
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeaminative coupling of benzylamines and arylboronic acids†
Giedre
Sirvinskaite
,
Julia C.
Reisenbauer
and
Bill
Morandi
 *
*
Laboratorium für Organische Chemie ETH Zürich, Vladimir-Prelog-Weg 3, HCI, Zürich 8093, Switzerland. E-mail: morandib@ethz.ch
First published on 13th January 2023
Abstract
A metal-free deaminative coupling of non-prefunctionalised benzylamines and arylboronic acids is reported. In this operationally simple reaction, a primary amine in benzylamine is converted into a good leaving group in situ using inexpensive and commercially available isoamyl nitrite as a nitrosating reagent. Lewis-acidic arylboronic acids are shown to replace mineral acids such as HCl or HBF4 that are conventionally used in the preparation of aryl diazonium salts. This unlocked the formation of the corresponding diarylmethanes by forging a new C–C bond in good yields.
Introduction
Although primary amines are highly attractive handles for late-stage modification and downstream functionalisation due to their prevalence in biologically active molecules and industrially relevant chemicals,1–5 their poor leaving group ability renders the cleavage of the C–N bond particularly challenging.6–8 A direct use of ubiquitous primary amines as coupling partners has been underexploited in deaminative cross-coupling reactions despite the potential to streamline organic synthesis. Among the limited examples addressing this challenge, the cleavage of unactivated C–N single bonds in primary amines has been largely achieved by acid-assisted transition metal-catalysed oxidative addition.7,8 Notably, the Tian group has developed Pd-catalysed deaminative couplings of non-prefunctionalised allylic amines with various nucleophiles such as boronic acids, boronates, sulfonate salts and phosphonium ylides (Fig. 1a).9–15 The proposed mechanism is suggested to proceed through a palladium allyl intermediate in analogy to the Tsuji–Trost reaction.9,16,17 An alternative, mechanistically distinct approach for the deaminative functionalization of primary amines relies on diazotisations. While such reactions using aromatic amines are commonly encountered in both academic and industrial settings, as highlighted by the plethora of reports employing aryldiazonium species for cross-coupling and other functionalisation reactions,18–21 successful utilisation of alkyl amines in deaminative reactions usually relies on the presence of a neighbouring electron-withdrawing group to form the alkyl diazo compound instead of the notoriously unstable, high-energy alkyl diazonium species.22–29 A notable exception is the deaminative functionalisation of electronically non-biased primary amines reported by the Lebel group (Fig. 1b).30–33 The reaction was suggested to proceed via a transient diazonium intermediate, however the transformation was limited to the use of carboxylic acids or electron-poor phenols as highly acidic coupling partners to form and trap the high-energy diazonium species. Thus, the reaction was restricted to the formation of C–O bonds, while the formation of C–C bonds has so far remained elusive. The scarce literature precedent for the direct deaminative cross-coupling of primary amines, as well as their limitations, calls for the development of new, efficient approaches to harness the underexplored potential of amines as carbon-centered coupling partners.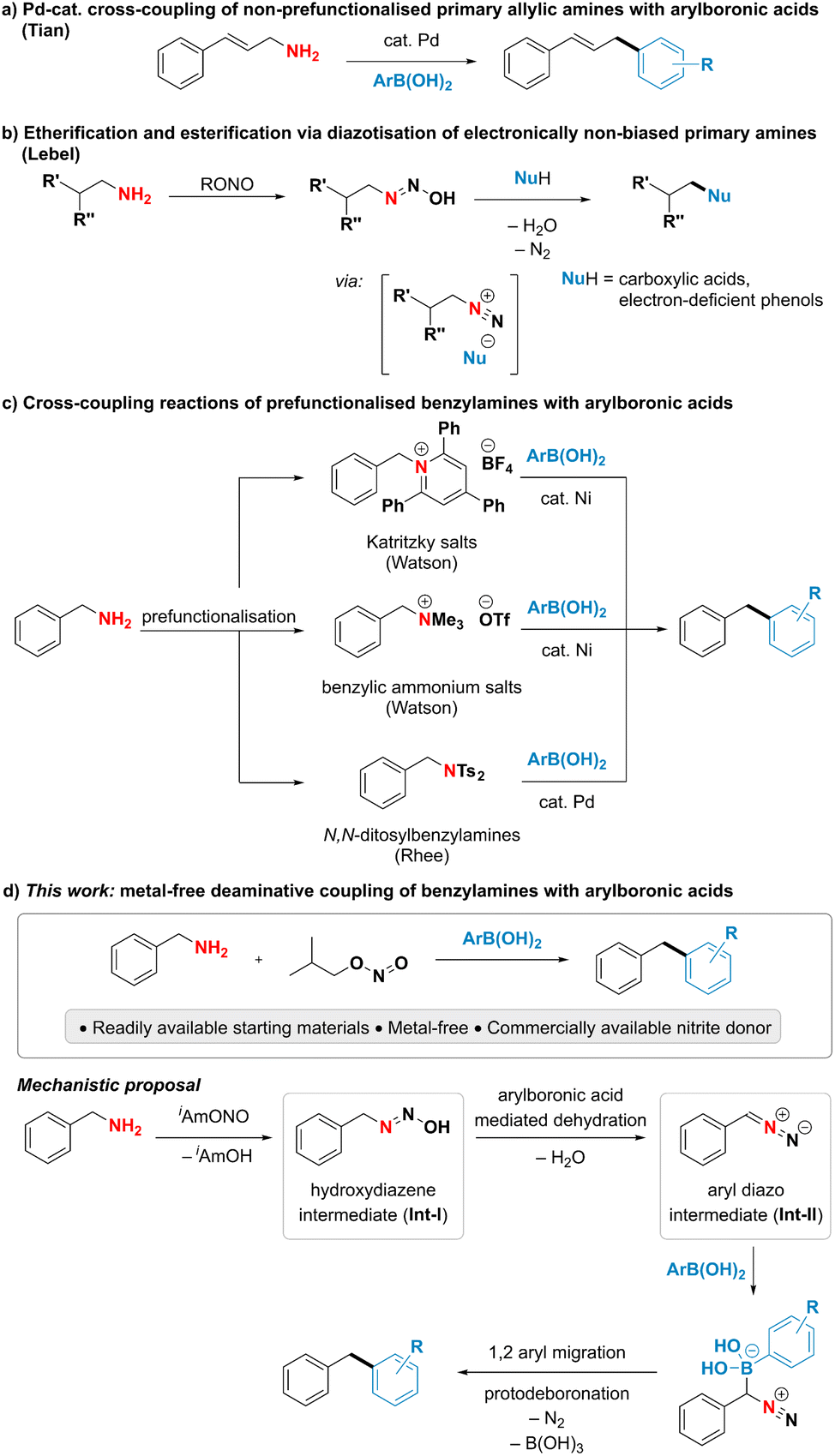 | ||
| Fig. 1 Context of this work. | ||
To overcome the difficulty of breaking a strong C(sp3)–N bond under mild conditions, existing indirect deaminative cross-coupling reactions of benzylamines with arylboronic acids require an additional prefunctionalisation step to transform the primary amine into a good leaving group and a transition metal to perform the desired reaction (Fig. 1c).34–36 The Watson group has developed a nickel-catalysed deaminative cross-coupling of arylboronic acids with Katritzky's salts along with an analogous reaction using methylated quaternary amines. Rhee and coworkers have disclosed a conceptually related approach using N,N-ditosylbenzylamines under palladium catalysis. In all these reports, an additional step for the conversion of an amine into a better leaving group was necessary, thus limiting the step and atom economy of the reactions.
In this work, we present an interrupted diazotisation strategy for the direct coupling of benzylamines with arylboronic acids to forge a new carbon–carbon bond (Fig. 1d). The reaction is operationally simple, proceeds under mild reaction conditions in the absence of any catalyst and tolerates a wide range of functional groups.
Results and discussion
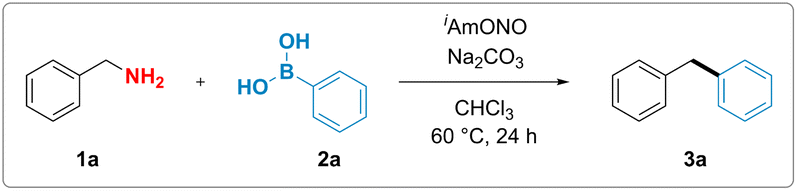
Although, in analogy to aromatic amines, there are numerous reports attempting to functionalise electronically non-biased primary aliphatic amines by diazotisation, formation of highly reactive alkyldiazonium intermediates rendered those reactions inefficient and made them suffer from poor functional group tolerance and low yields.37–42 We hypothesized that substitution of conventional mineral acids, such as HCl and HBF4, used for the synthesis of aromatic diazonium salts, by a mild organic Lewis acid might suppress the unproductive formation of the high-energy alkyl diazonium species and thus unlock a divergent reaction outcome (Fig. 1d). More specifically, we proposed that benzylamine could react with the nitrite donor, producing a hydroxy diazene intermediate (Int-I). Arylboronic acid-facilitated dehydration of this intermediate would form the corresponding semi-stabilised diazo compound (Int-II).43 This intermediate could then subsequently react with arylboronic acid as described by Barluenga and coworkers44 and furnish the desired diarylmethane along with dinitrogen and boric acid as benign stoichiometric byproducts.To test this hypothesis, benzylamine (1a) was reacted with commercially available isoamyl nitrite and phenylboronic acid (2a). We observed that the formation of the desired diphenylmethane product 3a occurred upon mixing stoichiometric amounts of the three reagents, thereby validating our hypothesis experimentally.
With these initial reaction conditions in hand, the deaminative coupling reaction between benzylamine and phenylboronic acid was further optimized by evaluating various reaction parameters. The highest yield for the formation of the desired diphenylmethane product was observed upon incorporation of one equivalent of sodium carbonate as a base relative to phenylboronic acid. Control experiments revealed that deaminative coupling is contingent upon addition of the organic nitrite (Table 1, entry 2). Using phenylboronic pinacol ester instead of phenylboronic acid completely shut down the desired reactivity (Table 1, entry 4). Lower loadings of either the isoamyl nitrite or benzylamine led to diminished yields (see ESI†). Substitution of isoamyl nitrite with either tert-butyl nitrite or nitrosonium tetrafluoroborate also resulted in lower yield or no formation of the product, respectively (see Table 1, entry 5 and ESI†). Interestingly, addition of 1.0 equivalent of boric acid (Table 1, entry 6), which is believed to be formed as a stoichiometric byproduct in the reaction, gave lower yields of the desired product, suggesting that byproduct formation might suppress the desired reaction pathway.
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yielda [%] |
| a GC yield of the crude reaction mixture using n-dodecane as an internal standard. b Isolated yield. | ||
| 1 | No deviation | 80 (70)b |
| 2 | No iAmONO | 0 |
| 3 | No base | 72 |
| 4 | Pinacol ester instead of phenylboronic acid | 0 |
| 5 | t BuONO instead of iAmONO | 23 |
| 6 | 1 equiv. of boric acid (B(OH)3) | 70 |
With the optimized conditions in hand, the benzylamine substrate scope of this synthetic transformation was explored (Fig. 2). Electron-rich p-tolyl- and 4-tert-butylbenzylamines gave the corresponding products 3b and 3c in moderate yields. 4-Phenylbenzylamine gave the product 3d in 60% yield. 4-Fluoro, 4-chloro, 4-bromo and 4-iodobenzylamines could be transformed to target diarylmethanes 3e–h in 51%, 76%, 85% and 89% yields, respectively. Furthermore, diarylmethanes bearing electron-withdrawing functional groups, such as nitrile (3i), nitro (3j), ester (3k) and methylsulfonyl (3l), were synthesised from the corresponding benzylamines in good to excellent yields. Fluorine-containing functional groups such as trifluoromethoxy (3m), difluoromethoxy (3n), trifluoro-methylsulfane (3o) and trifluoromethyl (3p) were also found to be tolerated under reaction conditions. Meta-substituted benzylamines have also been found to be good coupling partners in the transformation, affording products 3q–ab. Deaminative coupling between benzylamines and arylboronic acids was also shown to tolerate ortho substitution on the benzylamine partner, with yields of the corresponding diarylmethanes depending on both electronic and steric properties of the benzylamine substrate. 2-Methylbenzylamine (1ac) gave the desired product 3ac in 43% yield. Diarylmethanes 3ad, 3ag and 3ah bearing ortho-fluoro group were obtained in 70%, 83% and 85% yield, respectively. Introduction of 2-bromo or 2-trifluoromethyl group was also shown to furnish the corresponding products in good yields (3ae and 3af, respectively). Importantly, a pyridine heterocycle was also tolerated under reaction conditions, and 4-benzylpyridine (3aj) was obtained from 4-picolylamine (1aj) in 62% yield.
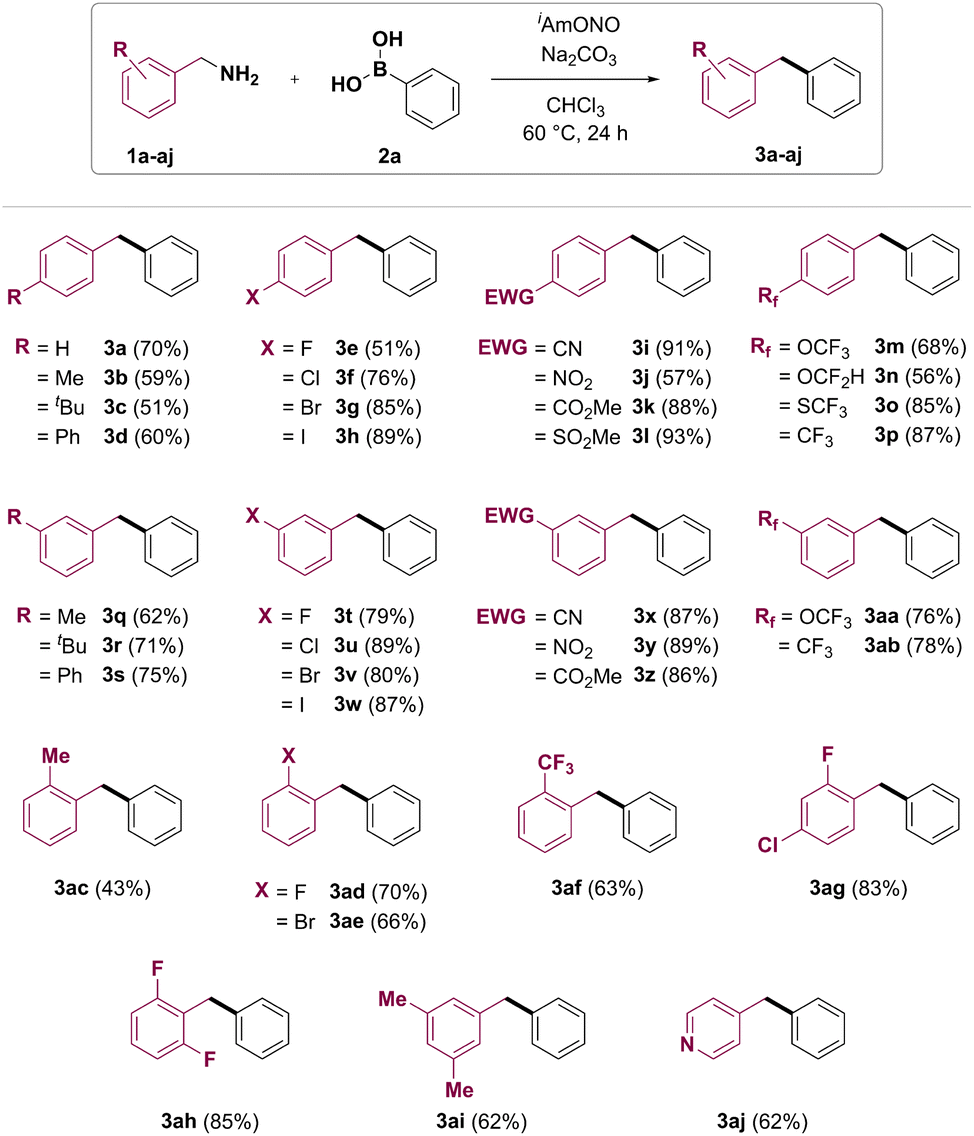 | ||
| Fig. 2 Benzylamine scope of the deaminative coupling between benzylamines and arylboronic acids. Reaction conditions: benzylamine (1a–aj, 2.0 mmol), phenylboronic acid (2a, 0.5 mmol), isoamyl nitrite (2.5 mmol), sodium carbonate (0.5 mmol), chloroform (1.25 mL), 60 °C, 24 h. Isolated yields after flash column chromatography. | ||
The substrate scope of arylboronic acids was then investigated (Fig. 3). A wide range of products were obtained in good yields and high selectivity. Unexpectedly, substituted arylboronic acids gave trace amounts of a minor regioisomeric product. Para-substituted arylboronic acids (2a–i) have been found to give a mixture of para- and meta-substituted diarylmethanes 4a–i in 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 to 91:9 p:m isomer ratio. Similar trend was also observed with meta-substituted arylboronic acids. Formation of a mixture of all three possible isomers was observed for diarylmethanes 4j, 4k, 4l and 4m, synthesized using 3-methyl, 3-fluoro, 3-chloro and 3-bromophenylboronic acids, respectively, with the desired meta isomeric product obtained in 89 to 96% selectivity. Introduction of an electron withdrawing group such as trifluoromethoxy (4n) or trifluoromethyl (4o) in the meta position led to the formation of meta and para isomers. Ortho-substituted phenylboronic acids 4p and 4q were also observed to furnish both the ortho and the meta isomer of diarylmethane product.
:4 to 91:9 p:m isomer ratio. Similar trend was also observed with meta-substituted arylboronic acids. Formation of a mixture of all three possible isomers was observed for diarylmethanes 4j, 4k, 4l and 4m, synthesized using 3-methyl, 3-fluoro, 3-chloro and 3-bromophenylboronic acids, respectively, with the desired meta isomeric product obtained in 89 to 96% selectivity. Introduction of an electron withdrawing group such as trifluoromethoxy (4n) or trifluoromethyl (4o) in the meta position led to the formation of meta and para isomers. Ortho-substituted phenylboronic acids 4p and 4q were also observed to furnish both the ortho and the meta isomer of diarylmethane product.
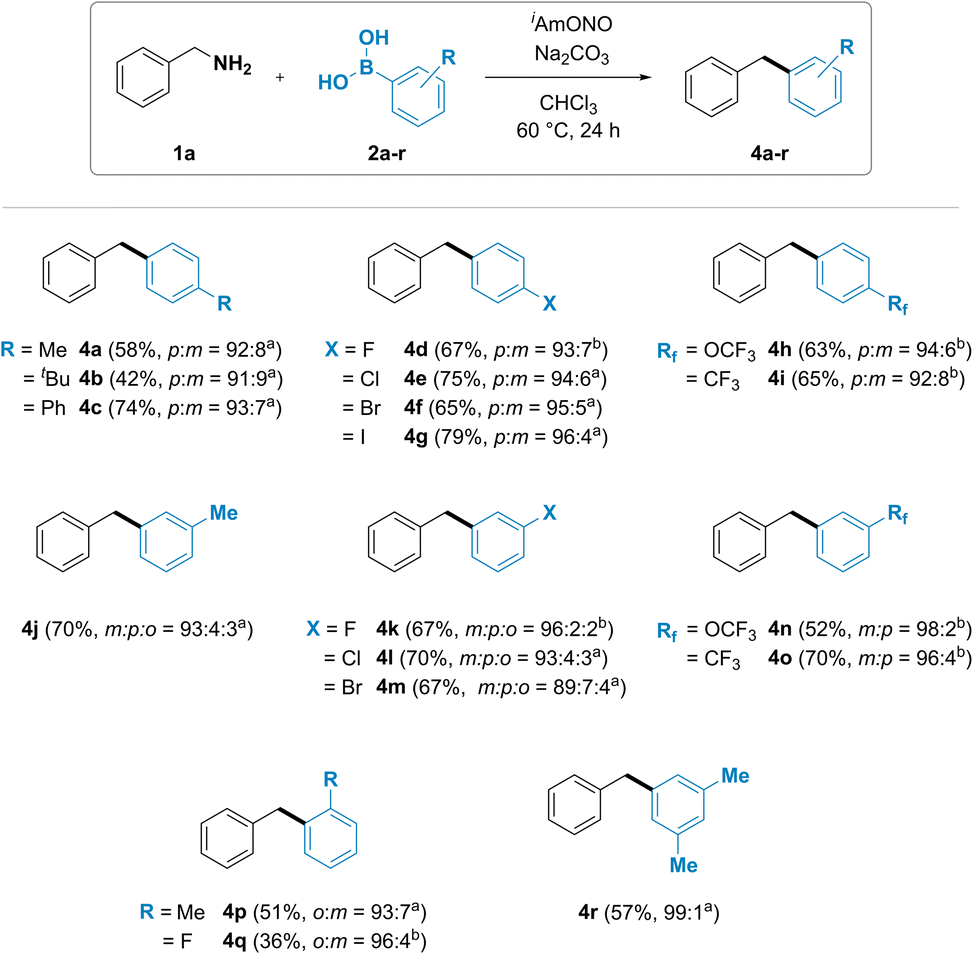 | ||
| Fig. 3 Arylboronic acid scope of the deaminative coupling between benzylamines and arylboronic acids. Reaction conditions: benzylamine (1a, 2.0 mmol), arylboronic acid (2a–r, 0.5 mmol), isoamyl nitrite (2.5 mmol), sodium carbonate (0.5 mmol), chloroform (1.25 mL), 60 °C, 24 h. Isolated yields after flash column chromatography. a Isomer ratio determined by 13C NMR. b Isomer ratio determined by 19F NMR. | ||
To better understand the unusual reactivity of substituted phenylboronic acids not only through the ipso but also through the ortho position, the mechanistic aspects of the deaminative coupling were investigated (Fig. 4). To probe whether the isomer formation is feasible under the originally proposed diazo pathway, 1-benzyl-4-methylbenzene (3b) was synthesised using conditions described by Barluenga and coworkers44 from benzaldehyde tosylhydrazone (6) and p-tolylboronic acid (2b) (Fig. 4a). The desired product 3b was obtained in 60% yield, however no formation of the minor meta isomer was observed. To gain further insight into the mechanism of the deaminative coupling, a p-tolylbenzylamine deuterated on the benzylic position (1b-d2) was subjected to the reaction conditions using phenylboronic acid (2a) as a coupling partner (Fig. 4b). A high extent of scrambling was observed in the benzylic position of the diarylmethane product 7. This suggests that the reaction likely proceeds through diazo species Int-II (Fig. 1d), however the deprotonation of the intermediate species might not be strictly necessary for the formation of the desired product. Formation of one isomer in the diazo pathway and incomplete hydrogen–deuterium scrambling in the benzylic position of benzylamine indicate that more than one mechanistically distinct pathway is accessible for the formation of diarylmethane products. This observation was indirectly supported by reacting 4-(trifluoromethyl)benzylamine (3p) with p-tolylboronic acid (2b) using optimized reaction conditions (Fig. 4c). In contrast to the unsubstituted benzylamine, formation of the diazo compound from 4-(trifluoro-methyl)benzylamine is expected to be favoured due to the ability of the neighbouring electron-withdrawing groups to stabilise diazo compounds.26–28 It was therefore anticipated that subjecting the benzylamine bearing electron-withdrawing group to the deaminative coupling with p-tolylboronic acid would almost exclusively furnish the desired para isomer. However, a mixture of both para and meta isomers was still obtained in 95:5 ratio, which is similar to that of unsubstituted benzylamine (93:7).
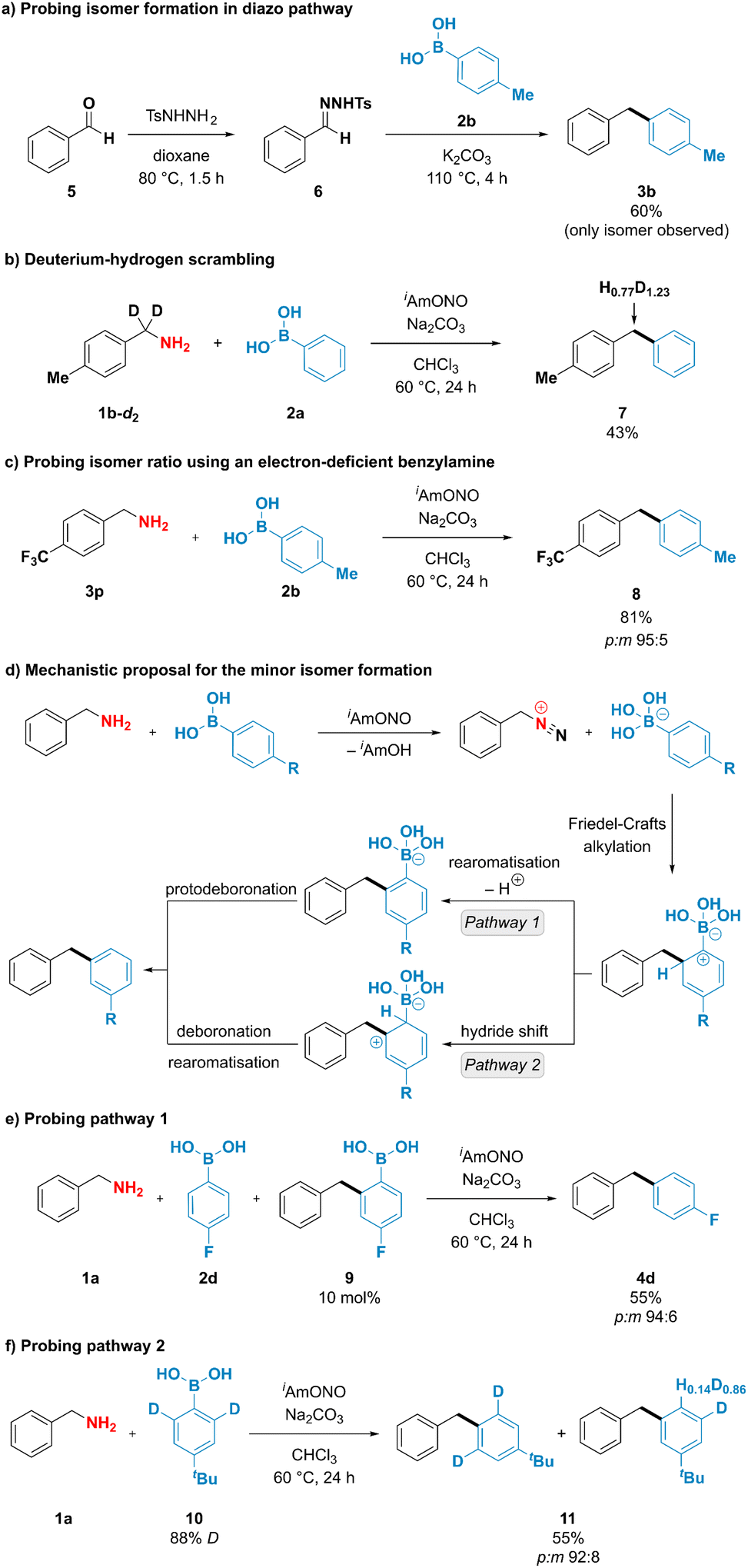 | ||
| Fig. 4 Mechanistic experiments and proposed mechanism for the formation of minor isomer. | ||
Exclusive formation of the major isomer in the diazo pathway, incomplete deuterium–hydrogen scrambling, and formation of both isomeric products when using electron-deficient benzylamines suggest that at least two mechanistically distinct pathways are operating under reaction conditions, with one being responsible for the formation of the minor product. Interestingly, boronate salts have been reported to engage in electrophilic aromatic substitution reactions if exposed to highly reactive species, such as NO2+, Cl+ or F+, among others.45–52 To explain the formation of the minor product, it could be envisioned that arylboronic acids participate in electrophilic substitution reaction through their ortho position with a subsequent loss of boron (Fig. 4d). After arylboronic acid-assisted elimination of the hydroxy group from the hydroxydiazene intermediate Int-I, a benzyldiazonium–arylboronate ion pair might be formed, and the aryl ring of the arylboronic acid could engage in an intramolecular Friedel–Crafts alkylation reaction, furnishing the minor isomer either after (1) rearomatisation and subsequent protodeboronation or after (2) hydride shift and subsequent deboronation. Mechanistic pathway (1) has been probed by adding 10 mol% of the putative intermediate arylboronic acid 9 to the deaminative coupling reaction between 4-fluorophenylboronic acid (2d) and benzylamine (1a), which under optimized conditions was reported to furnish the isomeric products in 67% yield and 94:6 p:m isomer ratio (Fig. 3, entry 4d). Upon spiking the reaction mixture with 10% of the putative intermediate 9, diarylmethane products were obtained in 55% yield and 94:6 p:m isomer ratio, suggesting that addition of the arylboronic acid 9 to the reaction mixture did not increase the amount of the minor isomer formed and that compound 9 is likely not an intermediate in the formation of the minor product. To check whether mechanistic pathway (2) is operative under the reaction conditions, deuterium-enriched arylboronic acid 10 was subjected to the deaminative coupling with benzylamine 1a. Deuterium enrichment was observed in the position para to the tert-butyl group in the minor isomer, suggesting that hydride/deuteride shift is likely to operate under the reaction conditions. Therefore, we propose that mechanistic pathway (2) is likely to be responsible for the formation of the minor isomer, thus providing the rationale for the formation of isomeric products when using substituted arylboronic acids as coupling partners.
Conclusions
In this report, a deaminative coupling between benzylamines and arylboronic acids under basic conditions is disclosed. Utilisation of inexpensive isoamyl nitrite for diazotisation of benzylamines along with arylboronic acids as Lewis-acidic coupling partners enabled the forging of a C–C bond to give diarylmethane products in good to excellent yields with a broad functional group tolerance. Mechanistic experiments supported the operation of at least two mechanistically distinct reaction pathways under optimised conditions to furnish desired deamination products. Intramolecular Friedel–Crafts reaction followed by hydride shift and deboronation was proposed to be the pathway responsible for the formation of isomeric diarylmethane products when using substituted arylboronic acids as deaminative coupling partners.Author contributions
B. M. and G. S. designed the project. G. S. and J. C. R. carried out the experiments. B. M., G. S. and J. C. R. analysed the data and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NMR and the Molecular and Biomolecular Analysis Service (MoBiAS) of ETH Zürich for technical assistance and the Morandi group for critical proofreading. Funding: This work was supported by the European Research Council under the European Union's Horizon 2020 research and innovation program (Shuttle Cat, project ID: 757608), by ETH Zürich and the Swiss National Science Foundation (SNSF 184658). J. C. R. acknowledges a fellowship from the Stipendienfonds der Schweizerischen Chemischen Industrie (SSCI).Notes and references
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- R. Vardanyan and V. Hruby, Synthesis of best-seller drugs, Academic press, 2016 Search PubMed.
- S. A. Lawrence, Amines: synthesis, properties and applications, Cambridge University Press, 2004 Search PubMed.
- K. Weissermel and H.-J. Arpe, Industrial organic chemistry, John Wiley & Sons, 2008 Search PubMed.
- A. Ricci, Amino group chemistry: from synthesis to the life sciences, John Wiley & Sons, 2008 Search PubMed.
- K. J. Berger and M. D. Levin, Org. Biomol. Chem., 2021, 19, 11–36 RSC.
- Q. Wang, Y. Su, L. Li and H. Huang, Chem. Soc. Rev., 2016, 45, 1257–1272 RSC.
- K. Ouyang, W. Hao, W.-X. Zhang and Z. Xi, Chem. Rev., 2015, 115, 12045–12090 CrossRef CAS PubMed.
- M.-B. Li, Y. Wang and S.-K. Tian, Angew. Chem., Int. Ed., 2012, 51, 2968–2971 CrossRef CAS PubMed.
- X.-S. Wu, Y. Chen, M.-B. Li, M.-G. Zhou and S.-K. Tian, J. Am. Chem. Soc., 2012, 134, 14694–14697 CrossRef CAS PubMed.
- X.-T. Ma, Y. Wang, R.-H. Dai, C.-R. Liu and S.-K. Tian, J. Org. Chem., 2013, 78, 11071–11075 CrossRef CAS PubMed.
- Y. Wang, J.-K. Xu, Y. Gu and S.-K. Tian, Org. Chem. Front., 2014, 1, 812–816 RSC.
- M.-B. Li, H. Li, J. Wang, C.-R. Liu and S.-K. Tian, Chem. Commun., 2013, 49, 8190–8192 RSC.
- T.-T. Wang, F.-X. Wang, F.-L. Yang and S.-K. Tian, Chem. Commun., 2014, 50, 3802–3805 RSC.
- X.-S. Wu, M.-G. Zhou, Y. Chen and S.-K. Tian, Asian J. Org. Chem., 2014, 3, 711–714 CrossRef CAS.
- J. Tsuji, H. Takahashi and M. Morikawa, Tetrahedron Lett., 1965, 6, 4387–4388 CrossRef.
- B. M. Trost and T. J. Fullerton, J. Am. Chem. Soc., 1973, 95, 292–294 CrossRef CAS.
- F. Mo, G. Dong, Y. Zhang and J. Wang, Org. Biomol. Chem., 2013, 11, 1582–1593 RSC.
- F. Mo, D. Qiu, L. Zhang and J. Wang, Chem. Rev., 2021, 121, 5741–5829 CrossRef CAS PubMed.
- S. S. Babu, P. Muthuraja, P. Yadav and P. Gopinath, Adv. Synth. Catal., 2021, 363, 1782–1809 CrossRef CAS.
- J. Chen, X. Xie, J. Liu, Z. Yu and W. Su, React. Chem. Eng., 2022, 7, 1247–1275 RSC.
- G. Maas, Angew. Chem., Int. Ed., 2009, 48, 8186–8195 CrossRef CAS PubMed.
- M. Rosenberger, P. Yates, J. B. Hendrickson and W. Wolf, Tetrahedron Lett., 1964, 5, 2285–2289 CrossRef.
- M. Regitz and J. Rüter, Chem. Ber., 1968, 101, 1263–1270 CrossRef CAS.
- R. L. Danheiser, R. F. Miller, R. G. Brisbois and S. Z. Park, J. Org. Chem., 1990, 55, 1959–1964 CrossRef CAS.
- M. Regitz, Diazo compounds: properties and synthesis, Elsevier, 2012 Search PubMed.
- M. P. Doyle, M. A. McKervey and T. Ye, Modern catalytic methods for organic synthesis with diazo compounds, Wiley, 1998 Search PubMed.
- H. Zollinger, Diazo Chemistry, Diazo Chemistry II: Aliphatic, Inorganic and Organometallic Compounds, Wiley, 1995 Search PubMed.
- G. Wu, Y. Deng, C. Wu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 10510–10514 CrossRef CAS PubMed.
- G. Reynard, E.-M. Joseph-Valcin and H. Lebel, Chem. Commun., 2020, 56, 10938–10941 RSC.
- G. Reynard, H. Mayrand and H. Lebel, Can. J. Chem., 2020, 98, 480–484 CrossRef CAS.
- C. Audubert and H. Lebel, Org. Lett., 2017, 19, 4407–4410 CrossRef CAS PubMed.
- R. M. Jacobson, Synth. Commun., 1978, 8, 33–37 CrossRef CAS.
- J. Liao, W. Guan, B. P. Boscoe, J. W. Tucker, J. W. Tomlin, M. R. Garnsey and M. P. Watson, Org. Lett., 2018, 20, 3030–3033 CrossRef CAS PubMed.
- P. Maity, D. M. Shacklady-McAtee, G. P. A. Yap, E. R. Sirianni and M. P. Watson, J. Am. Chem. Soc., 2013, 135, 280–285 CrossRef CAS PubMed.
- S. Yoon, M. C. Hong and H. Rhee, J. Org. Chem., 2014, 79, 4206–4211 CrossRef CAS PubMed.
- R. J. Baumgarten, J. Chem. Educ., 1966, 43, 398 CrossRef CAS.
- L. G. Cannell and R. W. Taft, J. Am. Chem. Soc., 1956, 78, 5812–5817 CrossRef CAS.
- A. T. Jurewicz, J. H. Bayless and L. Friedman, J. Am. Chem. Soc., 1965, 87, 5788–5790 CrossRef CAS.
- J. H. Bayless, F. D. Mendicino and L. Friedman, J. Am. Chem. Soc., 1965, 87, 5790–5791 CrossRef CAS.
- L. Friedman, A. T. Jurewicz and J. H. Bayless, J. Am. Chem. Soc., 1969, 91, 1795–1799 CrossRef CAS.
- J. H. Bayless and L. Friedman, J. Am. Chem. Soc., 1967, 89, 147–148 CrossRef CAS.
- A. Greb, J.-S. Poh, S. Greed, C. Battilocchio, P. Pasau, D. C. Blakemore and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 16602–16605 CrossRef CAS PubMed.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Nat. Chem., 2009, 1, 494–499 CrossRef CAS PubMed.
- T. Kamei, A. Ishibashi and T. Shimada, Tetrahedron Lett., 2014, 55, 4245–4247 CrossRef CAS.
- I. Vints, J. Gatenyo and S. Rozen, J. Org. Chem., 2013, 78, 11794–11797 CrossRef CAS PubMed.
- P. Appukkuttan, W. Dehaen and E. Van der Eycken, Chem.–Eur. J., 2007, 13, 6452–6460 CrossRef CAS PubMed.
- G. Wu, S. Xu, Y. Deng, C. Wu, X. Zhao, W. Ji, Y. Zhang and J. Wang, Tetrahedron, 2016, 72, 8022–8030 CrossRef CAS.
- G. Berionni, V. Morozova, M. Heininger, P. Mayer, P. Knochel and H. Mayr, J. Am. Chem. Soc., 2013, 135, 6317–6324 CrossRef CAS PubMed.
- G. A. Molander and L. N. Cavalcanti, J. Org. Chem., 2011, 76, 7195–7203 CrossRef CAS PubMed.
- J. Kim and M. Movassaghi, J. Am. Chem. Soc., 2011, 133, 14940–14943 CrossRef CAS PubMed.
- G. A. Olah, M. Piteau, K. Laali, C. B. Rao and O. Farooq, J. Org. Chem., 1990, 55, 46–48 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06055h |
| This journal is © The Royal Society of Chemistry 2023 |