
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring-opening terpolymerisation of phthalic thioanhydride with carbon dioxide and epoxides†
Merlin R.
Stühler
,
Cesare
Gallizioli
,
Susanne M.
Rupf
 and
Alex J.
Plajer
*
and
Alex J.
Plajer
*
Institut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstraße 34-36, 14195 Berlin, Germany. E-mail: plajer@zedat.fu-berlin.de
First published on 9th October 2023
Abstract
In seeking to expand the portfolio of accessible polymer structures from CO2 waste, we report the ring-opening terpolymerisation (ROTERP) of phthalic thioanhydride with CO2 and epoxides, forming statistical poly(ester-thioester-carbonates) by employing heterobimetallic catalysts. Both metal choice and ligand chemistry modulate the amount of CO2 incorporated into the polymer microstructure. Terpolymerisation occurs when maintaining polymerisation rates of the faster parent ring-opening copolymerisation and this finding led us to develop the formation of CO2-derived terpolymers with butylene oxide at low CO2 pressure under bicomponent catalysis. Tetrapolymerisation with added phthalic anhydride leads to the preferential polymerisation of phthalic anhydride before the polymerisation of sulfur derivatives with CO2 and epoxides. Finally, we show that the presence of sulfur-containing thioester links leads to polymers with degradability benefits compared to those from all-oxygen derivatives.
Introduction
Heteroatom-containing polymers have great potential as degradable replacements for polymers based on backbones made up of –C–C– bonds.1–4 An increasingly popular synthetic methodology for accessing heteroatom-containing polymers is the copolymerisation of three- or four-membered heterocycles A with heteroallenes or cyclic anhydrides B to generate alternating copolymers (AB)n in the so-called ring-opening copolymerisation (ROCOP).5 Prominent examples include CO2/epoxide ROCOP forming polycarbonates or cyclic anhydride/epoxide ROCOP forming polyesters.6–8 Consequently, ROCOP has been identified as a promising methodology for the utilization of globally accumulating CO2 waste.9 However, sulfur-containing variants also exist, such as cyclic thioanhydride/epoxide ROCOP forming poly(ester-alt-thioesters) or CS2/epoxide ROCOP yielding poly(thiocarbonates) with improved photochemical and oxidative degradability compared to their related all-oxygen analogues.10–22 Thermal properties can also be improved by formal sulfuration as recently reported for CS2/oxetane copolymers, which are semi-crystalline, whereas the CO2/oxetane analogues are amorphous.23–25 Moving to more complex monomer mixtures comprising three monomers A, B and C (e.g. epoxide A, phthalic anhydride (PA) B and CO2C) yield either statistical terpolymers (poly(ester-carbonates), [(AB)l-(AC)n]m) or block terpolymers (polyester-b-polycarbonates, (AB)n-(AC)m).26–30 In these cases, the monomer sequences usually depend on catalyst selection. In the cases of sulfurated monomers, further insights into new terpolymerisation processes have been made. We, for example, recently realised sequence selective ring-opening terpolymerisation (ROTERP) of ternary monomer mixtures comprising phthalic thioanhydride (PTA), CS2 or PhNCS and monosubstituted epoxide (propylene oxide PO and butylene oxide BO).31–34 Here, a simple lithium catalyst (e.g. lithium benzyloxide LiOBn) selectively forms poly(ester-alt-ester-alt-heterocarbonates). In another work directly relevant to this report, we achieved statistical terpolymerisation of CS2 and CO2 with epoxides by employing heterobimetallic Cr(III)Rb catalysts.35 Here, CO2 insertion into alkoxide intermediates formed from epoxide ring-opening appeared to be kinetically favourable over CS2 insertion, so that the resulting terpolymers mostly contained all-oxygen carbonates and few sulfur-containing thiocarbonate links. Although there was some control over the amount of CS2vs. CO2 incorporation by adjusting the amount of initially supplied CS2 in the starting monomer feed, the obtained molecular weights deteriorated particularly with higher CS2 loadings.Aiming to expand the monomer scope of CO2 terpolymerisation with sulfurated monomers, we hypothesized that PTA/CO2/epoxide ROTERP could be possible, which we investigate here, particularly in relation to the parent CO2/epoxide and PTA/epoxide ROCOPs (Fig. 1).
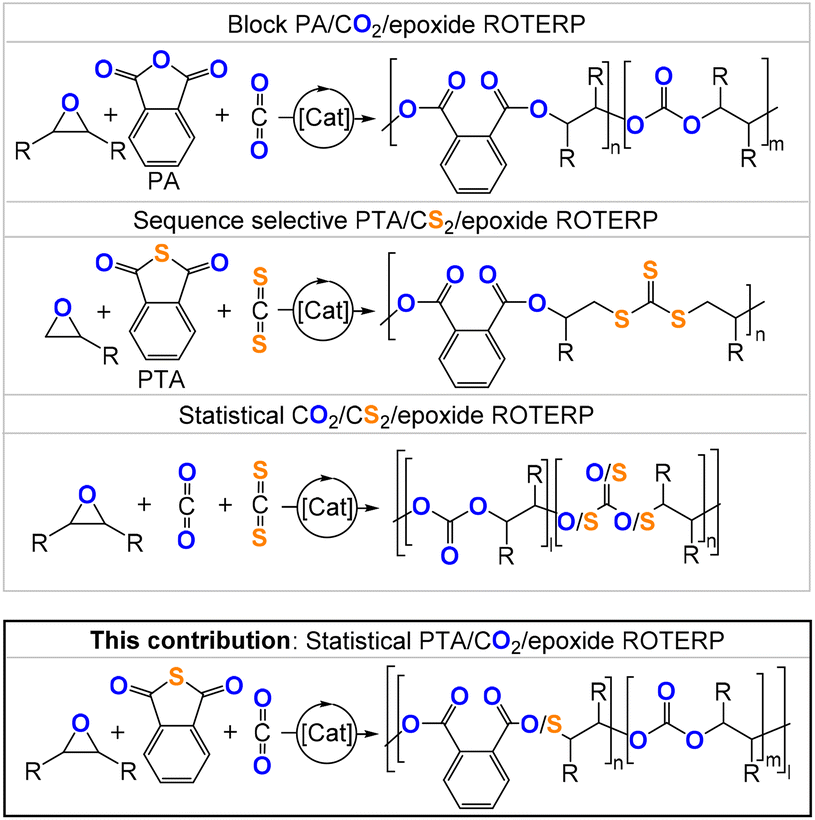 | ||
| Fig. 1 Comparison of the literature-reported ROTERP and the ROTERP presented in this contribution; [Li] = LiOCH2Ph (LiOBn), R = Me, Et, –(C4H8)–. | ||
Results and discussion
We hypothesized that in order to achieve the terpolymerisation of PTA, CO2 and epoxides, a catalyst needs to be active in the parent copolymerisations of CO2 with epoxides and PTA with epoxides, respectively. Given the precedence for heterobimetallic and Cr(III)-containing catalysts in CO2/epoxide and PTA/epoxide ROCOP, we decided to employ a series of Cr(III)AM (AM = Li, Na, K, Rb, Cs) LXCrAM complexes based on binucleating ligands LXH2 (Fig. 2) in an easy-to-perform, low-pressure PTA/CO2/cyclohexene oxide (CHO) ROTERP.36,37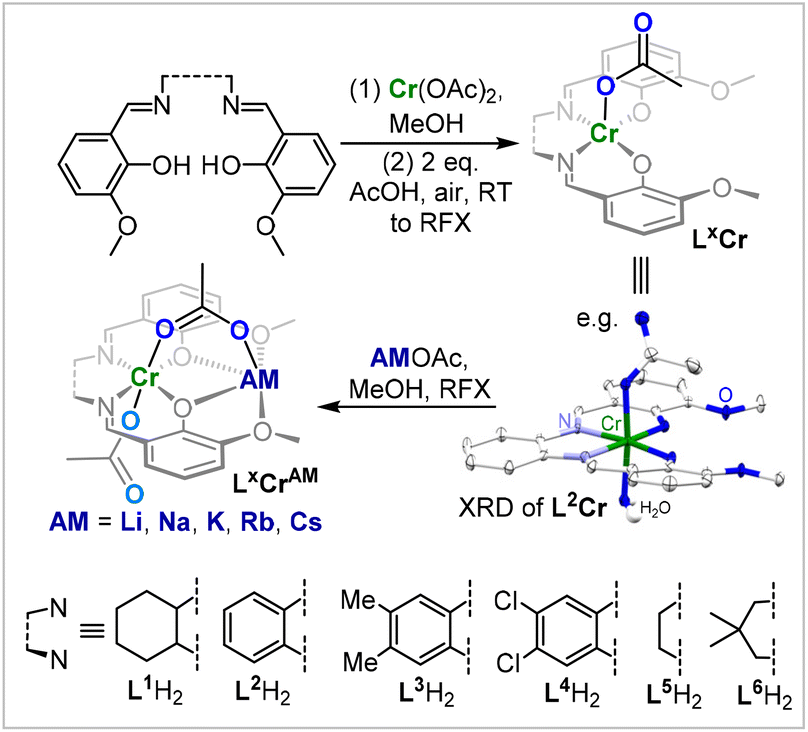 | ||
| Fig. 2 Synthesis of LXCrAM as well as the solid-state structure of L2Cr, with H atoms apart from co-crystallised H2O and co-crystallised MeOH omitted for clarity; ellipsoids set at 40% probability. | ||
To explore how catalyst selection influences the new ROTERP, we synthesized a range of different ligands LXH2 featuring aromatic backbones bearing electron-donating and electron-withdrawing substituents, and aliphatic backbones of varying rigidity. Metalation was achieved in a straightforward fashion by following our recently published procedure.35 The treatment of LXH2 with Cr(OAc)2 in acetonitrile at room temperature, followed by aerobic oxidation in the presence of additional acetic acid, yields LXCr, i.e., ligands LX coordinated to a CrOAc moiety, which we could crystallographically verify for L2Cr (Fig. 2). A consecutive reaction with alkali metal acetates AMOAc (AM = Li, Na, K, Rb, Cs) in refluxing MeOH yields LXCrAM (ESI section S2†) as brown or red powders after the removal of all solvents, which were characterised by HR-ESI MS, IR and elemental analyses confirming their heterobimetallic nature. With a series of LXCrAM catalysts in hand, we then turned towards their application in PTA/CO2/CHO ROTERP with 1 eq. of LXCrAM:100 eq. of PTA:1000 eq. of CHO, 4 bar CO2 pressure and 100 °C (ESI section S3†). Under these conditions, terpolymerisation can be performed in screw-cap glass vessels, which obviates the need for steel reactor set-ups as are commonplace in CO2 ROCOP. At the end of the reaction, ROTERP was terminated by the addition of MeOH to the cooled-down reaction mixture. All polymers were then isolated from DCM/MeOH and THF/pentane by repeated precipitation, yielding off-white colourless powders in typical yields of 50–60% with respect to the consumed CHO.
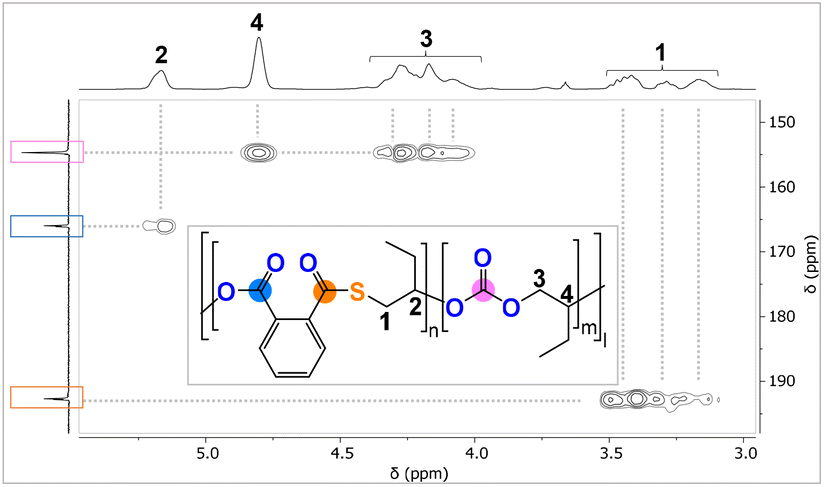
The analysis of the precipitated polymers by 1H, 13C and 2D NMR spectroscopy reveals the formation of terpolymers comprising carbonate, ester and thioester links, as exemplified in Fig. 3. The 1H NMR spectrum shows tertiary CH resonances between δ = 5.35 and 4.90 ppm, which correlate to the R–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O–R ester units at δ = 164.4 and 168.8 ppm in the 13C NMR spectrum, as determined by 1H–13C HMBC spectroscopy. Similarly, the tertiary CH resonances between δ = 4.90 and 4.50 ppm correlate to R–O–(CO)–O–R carbonate groups at δ = 151.0 and 156.0 ppm, while the resonances between δ = 4.10 and 3.80 ppm correlate to R–(CO)–S–R thioester groups at δ = 190.0 and 194.0 ppm. Only trace amounts of poly(thio)ether links from CHO (or CHS vide infra) homopolymerisation could be observed as being part of the polymers for all catalysts comprising Na–K. However, for the Li-containing catalyst (Table 1, run #7), ca. 21% (thio)ether links are formed in the terpolymerisation. Notably in all cases, the respective carbonyl regions in the 13C NMR spectrum for the quaternary ester, thioester, and carbonate carbons are highly complex, showing multiple, ill-defined, and overlapping resonances in the diagnostic regions, which suggest statistical terpolymer formation. This notion could be confirmed by following the polymerisation by 1H NMR analysis of aliquots removed at regular time intervals, which indeed revealed continuous PTA consumption and uniform generation of ester, carbonate and thioester links in the same ratio as observed in the final isolated polymers. Finally, IR spectroscopy of the isolated polymers further supports the presence of carbonate and ester (
O)–O–R ester units at δ = 164.4 and 168.8 ppm in the 13C NMR spectrum, as determined by 1H–13C HMBC spectroscopy. Similarly, the tertiary CH resonances between δ = 4.90 and 4.50 ppm correlate to R–O–(CO)–O–R carbonate groups at δ = 151.0 and 156.0 ppm, while the resonances between δ = 4.10 and 3.80 ppm correlate to R–(CO)–S–R thioester groups at δ = 190.0 and 194.0 ppm. Only trace amounts of poly(thio)ether links from CHO (or CHS vide infra) homopolymerisation could be observed as being part of the polymers for all catalysts comprising Na–K. However, for the Li-containing catalyst (Table 1, run #7), ca. 21% (thio)ether links are formed in the terpolymerisation. Notably in all cases, the respective carbonyl regions in the 13C NMR spectrum for the quaternary ester, thioester, and carbonate carbons are highly complex, showing multiple, ill-defined, and overlapping resonances in the diagnostic regions, which suggest statistical terpolymer formation. This notion could be confirmed by following the polymerisation by 1H NMR analysis of aliquots removed at regular time intervals, which indeed revealed continuous PTA consumption and uniform generation of ester, carbonate and thioester links in the same ratio as observed in the final isolated polymers. Finally, IR spectroscopy of the isolated polymers further supports the presence of carbonate and ester (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) ≈ 1734.0 cm−1) in addition to thioester ( ≈ 1676.0 cm−1) links. The materials are amorphous with Tg values between 118 and 128 °C with no clear trend between composition and glass transition. GPC analysis of the isolated polymers reveals molecular weights of Mn = 5.1–16.9 kg mol−1 and bimodal distributions with Đ = 1.2–1.4. We observed linearly increasing molecular weights with reaction progress, indicative of a controlled ROTERP process, albeit with bimodal molecular weight distributions formed from the beginning of the polymerisation (Fig. 4(a)). The bimodal distribution in each case comprises two narrow overlapping distributions of which one exhibits approximately half the molecular weight of the other, and the ratio of the two remains constant throughout the polymerisation. It is well established that bimodality in epoxide ROCOP catalysis stems chain transfer with protic impurities.38,39 Usually one distribution of chains is formed from an ROCOP process initiated by monofunctional initiators X, which are part of the catalyst, yielding α-X,ω-OH-chains, while the second distribution of chains is formed via ROCOP initiated from either H2O or diol impurities in the monomers via chain-transfer processes yielding α,ω-OH-telechelic chains. Indeed, in all our catalysts LXCrAM, acetate coligands, which can be expected to serve as monofunctional initiators, are introduced which would produce aliphatic ester chain ends and these were identified to be part of the isolated polymers by 13C NMR (δ(Cq) = 171 ppm). To test whether chain transfer with diol impurities is the origin of bimodality, we deliberately added the bifunctional alcohol 1,4-benzenedimethanol (BDM) to an ROTERP run (employing 1 eq. of L1CrCs:20 eq. of BDM:100 eq. of PTA:1000 eq. of CHO, 4 bar CO2 and identical reaction time and temperature). Compared to the run without BDM, we observed a clear decrease of the obtained Mn from 13.0 to 3.1 kg mol−1 and furthermore, a narrowed monomodal distribution in the latter case with Đ = 1.1. NMR analysis of this polymer unambiguously shows that BDM has been incorporated into the polymer structure. All findings support that BDM acts as a chain transfer agent by increasing the number of telechelic chains formed so that the polymer samples predominantly comprise α,ω-OH telechelic chains. This leads to apparent monomodality with decreased molecular weights at comparable monomer conversion, as now a similar number of monomers have to be distributed across more initiators. On a side note, our experiments also show that our new ROTERP tolerates chain-transfer agents to control the molecular weights and the chain-end chemistry. Our finding suggests that bimodality in the first place is caused by diol or H2O impurities in the liquid monomers at the beginning of the polymerization, impurities introduced during the setup of the reaction, or impurities in the CO2 supplied throughout the reaction since the degree of bimodality remains constant throughout ROTERP. This variable is intrinsically hard to control between monomer batches and reactions although all reaction parameters and purification methods are kept identical. Accordingly, the degree of bimodality varies between runs, as shown in Table 1. This variability furthermore extends to repeat runs although reaction rates, polymer compositions and molecular weights are reproducible. In terms of catalyst dependence, the precise ratio of ester
≈ 1734.0 cm−1) in addition to thioester ( ≈ 1676.0 cm−1) links. The materials are amorphous with Tg values between 118 and 128 °C with no clear trend between composition and glass transition. GPC analysis of the isolated polymers reveals molecular weights of Mn = 5.1–16.9 kg mol−1 and bimodal distributions with Đ = 1.2–1.4. We observed linearly increasing molecular weights with reaction progress, indicative of a controlled ROTERP process, albeit with bimodal molecular weight distributions formed from the beginning of the polymerisation (Fig. 4(a)). The bimodal distribution in each case comprises two narrow overlapping distributions of which one exhibits approximately half the molecular weight of the other, and the ratio of the two remains constant throughout the polymerisation. It is well established that bimodality in epoxide ROCOP catalysis stems chain transfer with protic impurities.38,39 Usually one distribution of chains is formed from an ROCOP process initiated by monofunctional initiators X, which are part of the catalyst, yielding α-X,ω-OH-chains, while the second distribution of chains is formed via ROCOP initiated from either H2O or diol impurities in the monomers via chain-transfer processes yielding α,ω-OH-telechelic chains. Indeed, in all our catalysts LXCrAM, acetate coligands, which can be expected to serve as monofunctional initiators, are introduced which would produce aliphatic ester chain ends and these were identified to be part of the isolated polymers by 13C NMR (δ(Cq) = 171 ppm). To test whether chain transfer with diol impurities is the origin of bimodality, we deliberately added the bifunctional alcohol 1,4-benzenedimethanol (BDM) to an ROTERP run (employing 1 eq. of L1CrCs:20 eq. of BDM:100 eq. of PTA:1000 eq. of CHO, 4 bar CO2 and identical reaction time and temperature). Compared to the run without BDM, we observed a clear decrease of the obtained Mn from 13.0 to 3.1 kg mol−1 and furthermore, a narrowed monomodal distribution in the latter case with Đ = 1.1. NMR analysis of this polymer unambiguously shows that BDM has been incorporated into the polymer structure. All findings support that BDM acts as a chain transfer agent by increasing the number of telechelic chains formed so that the polymer samples predominantly comprise α,ω-OH telechelic chains. This leads to apparent monomodality with decreased molecular weights at comparable monomer conversion, as now a similar number of monomers have to be distributed across more initiators. On a side note, our experiments also show that our new ROTERP tolerates chain-transfer agents to control the molecular weights and the chain-end chemistry. Our finding suggests that bimodality in the first place is caused by diol or H2O impurities in the liquid monomers at the beginning of the polymerization, impurities introduced during the setup of the reaction, or impurities in the CO2 supplied throughout the reaction since the degree of bimodality remains constant throughout ROTERP. This variable is intrinsically hard to control between monomer batches and reactions although all reaction parameters and purification methods are kept identical. Accordingly, the degree of bimodality varies between runs, as shown in Table 1. This variability furthermore extends to repeat runs although reaction rates, polymer compositions and molecular weights are reproducible. In terms of catalyst dependence, the precise ratio of ester![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :carbonate:thioester links depends on both the alkali metal and the backbone choice with regards to LXCrAM (Table 1), although no obvious trends could be identified. This suggests that an interplay, rather than a simple combination, of the electronic and steric nature of the chromium coordination site alongside the one imposed through alkali metal choice determines the respective insertion events. However, regardless of the catalyst employed, the ester:thioester ratio observed is always lower than the perhaps expected 1:1 ratio resulting from PTA insertion following propagation, implying the loss of sulfur centres and concurrent oxygen enrichment of the polymer during ROTERP. Confirming that “sulfur-loss” is an inherent side-process of the PTA-CHO insertion events, we performed a separate PTA/CHO ROCOP reaction (1 eq. of L1CrCs:1000 eq. of PTA:1000 eq. of CHO) and observed an ester to thioester ratio of ca. 1.8:1 deviating substantially from the ideal 1:1 ratio. The broadened polydispersity of the obtained polymer (Mn = 19.8 kg mol−1 and Đ = 1.6) also suggests that PTA/CHO insertion events during ROTERP could contribute to bimodality and the different extents to which this occurs for different catalysts could also cause different degrees of bimodality between different catalysts, as shown in Table 1. In fact, we observed a similar oxygen enrichment process of the polymer in our previous study on CS2/CHO ROCOP and were able to conclusively show that O/S exchange processes at the chain end and consecutive thiirane elimination cause this.18 Accordingly, 1H NMR aliquot analysis of the ROTERP process reveals steadily increasing amounts of thiirane cyclohexene sulfide (CHS) being generated alongside propagation, which accounts for the apparent sulfur loss and oxygen enrichment of the polymer. Given all our observations above, we propose the following mechanism (Fig. 4) in which initiation either happens from acetate coligands generating an alkoxide via CHO insertion or from deprotonated diol impurities after which two primary propagation cycles occur. Among them, CO2versus PTA insertion from the common alkoxide intermediate determines the (thio)ester:carbonate linkage ratio. Analogous to our previous report on CS2/CHO ROCOP, polymer oxygen enrichment is proposed to originate from an O/S rearrangement step from alkoxide chain ends adjacent to thioester links that eliminate CHS to create a carboxylate chain end, which then inserts CHO to generate a diester link.35 This effectively turns PTA and 2 equivalents of CHO into a diester link and CHS. Note that the O/S exchange process transforming alkoxides into thiolates could in principle also occur intermolecularly between a chain end and a thioester link of another polymer. Yet as such an intermolecular process would broaden dispersities substantially, we infer that the intramolecular pathway at the chain ends is dominant.
:carbonate:thioester links depends on both the alkali metal and the backbone choice with regards to LXCrAM (Table 1), although no obvious trends could be identified. This suggests that an interplay, rather than a simple combination, of the electronic and steric nature of the chromium coordination site alongside the one imposed through alkali metal choice determines the respective insertion events. However, regardless of the catalyst employed, the ester:thioester ratio observed is always lower than the perhaps expected 1:1 ratio resulting from PTA insertion following propagation, implying the loss of sulfur centres and concurrent oxygen enrichment of the polymer during ROTERP. Confirming that “sulfur-loss” is an inherent side-process of the PTA-CHO insertion events, we performed a separate PTA/CHO ROCOP reaction (1 eq. of L1CrCs:1000 eq. of PTA:1000 eq. of CHO) and observed an ester to thioester ratio of ca. 1.8:1 deviating substantially from the ideal 1:1 ratio. The broadened polydispersity of the obtained polymer (Mn = 19.8 kg mol−1 and Đ = 1.6) also suggests that PTA/CHO insertion events during ROTERP could contribute to bimodality and the different extents to which this occurs for different catalysts could also cause different degrees of bimodality between different catalysts, as shown in Table 1. In fact, we observed a similar oxygen enrichment process of the polymer in our previous study on CS2/CHO ROCOP and were able to conclusively show that O/S exchange processes at the chain end and consecutive thiirane elimination cause this.18 Accordingly, 1H NMR aliquot analysis of the ROTERP process reveals steadily increasing amounts of thiirane cyclohexene sulfide (CHS) being generated alongside propagation, which accounts for the apparent sulfur loss and oxygen enrichment of the polymer. Given all our observations above, we propose the following mechanism (Fig. 4) in which initiation either happens from acetate coligands generating an alkoxide via CHO insertion or from deprotonated diol impurities after which two primary propagation cycles occur. Among them, CO2versus PTA insertion from the common alkoxide intermediate determines the (thio)ester:carbonate linkage ratio. Analogous to our previous report on CS2/CHO ROCOP, polymer oxygen enrichment is proposed to originate from an O/S rearrangement step from alkoxide chain ends adjacent to thioester links that eliminate CHS to create a carboxylate chain end, which then inserts CHO to generate a diester link.35 This effectively turns PTA and 2 equivalents of CHO into a diester link and CHS. Note that the O/S exchange process transforming alkoxides into thiolates could in principle also occur intermolecularly between a chain end and a thioester link of another polymer. Yet as such an intermolecular process would broaden dispersities substantially, we infer that the intramolecular pathway at the chain ends is dominant.
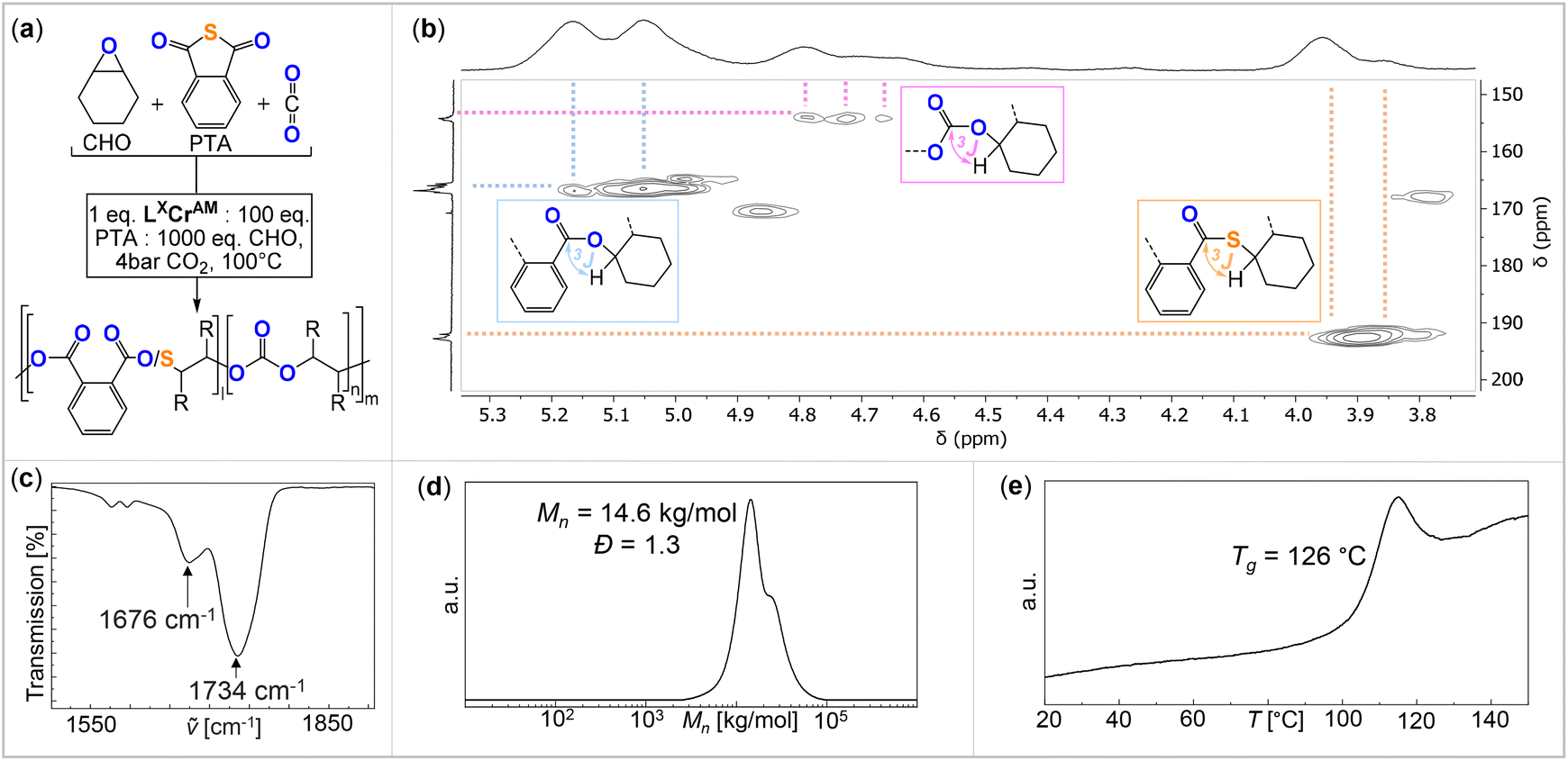 | ||
| Fig. 3 (a) Schematic representation of the ROTERP reaction. (b) 1H–13C HMBC NMR spectrum (25 °C, CDCl3), (c) IR spectrum, (d) SEC trace calibrated against a narrow polystyrene standard and (e) DSC thermogram of the polymer corresponding to Table 1, run #1. | ||
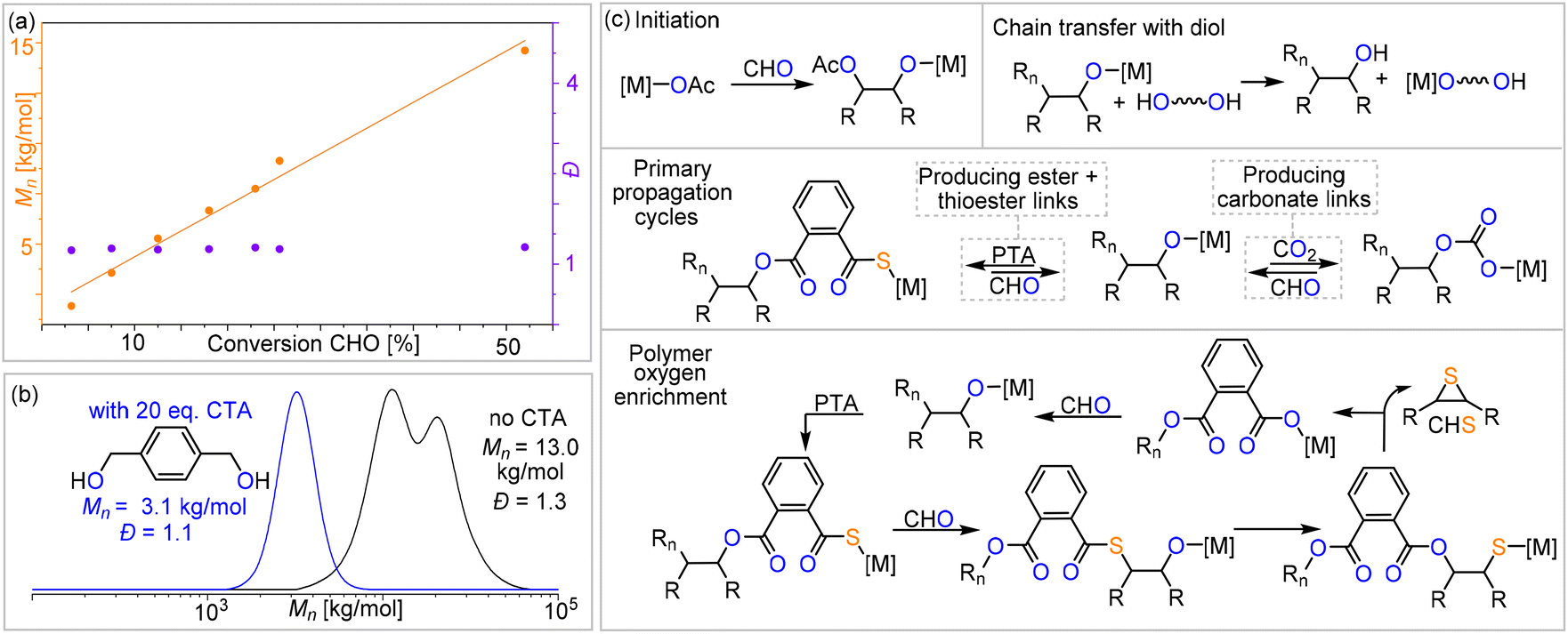 | ||
| Fig. 4 (a) Plot of conversion vs. Mn and Đ for Table 1 run #1 ROTERP; note that due to the viscosity of the reaction mixture, no aliquots could be taken between 25% and 55% conversions. (b) Overlaid GPC traces of Table 1 run #10 with and without the addition of 20 equiv. of 1,4-benzendimethanol as a chain transfer agent. (c) The proposed PTA/CO2/CHO ROTERP mechanism and intermediate speciation. [M] = metal catalyst, Rn = polymer chain, and R = –(C4H8)–. | ||
| Run | Cat.a | TOF ROTERPb [h−1] | Final TONc | Ester:thioester:carbonated (%) |
M
ne [kg mol−1] |
Đ | TOF (CHO/CO2)g [h−1] | TOF (CHO/PTA)h [h−1] | LCi |
|---|---|---|---|---|---|---|---|---|---|
| a Terpolymerisation at 1 eq. cat.:100 eq. PTA:1000 eq. CHO, T = 100 °C, 4 bar CO2. b Turnover frequency (TOF) determined by a linear fit to the eq. of CHO conversion per eq. of catalyst vs. time plot. Data points are excluded once conversion vs. time plots deviate from linearity. c Final turnover number (TON) at the specific endpoints of the polymerisations as depicted in the conversion vs. time plots of ESI section S3.† d Relative ratio of ester (4.9–5.2 ppm) versus thioester (3.7–4.2 ppm) versus carbonate (4.4–4.9 ppm) linkages assessed by the relative integrals of the tertiary CH resonances in the corresponding 1H NMR spectrum of the precipitated polymers. e Determined by SEC measurements conducted in THF using narrow MW polystyrene standards to calibrate the instrument. f Đ = Mw/Mn. g CHO/CO2 copolymerisation at 1 eq. of Cat.:1000 eq. of CHO, T = 100 °C, 4 bar CO2. h CHO/PTA copolymerisation at 1 eq. of Cat.:100 eq. of PTA:1000 eq. of CHO, T = 100 °C. i Proportionate linear combination (LC) of the TOFs of the parent ROCOPs. j According to ref. 35. k ca. 21% of ether and thioether links are part of the final polymer. | |||||||||
| #1 | L1CrRb | 200 ± 5 | 520 | 14:9:77 |
14.6 | 1.3 | 161 ± 1 | 51 ± 3 | 136 ± 1 |
| #2 | L2CrRb | 189 ± 7 | 570 | 16:9:75 |
10.3 | 1.2 | 149 ± 20 | 70 ± 6 | 129 ± 17 |
| #3 | L3CrRb | 122 ± 8 | 460 | 16:12:72 |
7.9 | 1.2 | 110 ± 2 | 85 ± 8 | 103 ± 4 |
| #4 | L4CrRb | 111 ± 9 | 280 | 52:19:23 |
5.1 | 1.3 | 40 ± 4 | 40 ± 5 | 40 ± 5 |
| #5 | L5CrRb | 367 ± 32 | 440 | 23:11:66 |
12.1 | 1.3 | 273 ± 17 | 144 ± 9 | 229 ± 14 |
| #6 | L6CrRb | 223 ± 13 | 550 | 17:7:76 |
13.3 | 1.3 | 261 ± 7 | 41 ± 1 | 208 ± 5 |
| #7 | L1CrLi | 84 ± 4 | 270 | 55:18:27k |
5.5 | 1.4 | 38 ± 4j | 84 ± 5 | 72 ± 5 |
| #8 | L1CrNa | 66 ± 2 | 260 | 16:13:71 |
6.4 | 1.2 | 76 ± 3j | 32 ± 4 | 63 ± 3 |
| #9 | L1CrK | 360 ± 15 | 500 | 15:20:65 |
16.9 | 1.2 | 127 ± 4j | 33 ± 1 | 94 ± 3 |
| #10 | L1CrCs | 195 ± 8 | 400 | 19:20:61 |
13.0 | 1.3 | 162 ± 15j | 58 ± 2 | 121 ± 10 |
With regards to polymerisation rates, it is reasonable to hypothesise that the reaction rate in terms of CHO conversion of PTA/CO2/CHO ROTERP should be in the first approximation of the proportionate linear combination of the rates of the parent PTA/CHO and CO2/CHO ROCOPs. For example, if a polymer comprises 50% ester and thioester (from PTA/CHO) and 50% carbonate links (from CO2/CHO), the TOF in the linear regime of the reaction kinetics where excess monomers are present should be 0.5 × TOF(PTA/CHO ROCOP) + 0.5 × TOF(CO2/CHO ROCOP) of the ROCOPs. To test this hypothesis, we monitored the ROTERP (1 eq. of LXCrAM:100 eq. of PTA:1000 eq. of CHO, 4 bar CO2, 100 °C) and all the parent CO2/CHO (1 eq. of LXCrAM:1000 eq. of CHO, 4 bar CO2, 100 °C) and PTA/CHO (1 eq. of LXCrAM:100 eq. of PTA:1000 eq. of CHO, 100 °C) ROCOPs by aliquot analysis to determine and compare the initial TOFs. As can be seen in Table 1, we observe that ROTERP at least maintains the TOF of the faster ROCOP. In some cases (runs #2, #4 and #9), we observe perhaps a counterintuitive modest rate enhancement when moving from the copolymerisations to terpolymerisation. Here, at every point in the conversion versus time plot, the CHO conversion for ROTERP exceeds those for the ROCOPs (ESI Fig. S27, S29 and S34†). At the current stage, we propose that this effect could originate from the fact that epoxide insertion is reversible during CO2/epoxide ROCOP, i.e., alkoxide intermediates eliminating epoxides to generate carbonates, a currently emerging notion in the literature.40,41 If PTA is present during the CO2/epoxide ROCOP process, i.e., the situation encountered during ROTERP, alkoxide intermediates are scavenged via PTA insertion, effectively suppressing the alkoxide-originated depropagation pathway. This proposition remains a mere hypothesis at the current stage and further mechanistic studies are underway. Motivated by the possibility that the presence of PTA could scavenge alkoxide intermediates, we turned towards the monosubstituted epoxide BO (ESI section S4†). ROCOP of CO2 with monosubstituted epoxides usually requires high CO2 pressures (>20 bar) to avoid backbiting pathways, which originate from alkoxide intermediates and yield small molecule cyclic carbonates instead of the desired polycarbonate products.42 Given the precedent of bicomponent catalysts comprising metal–salen complexes with μ-nitrido-bis(triphenylphosphan)-chloride (PPNCl) in the ROCOP of monosubstituted epoxides, we attempted PTA/CO2/BO ROTERP (1 eq. of SalcyCrCl:1 eq. of PPNCl:100 eq. of PTA:1000 eq. of BO, 4 bar CO2, 25 °C) and gratifyingly terpolymer (Table 2, run #1) formation was observed.43 However, both the 13C and 1H NMR spectra are very complex, which we attributed to regio-random polymer formation and O/S scrambling side-reaction links via a reaction pathway like the one outlined in Fig. 4. The latter was tentatively confirmed by 13C NMR showing more ester than thioester links by integration of the quaternary carbon atoms, which we previously established to be a good measure for linkage ratios in related polymers.33 Furthermore, previous reports on PTA/propylene oxide copolymerisation show that thiirane byproducts can themselves serve as monomers to re-enter the polymerisation forming thioether links.37 In fact, the 1H NMR spectra show signals centred around δ = 2.7 ppm in the polymer formed at an elevated temperature (Table 2, runs #2–#4), which increase in intensity with an increasing reaction temperature and these can be assigned to thioether links in reference to Ren and coworkers.37 Nevertheless, 13C NMR analysis unambiguously reveals the presence of polycarbonate linkages in all the produced polymers (δ(–O–(CqO)–O–) = 155 ppm). Importantly, in the absence of PTA (i.e. 1 eq. of SalcyCrCl:1 eq. of PPNCl:1000 eq. of BO, 4 bar CO2, 80 °C), no polycarbonate is present and only cyclic five-membered butylene carbonate is obtained supporting our initially formulated hypothesis of the limited lifetime of alkoxide intermediates due to the presence of PTA.
| Run | T [°C] | t [h] | Conv. PTA [%] | –OCO2–a [%] | M n [kg mol−1] (Đ)b |
|---|---|---|---|---|---|
| Copolymerisation at 1 eq. of SalcyCrCl:1 eq. of PPNCl:100 eq. of PTA:1000. BO, 4 bar CO2.a Relative ratio of carbonate versus carbonate, ester and thioester linkages assessed by the relative integrals of the corresponding 1H NMR spectrum.b Determined by SEC measurements conducted in THF using narrow MW polystyrene standards to calibrate the instrument. | |||||
| #1 | 25 | 40 | 80 | 53 | 8.7 (1.2) |
| #2 | 40 | 4 | 99 | 66 | 12.7 (1.1) |
| #3 | 60 | 1 | 99 | 36 | 9.8 (1.2) |
| #4 | 80 | 0.3 | 99 | 22 | 9.1 (1.3) |
Moving to lower reaction temperatures (Table 2, runs #1–#3) results in an increasing amount of CO2 incorporation into the final polymer, as evident from 13C NMR spectroscopy. Performing PTA/CO2/BO ROTERP at room temperature (Table 2, run #1) further minimizes the side-reactions resulting in e.g. sharp single rather than multiple split resonances for quaternary ester, thioester and carbonate 13C resonances and no thioether links. 2D NMR spectra show thioester units adjacent to CH2R2 (often referred to as the “tail” position of ring-opened epoxides) while ester units are adjacent to CH3R (or the “head” position), suggesting the formation of poly[(ester-alt-thioester)-carbonates] as shown in Fig. 5. In each case (Table 2, runs #1–#4), narrow bimodal molecular weight distributions were obtained similar to the above cases with CHO leading us to propose the same underlying reasons for this (vide supra). DSC shows glass transition temperatures of Tg = 17–27 °C for the obtained materials with no clear trends with regard to linkage composition.
 | ||
| Fig. 5 1H, 13C and 1H–13C HMBC NMR spectrum of polymer corresponding to Table 2, run #4. | ||
As PTA/CO2/CHO ROTERP yields statistical terpolymers while the all-oxygen combination of PA/CO2/CHO yields block polymers with many catalysts as outlined in the introduction, the question arises what quaternary monomer mixtures comprising PA/PTA/CO2/CHO would yield (ESI section S5†). To investigate this question, we conducted tetrapolymerisation by employing L1CrK at 1 eq. of Cat:50 eq. of PA:50 eq. of PTA:1000 eq. of CHO at 4 bar CO2 pressure and 100 °C. The reaction was monitored by 1H NMR analysis of aliquots removed at regular time intervals, which shows that statistical PA/CO2/CHO ROTERP producing poly(ester-carbonates) occurs until PA is consumed, followed by statistical PTA/CO2/CHO ROTERP producing poly(ester-thioester-carbonates), although some tapering was observed, producing a polymer with a final Mn = 12.6 kg mol−1, and Đ = 1.2 after 2 h. Our findings are in line with recent reports on PA/PTA/epoxide terpolymerisation employing related metal-based catalysts.44 There, slower PA ROCOP occurred after faster PTA ROCOP due to faster PA compared to PTA insertion into alkoxide chain ends. However, for L1CrK we additionally found that PA/CHO copolymerisation is approximately 3 times faster than PTA/CHO copolymerisation when the stand-alone ROCOPs were investigated. This might contribute to the preferential polymerisation of PA over PTA in the above tetrapolymerisation. We previously showed that the formal incorporation of sulfur-containing links into CO2/CHO copolymers for polymers obtained from CS2/CO2/epoxide ROTERP bestowed typical degradability benefits of sulfurated polymers upon the parent CO2/CHO polycarbonates.35,45 Therefore, we hypothesized that the thioester links introduced in our PTA/CO2/CHO ROTERP could serve a similar function. Accordingly, we subjected one of our terpolymers (Table 1 run #10) to broadband UV irradiation in CDCl3, conditions (see ESI section S6†) under which polycarbonates from CO2 or polyesters from PA/epoxide copolymerisation show no appreciable degradation.31,35 Gratifyingly, our terpolymer (starting Mn = 13.0) degraded into oligomers with Mn < 1.5 kg mol−1 under UV irradiation. NMR analysis of the sample after photolysis reveals that while ester and carbonate links remain mostly intact, thioester links are completely degraded supporting the theory that these represent photochemical breaking points in the polymer structure. Interestingly, PTA can be identified as a major degradation product hinting towards the potential of chemical recycling to the monomer of these PTA-derived polymers, which should be investigated in the future.46 Elsewhere, the statistical incorporation of thioester links into the polymer main chain of polyacrylates caused susceptibility to degradation by aminolysis and we found the same to be the case for our ROTERP terpolymers.47 Subjecting a PTA/CO2/CHO terpolymer (Table 1 run #10) to methanolic ammonia for 48 h leads to its complete degradation into oligomers with Mn < 1 kg mol−1. In contrast, the all-oxygen counterparts from either CO2/CHO or PA/CHO ROCOP do not degrade under identical conditions. This shows that again the thioester links in the ROTERP terpolymers act as breaking points for aminolysis.
Conclusions
In conclusion, we have investigated the ROTERP of alicyclic CHO with PTA and CO2 by employing a series of heterobimetallic Cr(III)-based catalysts to produce novel poly(ester-thioester-carbonates). Molecular weights can be controlled by monomer conversion and the addition of chain transfer agents, while the linkage ratios can be affected by the catalyst for which both ligand electronics and metal choice are important. Kinetics studies revealed a rate maintenance of terpolymerisation compared to the faster parent copolymerisation, which can be tentatively explained by the suppression of depolymerisation pathways. This realisation led us to produce terpolymers from BO, PTA and CO2 under conditions where the parent CO2/BO copolymerisation fails to produce polymers. Furthermore, the new terpolymerisation can be combined with PA/epoxide ROCOP in tetrapolymerisations. Our study indicates that there could be rate and selectivity benefits when moving to the polymerisation of more complex monomer mixtures, which should motivate further investigation of such, particularly as the introduced sulfur-containing links can be utilised as predetermined breaking points in the polymer main chain.Experimental details
General complex synthesis
Under inert conditions, the proligand LXH2 (1 eq.) and Cr(OAc)2 (1 eq.) were dissolved in degassed acetonitrile and stirred overnight at room temperature or under reflux. Glacial acetic acid (2 eq.) was added and the solution was refluxed for 8 h and then stirred overnight at room temperature. The solvent was removed under reduced pressure and the crude LXCr was washed three times with diethyl ether to obtain a brown powder. The intermediate LXCr (1 eq.) was suspended with AMOAc (1.05 eq., AM = Li, Na, K, Rb, Cs) in MeOH and refluxed briefly. All volatiles were removed under reduced pressure and the crude product was washed three times with diethyl ether to obtain LXCrAM as a brown powder.General polymerisation protocol
Under inert conditions, the catalyst and the monomers were added to an oven-dried vial sealed with a melamine cap containing a Teflon inlay or an oven-dried Schlenk flask equipped with a magnetic stirrer. The flask was brought outside the glovebox and placed in a pre-heated aluminium block or oil bath at the specified temperature for the specified time. If CO2 was used, the mixture was briefly degassed under vacuum until gas evolution ceased before supplying CO2 at 4 bar. Aliquots were taken by cooling the vial or Schlenk flask in an ice bath before removing 10 μL and then flushing the reaction system with CO2 for 3 seconds in the case of CO2 polymerisations and argon in the other cases. The polymerisation progress was monitored by 1H NMR analysis in CDCl3 of the crude aliquots. The polymerisations were terminated when mixtures became too viscous to be magnetically stirred or after 24 h. The highly viscous mixture was dissolved in 4 mL of CDCl3 and a final aliquot was taken from which the overall final conversion was determined. The CDCl3 solution was precipitated in 40 mL of MeOH and then further purified by precipitation from THF/pentane and DCM/methanol before drying the polymers under vacuum at 60 °C overnight.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The VCI is acknowledged for a Liebig Fellowship for A. J. P. and a Ph.D. studentship for C. G. Prof. Dr Christian Müller and Prof. Dr Rainer Haag are thanked for their continuous support and valuable discussions.References
- G. Becker and F. R. Wurm, Chem. Soc. Rev., 2018, 47, 7739–7782 RSC.
- N. M. Bingham, Q. un Nisa, P. Gupta, N. P. Young, E. Velliou and P. J. Roth, Biomacromolecules, 2022, 23, 2031–2039 CrossRef CAS PubMed.
- N. M. Bingham, Z. Abousalman-Rezvani, K. Collins and P. J. Roth, Polym. Chem., 2022, 13, 2880–2901 RSC.
- P. Yuan, Y. Sun, X. Xu, Y. Luo and M. Hong, Nat. Chem., 2022, 14, 294–303 CrossRef CAS PubMed.
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2022, 61, e202104495 CrossRef CAS PubMed.
- C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, ACS Catal., 2022, 12, 11037–11070 CrossRef CAS.
- S. Kernbichl and B. Rieger, in Engineering Solutions for CO2 Conversion, John Wiley & Sons, Ltd, 2021, pp. 385–406 Search PubMed.
- J. Xu, P. Zhang, Y. Yuan and N. Hadjichristidis, Angew. Chem., Int. Ed., 2023, 62, e2022188 Search PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- J. Diebler, H. Komber, L. Häußler, A. Lederer and T. Werner, Macromolecules, 2016, 49, 4723–4731 CrossRef CAS.
- L.-Y. Wang, G.-G. Gu, B.-H. Ren, T.-J. Yue, X.-B. Lu and W.-M. Ren, ACS Catal., 2020, 10, 6635–6644 CrossRef CAS.
- T.-J. Yue, M.-C. Zhang, G.-G. Gu, L.-Y. Wang, W.-M. Ren and X.-B. Lu, Angew. Chem., 2019, 131, 628–633 CrossRef.
- J.-L. Yang, Y. Wang, X.-H. Cao, C.-J. Zhang, Z. Chen and X.-H. Zhang, Macromol. Rapid Commun., 2021, 42, 2000472 CrossRef CAS PubMed.
- C. Fornacon-Wood, M. R. Stühler, C. Gallizioli, B. R. Manjunatha, V. Wachtendorf, B. Schartel and A. J. Plajer, Chem. Commun., 2023, 59, 11353–11356 RSC.
- E. M. López-Vidal, G. L. Gregory, G. Kociok-Köhn and A. Buchard, Polym. Chem., 2018, 9, 1577–1582 RSC.
- X.-L. Chen, B. Wang, D.-P. Song, L. Pan and Y.-S. Li, Macromolecules, 2022, 55, 1153–1164 CrossRef CAS.
- T.-J. Yue, L.-Y. Wang, W.-M. Ren and X.-B. Lu, Eur. Polym. J., 2023, 190, 111985 CrossRef CAS.
- C. Hardy, G. Kociok-Köhn and A. Buchard, Chem. Commun., 2022, 58, 5463–5466 RSC.
- D. K. Tran, A. N. Braaksma, A. M. Andras, S. K. Boopathi, D. J. Darensbourg and K. L. Wooley, J. Am. Chem. Soc., 2023, 145(33), 18560–18567 CrossRef CAS PubMed.
- M. Sengoden, G. A. Bhat and D. J. Darensbourg, Green Chem., 2022, 24, 2535–2541 RSC.
- X. Geng, Z. Liu, C. Zhang and X. Zhang, Macromolecules, 2023, 56, 4649–4657 CrossRef CAS.
- X.-F. Zhu, G.-W. Yang, R. Xie and G.-P. Wu, Angew. Chem., Int. Ed., 2022, 61, e202115189 CrossRef CAS PubMed.
- C. Fornacon-Wood, B. R. Manjunatha, M. R. Stühler, C. Gallizioli, C. Müller, P. Pröhm and A. J. Plajer, Nat. Commun., 2023, 14, 4525 CrossRef CAS PubMed.
- G. Feng, X. Feng, X. Liu, W. Guo, C. Zhang and X. Zhang, Macromolecules, 2023, 56, 6798–6805 CrossRef CAS.
- M. Luo, X.-H. Zhang and D. J. Darensbourg, Macromolecules, 2015, 48, 5526–5532 CrossRef CAS.
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2021, 60, 13372–13379 CrossRef CAS PubMed.
- A. J. Plajer and C. K. Williams, ACS Catal., 2021, 11, 14819–14828 CrossRef CAS.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS PubMed.
- S. Ye, Y. Ren, J. Liang, S. Wang, S. Huang, D. Han, Z. Huang, W. Liu, M. Xiao and Y. Meng, J. CO2 Util., 2022, 65, 102223 CrossRef CAS.
- C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS PubMed.
- S. Rupf, P. Pröhm and A. J. Plajer, Chem. Sci., 2022, 13, 6355–6365 RSC.
- D. Silbernagl, H. Sturm and A. J. Plajer, Polym. Chem., 2022, 13, 3981–3985 RSC.
- A. J. Plajer, ChemCatChem, 2022, 14, e202200867 CrossRef CAS.
- P. Deglmann, S. Machleit, C. Gallizioli, S. M. Rupf and A. J. Plajer, Catal. Sci. Technol., 2023, 13, 2937–2945 RSC.
- J. Stephan, M. R. Stühler, S. M. Rupf, S. Neale and A. J. Plajer, Cell Rep. Phys. Sci., 2023, 101510 CrossRef CAS.
- W. T. Diment, G. L. Gregory, R. W. F. Kerr, A. Phanopoulos, A. Buchard and C. K. Williams, ACS Catal., 2021, 11, 12532–12542 CrossRef CAS.
- L.-Y. Wang, G.-G. Gu, T.-J. Yue, W.-M. Ren and X.-B. Lu, Macromolecules, 2019, 52, 2439–2445 CrossRef CAS.
- D. J. Darensbourg, Green Chem., 2019, 21, 2214–2223 RSC.
- F. Della Monica and C. Capacchione, Asian J. Org. Chem., 2022, 11, e202200300 CrossRef CAS.
- (a) F. N. Singer, A. C. Deacy, T. M. McGuire, C. K. Williams and A. Buchard, Angew. Chem., Int. Ed., 2022, 61, e202201785 CrossRef CAS PubMed; (b) T. M. McGuire, A. C. Deacy, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2022, 144(40), 18444–18449 CrossRef CAS PubMed.
- (a) Y. Liu and X.-B. Lu, Macromolecules, 2023, 56, 1759–1777 CrossRef CAS; (b) Y. Yu, B. Gao, Y. Liu and X.-B. Lu, Angew. Chem., Int. Ed., 2022, 61, e202204492 CrossRef CAS PubMed.
- D. J. Darensbourg, P. Bottarelli and J. R. Andreatta, Macromolecules, 2007, 40, 7727–7729 CrossRef CAS.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- X.-L. Chen, B. Wang, D.-P. Song, L. Pan and Y.-S. Li, Macromolecules, 2022, 55, 1153–1164 CrossRef CAS.
- H. Li, S. M. Guillaume and J.-F. Carpentier, Chem. – Asian J., 2022, 17, e202200641 CrossRef CAS PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- R. Abu Bakar, K. S. Hepburn, J. L. Keddie and P. J. Roth, Angew. Chem., Int. Ed., 2023, 62, e202307009 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2291620. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3py01022h |
| This journal is © The Royal Society of Chemistry 2023 |