
Asymmetric modification of Anderson-type polyoxometalates towards organic–inorganic homo- and hetero-cluster oligomers†
Meng-Meng
Zhang
a,
Yi-An
Yin
a,
Wu-Ji
Chen
a,
Chang-Gen
Lin
 *a,
Yongge
Wei
*b and
Yu-Fei
Song
*a
*a,
Yongge
Wei
*b and
Yu-Fei
Song
*a
aState Key Laboratory of Chemical Resource Engineering, Beijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: linchg@mail.buct.edu.cn; songyf@mail.buct.edu.cn
bDepartment of Chemistry, Tsinghua University, Beijing 100084, P. R. China. E-mail: yonggewei@mail.tsinghua.edu.cn
First published on 7th December 2022
Abstract
The preparation of asymmetrically modified polyoxometalates (POMs) bearing two different functional sites is of great interest, since it allows for the rational design and controlled synthesis of novel POM-based hybrids and further development of multi-POM supramolecular architectures. Herein, we report a simple and powerful synthetic approach for the isolation of two asymmetric Anderson hybrids that can be sequentially functionalized to produce organic–inorganic cluster oligomers. It is found that the type of tripodal alcohol can significantly affect the modification products, highlighting the importance of amide-functionalized tris derivatives, which selectively generate the target asymmetric hybrids, while the other tripodal alcohols tried in this work inevitably lead to symmetric by-products. Based on the obtained asymmetric hybrids, we further develop a modular synthetic methodology for stepwise coupling of Anderson clusters to form linear cluster oligomers. Moreover, this modular coupling methodology provides versatile intermediates that can be used to bind different clusters together via covalent approaches. As a proof of concept, a hetero-cluster of a Keggin–Anderson trimer has been prepared and carefully characterized. As such, this work provides a promising approach for the development of functional asymmetric hybrids and controlled synthesis of metal-oxo cluster oligomers with precise cluster numbers and chain length.
Introduction
Polyoxometalates (POMs) are all-inorganic metal-oxo clusters with highly diverse, although well-defined, structures that range in size from nano- to micrometer dimensions.1,2 The remarkable physical–chemical properties of POMs make them potentially useful in a wide range of applications such as catalysis, photo-sensitizers, energy conversion and storage, and materials science.3–8 The integration of organic moieties and POMs to create complex hybrid assemblies is a fascinating and rapidly growing area in the POM field.9–11 Although the supramolecular assemblies of POM-based hybrids often rely on the electrostatic interactions of POM anions and cationic organo-components, the covalent modification of POMs through stronger chemical bonds has attracted considerable attention, owing to a better understanding of the local structures and the close control of the organic and inorganic components to achieve a synergistic marriage of properties inherent to each constituent.12–14Up to now, the covalent functionalization of POMs has overwhelmingly concentrated on symmetric systems,15–30 where two identical organic components are anchored onto an inorganic core. Only a few examples of asymmetrically modified POM hybrids have been reported,31–37 largely because of the significant challenges in their synthesis and purification. Asymmetric POM hybrids, compared with their symmetric counterparts, not only allow for the fine tuning of molecular structures to achieve suitable building blocks that can be incorporated into peptide chains38 or metal oxide oligomers,39 but also provide controlled modulation of solid surfaces to study selective cell adhesion.40 In 2008, Song and Cronin et al. reported the first example of an asymmetrically modified Anderson cluster via a traditional one-pot method in the presence of two different tripodal alcohols.31 The isolation of the target asymmetric compound was achieved by a method of fractional crystallization coupled with mass spectrometry, unambiguously verifying the purity of the obtained hybrids. Cronin et al. later developed a universal protocol for the purification of asymmetric Mn-Anderson hybrids through reverse-phase liquid chromatography.35 However, this method requires demanding and expensive instruments, and multi-step organic synthesis, including the protection and de-protection of functional groups.
Recently, using the primary Anderson cluster [X(OH)6M6O18]3− (X = CrIII, AlIII, MnIII, etc.) and tripodal alcohols, the preparation of covalently modified Anderson hybrids has been achieved under refluxing,41–45 hydrothermal,46–48 and even microwave49 conditions in aqueous solution. The modification mechanism involves an esterification reaction between tripodal alcohols and the protonated primary Anderson cluster. A distinctive advantage of this method lies in the fact that it allows for controlled modification of the Anderson cluster with only one side occupied. The other side, left unaffected, can further be accessed by a different tripodal alcohol to form an asymmetric hybrid. As such, a stepwise method for asymmetric modification of primary Anderson clusters has been proposed.45,46 However, after carefully checking the method reported in the literature, it is found that (1) the obtained asymmetric hybrids to some extent contain impurities, i.e., symmetric by-products, and the purification procedure is not carefully discussed and clearly demonstrated, (2) the effects of the selected tripodal alcohols on the purification process of asymmetric Anderson clusters have not been studied, and (3) the post-functionalization of the asymmetric Anderson hybrids towards versatile building blocks that could be integrated into homo- and hetero-cluster oligomers remains unexplored.
In this context, we report here the synthesis, purification, and characterization of two novel asymmetrically functionalized Anderson hybrids by varying the tripodal alcohol type and the modification sequence in pursuit of exploiting the synthetic parameter space for asymmetric molecules. To isolate the target asymmetric hybrids from symmetric by-products we systematically investigate the solubility of the modified hybrids and optimize the purification process by carefully selecting the anchoring tripodal alcohols. The obtained asymmetric hybrids, containing a reactive site on one side of the Anderson plate, are further transformed to amide-functionalized tris derivatives that are subsequently used as coupling building blocks to selectively generate linear Anderson dimers and trimers. A hetero-cluster trimer has also been successfully prepared using an NHS-activated asymmetric Anderson cluster and an amino-functionalized Keggin cluster.
Experimental
Materials
All the chemicals and solvents used were of analytical grade, purchased from Shanghai Energy Chemical and used without further purification. Na3[Al(OH)6Mo6O18]·(H2O)6·2H2O,50 [(C4H9)4N]3{Al-(OH)3Mo6O18[(OCH2)3CCH2OH]} (Al-OH),46 [(C4H9)4N]3{AlMo6-O18[(OCH2)3CCH2OH]2} (HO-Al-OH),51 [(C4H9)4N]4{SiW11O40[Si(CH2)3NH2]2} (SiW11-NH2),52 and 2-(hydroxyl-methyl)-2-(4-nitrophenyl)-propane-1,3-diol (tris-NO2)53 were prepared and characterized according to the literature methods.[(C4H9)4N]3{AlMo6O18[(OCH2)3CCH2OH][(OCH2)3CC6H4NO2]} (1)
Route 1: Al-OH (901.1 mg, 0.5 mmol) was dissolved in 30 mL ethanol. To this solution tris-NO2 (136.2 mg, 0.6 mmol) was added. The resulting solution was refluxed for 6 hours and then cooled down to r.t. The white precipitates formed during the reaction were filtered and dried in air to give the crude product. Yield: 85.0%. 1H-NMR (400 MHz, DMSO-d6): δ = 8.24 (s, 2H), 7.52 (s, 2H), 4.79 (s, 2H), 4.74 (s, 3H), 4.43 (s, 3H), 4.38 (s, 3H), 3.25–3.07 (m, 24H), 3.00 (d, J = 5.6 Hz, 1H), 2.96 (d, J = 5.5 Hz, 1H), 1.58 (p, J = 8.0 Hz, 24H), 1.32 (h, J = 7.3 Hz, 24H), 0.94 (t, J = 7.3 Hz, 36H).Route 2: Al-NO2 (946.5 mg, 0.5 mmol) was dissolved in 30 mL ethanol and then pentaerythritol (81.7 mg, 0.6 mmol) was added. The resulting solution was refluxed for 6 hours and then cooled down to r.t. The white precipitates formed during the reaction were filtered and dried in air to give the crude product. Yield: 80.0%. 1H-NMR (400 MHz, DMSO-d6): δ = 8.26 (d, J = 8.9 Hz, 1H), 7.51 (d, J = 9.0 Hz, 2H), 4.79 (s, 2H), 4.73 (s, 3H), 4.42 (s, 3H), 4.37 (s, 5H), 3.24–3.09 (m, 24H), 2.98 (d, J = 15.7, 5.6 Hz, 3H), 1.58 (p, J = 7.9 Hz, 24H), 1.32 (h, J = 7.3 Hz, 24H), 0.94 (t, J = 7.3 Hz, 36H).
The purification of asymmetric 1
First of all, the crude product (1.2 g, ca. 0.6 mmol) was refluxed in 150 mL acetone for 5 hours. After cooling down to r.t., the precipitate, which turned out to be symmetric HO-Al-OH by 1H-NMR, was removed by centrifugation. The resulting solution was evaporated to give the intermediate product that only contained asymmetric 1 and symmetric NO2-Al-NO2.Second, in order to remove the symmetric NO2-Al-NO2, the intermediate product (820 mg, ca. 0.4 mmol) was dissolved in 15 mL of acetonitrile and then cation-exchanged into sodium salts by adding NaClO4 (500 mg, ca. 10 equiv.) to the above solution. Thereafter, the sodium salts of the mixture of 1 and NO2-Al-NO2 were centrifuged and re-dissolved in 15 mL water. TBA-Br (210 mg, 0.65 mmol) in 0.5 mL H2O was added dropwise to the above solution. White precipitates began to form upon adding and were removed by centrifugation after adding. The obtained precipitates were proved to be the symmetric NO2-Al-NO2 by-product by 1H-NMR.
Finally, excessive TBA-Br was added into the filtrate to get the target asymmetric 1. Total yield: 28.3%. 1H-NMR (400 MHz, DMSO-d6): δ = 8.26 (d, J = 9.0 Hz, 2H), 7.51 (d, J = 9.1 Hz, 2H), 4.74 (s, 6H), 4.43 (s, 6H), 3.25–3.10 (m, 24H), 3.00 (d, J = 5.6 Hz, 2H), 1.58 (p, J = 8.0 Hz, 24H), 1.32 (h, J = 7.3 Hz, 24H), 0.94 (t, J = 7.3 Hz, 36H). FT-IR (KBr, cm−1): 3434 (br), 2961 (s), 2875 (s), 1660 (m), 1602 (w), 1524 (m), 1481 (s), 1382 (m), 1349 (m), 1133 (w), 1073 (m), 1022 (m), 940 (vs), 924 (vs), 852 (m), 669 (vs), 578 (m), 526 (m), 472 (m), 448 (m). ESI-TOF-MS (neg. mode, CH3CN): 1733.00 ({(TBA)2{AlMo6O18[(OCH2)3CCH2OH][(OCH2)3CC6H4 NO2]}}−, calcd 1732.97), 744.36 ({(TBA){AlMo6-O18[(OCH2)3C-CH2OH][(OCH2)3CC6H4NO2]}}2−, calcd 745.26). Elemental analysis (%) calcd for C63H127AlMo6N4O27: C 38.27, H 6.43, N 2.83; found: C 38.35, H 6.28, N 2.94. Single-crystals suitable for X-ray were obtained by diethyl ether diffusion into an acetonitrile solution of 1 for 3 days.
Results and discussion
The asymmetric modification of the Anderson cluster
Previous studies showed that tripodal alcohols such as tris(hydroxymethyl)-amino-methane (tris-NH2) and pentaerythritol (tris-OH) can be anchored onto Anderson clusters to generate single-side modified POM hybrids.41,42 A stepwise method was proposed to develop Anderson clusters with different tripodal appendants.45,46 Encouraged by these studies, we here used a synthesized tripodal alcohol, 2-(hydroxyl-methyl)-2-(4-nitro-phenyl)propane-1,3-diol (tris-NO2), besides tris-NH2 and pentaerythritol, to explore the effects of alcohol types and reaction sequences on the stepwise modification of Anderson clusters.As shown in Fig. 1a, tris-NO2 and pentaerythritol were examined. In route 1 the pentaerythritol modified Al-Anderson hybrid, [(C4H9)4N]3{Al(OH)3Mo6O18[(OCH2)3CCH2OH]} (Al-OH), was refluxed with tris-NO2 to give the crude product, which was precipitated from the reaction solution with a yield of ca. 85.0%. 1H-NMR spectra showed that the obtained compound contained two symmetric by-products, i.e., HO-Al-OH and NO2-Al-NO2, and it was difficult to get pure asymmetric 1 through this one-step reaction (Fig. 1b). As can be observed, the –CH2O– moieties of symmetric NO2-Al-NO2 showed a single peak at 4.78 ppm, and those of HO-Al-OH exhibited a peak at 4.38 ppm. In the case of the crude product, two other sets of peaks, which belonged to the target asymmetric 1, were observed at 4.74 and 4.43 ppm. The ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HO-Al-OH:NO2-Al-NO2 in the crude mixture was nearly 2:1:1 according to the integration of the 1H-NMR spectrum. ESI-TOF-MS, which provided a soft ionization of the POM hybrids and effectively detected the covalently modified POM anions in negative mode, verified this finding. As shown in Fig. S4,† the molecular ion peak observed at m/z 1733.00 belonged to the target asymmetric product 1 ({(TBA)2{AlMo6O18[(OCH2)3CCH2OH][(OCH2)3CC6H4NO2]}}−), while the peaks found at 1641.99 and 1825.01 were attributed to symmetric HO-Al-OH ({(TBA)2{AlMo6O18[(OCH2)3CCH2OH]2}}−) and NO2-Al-NO2 ({(TBA)2{AlMo6O18[(OCH2)3CC6H4NO2]2}}−), respectively. The crude product obtained via route 2 showed the same results (Fig. S5†). We followed the reaction by monitoring the precipitates formed during refluxing using 1H-NMR. It is found that the symmetric by-products exist once the precipitates begin to form, suggesting that the reaction intermediates were in solution and might be detected using in situ techniques. We therefore examined the stability of Al-OH in ethanol upon refluxing. It was found that the Al-OH had undergone solvolysis in ethanol and resulted in symmetric HO-Al-OH after refluxing for 3 hours (Fig. S6†). The decomposition or reassembly of the Anderson cluster might also take place as indicated by the newly formed proton peaks. Asymmetrically modified Anderson cluster 2, obtained from tris and Al-NO2, contained a symmetric by-product. However, in this case only symmetric NO2-Al-NO2 was observed (Fig. S9†). These findings gave us a clue that it was the type of tripodal alcohol rather than the modification sequence that affected the asymmetric functionalization (vide infra).
:HO-Al-OH:NO2-Al-NO2 in the crude mixture was nearly 2:1:1 according to the integration of the 1H-NMR spectrum. ESI-TOF-MS, which provided a soft ionization of the POM hybrids and effectively detected the covalently modified POM anions in negative mode, verified this finding. As shown in Fig. S4,† the molecular ion peak observed at m/z 1733.00 belonged to the target asymmetric product 1 ({(TBA)2{AlMo6O18[(OCH2)3CCH2OH][(OCH2)3CC6H4NO2]}}−), while the peaks found at 1641.99 and 1825.01 were attributed to symmetric HO-Al-OH ({(TBA)2{AlMo6O18[(OCH2)3CCH2OH]2}}−) and NO2-Al-NO2 ({(TBA)2{AlMo6O18[(OCH2)3CC6H4NO2]2}}−), respectively. The crude product obtained via route 2 showed the same results (Fig. S5†). We followed the reaction by monitoring the precipitates formed during refluxing using 1H-NMR. It is found that the symmetric by-products exist once the precipitates begin to form, suggesting that the reaction intermediates were in solution and might be detected using in situ techniques. We therefore examined the stability of Al-OH in ethanol upon refluxing. It was found that the Al-OH had undergone solvolysis in ethanol and resulted in symmetric HO-Al-OH after refluxing for 3 hours (Fig. S6†). The decomposition or reassembly of the Anderson cluster might also take place as indicated by the newly formed proton peaks. Asymmetrically modified Anderson cluster 2, obtained from tris and Al-NO2, contained a symmetric by-product. However, in this case only symmetric NO2-Al-NO2 was observed (Fig. S9†). These findings gave us a clue that it was the type of tripodal alcohol rather than the modification sequence that affected the asymmetric functionalization (vide infra).
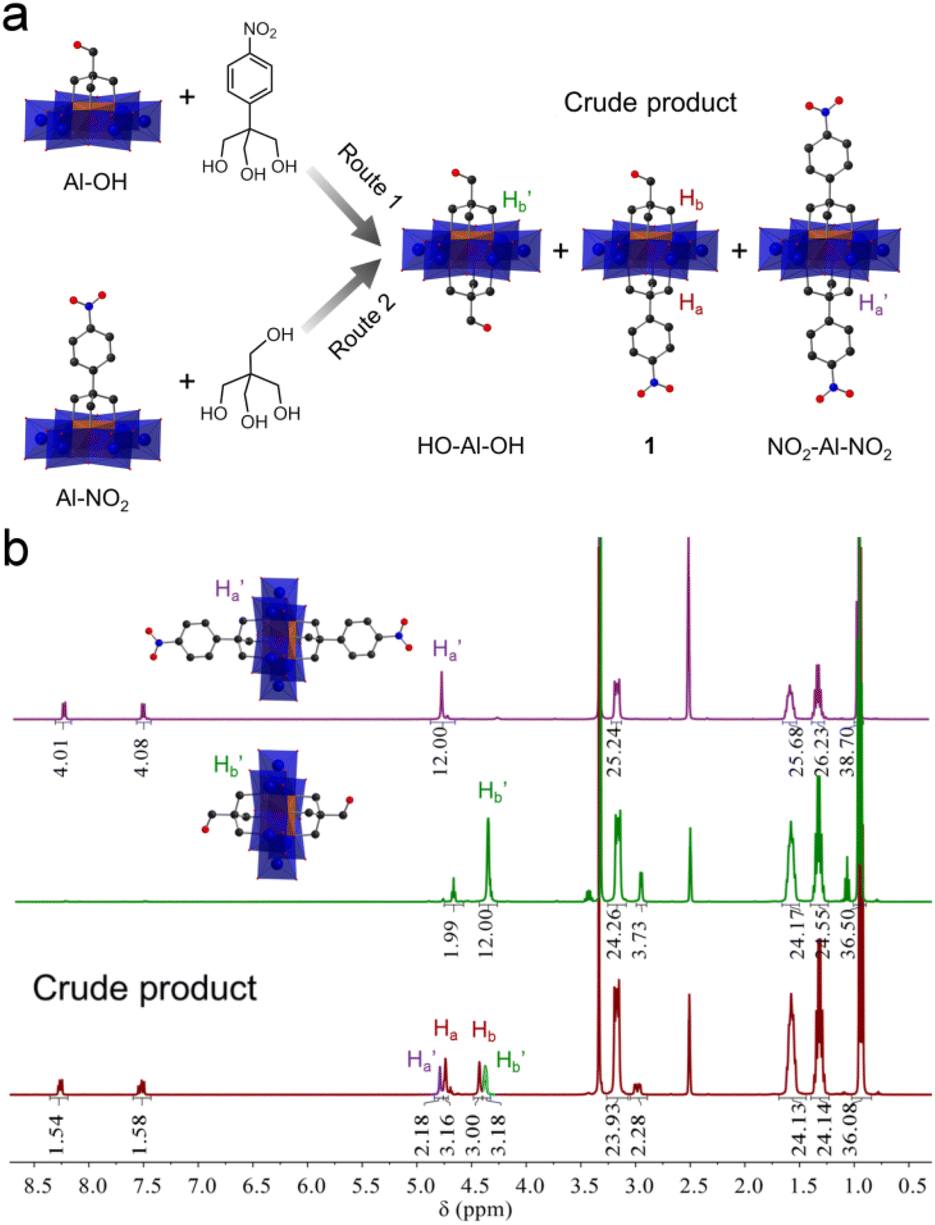 | ||
| Fig. 1 (a) Schematic representation of the synthetic routes to asymmetric Anderson hybrid 1. Color code: C, black; N, blue; O, red. The {MoO6} octahedron is show in blue and {AlO6} in brown. (b) 1H-NMR spectra of symmetric NO2-Al-NO2 (top, purple), symmetric HO-Al-OH (middle, green), and the crude product obtained via route 1 (bottom, red). | ||
In order to get the asymmetric product 1, a stepwise purification process was developed (Fig. 2a). In the first step, symmetric HO-Al-OH was removed from the crude product due to its poor solubility in acetone (Fig. 2b). Thereafter, the mixture of 1 and NO2-Al-NO2 (in a ratio of ca. 2:1) was cation-exchanged into sodium salts in the presence of excessive sodium perchlorate. After dissolving the obtained sodium salts in water, TBA-Br (3 equiv. of NO2-Al-NO2) was slowly added to the above solution to quantitatively precipitate NO2-Al-NO2. Finally, the pure asymmetric product 1 was obtained by adding excessive TBA-Br to the supernatant. The purity and composition of 1 were confirmed by 1H-NMR (Fig. 2b). Single crystals can be obtained by slow diethyl ether diffusion into an acetonitrile solution of 1. X-ray diffraction reveals that 1 crystallizes in a monoclinic system with the P21/c space group. The geometry of asymmetric 1 shows a common Anderson-type framework: six edge-sharing {MoO6} octahedra are arranged around a central {AlO6} unit and all the μ3-O have been replaced by tripodal alcohols (Fig. 3a). The Al–O distance is in the range of 1.878–1.886 Å, which is slightly shorter than the mean Al–O distance (1.896 Å) in the single-side modified Al-Anderson hybrid.42 It should be noted that the two tripodal appendants in the crystal structure exhibit position disorders (Fig. 3b) and the occupancy of each is fixed to 0.5 during refinement. ESI-TOF-MS also proves the purity of 1. The ion peaks observed at m/z 744.36 and 1733.00 can be ascribed to {(TBA){AlMo6O18[(OCH2)3CCH2OH][(OCH2)3CC6H4NO2]}}2− and {(TBA)2{AlMo6O18[(OCH2)3CCH2OH][(OCH2)3CC6H4NO2]}}−, respectively (Fig. 3e). The FT-IR spectrum of 1 shows typical Anderson cluster vibrations (Fig. S7†). The Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and Mo–O–Mo bonds are found at 925 and 670 cm−1, respectively, while the –NO2 and C–O bonds are seen at 1348 and 1094 cm−1, respectively. These results clearly indicate that the tripodal alcohols have been successfully tethered onto the Anderson cluster.
O and Mo–O–Mo bonds are found at 925 and 670 cm−1, respectively, while the –NO2 and C–O bonds are seen at 1348 and 1094 cm−1, respectively. These results clearly indicate that the tripodal alcohols have been successfully tethered onto the Anderson cluster.
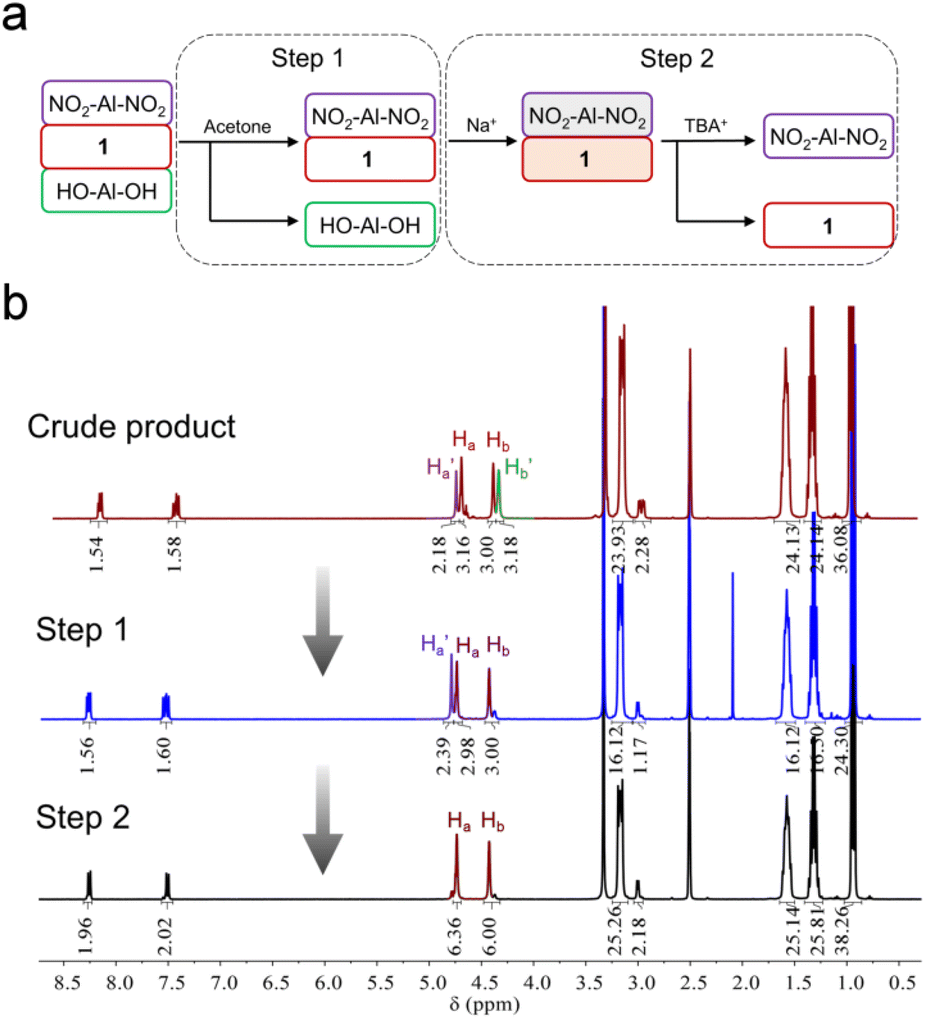 | ||
| Fig. 2 (a) Flow chart representation of the purification process of asymmetric 1 and (b) the corresponding 1H-NMR spectra of the compounds obtained at each purification step. | ||
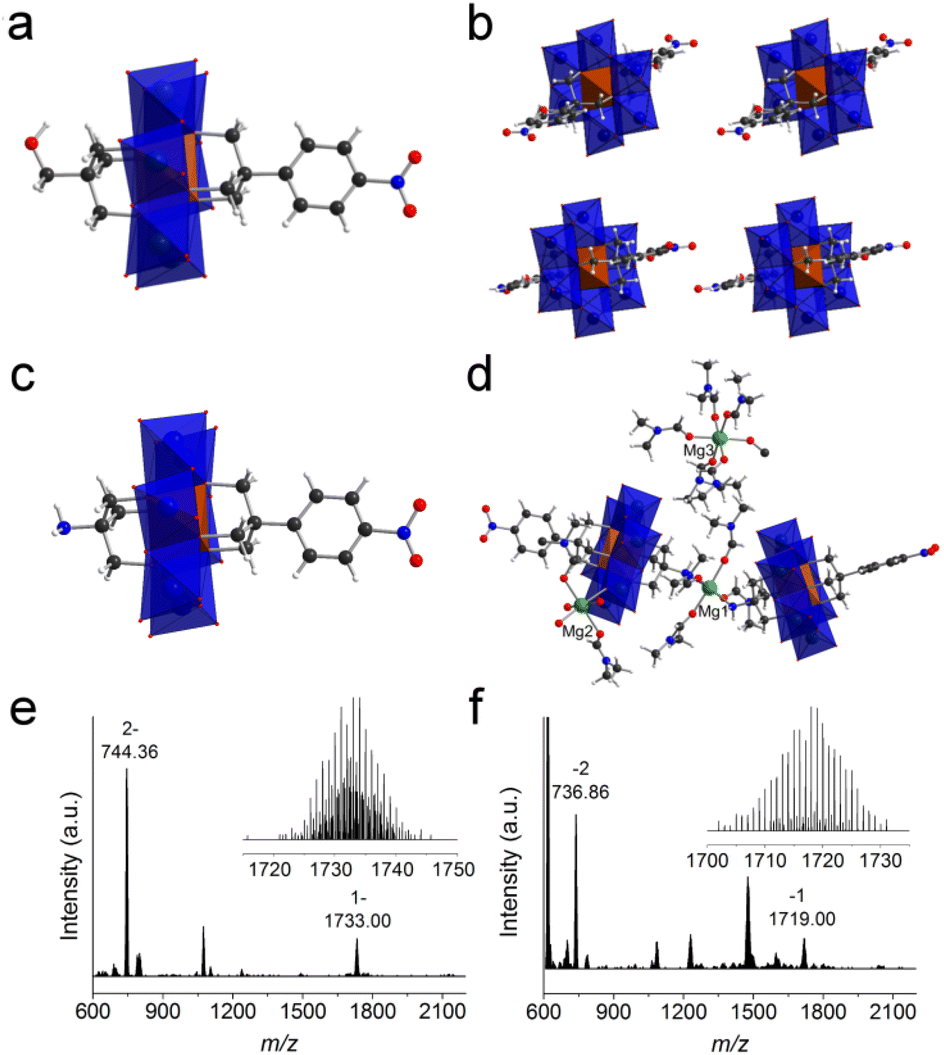 | ||
| Fig. 3 (a) The single-crystal structure of 1, (b) the crystal packing of 1 viewed from the b axis, (c) the single-crystal structure of 2′, (d) the asymmetric unit of 2′, (e) the full ESI-TOF-MS of 1 (inset: the enlarged ion peak at m/z 1733.00), and (f) the full ESI-TOF-MS of 2 (inset: the enlarged ion peak at m/z 1719.00). The {MoO6} is represented in blue and {AlO6} in brown. Color code: C, black; N, blue; O, red; H, white; Mg, green. | ||
Considering the fact that the step-wise method adopted here generates a mixture of asymmetric product and symmetric by-products, we have also performed one-pot synthesis by using (TBA)3[Al(OH)6Mo6O18] as the starting material in the presence of tris-NO2 and pentaerythritol (see the ESI† for details). The resulting crude products were purified similarly as stated above.
The purification of asymmetric 2 ((TBA)3{AlMo6O18[(OCH2)3CNH2][(OCH2)3CC6H4NO2]}) is similar to that of 1. The purity of 2 was examined by 1H-NMR (Fig. S10†). As can be observed, asymmetric 2 exhibits two groups of peaks in the range of 4–5 ppm while the crude product shows three groups of peaks in the same region (the other group belongs to symmetric NO2-Al-NO2). All the other peaks are well resolved and can be unambiguously assigned to the molecular structure. ESI-TOF-MS of 2 represents three groups of isotopic peaks at m/z 736.86, 1475.72, and 1719.00, which can be allocated to the ion peaks of {(TBA){AlMo6O18[(OCH2)3CNH2][(OCH2)3CC6H4-NO2]}}2−, {(TBA)H{AlMo6O18[(OCH2)3CNH2][(OCH2)3CC6H4NO2]}}−, and {(TBA)2{AlMo6O18[(OCH2)3CNH2][(OCH2)3CC6H4NO2]}}−, respectively (Fig. 3f). Diethyl ether diffusion into an acetonitrile solution of 2 gives block single crystals, which unfortunately show very weak X-ray diffraction. In order to get high-quality single crystals, we conducted a cation exchange experiment. The TBA cations of 2 are transformed to Mg2+, and the resulting magnesium salts are denoted as 2′. Acetone diffusion into a DMF solution of 2′ gives single crystals suitable for X-ray diffraction. It is found that 2′ crystallizes in the monoclinic space group P21/c. There are two Anderson polyanions and three charge-balancing Mg2+ cations in the asymmetric unit (Fig. 3c and d). Different from the position disorders in 1, the two tripodal appendants in 2′ are unambiguously allocated at both sides of the Anderson plate with full crystallographic occupancy. The –NH2 groups from the tris moieties are coordinated to Mg1 with a Mg–N distance of 2.266 Å, while the Mg–O distances attributed to four coordinated DMF molecules are in the range of 2.006–2.055 Å. The Mg2 cation is coordinated by two DMF molecules, three water molecules and one terminal oxygen from the Anderson cluster, while the Mg3 cation is fully ligated by six DMF molecules. Due to the weak X-ray diffraction at high angles, some of the DMF molecules cannot be fully identified and have been ‘squeezed’ during the refinement.
Anderson cluster oligomers
Following the successful isolation of 1 and 2, we explored the possibility of using them as platforms to construct large designed oligomers via the post-functionalization approach. Asymmetric 1 with a reactive –CH2OH moiety was chosen as an example. As depicted in Scheme 1, the synthetic methodology is to transform the –CH2OH group into an amide-functionalized tris derivative, which can further be anchored onto single-side Al-OH to create a reactive ‘asymmetric’ Anderson dimer. Repeating the post-functionalization process can lead to chain growth and transform the dimer into trimer and so forth. Cronin et al. recently reported the preparation of Anderson oligomers via a copper-catalyzed alkyne–azide cycloaddition reaction.39 However, this method, as mentioned above, needs reverse-phase HPLC for the purification of the monomer precursors and the resulting coupled oligomers. However, the methodology proposed here is more straightforward and the cluster oligomers can be selectively formed without the need for further purification.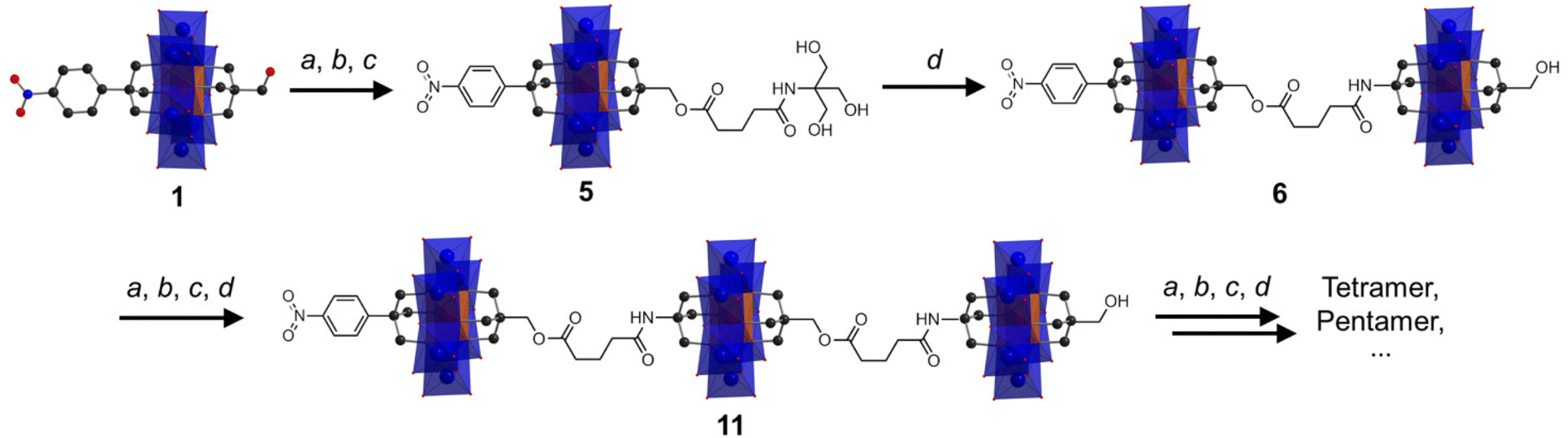 | ||
| Scheme 1 Schematic illustration of the post-functionalization approach for modular coupling of Anderson cluster oligomers: (a) glutaric anhydride, DIPEA, DMAP; (b) NHS, DCC; (c) tris, DIPEA; and (d) Al-OH. | ||
To verify this synthetic methodology, 1 was first reacted with glutaric anhydride to give the carboxylic acid functionalized 3. The structural composition of 3 has been fully characterized by 1H-NMR, FT-IR, ESI-TOF-MS and elemental analysis. In the 1H-NMR spectrum, an obvious distinction is that the peak of the –CH2O– group from the pentaerythritol part shifts from the 3.00 ppm of 1 to the 3.78 ppm of 3, and the peak type changes from doublet to singlet (Fig. S11†). The CO vibration of 3 in the FT-IR spectrum is observed at 1740 cm−1 (Fig. S12†). The C–O, MoO, and Mo–O–Mo bonds are found at 1092, 924, and 668 cm−1, respectively, which verifies the intactness of the Anderson cluster upon post-functionalization. The ESI-TOF-MS spectrum of 3 depicts four groups of isotopic peaks at m/z 802.37, 923.01, 1602.72, and 1846.03, which can be ascribed to the ion peaks of {(TBA){AlMo6O18[(OCH2)3CC6H4NO2][(OCH2)3CCH2OCO(CH2)3COOH]}}2−, {(TBA)2{AlMo6O18[(OCH2)3CC6H4NO2][(OCH2)3CCH2OCO(CH2)3C-OO]}}2−, {(TBA)H{AlMo6O18[(OCH2)3CC6H4NO2][(OCH2)3CCH2O-CO(CH2)3COOH]}}−, and {(TBA)2{AlMo6O18[(OCH2)3CC6H4NO2][(OCH2)3CCH2OCO(CH2)3COOH]}}2−, respectively (Fig. S13†). Activating the carboxylic acid group with N-hydroxy-succinimide (NHS) leads to the formation of 4, which has also been fully characterized using various techniques (see the ESI† for details). 4 reacting with tris generates the amide-functionalized tris derivative 5 in a moderate yield of 65.0%. In the 1H-NMR spectrum of 5, the –OH groups show a triplet peak at 4.69 ppm, while the –CH2O– groups from the tris moiety give a doublet peak at 3.56 ppm (Fig. 4a). The amide (–NH-CO–) proton Hj is found at 7.08 ppm. In the FT-IR spectrum, the –NH-CO– amide and the –O–CO– ester stretching vibrations are observed at 1642 and 1738 cm−1, respectively (Fig. S17†). These data all demonstrate the successful preparation of 5.
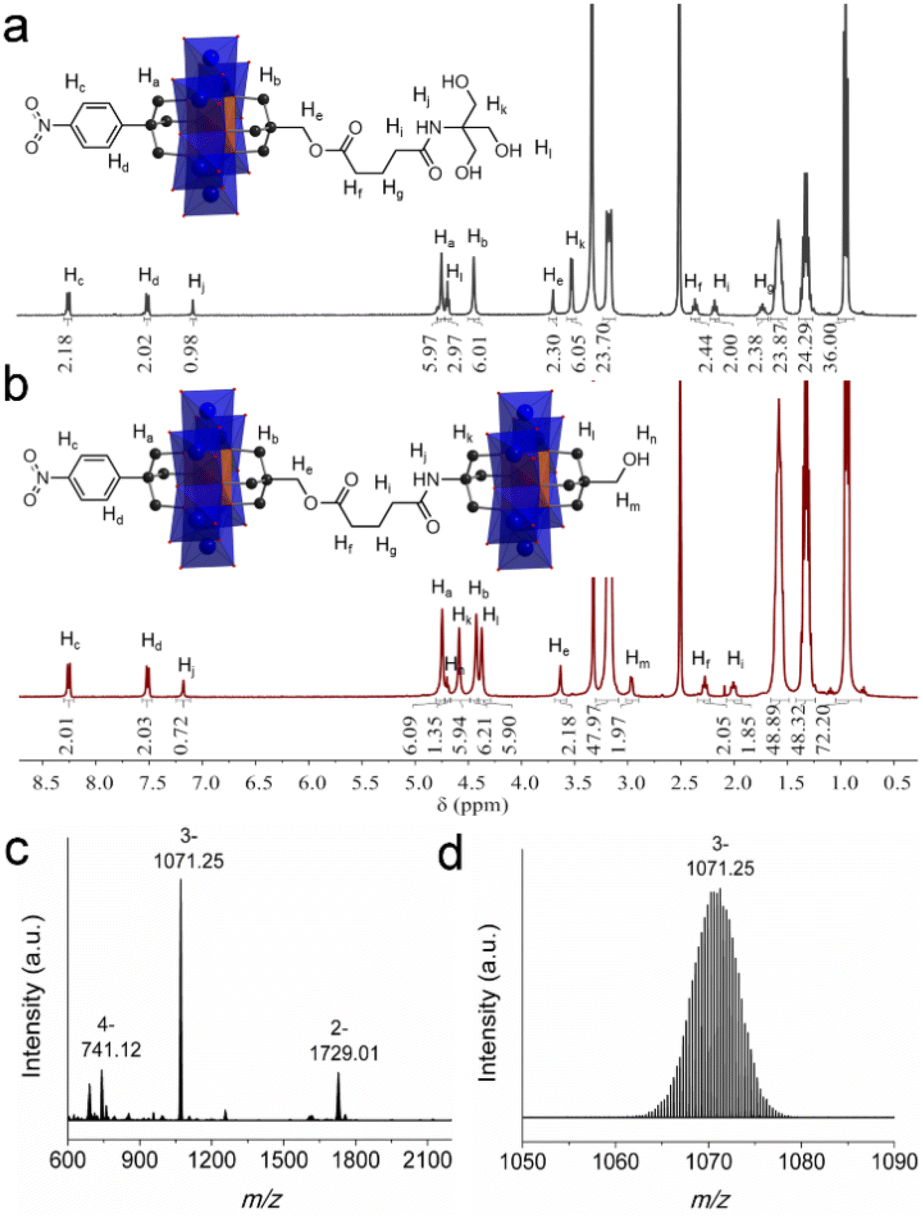 | ||
| Fig. 4 The 1H-NMR spectra of (a) 5 and (b) 6. (c) The full ESI-TOF-MS spectrum of 6 and (d) the enlarged ion peak at m/z 1071.25. | ||
Upon the preparation of dimer 6, we suspected that the reaction of 5 with Al-OH would also lead to a crude mixture like the abovementioned studies. However, to our delight the product contains only the target ‘asymmetric’ dimer. 1H-NMR was first adopted to investigate the structure of 6 (Fig. 4b). As observed, four sets of peaks are present in the region of 4–5 ppm in the 1H-NMR spectrum, which is in good accordance with the molecular structure. The proton integration numbers are also in good agreement with the structural composition. Typically, the peak observed at 4.74 ppm is assigned to the –CH2O– moieties of the tris-NO2 appendant, and the peak at 4.61 ppm is from the amide-tris derivative. The peak found at 4.45 ppm is attributed to the –CH2O– moieties of the pentaerythritol ester, and the peak at 4.37 ppm is due to the pentaerythritol appendant. ESI-TOF-MS further reveals the composition and purity of 6. As shown in Fig. 4c, three groups of isotopic peaks are observed and can be clearly assigned according to the molecular structure. For instance, the peak at m/z 741.12 is ascribed to {(TBA)2{(AlMo6O18)2[(OCH2)3CC6H4NO2][(OCH2)3C-CH2OCO(CH2)3CONHC(OCH2)3][(OCH2)3CCH2OH]}}4−, while the peaks at 1071.25 and 1729.01 can be assigned to {(TBA)3{(AlMo6O18)2[(OCH2)3CC6H4NO2][(OCH2)3CCH2OCO(CH2)3CONH-C(OCH2)3][(OCH2)3CCH2OH]}}3− and {(TBA)4{(AlMo6O18)2[(OC-H2)3CC6H4NO2][(OCH2)3CCH2OCO(CH2)3CONHC(OCH2)3][(OC-H2)3CCH2OH]}}2−, respectively. No isotopic peaks of the symmetric by-products are observed in the ESI-TOF-MS spectrum. The dynamic light scattering (DLS) experiment shows that the dimer 6 is monodisperse in acetonitrile with an average size diameter of 0.7482 nm (Fig. S19†).
To further prove that the amide-tris derivative can selectively generate the asymmetric product, dimer 7 was synthesized by using 5 and Al-NO2 as starting materials. The structural composition and purity of 7 have been examined by 1H-NMR and ESI-TOF-MS. Dimer 7 shows three sets of proton peaks in the range of 4–5 ppm in the 1H-NMR spectrum (Fig. S20†). The peak that appeared at 4.75 ppm is assigned to the –CH2O– moieties of the tris-NO2 appendants and is integrated to have 12 protons. The peak at 4.64 ppm is from the amide-tris derivative part and is integrated to 6 protons. The –CH2O– components of the pentaerythritol ester show a peak at 4.43 ppm and the proton integration number is 6. 2D total correlation spectroscopy (TOCSY) NMR of dimer 7 shows strong proton correlation between Hf, Hg, and Hi from the alkane chain (Fig. S21†). Similar to the ESI-TOF-MS spectrum of 6, three sets of isotopic peaks can be clearly distinguished in the case of 7 (Fig. S22†). The peaks at m/z 764.37, 1100.92, and 1773.51 can be indexed to anions of {(TBA)2{(AlMo6O18)2[(OCH2)3CC6H4NO2]2[(OCH2)3CCH2OCO(C-H2)3CONHC(OCH2)3]}}4−, {(TBA)3{(AlMo6O18)2[(OCH2)3CC6H4N-O2]2[(OCH2)3CCH2OCO(C-H2)3CONHC(OCH2)3]}}3−, and {(TBA)4{(AlMo6O18)2[(OCH2)3CC6H4NO2]2[(OCH2)3CCH2OCO(CH2)3CON-HC(OCH2)3]}}2−, respectively. We also tried to synthesize dimer 7 by using NHS-activated 4 and asymmetric monomer 2. However, probably due to the high steric hindrance of the Anderson cluster the attempts failed.
The successful preparation of the Anderson dimer encouraged us to go further to produce a trimer using the same synthetic methodology (see the ESI† for details). To this end, trimer 11 has been prepared and fully characterized. The 1H-NMR spectrum of 11 is very similar to that of dimer 6 with only differences in proton integration (Fig. S26†). ESI-TOF-MS of 11 is consistent with the molecular structure (Fig. S27†). For instance, the peaks observed at m/z 741.37, 1234.45, and 1726.03 are attributed to {(TBA)3{(AlMo6O18)3[(OCH2)3CC6H4NO2][(OCH2)3CCH2OCO-(CH2)3CONHC(OCH2)3]2[(OCH2)3CCH2OH]}}6−, {(TBA)5{(AlMo6-O18)3[(OCH2)3CC6H4NO2][(OCH2)3CCH2OCO(CH2)3CONC(OCH2)3]2[(OCH2)3CCH2OH]}}4−, and {(TBA)6{(AlMo6O18)3[(OCH2)3CC6H4-NO2][(OCH2)3CCH2OCO(CH2)3CONHC(OCH2)3]2[(OCH2)3CCH2O-H]}}3−, respectively. The successful preparation of trimer 11 further demonstrates the feasibility of the proposed method in controlled cluster coupling.
Keggin–Anderson cluster oligomer
The post-functionalization of the asymmetric Anderson cluster could also lead to useful intermediate building blocks that could be coupled onto other POMs to form large hetero-cluster assemblies. Taking the NHS-activated 4 and the amide-functionalized tris derivative 5 as examples: the latter can be anchored onto the Lindqvist {V6} or the Wells–Dawson {P2W15V3} cluster via esterification reactions (a similar study has recently been published by Parac-Vogt et al.30), while the former could be attached onto amino-functionalized clusters such as Keggin [(C4H9)4N]4{SiW11O40[Si(CH2)3NH2]2} (SiW11-NH2).54To achieve this, SiW11-NH2 was stirred overnight at room temperature in the presence of excessive 4 to form the hetero-cluster trimer 12 (Fig. 5a). The excessive 4 guaranteed the complete reaction of SiW11-NH2 and was removed by washing with acetone. In the 1H-NMR spectrum of 12, the appearance of the amide (–NH-CO–) proton at 7.83 ppm proves the successful linkage between Keggin and Anderson clusters (Fig. S29†). According to the integration of protons in the aliphatic area, only nine TBA counter-cations are present. All other peaks can be well resolved and are in good agreement with the molecular structure. In the FT-IR spectrum, the amide bond (–NH-CO–) is found at 1640 cm−1 (Fig. 5b). The strong vibration of the Si–O–Si bond in the Keggin hybrid is overlaid with the C–O bond of 4 and appears at 1044 cm−1. MALDI-TOF-MS was utilized to investigate the structural composition of trimer 12 (Fig. 5c). The peak observed at m/z 4002.58 with 2− charge is the molecular ion peak ({(TBA)10(M-2H)}2−, where M stands for poly-anions). The peaks at m/z 2432.04 and 2334.16 can be assigned to {(TBA)7M}3− and {(TBA)6HM}3−, respectively.
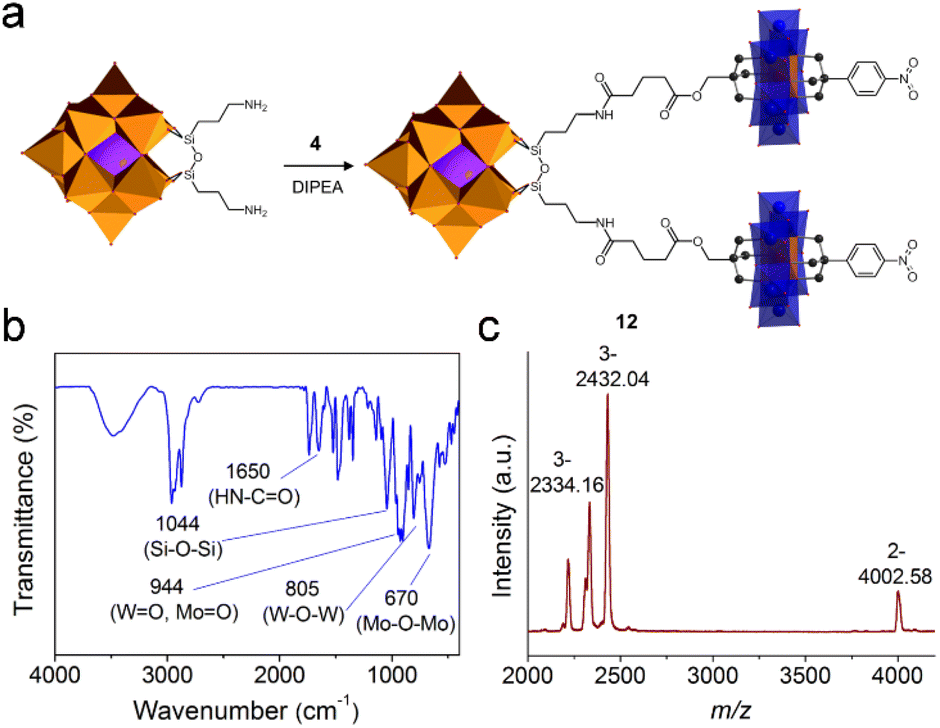 | ||
| Fig. 5 (a) Schematic representation of the synthetic process of Keggin–Anderson trimer 12, (b) the FT-IR spectrum of 12, and (c) the MALDI-TOF-MS spectrum of 12. | ||
Conclusions
In conclusion, we have presented the successful synthesis and purification of asymmetrically modified Anderson clusters, and further developed a coupling methodology based on the asymmetric Anderson platform to access precisely-designed organic–inorganic cluster oligomers. By varying the tripodal alcohols and changing the modification sequence, we were able to find that the type of tripodal alcohol plays an essential role in the asymmetric modification, which was somehow neglected previously. Although the reaction mechanism is still unclear, it seems that the acid–base properties of the tripodal alcohols can strongly affect the modification results. For example, neutral alcohols such as pentaerythritol and tris-NO2 inevitably generate two symmetric by-products upon asymmetric modification, while the basic alcohol such as tris leads to only one type of symmetric by-product. Upon decreasing the basicity of tris by amide-functionalization, the asymmetric hybrid is obtained as the product with satisfactory purity. Nevertheless, more efforts are still needed to verify this hypothesis and elucidate the reaction mechanism.The purification of the asymmetric Anderson hybrid involves a two-step process to sequentially remove the symmetric by-products based on their differences in solubility. Following the successful isolation of asymmetric Anderson hybrids, we have further developed a simple but robust methodology to modularly synthesize hybrid metal-oxo oligomers with controlled molecular length and cluster numbers. It is envisioned that Anderson tetramers, pentamers, etc. could also be achieved (Scheme 1). Another advantage of this coupling methodology is that it provides functional intermediates that can be used as building blocks to incorporate a range of other clusters. As an example, a Keggin–Anderson trimer has been prepared and fully characterized. Therefore, this work provides an easy and feasible strategy for the development of functional asymmetric clusters and the precision synthesis of cluster oligomers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (22178019, 21901016) and the Fundamental Research Funds for the Central Universities (XK1802-6, XK1803-05, XK1902, 12060093063).References
- D.-L. Long, R. Tsunashima and L. Cronin, Polyoxometalates: building blocks for functional nanoscale systems, Angew. Chem., Int. Ed., 2010, 49, 1736 CrossRef CAS PubMed.
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Hybrid polyoxometalates as post-functionalization platforms: from fundamentals to emerging applications, Chem. Soc. Rev., 2020, 49, 382 RSC.
- S.-S. Wang and G.-Y. Yang, Recent advances in polyoxometalate-catalyzed reactions, Chem. Rev., 2015, 115, 4893 CrossRef CAS PubMed.
- S. Amthor, S. Knoll, M. Heiland, L. Zedler, C. Li, D. Nauroozi, W. Tobaschus, A. K. Mengele, M. Anjass, U. S. Schubert, B. Dietzek-Ivanšić, S. Rau and C. Streb, A photosensitizer-polyoxometalate dyad that enables the decoupling of light and dark reactions for delayed on-demand solar hydrogen production, Nat. Chem., 2022, 14, 321 CrossRef CAS PubMed.
- Y. Ji, L. Huang, J. Hu, C. Streb and Y.-F. Song, Polyoxometalate-functionalized nanocarbon materials for energy conversion, energy storage and sensor systems, Energy Environ. Sci., 2015, 8, 776 RSC.
- M. Bonchio, Z. Syrgiannis, M. Burian, N. Marino, E. Pizzolato, K. Dirian, F. Rigodanza, G. A. Volpato, G. La Ganga, N. Demitri, S. Berardi, H. Amenitsch, D. M. Guldi, S. Caramori, C. A. Bignozzi, A. Sartorel and M. Prato, Hierarchical organization of perylene bisimides and polyoxometalates for photo-assisted water oxidation, Nat. Chem., 2019, 11, 146 CrossRef CAS PubMed.
- B. Rausch, M. D. Symes, G. Chisholm and L. Cronin, Decoupled catalytic hydrogen evolution from a molecular metal oxide redox mediator in water splitting, Science, 2014, 345, 1326 CrossRef CAS PubMed.
- S. Zhang, W. Shi and X. Wang, Locking volatile organic molecules by subnanometer inorganic nanowire-based organogels, Science, 2022, 377, 100 CrossRef CAS PubMed.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Hybrid organic-inorganic polyoxometalate compounds: from structural diversity to applications, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Functionalization and post-functionalization: a step towards polyoxometalate-based materials, Chem. Soc. Rev., 2012, 41, 7605 RSC.
- Y.-F. Song and R. Tsunashima, Recent advances on polyoxometalate-based molecular and composite materials, Chem. Soc. Rev., 2012, 41, 7384 RSC.
- J. M. Cameron, G. Guillemot, T. Galambos, S. S. Amin, E. Hampson, K. M. Haidaraly, G. N. Newton and G. Izzet, Supramolecular assemblies of organo-functionalised hybrid polyoxometalates: from functional building blocks to hierarchical nanomaterials, Chem. Soc. Rev., 2022, 51, 293 RSC.
- J. Zhang, F. Xiao, J. Hao and Y. Wei, The chemistry of organoimido derivatives of polyoxometalates, Dalton Trans., 2012, 41, 3599 RSC.
- A. Blazevic and A. Rompel, The Anderson-Evans polyoxometalate: from inorganic building blocks via hybrid organic-inorganic structures to tomorrows “bio-POM”, Coord. Chem. Rev., 2016, 307, 42 CrossRef CAS.
- B. Hasenknopf, R. Delmont, P. Herson and P. Gouzerh, Anderson-type heteropolymolybdates containing tris(alkoxo) ligands: synthesis and structural characterization, Eur. J. Inorg. Chem., 2002, 1081 CrossRef CAS.
- P. R. Marcoux, B. Hasenknopf, J. Vaissermann and P. Gouzerh, Developing remote metal binding sites in heteropoly-molybdates, Eur. J. Inorg. Chem., 2003, 2406 CrossRef CAS.
- Y.-F. Song, D.-L. Long and L. Cronin, Noncovalently connected frameworks with nanoscale channels assembled from a tethered polyoxometalate-pyrene hybrid, Angew. Chem., Int. Ed., 2007, 46, 3900 CrossRef CAS PubMed.
- Y.-F. Song, N. McMillan, D.-L. Long, J. Thiel, Y. Ding, H. Chen, N. Gadegaard and L. Cronin, Design of hydrophobic polyoxometalate hybrid assemblies beyond surfactant encapsulation, Chem. – Eur. J., 2008, 14, 2349 CrossRef CAS PubMed.
- Y.-F. Song, D.-L. Long and L. Cronin, Hybrid polyoxometalate clusters with appended aromatic platforms, CrystEngComm, 2010, 12, 109 RSC.
- Y. Yan, H. Wang, B. Li, G. Hou, Z. Yin, L. Wu and V. W. W. Yam, Smart self-assemblies based on a surfactant-encapsulated photoresponsive polyoxometalate complex, Angew. Chem., Int. Ed., 2010, 49, 9233 CrossRef CAS PubMed.
- P. Yin, P. Wu, Z. Xiao, D. Li, E. Bitterlich, J. Zhang, P. Cheng, D. V. Vezenov, T. Liu and Y. Wei, A double-tailed fluorescent surfactant with a hexavanadate cluster as the head group, Angew. Chem., Int. Ed., 2011, 50, 2521 CrossRef CAS PubMed.
- J. Thiel, D. Yang, M. H. Rosnes, X. Liu, C. Yvon, S. E. Kelly, Y.-F. Song, D.-L. Long and L. Cronin, Observing the hierarchical self-assembly and architectural bistability of hybrid molecular metal oxides using ion-mobility mass spectrometry, Angew. Chem., Int. Ed., 2011, 50, 8871 CrossRef CAS PubMed.
- U. Tong, W. Chen, C. Ritchie, X. Wang and Y.-F. Song, Chem. – Eur. J., 2014, 20, 1500 CrossRef CAS PubMed.
- S. Vanhaecht, J. Jacobs, L. Van Meervelt and T. N. Parac-Vogt, A versatile and highly efficient post-functionalization method for grafting organic molecules onto Anderson-type polyoxometalates, Dalton Trans., 2015, 44, 19059 RSC.
- X.-X. Li, Y.-X. Wang, R.-H. Wang, C.-Y. Cui, C.-B. Tian and G.-Y. Yang, Designed assembly of heterometallic cluster organic frameworks based on Anderson-type polyoxometalate clusters, Angew. Chem., Int. Ed., 2016, 55, 6462 CrossRef CAS PubMed.
- O. Linnenberg, A. Kondinski, C. Stöcker and K. Y. Monakhov, The Cu(I)-catalysed Huisgen 1,3-dipolar cycloaddition route to (bio-)organic functionalisation of polyoxovanadates, Dalton Trans., 2017, 46, 15636 RSC.
- C.-G. Lin, G. D. Fura, Y. Long, W. Xuan and Y.-F. Song, Polyoxometalate-based supramolecular hydrogels constructed through host-guest interactions, Inorg. Chem. Front., 2017, 4, 789 RSC.
- A. Boulmier, A. Vacher, D. Zang, S. Yang, A. Saad, J. Marrot, O. Oms, P. Mialane, I. Ledoux, L. Ruhlmann, D. Lorcy and A. Dolbecq, Anderson-type polyoxometalates functionalized by tetrathiafulvalene groups: synthesis, electrochemical studies, and NLO properties, Inorg. Chem., 2018, 57, 3742 CrossRef CAS PubMed.
- Z. Xia, C.-G. Lin, Y. Yang, Y. Wang, Z. Wu, Y.-F. Song, T. P. Russell and S. Shi, Polyoxometalate-surfactant assemblies: responsiveness to orthogonal stimuli, Angew. Chem., Int. Ed., 2022, 61, e202203741 CAS.
- D. E. S. Marcano, M. A. Moussawi, A. V. Anyushin, S. Lentink, L. Van Meervelt, I. Ivanović-Burmazović and T. N. Parac-Vogt, Versatile post-functionalisation strategy for the formation of modular organic-inorganic polyoxometalate hybrids, Chem. Sci., 2022, 13, 2891 RSC.
- Y.-F. Song, D.-L. Long, S. E. Kelly and L. Cronin, Sorting the assemblies of unsymmetrically covalently functionalised Mn-Anderson polyoxometalate, Inorg. Chem., 2008, 47, 9137 CrossRef CAS PubMed.
- M. H. Rosnes, C. Musumeci, C. P. Pradeep, J. S. Mathieson, D.-L. Long, Y.-F. Song, B. Pignataro, R. Cogdell and L. Cronin, Assembly of modular asymmetric organic-inorganic polyoxometalate hybrids into anisotropic nanostructures, J. Am. Chem. Soc., 2010, 132, 15490 CrossRef CAS PubMed.
- D. Li, J. Song, P. Yin, S. Simotwo, A. J. Bassler, Y. Aung, J. E. Roberts, K. I. Hardcastle, C. L. Hill and T. Liu, Inorganic-organic hybrid vesicles with counterion- and pH-controlled fluorescent properties, J. Am. Chem. Soc., 2011, 133, 14010 CrossRef CAS PubMed.
- O. Oms, K. Hakouk, R. Dessapt, P. Deniard, S. Jobic, A. Dolbecq, T. Palacin, L. Nadjo, B. Keita, J. Marrot and P. Mialane, Photo- and electrochromic properties of covalently connected symmetrical and unsymmetrical spiropyran/polyoxometalate dyads, Chem. Commun., 2012, 48, 12103 RSC.
- C. Yvon, A. Macdonell, S. Buchwald, A. J. Surman, N. Follet, J. Alex, D.-L. Long and L. Cronin, A collection of robust methodologies for the preparation of asymmetric hybrid Mn-Anderson polyoxometalates for multifunctional materials, Chem. Sci., 2013, 4, 3810 RSC.
- A. Saad, O. Oms, A. Dolbecq, C. Menet, R. Dessapt, H. Serier-Brault, E. Allard, K. Baczko and P. Mialane, A high fatigue resistant, photoswitchable fluorescent spiropyran-polyoxometalate-BODIPY single-molecule, Chem. Commun., 2015, 51, 16088 RSC.
- E. Hampson, J. M. Cameron, S. Amin, J. Kyo, J. A. Watts, H. Oshio and G. N. Newton, Asymmetric hybrid polyoxometalates: a platform for multifunctional redox-active nanomaterials, Angew. Chem., Int. Ed., 2019, 58, 18281 CrossRef CAS PubMed.
- C. Yvon, A. J. Surman, M. Hutin, J. Alex, B. O. Smith, D.-L. Long and L. Cronin, Polyoxometalate clusters integrated into peptide chains and as inorganic amino acids: solution- and solid-phase approaches, Angew. Chem., Int. Ed., 2014, 53, 3336 CrossRef CAS PubMed.
- A. Macdonell, N. A. B. Johnson, A. J. Surman and L. Cronin, Configurable nanosized metal oxide oligomers via precise “click” coupling control of hybrid polyoxometalates, J. Am. Chem. Soc., 2015, 137, 5662 CrossRef CAS PubMed.
- Y.-F. Song, N. McMillan, D.-L. Long, S. Kane, J. Malm, M. O. Riehle, C. P. Pradeep, N. Gadegaard and L. Cronin, Micropatterned surfaces with covalently grafted unsymmetrical polyoxometalate-hybrid clusters lead to selective cell adhesion, J. Am. Chem. Soc., 2009, 131, 1340 CrossRef CAS PubMed.
- P. Wu, P. Yin, J. Zhang, J. Hao, Z. Xiao and Y. Wei, Single-side organically functionalized Anderson-type polyoxometalates, Chem. – Eur. J., 2011, 17, 12002 CrossRef CAS PubMed.
- H. Ai, Y. Wang, B. Li and L. Wu, Synthesis and characterization of single-side organically grafted Anderson-type polyoxometalates, Eur. J. Inorg. Chem., 2014, 2766 CrossRef CAS.
- B. Zhang, L. Yue, Y. Wang, Y. Yang and L. Wu, A novel single-side azobenzene-grafted Anderson-type polyoxometalate for recognition-induced chiral migration, Chem. Commun., 2014, 50, 10823 RSC.
- Q. Li and Y. Wei, A series of unprecedented triol-stabilized [H3MW6O24]n−: the missing piece between A- and B-type Anderson-Evans polyoxometalates, Chem. Commun., 2018, 54, 1375 RSC.
- J. Zhang, J. Luo, P. Wang, B. Ding, Y. Huang, Z. Zhao, J. Zhang and Y. Wei, Step-by-Step strategy from achiral precursors to polyoxometalates-based organic-inorganic hybrids, Inorg. Chem., 2015, 54, 2551 CrossRef CAS PubMed.
- C.-G. Lin, W. Chen, D.-L. Long, L. Cronin and Y.-F. Song, Step-by-step covalent modification of Cr-templated Anderson-type polyoxometalates, Dalton Trans., 2014, 43, 8587 RSC.
- X.-X. Li, X. Ma, W.-X. Zheng, Y.-J. Qi, S.-T. Zheng and G.-Y. Yang, Composite hybrid cluster built from the integration of polyoxometalate and a metal halide cluster: synthetic strategy, structure, and properties, Inorg. Chem., 2016, 55, 8257 CrossRef CAS PubMed.
- N. I. Gumerova, T. C. Fraile, A. Roller, G. Giester, M. Pascual-Borras, C. A. Ohlin and A. Rompel, Direct single- and double-side triol-functionalization of the mixed type Anderson polyoxotungstate [Cr(OH)6W6O21]6−, Inorg. Chem., 2019, 58, 106 CrossRef CAS PubMed.
- W.-D. Yu, Y. Zhang, Y.-Y. Han, B. Li, S. Shao, L.-P. Zhang, H.-K. Xie and J. Yan, Microwave-assisted synthesis of tris-Anderson polyoxometalates for facile CO2 cycloaddition, Inorg. Chem., 2021, 60, 3980 CrossRef CAS PubMed.
- S. Manikumari, V. Shivaiah and S. K. Das, Identification of a near-linear supramolecular water dimer, (H2O)2, in the channel of an inorganic framework material, Inorg. Chem., 2002, 41, 6953 CrossRef CAS PubMed.
- C.-G. Lin, M. Hutin, C. Busche, N. L. Bell, D.-L. Long and L. Cronin, Elucidating the paramagnetic interactions of an inorganic-organic hybrid radical-functionalized Mn-Anderson cluster, Dalton Trans., 2021, 50, 2350 RSC.
- I. Bar-Nahum, H. Cohen and R. Neumann, Organometallic-polyoxometalate hybrid compounds: metallosalen compounds modified by Keggin type polyoxometalates, Inorg. Chem., 2003, 42, 3677 CrossRef CAS PubMed.
- T. K. Prasad, G. Poneti, L. Sorace, M. J. Rodriguez-Douton, A. L. Barra, P. Neugebauer, L. Costantino, R. Sessoli and A. Cornia, Magnetic and optical bistability in tetrairon(III) single molecule magnets functionalized with azobenzene groups, Dalton Trans., 2012, 41, 8368 RSC.
- C.-G. Lin, W. Chen, S. Omwoma and Y.-F. Song, Covalently grafting nonmesogenic moieties onto polyoxometalate for fabrication of thermotropic liquid-crystalline nanomaterials, J. Mater. Chem. C, 2015, 3, 15 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, 1H-NMR, FT-IR, ESI-TOF-MS, elemental analysis and single-crystal X-ray crystallographic data. CCDC 2212965 and 2212866. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qi02233h |
| This journal is © the Partner Organisations 2023 |