
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceδ-VOPO4 as a high-voltage cathode material for aqueous zinc-ion batteries†
Dong
Zhao
a,
Xiangjun
Pu
*b,
Shenglong
Tang
a,
Mingyue
Ding
 *a,
Yubin
Zeng
a,
Yuliang
Cao
b and
Zhongxue
Chen
*a
*a,
Yubin
Zeng
a,
Yuliang
Cao
b and
Zhongxue
Chen
*a
aKey Laboratory of Hydraulic Machinery Transients, Ministry of Education, School of Power and Mechanical Engineering, Wuhan University, Wuhan 430072, China. E-mail: zxchen_pmc@whu.edu.cn; dingmy@whu.edu.cn
bCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China. E-mail: joy_pu@whu.edu.cn
First published on 7th July 2023
Abstract
Aqueous zinc-ion batteries (AZIBs) with excellent safety, low-cost and environmental friendliness have great application potential in large-scale energy storage systems and thus have received extensive research interest. Layered oxovanadium phosphate dihydrate (VOPO4·2H2O) is an appealing cathode for AZIBs due to the unique layered framework and desirable discharge plateau, but bottlenecked by low operation voltage and unstable cycling. Herein, we propose delta-oxovanadium phosphate (δ-VOPO4) without conventional pre-embedding of metal elements or organics into the structure and paired it into AZIBs for the first time. Consequently, superior to the layered counterpart, δ-VOPO4 exhibits better performance with a prominent discharge voltage of 1.46 V and a higher specific capacity of 122.6 mA h g−1 at 1C (1C = 330 mA g−1), as well as an impressive capacity retention of 90.88 mA h g−1 after 1000 cycles under 10C. By investigation of structure resolution and theoretical calculation, this work well elucidates the structure–function relationship in vanadyl phosphates, offering more chances for exploration of new cathode materials to construct high performance AZIBs.
Introduction
Facing the shortage of fossil energy and greenhouse effect, development and utilization of sustainable energy is one of the global challenges.1,2 The successful application of lithium-ion batteries (LIBs) in industries for portable electronic devices and electric vehicles provides an electrochemical solution to this dilemma.3 However, the long-term utilization of LIBs is restricted by high cost, limited resources, poor thermal stability from organic electrolytes and strict assembly conditions.4 Aqueous multivalent metal-ion batteries especially aqueous zinc-ion batteries (AZIBs) inherent with non-flammability of water could merge this gap between high specific capacity and excellent safety.5,6 They have been widely studied due to the high volumetric energy density and abundant zinc reserves.7At present, the cathode material is the bottleneck limiting the energy density of AZIBs due to the limited specific capacity compared to that of Zn metal anodes. Typical cathode materials for aqueous zinc-ion batteries include manganese-based materials, Prussian blue analogs and vanadium-based materials. Among them, manganese-based compounds inevitably suffer from manganese dissolution and structure destruction due to the Jahn–Teller effect when Mn3+ is formed. In addition, the competition mechanism embedded by H+ and Zn2+ is still controversial.8,9 Prussian blue materials have low electrical conductivity, and limited first-cycle efficiency and cycling life, which restrict their further development. Vanadium-based materials are widely studied in virtue of multi-valence changes and compositional structures. However, vanadium oxides and vanadates have low operating voltages and always undergo a large irreversible deformation during the process of Zn2+ intercalation, which is detrimental to long-term cycling stability. Therefore, it is urgent and significant to exploit and develop novel cathode materials.
Among various chemistry systems, vanadium-based phosphates have advantages of high theoretical capacity, open structural framework and high operating voltage, and stand out as potential candidates for AZIBs.10,11 Indeed, they have been proved to have excellent performance in lithium and sodium ion batteries and have potential for large-scale commercial use.12 Notably, resulting from the aforementioned inductive effect, the higher operating voltage of phosphate vanadates than that of vanadium oxides is one attracting merit of phosphate vanadium. The most feasible homologue of vanadium phosphate is VOPO4, since it doesn't contain many more unwanted elements like Na or K when used in AZIBs (which means no pre-desodiation or pre-depotassiation is required). VOPO4·2H2O with high operating voltage has become a competitive material, and the interlayer spacing can be widened through pre embedding engineering to improve the zinc storage performance. However, despite measures like introducing metal ions (such as K+, Zn2+)13,14 or small organic molecules (such as phenylamine, polypyrrole)15,16 into the layers to adjust the interlayer spacing or stabilize the structure, the capacity of this material is far from reaching the theoretical capacity in the presented reports. Meanwhile, the dissolution of V is still a sticky issue even though high-cost super-concentration electrolytes or organic electrolytes are incorporated.17
Alternatively, multiple phases of vanadyl phosphates have been reported in addition to layered VOPO4·2H2O, including αI, αII, β, γ, δ, ε, ω (Fig. S1†), among which some phases have been studied tentatively as cathode materials in Li-ion and Na-ion batteries,18–26 yet their zinc storage properties are not clear in aqueous batteries. Herein, we successfully synthesize δ-VOPO4 and measured its electrochemical performance in aqueous zinc-ion batteries. Compared to the performance of other phases of VOPO4 in aqueous zinc-ion batteries, the delta-phase sample shows a higher operating voltage and satisfactory specific capacity (Fig. S2†). Results show that the δ-VOPO4 in platelet-like form displays a desirable capacity (90.88 mA h g−1 at 10C, 1C = 330 mA g−1) and high discharge voltage (1.46 V vs. Zn2+/Zn), laying a simple but strong foundation to exploit advanced ensuing AZIBs with high energy density without introducing complex doping or substitution groups.
Experimental
Synthesis
Various phases of vanadyl phosphates (VOPO4) were prepared according to the methods reported in the literature.17 In a typical process, δ-VOPO4 was prepared by a two-step method. First, VOPO4·2H2O (1 g) and isopropyl alcohol (14 mL) were first transferred to a Teflon-lined stainless-steel autoclave (50 mL) followed by keeping under 130 °C for 24 h. After washing with isopropyl alcohol several times and vacuum-drying at 110 °C, the powder was calcined at 600 °C for 14 h in a Muffle furnace to obtain δ-VOPO4.Characterization
The phases of powder materials were studied by X-ray diffraction (Rigaku MiniFlexll with Cu Kα radiation (k = 1.5408 Å)) at a scan rate of 2° min−1. The structure morphologies were studied by scanning electron microscopy (SEM, Zeiss Merlin Compact) and transmission electron microscopy (TEM, JEM-2100 Plus, JEOL). Energy dispersive spectroscopy (EDS) paired with SEM mapping was used to investigate the element distribution of the samples. An inductively coupled plasma optical emission spectrometer (ICP-OES, Agilent 5800) was used to determine the element ratio. An X-ray Photoelectron Spectrometer (XPS, ESCALAB250Xi, Thermo Fisher Scientific) was used to reveal the existing forms of element valence states within samples. Further information on the chemical bonding is revealed by means of a Fourier transform infrared spectrometer (FTIR, NICOLET 5700) and Raman spectrometer (DXR2, Thermo Fisher).Electrochemical measurements
Electrochemical tests of all the electrodes were performed with CR2032 coin-type cells with Zn foil as the counter electrode. The working electrode was fabricated using a slurry of active material, Super P, and PTFE (poly-tetrafluoroethylene) in a weight ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2 with isopropyl alcohol as a humectant. The slurry was coated onto a Ti mesh and dried at 80 °C in a vacuum oven for 12 h. A glass fiber membrane was used as the separator and 3 m Zn(CF3SO3)2 was prepared as the electrolyte. The electrochemical performance of the battery was evaluated using a LAND battery test system (CT2001A) including galvanostatic charge–discharge (GCD), rate and long-term cycling performance. The galvanostatic intermittent titration technique (GITT) was performed under a modified GCD mode, in which an operation period includes two parts: a charge/discharge procedure lasting for 10 min at 50 mA g−1 and a subsequent pause time for 30 min. Cyclic voltammetry (CV) tests at different scan rates were performed on an electrochemical workstation (Corrtest, CS350H).
:2:2 with isopropyl alcohol as a humectant. The slurry was coated onto a Ti mesh and dried at 80 °C in a vacuum oven for 12 h. A glass fiber membrane was used as the separator and 3 m Zn(CF3SO3)2 was prepared as the electrolyte. The electrochemical performance of the battery was evaluated using a LAND battery test system (CT2001A) including galvanostatic charge–discharge (GCD), rate and long-term cycling performance. The galvanostatic intermittent titration technique (GITT) was performed under a modified GCD mode, in which an operation period includes two parts: a charge/discharge procedure lasting for 10 min at 50 mA g−1 and a subsequent pause time for 30 min. Cyclic voltammetry (CV) tests at different scan rates were performed on an electrochemical workstation (Corrtest, CS350H).
Computational methods
First-principles calculations were carried out using density functional theory (DFT) with the projector augmented-wave method as implemented using the Vienna Ab initio Simulation Package (VASP) code. The Perdew–Burke–Ernzerhof (PBE) of generalized gradient approximation (GGA) was chosen for the exchange–correction functional. Hubbard corrections to intra-atomic coulombic repulsion were considered and the effective Hubbard U parameters to 4.5 eV for V were set to correct electron–electron interactions of V 3d electrons.27–29 The wave functions were expanded based on plane-waves with the energy cutoff set to 600 eV. All lattice parameters and atomic sites were fully optimized until the change of the total energy and residual forces were less than 10−5 eV and 0.02 eV Å−1, respectively. A 1 × 1 × 1 supercell containing 8 V and 8 P and 40 O atoms was considered for exploring the energies of different configurations taking up Zn ions. A Γ-centered 5 × 5 × 5 k-point mesh with a reciprocal space was employed in the Brillouin zone of the supercell. The equilibrium voltage is evaluated using the internal energies from the DFT calculations according to the following equation:where F is the Faraday constant and E(Znx1VOPO4), E(Znx2VOPO4) and E(Zn) are the internal energies at 0 K, respectively.
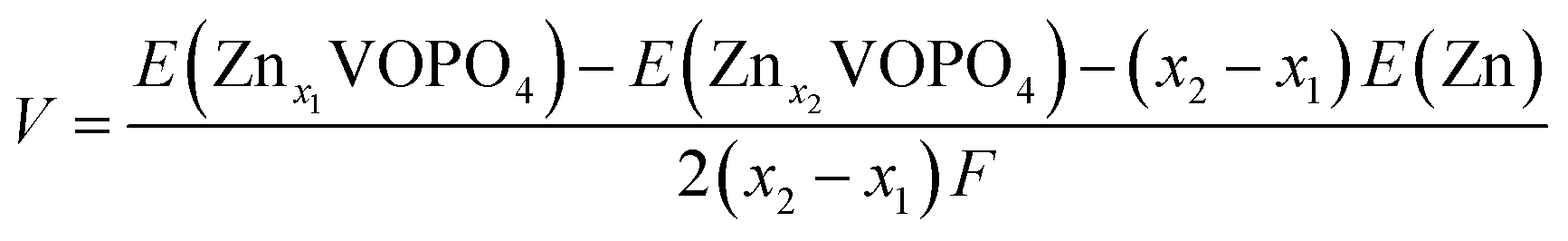
Results and discussion
Derivation and characterization of δ-VOPO4
Scanning electron microscopy (SEM) was performed to track the morphology evolution during the preparation process. δ-VOPO4 prepared herein can be described as aggregates of micron-sized platelets in Fig. 1a and b. Such a morphology is similar to the flaky appearance of its precursors (VOPO4·2H2O and VOHPO4·0.5H2O) shown in Fig. S3a and b.† Compared with VOPO4·2H2O consisted of dense massive flakes, the units of δ-VOPO4 are smaller in diameter and less tightly stacked with each other, and the gap in the framework can provide more contact area for the material and the electrolyte, which is beneficial for performance. The elemental mappings displayed in Fig. 1d–f show that the V, O and P elements are homogeneously distributed in the sample. The XRD pattern of δ-VOPO4 was refined according to the standard card (JCPDS no. 07-1760). The Rietveld refinement in Fig. 1g confirms that the crystal structure of δ-VOPO4 is indexed to a space group of tetragonal P42/mbc, and the unit cell parameters are identified as a = b = 9.2016 Å, and c = 8.4953 Å. The fitting was satisfactory (Rwp = 5.601%, Rp = 7.219%). The high-resolution transmission electron microscopy (HRTEM) image of δ-VOPO4 shown in Fig. 1h exhibits lattice fringes with a spacing of 0.312 nm corresponding to the (202) planes. Selected-area electron diffraction (SAED) patterns (Fig. 1i) observed from the flakes in a normal direction can be well indexed to the (212), (211) and (202) projection of δ-VOPO4. | ||
| Fig. 1 (a–c) SEM images, (d–f) elemental mappings, (g) Rietveld XRD pattern, (h) HR-TEM and (i) SAED images of δ-VOPO4. | ||
Fourier transform infrared spectroscopy (FTIR) and Raman spectroscopy are subsequently employed to clearly unveil the molecular structure and chemical composition of δ-VOPO4. As shown in Fig. 2a, the V–O–P bending vibration peak is located at 679 cm−1. The peaks located in the range of 1000 cm−1 to 1250 cm−1 can be attributed to P–O stretching vibration modes in the (PO4) group, while the peak at 1630 cm−1 of the sample corresponds to H–O–H bending vibrations due to adsorbed water on the surface of the material.30 Raman spectroscopy can also convey the same information as depicted in Fig. 2b. The peak at 941 cm−1 in δ-VOPO4 can be attributed to O–P–O stretching modes. The peaks of V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration and V–O–P coupled stretching in δ-VOPO4 are located respectively at 1015 cm−1 and 1084 cm−1.
O stretching vibration and V–O–P coupled stretching in δ-VOPO4 are located respectively at 1015 cm−1 and 1084 cm−1.
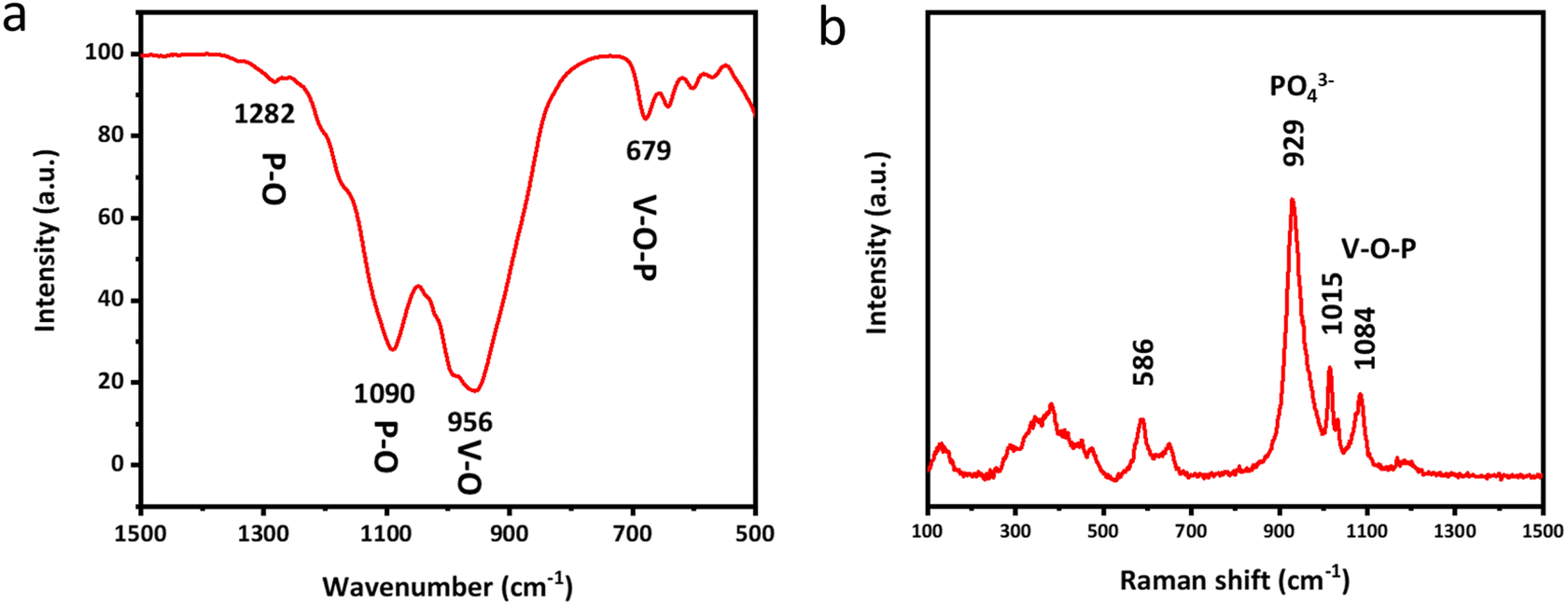 | ||
| Fig. 2 (a) FTIR spectra and (b) Raman spectra of δ-VOPO4 powder. | ||
Zn2+ storage performance of δ-VOPO4
In light of the merits described above, we paid special attention to the electrochemical properties of different phases in AZIBs. Compared to the layered dihydrate phase with average voltage only around 1.2 V vs. Zn2+/Zn, δ-VOPO4 has witnessed an elevated operating voltage of 1.46 V as shown in Fig. 3a. The charge–discharge behaviour of δ-VOPO4 can be seen through the comparison of the CV tests at the scan rate of 0.1 mV s−1 in the voltage range of 0.3–1.9 V as shown in Fig. S4a.†31 δ-VOPO4 has a pair of major oxidation/reduction peaks at 1.63 V and 1.51 V corresponding to the redox couple of V5+/V4+. A single major redox peak proves that the material has fewer phase transitions and the capacity is concentrated at high voltages, providing higher energy densities. Meantime, δ-VOPO4 exhibits a discharge capacity of 94.8 mA h g−1 at 0.1C (1C = 330 mA g−1) in the initial cycle and 117.8 mA h g−1 in the second cycle with nearly 100% coulombic efficiency. The average discharge voltage of the delta-phase is also in the leading position among the various reported vanadium phosphate materials as displayed in Fig. 3b. Owing to the micron-sized platelets with limited diameter and stronger induction effect, the as prepared δ-VOPO4 sample shows an excellent rate performance as revealed in Fig. 3c and d. In particular, it provides 126.6, 132.2, 125.3, 116.1, 97.1, 73.3 and 53 mA h g−1 at 0.2C, 0.5C, 1C, 2C, 5C, 10C and 20C respectively. When the current density comes back to 0.2C, the battery can still deliver the ideal capacity stably. The capacity of δ-VOPO4 slowly rises to 122.6 mA h g−1 at a current density of 1C and keeps stable after 940 cycles (Fig. 3e). The rise of the capacity is related to the typical activation process, which was commonly observed for multivalent metal-ion batteries in many previous studies.32–34 What is more impressive is the long-term cycle performance at the high current density displayed in Fig. 3f and g. When the current density is increased to 10C (3300 mA g−1), δ-VOPO4 still exhibits an inspiring high reversible capacity of 90.9 mA h g−1. The corresponding charge–discharge profiles of the δ-VOPO4 cathode are displayed in Fig. S5.† It can be seen that the average discharge voltage at 1 A g−1 drops from 1.46 V to 1.32 V after 300 cycles. The voltage decay is much smaller than that of other V-based polyanionic cathodes in ZIBs, such as Li3V2(PO4)3, Na3V2(PO4)3, and Na3V2(PO4)2F3.35,36 Note that the GCD profiles almost kept unchanged at 10 A g−1, and a high operating voltage of 1.36 V was still retained after 1000 cycles, demonstrating an outstanding operating voltage retention.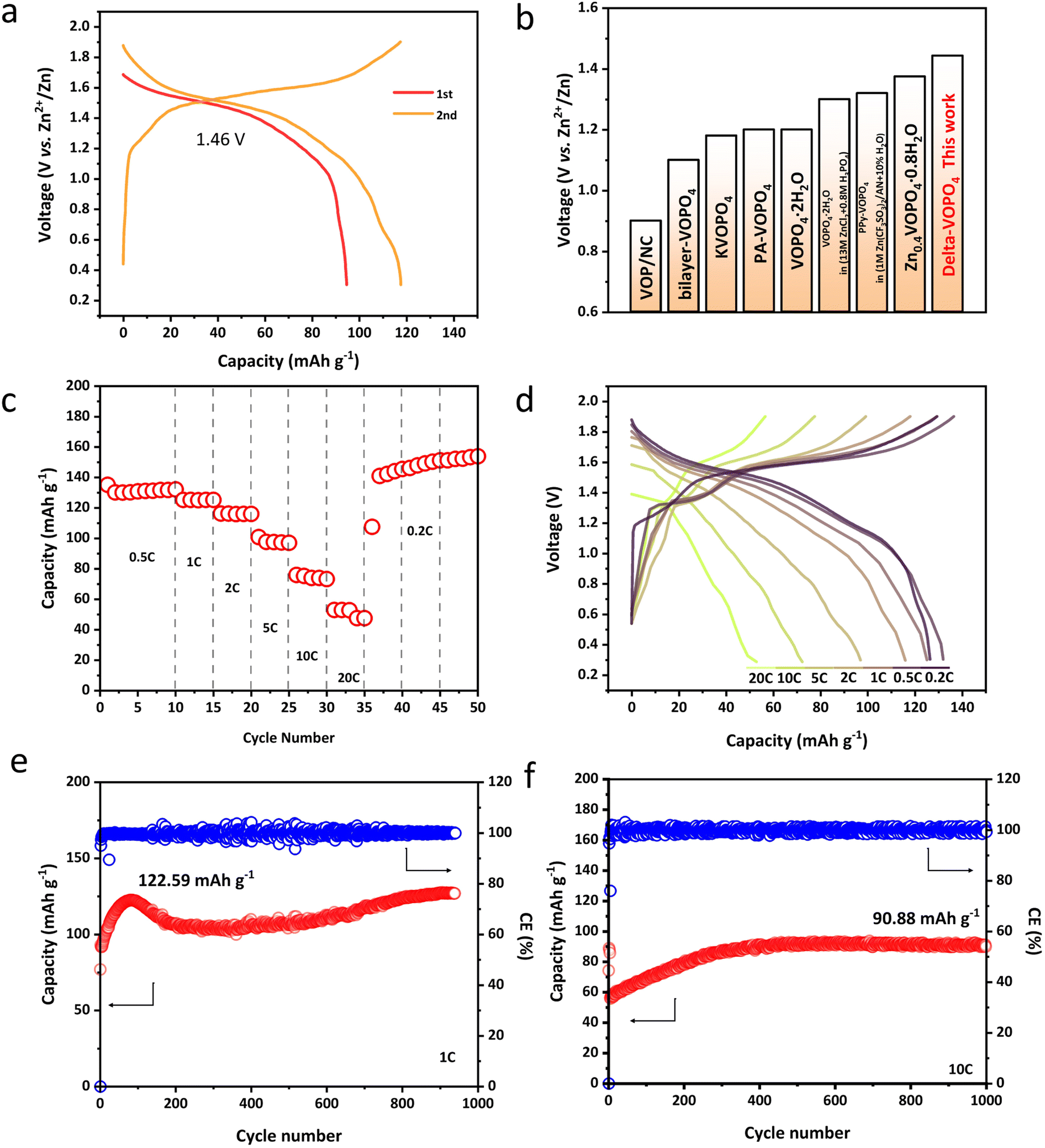 | ||
| Fig. 3 (a) The first two discharge/charge curves of δ-VOPO4; (b) the average discharge voltage of reported VOPO4 materials13–17,37,38 and this work; (c) rate performances of δ-VOPO4 at different current densities; (d) charge/discharge curves of δ-VOPO4 at different current densities; cycling performance of δ-VOPO4 at 1C (330 mA g−1) (e) and 10C (3300 mA g−1) (f). | ||
The reason behind such excellent kinetics can be explained by the galvanostatic intermittent titration technique (GITT) to distinguish the ion mobility in Fig. S6.† and as a result, an overwhelming Zn2+ diffusion coefficient of 10−10 cm−2 s−1 has been detected in the δ-VOPO4. To confirm the kinetics, Cyclic Voltammetry (CV) under various scan rates was performed to further explore the electrochemical kinetics of Zn2+ storage performance as depicted in Fig. S4b–d.† The energy storage process can be distinguished based on the power-law relationship between peak currents (i) and scan rate (ν) (i = aνb) just as described in the reported literature. Parameters a and b are constants that can be calculated from log(ν) vs. log(i). When b = 0.5 it is ascribed to the diffusion-controlled process and when b = 1 it corresponds to capacitive behaviour. The fitted slope values (b) for the two main redox peaks are calculated as 0.757 and 0.899, indicating fast Zn2+ diffusion kinetics in the δ-VOPO4 electrode.
Zn2+ storage mechanism of δ-VOPO4
The change of the vanadium valence state from the initial voltage to the cut-off voltage is analysed using XPS spectra for samples at different states of charge (SOC) as shown in Fig. 4a. It's clearly shown that the valence states of vanadium in the prepared samples are dominated by V5+, consistent with our designed formula. When discharged to 0.3 V, the valence state of V is reduced along with the intercalation of Zn2+ and thus both V3+ and V4+ are observed. Conversely, vanadium has been oxidized to V4+ and V5+ during Zn2+ extraction from the structure when recharging to 1.9 V. Notably, the combination of V4+ and V5+ at the charged state indicates there was Zn residual. This hypothesis could be validated by Inductively coupled plasma optical emission spectrometer (ICP-OES) measurements shown in Fig. 4b. When discharging from the open-circuit voltage of 1.68 V of the pristine state to 0.3 V, the ratio of Zn:V:P is calculated as 0.51:1:1. When charged to 1.9 V, the molar ratio of Zn:V:P is 0.13:1:1, indicating Zn2+ residual and agreeing with the above XPS test result.
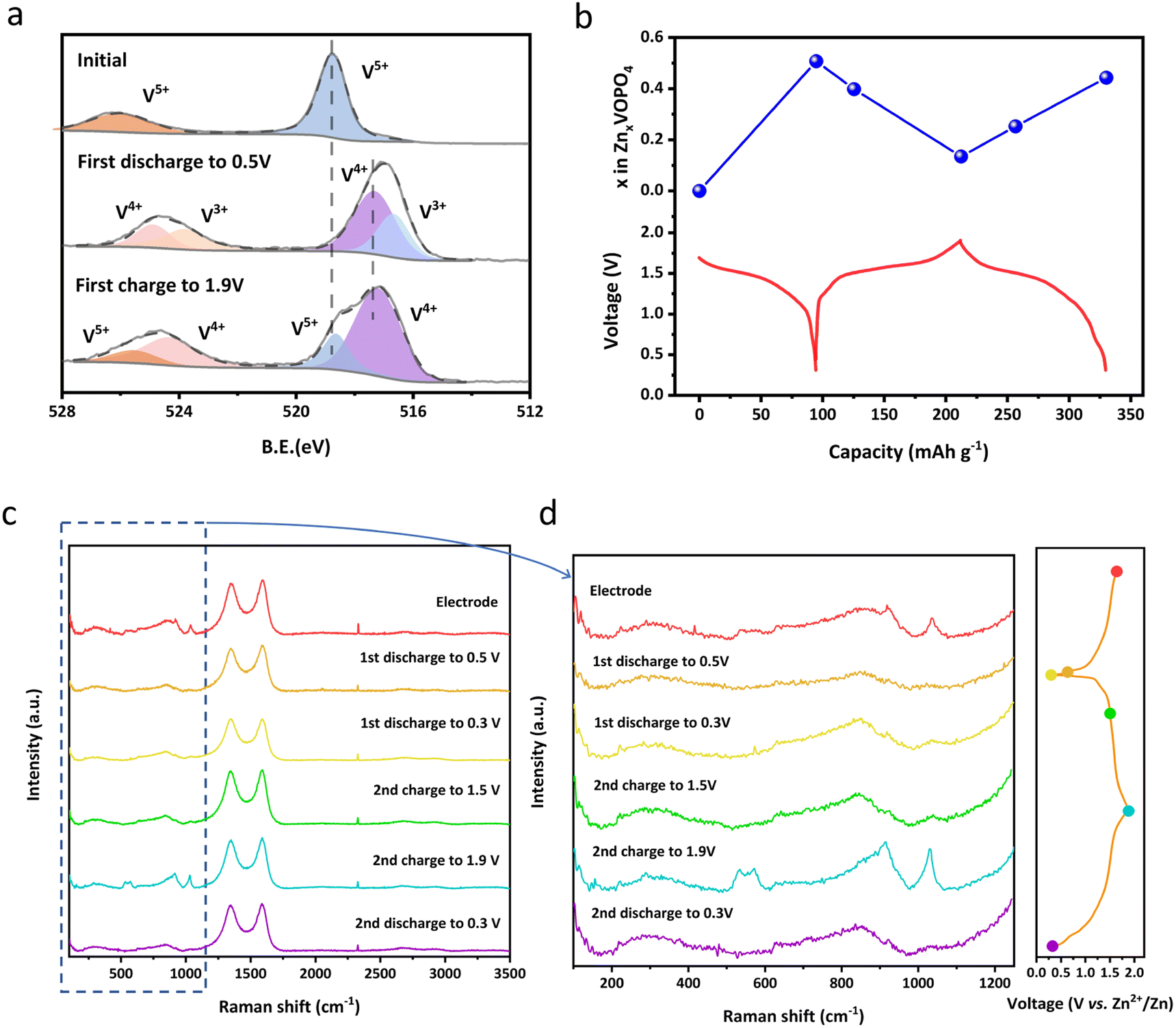 | ||
| Fig. 4 (a) Ex situ XPS spectra of V 2p of δ-VOPO4; (b) ex situ ICP measurement of δ-VOPO4 during the first two cycles; (c and d) ex situ Raman spectra of δ-VOPO4 during the first two cycles. | ||
Ex situ XRD was carried out to investigate the phase changes of δ-VOPO4 during the discharging and charging process. As displayed in Fig. S7,† the typical peaks such as (201) and (202) have experienced a reversible shift during the Zn2+ intercalation/deintercalation, indicating a topological Zn2+ insertion/extraction into/from the lattice. Calculated based on the ex situ XRD pattern, the volume change of δ-VOPO4 is only 1.1% after fully intercalating Zn2+, which is essential for its prolonged cycle life. Besides, two new peaks located at 31.2° and 32.8° emerged during discharging and disappeared upon charging, which was related to the formation of partially hydrated phosphate on the surface of the electrode. Generally, the main crystal structure of δ-VOPO4 was still maintained upon discharging and charging, which can account for the outstanding capacity and operating voltage retention in Fig. 3 and S5.†
Moreover, more information about the molecular structure can be concluded from ex situ Raman spectra displayed in Fig. 4c to unveil the Zn2+ storage mechanism in δ-VOPO4. In the process of Zn2+ insertion, Zn forms coordination with the O atom from the PO4 group, resulting in the deformation of the PO4 structure and the weakening and shift of the Raman intensity. When the Zn2+ is deintercalated, the P–O bond gets stronger and shorter due to the increased Coulomb force. The intensity of the Raman characteristic peak of PO43− recovers, indicating the stability of the structure of PO4 groups during the (de)intercalation of Zn2+.
To further reveal the fundamental mechanisms of δ-VOPO4, theoretical calculation with density functional theory (DFT) was used to establish the structural models of δ-VOPO4 before and after zincification as illustrated in Fig. 5a. When became fully zincified, each unit cell is able to store eight Zn atoms corresponding to the chemical formula ZnVOPO4, corresponding to a theoretical capacity of over 300 mA h g−1. The chemical coordination environment of all the Zn is the same. However, in fact, none of the reported vanadium phosphate (VOPO4) materials have achieved such high specific capacity due to the unstable structure. In this work, due to the ICP measurements, 0.5 Zn2+ can be intercalated per δ-VOPO4. As depicted in Fig. 5b, the DFT calculation results illuminate that the equilibrium voltage is 1.47 V at 0 ≤ x ≤ 0.5 and 1.45 V at 0.5 ≤ x ≤ 0.625 corresponding to the V5+/V4+ and V4+/V3+ redox couples, respectively, which agree well with the experimental results. First, the high potential is brought about by the “inductive effect” of strong P–O covalent bonds that drive the electron density away from the V center in the V–O–P layers, thus weakening the covalence of the V–O bonds, and leading to the higher operating voltage. Second, the unique structure and V⋯O contact are the key factors for its high voltage and excellent Zn2+ storage performance. The structure of δ-VOPO4 contains infinite polar chains of trans-corner sharing VO6 octahedra with the [VO⋯VO⋯]∞ backbone. Girgsdies et al. found that the V⋯O contact length of δ-VOPO4 reached 3.1 Å, which was significantly longer than that of other VOPO4 phases.39 In addition, the OV⋯O angle is 168°, which obviously deviates from more or less straight arrangement compared with other VOPO4 phases. The unique structural distortion indicates that vanadium in δ-VOPO4 has higher redox activity and potential. There is also a lower plateau at 0.69 V, which may be associated with the insertion of Zn2+ into different sites of δ-VOPO4.
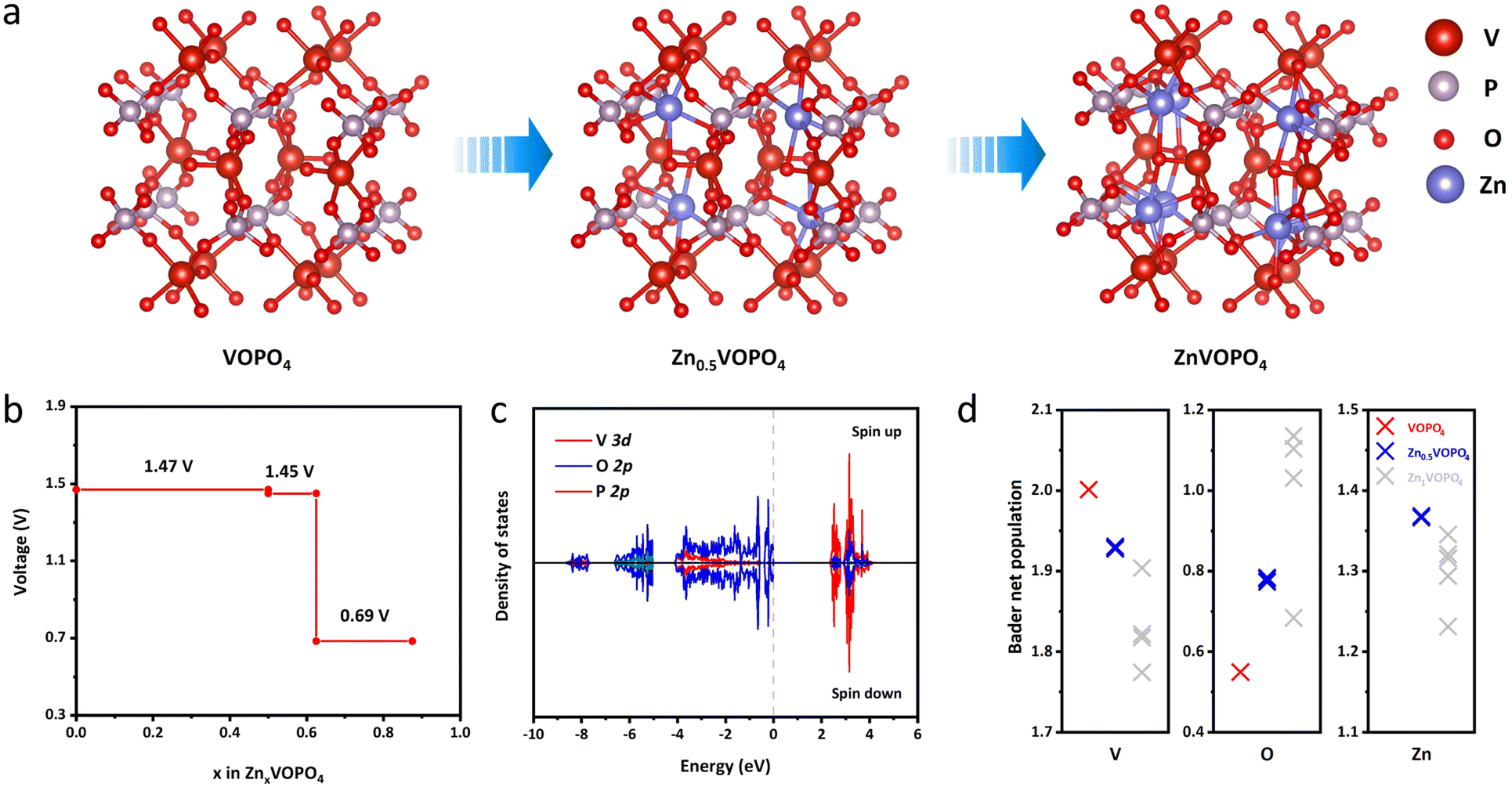 | ||
| Fig. 5 (a) The schematic illustration of structural models of δ-VOPO4 before and after dezincification; (b) the equilibrium reaction voltage profiles predicted with DFT calculations; (c) the density of states (DOS) of δ-VOPO4; the Fermi energy level is set to zero; (d) the Bader charge analysis of δ-VOPO4. | ||
The total density of states (DOS) of δ-VOPO4 was calculated to further illuminate the bonding condition of the structure (Fig. S8†). shows that the O-2p and P-2p orbitals are clearly overlapped corresponding to the (PO4)3− group. The strong overlapping of the V-3p orbitals and the O-2p orbitals shows the strong covalent bond between V and O atoms. It is observed at the valence band position in Fig. S9† that the area variation of state density before and after zinc insertion is mainly concentrated between −4.45 eV (ε0) and the Fermi level because the integration below −4.45 eV has no change before and after zincification. Through integration, it can be deduced that the intercalation of Zn mainly affects the density of states in this region, and only the integral value of V has been changed proving that the electrons supplied by Zn2+ are completely added to the V-3d orbital, so that V is the redox centre of the material.40 In order to elucidate the interaction between different atoms, the Bader charge population of δ-VOPO4 before and after zincification is calculated in Fig. 5d. During zincification, due to the charge compensation, the Bader net charge of the V atom has been reduced as the redox center. The net charge of the O atom has an evident increase close to the absolute value of formal charge (−2), implying that there is a strong ionic bond between Zn and O. Similarly, the net charge of Zn is close to the formal charge (+2), indicating that the Zn–O bond is more like an ionic bond. Obviously, the ionic character of the Zn–O bond may significantly raise the Vn+/V(n+1)+ redox potential as a primary inductive effect.
Conclusions
A novel polyanionic delta oxovanadium phosphate (δ-VOPO4) is synthesized without complex pre-intercalation of metal elements/small molecules, and employed as a cathode in aqueous zinc-ion batteries. Superior to various vanadyl phosphates with a similar chemical composition, it displays a high average discharge voltage of 1.46 V, and an impressive long-term cycling performance at 1C and 10C. This work assuredly illustrates that the performance could be enhanced by structure optimization and phase transition, which may shed new insights to exploit alternative cathodes out of vanadyl phosphate families for zinc-ion batteries.Data availability
The authors declare that all data supporting the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Dong Zhao: data curation, formal analysis, investigation, writing – original draft, writing – review & editing. Xiangjun Pu: investigation, writing – original draft, writing – review & editing. Shenglong Tang: data curation, methodology. Mingyue Ding: supervision, writing – original draft. Yubin Zeng: methodology, writing – review & editing. Yuliang Cao: funding acquisition, writing – review & editing. Zhongxue Chen: supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Intergovernmental International Science and Technology Innovation Cooperation Project (2019YFE010186), the National Natural Science Foundation of China (U22A20438, U22A20193), the Hubei Natural Science Foundation (2020CFB771), and the Fundamental Research Funds for the Central Universities. Also, we wish to thank the Supercomputing Center of Wuhan University for the calculation assistance.References
- Y. Cao, M. Li, J. Lu, J. Liu and K. Amine, Nat. Nanotechnol., 2019, 14, 200–207 CrossRef CAS PubMed.
- Z. Lin, T. Liu, X. Ai and C. Liang, Nat. Commun., 2018, 9, 5262 CrossRef CAS.
- D. Zhao, C. Wang, Y. Ding, M. Ding, Y. Cao and Z. Chen, ChemSusChem, 2022, 15, 202200479 Search PubMed.
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121, 1623–1669 CrossRef CAS PubMed.
- S. Chen, D. Zhao, L. Chen, G. Liu, Y. Ding, Y. Cao and Z. Chen, Small Struct., 2021, 2, 2100082 CrossRef CAS.
- C. Dong, F. Xu, L. Chen, Z. Chen and Y. Cao, Small Struct., 2021, 2, 2100001 CrossRef CAS.
- D. Zhao, S. Chen, Y. Lai, M. Ding, Y. Cao and Z. Chen, Nano Energy, 2022, 100, 107520 CrossRef CAS.
- H. Li, S. Liu, T. Yuan, B. Wang, P. Sheng, L. Xu, G. Zhao, H. Bai, X. Chen, Z. Chen and Y. Cao, Acta Phys.-Chim. Sin., 2021, 37, 1907049 Search PubMed.
- S. Tang, C. Wang, X. Pu, X. Gu and Z. Chen, Acta Phys.-Chim. Sin., 2023, 39, 2212037 Search PubMed.
- Z. Wu, F. Ye, Q. Liu, R. Pang, Y. Liu, L. Jiang, Z. Tang and L. Hu, Adv. Energy Mater., 2022, 12, 2200654 CrossRef CAS.
- F. Ye, R. Pang, C. Lu, Q. Liu, Y. Wu, R. Ma and L. Hu, Angew. Chem., Int. Ed., 2023, 62, e202303480 CrossRef CAS PubMed.
- X. Pu, H. Wang, D. Zhao, H. Yang, X. Ai, S. Cao, Z. Chen and Y. Cao, Small, 2019, 15, 1805427 CrossRef PubMed.
- K. Zhu, Z. Sun, P. Liu, H. Li, Y. Wang, K. Cao and L. Jiao, J. Energy Chem., 2021, 63, 239–245 CrossRef CAS.
- Z. Wu, Y. Wang, L. Zhang, L. Jiang, W. Tian, C. Cai, J. Price, Q. Gu and L. Hu, ACS Appl. Energy Mater., 2020, 3, 3919–3927 CrossRef CAS.
- L. Hu, Z. Wu, C. Lu, F. Ye, Q. Liu and Z. Sun, Energy Environ. Sci., 2021, 14, 4095–4106 RSC.
- V. Verma, S. Kumar, W. Manalastas, J. Zhao, R. Chua, S. Meng, P. Kidkhunthod and M. Srinivasan, ACS Appl. Energy Mater., 2019, 2, 8667–8674 CrossRef CAS.
- H. Y. Shi, Y. Song, Z. Qin, C. Li, D. Guo, X. X. Liu and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 16057–16061 CrossRef CAS PubMed.
- B. M. Azmi, T. Ishihara, H. Nishiguchi and Y. Takita, Electrochim. Acta, 2002, 48, 165–170 CrossRef CAS.
- B. M. Azmi, T. Ishihara, H. Nishiguchi and Y. Takita, J. Power Sources, 2003, 119–121, 273–277 CrossRef CAS.
- Z. Chen, Q. Chen, H. Wang, R. Zhang, H. Zhou, L. Chen and M. S. Whittingham, Electrochem. Commun., 2014, 46, 67–70 CrossRef CAS.
- N. Dupré, G. Wallez, J. Gaubicher and M. Quarton, J. Solid State Chem., 2004, 177, 2896–2902 CrossRef.
- Y.-C. Lin, M. F. V. Hidalgo, I.-H. Chu, N. A. Chernova, M. S. Whittingham and S. P. Ong, J. Mater. Chem. A, 2017, 5, 17421–17431 RSC.
- M. Zhang, S. Zhang, H. Gao, F. L. Meng and C. Deng, J. Electroanal. Chem., 2014, 713, 119–124 CrossRef CAS.
- Y. Fang, Q. Liu, L. Xiao, Y. Rong, Y. Liu, Z. Chen, X. Ai, Y. Cao, H. Yang, J. Xie, C. Sun, X. Zhang, B. Aoun, X. Xing, X. Xiao and Y. Ren, Chem, 2018, 4, 1–14 Search PubMed.
- L. Zhang, X. Hu, Y. Wu, Y. Gao, C. Lin, C. Dong, G. Li, F. Xu, S. Zhang and K. Zhang, Mater. Today Energy, 2021, 21, 100756 CrossRef CAS.
- H. Y. Shi, Z. Jia, W. Wu, X. Zhang, X. X. Liu and X. Sun, Chem.–Eur. J., 2020, 26, 8190–8204 CrossRef CAS PubMed.
- Q. Bai, L. Yang, H. Chen and Y. Mo, Adv. Energy Mater., 2018, 8, 1702998 CrossRef.
- A. Jain, G. Hautier, C. J. Moore, S. Ping Ong, C. C. Fischer, T. Mueller, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2011, 50, 2295–2310 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. Mei, Y. V. Kaneti, M. Pramanik, T. Takei, Ö. Dag, Y. Sugahara and Y. Yamauchi, Nano Energy, 2018, 52, 336–344 CrossRef CAS.
- X. Pu, D. Zhao, C. Fu, Z. Chen, S. Cao, C. Wang and Y. Cao, Angew. Chem., Int. Ed., 2021, 60, 21310–21318 CrossRef CAS PubMed.
- F. Ming, H. Liang, Y. Lei, S. Kandambeth, M. Eddaoudi and H. N. Alshareef, ACS Energy Lett., 2018, 3, 2602–2609 CrossRef CAS.
- C. E. M. Lewis, J. F. S. Fernando, D. P. Siriwardena, K. L. Firestein, C. Zhang, J. E. Treifeldt and D. V. Golberg, Adv. Mater. Technol., 2021, 7, 2100505 CrossRef.
- H. Liu, L. Jiang, B. Cao, H. Du, H. Lu, Y. Ma, H. Wang, H. Guo, Q. Huang, B. Xu and S. Guo, ACS Nano, 2022, 16, 14539–14548 CrossRef CAS.
- C. Li, W. Yuan, C. Li, H. Wang, L. Wang, Y. Liu and N. Zhang, Chem. Commun., 2021, 57, 4319–4322 RSC.
- W. Li, X. Jing, K. Jiang and D. Wang, ACS Appl. Energy Mater., 2021, 4, 2797–2807 CrossRef CAS.
- L. Ou, Z. Liu, Y. Zhou, H. Ou, J. Zhu, X. Cao, G. Fang, J. Zhou and S. Liang, Chem. Eng. J., 2021, 426, 131868 CrossRef CAS.
- Z. Wu, C. Lu, F. Ye, L. Zhang, L. Jiang, Q. Liu, H. Dong, Z. Sun and L. Hu, Adv. Funct. Mater., 2021, 31, 2106816 CrossRef.
- F. Girgsdies, M. Schneider, A. Brückner, T. Ressler and R. Schlögl, Solid State Sci., 2009, 11, 1258–1264 CrossRef CAS.
- R. Lian, D. Wang, X. Ming, R. Zhang, Y. Wei, J. Feng, X. Meng and G. Chen, J. Mater. Chem. A, 2018, 6, 16228–16234 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Structural models, SEM images, CV curves, GCD curves and DOS. See DOI: https://doi.org/10.1039/d3sc02382f |
| This journal is © The Royal Society of Chemistry 2023 |