
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInstallation of the adamantyl group in polystyrene-block-poly(methyl methacrylate) via Friedel–Crafts alkylation to modulate the microphase-separated morphology and dimensions†
Takuya
Isono
 *a,
Ema
Baba‡
b,
Shunma
Tanaka‡
b,
Ken
Miyagi
c,
Takahiro
Dazai
c,
Feng
Li
a,
Takuya
Yamamoto
a,
Kenji
Tajima
a and
Toshifumi
Satoh
*a
*a,
Ema
Baba‡
b,
Shunma
Tanaka‡
b,
Ken
Miyagi
c,
Takahiro
Dazai
c,
Feng
Li
a,
Takuya
Yamamoto
a,
Kenji
Tajima
a and
Toshifumi
Satoh
*a
aFaculty of Engineering, Hokkaido University, Sapporo 060-8628, Japan. E-mail: isono.t@eng.hokudai.ac.jp; satoh@eng.hokudai.ac.jp
bGraduate School of Chemical Sciences and Engineering, Hokkaido University, Sapporo 060-8628, Japan
cLithography Process Materials Research Group, R&D Department, Tokyo Ohka Kogyo Co., Ltd, Kanagawa 253-0114, Japan
First published on 29th March 2023
Abstract
Although polystyrene-block-poly(methyl methacrylate) (PS-b-PMMA) is the most widely employed template and scaffold for constructing nanoscale patterns, its relatively small Flory–Huggins interaction parameter makes it challenging to realize microphase-separated structures with a periodicity (or domain spacing) of less than 20 nm. The direct post-polymerization modification of PS-b-PMMA can resolve this issue. In this study, we present the PS block post-polymerization modification of commercially available PS-b-PMMA through a metal-free Friedel–Crafts alkylation reaction with adamantanols to modulate its microphase-separated morphology and dimensions. The adamantyl group can be easily installed on the PS benzene ring, which increases the incompatibility with the PMMA block, enabling ordered microphase-separated structures, including lamellar, gyroid and hexagonally close-packed cylindrical structures, from low-molecular-weight PS-b-PMMA. Notably, a periodicity as low as approximately 16 nm was realized with this strategy, which is much lower than the lowest accessible periodicity of pristine PS-b-PMMA. Furthermore, we observed additional benefits from the installation of the adamantyl group, such as increased thermal stability, glass transition temperature and etching contrast.
Introduction
Block copolymers (BCPs) consisting of two chemically incompatible polymer blocks form periodic nanostructures known as microphase-separated structures. Depending on the total degree of polymerization (N) and the volume fraction, BCPs show microphase-separated structures with different periodicities ranging from a few to hundreds of nanometers and different morphologies, such as lamellar, gyroid, hexagonally close-packed cylinders and body-centered cubic spheres.1–3 Such tunable nanostructures have a wide range of applications, with the most important one being their use as structural templates for nanofabrication.3–6 For example, applying perpendicularly orientated lamellar structures to a semiconductor substrate can allow fabrication of line-and-space patterns on the underlying substrate by the selective removal of one of the blocks and subsequent etching.7 Perpendicularly orientated cylinders can also be used for fabricating holes and pillars on an underlying substrate.8,9 Meanwhile, gyroid structures have been used as nanotemplates to create nanofiltration membranes,10 efficient photovoltaics,11 photonic crystals,12etc.13,14Among the wide variety of BCPs available,6 PS-b-PMMA has been the focus of intense research on nanotemplate applications. As the precise synthesis of PS-b-PMMA is achievable by living anionic polymerization, narrowly dispersed products with desirable molecular weights and compositions are made available on a commercial scale. This is extremely important to realize nanofabrications with high precision and reproducibility.15 Moreover, the reasonable etching contrast between the PS and PMMA domains makes PS-b-PMMA more attractive and useful for constructing nanostructures as templates and scaffolds.16 However, the major limitation of PS-b-PMMA is the difficulty in accessing microphase-separated structures with a periodicity of less than 20 nm, which limits its application in nanofabrications at the sub-10 nm resolution.17 Such limitation stems from the relatively low Flory–Huggins interaction (χ) parameter of PS-b-PMMA.18 Since the product of χ and N must be greater than 10.5 for microphase separation to occur in symmetrical BCPs, low-χ BCPs are required to have relatively high molecular weights to satisfy the “χN > 10.5” requirement. Meanwhile, the periodicity of the microphase-separated structure, that is, the domain spacing (d), is proportional to N2/3, so the lower limit of d is determined by the χ value of the BCP. In the case of PS-b-PMMA, the lowest theoretical molecular weight required for microphase separation is 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, resulting in the smallest accessible d of approximately 20 nm.18
000 g mol−1, resulting in the smallest accessible d of approximately 20 nm.18
To expand the accessible d and control morphology while maintaining the advantages of PS-b-PMMA, minor chemical modification of PS-b-PMMA should be an effective strategy. For example, the incorporation of short polar blocks, such as polymethacrylate derivatives and oligopeptides, between the PS and PMMA blocks was found to be effective for producing ordered microphase-separated structures with a d smaller than the smallest accessible one of unmodified PS-b-PMMA (Fig. 1a).19–21 Moreover, Kim reported a reduction in the line edge roughness of the PS-b-PMMA nanopattern by incorporating a hydrogen bond-forming urea functionality into the junction point (Fig. 1a).22 However, such modified PS-b-PMMAs require multistep synthesis or a custom-made PS-b-PMMA possessing a reactive functional group. Meanwhile, our group reported the direct post-polymerization modification of commercially available (anionically synthesized) PS-b-PMMA to install polar functional groups at either the PMMA side chain or at the chain end (Fig. 1b).23–25 We employed the ester–amide exchange reaction for the side-chain functionalization23 and the terminal-selective transesterification reaction for the PMMA chain end functionalization.24,25 In both cases, the installation of a small proportion of polar functional groups (e.g., amide and oligosaccharide segments) enhanced the polarity of the PMMA block, which drastically promoted segregation of the PS block, resulting in ordered microphase-separated structure formation from low-molecular-weight PS-b-PMMA. The biggest advantage of the post-polymerization modification strategy is the easy access to a wide variety of PS-b-PMMA derivatives, involving different functional groups and a desirable degree of functionalization, in addition to a fixed backbone length and dispersity from a single starting material. This can lead to rapid discovery of suitable materials for nanofabrication applications using emerging material informatics. However, the post-polymerization modifications of pristine PS-b-PMMA reported thus far are based on the reactivity of the PMMA ester group, which limits the accessible chemical space of the PS-b-PMMA derivatives.
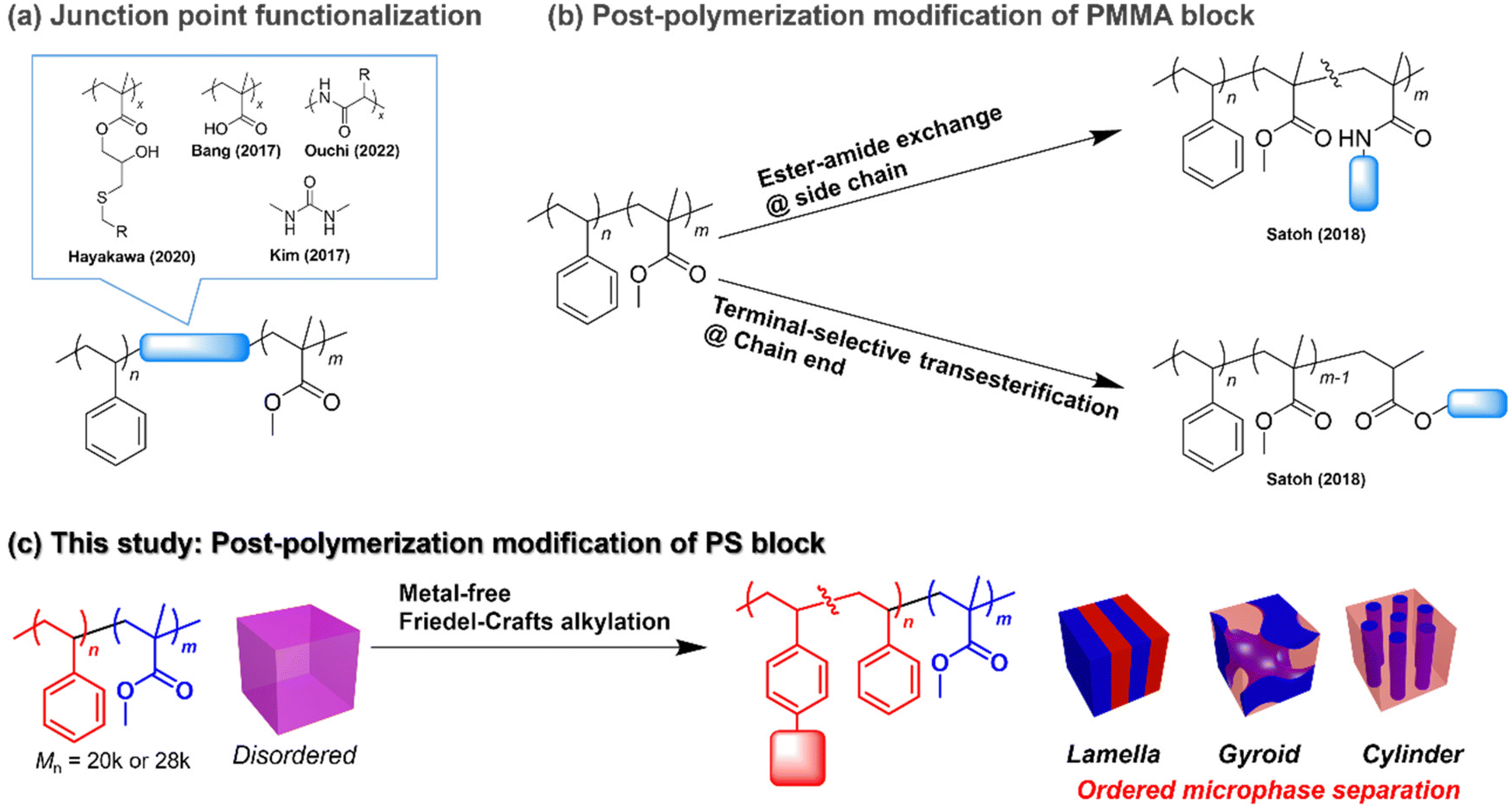 | ||
| Fig. 1 Chemical modification of PS-b-PMMA. (a) Previous works on the junction point functionalization. (b) Previous works on the PMMA side-chain and chain-end functionalization via post-polymerization modification of commercially available PS-b-PMMA. (c) This work, which focuses on post-polymerization modification of the PS block via the metal-free Friedel–Crafts alkylation reaction. | ||
Herein, we focus on the PS block post-polymerization modification of commercially available PS-b-PMMA to modulate its microphase-separated morphology and dimensions (Fig. 1c). Although the benzene ring is chemically stable and much less reactive than other functional groups, such as ester groups, PS benzene ring modifications have been reported and widely implemented on a commercial scale. For example, sulfonation and chloromethylation of crosslinked polystyrene are well-known important steps for producing ion-exchange resins.26 In addition, many other aromatic ring transformation reactions, such as hydrogenation (dearomatization),27 halogenation,28 nitration,29 borylation30 and acrylation reactions,31,32 have been applied to modify the PS benzene ring. Among them, the Friedel–Crafts alkylation reaction is a facile tool to install hydrophobic functional groups on the PS block, which endows the modified PS-b-PMMA with a higher polarity difference between the blocks, thus enhancing the χ value. Indeed, poly(4-tert-butylstyrene)-b-PMMA, which has an extra tert-butyl group on each PS benzene ring compared to conventional PS-b-PMMA, shows a higher χ value, thus allowing access to a smaller d value.33 Notably, metal-free Friedel–Crafts alkylation is readily realized by combining a 3° alcohol substrate with a strong Brønsted acid catalyst.34,35 Such metal-free reactions are highly advantageous for reducing metal contamination, which is a prerequisite for semiconductor lithographic applications.
In this study, we investigated the installation of an adamantyl group on the PS block of low-molecular-weight PS-b-PMMA via metal-free Friedel–Crafts alkylation to control its morphology and dimensions. Interestingly, we found that the installation of an adamantyl group on the PS block resulted in higher thermal stability and higher etching resistance of the modified PS block. The adamantyl group also served as an additional hydrophobic functional group, which increased the χ value of the modified PS-b-PMMA, thus allowing the formation of ordered microphase-separated structures with d values as low as 16 nm. Moreover, the volume fraction change through the adamantyl group installation led to a shift in the microphase-separated morphology from lamellar to gyroid and finally to a hexagonally close-packed cylinder structure. These findings contribute to the discovery of nanotemplate materials suitable for various nanofabrication applications.
Experimental
The general procedure for the Friedel–Crafts alkylation reaction is as follows: in an Ar-filled glovebox, PS10k-b-PMMA10k (300 mg, 1.47 mmol of styrene units, 1.00 eq.) and an adamantanol (0.735 mmol, 0.50 eq.) were placed in a Schlenk flask and dissolved in CH2Cl2 (10 mL). After sealing with a rubber septum, the Schlenk flask was taken out from the glovebox. Triflic acid solution (1.10 mmol, 0.75 eq. in 10 mL CH2Cl2) was added into the polymer solution to start the reaction. The reaction mixture was stirred at room temperature for 1 h, after which the solution was poured into methanol to precipitate the polymer. The resulting precipitate was filtered and dried under vacuum to give the adamantly-modified PS10k-b-PMMA10k as a white powder. See the ESI† for more details.Results and discussion
Post-polymerization modification of the polystyrene block via the Friedel–Crafts alkylation reaction with adamantanols
The preferred adamantane source was 1-adamantanol (Ad-OH) (Scheme 1). Thus, the metal-free Friedel–Crafts alkylation reaction was carried out by treating PS10k-b-PMMA10k with Ad-OH in the presence of triflic acid (TfOH) in CH2Cl2 with a [styrene unit]0/[Ad-OH]0/[TfOH] ratio of 1.00/0.50/0.75 (run 1 in Table 1). The resulting product (Ad-PS10k-b-PMMA10k) exhibited new signals at 1.77, 1.87 and 2.08 ppm in its 1H NMR spectrum due to the adamantyl group, supporting the progress of the reaction (Fig. S1b†). Based on 1H NMR analysis, the degree of side-chain functionalization (f: the average number of adamantyl groups attached per styrene unit) was determined to be 0.44. However, size exclusion chromatography (SEC) analysis revealed the presence of many high-molecular-weight by-products (line (b) in Fig. 2). This can be explained by crosslink formation through the adamantane moiety. Since multiple substitution reactions have been reported during the Lewis acid-catalyzed arylation of 1-bromoadamantane with benzene,36–38 it is reasonably expected that crosslinking could occur in the same manner. Although the mechanism of the multiple substitution reaction is not mentioned in the literature, the reaction most likely proceeds via pathways involving carbocation formation from the 3° carbons of the adamantyl group.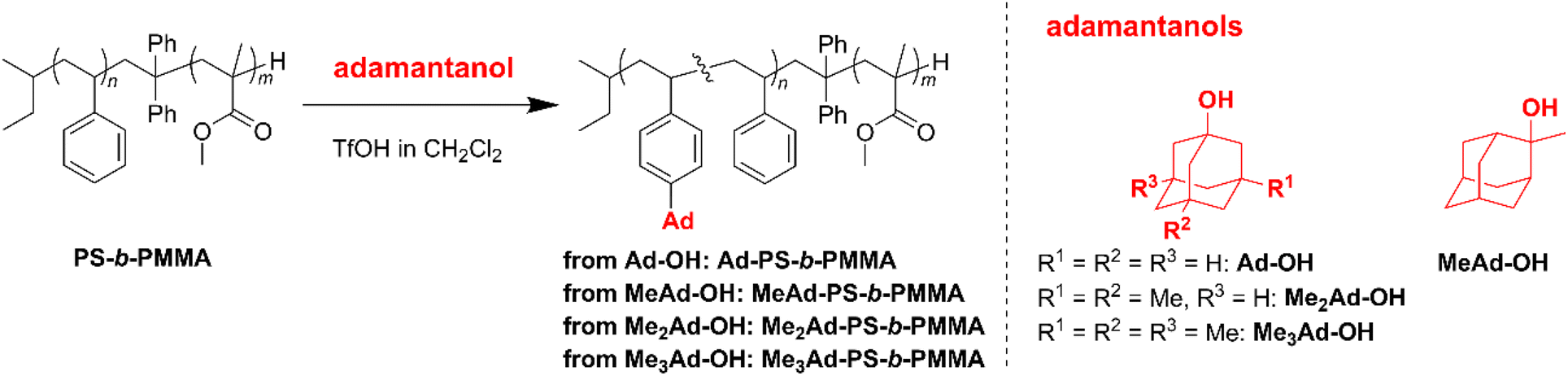 | ||
| Scheme 1 Metal-free Friedel–Crafts alkylation of PS-b-PMMA using adamantanols. | ||
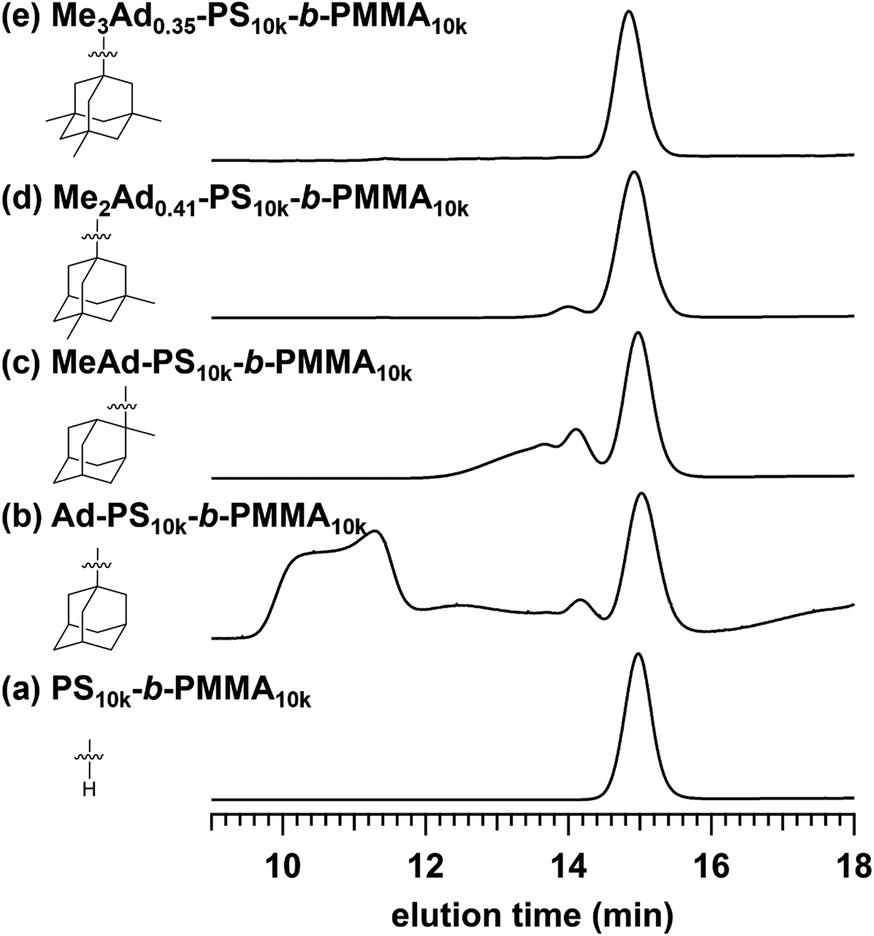 | ||
| Fig. 2 SEC traces of (a) PS10k-b-PMMA10k and PS10k-b-PMMA10k modified with (b) Ad-OH, (c) MeAd-OH, (d) Me2Ad-OH, and (e) Me3Ad-OH ([styrene unit]0/[adamantanol]0/[TfOH] = 1.00/0.50/0.75). | ||
| Run | Sample name | [Styreneb]0/[adamantanol]0/[TfOH] | f | M n | Đ | Yield (%) | ϕ PS |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: Ar atmosphere; solvent, dichloromethane; temperature, r.t.; and reaction time, 1 h. b Styrene units in PS-b-PMMA. c Degree of substitution (f) was determined by quantitative 13C NMR analysis of Me2Ad-PS-b-PMMA and by 1H NMR analysis of Me3Ad-PS-b-PMMA. d Determined by SEC in THF, using PS standards. e Volume fraction of the modified PS block. | |||||||
| 1 | Ad-PS10k-b-PMMA10k | 1.00/0.50/0.75 | — | — | — | 92.5 | — |
| 2 | MeAd-PS10k-b-PMMA10 | 1.00/0.50/0.75 | — | 26400 |
1.44 | 69.6 | — |
| 3 | Me2Ad0.41-PS10k-b-PMMA10k | 1.00/0.50/0.75 | 0.41 | 21400 |
1.06 | 90.1 | 0.65 |
| 4 | Me3Ad0.35-PS10k-b-PMMA10k | 1.00/0.50/0.75 | 0.35 | 22200 |
1.02 | 97.7 | 0.64 |
| 5 | Me2Ad0.07-PS10k-b-PMMA10k | 1.00/0.10/0.15 | 0.07 | 19300 |
1.02 | 92.1 | 0.55 |
| 6 | Me2Ad0.20-PS10k-b-PMMA10k | 1.00/0.20/0.30 | 0.20 | 20300 |
1.03 | 84.9 | 0.60 |
| 7 | Me2Ad0.29-PS10k-b-PMMA10k | 1.00/0.30/0.45 | 0.29 | 21600 |
1.07 | 87.1 | 0.62 |
| 8 | Me2Ad0.38-PS10k-b-PMMA10k | 1.00/0.40/0.60 | 0.38 | 24200 |
1.35 | 83.1 | 0.64 |
| 9 | Me3Ad0.63-PS10k-b-PMMA10k | 1.00/0.60/0.90 | 0.63 | 24000 |
1.06 | 96.5 | 0.70 |
| 10 | Me3Ad0.86-PS10k-b-PMMA10k | 1.00/0.80/1.20 | 0.86 | 25400 |
1.06 | 84.6 | 0.73 |
| 11 | Me2Ad0.08-PS14k-b-PMMA14k | 1.00/0.10/0.15 | 0.08 | 26600 |
1.02 | 86.6 | 0.56 |
| 12 | Me2Ad0.14-PS14k-b-PMMA14k | 1.00/0.20/0.30 | 0.14 | 28000 |
1.03 | 89.5 | 0.58 |
| 13 | Me2Ad0.28-PS14k-b-PMMA14k | 1.00/0.30/0.45 | 0.28 | 30300 |
1.09 | 92.4 | 0.62 |
| 14 | Me2Ad0.09-PS4.2k-b-PMMA4.3k | 1.00/0.10/0.15 | 0.09 | 9300 | 1.03 | 85.1 | 0.56 |
| 15 | Me2Ad0.41-PS4.2k-b-PMMA4.3k | 1.00/0.30/0.45 | 0.41 | 10200 |
1.05 | 88.1 | 0.64 |
| 16 | Me2Ad0.59-PS4.2k-b-PMMA4.3k | 1.00/0.60/0.90 | 0.59 | 13400 |
1.36 | 86.5 | 0.68 |
Using the preliminary result, we employed commercially available 2-methyl-2-adamantanol (MeAd-OH) and 3,5-dimethyl-1-adamantanol (Me2Ad-OH) as the adamantane sources to reduce the possibility of the carbocation migration. Friedel–Crafts alkylation reactions were performed using these two adamantanol derivatives under the same conditions established above (runs 2 and 3 in Table 1). The SEC trace of the product obtained with MeAd-OH (MeAd-PS10k-b-PMMA10k) exhibited high molecular weight elution peaks (line (c) in Fig. 2), indicating intermolecular crosslinking. In contrast, the product obtained with Me2Ad-OH indicated a suppressed crosslinking reaction, as evidenced by the clean SEC trend, even though a small high-molecular-weight elution peak was still visible (line (d) in Fig. 2). Nevertheless, the 1H NMR spectrum showed new signals corresponding to the dimethyladamantyl group (Fig. S1c†), supporting the success of the Friedel–Crafts alkylation reaction. A logical outcome from the above results is that the best source of adamantane is 3,5,7-trimethyl-1-adamantanol (Me3Ad-OH), in which all 3° carbons are blocked by methyl groups. Indeed, Friedel–Crafts alkylation reactions using Me3Ad-OH with a [styrene unit]0/[adamantanol]0/[TfOH] ratio of 1.00/0.50/0.75 successfully produced side-chain-functionalized PS-b-PMMA (Me3Ad-PS10k-b-PMMA10k) virtually without the high-molecular-weight elution peak due to intermolecular crosslinking (run 4 in Table 1, line (e) in Fig. 2 for SEC, and Fig. S3† for 1H NMR spectra). However, the limited commercial availability and the high price of Me3Ad-OH led us to use Me2Ad-OH for subsequent experiments.
Because the 1H NMR signals of the Me2Ad groups overlapped with those of the polymer main chain, we employed an inverse-gated decoupling 13C NMR spectrum to calculate the f value of Me2Ad-PS10k-b-PMMA10k (Fig. 3). By comparing the signal intensities of the aromatic carbons (128–124 ppm) and adamantyl carbons (31.6–30.0 ppm), the f value was calculated to be 0.41. Based on the adamantane source, f value and the starting PS-b-PMMA, this sample was named Me2Ad0.41-PS10k-b-PMMA10k.
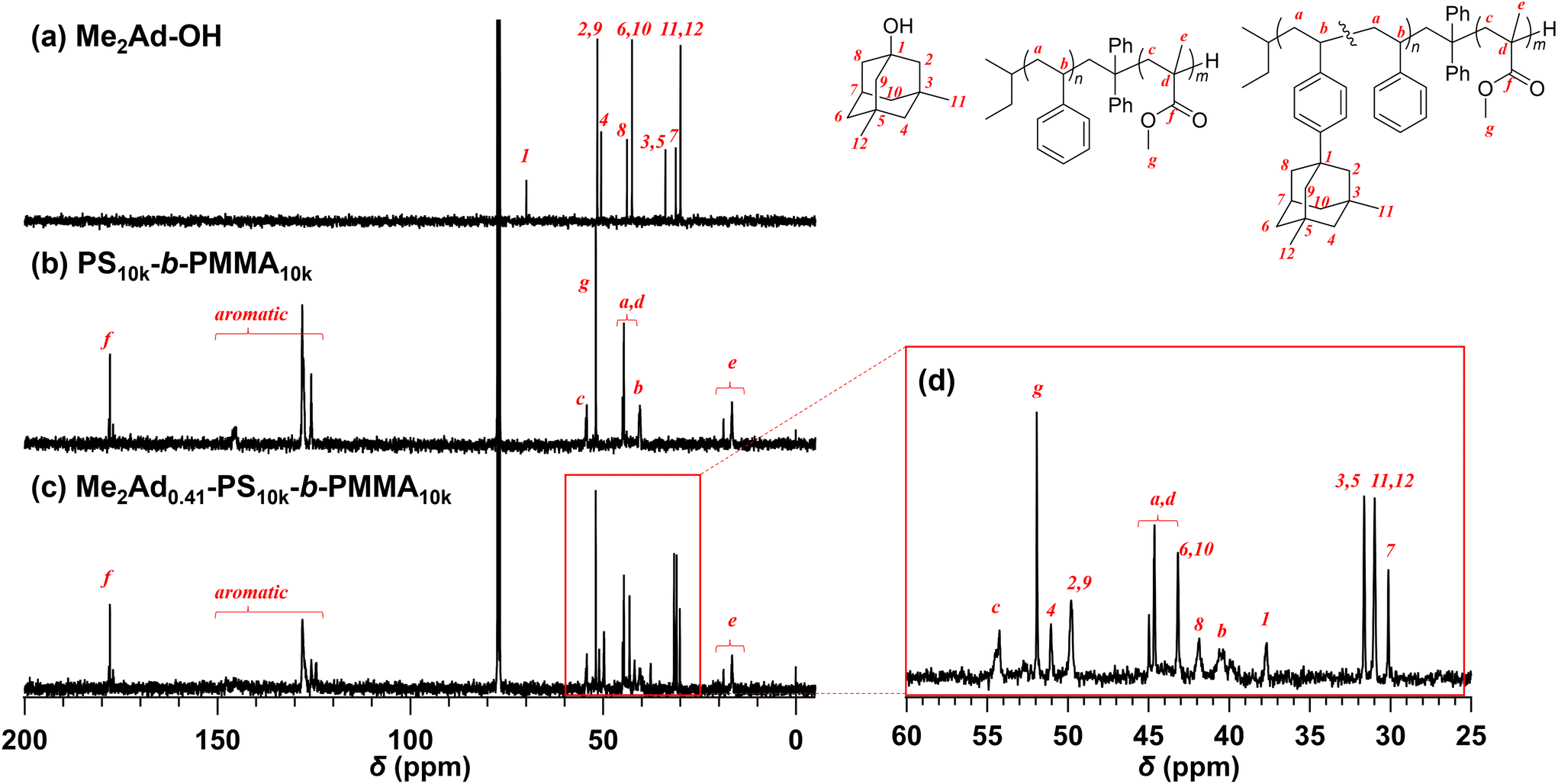 | ||
| Fig. 3 13C NMR spectra of (a) Me2Ad-OH and (b) PS10k-b-PMMA10k in CDCl3 (100 MHz), and (c) inverse-gated decoupling 13C NMR spectrum of Me2Ad0.41-PS10k-b-PMMA10k (lower) in CDCl3 (100 MHz). (d) The expansion from 60 to 25 ppm of the spectrum (c). | ||
Using the established protocol, we performed the Friedel–Crafts alkylation reactions with PS10k-b-PMMA10k using Me2Ad-OH at various [Me2Ad-OH]0/[styrene unit]0 ratios, from 0.10 to 0.50, to control the f value (runs 5–8 in Table 1). Note that a [TfOH]/[Me2Ad-OH]0 ratio of 1.5 was applied for all of the reactions. Importantly, f increased linearly from 0.07 to 0.41 with an increasing [Me2Ad-OH]0/[styrene unit]0 ratio, suggesting a high reaction yield and easy control of f (Fig. 4). The Fourier transform infrared (FT-IR) spectra of the products revealed a new characteristic absorption band at approximately 2900 cm−1 due to the C–H stretching of the adamantyl side chain, and the peak intensity increased with increasing f (Fig. S2†). The SEC traces of the resulting products showed a sharp elution peak with a dispersity (Đ) typically well below 1.1 (Fig. 5). However, the increase in the [Ad-OH]0/[styrene unit]0 ratio (typically >0.4) resulted in high-molecular-weight by-product formation. Friedel–Crafts alkylation reactions were also successfully applied to PS-b-PMMAs with different molecular weights, that is, PS14k-b-PMMA14k and PS4.2k-b-PMMA4.3k (runs 11–16 in Table 1).
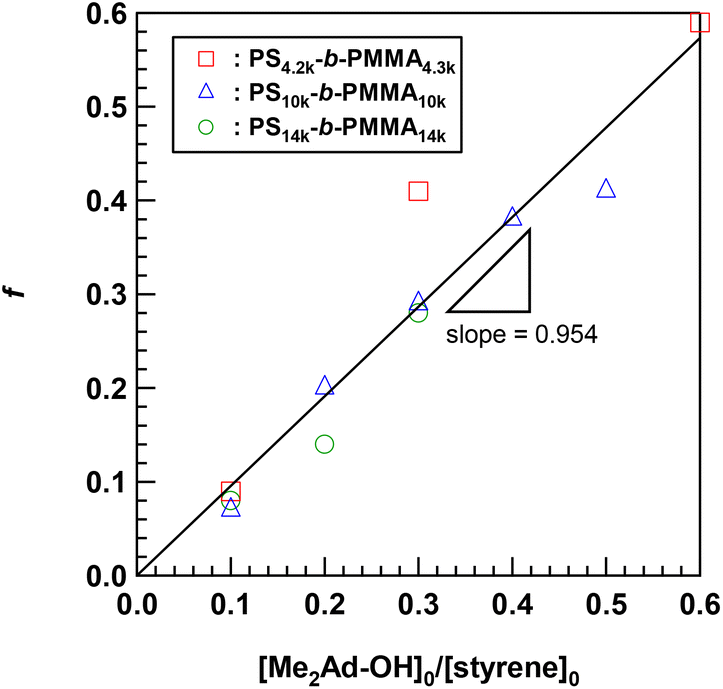 | ||
| Fig. 4 Linear dependence of the f value on the initial ratio of Me2Ad-OH to the styrene unit. | ||
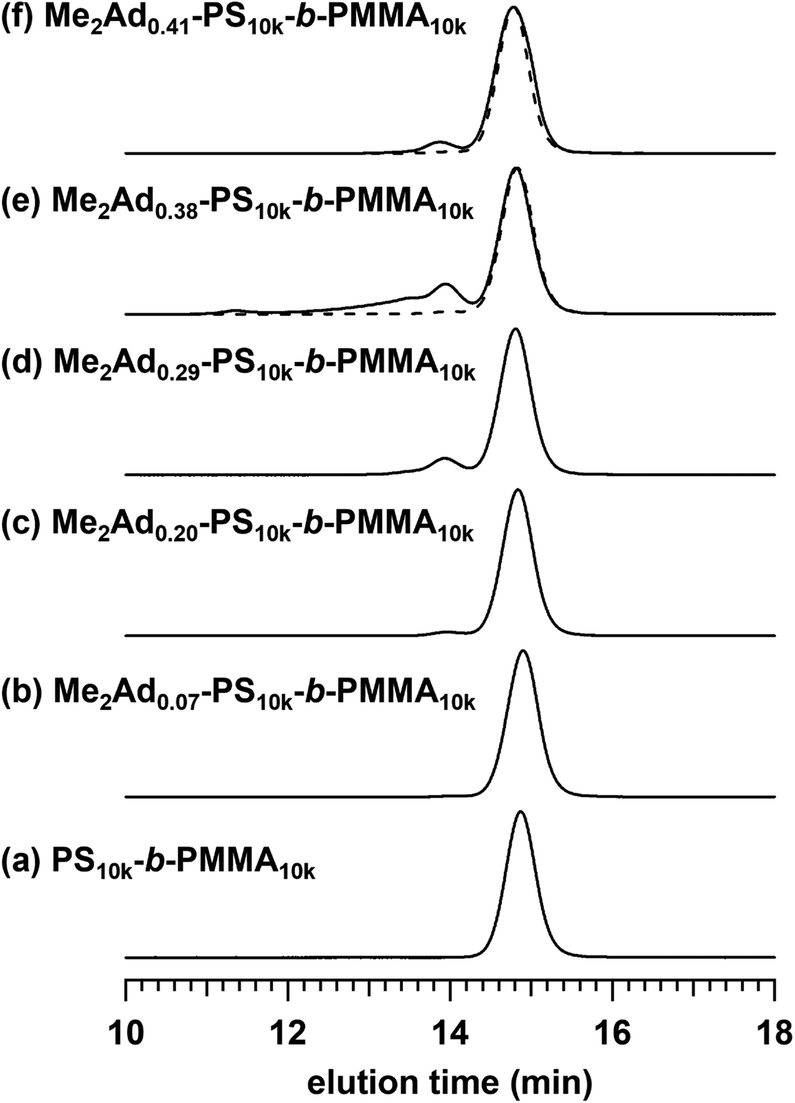 | ||
| Fig. 5 SEC traces of Me2Ad-PS10k-b-PMMA10k samples (b)–(f) and the corresponding PS10k-b-PMMA10k (a) (eluent, THF; flow rate, 1.0 mL min−1). The dashed curves represent the SEC traces after preparative SEC purification. | ||
In addition, we attempted to incorporate an adamantyl side chain with f = 0.5, or higher, using Me3Ad-OH. Owing to the absence of free 3° carbons, Friedel–Crafts alkylation reactions on PS10k-b-PMMA10k with [Me3Ad-OH]0/[styrene unit]0 ratios of 0.60 and 0.80 successfully produced the desired products with f values of 0.63 and 0.86, respectively (runs 9 and 10 in Table 1). Notably, gelation was not observed in these reactions. This was in contrast to the reaction using Me2Ad-OH at the same [Me2Ad-OH]0/[styrene unit]0 ratio, which produced a partially insoluble product (data not shown). However, even with Me3Ad-OH, a small measure of the high-molecular-weight elution peak became pronounced with an increasing [Me3Ad-OH]0/[styrene unit]0 ratio (Fig. S3†).
Overall, we successfully demonstrated an easy post-polymerization modification of the polystyrene block in PS-b-PMMAs via Friedel–Crafts alkylation. The synthesis results are summarized in Table 1. The volume fraction of the modified PS block (ϕPS) was calculated based on the density values of PMMA (1.18 g cm−3) and PS (1.05 g cm−3). Here, the density of the modified PS block was assumed to be the same as that of pristine PS because a previous study confirmed that there is no significant difference in density between PS and poly[4-(1-adamantyl)styrene] homopolymers.39
Thermal properties
To investigate the impact of the adamantyl side-chain modification, we assessed the thermal properties of a series of Me2Ad-PS10k-b-PMMA10k copolymers with varied f values by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). TGA revealed a slight increase in 5% and 10% weight loss temperatures (Td5 and Td10, respectively) with an increasing f value (Td5: 348 °C for f = 0 and 355 °C for f = 0.41; Td10: 356 °C for f = 0 and 364 °C for f = 0.41) (Fig. S4†). This confirmed that the adamantyl side chain rendered PS-b-PMMA more thermally stable. Ishizone reported a slightly increased Td10 in poly[4-(1-adamantyl)styrene] compared to that in pristine polystyrene,40 which is consistent with our results. Through DSC analysis, we observed an increase in the glass transition temperature (Tg) with increasing f (Fig. 6). A baseline shift was observed at 108 °C in the second heating DSC curve of the parent PS10k-b-PMMA10k, which was assignable to the composite of Tg values of the PMMA and PS blocks. When the f value increased from 0.07 to 0.29, the composite Tg increased from 116 to 133 °C. A further increase in the f value resulted in two separate Tg values, in which the lower Tg corresponding to the PMMA block was observed at 132–133 °C. Meanwhile, the higher Tg corresponding to the modified PS block increased from 145 to 167 °C with increasing f value. These results also demonstrate the increased thermal stability of the adamantyl-modified PS blocks. Such increased thermal stability is preferable for various nanotemplate applications.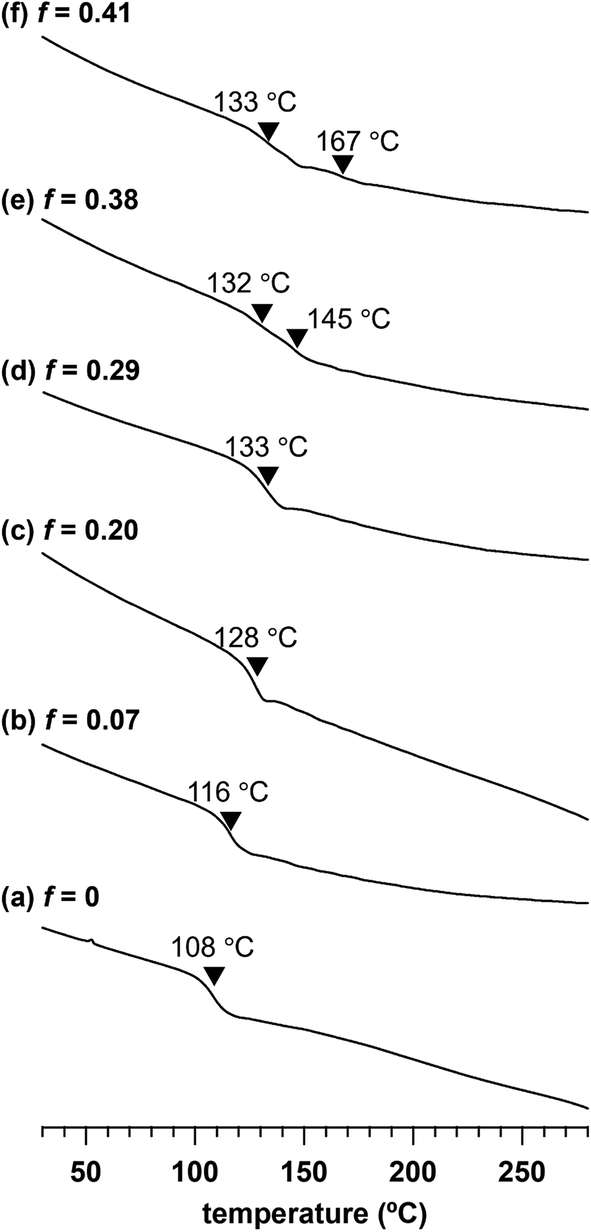 | ||
| Fig. 6 Second heating DSC traces of (a) PS10k-b-PMMA10k, (b) Me2Ad0.07-PS10k-b-PMMA10k, (c) Me2Ad0.20-PS10k-b-PMMA10k, (d) Me2Ad0.29-PS10k-b-PMMA10k, (e) Me2Ad0.38-PS10k-b-PMMA10k and (f) Me2Ad0.41-PS10k-b-PMMA10k. | ||
Etching resistance of adamantyl-modified PS
Since the adamantyl group is widely known as an etching-resistive functional group,40,41 we predicted that the installation of the adamantyl group on the PS block could improve the etching resistance. To test this hypothesis, we prepared a series of PS10k samples modified with Me2Ad-OH, that is, Me2Ad0.18-PS10k and Me2Ad0.62-PS10k, in the same manner as the PS-b-PMMA modification (Fig. S5 and S6†). Thin films of the modified PS, PS and PMMA homopolymers were prepared on a silicon substrate by spin coating, which was then subjected to O2 reactive ion etching for 15, 30, 60 and 120 s. The etching depth for all materials linearly increased with the etching time (Fig. 7). Importantly, the etching rate decreased with increasing adamantyl content. Notably, the etching resistance of Me2Ad0.62-PS was 7.7 and 2.0 times compared to PMMA and PS, respectively. These results confirm that the adamantyl group effectively enhanced the etching resistance of the PS block. This is another merit of installing an adamantyl group onto PS-b-PMMA for nanofabrication.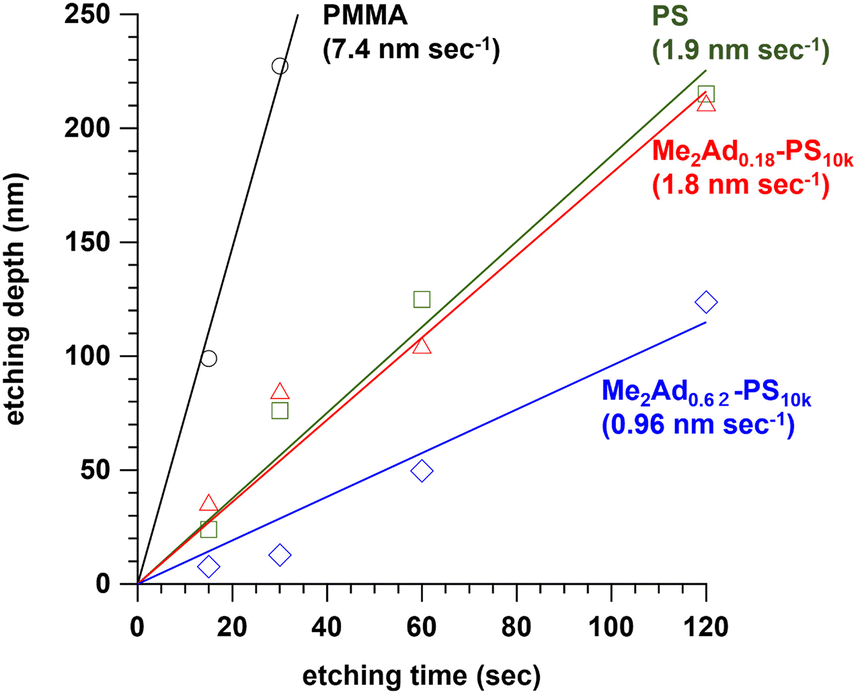 | ||
| Fig. 7 Plots of etching depth of PMMA, PS, Me2Ad0.18-PS and Me2Ad0.62-PS thin films with increasing etching time. The values in parentheses represent the etching rate determined by the slope. | ||
Microphase-separated structures
Using a series of Me2Ad-functionalized PS-b-PMMAs, we investigated their microphase-separated structures in the bulk material by small-angle X-ray scattering (SAXS) and transmission electron microscopy (TEM). To study the effects of f on the microphase separation behavior, the Me2Ad-PS10k-b-PMMA10k samples were thermally annealed at 200 °C for 24 h and then subjected to SAXS measurements at room temperature. As we previously reported,23 unmodified PS10k-b-PMMA10k did not show any ordered microphase-separated structures, as evidenced by the correlation of hole scattering in its SAXS profile, because χN was lower than the critical value owing to its low N content (line (a) in Fig. 8). Similarly, the 200 °C-annealed Me2Ad0.07-PS10k-b-PMMA10k and Me2Ad0.20-PS10k-b-PMMA10k samples did not show any higher-order scattering in their SAXS profiles, indicating the absence of ordered nanostructure formation. We also examined the SAXS profiles of the BCP annealed at 150 °C for 24 h (lines (b) and (c) in Fig. 8). Me2Ad0.07-PS10k-b-PMMA10k exhibited only a broad peak, confirming the absence of an ordered structure. In contrast, the Me2Ad0.20-PS10k-b-PMMA10k sample exhibited a sharp primary scattering peak (q*) at 0.380 nm−1 together with higher ordered scattering peaks at the 2q* and 3q* positions. This scattering pattern is assignable to the lamellar morphology, and the domain spacing (d) was calculated to be 16.5 nm using Bragg's equation (d = 2π/q*). This result successfully demonstrated that χN crossed the critical value (χN > 10.5) upon the installation of the adamantyl group with f > 0.20. More importantly, a d of 16.5 nm was well below the smallest d achievable in pristine PS-b-PMMA (approximately 20 nm).18 Thus, we can conclude that the introduction of the adamantyl group on the PS block is an easy and efficient strategy to realize smaller nanopatterns from low-molecular-weight PS-b-PMMA.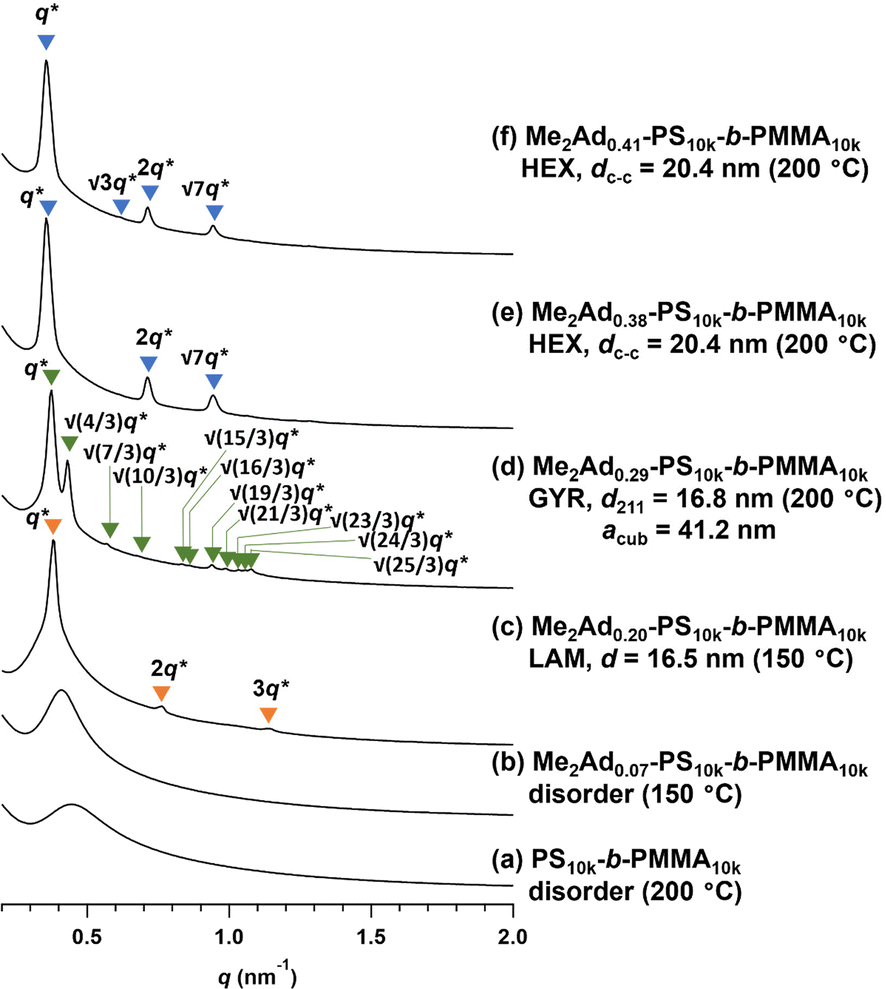 | ||
| Fig. 8 SAXS profiles of thermally annealed Me2Ad-PS10k-b-PMMA10k. (a) PS10k-b-PMMA10k, (b) Me2Ad0.07-PS10k-b-PMMA10k, (c) Me2Ad0.20-PS10k-b-PMMA10k, (d) Me2Ad0.29-PS10k-b-PMMA10k, (e) Me2Ad0.38-PS10k-b-PMMA10k and (f) Me2Ad0.41-PS10k-b-PMMA10k.The value in the parentheses represents the annealing temperature. | ||
A further increase in the f value resulted in a shift of the microphase-separated morphology from lamellar (LAM) to gyroid (GYR) and finally to hexagonally close-packed cylinder (HEX) structures. The 200 °C-annealed Me2Ad0.29-PS10k-b-PMMA10k samples exhibited a rather complicated scattering pattern, that is, q*, (4/3)1/2q*, (7/3)1/2q*, (8/3)1/2q*, (10/3)1/2q*…, which can be assigned to the GYR structure (Fig. 8d). The linear relationship in the plot for qhklvs. (h2 + k2 + l2)1/2, where h, k and l are the Miller indices, validates the successful peak assignment (Fig. S7†). The cubic lattice parameter, acub, was calculated to be 41.2 nm using acub = 2π(h2 + k2 + l2)1/2/qhkl. To further confirm the GYR structure, the nanostructures were examined using TEM (Fig. 9a). In the TEM images, double-wave morphologies, which are typical for GYR, were observed over a large area, which again confirmed the long-range order of GYR formation. Interestingly, the GYR structure was maintained for more than one year, as confirmed by SAXS measurements on the same sample left for one year under ambient conditions (Fig. S8†). This clearly demonstrates the highly stable nature of the GYR morphology. To date, ordered GYR structures have rarely been reported for PS-b-PMMA and solvent annealing or homopolymer blending are generally required to induce the GYR structure.42,43
 | ||
| Fig. 9 TEM micrographs of (a) Me2Ad0.29-PS10k-b-PMMA10k (annealed at 200 °C for 24 h) and (b) Me2Ad0.28-PS14k-b-PMMA14k (annealed at 250 °C for 24 h). | ||
For Me2Ad0.38-PS10k-b-PMMA10k and Me2Ad0.41-PS10k-b-PMMA10k, a sharp primary scattering peak (q*) together with higher-order scattering peaks at 2q* and √7q* were observed in the SAXS profiles (lines (e) and (f) in Fig. 8). In addition, the √3q* peak was barely observed. Considering the PS volume fraction, the scattering pattern could be reasonably assigned to the HEX morphology. The intercylinder distance (dc–c = (2π/q*)(4/3)1/2) was calculated to be 20.4 nm for both BCPs.
Thus, it was demonstrated that the installation of the adamantyl side chain is an efficient strategy to induce microphase separation even from the low-molecular-weight PS-b-PMMA, which originally did not form an ordered structure. Furthermore, the control of the f value enabled access to the desired morphologies involving LAM, HEX and even GYR. The shift in the morphology appeared to be the result of both the increase in N and χ upon PS side-chain functionalization.
To gain an insight into the effect of a higher f on the microphase-separated structures, we performed SAXS measurements on Me3Ad-PS10k-b-PMMA10k. For the 200 °C-annealed Me3Ad0.35-PS10k-b-PMMA10k, the SAXS profile was assigned to the HEX structure with a dc–c of 20.7 nm. Notably, the morphology and dimensions of this sample closely resemble those of Me2Ad0.38-PS10k-b-PMMA10k. This confirmed that f is the most important parameter for tuning the microphase-separated structure, whereas the difference between the Me2Ad and Me3Ad groups has no significant impact. Considering the elevated Tg for the high-f BCPs (Tg of the modified PS block:176 and 197 °C for f = 0.63 and 0.86, respectively; see Fig. S9†), the thermal annealing of Me3Ad0.63-PS10k-b-PMMA10k and Me3Ad0.86-PS10k-b-PMMA10k was conducted at 250 °C for 24 h. The SAXS profiles of the samples also showed a typical scattering pattern assignable to the HEX morphology (Fig. 10). This result reveals that a further increase in f did not alter the resulting morphology when starting from the symmetric PS-b-PMMA.
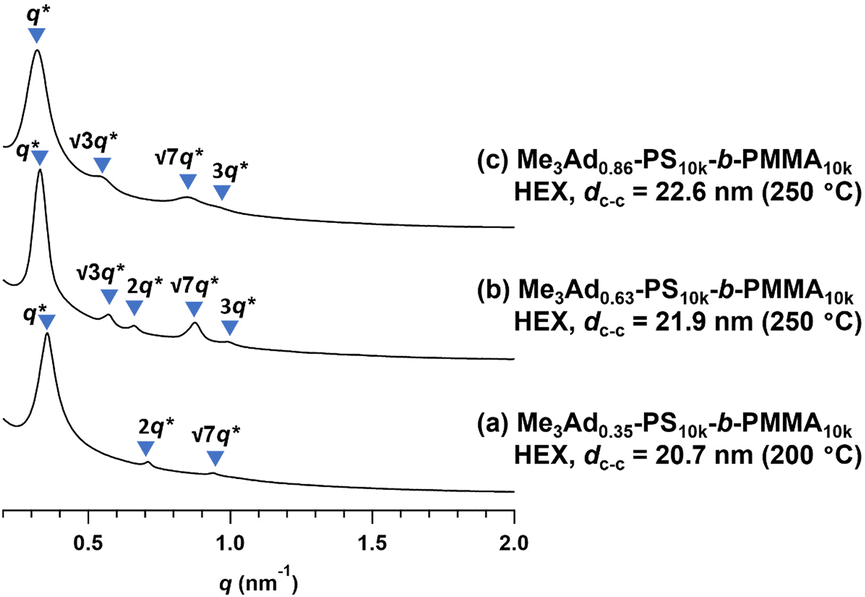 | ||
| Fig. 10 SAXS profiles of the thermally annealed Me3Ad-PS10k-b-PMMA10k. (a) Me3Ad0.35-PS10k-b-PMMA10k, (b) Me3Ad0.63-PS10k-b-PMMA10k and (c) Me3Ad0.86-PS10k-b-PMMA10k. The value in the parentheses represents the annealing temperature. | ||
SAXS measurements were also performed on the Me2Ad-PS14k-b-PMMA14k and Me2Ad-PS4.2k-b-PMMA4.3k series to verify the versatility of the proposed strategy. For Me2Ad-PS4.2k-b-PMMA4.3k, no nanostructure formation was observed, even after annealing at 150 °C, regardless of the f value, as indicated by the absence of a higher-order scattering peak (Fig. S10†). This suggests that the increments in N and χ upon side-chain modification are not sufficient to cross the critical value for microphase separation (χN > 10.5) for this low-molecular-weight PS-b-PMMA. A higher f value is necessary to induce the microphase separation. However, it is more challenging to synthesize such high-f value samples because the intermolecular crosslinking reaction occurs more easily when the [Me2Ad-OH]0/[styrene]0 ratio is increased, as mentioned before. In contrast, Me2Ad-PS14k-b-PMMA14k exhibited evidence of microphase separation even in the samples with f = 0.08. This indicates that the slight increase in χ by adamantyl side-chain installation is sufficient to surpass the critical χN for this relatively high-molecular-weight PS-b-PMMA. The SAXS profiles of 150 °C-annealed Me2Ad0.08-PS14k-b-PMMA14k and 200 °C-annealed Me2Ad0.14-PS14k-b-PMMA14k exhibited scattering patterns associated with LAM structures with a d value of 18.8 and 20.7 nm, respectively (lines (b) and (c) in Fig. 11). A further increase in the f value resulted in the formation of the GYR structure, and the SAXS profile of the 250 °C-annealed Me2Ad0.28-PS14k-b-PMMA14k exhibited the characteristic scattering pattern assignable to the GYR structure (line (d) in Fig. 11). The characteristic morphologies observed in the TEM images also confirm the formation of a highly ordered GYR structure (Fig. 9b). SAXS analysis revealed acub = 51.5 nm for Me2Ad0.29-PS10k-b-PMMA10k. This GYR structure was also stable for more than one year (Fig. S8†).
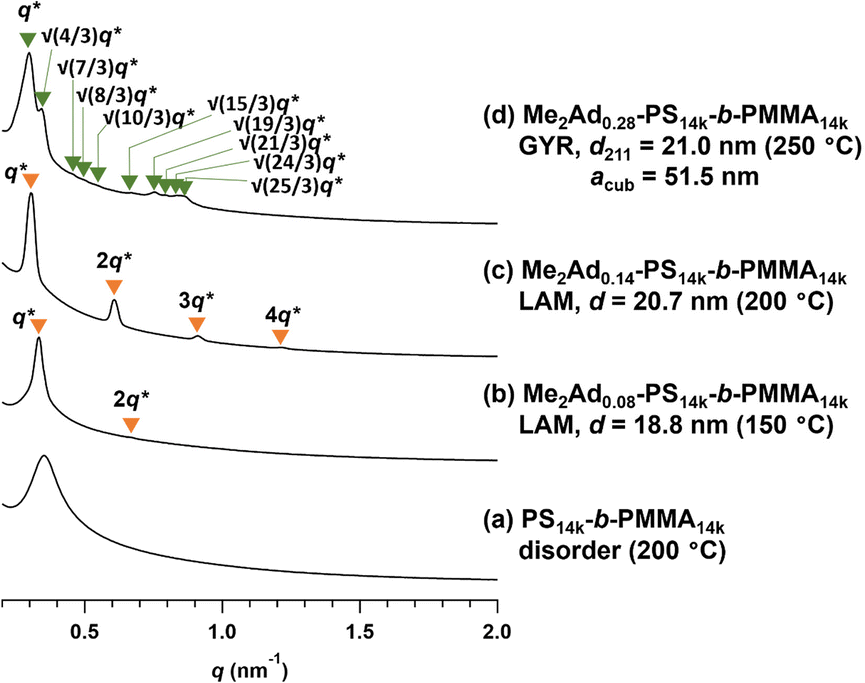 | ||
| Fig. 11 SAXS profiles of the thermally annealed Me2Ad-PS14k-b-PMMA14k. (a) PS14k-b-PMMA14k, (b) Me2Ad0.08-PS14k-b-PMMA14k, (c) Me2Ad0.14-PS14k-b-PMMA14k and (d) Me2Ad0.28-PS14k-b-PMMA14k. The value in the parentheses represents the annealing temperature. | ||
Phase behavior
The results described above clearly demonstrate the enhancement of the χ value by side-chain modification. To investigate the change in the χ value by changing the f value, we used the SAXS data of the disordered Me2Ad-PS4.2k-b-PMMA4.3k with different f values (f = 0.09, 0.41 and 0.59) and PS10k-b-PMMA10k (f = 0) acquired at 200 °C for fitting with Leibler's mean-field theory.44 These details are summarized in the ESI.† The result of the fitting for PS10k-b-PMMA10k yielded a χ value of 0.032 at 200 °C, which is close to the reported value for PS-b-PMMA (χ = 0.0282 + 4.46/T, where T is the absolute temperature; χ = 0.038 at 200 °C).18 As shown in Fig. 12a, the χ value increased with increasing f and reached an almost five-fold value (χ ∼0.20) of that for the original PS-b-PMMA when f = 0.59. This confirmed the versatile control of the χ value through optimization of the [adamantanol]0/[styrene unit]0 ratio. The increased hydrophobicity of the polystyrene block stemming from the additional adamantyl group was responsible for the increase in the χ value. As a consequence, poly(4-tert-butylstyrene)-b-PMMA also showed an increased χ value (0.043 at 200 °C) over that of PS-b-PMMA.33 Compared to the tert-butyl group, the damantly group appeared to contribute more efficiently to the increased χ value due to its higher carbon number.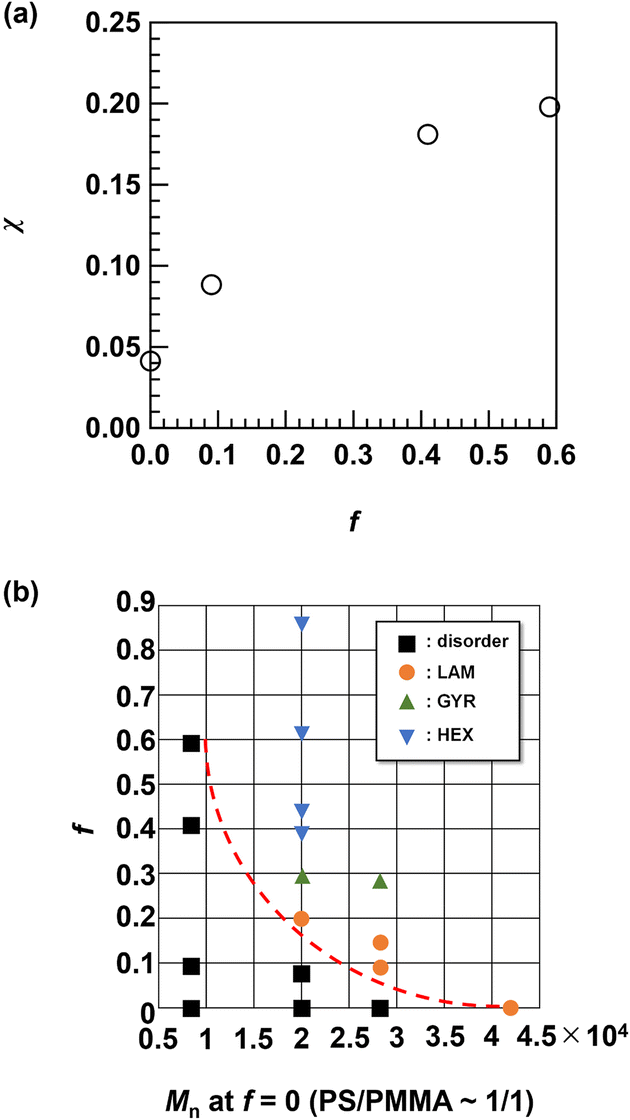 | ||
| Fig. 12 (a) Dependence of the χ value on f determined from the SAXS curve analysis of disordered Me2Ad-PS4.2k-b-PMMA4.3k at 200 °C. (b) Summary of the microphase-separated morphologies of the adamantyl-modified PS-b-PMMA. | ||
The shift in the morphology from LAM to GYR and finally to HEX is reminiscent of the conventional BCP phase diagram.2 As shown in Table 1, an increase in f results in an increase in ϕPS. Thus, it is reasonable to deduce that the morphology shift is a direct result of increased ϕPS. In this study, we did not observe a body-centered cubic (BCC) sphere morphology. To obtain the BCC morphology, ϕPS must be higher than 0.8. Given that the original PS-b-PMMA has a symmetric volume fraction, ϕPS > 0.8, this cannot be achieved even with f = 1.0. Nevertheless, we have demonstrated easy access to nanotechnologically interesting GYR and lithographically relevant LAM and HEX morphologies from a single PS-b-PMMA starting material. These results support the facile access to various nanotemplate structures by a judicious choice of the f value.
Conclusions
We have successfully demonstrated PS block post-polymerization modification of low-molecular-weight PS-b-PMMA via a Friedel–Crafts alkylation reaction to induce microphase separation. In this study, an adamantane moiety was installed on the benzene ring of PS, and the degree of functionalization was easily controlled. One of the important features of this post-polymerization modification is that the reaction proceeds without a metal catalyst, which is advantageous for microelectronic applications. The incorporation of the adamantyl group enhanced the incompatibility with the PMMA block, resulting in microphase separation into lamellar, gyroid and hexagonally close-packed cylindrical morphologies, even in the low-molecular-weight region. Notably, using this strategy, we achieved a domain spacing of approximately 16 nm, which was considerably lower than the lower limit of pristine PS-b-PMMA (∼20 nm). Interestingly, the adamantyl group increased the thermal stability, glass transition temperature and etching resistance of the modified PS block, which are all preferable for nanofabrication applications. Because the presented post-polymerization modification reaction proceeds under mild reaction conditions, this strategy should be applied to a wide variety of PS-containing BCPs as well as tertiary alcohol substrates. Moreover, by combining this approach with the previously established direct PMMA post-polymerization modifications, the accessible chemical space of PS-b-PMMA derivatives can be vastly expanded, creating a large BCP library with a fixed total degree of polymerization and dispersity from a single PS-b-PMMA starting material. With the aid of an emerging materials informatics approach, a novel BCP material that is best suited for future nanofabrication applications can be obtained.Author contributions
Takuya Isono: conceptualization, project administration, investigation, supervision, writing – review and editing, funding acquisition. Ema Baba: investigation, methodology. Shunma Tanaka: investigation, methodology. Ken Miyagi: investigation. Takahiro Dazai: investigation. Feng Li: investigation, supervision. Takuya Yamamoto: investigation, supervision. Kenji Tajima: investigation, supervision. Toshifumi Satoh: writing – review and editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by a JSPS Grant-in-Aid for Scientific Research (B) (No. 20H02792 and 19H02769), the Photoexcitonix Project (Hokkaido University), the project of junior scientist promotion in Hokkaido University, and the Creative Research Institute (Hokkaido University). This work was performed with the approval of the Photon Factory Program Advisory Committee (Proposal No.: 2022G555 and 2021G0531). The authors thank Mr Toshiaki Ito (Hokkaido University, Japan) for his assistance with the TEM experiment.References
- I. W. Hamely, The Physics of Block Copolymers, Oxford University Press, Oxford, 1988 Search PubMed.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- M. Lazzari, G. Liu and S. Lecommandoux, Block Copolymers in Nanoscience, Wiley-VCH Press, Weinheim, 2006 Search PubMed.
- H. Hu, M. Gopinadhan and C. O. Osuji, Soft Matter, 2014, 10, 3867–3889 RSC.
- H. Jung, W. H. Shin, T. W. Park, Y. J. Choi, Y. J. Yoon, S. H. Park, J. H. Lim, J. D. Kwon, J. W. Lee, S. H. Kwon, G. H. Seong, K. H. Kim and W. I. Park, Nanoscale, 2019, 11, 8433–8441 RSC.
- G. G. Yang, H. J. Choi, K. H. Han, J. H. Kim, C. W. Lee, E. I. Jung, H. M. Jin and S. O. Kim, ACS Appl. Mater. Interfaces, 2022, 14, 12011–12037 CrossRef CAS PubMed.
- H. Tsai, J. W. Pitera, H. Miyazoe, S. Bangsaruntip, S. U. Engelmann, C.-C. Liu, J. Y. Cheng, J. J. Bucchingnano, D. P. Klaus, E. A. Joseph, D. P. Sanders, M. E. Colburn and M. A. Guillorn, ACS Nano, 2014, 5, 5227–5232 CrossRef PubMed.
- R. Ruiz, H. Kang, F. A. Detcheverry, E. Dobisz, D. S. Kercher, T. R. Albrecht, J. J. de Pablo and P. F. Nealey, Science, 2008, 321, 936–939 CrossRef CAS PubMed.
- C. Tang, E. M. Lennon, G. H. Fredrickson, E. J. Kramer and C. Hawker, J. Sci., 2008, 322, 429–432 CrossRef CAS PubMed.
- S. Park, Y. Kim, H. Ahn, J. H. Kim, P. J. Yoo and D. Y. Ryu, Sci. Rep., 2016, 6, 36326 CrossRef CAS PubMed.
- E. J. W. Crossland, M. Kamperman, M. Nedelcu, C. Ducati, U. Wiesner, D.-M. Smilgies, G. E. S. Toombes, M. A. Hillmyer, S. Ludwigs, U. Steiner and H. A. Snaith, Nano Lett., 2009, 8, 2807–2812 CrossRef PubMed.
- E. L. Lin, W. L. Hsu and Y. W. Chiang, ACS Nano, 2018, 12, 485–493 CrossRef CAS PubMed.
- M. R. J. Scherer and U. Steiner, Nano Lett., 2013, 13, 3005–3010 CrossRef CAS PubMed.
- W.-C. Ma, W.-S. Huang, C.-S. Ku and R.-M. Ho, J. Mater. Chem. C, 2016, 4, 840–849 RSC.
- H. Lai, G. Huang, X. Tian, Y. Liu and S. Ji, Polymer, 2022, 249, 124853 CrossRef CAS.
- M. G. Gusmão Cacho, P. Pimenta-Barros, M. Argoud, C. Navarro, K. Sakavuyi, R. Tiron and N. Possémé, Microelectron. Eng., 2020, 230, 111369 CrossRef.
- L. Wan, R. Ruiz, H. Gao, K. C. Patel, T. R. Albrecht, J. Yin, J. Kim, Y. Cao and G. Lin, ACS Nano, 2015, 9, 7506–7514 CrossRef CAS PubMed.
- Y. Zhao, E. Sivaniah and T. Hashimoto, Macromolecules, 2008, 41, 9948–9951 CrossRef CAS.
- S. Woo, S. Jo, D. Y. Ryu, S. H. Choi, Y. Choe, A. Khan, J. Huh and J. Bang, ACS Macro Lett., 2017, 6, 1386–1391 CrossRef CAS PubMed.
- K. Wylie, L. Dong, A. Chandra, Y. Nabae and T. Hayakawa, Macromolecules, 2020, 53, 1293–1301 CrossRef CAS.
- T. Yoshimura, T. Morishita, Y. Agata, K. Nagashima, K. Wylie, Y. Nabae, T. Hayakawa and M. Ouchi, Macromolecules, 2022, 55, 2148–2159 CrossRef CAS.
- K. S. Lee, J. Lee, J. Kwak, H. C. Moon and J. K. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 31245–31251 CrossRef CAS PubMed.
- K. Yoshida, L. Tian, K. Miyagi, A. Yamazaki, H. Mamiya, T. Yamamoto, K. Tajima, T. Isono and T. Satoh, Macromolecules, 2018, 51, 8064–8072 CrossRef CAS.
- K. Yoshida, S. Tanaka, T. Yamamoto, K. Tajima, R. Borsali, T. Isono and T. Satoh, Macromolecules, 2018, 51, 8870–8877 CrossRef CAS.
- K. Yoshida, T. Yamamoto, K. Tajima, T. Isono and T. Satoh, Polym. Chem., 2019, 10, 3390–3398 RSC.
- A. Ravve, Principles of Polymer Chemistry, Springer, New York, 3rd edn, 2012 Search PubMed.
- Z. Siddiqi and D. Sarlah, J. Am. Chem. Soc., 2021, 143, 21264–21269 CrossRef CAS PubMed.
- G. A. Crosby, N. M. Weinshenker and H. S. Uh, J. Am. Chem. Soc., 1975, 97, 2232–2235 CrossRef CAS.
- A. Philippides, P. M. Budd, C. Price and A. V. Cuncliffe, Polymer, 1993, 34, 3509–3513 CrossRef CAS.
- J. Shin, S. M. Jensen, J. Ju, S. Lee, Z. Xue, S. K. Noh and C. Bae, Macromolecules, 2007, 40, 8600–8608 CrossRef CAS.
- R. L. Letsinger, M. J. Kornet, V. Mahadevan and D. M. Jerina, J. Am. Chem. Soc., 1964, 86, 5163–5165 CrossRef CAS.
- B. Xue, P. P. Huang, M. Z. Zhu, S. Q. Fu, J. H. Ge, X. Li and P. N. Liu, ACS Macro Lett., 2022, 11, 1252–1257 CrossRef CAS PubMed.
- J. G. Kennemur, M. A. Hillmyer and F. S. Bates, Macromolecules, 2012, 45, 7228–7236 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, Macromolecules, 2020, 53, 4722–4732 CrossRef CAS PubMed.
- J. Y. Jeon, Z. Umstead, G. N. Kangovi, S. Lee and C. Bae, Top. Catal., 2018, 61, 610–615 CrossRef CAS.
- H. Newman, Synthesis, 1972, 692–693 CrossRef CAS.
- V. R. Reichert and L. J. Mathias, Macromolecules, 1994, 27, 7015–7023 CrossRef CAS.
- H. Lim, M. C. Cha and J. Y. Chang, Polym. Chem., 2012, 3, 686–870 RSC.
- B. J. Ree, S. Kobayashi, K. Heo, T. J. Lee, T. Satoh, T. Ishizone and M. Ree, Polymer, 2019, 169, 225–233 CrossRef CAS.
- S. Kobayashi, T. Matsuzawa, S.-i. Matsuoka, H. Tajima and T. Ishizone, Macromolecules, 2006, 39, 5979–5986 CrossRef CAS.
- J. M. Klopp, D. Pasini, J. D. Byers, C. G. Willson and J. M. J. Fréchet, Chem. Mater., 2001, 13, 4147–4153 CrossRef CAS.
- S. Park, Y. Kim, W. Lee, S.-M. Hur and D. Y. Kim, Macromolecules, 2017, 50, 5033–5041 CrossRef CAS.
- J.-W. Hong, J.-H. Chang, I. C.-Y. Chang and Y.-S. Sun, Soft Mater., 2021, 17, 9189–9197 RSC.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and additional data and discussions. See DOI: https://doi.org/10.1039/d3py00113j |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |