
Molecular modification of planar four-coordinated cobalt active site for the electrochemical reduction of carbon dioxide: a density functional theory study†
Xu
Ding
,
Yucheng
Jin
,
Hailong
Wang
 * and
Dongdong
Qi
*
* and
Dongdong
Qi
*
Beijing Advanced Innovation Center for Materials Genome Engineering, Beijing KeyLaboratory for Science and Application of Functional Molecular and Crystalline Materials, Department of Chemistry and Chemical Engineering, School of Chemistry and Biological Engineering, University of Science and Technology Beijing, Beijing 100083, China. E-mail: hlwang@ustb.edu.cn; qdd@ustb.edu.cn; jianzhuang@ustb.edu.cn
First published on 17th October 2023
Abstract
Single atomic catalysts (SACs) with planar metal-N4 sites are promising electrocatalysts for CO2 reduction reaction (CO2RR) in which the coordination environment plays a crucial role in intrinsic catalytic activity. Cobalt porphyrin is one of the most intensely studied model compound towards CO2 reduction photocatalysts and electrocatalysts because of the central metal located in a well-defined planar four-coordinated (Co–N4) environment. Appropriate structural adjustment of tetraaza-macrocyclic ligands has been demonstrated to improve their thermodynamic reactivity towards CO2 reduction. Herein, we theoretically reveal that such approach enables to tune the electronic structure around active metallic sites due to the changed Co–N4 local structures with broken D4h symmetry and different conjugated ligands from density functional theory (DFT) calculations upon a series of model compounds. They contain cobalt corrole (Co2), cobalt octahydroporphyrin (Co3), and cobalt 1,5,9,13-tetraazacyclohexadecane (Co4), referring to the cobalt porphyrin (Co1) as the benchmark. In addition, the replacement of N atom(s) in Co–N4 sites in Co1 using O and S heteroatoms to form cobalt 21-oxaporphyrin (Co5), cobalt 21,23-dioxaporphyrin (Co6), 21,22-dioxaporphyrin (Co7), cobalt 21-thiaporphyrin (Co8), cobalt 21,23-dithiaporphyrin (Co9), and cobalt 21,22-dithiaporphyrin (Co10) has been evaluated for the rational design and synthesis of high efficiency Co–N–C (cobalt-based) catalysts made up of single metallic atoms coordinated by nonmetallic ligands. In particular, Co3 and Co8 show the lower limiting potentials for CO product as −0.61 and −0.58 eV, respectively, compared with that of Co1. Co2 and Co5 have promising activity for the CH3OH product with the limiting potentials of −1.30 and −1.04 eV and for CH4 product with the limiting potentials of −1.37 and −1.04 eV. This study not only provides a comprehensive view of Co-porphyrinoids for CO2RR but also presents a theoretical screening path for designing and searching efficient molecular catalysts toward electrochemical reactions.
Introduction
The lasting emissions of carbon dioxide from the combustion of fossil fuels have resulted in serious energy and environmental issues since industrial revolution.1–3 Among various CO2 conversion technologies, photocatalytic and electrocatalytic CO2 reduction reaction (CO2RR) are one of the most promising strategies as they can convert CO2 exhaust into synthetic fuels and industrial feedstocks using sustainable power.4–7 Because the electricity is capable of being substantially obtained from solar, wind, hydropower, and other sources, the electrochemical reduction of carbon dioxide becomes an important paradigm to close the carbon cycle.8–11 However, the practical implementation of this technology is prevented by the high activation CO2RR barrier and low catalyst stability. Thus, it is important to develop robust electrocatalysts with high performance.12Single atomic catalysts (SACs) with well-dispersed metal sites coordinated by non-metallic ligand possess excellent catalytic activity and atom utilization towards CO2RR.13–16 In particular, metal-coordinated N-doped carbon-supported SACs (M–N–C, M = transition metals) have emerged as promising CO2RR electrocatalysts, featuring long-term stability, reproducible performance, and potential for large-scale synthesis.17–21 Prior to these catalysts, metallic tetraazamacrocycles, including porphyrins, phthalocyanines, and biphenanthrolinic hexaazacyclophanes, with similar M–N4 coordination structures have attracted attention towards catalysis for the CO2RR in the 1970s.22 As early as 1974, Meshitsuka first observed that graphite-supported cobalt or nickel phthalocyanine electrodes showed the polarization curves in aqueous electrolytes with CO2; the reduction product, however, was not identified.23 In 1984, cobalt phthalocyanine on carbon was proven to reduce CO2 into CO with a current density of 0.98 mA cm−2 at 0.5 V and a faradaic efficiency of 50%.24 In 1979, Takahashi et al. first disclosed that the cobalt porphyrin was active, and the reduction product was confirmed to be formic acid by the resorcinol test.25 In even recent 2019, CoPPCl has been demonstrated to achieve a high CO selectivity of 98.3% at 0.49 V with an overall current density of 25.1 mA cm−2.26 Aside from the metal active centers with tetraazamacrocycles, the coordinated heteroatoms show a crucial effect on the catalytic activity of SACs. The introduction of O atom into N coordination environment improved the catalytic activity. Kim et al. observed that the O atom coordinated in Ni(–Cl)–N3O-TPP led to an improved catalytic activity in CO2RR than N4-coordinated Ni–N4-TPP.27 They also demonstrated the important role of O heteroatom in subordinating the reaction barriers for CO2RR compared with Ni–N4-TPP. Obviously, the change of tetraazamacrocycles and introduction of heteroatoms around the metal improves the CO2RR performances for SACs catalysts. However, thus far, it is still very challenging for the reduction of carbon dioxide into high value-added products including C1 and C2 products (such as CH3OH, CH4, and C2H2) with multi-electron mechanisms. The introduction of copper catalysts upon molecular catalysts and enhancement of the binding with CO intermediate are useful methods in this regard.28
In this work, we comprehensively investigated CO2 reduction to CO, CH3OH, and CH4 upon a series of Co-porphyrinoids (Co1–Co3 and Co5–Co10) and 1,5,9,13-tetraazacyclohexadecane (Co4), revealing the regulatory mechanism of the core-modification and ring-modification about its catalytic behaviors as well as the potential-determining step (PDS) by DFT calculations.29–32 First, the structure stability and electronic structure of all models were calculated. Second, the binding configurations and energies of CO2 intermediates on catalysts were determined, and the charge density difference and density of states were analyzed. Finally, the CO2RR mechanisms to possible C1 products via two-, six-, and eight-electron pathways were elucidated in turn. This work not only indicates the ring- and core-modification effect on SACs in the CO2RR mechanism but also shows a bright avenue to design excellent atomically-dispersed electrocatalysts at the molecular level based on theoretical simulations.33–36
Results and discussion
Chemical and electronic structures
In the present work, we tried to correlate the relationship between molecular structures and catalytic properties from the thermodynamic perspective to explore effective molecular catalysts.37–42 As a result, a series of molecular catalytic models containing Co–N4 and corresponding heteroatom-doped coordination environment have been selected to illustrate the two kinds of molecular catalyst design approaches, namely, ring-modification and core-modification approaches (Scheme 1). Herein, it is worth noting that the ring-modification approach is defined as removing or hydrogenating peripheral carbon atoms from the porphyrin ring of model compound Co1, generating Co2–Co4 with the similar Co–N4 core (Scheme 1). Typically, the resultant cobalt porphyrinoids and 1,5,9,13-tetraazacyclohexadecane derivatives possess different physicochemical properties in comparison to that of traditional porphyrin derivatives. On the other hand, core-modification approach is denoted as replacing one or two pyrrole N atoms with other heteroatoms such as O and S, leading to core-modificated porphyrins (Co5–Co10) with changed Co–NyX4−y (X = O, S; y = 2, 3) core (Scheme 1). The stable configurations of Co1–10 are shown in Fig. 1a–j. The optimized molecular size of Co1 is 12.10 Å × 12.10 Å × 4.00 Å with all the atoms in the same plane. After the ring-modification of the skeleton, the removal of one C atom at the meso-position of Co1 to generate Co2, the molecular size is slightly decreased to 11.42 Å × 11.49 Å × 4.00 Å, which still has a coplanar structure. As a consequence, the Co–N distance is shortened to 1.88 Å from 2.05 Å in Co1. The hydrogenation of the peripheral eight carbon atoms in Co1 gave rise to Co3; its optimized molecular size is 12.12 Å × 12.12 Å × 4.16 Å. The Co–N distance is almost not affected relative to that of Co1. For Co4, it is derived from the removal of peripheral pyrrole eight carbon atoms of Co1 and hydrogenation of residual unsaturated non-hydrogen atoms, possessing a much smaller molecule size of 10.08 Å × 10.17 Å × 6.42 Å. The configuration of Co4 is the distorted noncoplanar molecule, and the average Co–N distances are elongated to 2.17 Å from 2.05 Å in Co1. For core-modification species (Co5–Co10), the one or two pyrrole N atoms in Co1 are substituted by one/two S or O atoms to generate Co5–10. The detailed structural informations of Co-porphyrinoids are shown in Fig. 1e–j. The Co–N distances of Co5–Co8 are shortened as a result of the compression exerted on the cobalt atom by the heteroatoms. However, the S substitution of the two pyrrole N atoms (Co9 and 10) leads to an increased Co–N distance due to the deviation of cobalt atoms from the molecular N2S2 plane.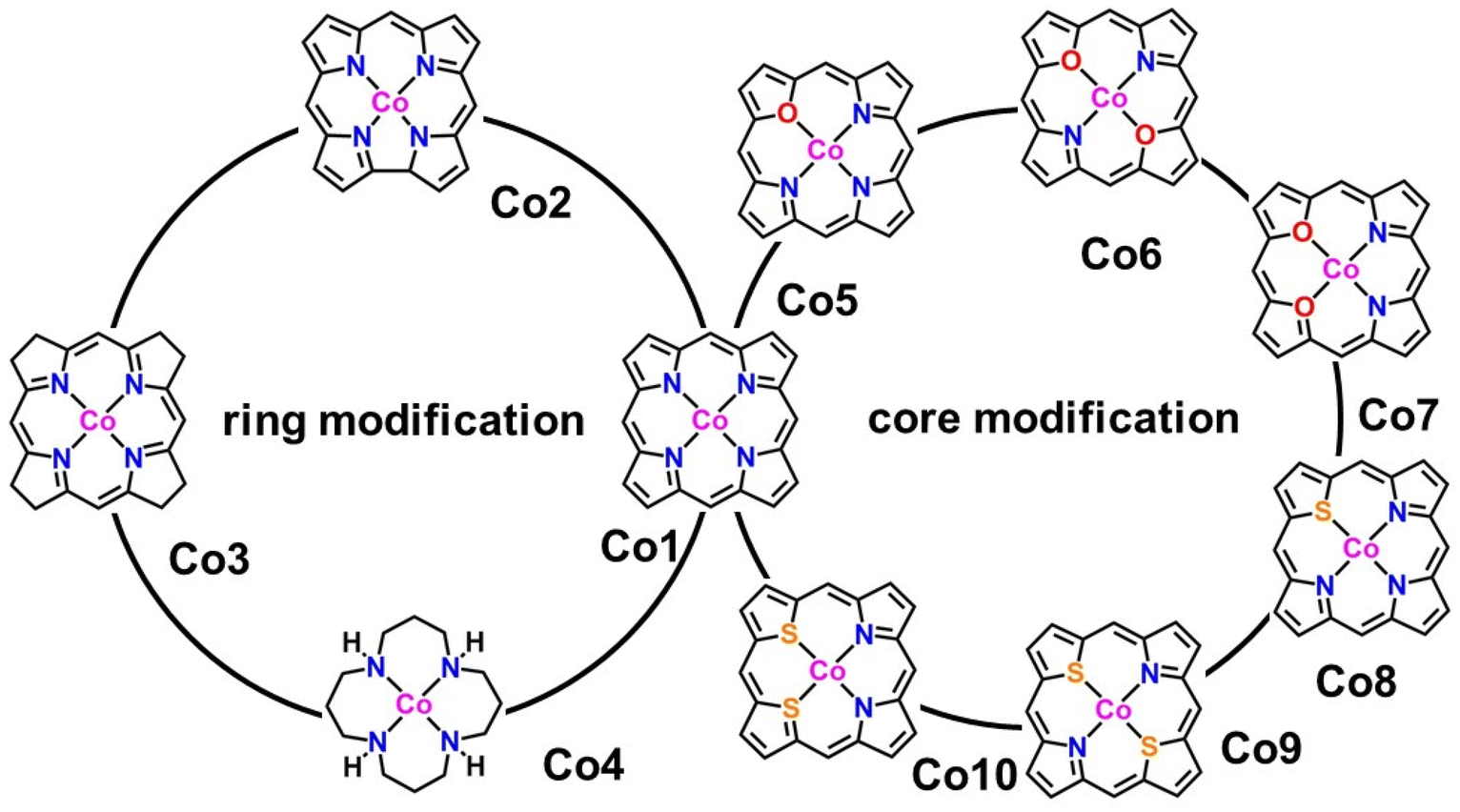 | ||
| Scheme 1 Geometric structure of cobalt complexes upon ring and core modification. | ||
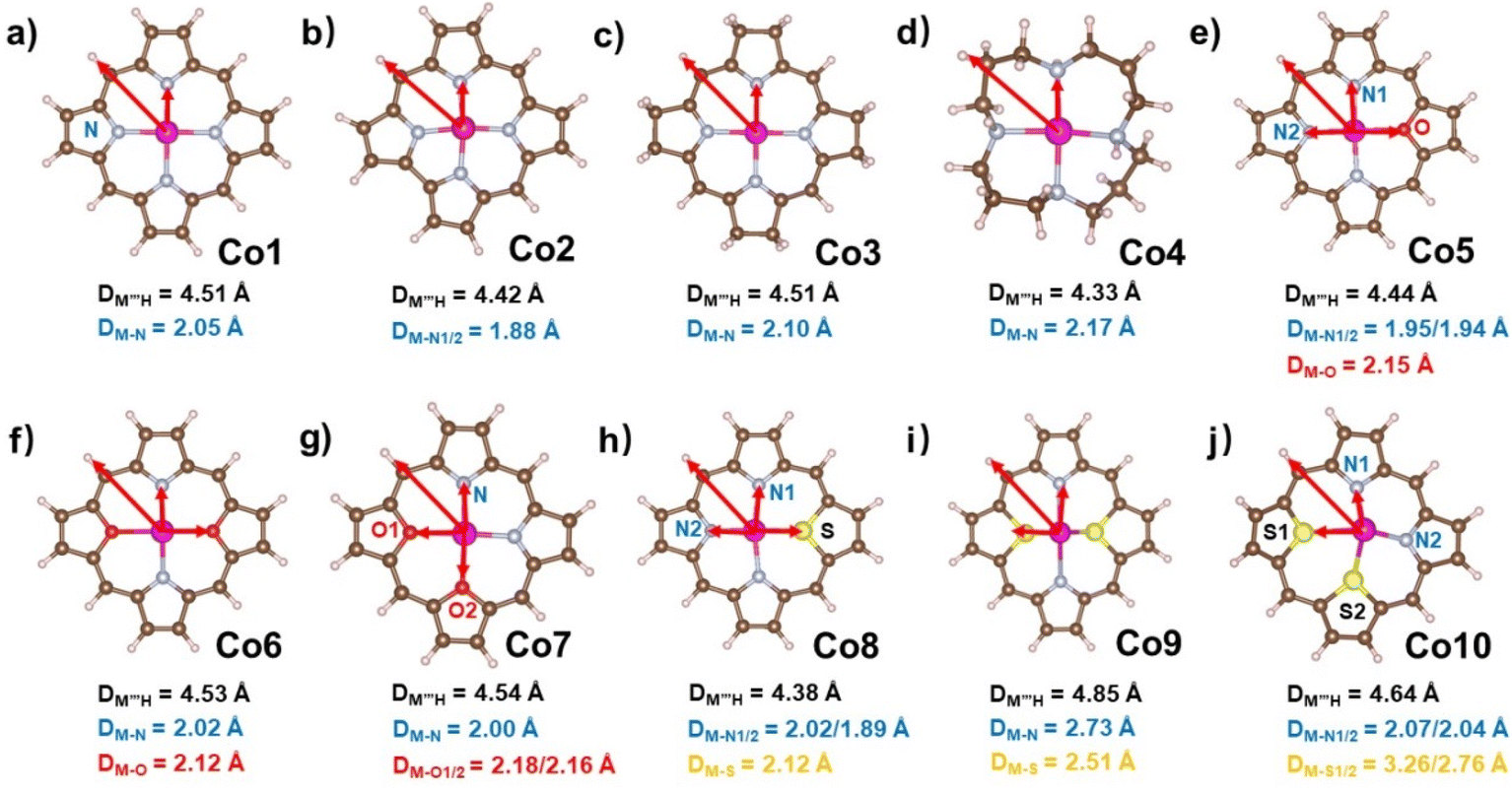 | ||
| Fig. 1 Optimized structures of (a–j) Co1–Co10 with average Co–X (X = N, O, S) bond lengths. All Co–X (X = N, O, S) bond lengths are listed in Table S1.† | ||
Meanwhile, the formation energy (Eformation) was calculated to evaluate the thermodynamic stability of Co-porphyrinoids and 1,5,9,13-tetraazacyclohexadecane (Table S2†). Eformation < 0 eV indicates that the cobalt compounds have good thermodynamic stability.43 Herein, the Eformation values of all cobalt complexes are calculated to be negative in the range between −0.44 and −11.2 eV, indicating that they are thermodynamically stable for the center cobalt ion during catalysis. Among these compounds, the Eformation values of Co4, Co6, Co7, and Co10 are much closer to zero than Co1. In addition, various Co1 derivatives have been reported and performed well in electrocatalysis;44 thus, Co2, Co3, Co5, Co8, and Co9 derivatives are expected to be available at the experimental level.
To understand the ring- and core-modification effect on this series of model complexes, the electronic structures of Co1 to Co10 were investigated by the electronic densities of states (DOS) and charge density differences.45 Co1 shows four distinct electron distribution peaks at −18.13, −13.55, −10.14, and −7.28 eV in Fig. 2a and Fig. S1,† and the band gap is 5.93 eV. The removal of a meso-positional C in Co1 to form Co2 induces four electron distribution peaks of Co2 at −16.95, −12.87, −9.39, and −6.25 eV, which slightly decreases the band gap to 5.61 eV. The peripheral hydrogenated Co3 has the four electron distribution peaks shifted to large energy values of −18.31, −15.59, −12.22, and −7.26 eV, and its band gap is further decreased to 4.95 eV. In contrast to these three compounds, the band gap of Co4 is suddenly increased to 9.22 eV.46,47 As shown in Fig. 2b and Fig. S2,† the replacement of N atom of Co1 with O atom to generate Co5 results in six electron distribution peaks at −18.26, −16.52, −13.74, −9.75, −6.99, and −1.12 eV with the smaller band gap of 5.40 eV. The replacement of two nitrogen atoms at either diagonal or adjacent position of N4 core with O atoms gives rise to six similar electron distribution peaks of Co6 and Co7 and the decreased band gaps of 5.57 eV. When one N atom in Co1 is replaced by S atom, there are six electron distribution peaks for Co8 observed at −18.19, −17.00, −13.71, −9.07, −4.98, and −1.19 eV, substantially decreasing the band gap to 3.40 eV. Consequently, the substitution of two N atoms in Co1 with S atoms shows five similar electron distribution peaks of Co9 and Co10 with S atoms at diagonal and neighboring positions, and the decreased band gaps are 5.55 and 5.53 eV, respectively.48 In the present case, both ring- and core-modification approach can decrease their band gaps, improving the intramolecular electron transfer of cobalt catalysts during the CO2RR process. Meanwhile, the LOL-π (localized orbital locator) analysis was calculated to evaluate the molecular conductivity of cobalt complexes in Fig. S3.† From the figure, it can be seen that the LOL-π map successfully reveals the delocalization of the π electrons around macrocycles with the exception of Co4.
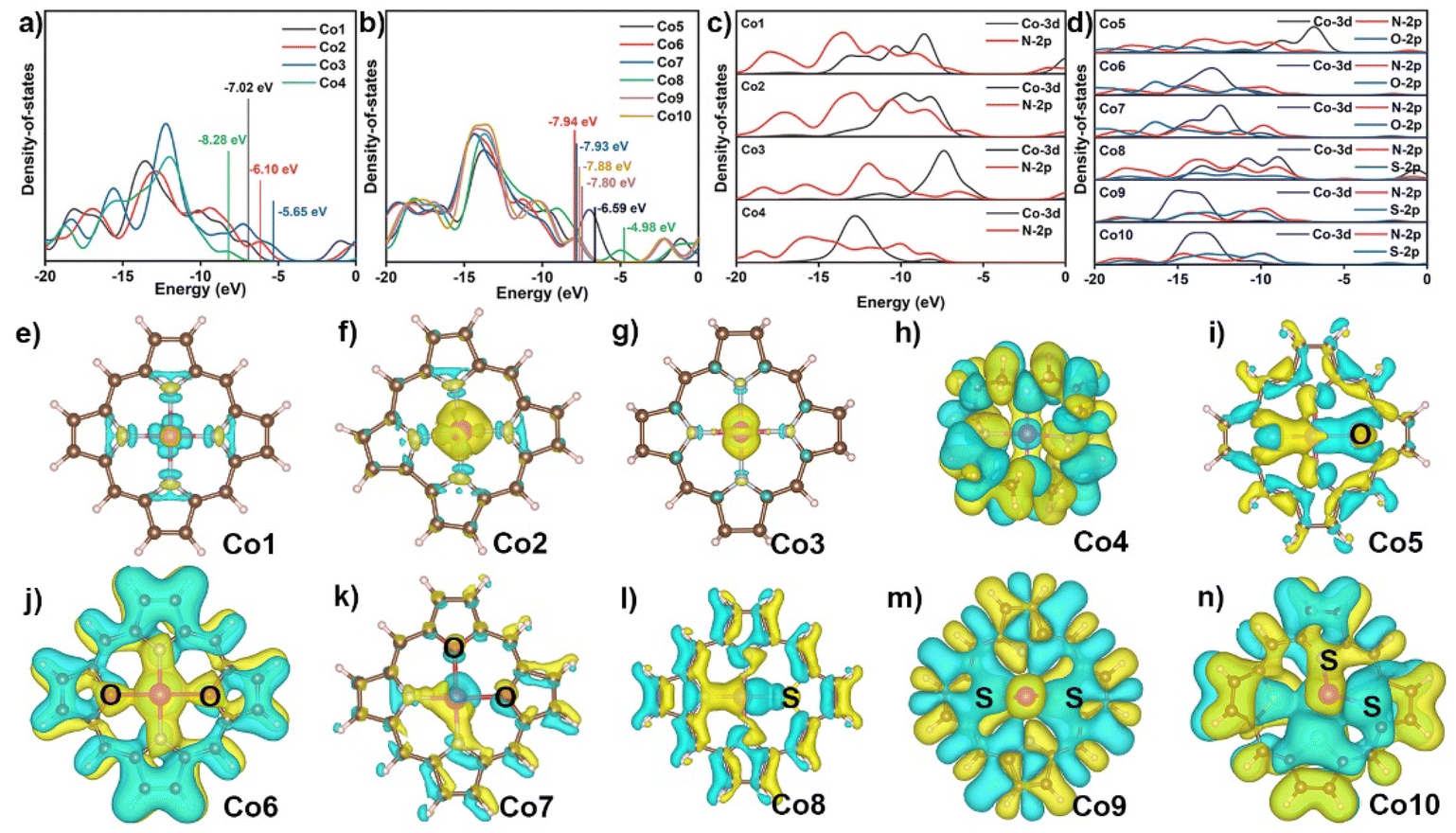 | ||
| Fig. 2 Electronic densities of states (DOS) of (a) Co1–Co4 and (b) Co5–Co10. The values in figures refer to the position of highest occupied molecular orbitals. Electronic projected densities of states (PDOS) of (c) Co1–Co4 and (d) Co5–Co10. Charge density difference of (e–n) Co1–Co10. Before and after cobalt atom anchored into ligand molecule, respectively. Yellow and blue regions represent increasing and decreasing electron densities, respectively. The isosurface values were 0.009. | ||
As shown in Fig. 2c and d, the projected DOS shows that the orbitals in Co1-Co10 compounds are in the range from −20 to −5 eV, consisting of Co 3d and N 2p or/and S/O 2p orbitals. For these compounds, the Co 3d orbital displays an obvious overlapping with N 2p and/or S, O 2p orbitals, implying the strongest interaction between the Co atom and the surrounding N atoms and other heteroatoms in all cobalt complexes. As displayed in Fig. 2e–n, the charge density differences show increased electron density on the metal surface in Co1 after the complexation with porphyrin, indicating the formation of stable coordination bonds. Simultaneously, the injection of electron occurred on the Co atom, which is beneficial for the enhancement of the reduction capability of the active metal site. The similar phenomena are observed in Co2 and Co3, albeit with a greater injection of electron than that of Co1. In Co4, electrons are distributed to C atoms and N atoms rather than transferring between Co atom and N atoms, indicating that a weak coordinate bond is formed in Co–N bonds in this compound. Following the core-modification, the majority of electrons are accumulated in the Co–O and Co–S bonds instead of Co–N bonds for Co5 and Co8, suggesting the presence of the stronger interactions between Co and O or S atoms rather than N atoms. The electron density on the upper surface of the Co atom increases in Co6, Co7, Co9, and Co10, while it decreases on the lower surface, Fig. 2j, k, m, and n. This phenomenon can be attributed to the different atomic radius of O and S atom compared with N atom, which induces a deviation of the cobalt atom from the molecular plane. The NPA charge in Table S3† and electrostatic potential (ESP) in Fig. S4† show that less electrons are transferred from divalent Co atom to macrocycles observed for C than that in Co1 (1.240), revealing the more electron density around the cobalt atom for Co2, Co3, Co5, and Co8. This evidence indirectly reflects the improved adsorption capability of active cobalt ion toward small substrate such as O2 and CO2. The high NPA charge of cobalt atom on Co4, Co6, Co7, Co9, and Co10 are 1.424, 1.530, 1.507, 1.757, and 1.750, respectively, lead to the more difficult electron transferred from the cobalt atom to the substrate. In a word, the systematic study on formation energy, DOS, charge density difference, NPA charge, and ESP of ten models preliminarily screens the stable and favorable electronic structure with potential to capture small molecule, certainly including CO2, for the next activation on metal sites with similar Co–NyX4−y coordination environment.
Screening adsorption capability
The possible CO2RR intermediates from CO2 to CO, CH3OH, and CH4 products along with corresponding reaction pathways are summarized in Fig. 3a. Adsorption thermodynamic energy of CO2 and important product/intermediate CO along various CO2RR pathways upon these compounds were elucidated to evaluate the core-modification and ring-modification strategy. It is well established that CO2 physical adsorption is the initial and crucial step for CO2RR.49 Therefore, a thermodynamics favorable CO2 adsorption configuration is significant for the CO2 activation and further reduction. For the Co1 compound (Fig. S5†), CO2 prefers to binding with Co atom via O-termination configuration with a ∠OCO of 178.45° and a Co–O distance of 3.42 Å, having the binding energy of −0.03 eV. The change of porphyrin with corrole (Co2) provides a Co–C distance of 2.05 Å and ∠OCO is 130.86° for the CO2 adsorbed on the cobalt site in Co2. In contrast to Co1, the larger binding energy of Co2 toward CO2 suggests the improved adsorption capacity for the latter one. On hydrogenated Co3, the C-termination configuration of CO2 adsorbed by cobalt site is also observed with a smaller distance of 2.18 Å and the ∠OCO of 135.75°, indicating the stronger interactions between the Co atom and CO2 molecule compared with that of Co1. This point is further confirmed by the relatively small value for CO2 adsorption energy of 0.17 eV. On the C-termination configuration of CO2 in Co4, Co–C distance of 3.28 Å and ∠OCO of 179.36° are detected. The positive adsorption energy of CO2 (0.14 eV) suggests a much weaker CO2 capture ability of this compound.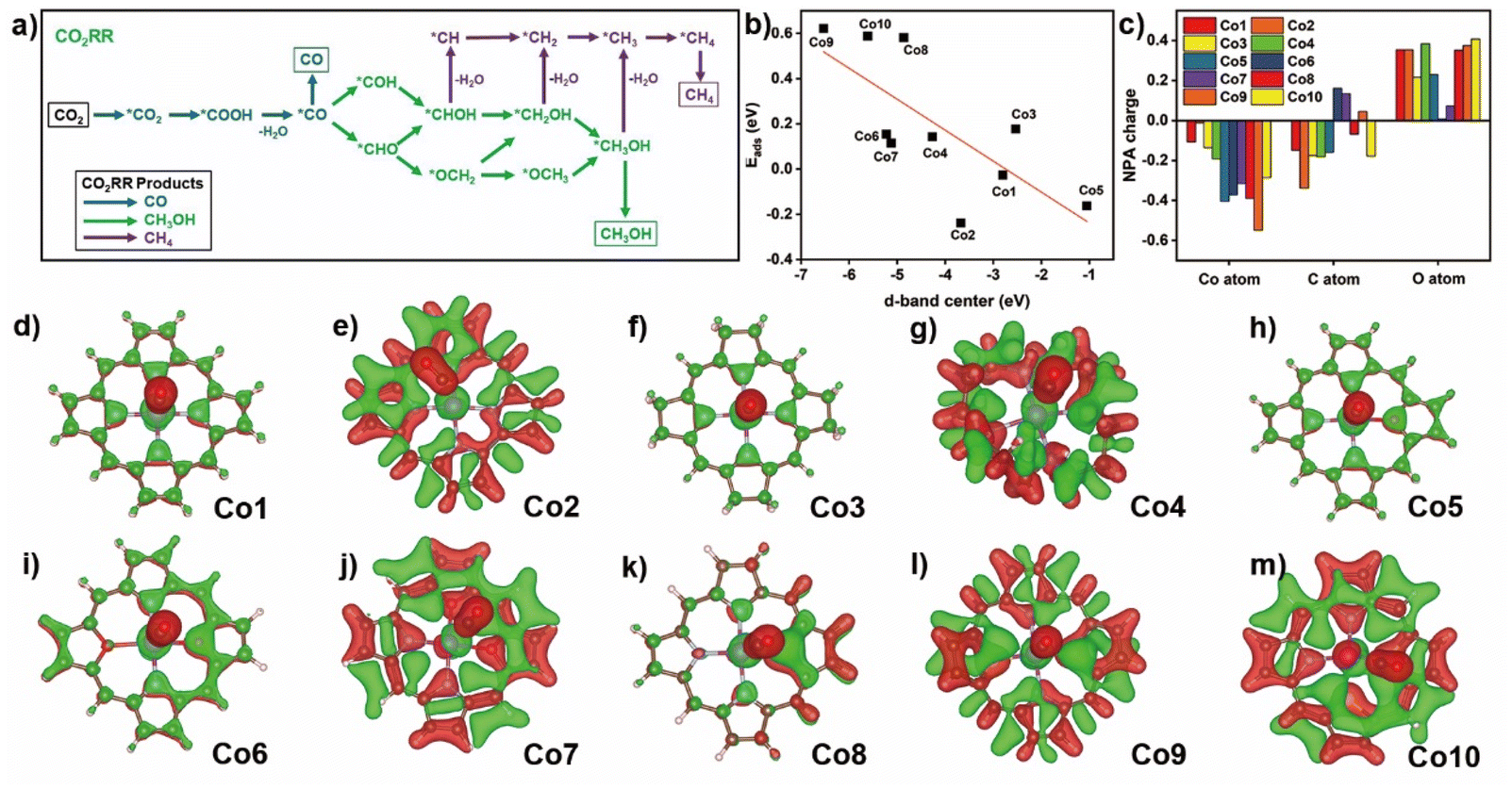 | ||
| Fig. 3 (a) Schematic of elementary reaction steps of CO2RR to CO, CH3OH, and CH4. (b) Relationship between the d-band center of Co1–Co10 and the adsorption energy of CO2 molecules. (c) NPA charge difference between C and O atoms of CO molecule and Co atom before and after CO binding on Co1–Co10 (The positive values represent increasing electron density). (d–m) Charge density difference (δρ = ρA+B − ρA − ρB) of CO binding on Co1–Co10 before and after CO binding, respectively. Red and green regions represent increasing and decreasing electron densities, respectively. | ||
In the core modification strategy, the C-termination configuration of CO2 is captured with a short Co–C distance of 1.94 Å for Co5. The adsorption energy of CO2 is −0.16 eV, much lower than that on Co1. This suggests an effective improvement of the CO2 capture ability on Co5. The substitution of two nitrogen atoms at either adjacent or diagonal position of N4 core with O atoms causes the relatively short distance of Co–C or Co–O of about 3.27 and 3.01 Å of CO2 adsorption configurations for Co6 and Co7, and the adsorption energy of CO2 is 0.15 and 0.11 eV, respectively. The replacement of O atom with S atom causes the long distance of Co–C of about 2.15 Å for C-termination configuration adsorbed on cobalt atom in Co8. The much larger positive adsorption energy of CO2 is 0.58 eV due to the S coordination weakening the CO2 capture ability. On Co9 and Co10 surface, in the adsorbed CO2 in C- and O-termination configuration, the distances of Co–C and Co–O are 3.29 and 2.96 Å, respectively. As a result, their values of adsorption energy of CO2 are further increased to 0.62 and 0.59 eV, respectively, hinting at the much weaker interaction between the CO2 molecule and divalent cobalt atom.
The changed CO2 adsorption energy on the same metal primarily relies on the occupancy weight of bonding and anti-bonding states formed by the hybridization of the wave functions of adsorbate and metallic d orbital electrons.50 The positive adsorption energies of a series of models (except Co1, Co2, and Co5) are due to the lower d-band of metal compared to the Fermi level (EF) accompanied by the higher occupancy of the antibonding state, leading to a certain repulsive interaction between the CO2 and cobalt atom and thereby a weak chemisorption. This hypothesis is overall correspondent with the calculation results about the position of the d-band center of the metal (Fig. 3b). The much closer position of the d-band toward Femi level (EF) seems to be helpful in the activity enhancement of the metallic d orbital electron, preferring to the formation of a strong chemisorption, as indicated by Co1, Co2, and Co5.
Since CO is one of the important intermediates during CO2RR, the binding configuration of CO on cobalt atom in Co1–Co10 were comparatively explored (Fig. S6†). In all the Co complexes, CO prefers to locate on the Co site in the form of C-termination configuration, and the calculated binding energy is 0.27, −1.36, −0.61, 0.32, −1.11, −0.36, −0.06, −0.53, 0.72, and 0.21 eV, respectively. Therefore, ring-modified Co1–Co3 together with core-modified Co5–Co8 could effectively improve the CO binding energy, making it favorable for CO2RR to C1 products (such as CH4 and CH3OH) with multi-electron mechanism, while Co4, Co9, and Co10 seem to favor the formation of only the CO product due to the small binding energy.
To further gain an insight into the effect of core and ring-modification on the important step of CO binding, the charge distribution in CO and Co on Co1–10 were analyzed in the unbinding and binding states (Fig. 3c and Table S4†). The electron density at the C atom of CO adsorbed by the Co site is increased obviously from Co1 (0.149 |e|) to Co2 (0.338 |e|), then decreased to the similar Co1 value of Co3–Co5, Co8, and Co10 (−0.046 |e| −0.182 |e|), implying the possibility of electron transfer from Co atom to binding CO. A similar phenomena is observed for the charge density of O atoms when CO is bound on Co compounds. For the change in the charge density in Co atom, there is 0.107 |e| electron transfer from Co to the adsorbed CO molecule, indicating the presence of interaction between Co and CO in Co1. On Co2, 0.013 |e| less electrons are transferred from the Co atom in Co2 after CO binding relative to that on Co1, suggesting the presence of a weaker interaction between Co atom and carbon monooxygen molecule for the former compound. After other model compounds, 0.135, 0.192, 0.404, 0.371, 0.315, 0.390, 0.549, and 0.285 |e| more electrons are shifted in compared with that on Co1 after CO attack, suggesting a stronger interaction between Co and CO on Co3–Co10 than Co1. The obvious electron transfer is indicated by the charge difference, showing either reduced or enhanced electron density for cobalt macrocyclic ligand and CO (Fig. 3d–m). This is in good agreement with the above mentioned NPA charge analytical result.
Screening CO2RR to CO via two-electron pathway
In CO2RR, the products involving C1 and C2 products have been obtained. Nevertheless, the generation of C2 products is difficult using porphyrin-based electrocatalysts owing to the absence of the binding center for C–C coupling to C2 intermediates.51,52 Two-electron, six-electron, and eight-electron pathways to C1 products are presented in Fig. 3a. In this paper, all the possible reaction pathways together with the involved intermediates on Co model compounds are provided in Fig. S7–S16 and Tables S6, S7.†CO is the main CO2RR product obtained via the two-electron pathway. As shown in Fig. 4 and Fig. S17,† CO2RR to form CO proceeds along the pathway CO2 → *COOH → *CO → CO.53–55 On Co1, CO2 adsorption is thermodynamically favorable due to the ΔG value of −0.03 eV, followed by a crucial protonation step from *CO2 to *COOH with a high ΔG value of 1.27 eV. Subsequently, the pathway contains the *COOH → *CO and CO desorption processes, having a ΔG value of −0.61 and −0.27 eV, respectively. On Co2, the spontaneous CO2 adsorption, *COOH formation, and *CO generation processes are supported from thermodynamics perspective by means of a ΔG value of −0.24, −0.38, and −0.38 eV, respectively. In contrast to the generation of *COOH upon Co1 as the potential determining step (PDS), Co2 has the PDS of CO desorption for CO2RR, as indicated by the high ΔG value of 1.36 eV. This high ΔG value for CO desorption seems to be helpful in the conversion into CH3OH and CH4 from the *CO intermediate. On Co3, the PDS for CO generation is still estimated as the desorption of CO (0.61 eV), significantly lower than that of Co1 (1.27 eV), indicating the much better activity to generate CO for the former compound. On Co4, the PDS is *COOH formation due to the highest ΔG value of 1.74 eV among these ten compounds, indicating the difficult protonation of CO2 to produce the *COOH intermediate. For the core-modification compounds, the PDS of Co5 is CO desorption due to the high ΔG value of 1.11 eV, indicating the promising catalyst for the further hydrogenation reactions of CO into others valuable products. For Co6 and Co7, high ΔG values of 2.12 and 1.89 eV, respectively, are seen for the production of *COOH. On Co8, the PDS for CO production is CO2 adsorption (0.58 eV), which is significantly lower than that of Co1 (1.27 eV), indicating the easy production of CO. This is consistent with the previously mentioned analytical result of the smallest band gaps (3.40 eV), implying the strongest reduction capability of Co8. For double sulfur-doped Co9 and Co10, the high ΔG, 1.17 and 1.41 eV, respectively, are seen for the production of *COOH. Notably, it is still challenging to rationally construct a highly efficient catalyst for special valuable products. Therefore, theoretical simulations were conducted on these compounds to screen promising catalysts for CH3OH and CH4 products from the *CO intermediate.
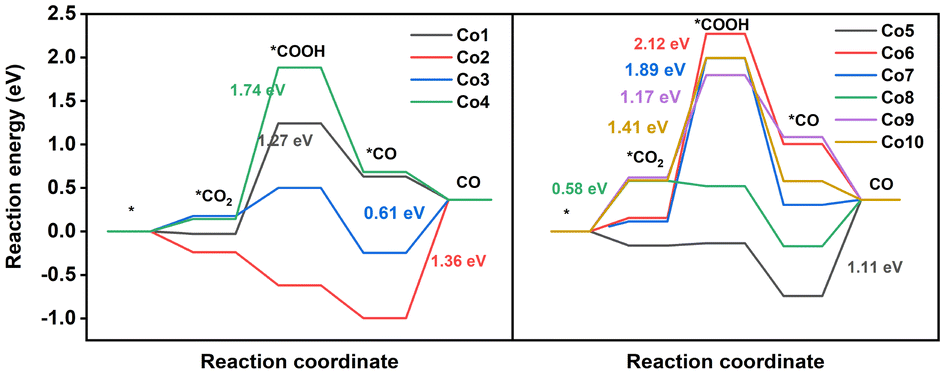 | ||
| Fig. 4 Free energy diagrams of CO2RR to CO and corresponding intermediates on Co1–Co10 complexes. The values in figures refer to the free energy change for the PDS. The asterisks mean intermediates bind at the active site. | ||
Screening CO2RR to CH3OH via the six-electron pathway
It is worth noting that both *COH and *CHO are the intermediates of CO2RR to generate CH4 and CH3OH. In com-parison with the free energy of CO desorption, the much higher free energy of *COH and *CHO on Co1, Co3, Co4, and Co6–Co10 are averse to corresponding intermediate formation (Table S5†). In great contrast, on Co2 and Co5, the *CHO formation by *CO protonation has a free energy barrier of 0.34 and 1.04 eV, respectively, smaller than the CO desorption barrier of 1.36 and 1.11 eV. Therefore, the formed *CO intermediate is expected to be further protonated to form the CH3OH and CH4 on Co2 and Co5. Fig. 5 introduces the preferred reaction pathways of CO2RR to CH3OH by means of the six-electron pathway on Co2 and Co5. On Co2, reactions proceed along the path CO2 → *COOH → *CO → *CHO → *OCH2 → *CH2OH → *CH3OH → CH3OH. After *CO formation through the first two steps with successive protonation, the third proton prefers to interacting with the C atom of *CO to form *CHO with the ΔG value of 0.34 eV, precluding the other pathway from *CO to *COH because of the high energy barrier of 3.62 eV. In the following step, *CHO → *OCH2 requires a negative ΔG value of −0.54 eV, representing a spontaneous reaction. In the next step, *OCH2 → *CH2OH needs a smaller ΔG of 0.40 eV than that for *OCH2 → *OCH3 (0.45 eV), identifying that CO2RR proceeds via the first pathway. It is followed by *CH2OH → *CH3OH with a very negative ΔG value of −1.09 eV. Then, the desorption of CH3OH requires a ΔG of 1.30 eV, being the PDS in the CH3OH generation pathway.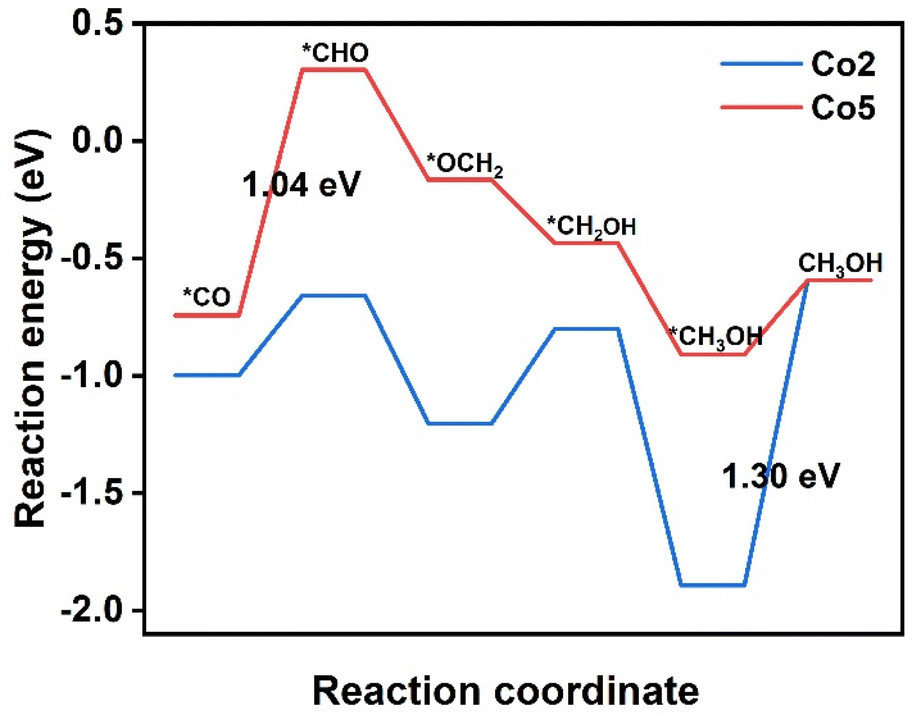 | ||
| Fig. 5 Free energy diagrams of CO2 reduction pathway to CH3OH on Co2 and Co5. The values in figure refers to the free energy change for the PDS. The asterisks mean that intermediates bind at the active site. | ||
On Co5, the proceeding *CO → *CHO reaction suffers a ΔG value of 1.04 eV, being assigned as the PDS in the CH3OH generation pathway. Then, the proton tends to attack the C atom instead of the O atom of *CHO to form *OCH2 with a ΔG value of −0.46 eV; the subsequent hydrogenation at the O atom of *OCH2 produces *CH2OH with a ΔG of −0.27 eV. The *CH2OH → *CH3OH step presents a spontaneous trend suggested by a ΔG value of −0.47 eV. After these steps, the CH3OH needs a small ΔG value of 0.32 eV for desorption, implying the easy escape of CH3OH from Co5. As a result, Co2 and Co5 may be promising catalytic candidates for CO2RR to CH3OH, although they encounter different PDS in the same six-electron pathway.
Screening CO2RR to CH4via the eight-electron pathway
The conversion CO2 to CH4 has been achieved via the eight-electron reaction pathway with the necessary *COH or *CHO.35,56,57 The reaction free energy profiles of CH4 production for the above selected Co2 and Co5 due to the easy formation of *CHO are calculated and shown in Fig. 6. In combination with the calculation results on CH3OH production, there are only two possible pathways for CO2RR, including CO2 → *CO2 → *COOH → *CO → *CHO → *OCH2 → *CH2OH → *CH2 → *CH3 → *CH4 → CH4(I) and CO2→*CO2 → *COOH → *CO → *CHO → *OCH2 → *CH2OH → *CH3OH → *CH3 → *CH4 → CH4(II). For Co2, the six reaction steps to form *CH2OH in CH4 generation are the same pathways as that in CH3OH generation. For the following step, *CH2OH suffers from the two possible pathways to form either *CH3OH or *CH2. The negative ΔG value of −1.09 eV allows the spontaneous protonation of *CH2OH to form *CH3OH. The *CH3OH → *CH3 pathway, rather than CH3OH desorption with a small ΔG of 0.06 eV, provides an opportunity to proceed *CH3 → *CH4 (ΔG = −1.40 eV) and produce CH4 (ΔG = 1.37 eV) (Fig. S8†). Therefore, methane desorption for CH4 generation is the PDS. The ΔG of CO2RR to prepare CH4 is similar to that for the CH3OH product (1.30 eV), predicting the presence of an intense competition between CH4 and CH3OH in CO2RR on Co2. For Co5, the calculation results reveal that it has the same CO2RR pathways as Co2. The *CHO → *OCH2, *OCH2 → *CH2OH, *CH2OH → *CH3, and *CH3 → *CH4 steps are spontaneous due to ΔG values in the range from −1.02 to −0.25 eV. In contrast, the energy barrier of CH4 desorption is still consistent with that of CH3OH desorption with small energy barriers (0.31 eV vs. 0.32 eV), indicating the potential of obtaining CH3OH and CH4 in CO2RR. As a result, its PDS is determined as *CO → *CHO (1.04 eV). The present results illustrate the CO2RR pathway to valuable CH3OH and CH4 and the possibility of achieving high efficiency catalysts.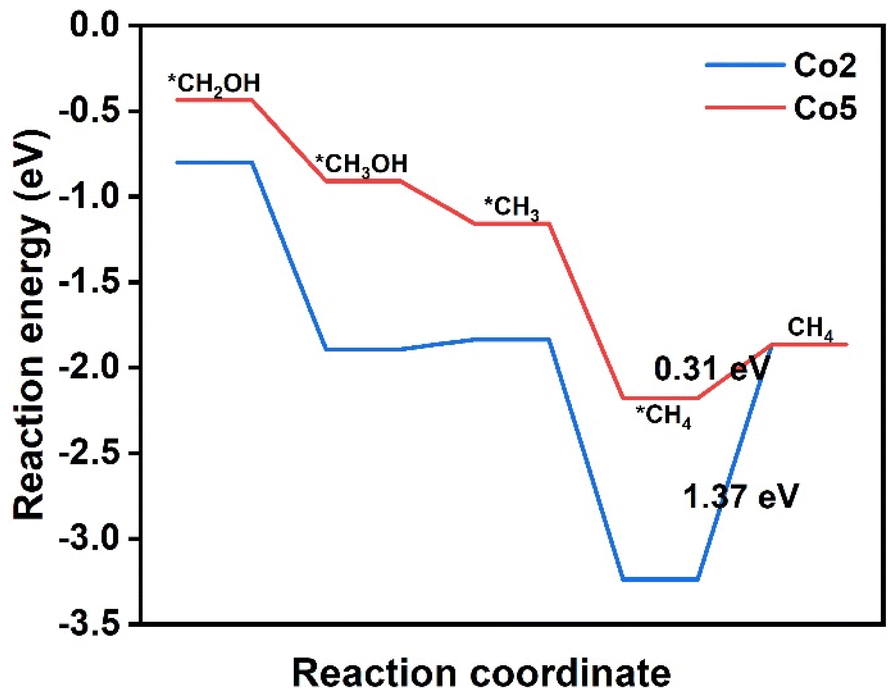 | ||
| Fig. 6 Free energy diagrams of the CO2 reduction pathway to CH4 on Co2 and Co5. The values in the figure refer to the free energy change for the PDS. The asterisks mean intermediates bind at the active site. | ||
In order to apply the electronic regulation strategy of the metal site to innovate molecular catalysts, substituent effects have also been introduced to regulate the electronic structure of cobalt porphyrin Co1.58 Inspired by the asymmetric oxygen and sulfur doping strategy, we introduce two electron-donating and electron-withdrawing substituents (–F, –CN and –NH2) on the pyrrole of porphyrin, and two electron-withdrawing substituents (–F) on the meso-site of porphyrin, such as the structure Co11–Co16, as displayed in Fig. 7. The NPA charge of the cobalt atom in all compounds was carried out (Table S3†). The findings indicate that the charge on the Co atom is minimally affected by asymmetric substitution. In the next step, eight substituents (–F and –OCH3) were introduced at the periphery of the porphyrin molecule, and the corresponding NPA charge was calculated; however, the observed impact remains negligible. As a result, the substitution effect has a little effect on the CO2 reduction properties. To verify our point, we calculate the energy barrier diagram, as shown in Fig. S20.† As expected, the formation of a step in *COOH still has a higher energy barrier.
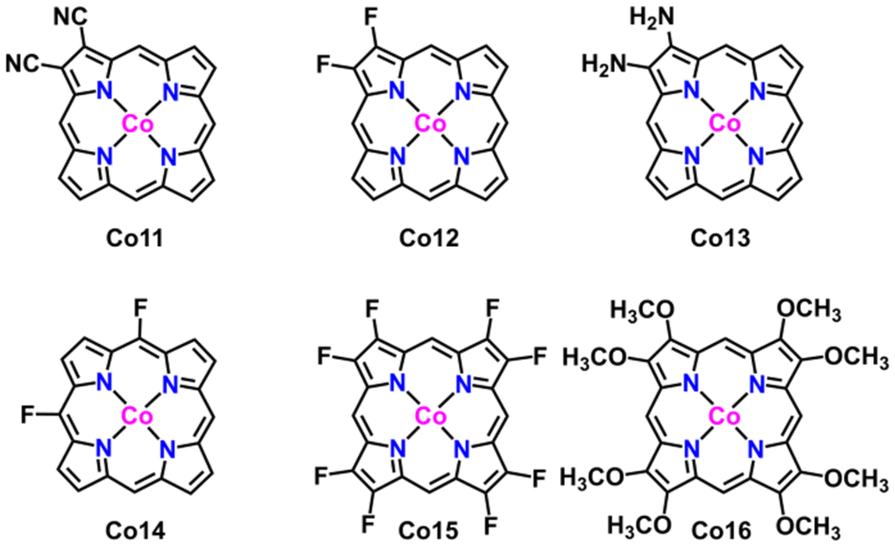 | ||
| Fig. 7 Geometric structure of cobalt complexes upon substituent effects. | ||
Conclusions
In summary, we have performed a comprehensive and systematic theoretical investigation about CO2RR thermodynamics behaviors over a series of cobalt porphyrin benchmark-derived model compounds based on the ring- and core-modification strategies as well as the substitution effect. The electronic densities of states (DOS), charge density difference, NPA charge, ESP mapping, d-band center, and adsorption capability toward CO2 and CO molecules of all the cobalt complexes were analyzed. Their different thermodynamics behaviors are mainly associated with the molecular structures rather than the substituent effect. In the present case, four cobalt complexes (Co3, Co5, Co8, and Co9) were identified as potential CO2RR catalysts with a high activity for the CO product. Co2 and Co5 have the opportunity to generate CH3OH and CH4 through more than two electron pathways. At the end of this section, it is worth noting that the molecular electrocatalysts have poor conductivity and thus require necessary immobilization with conductive supports for heterogeneous electrocatalytic CO2RR. Although some model compounds have been screened to exhibit a promising good performance on the basis of the thermodynamic calculations, however, the regulation of kinetics of electrocatalysts also plays a crucial role. For example, the good dispersion of molecular catalysts on the suitable conductive substrates is a determinant to improve the electron conductivity between interfaces toward high performance electrocatalysts. In addition, the new technology of electrolyzer is another important factor to enhance the efficiency of catalysts through the enhancement of the mass transport of the reactant gases to touch the electrodes.59 In the present case, this work is still surely helpful to guide the design and synthesis of new Co porphyrin catalysts for electrochemical CO2 reduction with good reactivity.Author contributions
Hailong Wang, Dongdong Qi and Xu Ding proposed the project. Xu Ding performed calculation under the guidance of Hailong Wang and Dongdong Qi. Yucheng Jin provided assistance for data analysis. All authors discussed the results and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (No. 22131005 and 22261132512), Xiaomi Young Scholars Program, and University of Science and Technology Beijing.References
- S. J. Davis, K. Caldeira and H. D. Matthews, Future CO2 emissions and climate change from existing energy infrastructure, Science, 2010, 329, 1330–1333 CrossRef CAS PubMed.
- F. Parrenin, V. Masson-Delmotte, P. Köhler, D. Raynaud, D. Paillard, J. Schwander, C. Barbante, A. Landais, A. Wegner and J. Jouzel, Synchronous change of atmospheric CO2 and antarctic temperature during the last deglacial warming, Science, 2013, 339, 1060–1063 CrossRef CAS PubMed.
- G. A. Olah, G. K. S. Prakash and A. Goeppert, Anthropogenic chemical carbon cycle for a sustainable future, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- S. Xu and E. A. Carter, Theoretical insights into heterogeneous (photo)electrochemical CO2 Reduction, Chem. Rev., 2019, 119, 6631–6669 CrossRef CAS PubMed.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Cocatalysts for selective photoreduction of CO2 into solar fuels, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. R. Cuenya, Rational catalyst and electrolyte design for CO2 electroreduction towards multicarbon products, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- J. Zhou, H. Liu and H. Wang, Photothermal catalysis for CO2 conversion, Chin. Chem. Lett., 2023, 34, 107420 CrossRef CAS.
- B. Han, Y. Jin, B. Chen, W. Zhou, B. Yu, C. Wei, H. Wang, K. Wang, Y. Chen, B. Chen and J. Jiang, Maximizing electroactive sites in a three-dimensional covalent organic framework for significantly improved carbon dioxide reduction electrocatalysis, Angew. Chem., Int. Ed., 2022, 61, e202114244 CrossRef CAS PubMed.
- Q. Fan, P. Hou, C. Choi, T.-S. Wu, S. Hong, F. Li, Y.-L. Soo, P. Kang, Y. Jung and Z. Sun, Activation of Ni particles into single Ni-N atoms for efficient electrochemical reduction of CO2, Adv. Energy Mater., 2020, 10, 1903068 CrossRef CAS.
- S. Chen, X. Li, C.-W. Kao, T. Luo, K. Chen, J. Fu, C. Ma, H. Li, M. Li, T.-S. Chan and M. Liu, Unveiling proton-feeding effect in sulfur-doped Fe-N-C single-atom catalyst for enhanced CO2 electroreduction, Angew. Chem., Int. Ed., 2022, 61, e202206233 CrossRef CAS PubMed.
- X. Wang, G. Chang, C. Liu, R. Li, Y. Jin, X. Ding, X. Liu, H. Wang, T. Wang and J. Jiang, Chemical conversion of metal-organic frameworks into hemi-covalent organic frameworks, Inorg. Chem. Front., 2022, 9, 4776 RSC.
- X. Chen, C. Wang, D. Qi and X. Xing, A theoretical approach for homogeneous CO2 reduction by Ni(cyclam): substituents with intra-molecular hydrogen-transfer, Inorg. Chem. Front., 2022, 9, 2691–2696 RSC.
- K. Li, S. Zhang, X. Zhang, S. Liu, H. Jiang, T. Jiang, C. Shen, Y. Yu and W. Chen, Atomic tuning of single-atom Fe-N-C catalysts with phosphorus for robust electrochemical CO2 reduction, Nano Lett., 2022, 22, 1557–1565 CrossRef CAS PubMed.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Heterogeneous single-atom catalysts for electrochemical CO2 reduction reaction, Adv. Mater., 2020, 32, 2001848 CrossRef CAS PubMed.
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Enhancing CO2 electroreduction to methane with a cobalt phthalocyanine and zinc-nitrogen-carbon tandem catalyst, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef CAS PubMed.
- Y. Liu, H. Yang, X. Fan, B. Shan and T. Meyer, Promoting electrochemical reduction of CO2 to ethanol by B/N-doped sp3/sp2 nanocarbon electrode, Chin. Chem. Lett., 2022, 33, 4691–4694 CrossRef CAS.
- Y. Han, Y.-G. Wang, W. Chen, R. Xu, L. Zheng, J. Zhang, J. Luo, R.-A. Shen, Y. Zhu, W.-C. Cheong, C. Chen, Q. Peng, D. Wang and Y. Li, Hollow N-doped carbon spheres with isolated cobalt single atomic sites: superior electrocatalysts for oxygen reduction, J. Am. Chem. Soc., 2017, 139, 17269–17272 CrossRef CAS PubMed.
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Well-defined materials for heterogeneous catalysis: from nanoparticles to isolated single-atom sites, Chem. Rev., 2020, 120, 623–682 CrossRef CAS PubMed.
- K. Xu, S. Zheng, Y. Li, H. Chu, Q. Xiong, Z. Mei, Q. Zhao, L. Yang, S. Li and F. Pan, Constructing low-valent Ni nanoparticles for highly selective CO2 reduction, Chin. Chem. Lett., 2022, 33, 424–427 CrossRef CAS.
- J. Xiong, M. Zhang, M. Lu, K. Zhao, C. Han, G. Cheng and Z. Wen, Achieving simultaneous Cu particles anchoring in meso-porous TiO2 nanofabrication for enhancing photo-catalytic CO2 reduction through rapid charge separation, Chin. Chem. Lett., 2022, 33, 1313–1316 CrossRef CAS.
- Z.-W. Huang, S.-W. An, K.-Q. Hu, X.-B. Li, Z.-N. Bin, Z.-H. Zhou, L. Mei, Z.-J. Guo, W.-S. Wu, Z.-F. Chai and W.-Q. Shi, Modulating the coordination microenvironment of uranyl compounds to enhance photocatalytic CO2 reduction, Inorg. Chem. Front., 2023, 10, 4754 RSC.
- K. S. Udupa, G. S. Subramanian and H. V. K. Udupa, The electrolytic reduction of carbon dioxide to formic acid, Electrochim. Acta, 1971, 16, 1593–1598 CrossRef CAS.
- S. Shunsuke, M. Ichikawa and K. Tamaru, Electrocatalysis by metal phthalocyanines in the reduction of carbon dioxide, J. Chem. Soc., Chem. Commun., 1974, 158–159 Search PubMed.
- C. M. Lieber and N. S. Lewis, Catalytic reduction of C02 at carbon electrodes modified with cobalt phthalocyanine, J. Am. Chem. Soc., 1984, 106, 5034–5035 CrossRef.
- K. Takahashi, K. Hiratsuka, H. Sasaki and S. Toshima, Electrocatalytic behavior of metal porphyrins in the reduction of carbon dioxide, Chem. Lett., 1979, 8, 305–308 CrossRef.
- M. Zhu, J. Chen, L. Huang, R. Ye, J. Xu and Y.-F. Han, Covalently grafting cobalt porphyrin onto carbon nanotubes for efficient CO2 electroreduction, Angew. Chem., Int. Ed., 2019, 58, 6595–6599 CrossRef CAS PubMed.
- H. Kim, D. Shin, W. Yang, D. H. Won, H.-S. Oh, M. W. Chung, D. Jeong, S. H. Kim, K. H. Chae, J. Y. Ryu, J. Lee, S. J. Cho, J. Seo, H. Kim and C. H. Choi, Identification of single-atom Ni site active toward electrochemical CO2 conversion to CO, J. Am. Chem. Soc., 2021, 143, 925–933 CrossRef CAS PubMed.
- E. Boutin, M. Wang, J. C. Lin, M. Mesnage, D. Mendoza, B. Lassalle-Kaiser, C. Hahn, T. F. Jaramillo and M. Robert, Aqueous electrochemical reduction of carbon dioxide and carbon monoxide into methanol with cobalt phthalocyanine, Angew. Chem., Int. Ed., 2019, 58, 16172–16176 CrossRef CAS PubMed.
- C. Wang, X. Chen, H. Pan, D. Qi and J. Jiang, Towards developing efficient aminopyridine-based electrochemical catalysts for CO2 reduction. A density functional theory study, J. Catal., 2019, 373, 75–80 CrossRef CAS.
- C. Wang, X.-M. Liu, M. Zhang, Y. Geng, L. Zhao, Y.-G. Li and Z.-M. Su, Two-dimensional cobaltporphyrin-based cobalt-organic framework as an efficient photocatalyst for CO2 reduction reaction: A computational study, ACS Sustainable Chem. Eng., 2019, 7, 14102–14110 CrossRef CAS.
- L. Yu, F. Li, J. Huang, B. G. Sumpter, W. E. Mustain and Z. Chen, Double-atom catalysts featuring inverse sandwich structure for CO2 reduction reaction: A synergetic first-principles and machine learning investigation, ACS Catal., 2023, 13, 9616–9628 CrossRef CAS.
- Y. Zhang, S. Chen, Y. Zhang, R. Li, B. Zhao and T. Peng, Hydrogen-bond regulation of the microenvironment of Ni(II)-porphyrin bifunctional electrocatalysts for efficient overall water splitting, Adv. Mater., 2023, 35, 2210727 CrossRef CAS PubMed.
- J. Zhao and Z. Chen, Single Mo atom supported on defective boron nitride monolayer as an efficient electrocatalyst for nitrogen fixation: A computational study, J. Am. Chem. Soc., 2017, 139, 12480–12487 CrossRef CAS PubMed.
- N. Wang, L. Feng, Y. Shang, J. Zhao, Q. Cai and P. Jin, Two-dimensional iron-tetracyanoquinodimethane (Fe-TCNQ) monolayer: an efficient electrocatalyst for the oxygen reduction reaction, RSC Adv., 2016, 6, 72952 RSC.
- S. Cao, S. Wei, X. Wei, S. Zhou, H. Chen, Y. Hu, Z. Wang, S. Liu, W. Guo and X. Lu, Can N, S cocoordination promote single atom catalyst performance in CO2RR? Fe-N2S2 porphyrin versus Fe-N4 porphyrin, Small, 2021, 17, 2100949 CrossRef CAS PubMed.
- X. Chen and Y. Bu, Cation-modulated electron-transfer channel: H-atom transfer vs proton-coupled electron transfer with a variable electron-transfer channel in acylamide units, J. Am. Chem. Soc., 2007, 129, 9713–9720 CrossRef CAS PubMed.
- H. Li, L. Yang, Z. Wang, P. Jin, J. Zhao and Z. Chen, N-heterocyclic carbene as a promising metal-free electrocatalyst with high efficiency for nitrogen reduction to ammonia, J. Energy Chem., 2020, 46, 78–86 CrossRef.
- J. Wang, M. Zheng, X. Zhao and W. Fan, Structure-performance descriptors and the role of the axial oxygen atom on M-N4-C single-atom catalysts for electrochemical CO2 reduction, ACS Catal., 2022, 12, 5441–5454 CrossRef CAS.
- J. L. Hitt, Y. C. Li, S. Tao, Z. Yan, Y. Gao, S. J. L. Billinge and T. E. Mallouk, A high throughput optical method for studying compositional effects in electrocatalysts for CO2 reduction, Nat. Commun., 2021, 12, 1114 CrossRef CAS PubMed.
- C. Granier and R. Guilard, Acidity constants, dioxygen affinity, UV-Visible, ESR, and electrochemical studies of some cobalt(II) (N- or C-monosubstituted) tetraazamacrocycles complexes: development of an electrochemical dioxygen generator, Microchem. J., 1996, 53, 109 CrossRef CAS.
- Y. Matano, T. Nakabuchi, S. Fujishige, H. Nakano and H. Imahori, Redox-coupled complexation of 23-Phospha-21-thiaporphyrin with Group 10 Metals: a convenient access to stable core-Modified isophlorin-metal complexes, J. Am. Chem. Soc., 2008, 130, 16446–16447 CrossRef CAS PubMed.
- L. Lin, Y. Ni, L. Shang, H. Sun, Q. Zhang, W. Zhang, Z. Yan, Q. Zhao and J. Chen, Atomic-level modulation-induced electron redistribution in Co coordination polymers elucidates the oxygen reduction mechanism, ACS Catal., 2022, 12, 7531–7540 CrossRef CAS.
- L. Zhang, X. Guo, S. Zhang and S. Huang, Building up the “Genome” of bi-atom catalysts toward efficient HER/OER/ORR, J. Mater. Chem. A, 2022, 10, 11600–11612 RSC.
- J. S. Derrick, M. Loipersberger, S. K. Nistanaki, A. V. Rothweiler, M. Head-Gordon, E. M. Nichols and C. J. Chang, Templating bicarbonate in the second coordination sphere enhances electrochemical CO2 reduction catalyzed by iron porphyrins, J. Am. Chem. Soc., 2022, 144, 11656–11663 CrossRef CAS PubMed.
- S. Manzetti, T. Lu, H. Behzadi, M. D. Estrafili, H.-L. T. Le and H. Vach, Intriguing properties of unusual silicon nanocrystals, RSC Adv., 2015, 5, 78192–78208 RSC.
- Y. Zhang, B. Liu and M.-H. Zeng, Theoretical insights on “Stable Triheteroarylmethyl Radical”: Nature, electronic structure, and semiconductor property, Chem. Phys. Lett., 2020, 759, 138046 CrossRef CAS.
- Q. Liu, B. Xiao, J.-B. Cheng, Y.-C. Li, Q.-Z. Li, W.-Z. Li, X.-F. Xu and X.-F. Yu, Carbon excess C3N: a potential candidate as Li-ion battery material, ACS Appl. Mater. Interfaces, 2018, 10, 37135–37141 CrossRef CAS PubMed.
- X. Liu, X. Yang, X. Ding, H. Wang, W. Cao, Y. Jin, B. Yu and J. Jiang, Covalent organic frameworks with imine proton acceptors for efficient photocatalytic H2 production, Chin. Chem. Lett., 2023, 34, 1081448 Search PubMed.
- S. Fernández, F. Franco, C. Casadevall, V. Martin-Diaconescu, J. M. Luis and J. Lloret-Fillol, A unified electro- and photocatalytic CO2 to CO reduction mechanism with aminopyridine cobalt complexes, J. Am. Chem. Soc., 2020, 142, 120–133 CrossRef PubMed.
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Density functional theory in surface chemistry and catalysis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, Electrochemical CO2 reduction: classifying Cu facets, ACS Catal., 2019, 9, 7894–7899 CrossRef CAS.
- F. Chang, Y. Liu, J. Wei, L. Yang and Z. Bai, In situ surface/interface generation on Cu2O nanostructures toward enhanced electrocatalytic CO2 reduction to ethylene using operando spectroscopy, Inorg. Chem. Front., 2023, 10, 240–249 RSC.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, A scalable general synthetic approach toward ultrathin imine-Linked two-dimensional covalent organic framework nanosheets for photocatalytic CO2 reduction, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- B. Yu, T. Meng, X. Ding, X. Liu, H. Wang, B. Chen, T. Zheng, W. Li, Q. Zeng and J. Jiang, Hydrogen-bonded organic Framework ultrathin nanosheets for efficient visible-light photocatalytic CO2 reduction, Angew. Chem., Int. Ed., 2022, 61, e202211482 CrossRef CAS PubMed.
- L. Gong, B. Chen, Y. Gao, B. Yu, Y. Wang, B. Han, C. Lin, Y. Bian, D. Qi and J. Jiang, Covalent organic frameworks based on tetraphenyl-p-phenylenediamine and metalloporphyrin for electrochemical conversion of CO2 to CO, Inorg. Chem. Front., 2022, 9, 3217–3223 RSC.
- L. Xu, F. Zhang, X. Song, Z. Yin and Y. Bu, Construction of reduced graphene oxide supported Ag-Cu2O composites with hierarchical structures for enhanced photocatalytic activities and recyclability, J. Mater. Chem. A, 2015, 3, 5923 RSC.
- B. Xiao and S. Watanabe, Oxygen vacancy effects on an amorphous-TaOx-based resistance switch: a first principles study, Nanoscale, 2014, 6, 10169 RSC.
- Z. Zhou, F. Yu, Y. You, J. Zhan and L.-H. Zhang, Tunable functional groups on MXene regulating the catalytic property of anchored cobalt phthalocyanine for electrochemical CO2 reduction, Inorg. Chem. Front., 2023, 10, 5371 RSC.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Gas diffusion electrodes, reactor designs and key metrics of low-temperature CO2 electrolysers, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational detail and additional figures and tables. See DOI: https://doi.org/10.1039/d3qi01643a |
| This journal is © the Partner Organisations 2023 |