
Accelerated rates of proton coupled electron transfer to oxygen deficient polyoxovanadate–alkoxide clusters†
Shannon E.
Cooney‡
 ,
Eric
Schreiber‡
,
William W.
Brennessel
and
Ellen M.
Matson
*
,
Eric
Schreiber‡
,
William W.
Brennessel
and
Ellen M.
Matson
*
Department of Chemistry, University of Rochester, Rochester, NY 14627, USA. E-mail: matson@chem.rochester.edu
First published on 4th April 2023
Abstract
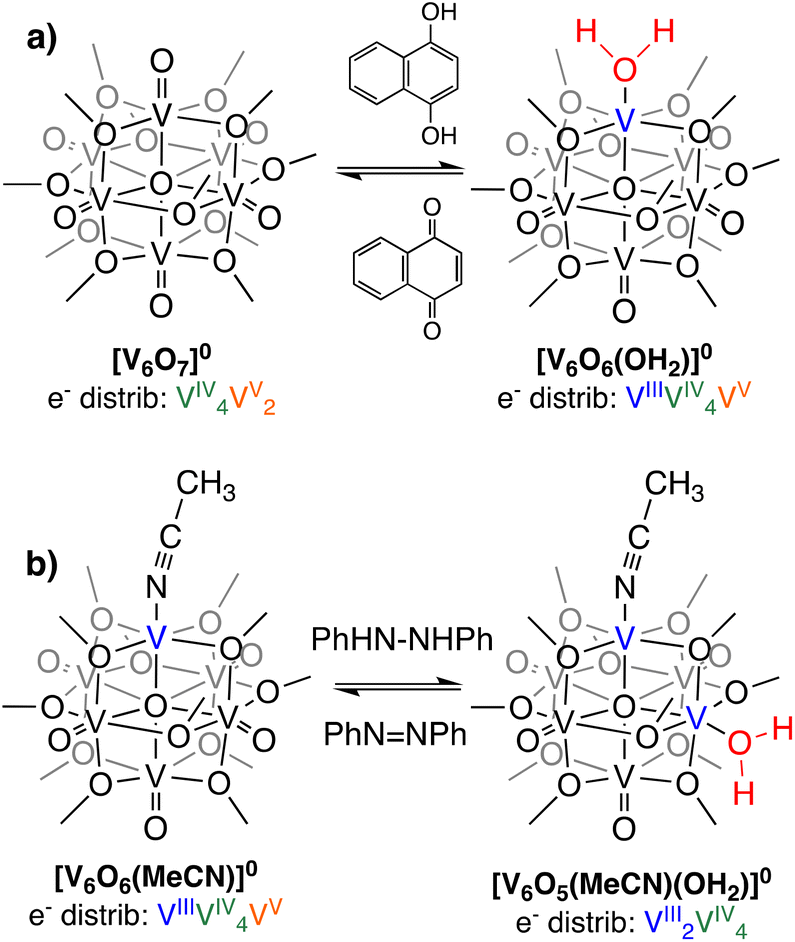
Anionic dopants, such as O-atom vacancies, alter the thermochemical and kinetic parameters of proton coupled electron transfer (PCET) at metal oxide surfaces; understanding their impact(s) is essential for informed material design for efficient energy conversion processes. To circumvent challenges associated with studying extended solids, we employ polyoxovanadate–alkoxide clusters as atomically precise models of reducible metal oxide surfaces. In this work, we examine net hydrogen atom (H-atom) uptake to an oxygen deficient vanadium oxide assembly, [V6O6(MeCN)(OCH3)12]0. Addition of two H-atom equivalents to [V6O6(MeCN)(OCH3)12]0 results in formation of [V6O5(MeCN)(OH2)(OCH3)12]0. Assessment of the bond dissociation free energy of the O–H bonds of the resultant aquo moiety reveals that the presence of an O-atom defect weakens the O–H bond strength. Despite a decreased thermodynamic driving force for the reduction of [V6O6(MeCN)(OCH3)12]0, kinetic investigations show the rate of H-atom uptake at the cluster surface is ∼100× faster than its oxidized congener, [V6O7(OCH3)12]0. Electron density derived from the O-atom vacancy is shown to play an important role in influencing H-atom uptake at the cluster surface, lowering activation barriers for H-atom transfer.
Introduction
The last several decades have seen significant advances in renewable energy technologies,1 driving rapid, global adoption of low-carbon electricity.2,3 However, continued progress toward a sustainable energy economy requires a transition from our dependence on fossil fuels. Electrochemistry can play a key role, namely through the development of electrochemical catalysts that recycle low-value precursors into chemical fuels.4 In the context of electrosynthetic hydrogenations, the concerted transfer of protons and electrons (i.e. proton coupled electron transfer, PCET) can uniquely enable the activation of energy-poor substrates by bypassing intermediates.5,6 While proton and electron transfer at metal oxides have been studied extensively in the field of electrochemical energy storage,7,8 researchers have only recently started to understand these processes in the presence of substrate.9,10 As such, further progress in the effective use of these materials as hydrogenation catalysts requires atomistic understanding of surface-mediated PCET reaction mechanisms.In materials science, O-atom deficiencies are considered a form of anionic dopant (Fig. 1). Their presence increases carrier density within a material, as the lack of an oxide ligand necessitates the presence of a reduced metal center.10,11 Accordingly, the Fermi energy of materials possessing O-atom defects is often disparate from that of their oxidized congeners; increased electron density in the extended solid modifies the driving force of electron transfer.12 This change in effective reduction potential of the material has been demonstrated via theoretical calculations to result in dramatic differences in bond dissociation free energies, BDFE(O–H), of surface hydroxide moieties in these materials.13
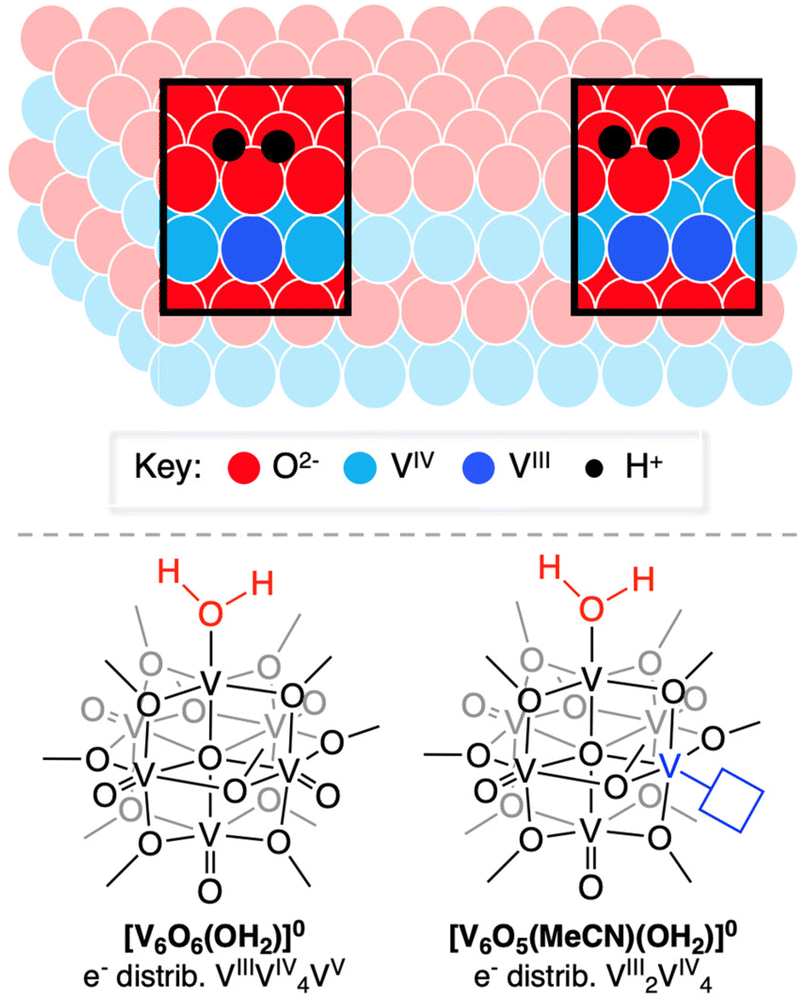 | ||
Fig. 1 PCET to the surface of pristine and O-deficient metal oxide surfaces (top). 2e−/2H+ reduced POV–alkoxide clusters studied in this work, where V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond reduction occurs at the surface of a fully oxygenated vanadium oxide assembly, and one bearing a preformed O-atom vacancy to generate aquo-ligated cluster complexes. O bond reduction occurs at the surface of a fully oxygenated vanadium oxide assembly, and one bearing a preformed O-atom vacancy to generate aquo-ligated cluster complexes. | ||
Our research team is interested in understanding factors that influence PCET reactivity that yields net hydrogen atom (H-atom) uptake at the surface of a series of vanadium oxide assemblies.14–20 The polyoxovanadate–alkoxide (POV–alkoxide) clusters studied by our team possess bridging alkoxide ligands, blocking traditionally nucleophilic sites at the surface of molecular metal oxide assemblies that dictate their reactivity with protons in solution.20–28 In some cases, saturation of bridging sites by organic groups has been studied as a means to isolate reactivity of protons and H-atom equivalents to terminal oxide sites, resulting in the formation of an O-atom vacancy and water under appropriate reaction conditions.
One advantage to studying molecular systems as models for extended solids is the ability to manipulate the composition of the assemblies with atomic precision.19 Indeed, our team has demonstrated formation of a POV–alkoxide cluster with two O-atom defect sites.29 In that work, we established that surface O-atoms are removed sequentially from the cluster surface. Our ability to isolate and handle the “intermediate”, [V6O6(MeCN)(OCH3)12]0 ([V6O6(MeCN)]0), allows for investigation of the impact embedded O-atom defects have on PCET in oxygen-deficient metal oxide assemblies.
Herein, we report the synthesis of two reduced forms of POV–alkoxide clusters with water ligated to surface of the assembly (Fig. 1). [V6O6(OH2)(OCH3)12]0 ([V6O6(OH2)]0) and [V6O5((MeCN)(OH2)(OCH3)12]0 ([V6O5(MeCN)(OH2)]0) are accessed via net multi H-atom transfer (HAT) to the fully oxygenated, neutral POV–alkoxide cluster, [V6O7(OCH3)12]0 ([V6O7]0), in tetrahydrofuran (THF). When HAT is performed in THF, the aquo ligand generated via PCET remains coordinated to the reduced VIII ion(s). This distinct attribute allows for experimental assessment of the BDFE(O–H)avg of the aquo O–H bonds. Analysis of the kinetics of net H-atom uptake reveals that while the mechanism of PCET proceeds via a concerted pathway in both examples, the presence of an O-atom defect accelerates the rate of net H-atom uptake by two orders of magnitude. Overall, this study reveals the impact O-atom defects have on PCET at metal oxides, providing insight into alterations of structural parameters that impact the reactivity of the surface with relevance to H-atom transfer chemistry.
Results & discussion
Initial studies probed addition of an excess of a potent H-atom transfer reagent to the neutral POV–alkoxide cluster, [V6O7]0, to confirm that formation of the di-vacant POV–alkoxide cluster is possible via PCET. Upon addition of 2 equiv. of 5,10-dihydrophenazine (H2Phen; BDFE(N–H)avg = 58.7 kcal mol−1)30 in acetonitrile (MeCN), a colour change from green to red is observed (Scheme 1). Conversion to [V6O5(MeCN)2(OCH3)12]0 ([V6O5(MeCN)2]0) is completed within 3 min, as confirmed by 1H NMR spectroscopy (Fig. S1;† see Experimental section for additional details). Formation of the oxidized H-atom transfer reagent, phenazine (Phen), is also observed in the 1H NMR spectrum of the crude reaction mixture.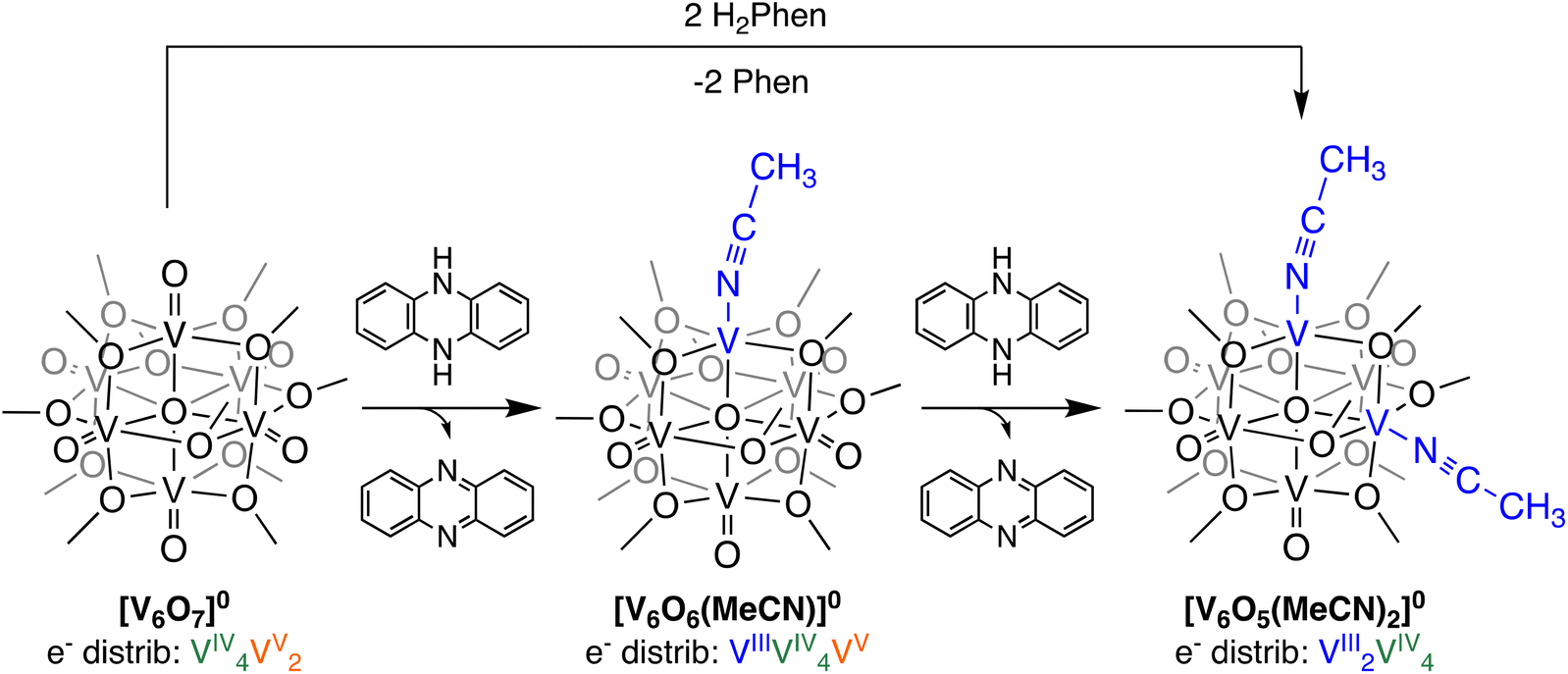 | ||
| Scheme 1 Synthesis of O-atom vacancies in POV–alkoxide clusters via net H-atom uptake in MeCN; formation of [V6O6(MeCN)]0 and [V6O5(MeCN)2]0. | ||
Formation of [V6O5(MeCN)2]0 is similarly possible from the oxygen-deficient POV–alkoxide starting material, [V6O6(MeCN)]0. Addition of 1 equiv. of H2Phen to [V6O6(MeCN)]0 results in an instantaneous colour change from brown to red (Scheme 1). Analysis of the product distribution via1H NMR spectroscopy confirms formation of the di-vacant POV–alkoxide cluster, [V6O5(MeCN)2]0 and Phen (Fig. S2;† see Experimental section for additional details).
Thermodynamics of H-atom uptake in O-deficient POV–alkoxides
Our previous work summarizing the synthesis and characterization of O-atom deficient POV–alkoxide clusters includes electrochemical analysis of these assemblies. Cyclic voltammetry (CV) reveals a cathodic shift in open circuit potential of the assembly upon vacancy formation (Fig. 2). This observation is consistent with the formation of an electron-rich cluster core upon removal of an O-atom from the cluster surface. Accordingly, generation of a defect site results in a change in driving force for electron transfer to the assembly (ΔE1/2 ∼ 0.3 V).29,31 We hypothesized that this change in redox potential would impact the thermodynamics of H-atom uptake and transfer at the surface of the cluster.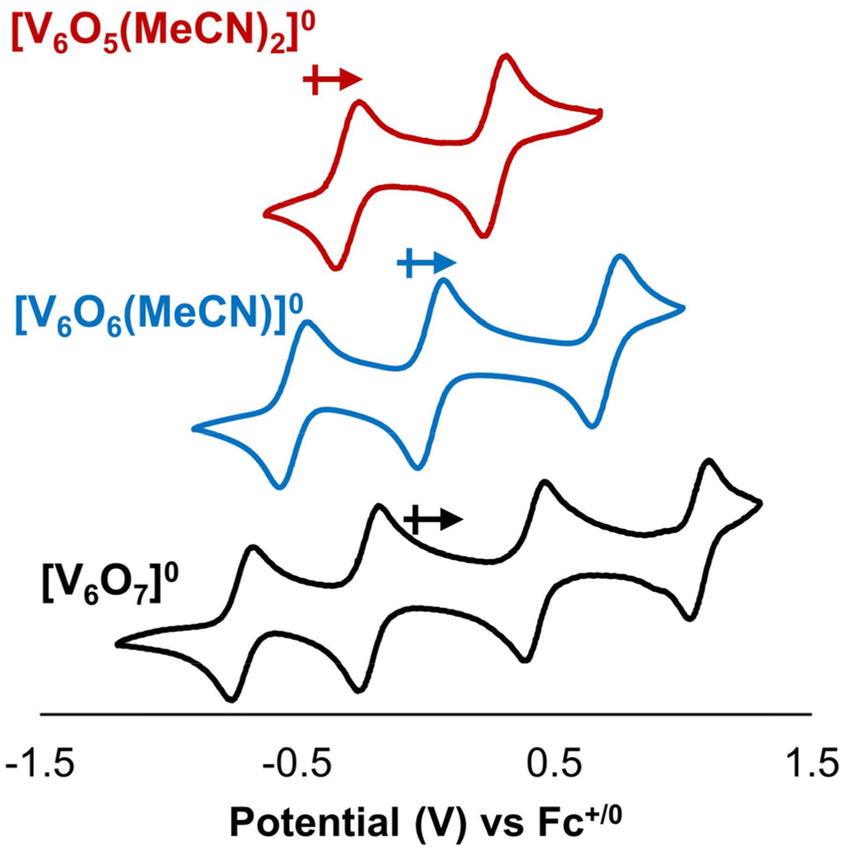 | ||
| Fig. 2 Cyclic voltammograms of 1 mM [V6O7]0 (black), [V6O6(MeCN)]0 (blue), and [V6O5(MeCN)2]0 (red) in DCM with 100 mM [nBu4N][PF6] supporting electrolyte. Arrows indicate open circuit voltages of the individual cluster complexes in their neutral charge state. Figure reproduced with permission from B. E. Petel et al., Inorg. Chem. 2019, 58, 10462–10471. Copyright 2019 American Chemical Society. | ||
To understand the impact of an O-atom vacancy on the thermodynamics of H-atom uptake and transfer in POV–alkoxide clusters, we performed a series of equilibrium studies to determine BDFE(O–H)avg of surface aquo moieties. Previous work from our group has demonstrated that addition of H-atom transfer reagents to calix[4]arene-substituted POV–alkoxide clusters in tetrahydrofuran (THF) results in retention of the aquo ligand at the surface of the assembly.15 We anticipate that similar stabilization of the surface aquo should occur in this system; therefore, all in situ equilibrium experiments reported here were conducted in THF-d8.
Confirmation of H-atom installation and aquo-stabilization at the surface of the cluster was obtained through stoichiometric reactions with the parent POV–alkoxide cluster in THF. Exposure of [V6O7]0 to 1 equiv. of hydrazobenzene (Hydz, BDFE(N–H)avg = 60.4 kcal mol−1) in THF results in a rapid colour change from green to brown.15 The 1H NMR spectrum of the product in THF-d8 reveals a reduction in symmetry of the vanadium-containing product (Oh → C4v) in analogy to previous results on VO bond reduction in POV–alkoxides (Fig. S3†).16,32 The three signals observed in the 1H NMR spectrum of the product are slightly shifted (29.6, 16.2, −11.5 ppm) in comparison with the O-atom deficient analogue bearing a MeCN ligand (29.5, 15.7, −12.1 ppm; Fig. S4†). Analysis of the crude reaction mixture did not indicate formation of water, in contrast to results obtained previously in PCET reactions to POV–alkoxides in MeCN (Fig. S4†). This suggests that the aquo moiety formed upon addition of 2 H-atom equivalents to the VO site remains coordinated to the surface of the cluster. Additional support for the formation of the aquo adduct, [V6O6(OH2)]0, was obtained from elemental analysis.
Recrystallization of the reduced POV–alkoxide was achieved by vapor diffusion of pentane directly into the reaction medium. The resultant crystals were suitable for analysis via single crystal X-ray diffraction (Fig. 3, and Table 1; see Table S1† for crystallographic parameters). Refinement of the data confirmed the identity of the reduced assembly to be the anticipated aquo-bound product, [V6O6(OH2)]0. The V1–O1 bond distance of 2.0671(19) Å is significantly elongated from that of the starting material, [V6O7]0 (VOt = 1.60 Å33), and consistent with V–O distances reported for vanadium(III) aquo complexes (1.967(3)–2.086(2) Å).15,34,35 In particular, the V1–O1 distance resembles that reported by our research group for the calix[4]arene-substituted POV–alkoxide cluster featuring an aquo ligand, [(calix)V6O5(OH2)(MeCN)(OCH3)8]0 (V(III)-O(aquo) = 2.052(2) Å).15 Further supporting our assignment of the aquo ligand are the identification of two THF molecules which serve as H-bond acceptors based on their orientation and proximity to the corresponding terminal O-atom (O1⋯O(THF) = 2.640(14), 2.700(3) Å; O1–Hn⋯O(THF) = 168(4), 175(5)°, n = 1A, 1B).
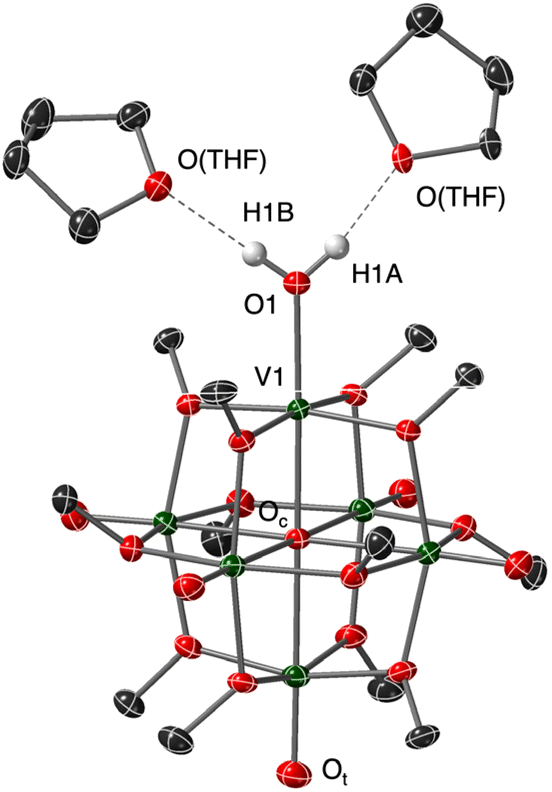 | ||
| Fig. 3 Molecular structure of [V6O6(OH2)]0 shown with 50% probability ellipsoids. Selected hydrogen atoms have been omitted for clarity. Key: dark green ellipsoids: V; red ellipsoids: O; dark grey ellipsoids: C; white spheres: H. | ||
| Bond | [V6O6(OH2)]0 | [V6O7]0 |
|---|---|---|
| V1–O1 | 2.0671(19) Å | — |
| V1–Oc | 2.1158(16) Å | — |
| VOt (avg) |
1.5998 Å | 1.60 Å |
| V–Oc (avg) | 2.3177 Å | 2.28 Å |
| O1⋯O(THF) | 2.640(14), 2.700(3) Å | — |
| O1–Hn⋯O(THF) | 168(4), 175(5)° | — |
The formal transfer of two H-atom equivalents to the surface of [V6O7]0 results in the reduction of the vanadium oxide core (VIV4VV2 → VIIIVIV4VV). Fortunately, within the unit cell of [V6O6(OH2)]0, all V centres are located in general positions, providing an opportunity to interrogate the electronic structure of the cluster using bond valence sum calculations (Table S2†). Indeed, the oxidation state distribution of the reduced complex was found to be VIIIVIV4VV. We note that this formal assignment is relevant at low temperature (100 K) and in the solid state; in solution at elevated temperatures the VV centre is expected to be delocalized about the fully oxygenated vanadium centres. This is evidenced by the observation of an intervalence charge transfer (IVCT) band diagnostic of VIV → VV electron transfer in the near infrared region of the electronic absorption spectrum of [V6O6(OH2)]0 (Fig. S5†).
With confirmation that water is stabilized at the surface of [V6O6(OH2)]0, we next sought to determine the strength of these surface O–H bonds. Determination of the experimental BDFE(O–H)avg of the surface H-atoms of [V6O6(OH2)]0 was achieved by exposure of the fully-oxygenated cluster to a reductant that does not react quantitatively with the complex in THF (Scheme 2). The generation of an equilibrium state allows us to invoke a modified version of the Nernst equation to find the adjusted BDFE(E–H) for the reductant, which under equilibrium conditions would be equivalent to the analogous parameter for the reduced cluster (Eqn (1)). Such analysis has been employed for studying surface O–H bonds on nanocrystalline ceria by Mayer and co-workers, as well as reduced POV–alkoxides by our group.13,15,18 Equilibrium is reached after exposure of the cluster to various equivalents (0.5 to 2) of 1,4-dihydroxynaphthalene (H2Naphth; BDFE(O–H)avg: 62.6 kcal mol−1) for 7 days in THF-d8 (Fig. S6, and S7†).30 Using the concentrations of the reductant and its oxidized partner, 1,4-naphthoquinone (Naphth), a value of n = 2 for the two H-atom equivalents transferred from the substrate, alongside Eqn (1), we determined the BDFE(O–H)avg of [V6O6(OH2)]0 to be 62.3 ± 0.1 kcal mol−1 (Table S3†).
 | (1) |
 | ||
| Scheme 2 Equilibrium reactions employed in the determination of BDFE(O–H)avg for (a) [V6O6(OH2)]0 and (b) [V6O5(MeCN)(OH2)]0. | ||
Next, we set out to determine the BDFE(O–H)avg of the aquo ligand bound to a second O-atom deficient site at the surface of the assembly (Scheme 2). Despite the quantitative transfer of the H-atoms from Hydz (BDFE(N–H)avg = 60.4 kcal mol−1)15,30 to [V6O7]0, exposure of this reductant to [V6O6(MeCN)]0 in THF-d8 results in only partial conversion to the di-vacant cluster, [V6O5(MeCN)(OH2)(OCH3)12]0 ([V6O5(MeCN)(OH2)]0) (Fig. S9†). After 15 days, the concentrations of Hydz and its oxidized counterpart, azobenzene (Azo) remain static, indicating the system has reached equilibrium (Fig. S10†). Measuring the respective concentrations of reduced and oxidized substrate at equilibrium, the BDFE(O–H)avg of the aquo ligand bound to [V6O5(MeCN)(OH2)]0 was determined to be 60.7 ± 0.1 kcal mol−1 (Table S4†).
The observed decrease in BDFE(O–H)avg from [V6O6(OH2)]0 to [V6O5(MeCN)(OH2)]0 highlights the impact of the oxidation state distribution of distal vanadium ions on the thermodynamics of PCET at the cluster surface. This is consistent with results published by Agapie and coworkers for a “Fe3Mn” cluster, in which electron-rich variants of these multimetallic compounds exhibit weak Mn(O–H) bond strengths in comparison to their oxidized derivatives.36 Our group has corroborated this trend, observing that the strength of surface O–H bonds located at both terminal and bridging positions of Lindqvist-type POV–alkoxide clusters is dependent on the degree of reduction of the constituent metal ions; in all examples, reduced forms of the vanadium oxide assemblies possess the lowest BDFE(O–H) values.14,16,20 The influence of oxidation state of metal centres in multimetallic configurations on the thermodynamics of PCET is also observed in nanocrystalline metal oxides, as shown in recent reporting from Mayer and coworkers.13
Kinetic analysis of PCET in oxygen-deficient POV–alkoxides
With an understanding of the impact of an O-atom defect on the thermodynamics of H-atom uptake in POV–alkoxide clusters, our attention turned to observed discrepancies in the rates of PCET at the surface of [V6O7]0 and [V6O6(MeCN)]0. According to Marcus Theory, relative rates of net H-atom transfer between two substrates are directly proportional to the ΔBDFE(E–H) between H-atom donor and acceptor; ΔBDFE(E–H), or driving force, influences equilibrium constants (Keq) in H-atom transfer reactions, thus impacting the observed rate of reaction for a system.37 Based on these considerations alone, we hypothesized that the rate of H-atom uptake and O-atom vacancy formation in [V6O6(MeCN)]0 would be slower than that of its fully oxygenated congener, [V6O7]0, given the resultant O–H bonds are weaker in the further reduced, O-atom deficient assembly.To evaluate our hypothesis, a series of kinetic analyses were performed in which electronic absorption spectroscopy was used to monitor cluster reduction. This approach leverages the differentiated spectra of [V6O6(MeCN)]0 and [V6O5(MeCN)(OH2)]0. The more oxidized cluster, [V6O6(MeCN)]0 (oxidation state distribution: VIIIVIV4VV) features an intervalence charge transfer (IVCT) band, corresponding to electron transfer between the VIV and VV ions in the core (λ = 900 nm (ε = 413 M−1 cm−1.29 The reduced species lacks VV ions (oxidation state distribution: VIII2VIV4), dramatically reducing its absorptivity at 900 nm (ε < 100 M−1 cm−1, Fig. S13, and S14†).29 As such, the change in absorbance at 900 nm was monitored upon addition of an excess of H2Phen to [V6O6(MeCN)]0 (Fig. S15–20,† see Experimental section for details). Fitting of the generated traces produces a linear plot when correlating the observed pseudo first-order rate constant, kobs to the reductant concentration (Fig. 4). This observation indicates that the rate-determining step is first order in both reductant and cluster (Eqn (2)). Notably, kinetic analyses on net H-atom uptake at the surface of the fully-oxygenated cluster, [V6O7]0, similarly reveal a second-order rate-limiting process.16 The analogous rate expressions provide an initial indication that both clusters undergo net H-atom uptake via similar mechanisms.
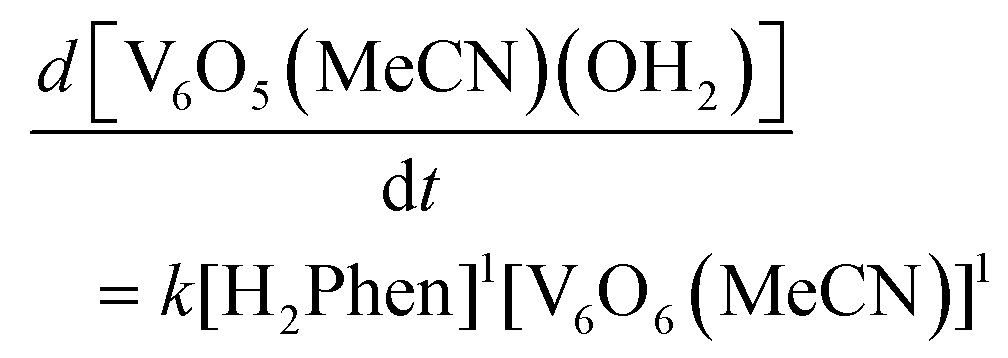 | (2) |
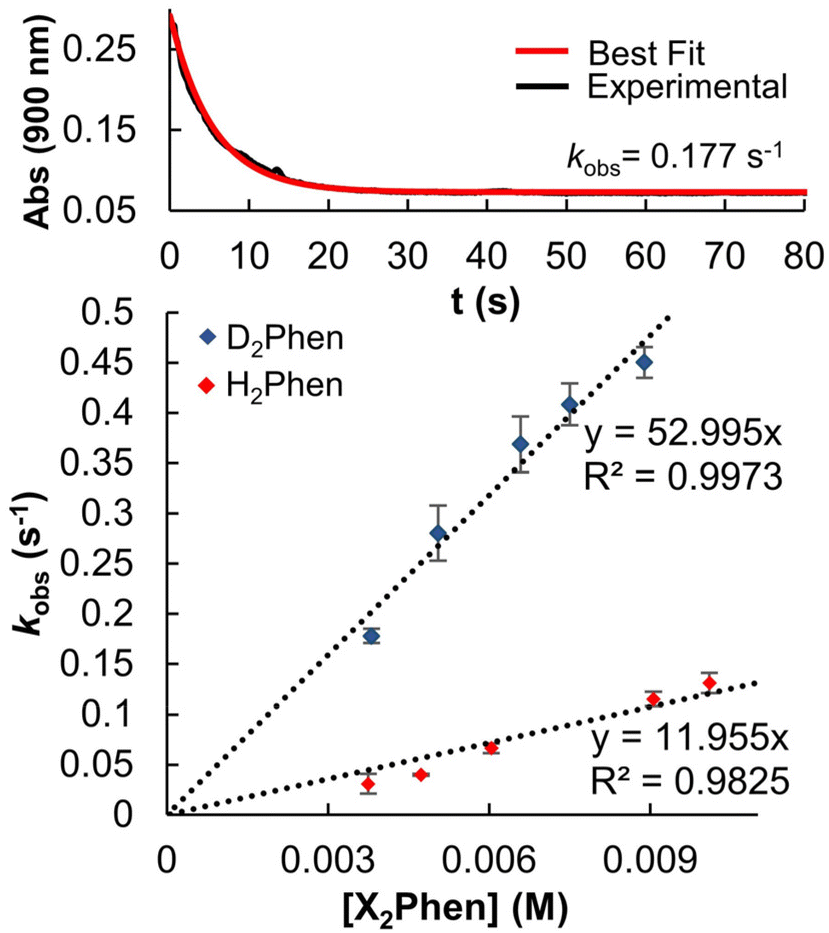 | ||
| Fig. 4 Determination of the observed pseudo first-order rate constant, kobs, for the reduction of [V6O6(MeCN)]0 (0.75 mM) by H2Phen (3.81 mM) in MeCN at −15 °C by monitoring absorbance at 900 nm over the reaction coordinate (top). Plot of kobs as a function of [X2Phen], where X = H, D. Concentration of [V6O6(MeCN)]0 was held constant at 0.75 mM, and reductant concentration was varied between 3.8 and 11.5 mM. The slope of the resultant lines * ¼ vanadyls possible for accepting the H-atom * ½ for the two chemically equivalent H-atoms transferred provides the experimentally derived second-order rate constants, kPCET. Comparison of kPCET for H2Phen and D2Phen yields a KIE of 4.4. | ||
In our previous study, the formation of an O-atom defect at [V6O7]0 by PCET was determined to proceed via two concerted proton–electron transfer (CPET) reactions.16 We thus hypothesized that a CPET mechanism is similarly operative for the reduction of [V6O6(MeCN)]0. Kinetic isotope effect (KIE) experiments, employing the deuterium-labelled reductant, D2Phen, reveal a substantial KIE (4.4). This finding disqualifies a potential electron transfer–proton transfer (ET–PT) mechanism (Fig. 4 and Table 2). This is supported by electrochemistry experiments, which found that the oxidation potential of H2Phen (E1/2 = −0.387 V vs. Fc+/0, Fig. S11†) is not sufficiently reducing to transfer an electron to [V6O6(MeCN)]0 (E1/2 = −0.646 V vs. Fc+/0, Fig. S12.†)
[V6O6(OH2)]0
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16
16
|
[V6O5(MeCN)(OH2)]0 | |
|---|---|---|
| BDFE(O–H)avg | 62.3 ± 0.1 kcal mol−1 | 60.7 ± 0.1 kcal mol−1 |
| k PCET@298 K | 0.14 ± 0.05 M−1 s−1 | 19.6 ± 3.2 M−1 s−1 |
| KIE | 2.1 | 4.4 |
| ΔH‡ (kcal mol−1) | 7.8 ± 0.8 | 4.3 ± 1.2 |
| ΔS‡ (cal mol−1 K−1) | −31.0 ± 3.3 | −33.9 ± 4.8 |
| ΔG‡ (kcal mol−1) | 18.7 ± 1.7 | 14.4 ± 2.6 |
The proton transfer–electron transfer (PT–ET) pathway is similarly eliminated due to the weak acidity of the organic substrate, as the deprotonation of a neutral amine is unlikely to occur in the absence of a strong base (e.g. Na metal).38 To determine the relative basicities of each reactive species, we employ the following equation popularized by Bordwell,39–41
| BDFE(E–H) = 1.37(pKa) + 23.06(E°) + CG | (3) |
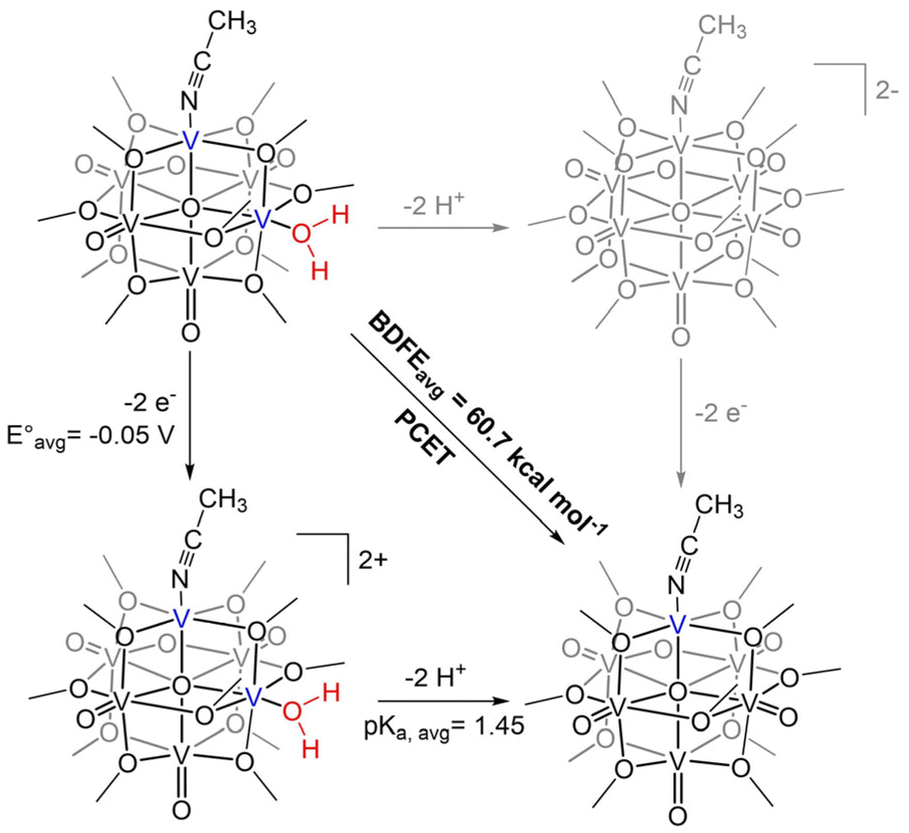 | ||
| Scheme 3 Square scheme of 2e−/2H+ reduction of [V6O6(OH2)]0 to [V6O5(MeCN)(OH2)]0. | ||
Using the established BDFE(N–H)avg of H2Phen in THF (59.2 kcal mol−1), the average reduction potential for 2e− reduction of Phen (−2.15 V vs. Fc+/0), and CG for THF (59.9 kcal mol−1), we can apply this function to find the average pKa for the protons bound to reduced [Phen]2− to be 35.6. Similarly, using the BDFE(O–H)avg of [V6O5(MeCN)(OH2)]0 from equilibrium experiments (60.7 ± 0.1 kcal mol−1), oxidation potentials of this compound (−0.325 V, 0.222 V vs. Fc+/0, Fig. S12†), and CG, we find the average pKa for the protonated intermediate of [V6O6(MeCN)]0 (“[V6O5(MeCN)(OH2)]2+”) to be 1.45. This indicates that the complex is a significantly weaker base than [Phen]2− and thus will not deprotonate H2Phen.
Additional evidence in support of a CPET mechanism is provided in the assessment of activation parameters obtained from variable temperature kinetics experiments. Construction of an Eyring plot allows for the determination of activation parameters for the rate-determining step of the PCET reaction (Fig. 5, and Table 2). We find that the activation entropy, ΔS‡, of net H-atom uptake reactions at the surface of [V6O7]0 and [V6O6(MeCN)]0 are similarly large in magnitude and negative in sign, while the enthalpic contributions to the overall activation barrier, ΔH‡, are comparatively small. These results suggest that the rate-determining step involves an inner-sphere process with a highly ordered transition state. Similar observations have been made in previous examples of net H-atom transfer reactivity invoked to occur through a CPET-type mechanism.14–16,18,20,42,43 Collectively, these findings support the hypothesis that generation of the aquo moiety in [V6O5(MeCN)(OH2)]0 occurs via a rate-determining CPET process.
 | ||
| Fig. 5 Eyring plot for the reaction of [V6O6(MeCN)]0 (0.75 mM) with H2Phen (7.3 mM) in MeCN between −35 and 15 °C. Y-Axis values were determined by dividing kobs by the concentration of H2Phen, providing the rate constant, kPCET. Activation parameters are labelled on the plot. | ||
With mechanistic insights in hand, we sought to evaluate how the O-atom defect site alters the rate of PCET to the cluster surface. The rate constant for the reduction of [V6O6(MeCN)]0 from H2Phen can be extrapolated from the Eyring plot at a given temperature, which was determined to be 19.6 ± 3.2 M−1 s−1 at 25 °C (Fig. 5 and Table 2). This corresponds with a 100-fold increase in reaction rate over that reported for [V6O7]0 at the same temperature (0.14 ± 0.05 M−1 s−1).16
The acceleration in rate of net H-atom uptake at [V6O6(MeCN)]0 is striking considering the reduction of thermodynamic driving force for PCET (smaller ΔBDFE(O–H), vide supra). To justify this observation, we considered the activation parameters derived from Eyring analysis (Table 2). As described above, the entropy of activation (ΔS‡) in both reactions are large, negative values. Our lab and others have shown that ΔS‡ values of this magnitude are consistent with a well-ordered intermediate, suggesting the existence of a hydrogen bonded complex between the H-atom donor and acceptor in the transition state.14–16,18,20,42,43 The observation of comparable values of ΔS‡ is unsurprising, as both transition states involve the preorganization of reductant and cluster in close proximity. However, in comparing the enthalpies of activation (ΔH‡) of H-atom transfer to fully oxygenated and O-atom deficient POV–alkoxide clusters, we note a significant discrepancy; while both values are consistent with previously reported CPET reactions to metal oxide surfaces, the small ΔH‡ associated with formation of [V6O5(MeCN)(OH2)]0 indicates that less energy is required for bond weakening processes relevant to the formation of the transition state.14–16,18,20,42,43
We attribute the reduction in ΔH‡ to the increased electron density in the core of the oxygen-deficient POV–alkoxide cluster, [V6O6(MeCN)]0. Injection of reducing equivalents into POV–alkoxide clusters has been previously shown to increase the basicity of terminal VO units;44 similarly, the electron donor character of the O-deficient VIII centre should lead to greater nucleophilicity of the remaining oxygenated sites. Indeed, determination of the average pKa's of the corresponding acids (represented as “[V–OH2]2+” in the thermochemical square scheme (Scheme 3)) of [V6O7]0 (pKa = −1.98) and [V6O6(MeCN)]0 (pKa = 1.45) found that the defect-containing species was more basic than its fully oxygenated congener (Table S6†). The rate of CPET has been shown in several reports to be tuneable with the pKa of the proton or H-atom acceptor, with more basic acceptors producing accelerated reaction rates.45–48 This is due to the influence of pKa on ΔBDFE(E–H), as well as transition state thermodynamics. In a variety of cases, donor/acceptor pKa has been shown to correlate more strongly to PCET rate constants than ΔBDFE(E–H), describing an imbalanced transition state with more proton transfer character and an asynchronous CPET mechanism.47,49–55 An example from Hammes–Schiffer and coworkers describes the influence of proton transfer free energy 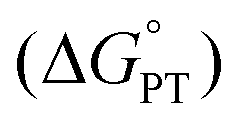 , related to pKa, on CPET in fluorenyl-benzoates, revealing that this thermodynamic parameter contributes to the proton donor–acceptor distance in the transition state.56 By increasing the relative basicity of the acceptor, this distance is minimized, decreasing the energetic requirement to facilitate proton tunnelling. In the present study, the increased surface basicity of the O-deficient POV–alkoxide is then expected to produce an H-bond with H2Phen with lower energetic cost than its fully-oxygenated partner, supported by a lower ΔH‡. Therefore, despite the greater driving force exhibited for PCET to [V6O7]0, the more basic surface of the O-atom deficient POV–alkoxide results in an accelerated rate of PCET to [V6O6(MeCN)]0.
, related to pKa, on CPET in fluorenyl-benzoates, revealing that this thermodynamic parameter contributes to the proton donor–acceptor distance in the transition state.56 By increasing the relative basicity of the acceptor, this distance is minimized, decreasing the energetic requirement to facilitate proton tunnelling. In the present study, the increased surface basicity of the O-deficient POV–alkoxide is then expected to produce an H-bond with H2Phen with lower energetic cost than its fully-oxygenated partner, supported by a lower ΔH‡. Therefore, despite the greater driving force exhibited for PCET to [V6O7]0, the more basic surface of the O-atom deficient POV–alkoxide results in an accelerated rate of PCET to [V6O6(MeCN)]0.
Conclusions
Here, we present an alternative approach for tuning the thermodynamics and kinetics of net H-atom uptake and transfer at metal oxide surfaces by introducing a structurally distinct, redox-inactive VIII ion into the cluster structure via defect engineering. The reduced site in [V6O6(MeCN)]0 functions as an anionic dopant, injecting electron density into the cluster core.29,31 Comparative studies on the strength of surface O–H bonds formed upon reduction of a VV-oxo to a VIII-aquo in this O-deficient assembly with its more oxidized congener, [V6O7]0, find that this additional electron density serves to weaken these bonds by ∼2 kcal mol−1. Kinetic analyses of these systems reveal, however, that despite this decrease in driving force, the O-deficient species undergoes PCET at rates two orders of magnitude higher than the respective oxygenated complex. This system breaks the expected driving force/rate scaling relationship predicted by Marcus Theory as a consequence of the increased electron density in this cluster, which imparts higher relative basicity to the remaining VO moieties of [V6O6(MeCN)]0 and allows for facile access to the transition state. Taken together, this work reveals how doping can serve to tune PCET processes at molecular metal oxides, furthering our understanding of these systems and providing new perspectives on analogous processes in reducible metal oxide materials.
Experimental
General considerations
All manipulations were carried out in the absence of water and oxygen using standard Schlenk techniques or in a UniLab MBraun inert atmosphere drybox under a dinitrogen atmosphere. All glassware was oven-dried for a minimum of 4 h and cooled in an evacuated antechamber prior to use in the drybox. Solvents were dried and deoxygenated on a glass contour system (Pure Process Technology, LLC) and stored over 3 Å molecular sieves purchased from Fisher Scientific and activated prior to use. 5,10-Phenazine (Phen), hydrazobenzene (Hydz), 1–4-naphthoquinone (Naphth), and 1.6 M nbutyllithium in hexanes were purchased from Sigma-Aldrich and used as received. D2O was purchased from Cambridge Isotope Laboratories and used as received. [V6O7(OCH3)12]0,33 [V6O6(OCH3)12]0,29 1,4-dihydroxynaphthalene (H2Naphth),57 5,10-dihydrophenazine (H2Phen),58 and its deuterated analogue (D2Phen)16 were generated according to literature precedent.1H NMR spectra were recorded at 500 MHz on a Bruker DPX500 spectrometer locked on the signal of deuterated solvents. All chemical shifts were reported relative to the peak of the residual H signal in deuterated solvents. CD3CN and THF-d8 were purchased from Cambridge Isotope Laboratories, degassed by three freeze–pump–thaw cycles, or received in a glass ampule, and stored over fully activated 3 Å molecular sieves. UV-Vis-NIR spectroscopy was collected using an Agilent Cary 6000i spectrophotometer at 21 °C and −35 °C. Samples were prepared in the drybox in MeCN or THF and added to airfree cuvettes and sealed prior to removing from the drybox. All molar absorptivity values were determined by averaging spectra collected in triplicate at different concentrations. Kinetics experiments were carried out on an Agilent Cary 60 UV-Vis-NIR spectrophotometer held at desired temperatures using an Unisoku CoolSpek UV cryostat.
Cyclic voltammetry (CV) was performed using a BioLogic SP 150 potentiostat/galvanostat and the EC-Lab software suite. Glassy carbon discs (3 mm, CH Instruments, USA) were used as working electrodes. Working electrodes were polished using a micro cloth pad and 0.05 μM alumina powder. Potentials recorded during CV were measured relative to a nonaqueous Ag/Ag+ reference electrode with 1 mM AgNO3 and 100 mM [nBu4N][PF6] in MeCN (BASi) and ultimately referenced against the Fc+/0 couple using an internal reference. A platinum wire served as the counter electrode. All experiments were carried out at room temperature inside a nitrogen-filled glove box (MBraun, USA). All CV measurements were iR compensated at 85% with impedance taken at 100 kHz using the ZIR tool included with the EC-Lab software. CV experiments were conducted at 100 mV s−1 on solutions of either 2 or 1 mM analyte and 200 mM [nBu4N][PF6] supporting electrolyte in THF.
A single crystal of [V6O6(OH2)]0 was placed onto a thin glass optical fibre and mounted on a Rigaku XtaLAB Synergy-S Dualflex diffractometer equipped with a HyPix-6000HE HPC area detector for data collection at 100.00(10) K. A preliminary set of cell constants and an orientation matrix were calculated from a small sampling of reflections.59 A short pre-experiment was run, from which an optimal data collection strategy was determined. The full data collection was carried out using a PhotonJet (Cu) X-ray source with frame times of 0.55 and 2.20 seconds and a detector distance of 34 mm. Series of frames were collected in 0.50° steps in ω at different 2θ, κ, and ϕ settings. After the intensity data were corrected for absorption, the final cell constants were calculated from the xyz centroids of 26024 strong reflections from the actual data collection after integration.39 See Table S1 (ESI†) for additional crystal and refinement information. The structure was solved using SHELXT60 and refined using SHELXL.61 The space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) was determined based on intensity statistics. All non-hydrogen atoms were refined with anisotropic displacement parameters. All other hydrogen atoms were placed in ideal positions and refined as riding atoms with relative isotropic displacement parameters. The final full matrix least squares refinement converged to R1 = 0.0360 (F2, I > 2σ(I)) and wR2 = 0.1030 (F2, all data). Elemental analysis was performed on a PerkinElmer 2400 Series II Analyzer, at the CENTC Elemental Analysis Facility, University of Rochester.
was determined based on intensity statistics. All non-hydrogen atoms were refined with anisotropic displacement parameters. All other hydrogen atoms were placed in ideal positions and refined as riding atoms with relative isotropic displacement parameters. The final full matrix least squares refinement converged to R1 = 0.0360 (F2, I > 2σ(I)) and wR2 = 0.1030 (F2, all data). Elemental analysis was performed on a PerkinElmer 2400 Series II Analyzer, at the CENTC Elemental Analysis Facility, University of Rochester.
Synthesis of [V6O5(MeCN)2]0
(Method A) A 20 mL scintillation vial was charged with [V6O7]0 (0.034 g, 0.042 mmol), H2Phen (0.002 g, 0.085 mmol, 2 equiv.), and 6 mL of MeCN. The reaction was stirred for 30 min to ensure completion, after which the solvent was removed under reduced pressure to yield a brown solid. The solid was stirred in pentane (10 mL) for 30 min. The solid was then filtered and continuously washed with pentane until the filtrate ran clear (∼15 mL). The solid was then extracted with MeCN, and any volatiles were removed under vacuum to yield [V6O5(MeCN)2]0 (0.027 g, 0.033 mmol, 79%). Characterization of the product, [V6O5(MeCN)2]0, matched that previously reported by our research group.29(Method B) A 20 mL scintillation vial was charged with [V6O6(MeCN)]0 (0.018 g, 0.022 mmol), H2Phen (0.005 g, 0.024 mmol, 1.1 eq.) and 6 mL of MeCN. The reaction was stirred 30 min to ensure completion and the solvent was removed under reduced pressure to yield a red solid. The solid was stirred in pentane (10 mL) for 30 min to remove phenazine byproduct. The solid was then extracted in MeCN and volatiles were removed under vacuum to yield [V6O5(MeCN)2]0 (0.031 g, 0.016 mmol, 73%) Characterization of the product, [V6O5(MeCN)2]0, matched that previously reported by our research group.29
Synthesis of [V6O6(OH2)]0
A 20 mL scintillation vial was charged with [V6O7]0 (0.010 g, 0.013 mmol), Hydz (0.003 g, 0.014 mmol, 1 equiv.), and 6 mL of THF. The reaction was stirred for 2 h, after which the solvent was removed under reduced pressure to yield a brown solid. The solid was stirred in pentane (10 mL) for 30 min, and subsequently filtered and continuously washed with pentane until the filtrate ran clear. The solid was then extracted with THF, and any volatiles were removed under vacuum to yield the product, [V6O6(OH2)]0, in good yield (0.007 g, 0.009 mmol, 69%). Crystals suitable for X-ray analysis were grown from the vapour diffusion of pentane into a crude reaction mixture in THF. 1H NMR (500 MHz, THF-d8): δ = 29.6, 16.2, −11.5 ppm. UV–vis (THF): 385 nm (ε = 2748 M−1 cm−1), 544 nm (ε = 306 M−1 cm−1), 1029 nm (ε = 533 M−1 cm−1). Elemental analysis for V6O18C12H36·1OH2·0.15OC4H8 (MW: 802.88 g mol−1) Calc'd (%): C, 18.85; H, 4.92. Found (%): C, 18.920; H, 4.739.Synthesis of [V6O5(MeCN)(OH2)]0
A 20 mL scintillation vial was charged with [V6O6(MeCN)]0 (0.024 g, 0.029 mmol), H2Phen (0.006 g, 0.030 mmol, equiv), and 6 mL of THF. The reaction was stirred for 2 h to ensure completion, after which the solvent was removed under reduced pressure to yield a red solid. The solid was stirred in pentane (10 mL) for 30 min. The solid was then filtered and continuously washed with pentane until the filtrate ran clear. The solid was then extracted with THF, and any volatiles were removed under vacuum to yield [V6O5(MeCN)(OH2)]0 (0.021 g, 0.025 mmol, 87%). 1H NMR (500 MHz, THF-d8): δ 27.43, −2.35, −3.37, −7.17, −21.52 ppm. UV–vis (THF): 440 nm (ε = 625 M−1 cm−1), 535 nm (ε = 361 M−1 cm−1), 950 nm (ε = 98 M−1 cm−1). Elemental analysis for V6O17C12H36·1OH2·1OC4H8·0.1C2H3N (MW: 852.28 g mol−1) Calc'd (%): C, 22.83; H, 5.48, N, 0.16. Found (%): C, 22.732; H, 5.086; N, 0.250.Procedure for thermochemical analysis of the BDFE(O–H) of [V6O6(OH2)]0 and [V6O5(MeCN)(OH2)]0
Determination of the BDFE(O–H)avg of [V6O6(OH2)]0 was performed using reactions between [V6O7]0 and 0.5–2 equivalents of 1,4-dihydroxynaphthalene (H2NQ). 300 μL of cluster stock solution (14.2 mM) and the appropriate volume (116–348 μL) of reductant solution (24.4 mM) in THF-d8 were combined in a J. Young tube and sealed prior to removal from the glovebox for analysis. Reactions were allowed to equilibrate over 7 days at room temperature, tracking progress by 1H NMR (Fig. S6†). Upon equilibration, the relative concentrations of naphthoquinone (NQ) to H2NQ were determined by using the integrations of resonances corresponding with each compound and normalizing for the number of protons each signal represents (Table S3†). Upon determination of [H2NQ]/[NQ], the adjusted BDFE of the reductant was determined for each reaction using the following equation:where BDFEH2NQ is the average BDFE(O–H) of the O–H bonds in H2NQ (62.6 kcal mol−1),30n is the number of H-atom equivalents transferred (n = 2), and BDFEadj is the adjusted BDFE of the reductant O–H bonds under the reaction conditions.13 Averaging the observed BDFEadj values provides the equilibrium BDFEadj for the reductant, which is equivalent to that of the O–H bond of [V6O6(OH2)]0. This was found to be 62.3 ± 0.1 kcal mol−1 (Table S3†).
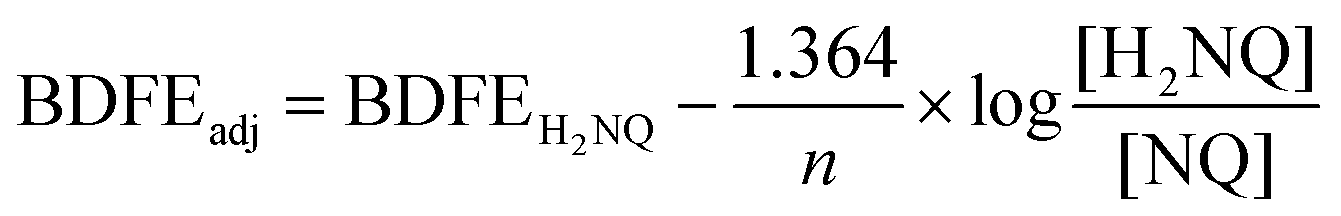
Determination of the BDFE(O–H)avg of [V6O5(MeCN)(OH2)]0 was performed using reactions between [V6O6(MeCN)]0 and 1 equivalent of Hydz in triplicate. 207 μL of cluster stock solution (7.73 mM) and 115 μL of reductant (13.9 mM) in THF-d8 and 78 μL of THF-d8 were combined in a J. Young tube and sealed prior to removal from the glovebox for analysis. Reactions were allowed to equilibrate over 15 days at room temperature, tracking progress by 1H NMR (Fig. S9†). Upon equilibration, the relative concentrations of azobenzene (Azo) to Hydz were determined by using the integrations of resonances corresponding with each compound and normalizing for the number of protons each signal represents (Table S4†). Upon determination of [Hydz]/[Azo], the adjusted BDFE of the reductant was determined for each reaction as above, where BDFEHydz = 60.4 kcal mol−130 and n = 2. Averaging the observed BDFEadj values finds the BDFE(O–H)avg of [V6O5(OH2)]0 to be 60.7 ± 0.1 kcal mol−1 (Table S4†).
Procedure for kinetic analyses of PCET at [V6O5(MeCN)(OH2)]0
Pseudo-first order reaction conditions were used to establish the rate constant of the PCET reaction between [V6O6(MeCN)]0 and H2Phen at −15 °C in MeCN. Using a UV-Vis-NIR spectrometer with an N2-cooled cryostat set to −15 °C, reactions between [V6O6(MeCN)]0 and excess H2Phen (5.0–13.5 equivalents) were tracked by monitoring the absorbance at 900 nm. Final reductant concentrations were varied from 3.75 to 10.16 mM, with a constant concentration of cluster of 0.75 mM. A 3 mL sample of [V6O6(MeCN)]0 was loaded in a quartz cuvette, sealed with a septum, and was allowed to equilibrate to −15 °C, at which time the spectrometer began collecting absorbance data. After ∼20 seconds, 0.3 mL of reductant stock solution (38.1 mM) was forcefully injected to ensure homogeneity in the sample. As the PCET reaction progressed, the absorbance decayed until the reaction reached completion, levelling to the absorbance for [V6O5(MeCN)2]0. The plots of absorbance over time were fit to the following equation by least squares fitting (Fig. S15–19†):| At = Af + (Ai − Af)e−kobst |
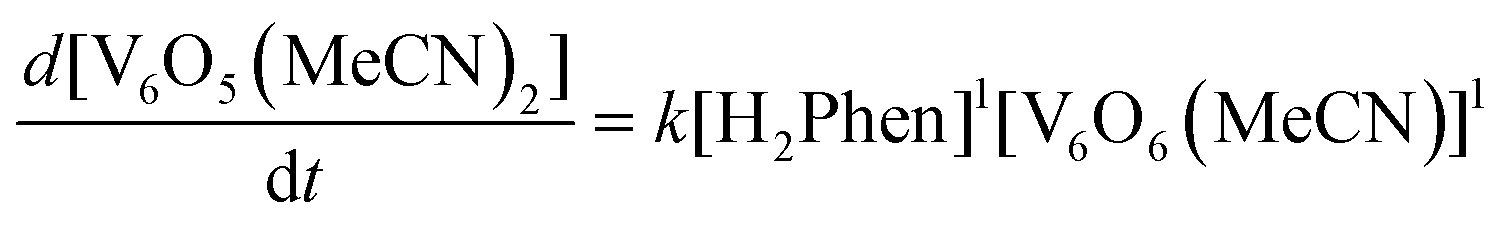
The slope of the line, when held to a y-intercept of 0, of 52.99 (R2 = 0.9973), provides the experimentally determined rate constant, kPCET at −15 °C of 6.6 ± 0.3 M−1 s−1, which accounts for the two H-atoms from the H2Phen and four possible VO reactive sites. To determine the kinetic isotope effect (KIE), analogous reactions were carried out at −15 °C using 3 mL samples of D2Phen solution between 3.75 and 10.1 mM, and 0.44 mL of cluster stock solution (5.14 mM) for a 0.75 mM final concentration (Fig. S20–24†). Experiments were repeated in triplicate. Similar treatment of the data produced a kPCET for the formation of deuterium-labelled species of 1.49 ± 0.05 M−1 s−1.
Procedure for determination of activation parameters
Eyring analysis was performed by collecting absorbance vs. time data at temperatures between −35 and 5 °C. Reactions were assembled in an analogous fashion to the above kinetics experiments, with constant reductant and cluster concentrations of 0.75 mM and 7.26 mM, respectively. Experiments were repeated in triplicate. Conversion of kobs to kPCET was done by dividing kobs by the reductant concentration and number of sites available for PCET (4 equatorial vanadyls) and number of H-atoms (2). Plotting ln(kPCET/T) as a function of 1/T (temperature converted to K), the linear plot was used to solve for activation parameters using the below equations where R is the gas constant in units of cal/(mol K), kBoltz is Boltzmann's constant, and hPlanck is Planck's constant. The activation parameters for the reduction of [V6O6(MeCN)]0 are listed in Fig. 5 and S25–28.†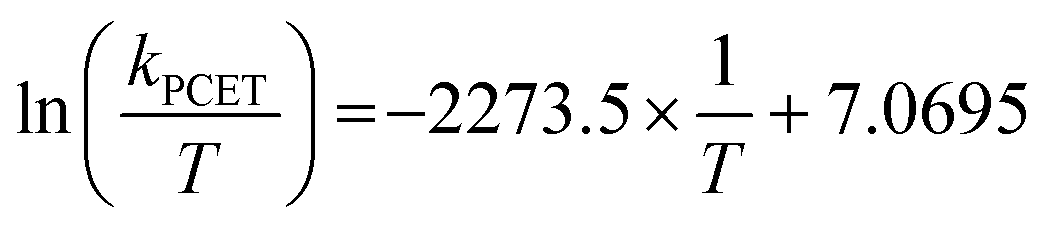
| ΔH‡ = −2273.5 × R |
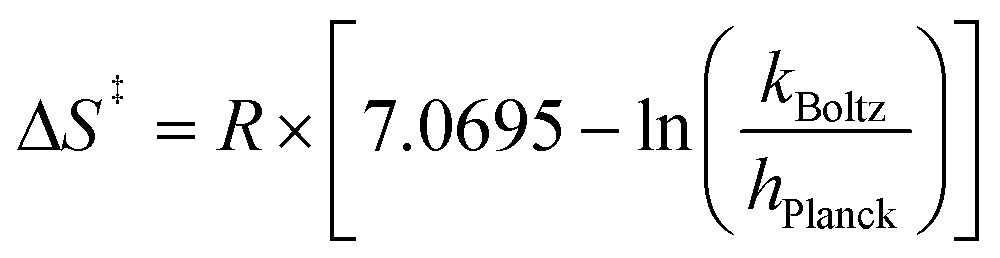
| ΔG‡ = ΔH‡ − TΔS‡. |
Author contributions
S.E.C, E.S., and E.M.M conceived all experiments. E.S. performed the synthesis and crystallization of [V6O6(OH2)]0, as well as all cyclic voltammetry and scanning kinetics experiments. All remaining syntheses and experimental data were performed and collected by S.E.C, except those involving X-Ray crystallography. W.W.B. collected single crystal X-Ray diffraction data and solved the crystal structure. All authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Science Foundation (CHE-2154727). E. M. M. is also a recipient of a Camille Dreyfus Teacher-Scholar Award from The Camille & Henry Dreyfus Foundation which has provided additional resources to support this work. E. S. is a recipient of the Messersmith Fellowship from the University of Rochester which has provided support for this study.References
- H. Ritchie and M. Roser, Our World in Data: Renewable Energy https://ourworldindata.org/renewable-energy.
- R. F. Service, Solar plus batteries is now cheaper than fossil power, Science, 2019, 365, 108 CrossRef CAS PubMed.
- U. S. E. I. Adminitration, International Energy Outlook 2020 https://www.eia.gov/outlooks/ieo/.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, Proton-Coupled Electron Flow in Protein Redox Machines, Chem. Rev., 2010, 110, 7024 CrossRef CAS PubMed.
- M. H. V. Huynh and T. J. Meyer, Proton-Coupled Electron Transfer, Chem. Rev., 2007, 107, 5004–5064 CrossRef CAS PubMed.
- B. E. Conway, Transition from “Supercapacitor” to “Battery” Behavior in Electrochemical Energy Storage, J. Electrochem. Soc., 1991, 138, 1539 CrossRef CAS.
- S. Fleischmann, J. B. Mitchell, R. Wang, C. Zhan, D.-e. Jiang, V. Presser and V. Augustyn, Pseudocapacitance: From Fundamental Understanding to High Power Energy Storage Materials, Chem. Rev., 2020, 120, 6738 CrossRef CAS PubMed.
- J. W. Darcy, B. Koronkiewicz, G. A. Parada and J. M. Mayer, A Continuum of Proton-Coupled Electron Transfer Reactivity, Acc. Chem. Res., 2018, 51, 2391 CrossRef CAS PubMed.
- J. L. Peper and J. M. Mayer, Manifesto on the Thermochemistry of Nanoscale Redox Reactions for Energy Conversion, ACS Energy Lett., 2019, 4, 866 CrossRef CAS.
- A. Ruiz Puigdollers, P. Schlexer, S. Tosoni and G. Pacchioni, Increasing Oxide Reducibility: The Role of Metal/Oxide Interfaces in the Formation of Oxygen Vacancies, ACS Catal., 2017, 7, 6493 CrossRef CAS.
- M. V. Ganduglia-Pirovano, A. Hofmann and J. Sauer, Oxygen vacancies in transition metal and rare earth oxides: Current state of understanding and remaining challenges, Surf. Sci. Rep., 2007, 62, 219 CrossRef CAS.
- R. G. Agarwal, H.-J. Kim and J. M. Mayer, Nanoparticle O–H Bond Dissociation Free Energies from Equilibrium Measurements of Cerium Oxide Colloids, J. Am. Chem. Soc., 2021, 143, 2896 CrossRef CAS PubMed.
- A. A. Fertig, W. W. Brennessel, J. R. McKone and E. M. Matson, Concerted Multiproton–Multielectron Transfer for the Reduction of O2 to H2O with a Polyoxovanadate Cluster, J. Am. Chem. Soc., 2021, 143, 15756 CrossRef CAS PubMed.
- S. E. Cooney, A. A. Fertig, M. R. Buisch, W. W. Brennessel and E. M. Matson, Coordination-induced bond weakening of water at the surface of an oxygen-deficient polyoxovanadate cluster, Chem. Sci., 2022, 13, 12726 RSC.
- E. Schreiber, A. A. Fertig, W. W. Brennessel and E. M. Matson, Oxygen-Atom Defect Formation in Polyoxovanadate Clusters via Proton-Coupled Electron Transfer, J. Am. Chem. Soc., 2022, 144, 5029 CrossRef CAS PubMed.
- S. Chakraborty, E. Schreiber, K. R. Sanchez-Lievanos, M. Tariq, W. W. Brennessel, K. E. Knowles and E. M. Matson, Modelling local structural and electronic consequences of proton and hydrogen-atom uptake in VO2 with polyoxovanadate clusters, Chem. Sci., 2021, 12, 12744 RSC.
- E. Schreiber, W. W. Brennessel and E. Matson, Regioselectivity of Concerted Proton-Electron Transfer at the Surface of a Polyoxovanadate Cluster, Chem. Sci., 2023, 14, 1386 RSC.
- S. Chakraborty, B. E. Petel, E. Schreiber and M. Ellen, Matson, Atomically precise vanadium-oxide clusters, Nanoscale Adv., 2021, 3, 1293 RSC.
- A. A. Fertig and E. M. Matson, Connecting Thermodynamics and Kinetics of Proton Coupled Electron Transfer at Polyoxovanadate Surfaces Using the Marcus Cross Relation, Inorg. Chem., 2023, 62, 1958 CrossRef CAS PubMed.
- C. Baffert, S. W. Feldberg, A. M. Bond, D.-L. Long and L. Cronin, pH-Dependence of the aqueous electrochemistry of the two-electron reduced α-[Mo18O54(SO3)] sulfite Dawson-like polyoxometalate anion derived from its triethanolammonium salt, Dalton Trans., 2007, 4599 RSC.
- P. J. S. Richardt, R. W. Gable, A. M. Bond and A. G. Wedd, Synthesis and Redox Characterization of the Polyoxo Anion, γ*-[S2W18O62]4−: A Unique Fast Oxidation Pathway Determines the Characteristic Reversible Electrochemical Behavior of Polyoxometalate Anions in Acidic Media, Inorg. Chem., 2001, 40, 703 CrossRef CAS PubMed.
- S.-X. Guo, S. W. Feldberg, A. M. Bond, D. L. Callahan, P. J. S. Richardt and A. G. Wedd, Systematic Approach to the Quantitative Voltammetric Analysis of the FeIII/FeII Component of the [α2-Fe(OH2)P2W17O61]7−/8− Reduction Process in Buffered and Unbuffered Aqueous Media, J. Phys. Chem. B, 2005, 109, 20641 CrossRef CAS PubMed.
- P. D. Prenzler, C. Boskovic, A. M. Bond and A. G. Wedd, Coupled Electron- and Proton-Transfer Processes in the Reduction of α-[P2W18O62]6− and α-[H2W12O40]6− As Revealed by Simulation of Cyclic Voltammograms, Anal. Chem., 1999, 71, 3650 CrossRef CAS PubMed.
- T. Ueda, K. Kodani, H. Ota, M. Shiro, S.-X. Guo, J. F. Boas and A. M. Bond, Voltammetric and Spectroscopic Studies of α- and β-[PW12O40]3− Polyoxometalates in Neutral and Acidic Media: Structural Characterization as Their [(n-Bu4N)3][PW12O40] Salts, Inorg. Chem., 2017, 56, 3990 CrossRef CAS PubMed.
- S. Himeno, M. Takamoto, R. Santo and A. Ichimura, Redox Properties and Basicity of Keggin-Type Polyoxometalate Complexes, Bull. Chem. Soc. Jpn., 2005, 78, 95 CrossRef CAS.
- T. Ueda, J.-i. Nambu, J. Lu, S.-X. Guo, Q. Li, J. F. Boas, L. L. Martin and A. M. Bond, Structurally characterised vanadium(V)-substituted Keggin-type heteropolysulfates [SVM11O40]3− (M = Mo, W): voltammetric and spectroscopic studies related to the V(V)/V(IV) redox couple, Dalton Trans., 2014, 43, 5462 RSC.
- V. W. Day, W. G. Klemperer and D. J. Maltbie, Where are the protons in H3V10O283−?, J. Am. Chem. Soc., 1987, 109, 2991 CrossRef CAS.
- B. E. Petel, A. A. Fertig, M. L. Maiola, W. W. Brennessel and E. M. Matson, Controlling Metal-to-Oxygen Ratios via MO Bond Cleavage in Polyoxovanadate Alkoxide Clusters, Inorg. Chem., 2019, 58, 10462 CrossRef CAS PubMed.
- R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. M. Mayer, Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications, Chem. Rev., 2022, 122, 1 CrossRef CAS PubMed.
- C. Daniel and H. Hartl, Neutral and Cationic VIV/VV Mixed-Valence Alkoxo-polyoxovanadium Clusters [V6O7(OR)12]n+ (R = −CH3, −C2H5): Structural, Cyclovoltammetric and IR-Spectroscopic Investigations on Mixed Valency in a Hexanuclear Core, J. Am. Chem. Soc., 2005, 127, 13978 CrossRef CAS PubMed.
- B. E. Petel, W. W. Brennessel and E. M. Matson, Oxygen-Atom Vacancy Formation at Polyoxovanadate Clusters: Homogeneous Models for Reducible Metal Oxides, J. Am. Chem. Soc., 2018, 140, 8424 CrossRef CAS PubMed.
- J. Spandl, C. Daniel, I. Brüdgam and H. Hartl, Synthesis and Structural Characterization of Redox-Active Dodecamethoxoheptaoxohexavanadium Clusters, Angew. Chem., Int. Ed., 2003, 42, 1163 CrossRef CAS PubMed.
- D. X. Ren, N. Xing, H. Shan, C. Chen, Y. Z. Cao and Y. H. Xing, Novel vanadium(III) complexes with rigid phenylpolycarboxylate ligands: synthesis, structures and application in C–H bond activation, Dalton Trans., 2013, 42, 5379 RSC.
- I. E. Grey, I. C. Madsen, K. Sirat and P. W. Smith, Structure of trans-diaquabis(oxalato)vanadate(III) complexes: A[V(C2O4)2(H2O)2]•xH2O, A = Cs (x = 4) and A = CH3NH3 (x = 4.5), Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 681 CrossRef.
- C. J. Reed and T. Agapie, A Terminal FeIII–Oxo in a Tetranuclear Cluster: Effects of Distal Metal Centers on Structure and Reactivity, J. Am. Chem. Soc., 2019, 141, 9479 CrossRef CAS PubMed.
- A. J. Ward, A. Ruseckas, M. M. Kareem, B. Ebenhoch, L. A. Serrano, M. Al-Eid, B. Fitzpatrick, V. M. Rotello, G. Cooke and I. D. W. Samuel, The Impact of Driving Force on Electron Transfer Rates in Photovoltaic Donor–Acceptor Blends, Adv. Mater., 2015, 27, 2496 CrossRef CAS PubMed.
- G. M. Loudon, Organic Chemistry, Roberts and Company, 2009 Search PubMed.
- F. G. Bordwell and M. J. Bausch, Acidity-oxidation-potential (AOP) values as estimates of relative bond dissociation energies and radical stabilities in dimethyl sulfoxide solution, J. Am. Chem. Soc., 1986, 108, 1979 CrossRef CAS.
- F. G. Bordwell, J. P. Cheng and J. A. Harrelson, Homolytic bond dissociation energies in solution from equilibrium acidity and electrochemical data, J. Am. Chem. Soc., 1988, 110, 1229 CrossRef CAS.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications, Chem. Rev., 2010, 110, 6961 CrossRef CAS PubMed.
- T. G. Carrell, P. F. Smith, J. Dennes and G. C. Dismukes, Entropy and enthalpy contributions to the kinetics of proton coupled electron transfer to the Mn4O4(O2PPh2)6 cubane, Phys. Chem. Chem. Phys., 2014, 16, 11843 RSC.
- N. Kindermann, C.-J. Günes, S. Dechert and F. Meyer, Hydrogen Atom Abstraction Thermodynamics of a μ-1,2-Superoxo Dicopper(II) Complex, J. Am. Chem. Soc., 2017, 139, 9831 CrossRef CAS PubMed.
- E. Schreiber, W. W. Brennessel and E. M. Matson, Charge-State Dependence of Proton Uptake in Polyoxovanadate-alkoxide Clusters, Inorg. Chem., 2022, 61, 4789 CrossRef CAS PubMed.
- C. J. Fecenko, H. H. Thorp and T. J. Meyer, The Role of Free Energy Change in Coupled Electron−Proton Transfer, J. Am. Chem. Soc., 2007, 129, 15098 CrossRef CAS PubMed.
- M. Bourrez, R. Steinmetz, S. Ott, F. Gloaguen and L. Hammarström, Concerted proton-coupled electron transfer from a metal-hydride complex, Nat. Chem., 2015, 7, 140 CrossRef CAS PubMed.
- J. W. Darcy, S. S. Kolmar and J. M. Mayer, Transition State Asymmetry in C–H Bond Cleavage by Proton-Coupled Electron Transfer, J. Am. Chem. Soc., 2019, 141, 10777 CrossRef CAS PubMed.
- T. Huang, E. S. Rountree, A. P. Traywick, M. Bayoumi and J. L. Dempsey, Switching between Stepwise and Concerted Proton-Coupled Electron Transfer Pathways in Tungsten Hydride Activation, J. Am. Chem. Soc., 2018, 140, 14655 CrossRef CAS PubMed.
- S. K. Barman, J. R. Jones, C. Sun, E. A. Hill, J. W. Ziller and A. S. Borovik, Regulating the Basicity of Metal–Oxido Complexes with a Single Hydrogen Bond and Its Effect on C–H Bond Cleavage, J. Am. Chem. Soc., 2019, 141, 11142 CrossRef CAS PubMed.
- D. Usharani, D. C. Lacy, A. S. Borovik and S. Shaik, Dichotomous Hydrogen Atom Transfer vs Proton-Coupled Electron Transfer During Activation of X–H Bonds (X = C, N, O) by Nonheme Iron–Oxo Complexes of Variable Basicity, J. Am. Chem. Soc., 2013, 135, 17090 CrossRef CAS PubMed.
- M. K. Goetz and J. S. Anderson, Experimental Evidence for pKa-Driven Asynchronicity in C–H Activation by a Terminal Co(III)–Oxo Complex, J. Am. Chem. Soc., 2019, 141, 4051 CrossRef CAS PubMed.
- S. K. Barman, M.-Y. Yang, T. H. Parsell, M. T. Green and A. S. Borovik, Semiempirical method for examining asynchronicity in metal–oxido-mediated C–H bond activation, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2108648118 CrossRef CAS PubMed.
- J. E. Schneider, M. K. Goetz and J. S. Anderson, Statistical analysis of C–H activation by oxo complexes supports diverse thermodynamic control over reactivity, Chem. Sci., 2021, 12, 4173 RSC.
- C. E. Elwell, M. Mandal, C. J. Bouchey, L. Que, Jr., C. J. Cramer and W. B. Tolman, Carboxylate Structural Effects on the Properties and Proton-Coupled Electron Transfer Reactivity of [CuO2CR]2+ Cores, Inorg. Chem., 2019, 58, 15872 CrossRef CAS PubMed.
- S. C. Coste, A. C. Brezny, B. Koronkiewicz and J. M. Mayer, C–H oxidation in fluorenyl benzoates does not proceed through a stepwise pathway: revisiting asynchronous proton-coupled electron transfer, Chem. Sci., 2021, 12, 13127 RSC.
- E. R. Sayfutyarova, Y.-C. Lam and S. Hammes-Schiffer, Strategies for Enhancing the Rate Constant of C–H Bond Cleavage by Concerted Proton-Coupled Electron Transfer, J. Am. Chem. Soc., 2019, 141, 15183 CrossRef CAS PubMed.
- T. Yamakado, S. Takahashi, K. Watanabe, Y. Matsumoto, A. Osuka and S. Saito, Conformational Planarization versus Singlet Fission: Distinct Excited-State Dynamics of Cyclooctatetraene-Fused Acene Dimers, Angew. Chem., Int. Ed., 2018, 57, 5438 CrossRef CAS PubMed.
- J. Lee, K. Shizu, H. Tanaka, H. Nakanotani, T. Yasuda, H. Kaji and C. Adachi, Controlled emission colors and singlet–triplet energy gaps of dihydrophenazine-based thermally activated delayed fluorescence emitters, J. Mater. Chem. C, 2015, 3, 2175 RSC.
- CrysAlisProf, version 171.41.97a, Rigaku Corporation, Oxford, UK, 2021 Search PubMed.
- G. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR of complexes and reactions, X-ray crystal structure parameters, bond-valence sum calculations, and electronic absorption spectrum of [V6O6(OH2]0, and kinetic analyses for the reduction of [V6O6(MeCN)]0. CCDC 2234804. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi00129f |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2023 |