
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRing-opening polymerization of functionalized aliphatic bicyclic carbonates†
Jixiang
Ni
ab,
Matteo
Lanzi
*a,
David H.
Lamparelli
a and
Arjan W.
Kleij
 *ac
*ac
aInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science & Technology (BIST), Av. Països Catalans 16, 43007 – Tarragona, Spain. E-mail: akleij@iciq.es; mlanzi@iciq.es
bUniversitat Rovira i Virgili (URV), Marcel·lí Domingo s/n, 43007 Tarragona, Spain
cCatalan Institute of Research and Advanced Studies (ICREA), Pg. Lluis Companys 23, 08010 – Barcelona, Spain
First published on 6th October 2023
Abstract
A series of six-membered bicyclic carbonates has been prepared from an alcohol precursor through esterification, etherification and silyl-etherification methods to afford functional monomers with synthetically useful vinyl, allyl, alkynyl and polymerizable groups that allow for orthogonal polymerization strategies. Anionic ring-opening polymerization (AROP) of these monomers allows us to access densely functional aliphatic polycarbonates with Mn's and Tg's in the range of 3.2–11.7 kg mol−1 and 11–106 °C, respectively. Bifunctional monomers comprising two separate polymerizable groups can be selectively polymerized via AROP or radical based/ring-opening metathesis polymerization methods. Selected polycarbonates were post-modified via thiol–ene chemistry to further modulate their properties. Finally, base-mediated depolymerization of these new polycarbonates was attempted giving rise to an unusual bicyclic oxetane product, which may be upcycled through appropriate copolymerization methods.
Environmentally friendly and modular approaches to functional polymers are key to the development of sustainable materials.1 Among the functional polymers developed to date, aliphatic polycarbonates (APCs) have received considerable consideration as a result of their low toxicity, high biocompatibility, and degradability.2,3 These features are especially attractive in the context of applications that focus on medical devices and drug delivery systems.4 Whereas the conventional way of polycarbonate synthesis relies on the use of phosgene derivatives, alternative strategies based on the use of carbon dioxide (CO2) have recently emerged. The utilization of CO2 as an abundant, cheap and safe reagent is considered to be a key factor for innovative process design towards APCs with biodegradation potential. APCs are typically prepared via two main approaches, either via ring-opening copolymerization (ROCOP) of epoxides and carbon dioxide5,6 or through anionic ring-opening polymerization (AROP) of suitable cyclic carbonate monomers. Although the use of CO2 as a monomer is attractive, the ROCOP approach using (acyclic) epoxides typically suffers from limitations in functional group diversity or compatibility, and is frequently accompanied by the formation of “ether” linkages (i.e., structural defects) and the concomitant formation of five-membered cyclic carbonates as by-products affecting the overall chemo-selectivity.
The AROP reaction of 6- and 7-membered cyclic carbonates (abbreviated as 6MCCs and 7MCCs) has emerged as a more robust and versatile approach for the preparation of functional polycarbonates.7,8 Larger ring cyclic carbonates such as 6MCCs over time have become popular monomers but are often prepared using harmful and low-atom economy reagents such as phosgene and its derivatives (Scheme 1a).9
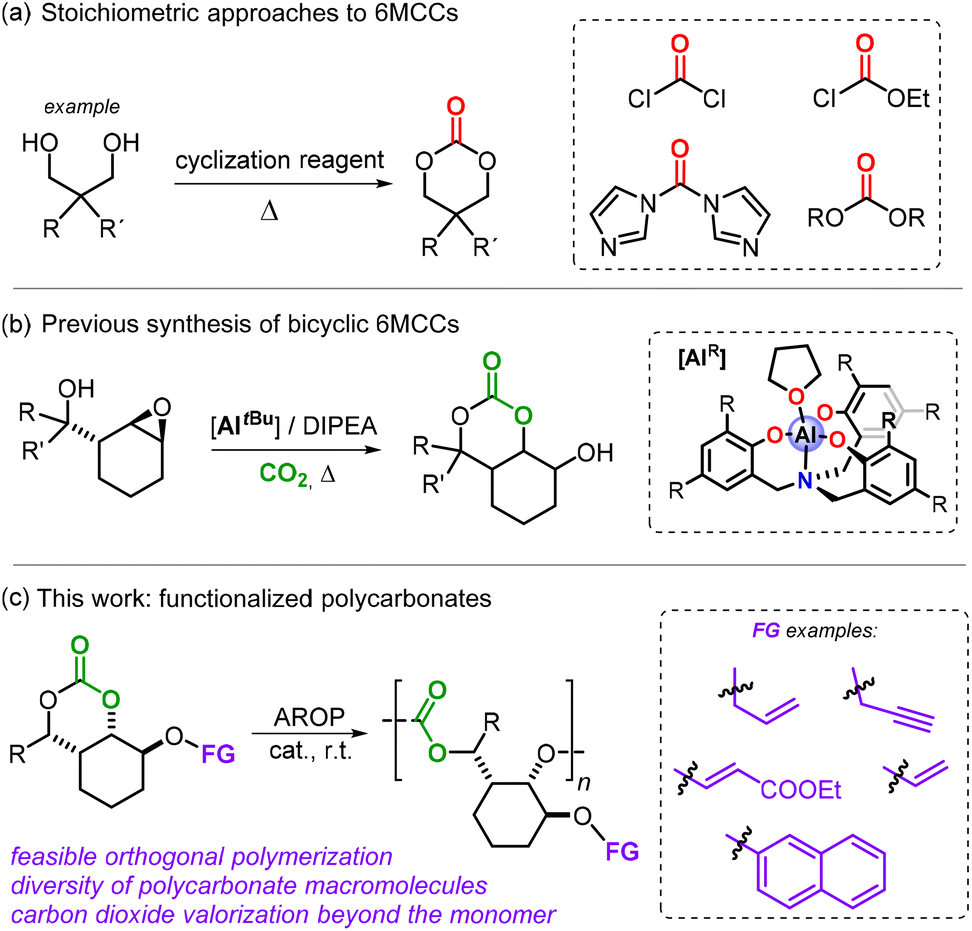 | ||
| Scheme 1 (a) Common approach towards polymerizable six-membered cyclic carbonates. (b) Previously reported bicyclic monomer comprising a secondary alcohol. (c) Current approach towards functional polycarbonates through ROP of functional six-membered cyclic carbonates. 6MCCs stands for six-membered cyclic carbonates. | ||
Our group recently reported a simple catalytic CO2-based approach towards bicyclic 6MCCs (Scheme 1b).10,11 This approach benefits from a double and cooperative activation strategy with a pendent alcohol group activating CO2 while an Al-complex promotes intramolecular ring-opening of the oxirane in the substrate through coordination. This approach allowed us to devise a structurally diverse range of novel bicyclic 6MCCs under mild conditions, and importantly enabled the isolation of compounds featuring a synthetically useful hydroxyl group. We envisioned that these –OH groups could be exploited such that different functionalities could be introduced in these 6MCC monomers. This would then offer a simple protocol to form functional carbonates for AROP leading ultimately to functionalized APCs (Scheme 1c), and herein we describe the advances we have made in the area.
Taking advantage of the residual hydroxy group in A–D, we began our investigation with the synthesis of a small library of 6MCCs decorated with various synthetically useful moieties (Scheme 2). The silyl-ether derivatives 1–5 were all prepared using the respective silyl-chloride reagent and performing the coupling reaction in the presence of an excess of triethylamine in dry CH2Cl2 at r.t. (see the ESI† for details). This approach allowed the introduction of either allyl (products 1–4) or vinyl groups (compound 5) in the different parent scaffolds (A–D) with appreciable yields of the isolated bicyclic 6MCCs. Monomers 3 and 4 are additionally equipped with an aryl/alkyl substituent at the other side of the bicycle allowing us to scrutinize their influence on the polymerizability of these 6MCCs. An oxo-Michael addition of precursor A to ethyl propiolate in the presence of N-methyl morpholine (NMM, 20 mol%) in CH3CN at r.t. afforded monomer 6 in 56% yield, and this shows that also acrylic esters are feasible functional groups.
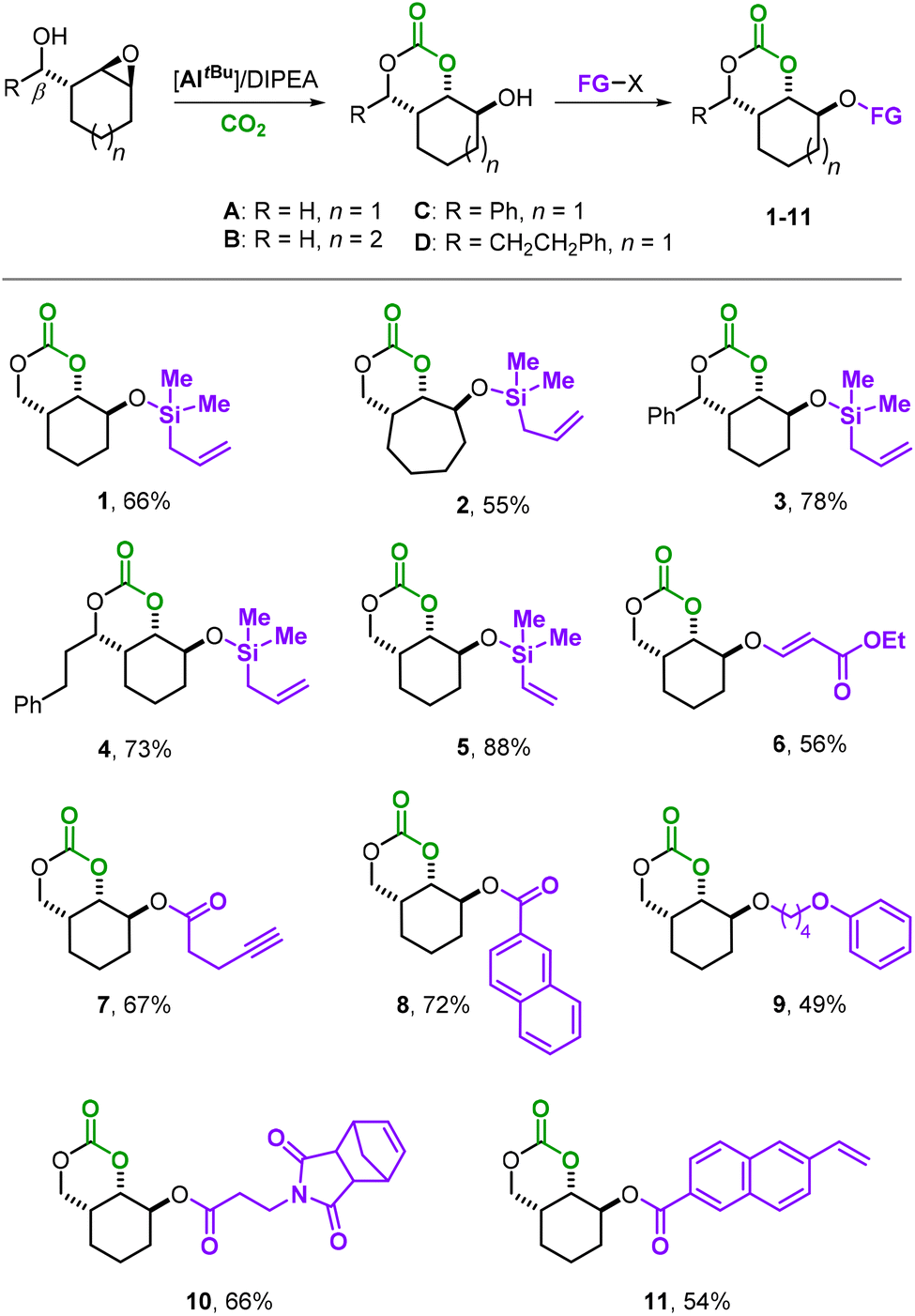 | ||
| Scheme 2 General synthetic scheme for the functionalized bicyclic six-membered cyclic carbonate monomers 1–11 derived from precursor 6MCCs A–D. FG-X stands for the coupling agent used to produce the functionalized bicyclic 6MCC. | ||
Steglich esterification (using DCC, dicyclohexylcarbodiimide; see the ESI† for details) enables the coupling of various functional carboxylic acids with the secondary alcohol group of A. Homopropargylic carboxylic acid is smoothly coupled with A providing 7 in 67% yield, expanding the possible functionality pool to terminal alkynes. At this stage, we believed that the introduction of a rigid naphthyl moiety in the bicyclic 6MCC scaffold could be a way to produce, after AROP, APCs with larger aromatic groups favorable towards higher thermal resistance. Product 8 that comprises naphthyl ester groups could be isolated in 72% yield. To further modulate the thermal properties of the target APCs, a more flexible aliphatic chain could also be introduced (9, 49%) by performing a coupling between A and Kobayashi aryne precursors in THF.12,13
Finally, we aimed at designing bifunctional monomers that contain two distinct and polymerizable units (10 and 11) so that orthogonal polymerization strategies could be enabled. Monomer 10 was attained in 66% yield by esterification of A with 3-(1,3-dioxo-1,3,3a,4,7,7a-hexahydro-2H-4,7-methano-isoindol-2-yl)-propanoic acid, whereas 11 could be prepared in 54% yield by the treatment of A with 6-vinyl-2-naphthoic acid in the presence of DCC (see the ESI† for details). All new monomers 1–11 were fully characterized by 1H NMR, 13C NMR, high-resolution ESI(+)-MS, and IR spectroscopy.
To study the reactivity of the monomers 1–9, we then directed our attention towards AROP using 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as a benchmark organocatalyst in the presence of an equimolar quantity of benzyl alcohol as an initiator at r.t. (Table 1, see the ESI† for experimental details).14 These preferable mild temperature conditions were chosen as to avoid the possible depolymerization of the formed polycarbonates, which is feasible with TBD at elevated temperatures.15 In nearly all cases, reasonable control of the polymerization process was achieved (Đ values in the range 1.29–1.64) delivering the functionalized APCs with low to moderately high molecular weights (Mn's 3.2–11.7 kg mol−1). Allyl-functionalized monomers 1 and 2 were both ring-open polymerized to their APCs P1 and P2 with around 90% conversion (Table 1, entries 1 and 2). In contrast to these promising results, monomers 3 and 4 proved to be completely unreactive towards the formation of P3 and P4 even when raising the reaction temperature to 60 °C and extending the reaction time to 48 h (entries 3 and 4). This can be explained by the additional substitution on the carbonate ring, which apparently creates a too large steric impediment for the incoming TBD hydrogen-bond activator and BnOH initiator.
|
|
|||||
|---|---|---|---|---|---|
| Entry | M | Conv.b (%) |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (kg mol−1) c (kg mol−1) |
Đ |
T
gd (°C) |
| a Unless otherwise specified, the polymerization reactions were performed using 0.30 mmol of monomer, 1 mol% of TBD, and 1 mol% of BnOH in toluene (1 M) for 20 h at r.t. (20 °C). b Conversion of the monomer was determined by 1H NMR (CDCl3) spectroscopy. c Determined by GPC in THF calibrated with PPO standards. d Obtained from DSC analysis, the data refer to the second heating. e Reaction was performed at a 0.60 mmol scale. f Polymerization was performed both at 45 °C and 60 °C for 48 h with the same results. M stands for monomer. g TBD/BnOH loading was 0.5/0.5 mol%. h TBD/BnOH loading was 2.0/2.0 mol%. i TBD/BnOH loading was 2.0/1.0 mol%. | |||||
| 1e | 1 | 91 | 5.6 | 1.54 | 19 |
| 2 | 2 | 86 | 6.8 | 1.64 | 11 |
| 3f | 3 | — | — | — | — |
| 4f | 4 | — | — | — | — |
| 5e | 5 | 95 | 11.7 | 1.44 | 57 |
| 6 | 6 | 87 | 6.8 | 1.50 | 70 |
| 7g | 6 | 90 | 3.5 | 1.59 | — |
| 8h | 6 | 89 | 5.0 | 1.36 | — |
| 9i | 6 | 88 | 4.3 | 1.71 | — |
| 10 | 7 | 98 | 6.9 | 1.29 | 41 |
| 11 | 8 | 90 | 3.2 | 1.61 | 106 |
| 12 | 9 | 84 | 7.0 | 1.44 | 15 |
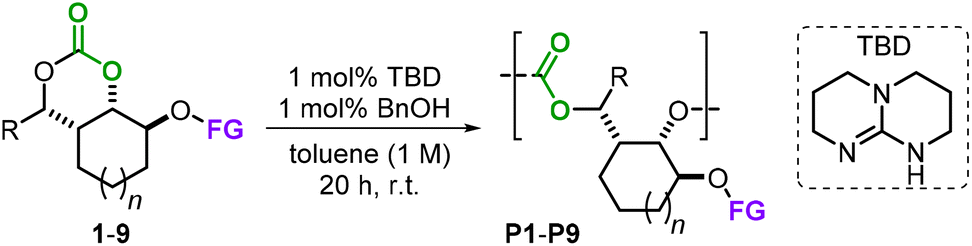
The AROP of the vinyl derivative 5 (entry 5) gave P5 with the highest molecular weight (Mn = 11.7 kg mol−1, Đ = 1.44) among the series. Other functional groups such as an acrylic ester (6) and a terminal alkyne (7) were compatible with the reaction conditions delivering P6 and P7 with modest Mn values and somewhat lower dispersities (entries 6 and 10). We used monomer 6 to examine the effect of the catalyst and initiator loading (cf., entries 6–9), but found that the best conditions (1 mol% of each) are most beneficial for these types of bicyclic carbonate monomers. Macromolecule P8 showed a substantial decrease in molecular weight (Mn = 3.2 kg mol−1, entry 11), which we ascribe to the more hindered carbonate ring upon activation by TBD following ring-opening by the initiator.
Though a low(er) molecular weight was attained for P8, the measured glass transition temperature (Tg = 106 °C) was significantly higher compared to those of the other APCs, which is likely due to an increase in pi–pi stacking in the solid state. The polymerization of monomer 9 provided a molecular weight, polymer dispersity and Tg (entry 12) similar to those of polymers P1 and P2. Purified samples of all the APCs (white powders) reported in Table 1 were obtained through simple precipitation of the crude reaction mixtures from methanol, and the analytical data and spectra are provided in the ESI.† All PCs reported here are likely regio-irregular on the basis of their 13C NMR spectra (see the ESI†) and display monomodal peak distributions in their GPCs. On the basis of our previous experience with ring-opening of substituted cyclic carbonates, the initiator (BnOH) most likely approaches from the least hindered side, thereby leaving a primary alcohol as an end-group.16
Additionally, bifunctional monomers 10 and 11 bearing orthogonal polymerizable groups were tested (Schemes 3 and 4). Under the AROP reaction conditions reported in Table 1, the monomers 10 and 11 are smoothly transformed delivering P10a and P11a with appreciable molecular weight (Mn = 7.23 and 7.18 kg mol−1, respectively) and reasonable polydispersities (Đ = 1.33 and 1.59, respectively).
 | ||
| Scheme 3 Synthesis of polymers P10a and P10b. | ||
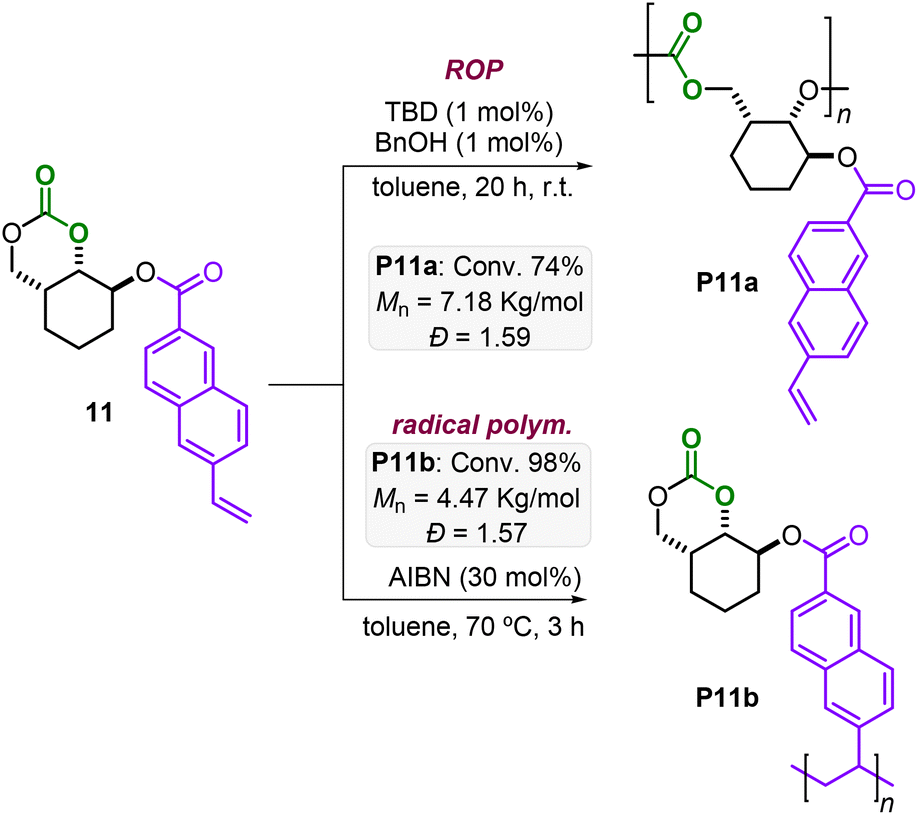 | ||
| Scheme 4 Synthesis of polymers P11a and P11b. | ||
Monomer 10 has, apart from the 6MCC fragment, an additional strained cyclohexene moiety, which is suitable for ring-opening metathesis polymerization (ROMP). ROMP is nowadays a robust and selective approach for the controlled preparation of polyolefins, especially starting from strained structures.7b,17–19 By treatment of 10 with a Grubbs 3rd generation catalyst, a complete conversion was obtained within 90 min at r.t. yielding the desired polyolefin P10b (Scheme 3) with a low polydispersity (Đ = 1.10) and a modest molecular weight (Mn = 4.02 kg mol−1). It should be noted that lower amounts of Ru-catalyst also worked well leading to gel-like products. However, in these cases, solubility issues did not allow further characterization. Azobisisobutyronitrile (AIBN) initiated radical polymerization of the vinyl moiety present in monomer 11 led to full conversion in 3 h at 70 °C yielding the poly-naphthalene product P11b (Mn = 4.47 kg mol−1, Đ = 1.57). Noteworthily, complete chemoselectivity is observed in all transformations of P10 and P11 preserving the other functional groups (i.e., the 6MCC, norbornene and vinyl moieties) as additional handles for further transformations.
Post-polymerization modification of P1 was also conducted to show the robustness of the polycarbonate macromolecules (Scheme 5). Two approaches were considered: a “click” reaction using a mono-thiol and a cross-linking reaction using a di-thiol reagent.20 First, P1 was subjected to cross-linking in the presence of 1,3-propanedithiol and AIBN as the radical initiator at 70 °C. Under these conditions, full conversion of the alkene moieties was observed after 12 h. Due to the high level of crosslinking, P1a was insoluble in the typical organic solvents, thus preventing further characterization by NMR and GPC. IR analysis of P1a showed a disappearance of the initial ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H and CC (stretch) absorptions for P1 at 3077 and 1631 cm−1, respectively, suggesting that most of the double bonds had reacted. Differential scanning calorimetry (DSC) analysis showed that P1a has a Tg of 32 °C, which is 12° higher than that of the parent polymer P1, thus implying that at least a part of the silyl-allyl fragments took part in the crosslinking event. The same silyl-allyl groups were also allowed to react with trifluorobenzyl thiol under similar reaction conditions to quantitatively afford (soluble) P1b. Gel-permeation chromatography analysis (GPC) of P1b displayed a significant increase in the polymer weight (the Mn rose to 12.2 kg mol−1) while the polydispersity remained virtually the same. This seems to corroborate with a high functionalization degree of P1 and a substantial mass increase of the repeat units (270 → 462 g mol−1, ΔM = 42%), and thus the polymer as a whole.
C–H and CC (stretch) absorptions for P1 at 3077 and 1631 cm−1, respectively, suggesting that most of the double bonds had reacted. Differential scanning calorimetry (DSC) analysis showed that P1a has a Tg of 32 °C, which is 12° higher than that of the parent polymer P1, thus implying that at least a part of the silyl-allyl fragments took part in the crosslinking event. The same silyl-allyl groups were also allowed to react with trifluorobenzyl thiol under similar reaction conditions to quantitatively afford (soluble) P1b. Gel-permeation chromatography analysis (GPC) of P1b displayed a significant increase in the polymer weight (the Mn rose to 12.2 kg mol−1) while the polydispersity remained virtually the same. This seems to corroborate with a high functionalization degree of P1 and a substantial mass increase of the repeat units (270 → 462 g mol−1, ΔM = 42%), and thus the polymer as a whole.
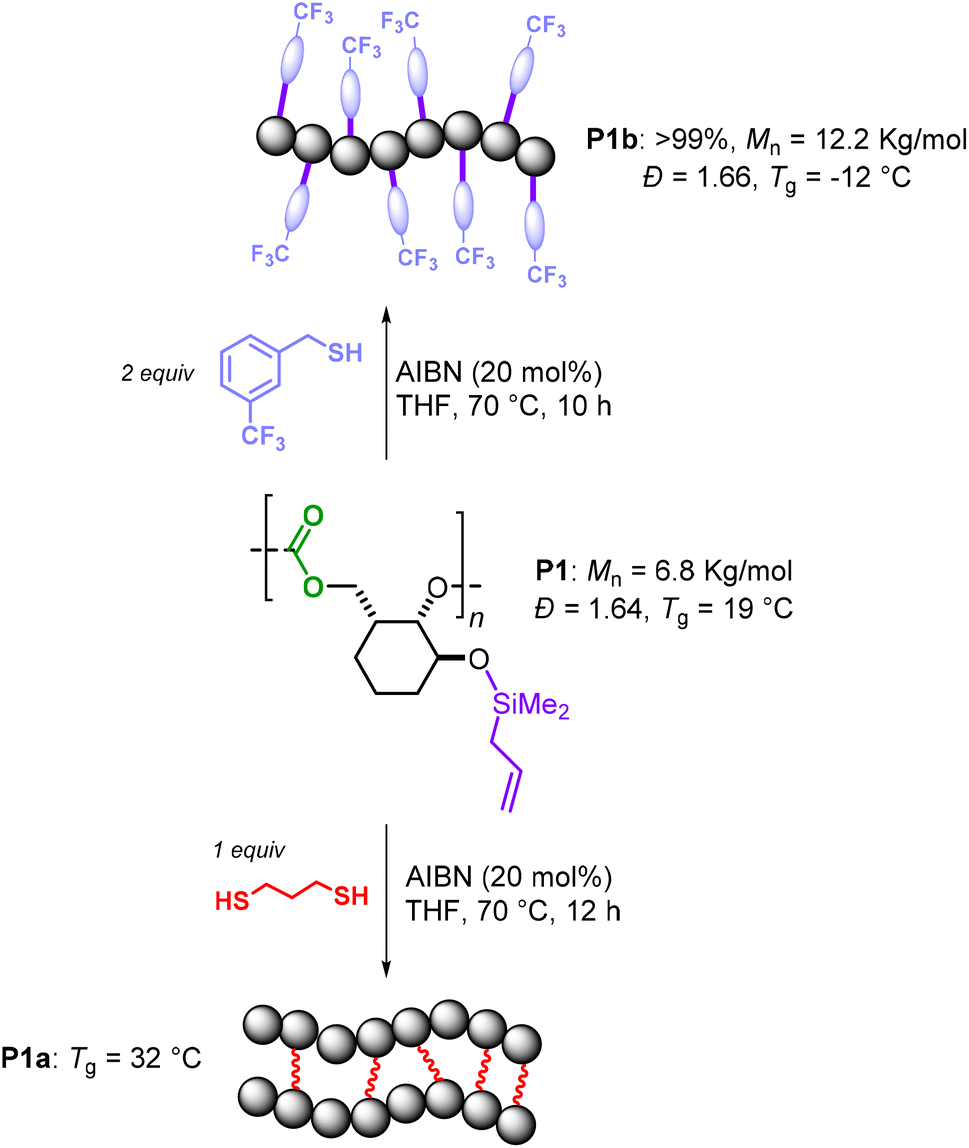 | ||
| Scheme 5 Thiol–ene based functionalization and cross-linking of P1. | ||
Finally, chemical degradation studies were conducted to examine whether these new polycarbonates could be selectively depolymerized. Depolymerization of P8 was assessed using TBD as a catalyst in toluene at 110 °C (Scheme 6), as previously a bio-based polycarbonate was reported to fully decompose towards its original monomers under such conditions.15 In the present case, a similar catalytic degradation was performed and the reaction delivered two new products: a fused bicyclic oxetane 8a and the diol 8b as an inseparable 85:15 mixture in a combined 56% yield. Whilst basic conditions for the degradation of polycarbonates often release the corresponding diol as a byproduct,14 to the best of our knowledge, the formation of a fused bicyclic oxetane 8a from the decomposition of polycarbonate is a new observation.21 Recently, both Buchard and Wooley showed that similar types of bicyclic oxetanes can be used to prepare different kinds of polymers including polycarbonates and polyethers,22 thus providing a potential recycling or repurposing route for 8a.
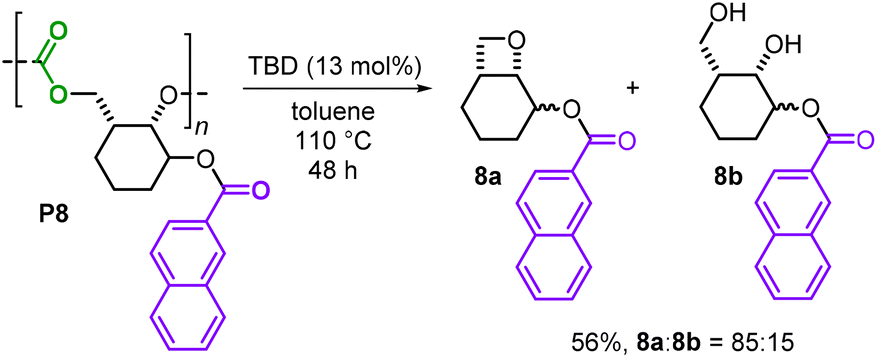 | ||
| Scheme 6 Depolymerization of P8 using TBD as a catalyst. | ||
Conclusions
In summary, we have prepared a family of functional, bicyclic 6MCC monomers bearing synthetically useful groups. The catalytic AROP provides straightforward access to densely substituted macromolecular aliphatic carbonates. Bifunctional monomers were also designed allowing the triggering of two different types of polymerizations and offering orthogonal polymerization/crosslinking strategies. Post-synthetic functionalization of these APCs was also demonstrated, which permits the change and modulation of the (thermal) properties. A TBD-catalyzed degradation of one selected oligocarbonate led to a new fused bicyclic oxetane, which had the potential to be recycled or repurposed to different kinds of polymeric materials. This work exemplifies that properly equipped carbonate monomers can be designed to extend the pool of functionality eventually present in APCs. We expect that this gives new impetus to different curing opportunities and post-polymerization adjustment of relevant physical properties such as hydrophobicity/philicity, polymer processability and miscibility.Author contributions
JN, ML and DHL carried out all the experiments and the analytical interpretation, ML assisted JN during the project with feedback and scientific input, and AWK and ML designed the project, provided continuous feedback on the results and wrote the manuscript in collaboration with JN and DHL.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Cerca program/Generalitat de Catalunya, ICREA, MINECO (PID2020-112684GB-100 and PDC2021-120952-I00), the Ministerio de Ciencia e Innovación (Severo Ochoa Excellence Accreditation 2020–2023 CEX2019-000925-S) and AGAUR (2021-SGR-00853) for support. JN thanks the Chinese Research Council for a predoctoral fellowship (2019-06180075), and ML acknowledges AGAUR for a Beatriu de Pinós Grant postdoctoral fellowship (BP-2021-00162).References
- (a) K. Wang, K. Amin, Z. An, Z. Cai, H. Chen, H. Chen, Y. Dong, X. Feng, W. Fu, J. Gu, Y. Han, D. Hu, R. Hu, D. Huang, F. Huang, F. Huang, Y. Huang, J. Jin, X. Jin, Q. Li, T. Li, Z. Li, Z. Li, J. Liu, J. Liu, S. Liu, H. Peng, A. Qin, X. Qing, Y. Shen, J. Shi, X. Sun, B. Tong, B. Wang, H. Wang, L. Wang, S. Wang, Z. Wei, T. Xie, C. Xu, H. Xu, Z.-K. Xu, B. Yang, Y. Yu, X. Zeng, X. Zhan, G. Zhang, J. Zhang, M. Q. Zhang, X.-Z. Zhang, X. Zhang, Y. Zhang, Y. Zhang, C. Zhao, W. Zhao, Y. Zhou, Z. Zhou, J. Zhu, X. Zhu and B. Z. Tang, Advanced functional polymer materials, Mater. Chem. Front., 2020, 4, 1803–1915 RSC; (b) G. Chyr and J. M. DeSimone, Review of high-performance sustainable polymers in additive manufacturing, Green Chem., 2023, 25, 453–466 RSC.
- (a) B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC; (b) W. Yu, E. Maynard, V. Chiaradia, M. C. Arno and A. P. Dove, Aliphatic Polycarbonates from Cyclic Carbonate Monomers and Their Application as Biomaterials, Chem. Rev., 2021, 121, 10865–10907 CrossRef CAS PubMed; (c) J. Xu, E. Feng and J. Song, Renaissance of Aliphatic Polycarbonates: New Techniques and Biomedical Applications, J. Appl. Polym. Sci., 2014, 131, 39822 CrossRef PubMed.
- (a) K. J. Zhu, R. W. Hendren, K. Jensen and C. G. Pitt, Synthesis, properties, and biodegradation of poly(1,3-trimethylene carbonate), Macromolecules, 1991, 24, 1736–1740 CrossRef CAS; (b) G. A. Bhat, M. Luo and D. J. Darensbourg, Catalysis of carbon dioxide and oxetanes to produce aliphatic polycarbonates, Green Chem., 2020, 22, 7707–7724 RSC.
- H. Tian, Z. Tang, X. Zhuang, X. Chen and X. Jing, Biodegradable synthetic polymers: Preparation, functionalization and biomedical application, Prog. Polym. Sci., 2012, 37, 237–280 CrossRef CAS.
- (a) S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Ring-opening copolymerization (ROCOP): synthesis and properties of polyesters and polycarbonates, Chem. Commun., 2015, 51, 6459–6479 RSC; (b) C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, Multifunctional Catalysts for Ring-Opening Copolymerizations, ACS Catal., 2022, 12, 11037–11070 CrossRef CAS; (c) X. Liang, F. Tan and Y. Zhu, Recent Developments in Ring-Opening Copolymerization of Epoxides With CO2 and Cyclic Anhydrides for Biomedical Applications, Front. Chem., 2021, 9, 647245 CrossRef CAS PubMed.
- For recent examples: (a) D. H. Lamparelli, I. Grimaldi, A. Martínez-Carrión, F. Bravo and A. W. Kleij, Supercritical CO2 as an Efficient Medium for Macromolecular Carbonate Synthesis through Ring-Opening Co- and Teroligomerization, ACS Sustainable Chem. Eng., 2023, 11, 8193–8198 CrossRef CAS; (b) K. A. Andrea, H. Plommer and F. M. Kerton, Ring-opening polymerizations and copolymerizations of epoxides using aluminum- and boron-centered catalysts, Eur. Polym. J., 2019, 120, 109202 CrossRef CAS; (c) Y. Wang and D. J. Darensbourg, Carbon dioxide-based functional polycarbonates: Metal catalyzed copolymerization of CO2 and epoxides, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS.
- (a) S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Synthesis and post-polymerisation modifications of aliphatic poly(carbonate)s prepared by ring-opening polymerisation, Chem. Soc. Rev., 2013, 42, 1312–1336 RSC; (b) T. M. McGuire, C. Pérale, R. Castaing, G. Kociok-Köhn and A. Buchard, Divergent Catalytic Strategies for the Cis/Trans Stereoselective Ring-Opening Polymerization of a Dual Cyclic Carbonate/Olefin Monomer, J. Am. Chem. Soc., 2019, 141, 13301–13305 CrossRef CAS PubMed; (c) W. Guerin, A. Khadri Diallo, E. Kirilov, M. Helou, M. Slawinski, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Enantiopure Isotactic PCHC Synthesized by Ring-Opening Polymerization of Cyclohexene Carbonate, Macromolecules, 2014, 47, 4230–4235 CrossRef CAS.
- (a) J. L. Hedrick, V. Piunova, N. H. Park, T. Erdmann and P. L. Arrechea, ACS Macro Lett., 2022, 11, 368–375 CrossRef CAS PubMed; (b) P. Brignou, M. Priebe Gil, O. Casagrande, J.-F. Carpentier and S. M. Guillaume, Polycarbonates Derived from Green Acids: Ring-Opening Polymerization of Seven-Membered Cyclic Carbonates, Macromolecules, 2010, 43, 8007–8017 CrossRef CAS; (c) W. Zhang, J. Dai, Y.-C. Wu, J.-X. Chen, S.-Y. Shan, Z. Cai and J.-B. Zhu, Highly Reactive Cyclic Carbonates with a Fused Ring toward Functionalizable and Recyclable Polycarbonates, ACS Macro Lett., 2022, 11, 173–178 CrossRef CAS PubMed.
- Y. Watanabe, S. Takaoka, Y. Haga, K. Kishi, S. Hakozaki, A. Narumi, T. Kato, M. Tanaka and K. Fukushima, Organic carboxylate salt-enabled alternative synthetic routes for bio-functional cyclic carbonates and aliphatic polycarbonates, Polym. Chem., 2022, 13, 5193–5199 RSC.
- C. Qiao, W. Shi, A. Brandolese, J. Benet-Buchholz, E. C. Escudero-Adán and A. W. Kleij, A Novel Catalytic Route to Polymerizable Bicyclic Cyclic Carbonate Monomers from Carbon Dioxide, Angew. Chem., Int. Ed., 2022, 61, e202205053 CrossRef CAS PubMed.
- C. Qiao, A. Villar-Yanez, J. Sprachmann, B. Limburg, C. Bo and A. W. Kleij, Organocatalytic Trapping of Elusive Carbon Dioxide based Heterocycles through a Kinetically Controlled Cascade Process, Angew. Chem., Int. Ed., 2020, 59, 18446–18451 CrossRef CAS PubMed.
- S. S. Bhojgude, A. Bhunia and A. T. Biju, Employing Arynes in Diels–Alder Reactions and Transition-Metal-Free Multicomponent Coupling and Arylation Reactions, Acc. Chem. Res., 2016, 49, 1658–1670 CrossRef CAS PubMed.
- M. Thangaraj, S. S. Bhojgude, M. V. Mane and A. T. Biju, From insertion to multicomponent coupling: temperature dependent reactions of arynes with aliphatic alcohols, Chem. Commun., 2016, 52, 1665–1668 RSC.
- C. Maquilón, F. Della Monica, B. Limburg and A. W. Kleij, Photocatalytic Synthesis of Substituted Cyclic Carbonate Monomers for Ring-Opening Polymerization, Adv. Synth. Catal., 2021, 363, 4033–4040 CrossRef.
- (a) C. Li, R. J. Sablong, R. A. T. M. van Benthem and C. E. Koning, Unique Base-Initiated Depolymerization of Limonene-Derived Polycarbonates, ACS Macro Lett., 2017, 6, 684–688 CrossRef CAS PubMed. See also: (b) F. N. Singer, A. C. Deacy, T. M. McGuire, C. K. Williams and A. Buchard, Chemical Recycling of Poly(Cyclohexene Carbonate) Using a Di-MgII Catalyst, Angew. Chem., Int. Ed., 2022, 61, e202201785 CrossRef CAS PubMed.
- (a) W. Guo, J. González-Fabra, N. A. G. Bandeira, C. Bo and A. W. Kleij, Angew. Chem., Int. Ed., 2015, 54, 11686–11690 CrossRef CAS PubMed; (b) S. Sopeña, V. Laserna, W. Guo, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Adv. Synth. Catal., 2016, 358, 2172–2178 CrossRef.
- S. Sutthasupa, M. Shiotsuki and F. Sanda, Recent advances in ring-opening metathesis polymerization, and application to synthesis of functional materials, Polym. J., 2010, 42, 905–915 CrossRef CAS.
- M. Yasir, P. Liu, I. K. Tennie and A. F. M. Kilbinger, Catalytic living ring-opening metathesis polymerization with Grubbs’ second- and third-generation catalysts, Nat. Chem., 2019, 11, 488–494 CrossRef CAS PubMed.
- Y.-C. Wu, H.-Z. Fan, W. Zhang, M.-Y. Wang, Z. Cai and J.-B. Zhu, Biobased Bifunctional Monomers toward Functionalizable Polycarbonates and Poly(cyclic olefin)s with Tunable Properties, Macromolecules, 2022, 55, 9232–9241 CrossRef CAS.
- (a) J. G. Kim and G. W. Coates, Macromolecules, 2012, 45, 7878–7883 CrossRef CAS; (b) G. A. Bhat, A. Z. Rashad and D. J. Darensbourg, Polym. Chem., 2020, 11, 4699–4705 RSC.
- Comparison of the NMR data for 8a with the literature combined with MS studies allowed us to propose the bicyclic oxetane structure, see: (a) T. M. McGuire, J. Bowles, E. Deane, E. H. E. Farrar, M. N. Grayson and A. Buchard, Angew. Chem., Int. Ed., 2021, 60, 4524–4528 CrossRef CAS PubMed; (b) C. Qiao, A. Villar-Yánez, D. Garay-Ruiz, J. Benet Buchholz, C. Bo and A. W. Kleij, ACS Catal., 2022, 12, 5464–5469 CrossRef CAS.
- (a) D. K. Tran, A. Z. Rashad, D. J. Darensbourg and K. L. Wooley, Sustainable synthesis of CO2-derived polycarbonates from D-xylose, Polym. Chem., 2021, 12, 5271–5278 RSC; (b) T. M. McGuire, J. Bowles, E. Deane, E. H. E. Farrar, M. N. Grayson and A. Buchard, Control of Crystallinity and Stereocomplexation of Synthetic Carbohydrate Polymers from D- and L-Xylose, Angew. Chem., Int. Ed., 2021, 60, 4524–4528 CrossRef CAS PubMed; (c) T. M. McGuire and A. Buchard, Polymers from sugars and CS2: ring opening copolymerisation of a D-xylose anhydrosugar oxetane, Polym. Chem., 2021, 12, 4253–4261 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data for all new and relevant compounds, monomers and polymers, and copies of relevant spectra and chromatograms. See DOI: https://doi.org/10.1039/d3py00860f |
| This journal is © The Royal Society of Chemistry 2023 |