyte and the formation of a robust SEI layer. Meanwhile, abundant epoxy groups impart strong binding of K adatoms via orbital hybridization but weak binding of electrolyte molecules, which can effectively promote K-ion desolvation and uniform deposition. As a result, it exhibited ultra-long stable cycling for over 5500 h at 0.5 mA cm−2/0.5 mA h cm−2 with a low polarization voltage of about 29 mV. Even at a high current density of 1 mA cm−2/0.5 mA h cm−2, O-CC also exhibits a stable cycling performance over 3500 h at a low polarization voltage of 41 mV for K plating/stripping, surpassing the cycle life of all previously reported K metal anodes. Additionally, high average CEs of over 99.3% during 210 cycles for the O-CC//K asymmetric cell and capacity retention of 73.3% during 300 cycles for the O-CC@K//PTCDA full cell are also achieved. Finally, the stable electrochemical performance of O-CC@K in full cells coupled with PTCDA cathodes is demonstrated, indicating great application potential.Data availability
All experimental supporting data and procedures are available in the ESI.†Author contributions
Z. Li: methodology, formal analysis, investigation and writing – original draft; L. Ma: methodology, formal analysis, investigation and writing – original draft; Han Kai: methodology; Y. Ji: investigation; J. Xie: investigation; L. Pan: formal analysis; J. Li: supervision, funding acquisition, and writing – review & editing; W. Mai: supervision, funding acquisition, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the financial support from the National Natural Science Foundation of China (52172202), Fundamental Research Funds for the Central Universities (11619103, 21621406), Science and Technology Program of Guangzhou, China (202102020737), and Shenzhen Science and Technology Program (JCYJ20200109113606007). We also thank Prof. Zhong Lin Wang from Beijing Institute of Nanoenergy and Nanosystems for giving us some very valuable suggestions.References
- R. Herrington, Mining our green future, Nat. Rev. Mater., 2021, 6, 456–458 CrossRef.
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Research Development on K-Ion Batteries, Chem. Rev., 2020, 120, 6358–6466 CrossRef CAS PubMed.
- M. Zhou, P. Bai, X. Ji, J. Yang, C. Wang and Y. Xu, Electrolytes and interphases in potassium ion batteries, Adv. Mater., 2021, 33, 2003741 CrossRef CAS PubMed.
- H. Lei, J. Li, X. Zhang, L. Ma, Z. Ji, Z. Wang, L. Pan, S. Tan and W. Mai, A review of hard carbon anode: Rational design and advanced characterization in potassium ion batteries, InfoMat, 2022, 4, e12272 CrossRef CAS.
- Q. Sun, D. Li, J. Cheng, L. Dai, J. Guo, Z. Liang and L. Ci, Nitrogen-doped carbon derived from pre-oxidized pitch for surface dominated potassium-ion storage, Carbon, 2019, 155, 601–610 CrossRef CAS.
- J. Xie, J. Li, X. Li, H. Lei, W. Zhuo, X. Li, G. Hong, K. N. Hui, L. Pan and W. Mai, Ultrahigh “relative energy density” and mass loading of carbon cloth anodes for K-ion batteries, CCS Chem., 2021, 3, 791–799 CrossRef CAS.
- Q. Li, Y. Zhang, Z. Chen, J. Zhang, Y. Tao and Q. H. Yang, Discrete Graphitic Crystallites Promise High-rate Ion Intercalation for KC8 Formation in Potassium Ion Batteries, Adv. Energy Mater., 2022, 12, 2201574 CrossRef CAS.
- L. Ma, Z. Li, J. Li, Y. Dai, C. Qian, Y. Zhu, H. Wang, K. N. Hui, L. Pan, M. A. Amin, Y. Yamauchi and W. Mai, Phytic acid-induced nitrogen configuration adjustment of active nitrogen-rich carbon nanosheets for high-performance potassium-ion storage, J. Mater. Chem. A, 2021, 9, 25445–25452 RSC.
- Q. Peng, S. Zhang, H. Yang, B. Sheng, R. Xu, Q. Wang and Y. Yu, Boosting potassium storage performance of the Cu2S anode via morphology engineering and electrolyte chemistry, ACS Nano, 2020, 14, 6024–6033 CrossRef CAS PubMed.
- X. Wu, D. P. Leonard and X. Ji, Emerging Non-Aqueous Potassium-Ion Batteries: Challenges and Opportunities, Chem. Mater., 2017, 29, 5031–5042 CrossRef CAS.
- H. Kim, J. C. Kim, M. Bianchini, D. H. Seo, J. Rodriguez-Garcia and G. Ceder, Recent Progress and Perspective in Electrode Materials for K-Ion Batteries, Adv. Energy Mater., 2017, 8, 1702384 CrossRef.
- C. Wei, Y. Tao, H. Fei, Y. An, Y. Tian, J. Feng and Y. Qian, Recent advances and perspectives in stable and dendrite-free potassium metal anodes, Energy Stor. Mater., 2020, 30, 206–227 Search PubMed.
- P. Liu and D. Mitlin, Emerging Potassium Metal Anodes: Perspectives on Control of the Electrochemical Interfaces, Acc. Chem. Res., 2020, 53, 1161–1175 CrossRef CAS PubMed.
- W. Liu, P. Liu and D. Mitlin, Tutorial review on structure–dendrite growth relations in metal battery anode supports, Chem. Soc. Rev., 2020, 49, 7284–7300 RSC.
- P. Liu, Y. Wang, Q. Gu, J. Nanda, J. Watt and D. Mitlin, Dendrite-Free Potassium Metal Anodes in a Carbonate Electrolyte, Adv. Mater., 2020, 32, e1906735 CrossRef PubMed.
- M. Zhou, W. Qi, Z. Hu, M. Cheng, X. Zhao, P. Xiong, H. Su, M. Li, J. Hu and Y. Xu, Highly Potassiophilic Carbon Nanofiber Paper Derived from Bacterial Cellulose Enables Ultra-Stable Dendrite-Free Potassium Metal Anodes, ACS Appl. Mater. Interfaces, 2021, 13, 17629–17638 CrossRef CAS PubMed.
- J. Wang, W. Yan and J. Zhang, High area capacity and dendrite-free anode constructed by highly potassiophilic Pd/Cu current collector for low-temperature potassium metal battery, Nano Energy, 2022, 96, 107131 CrossRef CAS.
- S. Ye, L. Wang, F. Liu, P. Shi and Y. Yu, Integration of homogeneous and heterogeneous nucleation growth via 3D alloy framework for stable Na/K metal anode, E-Science, 2021, 1, 75–82 Search PubMed.
- N. Xiao, W. D. McCulloch and Y. Wu, Reversible dendrite-free potassium plating and stripping electrochemistry for potassium secondary batteries, J. Am. Chem. Soc., 2017, 139, 9475–9478 CrossRef CAS PubMed.
- S. Li, H. Zhu, Y. Liu, Z. Han, L. Peng, S. Li, C. Yu, S. Cheng and J. Xie, Codoped porous carbon nanofibres as a potassium metal host for nonaqueous K-ion batteries, Nat. Commun., 2022, 13, 4911 CrossRef CAS PubMed.
- Y. Xie, J. Hu, Z. Han, H. Fan, J. Xu, Y. Lai and Z. Zhang, Ultra-stable K metal anode enabled by oxygen-rich carbon cloth, Nano Res., 2020, 13, 3137–3141 CrossRef CAS.
- M. Ye, J. Y. Hwang and Y. K. Sun, A 4 V Class Potassium Metal Battery with Extremely Low Overpotential, ACS Nano, 2019, 13, 9306–9314 CrossRef CAS PubMed.
- X. Zhao, F. Chen, J. Liu, M. Cheng, H. Su, J. Liu and Y. Xu, Enhanced surface binding energy regulates uniform potassium deposition for stable potassium metal anodes, J. Mater. Chem. A, 2020, 8, 5671–5678 RSC.
- J. Zhang, Y. Li, L. Zhu, X. Wang and J. Tu, Potassiophilic skeleton achieving highly stable potassium metal anode, Chem. Eng. J., 2022, 449, 137659 CrossRef CAS.
- A. Bagri, C. Mattevi, M. Acik, Y. J. Chabal, M. Chhowalla and V. B. Shenoy, Structural evolution during the reduction of chemically derived graphene oxide, Nat. Chem., 2010, 2, 581–587 CrossRef CAS PubMed.
- K. Bagani, M. K. Ray, B. Satpati, N. R. Ray, M. Sardar and S. Banerjee, Contrasting magnetic properties of thermally and chemically reduced graphene oxide, J. Phys. Chem. C, 2014, 118, 13254–13259 CrossRef CAS.
- W. Qiu, J. E. Leisen, Z. Liu, W. Quan and W. J. Koros, Key Features of Polyimide-Derived Carbon Molecular Sieves, Angew. Chem., Int. Ed., 2021, 60, 22322–22331 CrossRef CAS PubMed.
- F. Yang, X. Ma, W.-B. Cai, P. Song and W. Xu, Nature of oxygen-containing groups on carbon for high-efficiency electrocatalytic CO2 reduction reaction, J. Am. Chem. Soc., 2019, 141, 20451–20459 CrossRef CAS PubMed.
- C. Liang, Y. Chen, M. Wu, K. Wang, W. Zhang, Y. Gan, H. Huang, J. Chen, Y. Xia, J. Zhang, S. Zheng and H. Pan, Green synthesis of graphite from CO2 without graphitization process of amorphous carbon, Nat. Commun., 2021, 12, 119 CrossRef CAS PubMed.
- K. Tian, J. Wang, L. Cao, W. Yang, W. Guo, S. Liu, W. Li, F. Wang, X. Li, Z. Xu, Z. Wang, H. Wang and Y. Hou, Single-site pyrrolic-nitrogen-doped sp2-hybridized carbon materials and their pseudocapacitance, Nat. Commun., 2020, 11, 3884 CrossRef CAS PubMed.
- Y. Xu, T. Li, L. Wang and Y. Kang, Interlayered Dendrite-Free Lithium Plating for High-Performance Lithium-Metal Batteries, Adv. Mater., 2019, 31, 1901662 CrossRef PubMed.
- P. Shi, T. Li, R. Zhang, X. Shen, X.-B. Cheng, R. Xu, J.-Q. Huang, X.-R. Chen, H. Liu and Q. Zhang, Lithiophilic LiC6 Layers on Carbon Hosts Enabling Stable Li Metal Anode in Working Batteries, Adv. Mater., 2019, 31, 1807131 CrossRef PubMed.
- M. S. Kim, Z. Zhang, P. E. Rudnicki, Z. Yu, J. Wang, H. Wang, S. T. Oyakhire, Y. Chen, S. C. Kim, W. Zhang, D. T. Boyle, X. Kong, R. Xu, Z. Huang, W. Huang, S. F. Bent, L.-W. Wang, J. Qin, Z. Bao and Y. Cui, Suspension electrolyte with modified Li+ solvation environment for lithium metal batteries, Nat. Mater., 2022, 21, 445–454 CrossRef CAS PubMed.
- S. Hou, X. Ji, K. Gaskell, P.-f. Wang, L. Wang, J. Xu, R. Sun, O. Borodin and C. Wang, Solvation sheath reorganization enables divalent metal batteries with fast interfacial charge transfer kinetics, Science, 2021, 374, 172–178 CrossRef CAS PubMed.
- A. Hu, W. Chen, X. Du, Y. Hu, T. Lei, H. Wang, L. Xue, Y. Li, H. Sun and Y. Yan, An artificial hybrid interphase for an ultrahigh-rate and practical lithium metal anode, Energy Environ. Sci., 2021, 14, 4115–4124 RSC.
- J. Fawdon, J. Ihli, F. L. Mantia and M. Pasta, Characterising lithium-ion electrolytes via operando Raman microspectroscopy, Nat. Commun., 2021, 12, 4053 CrossRef CAS PubMed.
- L. Su, X. Zhao, M. Yi, H. Charalambous, H. Celio, Y. Liu and A. Manthiram, Uncovering the Solvation Structure of LiPF6-Based Localized Saturated Electrolytes and Their Effect on LiNiO2-Based Lithium-Metal Batteries, Adv. Energy Mater., 2022, 12, 2201911 CrossRef CAS.
- S.-K. Otto, Y. Moryson, T. Krauskopf, K. Peppler, J. Sann, J. Janek and A. Henss, In-Depth Characterization of Lithium-Metal Surfaces with XPS and ToF-SIMS: Toward Better Understanding of the Passivation Layer, Chem. Mater., 2021, 33, 859–867 CrossRef CAS.
- Y. Ouyang, W. Zong, J. Wang, Z. Xu, L. Mo, F. Lai, Z.-L. Xu, Y.-E. Miao and T. Liu, Multi-scale uniform Li regulation triggered by tunable electric field distribution on oxygen-functionalized porous framework for flexible Li-S full batteries, Energy Stor. Mater., 2021, 42, 68–77 Search PubMed.
- R. Lin, Y. He, C. Wang, P. Zou, E. Hu, X.-Q. Yang, K. Xu and H. L. Xin, Characterization of the structure and chemistry of the solid–electrolyte interface by cryo-EM leads to high-performance solid-state Li-metal batteries, Nat. Nanotechnol., 2022, 17, 768–776 CrossRef CAS PubMed.
- J. Xie, X. Li, H. Lai, Z. Zhao, J. Li, W. Zhang, W. Xie, Y. Liu and W. Mai, A Robust Solid Electrolyte Interphase Layer Augments the Ion Storage Capacity of Bimetallic-Sulfide-Containing Potassium-Ion Batteries, Angew. Chem., Int. Ed., 2019, 58, 14740–14747 CrossRef CAS PubMed.
- K. Han, J. Luo, J. Chen, Y. Liu, J. Li, Z. L. Wang and W. Mai, Plastic film based lightweight thruster driven by triboelectric nanogenerator for multi-purpose propulsion applications, Nano Energy, 2022, 101, 107558 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03203e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |