
Synthesis and reactivity of a uranium(IV) complex supported by a monoanionic nitrogen–phosphorus ligand†
Kai
Li
,
Jialu
He
,
Yue
Zhao
 and
Congqing
Zhu
*
and
Congqing
Zhu
*
State Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: zcq@nju.edu.cn
First published on 17th August 2023
Abstract
A monoanionic nitrogen–phosphorus ligand (CH3)2NCH2CH2NHPiPr2 (L3) was designed and the corresponding U(IV) chloride complex {[(CH3)2NCH2CH2NPiPr2]2UCl2} (1) and U(IV) iodide complex {[(CH3)2NCH2CH2NPiPr2]2UI2} (2) were readily synthesized. Complexes 1 and 2 were fully characterized and the reactivity of complex 1 was further investigated. Complex 3 {[(CH3)2NCH2CH2NPiPr2]2U(C12H8)} with a uranium cyclopentadiene unit was constructed by the reaction of 1 with 2,2′-dilithiobiphenyl, which is a rare example of a homoleptic metallafluorene containing an actinide element. In addition, a U(IV) bi-alkyl complex {[(CH3)2NCH2CH2NPiPr2]2U[o-N(CH3)(CH2)C6H4CH2]} (4) was isolated by the reaction of complex 1 with two equivalents of o-N(CH3)2C6H4CH2K. Moreover, the reactivity of 1 with a series of transition metal precursors was also investigated, from which heterometallic clusters {[(CH3)2NCH2CH2NPiPr2]2UCl2(μ-Cl)RuCl}2 (5), {[(CH3)2NCH2CH2NPiPr2]2UCl2Rh(μ-Cl)}2 (6) and {[(CH3)2NCH2CH2NPiPr2]2UCl2Ir(μ-Cl)}2 (7) were isolated. The U–Rh and U–Ir single bonds were observed in complexes 6 and 7, respectively. This study further confirms that the nitrogen–phosphorus ligand is an effective platform for the construction of species with U–M bonds.
Introduction
Ligands are important in organometallic chemistry because of their significant influence on the structure and properties of organometallic complexes.1,2 The synthesis of ferrocene supported by the cyclopentadienyl (Cp) ligand was considered to be a starting point of modern organometallic chemistry.3 The first example of a uranium organometallic complex, Cp3UCl, was also stabilized by the Cp ligand.4 Based on hard–soft acid–base theory, the N and O atoms are good donors to actinide elements. Consequently, a series of new ligands with N and O coordination sites were designed and used in the development of actinide organometallic chemistry.5–18 For instance, since the first example of an actinide complex supported by a triamidoamine ([(RNCH2CH2)3N]3−) ligand (Tren) was reported by Scott and co-workers in 1994,19 actinide organometallic chemistry with Tren ligands has flourished.18,20–26 With these trianionic ligands, a series of trivalent uranium complexes were isolated.24,25,27,28In 2019, we found that a novel phosphine-substituted Tren ligand (L1, Fig. 1), namely a double-layer N–P ligand, can also be used to stabilize uranium(III) complexes and construct a series of heterometallic clusters with multiple U–M single or triple bonds.29–31 In a subsequent study, we found that the dianionic double-layer N–P ligand (L2, Fig. 1) is an effective platform for the construction of heterometallic clusters with multiple U–M single or double bonds and could be used to synthesize neutral uranium(II) species.32–34 In 2022, Layfield and co-workers reported the first example of a uranium(I) complex supported by a monoanionic ligand, η5-C5iPr5.35 Very recently, Mazzanti and co-workers found that the –OSiPh3 ligand could be used to stabilize uranium(I) synthons.36 These U(I) complexes are ionic-type species supported by multiple monoanionic ligands. Therefore, we are curious if a neutral U(I) species could be stabilized by a monoanionic ligand with additional donor coordination sites. Based on our continuing interest in uranium chemistry supported by N–P ligands,37–43 herein, we have designed a new monoanionic double-layer N–P ligand with a pendant dimethylamino group as an additional coordination site (L3, Fig. 1). However, an unanticipated uranium precursor (1) supported by two L3 ligands was isolated. The reactivity of complex 1 was investigated in this study. Interestingly, the two P atoms from two L3 ligands in complex 1 could be coordinated with the same transition metal, which is totally different from the heterometallic clusters with multiple U–M bonds supported by L1 and L2.
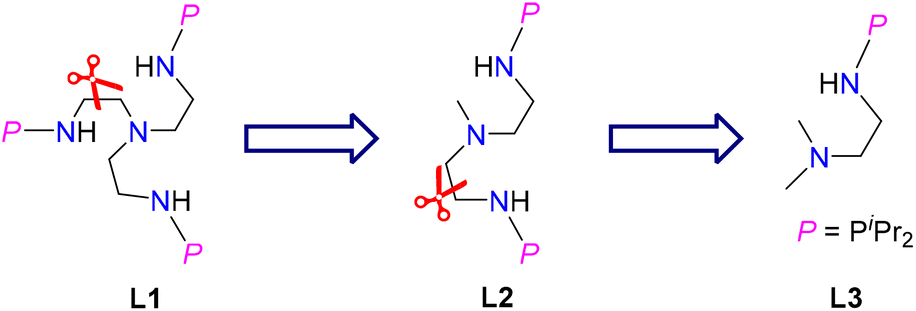 | ||
| Fig. 1 Design of the monoanionic N–P ligand L3. | ||
Results and discussion
Synthesis of U(IV) complexes 1 and 2
The monoanionic ligand L3 was prepared from the reaction of N,N-dimethylethylenediamine with one equivalent of iPr2PCl in the presence of excess Et3N (see the ESI for details†). L3 was deprotonated with nBuLi at 25 °C in THF for 3 h and then treated with UCl4 overnight at room temperature (RT) (Scheme 1), from which complex 1 was isolated as a bright-green solid in 78% yield after recrystallization from toluene at −30 °C. We found that different equivalents of L3 do not change the product in this reaction.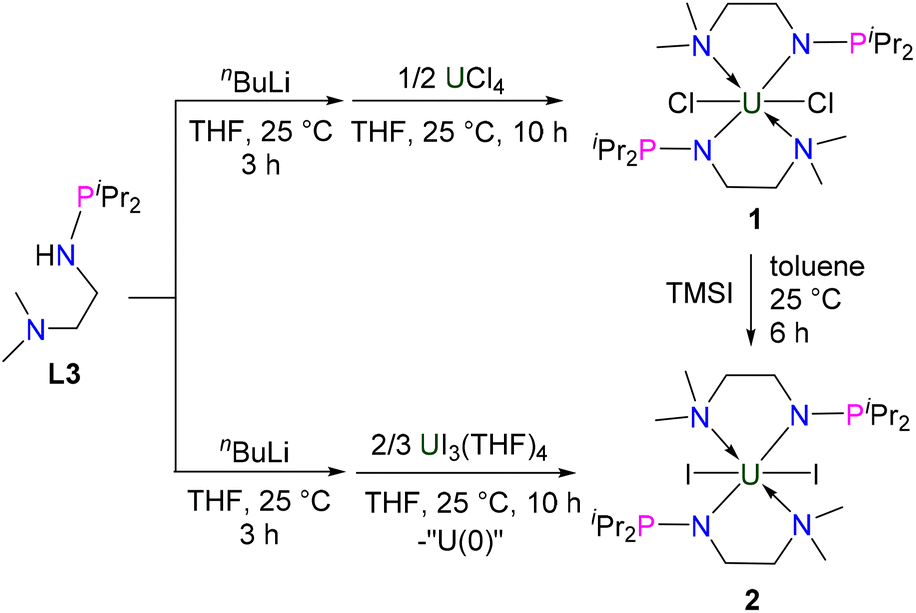 | ||
| Scheme 1 Synthesis of complexes 1 and 2. | ||
Under similar conditions, deprotonated L3 could also react with UI3(THF)4,44 leading to the formation of U(IV) complex 2 in 37% yield as a crystalline product (Scheme 1). We proposed that a U(III) complex supported by L3 was formed and disproportionated into U(IV) complex 2 and U(0) species, a phenomenon which has been observed previously.45–48 Complex 2 could also be synthesized in 91% yield by the reaction of complex 1 with TMSI. The reactions forming complexes 1 and 2 are different from the reactions leading to tri- and tetra-substituted U(IV) products containing isopropyl (iPr) and mestrimethylphenyl (Mes)-substituted monoanionic N–P ligands, iPrNHPPh2 and MesNHPiPr2, which were reported by Bart, Thomas and co-workers.49 This difference is probably due to the additional NMe2 group that could be coordinated with the U center, thus avoiding the formation of tri- and tetra-substituted U(IV) products.
The 1H NMR spectra of complexes 1 and 2 exhibit five signals between +58.0 and −74.4 ppm, consistent with reported U(IV) complexes supported by dianionic N–P ligands.32,33 The signals for N–CH3 in complexes 1 and 2 were observed at −70.5 and −74.4 ppm, respectively. No phosphorus signals were observed between +1000 and −1000 ppm, probably due to the paramagnetic nature of U(IV) species.
The molecular structure of complexes 1 and 2 was confirmed by single-crystal X-ray diffraction. As shown in Fig. 2 and 3, the uranium centre of these species is octa-coordinated with four N atoms and two P atoms from two monoanionic N–P ligands, and two halogen atoms. In complex 1, the bond lengths of U–Namine (U1–N1: 2.698(4) Å, U1–N3: 2.691(4) Å) are obviously longer than those of U–Namido (U1–N2: 2.249(4) Å, U1–N4: 2.248(4) Å), reflecting the dative bonds between the U center and N1 and N3. The bond lengths of U1–P1 (3.0042(14) Å) and U1–P2 (2.9589(14) Å) are longer than the sum of the covalent single-bond radii of U and P (2.80 Å),50 suggesting a weak coordination between the two P atoms and the U center. The structural parameters of complex 2 are comparable to those of complex 1.
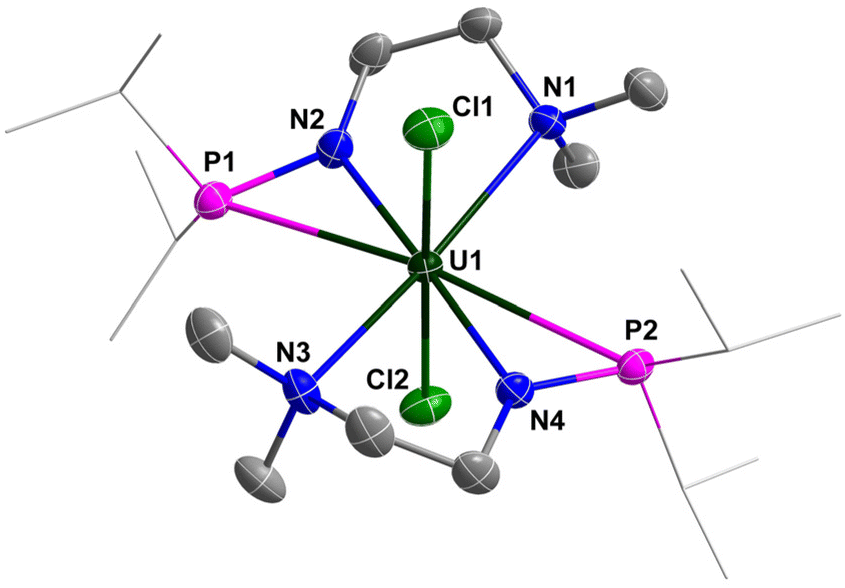 | ||
| Fig. 2 Molecular structure of 1 with thermal ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity. The iPr groups on P atoms are simplified into lines. Selected bond distances (Å): U1–N1 2.698(4), U1–N2 2.249(4), U1–N3 2.691(4), U1–N4 2.248(4), U1–P1 3.0042(14), U1–P2 2.9589(14), U1–Cl1 2.6890(13), U1–Cl2 2.6856(13). | ||
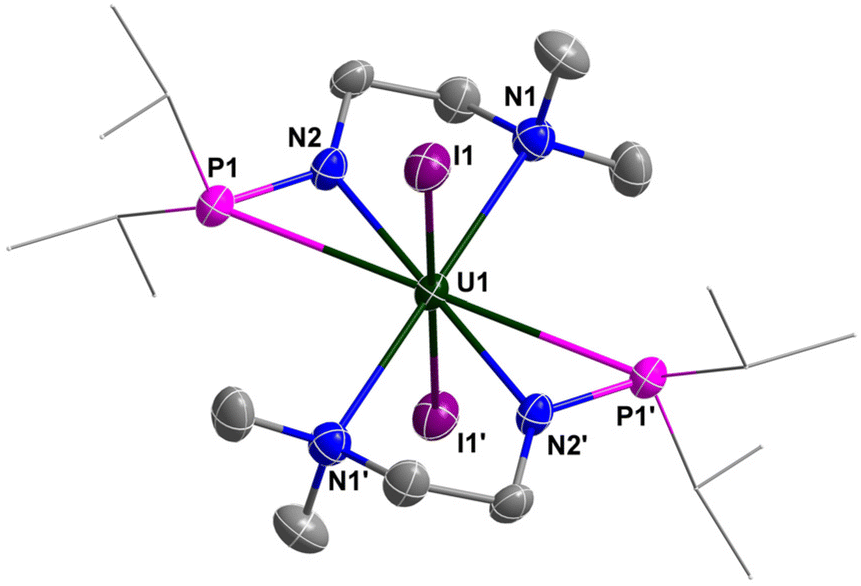 | ||
| Fig. 3 Molecular structure of 2 with thermal ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity. The iPr groups on P atoms are simplified into lines. Selected bond distances (Å): U1–N1 2.737(4), U1–N2 2.237(4), U1–P1 2.9694(11), U1–I1 3.1109(3). | ||
Synthesis of uranium bi-aryl complex 3 and uranium bi-alkyl complex 4
Compared with uranium alkyl complexes, uranium aryl complexes remain extremely rare.51–54 The first example of a homoleptic uranium aryl complex was reported by Arnold in 2016.55 Reported uranium aryl complexes feature one or more mono-aryl units, whereas uranium bi-aryl complexes have not been reported. Heterofluorenes, in which the CH2 unit in fluorene was replaced by a heteroatom, have been reported for several decades. Heteroatoms in the heterofluorenes could be main group elements,56,57 transition metals,58,59 and rare-earth metals.60,61 With complex 1 in hand, we attempted to synthesize a uranium-containing metallafluorene species via salt metathesis. Treatment of complex 1 with one equivalent of 2,2′-dilithiobiphenyl62 in THF at RT overnight afforded a brown turbid solution, from which complex 3 was isolated in 76% yield as a yellow crystalline solid (Scheme 2). The 1H NMR spectrum of complex 3 has fourteen peaks between +36.0 and −53.3 ppm, suggesting relatively tight binding of the dimethylamino pendant donor with the U center. The 31P{1H} NMR spectrum of complex 3 showed a single peak at 540.0 ppm, which suggested that the two P atoms are equivalent in solution. | ||
| Scheme 2 Synthesis of uranium bi-aryl complex 3. | ||
The metallafluorene unit in complex 3 was confirmed by X-ray diffraction (Fig. 4). The two six-membered rings and the five-membered ring of the uranium-containing fluorene unit are co-planar. The bond lengths of U1–C1 (2.50(2) Å) and U1–C2 (2.482(17) Å) are shorter than the U–Caryl bond lengths in [Li]2[U-(2,3-C6H3CH2NMe2)2(2-C6H4CH2NMe2)2] (2.609(4) Å),51 and [Li][U(2,3-C6H3CH2NMe2)(2-C6H4CH2NMe2)3] (2.604(4), 2.650(7), 2.615(8) Å),53 but are comparable to U–Caryl bond lengths in [Li(Et2O)3][UCl2(C6Cl5)3] (2.497(13), 2.504(13), 2.505(14) Å).52 They are also close to those of the U–Calkyl single bond in [Li(THF)4][U(CH2tBu)5] (2.47(1)–2.51(1) Å),63 [Li(THF)4][Li(THF)2U(CH3)6] (2.500(5)–2.615(5) Å),64 UCp*(TMTAA)(CH2TMTS) (TMTAA = tetramethyl-tetra-aza-annulene) (2.48(1) Å),65 [fc(NSitBuMe2)2U(CH2Ph)(OEt2)][BPh4] (2.482(12) Å),47 and [η5-1,2,4-(Me3C)3C5H2]2U(η2-C4Ph2) (2.448(5)–2.475(5) Å).66 The bond angle of C1–U1–C2 is 69.1(7)°, which is close to the C–Ln–C angles in [Li(DME)3][(C5Me5)2Ln(biphen)] (Ln = Ce, 69.0(7)°; Ln = La, 68.55(8)°)60 but is smaller than the C–M–C angles in other reported metallafluorenes (74°–130°).56–61,67,68 The bond lengths of U–Namine (average of 2.735 Å) and U–Namido (average of 2.316 Å) are longer than those observed in complex 1 (2.695 and 2.249 Å, respectively), which is probably due to the steric hindrance of the biphenyl group. Complex 3 represents a new example of a heterofluorene containing an actinide element.
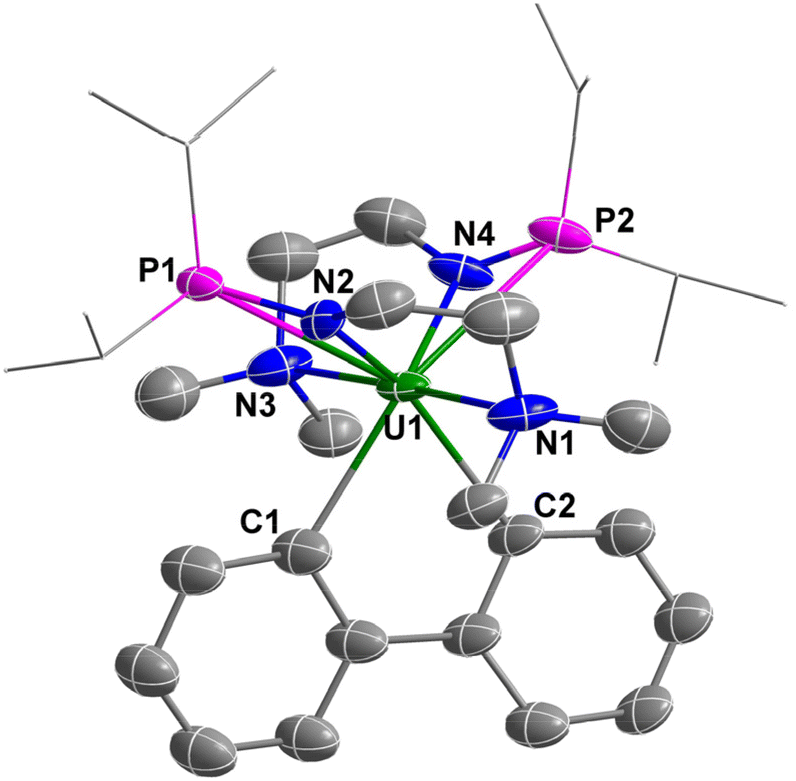 | ||
| Fig. 4 Molecular structure of 3 with thermal ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity. The iPr groups on P atoms are simplified into lines. Selected bond distances (Å): U1–N1 2.73(2), U1–N2 2.260(17), U1–N3 2.739(18), U1–N4 2.37(2), U1–P1 3.15(3), U1–P2 3.226(12), U1–C1 2.50(2), U1–C2 2.482(17). | ||
We further investigated the reactivity of complex 1 with o-NMe2C6H4CH2K via a salt metathesis reaction. Treatment of complex 1 with two equivalents of o-NMe2C6H4CH2K in THF afforded orange crystalline complex 4 in 68% yield (Scheme 3). The 1H NMR spectrum of complex 4 has twenty-nine peaks between +119.3 and −125.1 ppm, which suggested that complex 4 has a low-symmetric structure. The 31P{1H} NMR spectrum of complex 4 was not observed between +1000 and −1000 ppm.
 | ||
| Scheme 3 Synthesis of uranium bi-alkyl complex 4. | ||
The molecular structure of complex 4 was determined by single-crystal X-ray diffraction, which was revealed to be a unique example of a uranium bi-alkyl metallacycle stabilized by an N-donor (N5) (Fig. 5). The lengths of U–C bonds are 2.461(4) and 2.553(4) Å, which are within the reported range for U–C single bonds (2.29–2.78 Å).63–66,69–75 The bond lengths of U–Namine (U1–N1 2.734(4) Å, U1–N3 2.813(4) Å, U1–N5 2.568(3) Å) were obviously longer than those of U–Namido (U1–N2 2.297(3) Å, U1–N4 2.285(4) Å), reflecting dative bonding of N1, N3 and N5 to the U center. The bond distances of U1–P1 (3.2145(11) Å) and U1–P2 (3.1286(11) Å) are longer than the sum of the covalent single-bond radii of U and P (2.80 Å), suggesting weak coordination between the two P atoms and the U center.
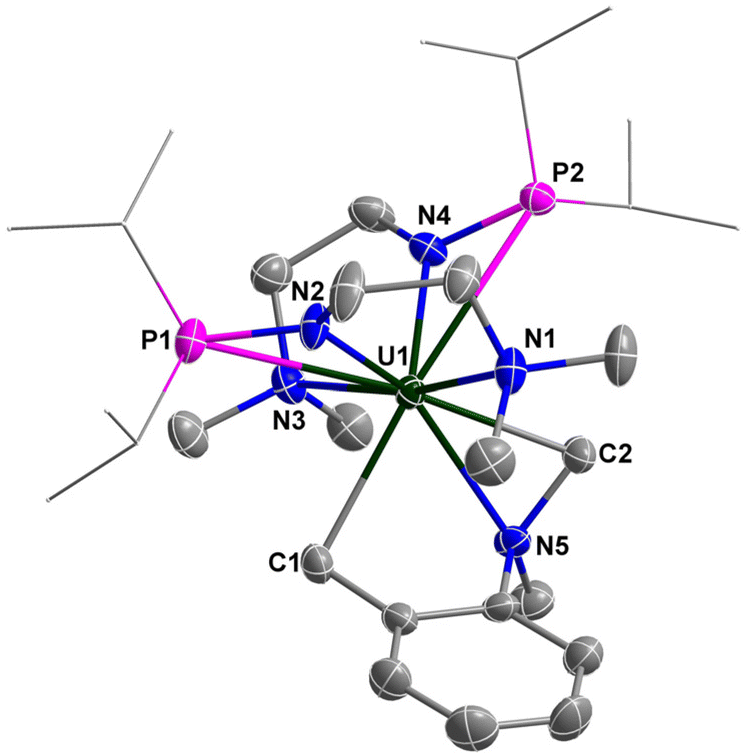 | ||
| Fig. 5 Molecular structure of 4 with thermal ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity. The iPr groups on P atoms are simplified into lines. Selected bond distances (Å): U1–N1 2.734(4), U1–N2 2.297(3), U1–N3 2.813(4), U1–N4 2.285(4), U1–N5 2.568(3), U1–P1 3.2145(11), U1–P2 3.1286(11), U1–C1 2.553(4), U1–C2 2.461(4). | ||
The sp3 C–H bond in the dimethylamino moiety of 4 is activated by the uranium centre. This phenomenon was observed previously in actinide organometallic chemistry,76–78 such as for the formation of uranium hexamethyldisilazide metallaycles.79,80 Inspired by the mechanism of generating {[N(CH2CH2NSiMe2tBu)]2U[η2-(CH2CH2NSitBu(CH3)(CH2))]},81 two possible routes were proposed for the formation of complex 4 (Scheme 3). Firstly, complex 1 reacts with two equivalents of o-NMe2C6H4CH2K to produce intermediate A with the release of KCl. Then, the “U-CH2” fragment in intermediate B coordinates with the C–H bond in the “NMe2” unit, allowing the σ-bond metathesis to contribute to the production of 4 with the release of ortho-dimethylaminotoluene (o-NMe2C6H5CH3). On the other hand, complex 1 could react with one equivalent of o-NMe2C6H4CH2K to produce intermediate C, which could further react with another equivalent of o-NMe2C6H4CH2K to produce D with the release of o-NMe2C6H5CH3. Finally, complex 4 was formed by releasing KCl from intermediate D.
The UV-Vis-NIR electronic absorption spectra of 1–4 were measured in THF at RT (Fig. 6). Complexes 1 and 4 showed intense absorption peaks at 216 and 220 nm, respectively. Complexes 2 and 3 showed two absorption peaks at 217 and 233 nm and 214 and 247 nm, respectively. In addition, a weak absorption peak at 420 nm in the visible region was observed for complex 1. A set of low intensity absorptions were observed in the NIR region for these complexes. For instance, the peaks at 1112 and 1310 nm were found for complex 1, 1076 and 1305 nm for complex 2, 1037 and 1294 nm for complex 3, and 1114 and 1300 nm for complex 4. These low-intensity absorptions (ε < 150 M−1 cm−1) for complexes 1–4 in the NIR region were attributed to the 5f–5f transitions, which are typical features of U(IV) species.
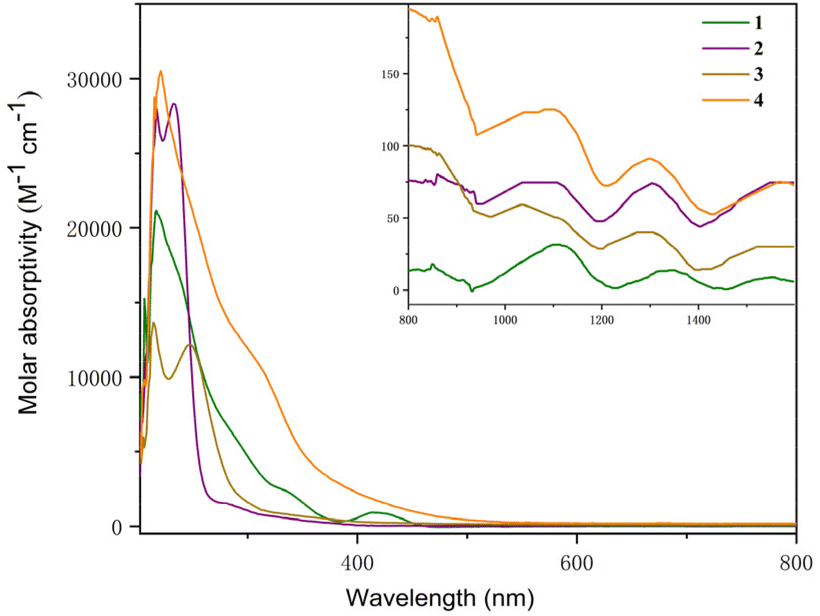 | ||
| Fig. 6 UV-visible absorption spectra of complexes 1–4 measured in THF at RT (inset: near-infrared absorption spectra). | ||
Synthesis of heterometallic clusters 5–7
Clusters with U–M bonds involving transition metals or even main-group metal elements have flourished in recent decades.82–94 Inspired by our previous isolation of heterometallic clusters with the U–TM bond supported by N–P ligands,37 we attempted to synthesize heterometallic clusters from complex 1. Treatment of 1 with one equivalent of RuCl2(PPh3)3 at RT in THF afforded a red-brown solution, from which a crystalline complex 5 was isolated in 42% yield (Scheme 4). Complex 5 is an example of a chlorine-bridged multimetallic cluster with U and Ru. The formal oxidation states of U and Ru in complex 5 are +4 and +2, respectively. The NMR characterization of complex 5 was hindered by the poor solubility of its crystalline species. Attempts to synthesize a heterometallic cluster with U–Ru bonds by the reduction of complex 5 with Na, Li, K or KC8 were unsuccessful.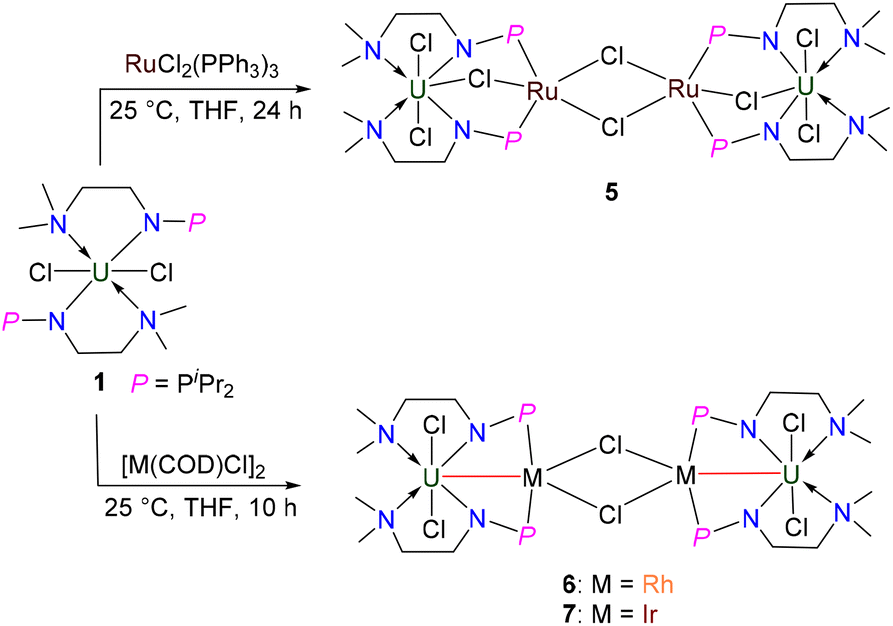 | ||
| Scheme 4 Synthesis of heterometallic clusters 5–7. | ||
The solid-state structure of 5 was confirmed by X-ray crystallographic analysis (Fig. 7). The centrosymmetric structure contains a heterometallic U–Ru–Ru–U core with four Cl atom substituents. The bond length of U–Cl3 in the U–Cl–Ru moiety (2.723(2) Å) is longer than the terminal U1–Cl1 (2.707(2) Å) and U1–Cl2 (2.674(3) Å) bond lengths, suggesting the bridged nature of the Cl3 atom. The two P atoms (P1 and P2) are coordinated to the same Ru centre in this species, which is similar to the formation of the U–M bond supported by a dianionic N–P ligand {[CH2O(CH2)2NHPiPr2]2}.32 The penta-coordinated Ru centre has an approximately square pyramidal geometry with one of the P atoms at the apical position. The U–Ru distances (3.6349(8) Å) are considerably longer than the sum of the covalent single-bond radii of U and Ru (2.95 Å),50 suggesting the absence of any bonding interaction between these metal atoms.
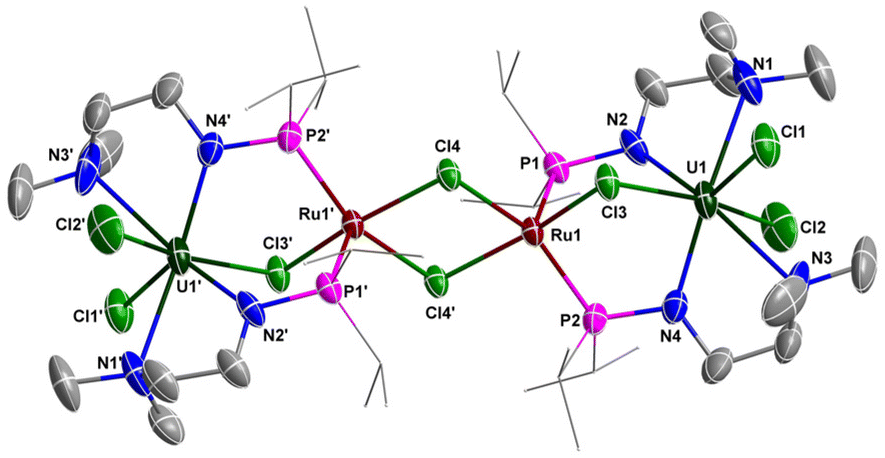 | ||
| Fig. 7 Molecular structure of 5 with thermal ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity. The iPr groups on P atoms are simplified into lines. Selected bond distances (Å): U1–N1 2.670(10), U1–N2 2.341(9), U1–N3 2.772(11), U1–N4 2.291(9), U1–Cl1 2.707(2), U1–Cl2 2.674(3), U1–Cl3 2.723(2), Ru1–Cl3 2.3769(19), Ru1–Cl4 2.3899(19), Ru1′–Cl4 2.498(2). | ||
To synthesize heterometallic clusters containing U–M bonds, we further attempted the reaction of complex 1 with [M(COD)Cl]2 (M = Rh, Ir). Treatment of 1 with 0.5 equivalents of [Rh(COD)Cl]2 at RT in THF for 10 h afforded complex 6, which was isolated as a yellow crystalline solid in 57% yield (Scheme 4). Under the same conditions, complex 7 was synthesized by the reaction of complex 1 with 0.5 equivalents of [Ir(COD)Cl]2, and was isolated as a yellow crystalline solid in 45% yield after storing at −30 °C (Scheme 4). Complexes 6 and 7 exhibited extremely low solubility after crystallization and no signals could be observed in the 1H NMR spectra of these clusters even when different deuterated solvents were used.
The molecular structures of 6 and 7 in the solid state were determined by single-crystal X-ray diffraction (Fig. 8 and 9). The crystal systems and space groups of 6 and 7 are monoclinic P21/n. The coordination environment of the U centers in 6 and 7 is very similar, and comprises a hepta-coordinated geometry with one Rh/Ir atom, two Cl atoms, and four N atoms. The Rh and Ir atoms in 6 and 7 adopt almost the same coordination geometry with one U atom, two P atoms, and two Cl atoms. The most remarkable feature of these complexes is the two U–Rh/Ir bonds bridged by two Cl atoms. In complex 6, the U–Rh bond length is 2.7910(6) Å, which is obviously shorter than the sum of the covalent single-bond radii of U and Rh (2.95 Å).50 The U–Rh bond length in 6 is longer than the U–Rh single-bond lengths reported in [UIVI2(μ-OArP-1κ1O,2κ1P)2RhI(μ-I)]2 (ArPO− = 2-(diphenylphosphino)-6-tert-butyl-4-methylphenoxide) (2.7601(5) Å), UIVI(μ-I)(μ-OArP-1κ1O,2κ1P)3RhI (2.7630(5) Å)95 and U[N(CH3)(CH2CH2NPiPr2)2](μ-Me)2Rh2(μ-Me)4Mg(C4H8O) (2.6612(5) Å).34 The U–Ir bond length (2.8656(3) Å) is shorter than the sum of the covalent single-bond radii of U and Ir (2.92 Å), but longer than the U–Ir single-bond length in complex U[N(CH3)(CH2CH2NPiPr2)2] [(μ-Me)2Ir2(μ-Me)4Mg(C4H8O)] (2.6968(4) Å).34 Nevertheless, the U–Rh/Ir bond lengths observed in 6 and 7 are consistent with the presence of a U–M bonding interaction in these clusters.
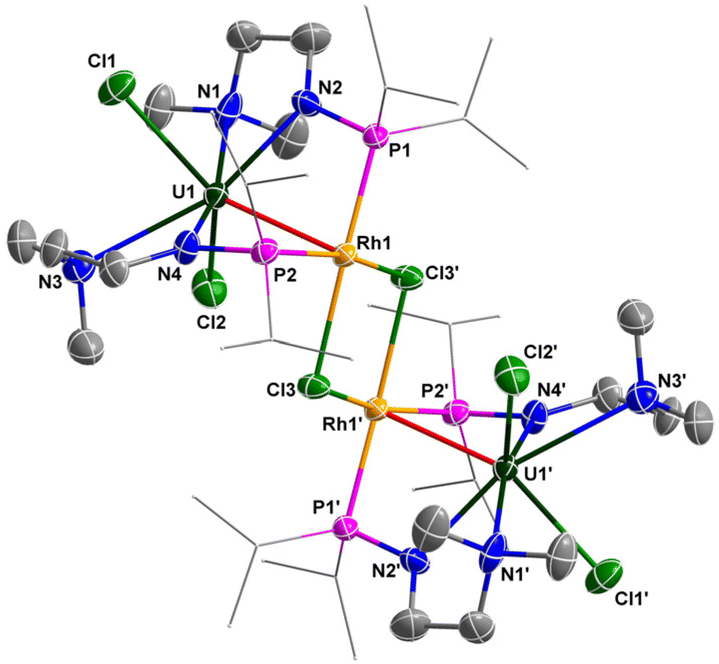 | ||
| Fig. 8 Molecular structure of 6 with thermal ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity. The iPr groups on P atoms are simplified into lines. Selected bond distances (Å): U1–N1 2.700(8), U1–N2 2.348(7), U1–N3 2.793(8), U1–N4 2.298(7), U1–Cl1 2.651(2), U1–Cl2 2.696(2), U1–Rh1 2.7910(6), Rh1–Cl3 2.4487(19), Rh1–Cl3′ 2.473(2). | ||
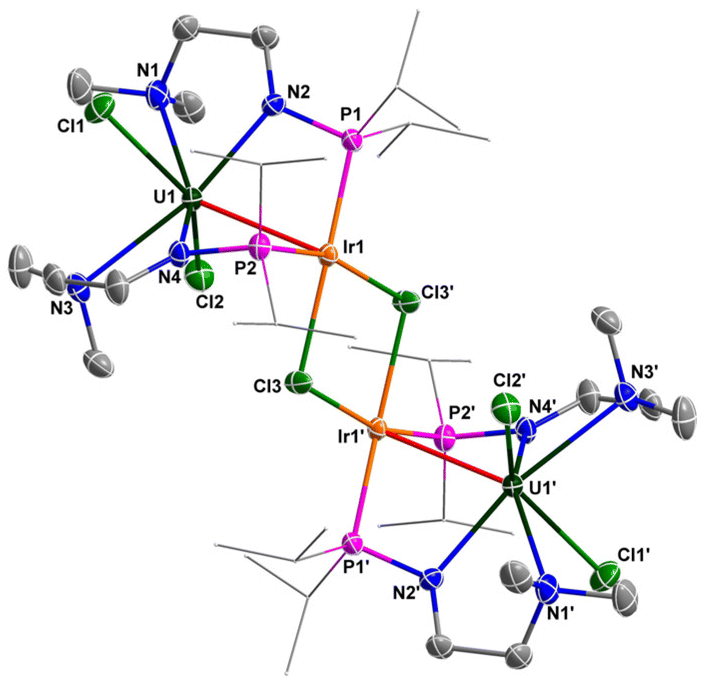 | ||
| Fig. 9 Molecular structure of 7 with thermal ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity. The iPr groups on P atoms are simplified into lines. Selected bond distances (Å): U1–N1 2.715(4), U1–N2 2.293(4), U1–N3 2.763(5), U1–N4 2.329(4), U1–Cl1 2.7378(14), U1–Cl2 2.6419(13), U1–Ir1 2.8656(3), Ir1–Cl3 2.4019(12), Ir1–Cl3′ 2.4377(12). | ||
The UV-Vis-NIR electronic absorption spectra of 5–7 were measured in THF at RT (Fig. 10). Complex 5 exhibited an intense peak at 223 nm and a weak peak at 313 nm in the UV-Vis region. Two peaks at 215 and 326 nm were observed for complex 6 and one intense peak at 219 nm was observed for complex 7. In the NIR region, complexes 5, 6 and 7 exhibited similar absorption peaks at 1070 and 1286 nm, 1072 and 1293 nm, and 1067 and 1294 nm, respectively. These weak absorptions (ε < 100 M−1 cm−1) were attributed to the f–f transitions expected for U(IV) complexes.
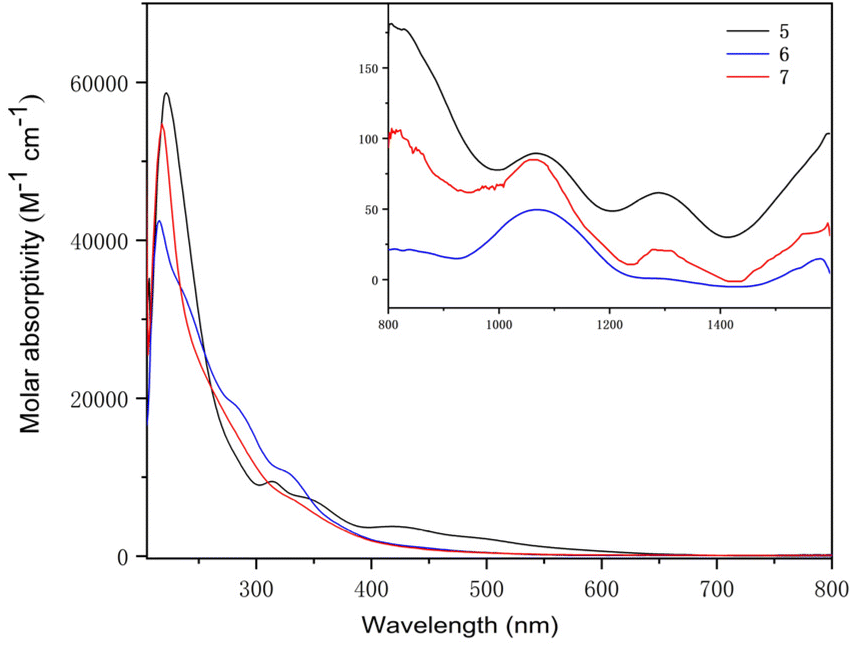 | ||
| Fig. 10 UV-visible absorption spectra of heterometallic clusters 5–7 measured in THF at RT (inset: near-infrared absorption spectra). | ||
Variable-temperature magnetic data of 5–7 in the solid state were collected using a superconducting quantum interference device (SQUID) (Fig. 11). The effective moments of 5, 6 and 7 at 300 K are 4.38, 4.77 and 4.54 μB per molecule, respectively, which are lower than the theoretical value (5.06 μB) for two independent 5f2 U(IV) ions. This phenomenon was probably due to the quenching of spin–orbit coupling and was observed for the reported U(IV) complexes.96 The magnetic moments of 6 and 7 decline slowly from 300 K to 50 K, and then drop sharply to 0.74 μB and 0.76 μB at 1.8 K with a trend toward zero. However, the magnetic moment of 5 declines much more obviously and starts to decline from 300 K persistently. At 1.8 K, the magnetic moment of 5 is 0.39 μB per molecule. The magnitude of μeff and temperature dependence of complexes 5, 6 and 7 are comparable to those observed for the reported U(IV) complexes.
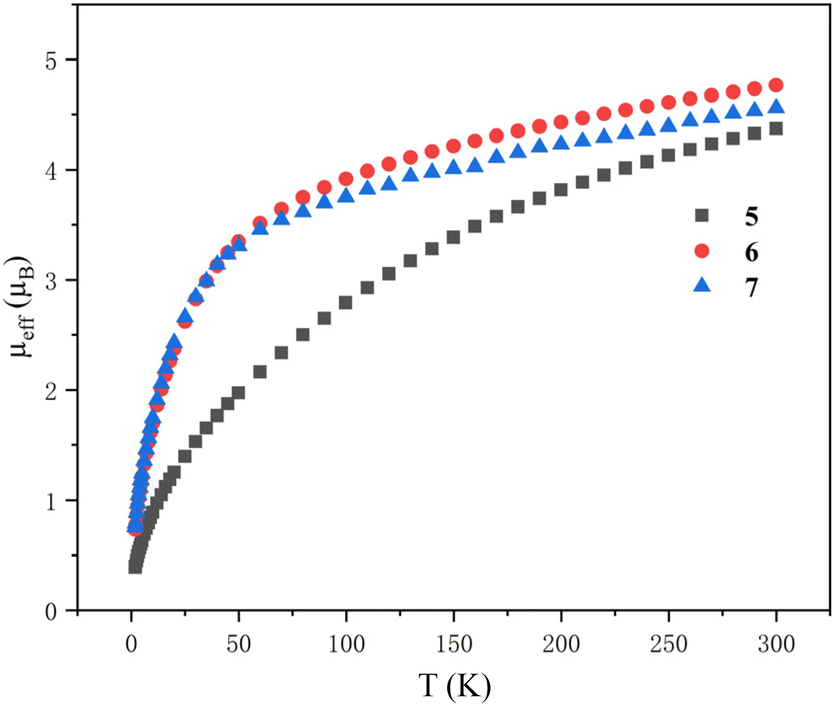 | ||
| Fig. 11 Variable-temperature effective magnetic moment data. Magnetic moment per molecule for clusters 5–7. | ||
Conclusions
A novel monoanionic N–P ligand L3 and the corresponding U(IV) complexes 1 and 2 were synthesized. An actinide-containing metallafluorene 3 was synthesized by the reaction of complex 1 and 2,2′-dilithiobiphenyl by a salt metathesis method. Using a similar strategy, U(IV) bi-alkyl complex 4 was isolated by the reaction of complex 1 with o-NMe2C6H4CH2K. The monoanionic N–P ligand L3 also proved to be an effective platform for the construction of heterometallic clusters with U–M bonds. For example, the heterometallic clusters 6 and 7 containing U–Rh or U–Ir bonds were synthesized by the reaction of complex 1 with [M(COD)Cl]2 (M = Rh, Ir). Further studies on the reactivity of the U(IV) bi-aryl complex 3, the bi-alkyl complex 4 and the heterometallic clusters 5–7 are in progress.Experimental section
All manipulations were performed under an N2 atmosphere using standard Schlenk techniques or in an N2 glovebox (<1 ppm O2/H2O). Solvents were dried and deoxygenated by distillation under nitrogen and further dried over 4 Å molecular sieves before use. Benzene-d6 and THF-d8 were dried over Na/K and stored under an N2 atmosphere prior to use. 2,2′-Dilithiobiphenyl,62o-NMe2C6H4CH2K,97 UCl4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 98 and UI3(THF)444 were prepared using published procedures. Other reagents were purchased and used without further purification. Nuclear magnetic resonance (NMR) spectroscopy was performed using a Bruker AVIII-400 or a Bruker AVIII-500 spectrometer at room temperature (RT). Absolute values of the coupling constants (δ) are provided in Hertz (Hz). Multiplicities of peaks are abbreviated as singlet (s), doublet (d), triplet (t), multiplet (m) or broad (br). Elemental analyses (C, H, N) were performed on a Vario MICRO elemental analyzer at the Center of Modern Analysis of Nanjing University.
98 and UI3(THF)444 were prepared using published procedures. Other reagents were purchased and used without further purification. Nuclear magnetic resonance (NMR) spectroscopy was performed using a Bruker AVIII-400 or a Bruker AVIII-500 spectrometer at room temperature (RT). Absolute values of the coupling constants (δ) are provided in Hertz (Hz). Multiplicities of peaks are abbreviated as singlet (s), doublet (d), triplet (t), multiplet (m) or broad (br). Elemental analyses (C, H, N) were performed on a Vario MICRO elemental analyzer at the Center of Modern Analysis of Nanjing University.
Synthesis of L3
A solution of iPr2PCl (4.20 g, 27.5 mmol) in THF (10 mL) was added dropwise to a solution of (CH3)2NCH2CH2NH2 (2.40 g, 27.5 mmol) and Et3N (15 mL, 110.0 mmol) in THF (20 mL), resulting in the immediate formation of a white precipitate. The mixture was stirred overnight before being dried in vacuo. The white solid was extracted with n-hexane (20 mL) and then filtered through Celite. Volatiles were removed under reduced pressure to give L3 as a colorless oil. Yield: 5.00 g (89%). 1H NMR (C6D6, 500 MHz, ppm) 2.91–2.95 (m, 2H, NCH2CH2), 2.18 (t, 3JHH = 6.1 Hz, 2H, NCH2CH2), 2.03 (s, 6H, N(CH3)2), 1.43–1.52 (m, 2H, CH(CH3)2), 0.98–1.03 (m, 12H, CH(CH3)2). 13C{1H} NMR (C6D6, 125 MHz, ppm) 61.86 (d, JPC = 6.1 Hz, NCH2CH2), 46.03 (d, JPC = 23.9 Hz, NCH2CH2), 45.07 (s, N(CH3)2), 26.42 (d, JPC = 13.0 Hz, CH(CH3)2), 19.10 (d, JPC = 21.0 Hz, CH(CH3)2), 17.29 (d, JPC = 8.2 Hz, CH(CH3)2). 31P{1H} NMR (C6D6, 202 MHz, ppm) 63.68. HRMS (ESI) calcd for C10H26N2P, [M + H]+, m/z = 205.1828. Found 205.1837.Synthesis of complex 1
Synthesis of complex 2
Synthesis of complex 3
Complex 1 (143.1 mg, 0.2 mmol) and 2,2′-dilithiobiphenyl (33.2 mg, 0.2 mmol) were added to a 5 mL flask and dissolved in THF (2 mL). The solution immediately changed from green to red-orange. The mixture was stirred at RT for 10 h and then the solvent was removed under reduced pressure and the residue was extracted with ether (3 mL). The filtrate was dried in vacuo to afford complex 3 as a pure yellow solid (121.1 mg, 76%). Crystals of 3 compatible for X-ray diffraction were grown from a saturated solution in ether (2 mL) stored at RT. 1H NMR (C6D6, 400 MHz, ppm) 36.0 (d, J = 18.7 Hz, 4H, CH2), 26.6 (d, J = 8.7 Hz, 2H, CH(CH3)2), 25.8 (d, J = 34.1 Hz, 4H, CH2), 21.3 (s, 2H, biphenyl), 17.5 (t, J = 6.5 Hz, 2H, CH(CH3)2), 15.7 (s, 2H, biphenyl), 13.0 (s, 6H, CH(CH3)2), 9.4 (s, 6H, CH(CH3)2), 8.9 (s, 6H, CH(CH3)2), 6.8 (s, 6H, CH(CH3)2), −24.5 (s, 2H, biphenyl), −26.0 (s, 6H, N(CH3)2), −43.7 (s, 2H, biphenyl), −53.3 (s, 6H, N(CH3)2). 31P{1H} NMR (C6D6, 162 MHz, ppm) 540.0. Anal. calcd (%) for C32H56N4P2U: C, 48.24; H, 7.08; N, 7.03. Found: C, 47.01; H, 6.91; N, 6.87. No satisfactory result for the C% value was obtained despite repeated attempts, which is probably due to the inadequate combustion of this air- and moisture-sensitive species.Synthesis of complex 4
Complex 1 (71.5 mg, 0.1 mmol) and o-NMe2C6H4CH2K (34.7 mg, 0.2 mmol) were added to a 5 mL flask and dissolved in THF (2 mL). The solution immediately changed from green to orange. The mixture was stirred at RT overnight. Then the solvent was removed under reduced pressure and the residue was extracted with n-hexane (3 mL). Yellow crystals of 4 were grown from a solution of ether (2 mL) at room temperature. Yield: 4 (52.9 mg, 68%). 1H NMR (d8-THF, 400 MHz, ppm) 119.3 (s, 1H), 98.4 (s, 1H), 95.2 (s, 3H), 88.7 (s, 1H), 67.4 (s, 1H), 48.5 (s, 1H), 43.5 (s, 3H), 38.9 (s, 3H), 31.4 (s, 4H), 23.2 (s, 4H), 20.8 (s, 1H), 8.2 (s, 3H), −1.3 (s, 2H), −1.9 (s, 3H), −6.9 (s, 3H), −12.5 (s, 1H), −16.8 (s, 4H), −28.4 (s, 1H), −30.7 (s, 1H), −34.5 (s, 1H), −41.1 (s, 1H), −45.8 (s, 3H), −48.0 (s, 1H), −50.8 (s, 1H), −63.2 (s, 3H), −67.4 (s, 3H), −74.1 (s, 1H), −96.2 (s, 3H), −125.1 (s, 1H). 31P{1H} NMR (C6D6, 162 MHz, ppm) was not observed in the range of +1000 to −1000 ppm. Anal. calcd (%) for C29H59N5P2U: C, 44.78; H, 7.65; N, 9.00. Found: C, 44.70; H, 7.55; N, 8.91.Synthesis of complex 5
A solution of RuCl2(PPh3)3 (95.9 mg, 0.1 mmol) in THF (2 mL) was added to a solution of complex 1 (71.5 mg, 0.1 mmol) in THF (2 mL) at RT. The mixture was left undisturbed for 1 day at RT. Then, black block crystals of 5 (74.6 mg, 42%) were obtained. NMR characterization of this species was prevented by its poor solubility after recrystallization. Anal. calcd (%) for C40H96Cl8N8P4Ru2U2·2C4H8O: C, 30.04; H, 5.88; N, 5.84. Found: C, 30.27; H, 5.65; N, 5.97.Synthesis of complex 6
A solution of [Rh(COD)Cl]2 (33.7 mg, 0.1 mmol) in THF (2 mL) was added to a solution of complex 1 (143.1 mg, 0.2 mmol) in THF (2 mL) at RT. The solution immediately changed from green to orange. The mixture was stirred at RT overnight and then the solvents were removed. The residue was extracted with dichloromethane (2 mL). Storing the filtrate at −30 °C for 18 h yielded product 6 as orange block crystals (97.3 mg, 57%). NMR characterization of this species was prevented by its poor solubility after recrystallization. Anal. calcd (%) for C40H96Cl6N8P4Rh2U2: C, 28.13; H, 5.67; N, 6.56. Found: C, 28.34; H, 5.73; N, 6.44.Synthesis of complex 7
A solution of [Ir(COD)Cl]2 (51.5 mg, 0.1 mmol) in THF (2 mL) was added to a solution of complex 1 (143.1 mg, 0.2 mmol) in THF (3 mL). The color of the solution immediately changed from green to orange. The mixture was stirred at RT overnight and then the solvents were removed. The residue was extracted with dichloromethane (2 mL). Storing the filtrate at −30 °C for 24 h yielded product 7 as orange block crystals (84.9 mg, 45%). NMR characterization of this species was prevented by its poor solubility after recrystallization. Anal. calcd (%) for C40H96Cl6N8P4Ir2U2: C, 25.47; H, 5.13; N, 5.94. Found: C, 25.76; H, 5.14; N, 5.87.Single-crystal X-ray diffraction
The crystallographic data of complexes 1, 2, 3, 4, 5, 6 and 7 were collected on Bruker D8 venture photon II detectors at 193 K or 296 K with a radiation source of Ga(Kα) (λ = 1.34139 Å) or Mo(Kα) (λ = 0.71073 Å) using the ω-scan technique. Multiscan or empirical absorption corrections (SADABS) were applied.99 The structures were solved by direct methods, expanded by difference Fourier syntheses, and refined by full-matrix least squares on F2 using Olex2.100 All non-hydrogen atoms were refined on F2 by full-matrix least-squares procedures with the use of anisotropic displacement parameters.101 Hydrogen atoms were introduced at their geometric positions and refined as riding atoms. In complex 3, the “CH2NPiPr2” unit was split into two parts with 43.3% and 56.7% occupancies. The X-ray crystal structures have been deposited in the Cambridge Crystallographic Data Centre (CCDC 2254129 (1), 2254126 (2), 2254132 (3), 2254127 (4), 2254131 (5), 2254128 (6), and 2254130 (7)†). Details of the data collection and refinement for complexes 1, 2, 3, 4, 5, 6 and 7 are given in Tables S1, S2 and S3 in the ESI.†Author contributions
Kai Li: investigation, writing – original draft. Jialu He: investigation. Yue Zhao: software. Congqing Zhu: resources, supervision, funding acquisition, writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by the National Key R&D Program of China (2021YFA1502500), the National Natural Science Foundation of China (No. 91961116 and 22271138), and the Natural Science Foundation of Jiangsu Province (BK20220065), Programs for High-Level Entrepreneurial and Innovative Talents Introduction of Jiangsu Province.References
- T. Storr, Ligand design in medicinal inorganic chemistry, John Wiley & Sons, 2014 Search PubMed.
- M. Stradiotto, Ligand design in metal chemistry: reactivity and catalysis, John Wiley & Sons, 2016 Search PubMed.
- T. J. Kealy and P. L. Pauson, A new type of organo-iron compound, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- L. T. Reynolds and G. Wilkinson, π-cyclopentadienyl compounds of uranium-IV and thorium-IV, J. Inorg. Nucl. Chem., 1956, 2, 246–253 CrossRef CAS.
- G. I. Vargas-Zuniga, M. A. Boreen, D. N. Mangel, J. Arnold and J. L. Sessler, Porphyrinoid actinide complexes, Chem. Soc. Rev., 2022, 51, 3735–3758 RSC.
- K. Li, W. Liu, H. Zhang, L. Cheng, Y. Zhang, Y. Wang, N. Chen, C. Zhu, Z. Chai and S. Wang, Progress in solid state and coordination chemistry of actinides in China, Radiochim. Acta, 2023, 111(1), 1–42 CrossRef CAS.
- Y. Wang, S. Hu, L. Cheng, C. Liang, X. Yin, H. Zhang, A. Li, D. Sheng, J. Diwu, X. Wang, J. Li, Z. Chai and S. Wang, Stabilization of plutonium(V) within a crown ether inclusion complex, CCS Chem., 2020, 2, 425–431 CrossRef CAS.
- Q. Liu and L. Zhao, Low valent palladium clusters: synthesis, structures and catalytic applications, Chin. J. Chem., 2020, 38, 1897–1908 CrossRef CAS.
- Y. Liu, L. Cui, W. Fang and J. Zhang, Design and synthesis of Al7Ni2 heterometallic clusters based on metal substitution and ligands protection strategies, Chin. J. Chem., 2023, 41, 521–526 CrossRef CAS.
- W. Fang, S. Pan, W. Su, S. Wang, L. Zhao, G. Frenking and C. Zhu, Complex featuring two double dative bonds between carbon(0) and uranium, CCS Chem., 2022, 4, 1921–1929 CrossRef CAS.
- W. Su, S. Pan, X. Sun, S. Wang, L. Zhao, G. Frenking and C. Zhu, Double dative bond between divalent carbon(0) and uranium, Nat. Commun., 2018, 9, 4997 CrossRef PubMed.
- M. Falcone, L. Barluzzi, J. Andrez, F. F. Tirani, I. Živković, A. Fabrizio, C. Corminboeuf, K. Severin and M. Mazzanti, The role of bridging ligands in dinitrogen reduction and functionalization by uranium multimetallic complexes, Nat. Chem., 2019, 11, 154–160 CrossRef CAS PubMed.
- M. Falcone, L. Chatelain, R. Scopelliti, I. Živković and M. Mazzanti, Nitrogen reduction and functionalization by a multimetallic uranium nitride complex, Nature, 2017, 547, 332–335 CrossRef CAS PubMed.
- C. D. Tutson and A. E. V. Gorden, Thorium coordination: A comprehensive review based on coordination number, Coord. Chem. Rev., 2017, 333, 27–43 CrossRef CAS.
- S. T. Liddle, The renaissance of non-aqueous uranium chemistry, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed.
- M. Ephritikhine, Recent advances in organoactinide chemistry as exemplified by cyclopentadienyl compounds, Organometallics, 2013, 32, 2464–2488 CrossRef CAS.
- J. L. Sessler, P. J. Melfi and G. D. Pantos, Uranium complexes of multidentate N-donor ligands, Coord. Chem. Rev., 2006, 250, 816–843 CrossRef CAS.
- J. Yu, K. Liu, Q. Wu, B. Li, X. Kong, K. Hu, L. Mei, L. Yuan, Z. Chai and W. Shi, Facile access to uranium and thorium phosphaethynolate complexes supported by tren: experimental and theoretical study, Chin. J. Chem., 2021, 39, 2125–2131 CrossRef CAS.
- P. B. Hitchcock and P. Scott, Synthesis of tripodal amido complexes of the early actinides; molecular structure of {N[CH2CH2N(SiMe3)]3UCl}2, Polyhedron, 1994, 13, 1651–1653 CrossRef.
- W. Su, T. Rajeshkumar, L. Xiang, L. Maron and Q. Ye, Facile synthesis of uranium complexes with a pendant borane lewis acid and 1,2-insertion of CO into a U-N bond, Angew. Chem., Int. Ed., 2022, 61, e202212823 CrossRef CAS PubMed.
- Q. Wu, C. Wang, J. Lan, Z. Chai and W. Shi, Electronic structures and bonding of the actinide halides An(TRENTIPS)X (An=Th-Pu; X=F-I): a theoretical perspective, Dalton Trans., 2020, 49, 15895–15902 RSC.
- K. Liu, J. Yu, Q. Wu, X. Tao, X. Kong, L. Mei, K. Hu, L. Yuan, Z. Chai and W. Shi, Rational design of a tripodal ligand for U(IV): synthesis and characterization of a U-Cl species and insights into its reactivity, Organometallics, 2020, 39, 4069–4077 CrossRef CAS.
- J. T. Boronski, J. A. Seed, D. Hunger, A. W. Woodward, J. Slageren, A. J. Wooles, L. S. Natrajan, N. Kaltsoyannis and S. T. Liddle, A crystalline tri-thorium cluster with σ-aromatic metal-metal bonding, Nature, 2021, 598, 72–75 CrossRef CAS PubMed.
- B. M. Gardner, C. E. Kefalidis, E. Lu, D. Patel, E. J. L. Mclnnes, F. Tuna, A. J. Wooles, L. Maron and S. T. Liddle, Evidence for single metal two electron oxidative addition and reductive elimination at uranium, Nat. Commun., 2017, 8, 1898 CrossRef PubMed.
- D. M. King, F. Tuna, E. J. L. Mclnnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Synthesis and structure of a terminal uranium nitride complex, Science, 2012, 337, 717–720 CrossRef CAS PubMed.
- D. M. King, F. Tuna, E. J. L. Mclnnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Isolation and characterization of a uranium(VI)-nitride triple bond, Nat. Chem., 2013, 5, 482–488 CrossRef CAS PubMed.
- P. Roussel and P. Scott, Complex of dinitrogen with trivalent uranium, J. Am. Chem. Soc., 1998, 120, 1070–1071 CrossRef CAS.
- S. C. Bart, C. Anthon, F. W. Heinemann, E. Bill, N. M. Edelstein and K. Meyer, Carbon dioxide activation with sterically pressured mid- and high-valent uranium complexes, J. Am. Chem. Soc., 2008, 130, 12536–12546 CrossRef CAS PubMed.
- G. Feng, M. Zhang, D. Shao, X. Wang, S. Wang, L. Maron and C. Zhu, Transition-metal-bridged bimetallic clusters with multiple uranium-metal bonds, Nat. Chem., 2019, 11, 248–253 CrossRef CAS PubMed.
- G. Feng, M. Zhang, P. Wang, S. Wang, L. Maron and C. Zhu, Identification of a uranium-rhodium triple bond in a heterometallic cluster, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17654–17658 CrossRef CAS PubMed.
- P. Wang, I. Douair, Y. Zhao, R. Ge, J. Wang, S. Wang, L. Maron and C. Zhu, Selective hydroboration of terminal alkynes catalyzed by heterometallic clusters with uranium-metal triple bonds, Chem, 2022, 8, 1361–1375 CAS.
- G. Feng, K. McCabe, S. Wang, L. Maron and C. Zhu, Construction of heterometallic clusters with multiple uranium-metal bonds by using dianionic nitrogen-phosphorus ligands, Chem. Sci., 2020, 11, 7585–7592 RSC.
- X. Xin, I. Douair, Y. Zhao, S. Wang, L. Maron and C. Zhu, Dinitrogen cleavage by a heterometallic cluster featuring multiple uranium-rhodium bonds, J. Am. Chem. Soc., 2020, 142, 15004–15011 CrossRef CAS PubMed.
- J. Shen, T. Rajeshkumar, G. Feng, Y. Zhao, S. Wang, L. Maron and C. Zhu, Complexes featuring a cis-[M⇉U⇇M] core (M=Rh, Ir): a new route to uranium-metal multiple bonds, Angew. Chem., Int. Ed., 2023, 62, e202303379 CrossRef CAS PubMed.
- L. Barluzzi, S. R. Giblin, A. Mansikkamaki and R. A. Layfield, Identification of oxidation state +1 in a molecular uranium complex, J. Am. Chem. Soc., 2022, 144, 18229–18233 CrossRef CAS PubMed.
- R. A. K. Shivaraam, M. Keener, D. K. Modder, T. Rajeshkumar, I. Živković, R. Scopelliti, L. Maron and M. Mazzanti, A route to stabilize uranium(II) and uranium(I) synthons in multimetallic complexes, Angew. Chem., Int. Ed., 2023, 62, e202304051 CrossRef PubMed.
- Q. Zhu, W. Fang, L. Maron and C. Zhu, Heterometallic clusters with uranium-metal bonds supported by double-layer nitrogen-phosphorus ligands, Acc. Chem. Res., 2022, 55, 1718–1730 CrossRef CAS PubMed.
- X. Xin, I. Douair, Y. Zhao, S. Wang, L. Maron and C. Zhu, Dinitrogen cleavage and hydrogenation to ammonia with a uranium complex, Natl. Sci. Rev., 2023, 10, nwac144 CrossRef PubMed.
- X. Xin, I. Douair, T. Rajeshkumar, Y. Zhao, S. Wang, L. Maron and C. Zhu, Photochemical synthesis of transition metal-stabilized uranium(VI) nitride complexes, Nat. Commun., 2022, 13, 3809 CrossRef CAS PubMed.
- P. Wang, Y. Zhao and C. Zhu, Photolysis, thermolysis, and reduction of a uranium azide complex supported by a double-layer N-P ligand, Organometallics, 2022, 41, 2448–2454 CrossRef CAS.
- X. Sun, X. Gong, Z. Xie and C. Zhu, A uranium(IV) alkyl complex: synthesis and catalytic property in carbonyl hydroboration, Chin. J. Chem., 2022, 40, 2047–2053 CrossRef CAS.
- P. Wang, I. Douair, Y. Zhao, S. Wang, J. Zhu, L. Maron and C. Zhu, Facile dinitrogen and dioxygen cleavage by a uranium(III) complex: cooperativity between the non-innocent ligand and the uranium center, Angew. Chem., Int. Ed., 2021, 60, 473–479 CrossRef CAS PubMed.
- W. Fang, I. Douair, A. Hauser, K. Li, Y. Zhao, P. W. Roesky, S. Wang, L. Maron and C. Zhu, Uranium(III)-phosphorus(III) synergistic activation of white phosphorus and arsenic, CCS Chem., 2022, 4, 2630–2638 CrossRef CAS.
- T. V. Fetrow, J. P. Grabow, J. Leddy and S. R. Daly, Convenient syntheses of trivalent uranium halide starting materials without uranium metal, Inorg. Chem., 2021, 60, 7593–7601 CrossRef CAS PubMed.
- A. L. Odom, P. L. Arnold and C. C. Cummins, Heterodinuclear Uranium/molybdenum dinitrogen complexes, J. Am. Chem. Soc., 1998, 120, 5836–5837 CrossRef CAS.
- P. L. Diaconescu and C. C. Cummins, Diuranium inverted sandwiches involving naphthalene and cyclooctatetraene, J. Am. Chem. Soc., 2002, 124, 7660–7661 CrossRef CAS PubMed.
- M. J. Monreal and P. L. Diaconescu, A weak interaction between iron and uranium in uranium alkyl complexes supported by ferrocene diamide ligands, Organometallics, 2008, 27, 1702–1706 CrossRef CAS.
- S. Duhović, S. Khan and P. L. Diaconescu, In situ generation of uranium alkyl complexes, Chem. Commun., 2010, 46, 3390–3392 RSC.
- J. W. Napoline, S. J. Kraft, E. M. Matson, P. E. Fanwick, S. C. Bart and C. M. Thomas, Tris(phosphinoamide)-supported uranium-cobalt heterobimetallic complexes featuring Co→U dative interactions, Inorg. Chem., 2013, 52, 12170–12177 CrossRef CAS PubMed.
- P. Pyykko, Additive covalent radii for single, double, and triple-bonded molecules and tetrahedrally bonded crystals: a summary, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef CAS PubMed.
- E. A. Pedrick, L. A. Seaman, J. C. Scott, L. Griego, G. Wu and T. W. Hayton, Synthesis and reactivity of a U(IV) dibenzyne complex, Organometallics, 2016, 35, 494–502 CrossRef CAS.
- O. Ordonez, X. Yu, G. Wu, J. Autschbach and T. W. Hayton, Homoleptic perchlorophenyl “ate” complexes of thorium(IV) and uranium(IV), Inorg. Chem., 2021, 60, 12436–12444 CrossRef CAS PubMed.
- L. A. Seaman, E. A. Pedrick, T. Tsuchiya, G. Wu, E. Jakubikova and T. W. Hayton, Comparison of the reactivity of 2-Li-C6H4CH2NMe2 with MCl4 (M=Th, U): isolation of a thorium aryl complex or a uranium benzyne complex, Angew. Chem., Int. Ed., 2013, 52, 10589–10592 CrossRef CAS PubMed.
- S. Fortier, B. C. Melot, G. Wu and T. W. Hayton, Homoleptic uranium(IV) alkyl complexes: synthesis and characterization, J. Am. Chem. Soc., 2009, 131, 15512–15521 CrossRef CAS PubMed.
- M. A. Boreen, B. F. Parker, T. D. Lohrey and J. Arnold, A homoleptic uranium(III) tris(aryl) complex, J. Am. Chem. Soc., 2016, 138, 15865–15868 CrossRef CAS PubMed.
- T. Delouche, M. Hissler and P. Bouit, Polycyclic aromatic hydrocarbons containing heavy group 14 elements: from synthetic challenges to optoelectronic devices, Coord. Chem. Rev., 2022, 464, 214553 CrossRef CAS.
- X. Su, T. A. Bartholome, J. R. Tidwell, A. Pujol, S. Yruegas, J. J. Martinez and C. D. Martin, 9-Borafluorenes: synthesis, properties, and reactivity, Chem. Rev., 2021, 121, 4147–4192 CrossRef CAS PubMed.
- W. Ma, C. Yu, T. Chen, L. Xu, W.-X. Zhang and Z. Xi, Metallacyclopentadienes: synthesis, structure and reactivity, Chem. Soc. Rev., 2017, 46, 1160–1192 RSC.
- A. Steffen, R. M. Ward, W. D. Jones and T. B. Marder, Dibenzometallacyclopentadienes, boroles and selected transition metal and main group heterocyclopentadienes: synthesis, catalytic and optical properties, Coord. Chem. Rev., 2010, 254, 1950–1976 CrossRef CAS.
- P. Pandey, X. Yu, G. B. Panetti, E. Lapsheva, M. R. Gau, P. J. Carroll, J. Autschbach and E. J. Schelter, Synthesis, electrochemical, and computational studies of organocerium(III) complexes with Ce-aryl sigma bonds, Organometallics, 2023, 42, 1267–1277 CrossRef CAS.
- M. Zhu, Z. Chai, Z.-J. Lv, T. Liu, W. Liu, J. Wei and W.-X. Zhang, Selective cleavage of the strong or weak C-C bonds in biphenylene enabled by rare-earth metals, J. Am. Chem. Soc., 2023, 145, 6633–6638 CrossRef CAS PubMed.
- Z. Huang, Y. Zhang, W.-X. Zhang, J. Wei, S. Ye and Z. Xi, A tris-spiro metalla-aromatic system featuring Craig-Möbius aromaticity, Nat. Commun., 2021, 12, 1319 CrossRef CAS PubMed.
- S. Fortier, B. Melot, G. Wu and T. W. Hayton, Homoleptic uranium(IV) alkyl complexes: synthesis and characterization, J. Am. Chem. Soc., 2009, 131, 15512–15521 CrossRef CAS PubMed.
- J. D. Sears, D. Sergentu, T. M. Baker, W. W. Brennessel, J. Autschbach and M. L. Neidig, The exceptional diversity of homoleptic uranium-methyl complexes, Angew. Chem., Int. Ed., 2020, 59, 13586–13590 CrossRef CAS PubMed.
- S. Hohloch, M. E. Garner, B. F. Parker and J. Arnold, New supporting ligands in actinide chemistry: tetramethyltetraazaannulene complexes with thorium and uranium, Dalton Trans., 2017, 46, 13768–13782 RSC.
- D. Wang, W. Ding, G. Hou, G. Zi and M. D. Walter, Uranium versus thorium: synthesis and reactivity of [η5-1,2,4(Me3C)3C5H2]2U[η2-C2Ph2], Chem. – Eur. J., 2021, 27, 6767–6782 CrossRef CAS PubMed.
- Y. Du, Y. Xiao, F. Tian, L. Han, Y. Gu and N. Zhu, Progress in annulative transformation from indoles to carbazoles: state of the art, Chin. J. Org. Chem., 2021, 41, 521–528 CrossRef CAS.
- W. Cao and X. Liu, Electrochemical 1,2-diarylation of alkenes enabled by direct dual C-H functionalization of elextron-rich aromatic hydrocarbons, Chin. J. Org. Chem., 2021, 41, 857–858 CrossRef CAS.
- P. L. Diaconescu, Reactions of aromatic N-heterocycles with d0fn-metal alkyl complexes supported by chelating diamide ligands, Acc. Chem. Res., 2010, 43, 1352–1363 CrossRef CAS PubMed.
- N. R. Andreychuk, S. Lango, B. Vidjayacoumar, D. J. Emslie and H. A. Jenkins, Uranium(IV) alkyl complexes of a rigid dianionic NON-donor ligand: synthesis and quantitative alkyl exchange reactions with alkyllithium reagents, Organometallics, 2013, 32, 1466–1474 CrossRef CAS.
- S. A. Johnson and S. C. Bart, Achievements in uranium alkyl chemistry: celebrating sixty years of synthetic pursuits, Dalton Trans., 2015, 44, 7710–7726 RSC.
- L. Zhang, G. Hou, G. Zi, W. Ding and M. D. Walter, Influence of the 5f orbitals on the bonding and reactivity in organoactinides: experimental and computational studies on a uranium metallacyclopropene, J. Am. Chem. Soc., 2016, 138, 5130–5142 CrossRef CAS PubMed.
- L. Zhang, B. Fang, G. Hou, G. Zi, W. Ding and M. D. Walter, Experimental and computational studies of a uranium metallacyclocumulene, Organometallics, 2017, 36, 898–910 CrossRef CAS.
- G. Zi, Recent developments in actinide metallacycles, Chem. Commun., 2018, 54, 7412–7430 RSC.
- S. A. Johnson, R. F. Higgins, M. M. Abu-Omar, M. P. Shores and S. C. Bart, Mechanistic insights into concerted C-C reductive elimination from homoleptic uranium alkyls, Organometallics, 2017, 36, 3491–3497 CrossRef CAS.
- J. A. Pool, B. L. Scott and J. L. Kiplinger, A new mode of reactivity for pyridine N-oxide: C-H activation with uranium(IV) and thorium(IV) bis(alkyl) complexes, J. Am. Chem. Soc., 2005, 127, 1338–1339 CrossRef CAS PubMed.
- W. Evans, J. Walensky and J. Ziller, Synthesis of a thorium tuck-in complex, [(η5:η1-C5Me4CH2) (η5-C5Me5)Th{iPrNC(Me)NiPr}], by C-H bond activation initiated by (C5Me5)−, Chem. – Eur. J., 2009, 15, 12204–12207 CrossRef CAS PubMed.
- B. M. Gardner, P. A. Cleaves, C. E. Kefalidis, J. Fang, L. Maron, W. Lewis, A. J. Blake and S. T. Liddle, The role of 5f-orbital participation in unexpected inversion of the σ-bond metathesis reactivity trend of triamidoamine thorium(IV) and uranium(IV) alkyls, Chem. Sci., 2014, 5, 2489–2497 RSC.
- J. M. Berg, D. L. Clark, J. C. Huffman, D. E. Morris, A. P. Sattelberger, W. E. Streib, W. G. Van Der Sluys and J. G. Watkin, Early actinide alkoxide chemistry. synthesis, characterization, and molecular structures of Th(IV) and U(IV) aryloxide complexes, J. Am. Chem. Soc., 1992, 114, 10811–10821 CrossRef CAS.
- M. J. Monreal, R. K. Thomson, T. Cantat, N. E. Travia, B. L. Scott and J. L. Kiplinger, UI4(1,4-dioxane)2, [UCl4(1,4-dioxane)]2, and UI3(1,4-dioxane)1.5: stable and versatile starting materials for low- and high-valent uranium chemistry, Organometallics, 2011, 30, 2031–2038 CrossRef CAS.
- R. Boaretto, P. Roussel, N. W. Alcock, A. J. Kingsley, I. J. Munslow, C. J. Sanders and P. Scott, Synthesis of a highly strained uranacycle: molecular structures of organometallic products arising from reduction, oxidation and protonolysis, J. Organomet. Chem., 1999, 591, 174–184 CrossRef CAS.
- R. S. Sternal and T. J. Marks, Actinide-to-transition metal bonds. synthesis, characterization, and properties of metal-metal bonded systems having the tris(cyclopentadlenyl)actinide fragment, Organometallics, 1987, 6, 2621–2623 CrossRef CAS.
- S. G. Minasian, J. L. Krinsky, V. A. Williams and J. Arnold, A heterobimetallic complex with an unsupported uranium(III)-aluminum(I) bond: (CpSiMe3)3U-AlCp*(Cp*=C5Me5), J. Am. Chem. Soc., 2008, 130, 10086–10087 CrossRef CAS PubMed.
- S. G. Minasian, J. Krinsky, J. D. Rinehart, R. Copping, T. Tyliszczak, M. Janousch, D. K. Shuh and J. Arnold, A comparison of 4f vs 5f metal-metal bonds in (CpSiMe3)3M-ECp* (M=Nd, U; E=Al, Ga; Cp*=C5Me5): synthesis, thermodynamics, magnetism, and electronic structure, J. Am. Chem. Soc., 2009, 131, 13767–13783 CrossRef CAS PubMed.
- S. T. Liddle, J. McMaster, D. P. Mills, A. J. Blake, C. Jones and W. D. Woodul, σ and π donation in an unsupported uranium-gallium bond, Angew. Chem., Int. Ed., 2009, 48, 1077–1080 CrossRef CAS PubMed.
- S. T. Liddle and D. P. Mills, Metal-metal bonds in f-element chemistry, Dalton Trans., 2009, 5592–5605 RSC.
- S. T. Liddle, Non-traditional ligands in f-block chemistry, Proc. R. Soc. A, 2009, 465, 1673–1700 CrossRef CAS.
- D. Patel, F. Moro, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, A formal high oxidation state inverse-sandwich diuranium complex: a new route to f-block-metal bonds, Angew. Chem., Int. Ed., 2011, 50, 10388–10392 CrossRef CAS PubMed.
- B. Oelkers, M. V. Butovskii and R. Kempe, F-element-metal bonding and the use of the bond polarity to build molecular intermetalloids, Chem. – Eur. J., 2012, 18, 13566–13579 CrossRef CAS PubMed.
- A. L. Ward, W. W. Lukens, C. C. Lu and J. Arnold, Photochemical route to actinide-transition metal bonds: synthesis, characterization and reactivity of a series of thorium and uranium heterobimetallic complexes, J. Am. Chem. Soc., 2014, 136, 3647–3654 CrossRef CAS PubMed.
- J. A. Hlina, J. R. Pankhurst, N. Kaltsoyannis and P. L. Arnold, Metal-metal bonding in uranium-group 10 complexes, J. Am. Chem. Soc., 2016, 138, 3333–3345 CrossRef CAS PubMed.
- S. Fortier, J. R. Aguilar-Calderon, B. Vlaisavljevich, A. J. Metta-Magana, A. G. Goos and C. E. Botez, An N-tethered uranium(III) arene complex and the synthesis of an unsupported U-Fe bond, Organometallics, 2017, 36, 4591–4599 CrossRef CAS.
- E. Lu, A. J. Wooles, M. Gregson, P. J. Cobb and S. T. Liddle, A very short uranium(IV)-rhodium(I) bond with net double-dative bonding character, Angew. Chem., Int. Ed., 2018, 57, 6587–6591 CrossRef CAS PubMed.
- W. Fang, Q. Zhu and C. Zhu, Recent advances in heterometallic clusters with f-block metal-metal bonds: synthesis, reactivity and applications, Chem. Soc. Rev., 2022, 51, 8434–8449 RSC.
- J. A. Hlina, J. A. L. Well, J. R. Pankhurst, J. Love and P. L. Arnold, Uranium rhodium bonding in heterometallic complexes, Dalton Trans., 2017, 46, 5540–5545 RSC.
- O. P. Lam, C. Anthon, F. W. Heinemann, J. M. O'Connor and K. Meyer, Structural and spectroscopic characterization of a charge-separated uranium benzophenone ketyl radical complex, J. Am. Chem. Soc., 2008, 130, 6567–6576 CrossRef CAS PubMed.
- S. Harder and F. Feil, Dimeric benzylcalcium complexes: influence of THF in stereoselective styrene polymerization, Organometallics, 2002, 21, 2268–2274 CrossRef CAS.
- J. A. Hermann and J. F. Suttle, Uranium(IV) chloride, Inorg. Synth., 1957, 5, 143–144 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment-Olex2 dissected, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–75 CrossRef CAS PubMed.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2254126–2254132. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi01447a |
| This journal is © the Partner Organisations 2023 |