
Borane catalysis for epoxide (co)polymerization
Stefan
Naumann

University of Stuttgart, Institute of Polymer Chemistry, 70569 Stuttgart, Germany. E-mail: stefan.naumann@ipoc.uni-stuttgart.de
First published on 14th March 2023
Abstract
Since 2016, when simple triethylborane (Et3B) was revealed as a very capable catalyst for the copolymerization of epoxides and carbon dioxide, research effort regarding this type of organic Lewis acid has soared. This interest is well-founded and connected to some striking features of Et3B-based polymerization catalysis. Especially for the homo- and copolymerization of epoxides, this has enabled a step change in functional group tolerance and polymerization control, also including beneficial practical aspects such as rapid monomer consumption, fully metal-free setups and very low catalyst loadings. As a result, polyethers, polyether-containing complex architectures and various epoxide-based copolymers can now be addressed with unprecedented precision, in turn unlocking extended or even novel applications for these polymers. This review intends to highlight the advances made in the past few years, focusing on the catalytic performance of Et3B and emerging strategies for (multi)borane catalyst design, alongside mechanistic considerations and how these are reflected in the respective polymers. Thereby, it is hoped to raise awareness for borane catalysis as a disruptive tool with significant technological potential for polyether chemistry and related fields.
 Stefan Naumann | Stefan Naumann received his PhD in 2014 (Buchmeiser Group, University of Stuttgart, Germany). In 2014–2015 he joined Prof. Andrew P. Dove (University of Warwick, UK) as a PostDoc. After returning to Stuttgart, he worked as Research Group Leader and successfully concluded his habilitation in polymer chemistry in 2021. Since 2018 he is PI in the collaborative research council 1333 (“Molecular Heterogeneous Catalysis in Confined Geometries”). In 2019, he was awarded with an advancement stipend by the Macromolecular Division of the German Chemical Society (GDCh). His current research interests encompass the development of polymerization catalysts and their impact on material design. |
Introduction
Epoxides (Fig. 1), in particular ethylene oxide (EO) and propylene oxide (PO), are among the most important monomers in polymer chemistry.1,2 The aliphatic polyethers resulting from their homo- or copolymerization display extraordinarily wide-ranging applicability, encompassing polyols for the preparation of polyurethanes,3,4 additives for lubrication,5 rheology control6 or as dispersants,7 and also a broad employment in skin care and related daily use products (tooth paste, lip sticks).8,9 Further examples, where a particularly rapid scientific development can be witnessed, include the use of polyethers for drug delivery,10,11 electrochemical devices (electrolyte, membranes)12–14 or the templating of nanoporous materials.15–17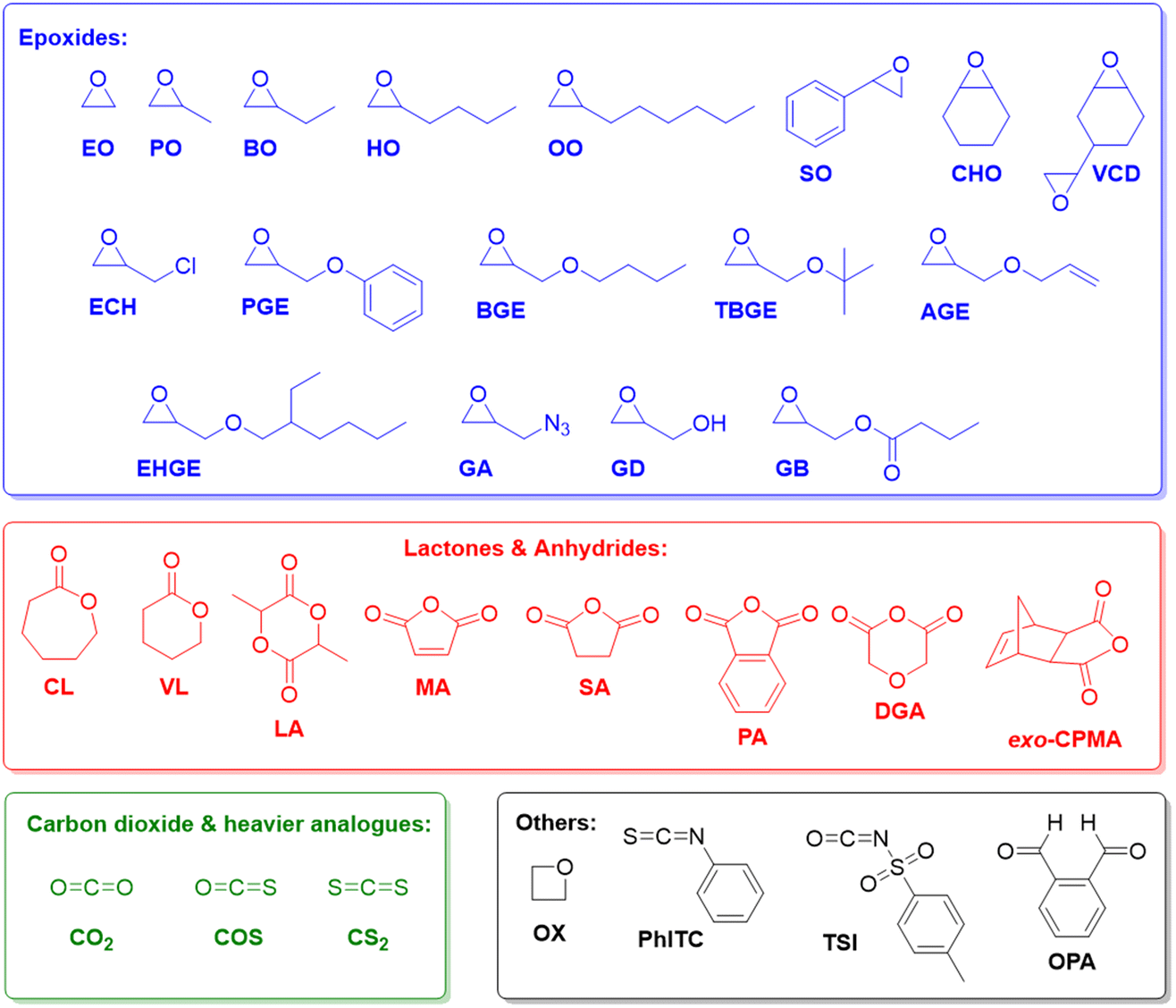 | ||
| Fig. 1 Monomers as discussed in this review and respective abbreviations. | ||
This broad applicability profile is rooted in the dualism of poly(ethylene oxide) (PEO) /poly(ethylene glycol) (PEG) on the one hand and poly(propylene oxide) (PPO) on the other. While PEG is water-soluble, semi-crystalline and of very low toxicity,18 PPO is usually a fully amorphous, viscous liquid with moderate lipophilicity.1 Properties can be further adjusted if different macromolecular architectures are considered (i.e., block or statistical copolymers, networks, hyperbranched systems)19–23 or if more complex epoxide monomers are employed (Fig. 1). With regard to the latter, especially 1-butylene oxide (BO) and other heavier alkylene oxides should be mentioned, alongside various glycidyl ethers and styrene oxide (SO), which can be used to introduce additional functionality to the polymer.24–27 The oxygen-carbon bond in the polyether backbone is stable and flexible, imparting the ability to interact with suitable solvents or Lewis acids and a high main chain mobility (low glass transition temperature) to the polymer.2 While the C–O–C motif is chemically relatively robust, aliphatic polyethers are, however, susceptible to radical degradation. This has been put forward as an attractive feature to replace other polymers in specific situations where marine littering occurs.28
Copolymerization with non-epoxide comonomers is also possible and well-established, further extending the structural diversity of the accessible macromolecules. Especially copolymerization with CO2, generating polycarbonates,29–33 and with anhydrides, yielding polyesters,34,35 will be a point of interest in this review, but many more examples could be mentioned.36–41
The chemical constitution of the epoxide functionality, as smallest cyclic ether, renders a broad range of catalysts applicable: the endocyclic oxygen atom is Lewis basic42 and thus primes the monomer for activation (i.e., by coordination to a Lewis acid or protonation). Consequently, epoxides can be polymerized along cationic or “coordination–insertion” pathways.43 Much in contrast to their heavier cyclic ether homologues, however, epoxides can also be readily polymerized following anionic routes, helped along by the high ring strain in the monomer (112 kJ mol−1).1,2 As a result, epoxide homopolymerization can, in principle, be catalysed by a plethora of compounds, including organocatalytic44–47 methods; in practical terms (and large-scale processes) only a few of those found wide-spread use. In particular, conventional anionic polymerization mediated by potassium hydroxide or potassium alkoxides is a still broadly employed technology.48 Additionally, the so-called double-metal cyanide (DMC) catalysts are increasingly popular for industrial purposes.1,49–51 For some polyether niche products alternative, Al-based organometallic compounds also play a role.52–54 All of these catalysts come with a number of significant downsides. Since a detailed discussion of the specific limitations of each type of catalyst would lead too far, the interested reader is referred to excellent review publications for epoxide homo- and copolymerization.1,2,29–35,45–48 It is, however, the ambition of the overview presented here to demonstrate that borane catalysis has contributed on several frontiers to overcome these limitations and in sum provides the field with a combination of features that is unparalleled. In short, these advantages include:
(a) Polymerization control. This relates to control over molar mass, molar mass distribution, end group fidelity and polymerization rates. This is by no means a trivial aspect. As an example, the seemingly simple homopolymerization of PO is impaired by well-known transfer to monomer side reactions. This has notoriously limited achievable molar masses (typically to less than 5 kg mol−1) via conventional anionic polymerization and prohibits the incorporation of PPO in more complex polymer architectures this way.1 To suppress this undesired reaction pathway, the basicity of the propagating chain end (secondary alkoxide) has to be managed without lowering the polymerization rates to impractical levels.55 Approaches to solve this by coordinative polymerization mechanisms (thereby avoiding free anionic species),56,57 by specifically adapted organocatalysts58,59 or via monomer-activation strategies60,61 have been successful in providing, i.e., high-Mn PPO,62 but at the cost of either broad molar mass distributions, slow polymerization rates or sharply limited functional group tolerance. As will be shown in the following, simple borane catalysts can provide high molar masses while retaining fast kinetics, narrow molar mass distributions and a broad scope of tolerated functionalities.
(b) Functional group tolerance. The ability to include functionalized epoxides, non-epoxide comonomers or more complex (macro)initiators is a key feature if polyethers or related polymers are to be tailored for specific advanced applications. Accordingly, there is an increasing demand which is, however, incompatible with the frequently employed conventional anionic polymerization where conditions are unsuitable for functionalities susceptible to nucleophilic attack.63 Similar limits apply to many metal-based catalysts, which can degrade or be deactivated by the desired moieties (i.e. DMC catalysts are deactivated by polyols based on sugars1). Suitable organocatalysts for epoxide conversion are usually strong bases, entailing comparable limitations.59,64 In this regard, borane (co)catalysis has enabled a true step change: triethylborane (Et3B) and modified variants thereof are capable of consuming sensitive epoxide monomers (such as epichlorohydrin or glycidyl esters) in a controlled manner and have even been employed for the preparation of well-defined poly(ester-ether) multiblock copolymers.63 Likewise, precisely constructed copolymers of epoxides with CO2 or anhydrides profit from this functional group tolerant behaviour. The following discussion will map out the current state of knowledge regarding this intriguing feature of borane-mediated epoxide conversion.
(c) Polymer Tacticity. Polymerization of rac-epoxides (i.e., PO, BO) to result in stereocontrolled (isotactic) aliphatic polyethers has long been a domain of metal-based setups.65 Chiral diborane catalysts now offer a first glimpse of what might be possible by using organocatalytic approaches.66 While selectivity is still inferior to the best bimetallic catalysts, it is especially the combination of controlled polymerization, functional group tolerance and stereoselectivity which might render this strategy useful for future applications.
In the following, the development of borane-mediated epoxide (co)polymerization will be described, looking first at the catalytic performance of Et3B. In accordance with the chronology of developments, this overview will start with the copolymerization of epoxides and CO2, followed by epoxide homopolymerization and epoxide/anhydride copolymerization and other examples.
In the second section, different types of modified borane catalysts for these polymerization applications will be described, including trialkyl- and triarylmonoboranes, bifunctional (multi)borane catalysts and chiral diborane structures. Recent efforts for rationally guided catalyst optimization alongside a detailed discussion of the polymerization mechanisms are provided. A recently published, excellent review article covering several of these aspects is also recommended to readers.67
Triethylborane
Copolymerization with CO2, COS and CS2
Triethylborane (Et3B) is a commercially available, colourless liquid which is usually sold as a solution in THF or hexane. It is also pyrophoric (hence its use as rocket/jet engine fuel ignition additive),68 moderately toxic (LD50 = 235 mg kg−1)69 and somewhat prone to hydrolysis, depending on conditions.70 The molecule does not dimerize and the core motif is planar.71 It is significantly less Lewis acidic than its well-known analogue tris(pentafluoro)phenylborane (BCF),72–74 and consequently shows a very different range of applicability in polymer chemistry. While BCF has been used in cationic-type polymerizations (including of epoxides, where relatively ill-controlled and regioirregular polyethers result75), Et3B has been previously employed to initiate radical polymerizations (Et3B/O2 is suitable for low-temperature initiation76) or to attenuate aggressive anionic propagating species (i.e. enolates in the polymerization of N,N-dialkylacrylamides77).This distinct reactivity profile rendered Et3B an attractive choice when it was first introduced to the field of epoxide copolymerization in 2016 in a key paper by Feng, Gnanou and Hadjichristidis.78 This work aimed for the first fully metal-free anionic copolymerization of epoxides (PO and cyclohexene oxide (CHO)) with carbon dioxide, whereby a strictly alternating monomer incorporation was targeted (Scheme 1). This was achieved by judicious choice of metal-free activators, in analogy to an earlier publication by the same group where lithium salts (to activate CO2) in combination with (iBu)3Al (to activate the epoxide) were employed.79 Taking inspiration from this, the authors opted for several onium and phosphazenium salts to provide bulky cations capable of slight interaction with CO2. Et3B was selected for metal-free epoxide activation, specifically because it was assumed that this compound was Lewis acidic enough to provide a weak interaction with PO or CHO but not strong enough to favour excessive epoxide homopolymerization.
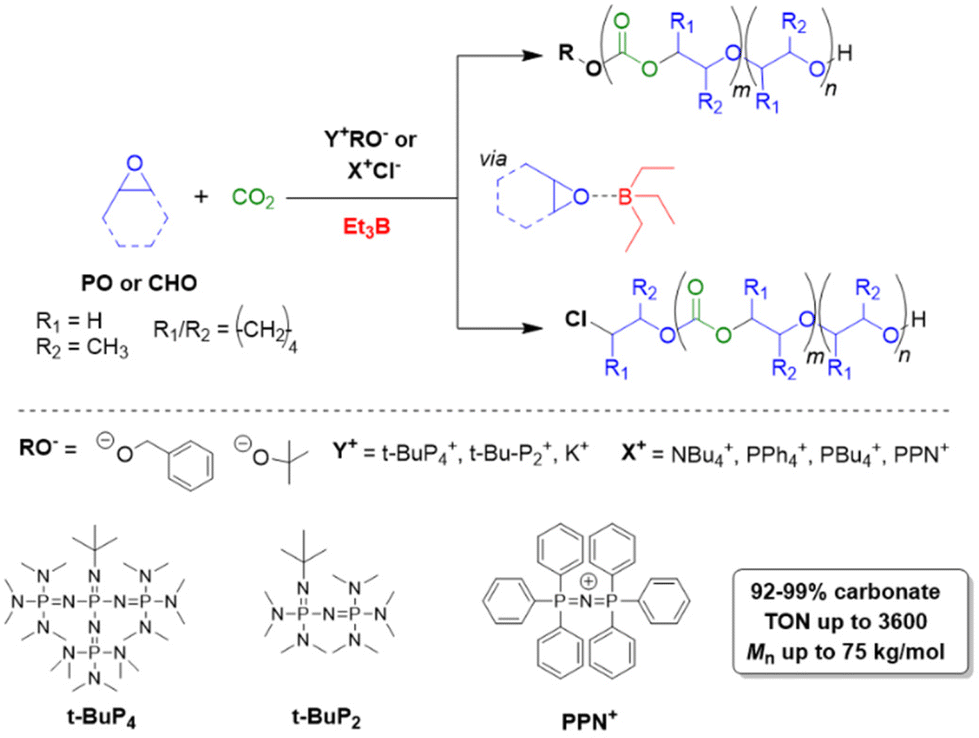 | ||
| Scheme 1 The first metal-free copolymerization of epoxides and CO2, enabled by Et3B as epoxide activator. Note that initiation can be achieved via alkoxides or chloride. | ||
A screening quickly revealed that the best results were obtained using NBu4Cl/Et3B for PO/CO2 copolymerizations (with a TON of up to 500), while a similar setup achieved a TON of up to 3600 for CHO/CO2 copolymerizations. Characterization via GPC, NMR and MALDI-ToF MS showed that, at low molar masses, the generated polycarbonates displayed monomodal and well-defined molar mass distributions (ĐM < 1.20) alongside a high carbonate content (92–99%) and end group fidelity. 13C NMR revealed 82% head-to-tail linkages for the polymer based on PO. Selectivity regarding linear vs. cyclic product was generally good. When higher molar masses were targeted, a broadening of the PDI and in some cases bimodal GPC results were received. Careful CO2 purification helps in this regard (see below).
Importantly, regarding the involvement of Et3B in the polymerization mechanism, 1H NMR experiments suggested a weak interaction between borane and epoxide (ca. 0.06 ppm difference for the PO methine proton (CDCl3, 20 °C), relative to free monomer). Secondly, it was found that the polymerization worked best if two equivalents of Et3B per propagating chain end are present; less than that resulted in loss of selectivity (linear vs. cyclic) or even a full suppression of polymerization activity when using only one equivalent of borane. These two observations, later reproduced by several studies, indicated that the role of borane exceeds that of a simple epoxide activator and fostered research activity regarding elucidation of mechanistic details as well as novel borane catalyst designs (see below). Indeed, in 2018, Feng, Gnanou and Huang investigated the influence of Et3B on the copolymerization mechanism of PO/CO2 and CHO/CO2 using detailed DFT calculations.80 It was convincingly demonstrated that the presence of borane strongly lowers the respective energy barriers for alternating CO2 and epoxide enchainment, and both the preferred stoichiometry (two equivalents of Et3B) as well as the advantageous behaviour of CHO relative to PO could be reproduced. It was also found that formation of cyclic carbonates via “backbiting” is massively hindered by the coordination between Et3B and the carbonate anion, which readily explains the excellent selectivity for polycarbonate formation.
Interest in borane-mediated CO2/epoxide copolymerization quickly gained momentum in the following years. In 2019, Gnanou and Feng investigated the suitability of carboxylate initiators for this type of catalysis, based on the serendipitous finding that tetrabutylammonium carbonate (TBAC) serves as a very potent, cheap and recyclable initiator (Scheme 2).81 Investigations in this direction were motivated by the fact that halide end groups render the resulting polycarbonate unsuitable as polyol component in polyurethane production (i.e. Bu4NCl/Et3B, see Scheme 1 above). Di- or multifunctional initiators, on the other hand, could provide access to exclusively –OH end-capped linear or star-shaped polyols, respectively. Indeed, using TBAC or two-, tri- or tetrafunctional ammonium carboxylates, PO and carbon dioxide were copolymerized to result in well-defined (ĐM = 1.1–1.2) polycarbonates with high carbonate content (up to 99%) and high selectivity of linear versus cyclic carbonate species (up to >99%); if the carboxylate initiator was introduced via poly(diallyl dimethylammonium) as polycationic counterion, also facile separation and recycling of the polyammonium species was possible.82
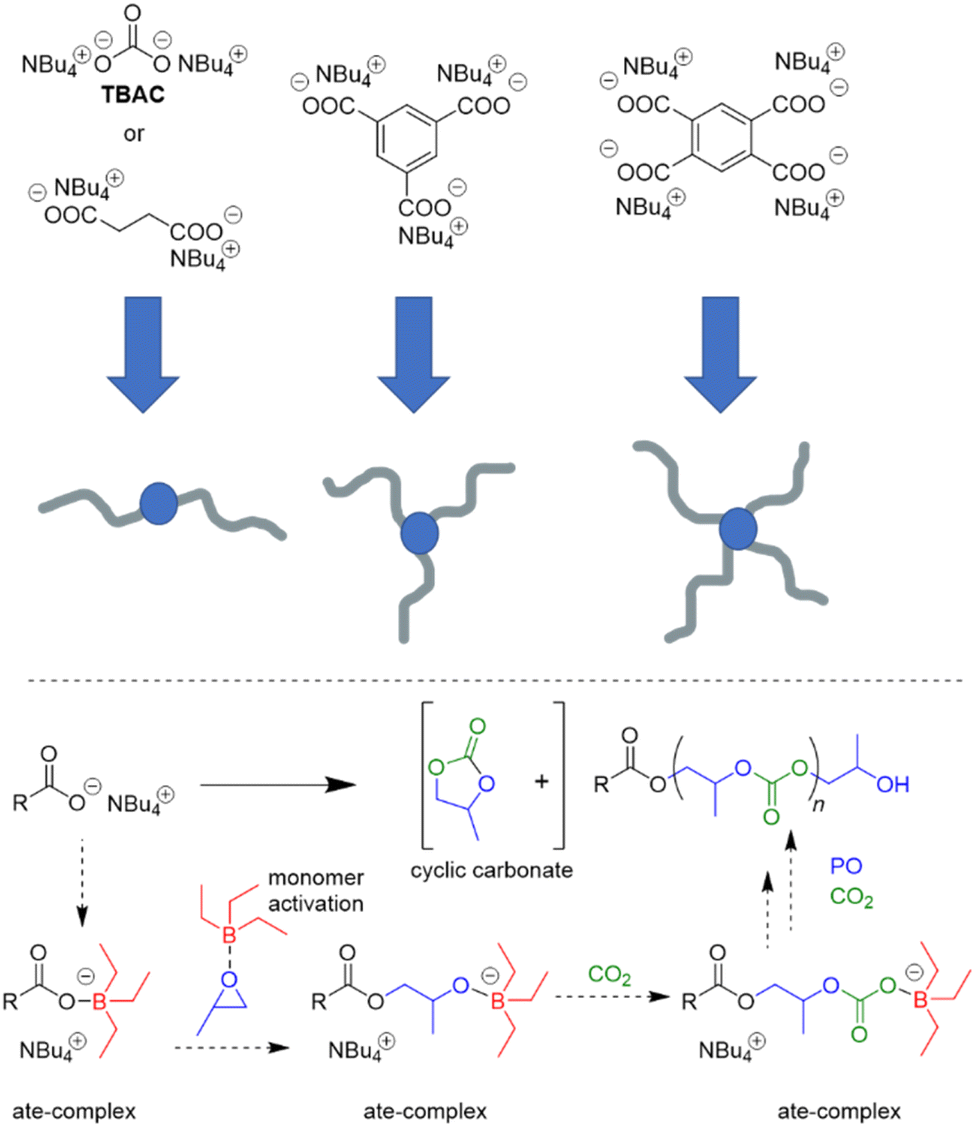 | ||
| Scheme 2 Top: ammonium carboxylate initiators and resulting hydroxyl-capped linear and star-shaped polycarbonates. Bottom: proposed polymerization mechanism including several ate-complexes and activated monomer. | ||
That same year, the group of Zhang used bis(triphenylphosphine) iminium chloride (PPNCl)/Et3B to successfully copolymerize phenyl glycidyl ether (PGE) or SO with CO2.83 Crucially, both research groups stressed the importance of the stoichiometry of borane relative to the propagating chain ends. Thus, in the latter case of PGE, the product spectrum could be tailored from yielding 100% cyclic carbonate to yielding 99% linear product (polycarbonate) by adjusting the molar ratio of Et3B/PPNCl from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 6:1. An even higher excess of borane led to a decrease of the carbonate content in the polymer, that is, a growing proportion of polyether linkages in the product copolymer. Similarly, for PO/CO2 copolymerization using carboxylate initiators as discussed above, it was found that higher Et3B loading relative to carboxylate favoured polyether linkages, while a lower loading favoured alternating copolymerization (higher carbonate content); for PO, however, much less of an excess was necessary to engender polyether linkages, supporting the occurrence of epoxide-dependent interactions with the Et3B. It should be noted that such behaviour, where the relative catalyst loading can be used to impact the product spectrum to such a fundamental degree, is very different from typical metal-based approaches to generate polycarbonates from carbon dioxide and epoxides.
:1 to 6:1. An even higher excess of borane led to a decrease of the carbonate content in the polymer, that is, a growing proportion of polyether linkages in the product copolymer. Similarly, for PO/CO2 copolymerization using carboxylate initiators as discussed above, it was found that higher Et3B loading relative to carboxylate favoured polyether linkages, while a lower loading favoured alternating copolymerization (higher carbonate content); for PO, however, much less of an excess was necessary to engender polyether linkages, supporting the occurrence of epoxide-dependent interactions with the Et3B. It should be noted that such behaviour, where the relative catalyst loading can be used to impact the product spectrum to such a fundamental degree, is very different from typical metal-based approaches to generate polycarbonates from carbon dioxide and epoxides.
Mechanistically, in both cases, the formation of “ate-complexes” was put forward to rationalize the catalytic performance (Scheme 2, bottom). In effect, the Lewis acid was thus proposed to fulfil a dual role of activating the monomer and stabilizing the propagating chain end. This proposal seems to explain nicely why an excess of Et3B is particularly useful for epoxide enchainment and why more than one equivalent is usually needed (one equivalent of borane being consumed by the propagating chain end, which competes with the monomer for borane coordination). Also, it should be noted that formation of an ate-complex renders the propagating chain end less reactive and could thus plausibly reduce side reactions or selectivity issues like backbiting. Formation of a true ate-complex is accompanied by a notable shift in 11B NMR, whereby free Et3B appears at δ = 86 ppm, while the complexed borate is found at δ = 54 ppm.82 Accordingly, in the presence of severely dried CO2, very high-Mn polycarbonates can be prepared (Mn ≫ 100 kg mol−1).84
Overall, this proposed mechanism is strongly reminiscent of Lewis pair polymerization (LPP, see discussion further below).85
In 2020, the beneficial aspects of Et3B-mediated epoxide/CO2 copolymerization were taken a step further by mapping out the epoxide scope and using this knowledge to create all-polycarbonate thermopolastic elastomers.86 In this large study, BO, 1-hexene oxide (HO), 1-octene oxide (OO), butyl glycidyl ether (BGE), 2-ethylhexyl glycidyl ether (EHGE) and allyl glycidyl ether (AGE) were copolymerized, using Et3B in conjunction with halide (i.e., NBu4Cl, PPNCl) or oxyanionic initiators. Reactivity of the epoxides under identical conditions was found to be BO > HO > OO > BGE > EHGE > AGE, thus showing a clear correlation with the monomer structure: the reactivity decreases as the borane-epoxide interactions are increasingly hindered by growing steric bulk or competing interactions (BGE, EHGE and AGE all possess a second oxygen atom, which can coordinate to Et3B, while AGE also carries an olefinic functionality which was shown to very slightly interact with the borane). In accordance with previous work as discussed above, the ideal Et3B loading was epoxide-dependent. Using more equivalents of borane relative to the propagating chain ends could partially compensate for a lower epoxide reactivity. Nonetheless, in all cases polycarbonates were received, underlining the unusually broad applicability of the Et3B-based catalytic setup.
With this range of different polycarbonates in hand, all-polycarbonate block copolymers were prepared, starting from a difunctional initiator. First, a soft block based on OO/CO2 was prepared, aiming at high molar masses (Mn = 280 kg mol−1, ĐM = 1.2). Then, two hard, high-Tg blocks based on CHO were grown on either termini of the polymer, taking the overall molar mass up to >350 kg mol−1 (ĐM = 1.3–1.4). The high molar masses were crucial in increasing the segregation strength of the blocks (targeting χN ≫ 10.5, with χ = Flory–Huggins parameter and N = degree of polymerization) to enable microphase separation and thus the formation of hard-block, high-Tg segments in a soft amorphous matrix. The resulting polymer displayed two distinct glass transition temperatures (Tg = −20 °C and 107–119 °C, respectively) and atomic force microscopy (AFM) substantiated microphase separation by revealing spherical or cylindrical hard segments (Fig. 2). Tensile strengths in the range of 2.04–3.24 MPa were found, while the elongation at break was between 330–1050%, overall characterizing the materials as soft rubbers, which might find application in tissue engineering. These materials represent the first example of the metal-free synthesis of all-polycarbonate thermoplastic elastomers. A similar methodology was employed to generate star-shaped polymers with a degradable polycarbonate core (using 4-vinylcyclohexene dioxide, VCD), of interest for, i.e., drug-delivery.87 Additionally, all-polycarbonate graft copolymers were constructed by a grafting-from approach, starting from polycarbonate based on vinyl-CHO and CO2.88 After conversion of the dangling olefinic groups into ammonium carboxylates, poly(propylene carbonate) (PPC) chains were grown from the backbone. Depending on the relative sizes of backbone and graft, different self-assembled morphologies could be created this way.
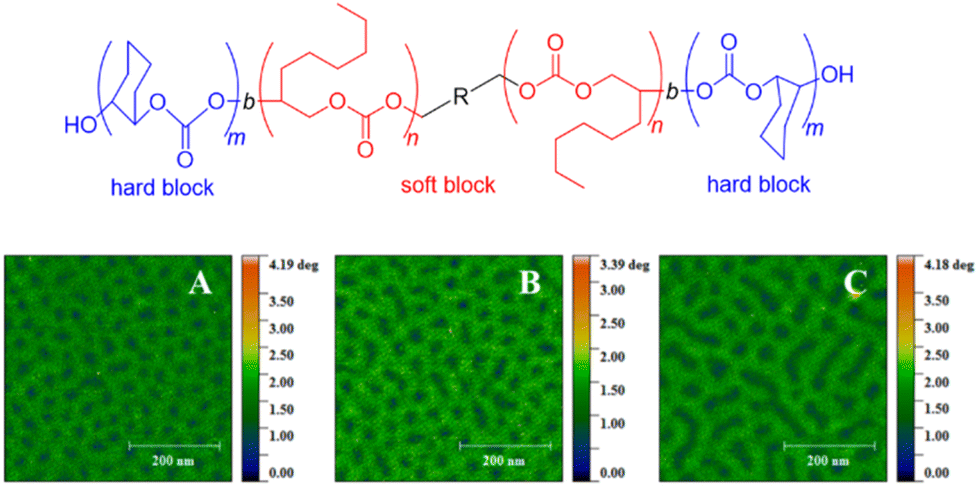 | ||
| Fig. 2 All-polycarbonate thermoplastic elastomer and AFM results showing microphase separation thereof, with A, B, C = polymers with same soft block but increasing hard block size. Reproduced from ref. 86 with permission from American Chemical Society, copyright 2020. | ||
In 2021, the set of initiators that can be combined with Et3B to produce PPC via copolymerization of PO with carbon dioxide was extended to include simple, neutral tertiary amines (i.e., triethylamine, Et3N).89 In contrast to halide or oxyanionic initiators, this results in a zwitterionic propagating species (Scheme 3). The Et3N/Et3B-pair proved to be effective in generating high molar-mass PPC (up to 56 kg mol−1), whereby strictly alternating comonomer incorporation was achieved. The selectivity for PPC over the cyclic propylene carbonate was generally good and could be further improved by using tributylborane (Bu3B) as Lewis acidic component: sitting on the oxyanionic chain end, the increased steric demand of this compound plausibly hinders the backbiting-type reaction forming the undesired small molecule carbonate. Application of triphenylborane (Ph3B) resulted in a complete loss of polymerization activity. Perhaps most strikingly, it was observed that the Et3N/Et3B-pair worked fine at a 1:1 or even 2:1 molar ratio. This contrasts earlier reports on the PPNCl/Et3B system where a 1:1 stoichiometry resulted in a sharply rising content of cyclic carbonate side product.83 The involvement of the intramolecular ion pair (Scheme 3) could potentially be responsible for this behaviour; in a DFT-based interpretation of the polymerization mechanism, the rate determining step is still the attack of Et3B-masked carbonate on Et3B-activated PO, thus dependent on the involvement of two equivalents of borane.
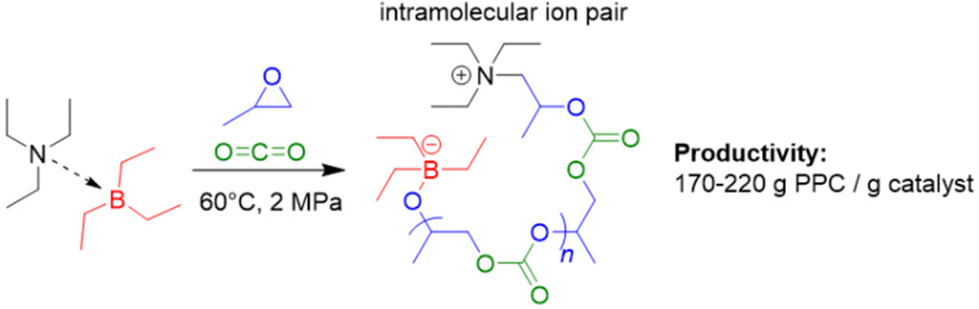 | ||
| Scheme 3 Preparation of polycarbonate via Et3N/Et3B and zwitterionic species. | ||
In 2022, Liu, Li and co-workers focused on the impact of CO2 pressure when using organic superbases to generate the initiating alkoxides species from alcohols in situ.90 Thus, a specific type of phosphazene (C3N3-Py-P3, Scheme 4) was employed in conjunction with diol initiators and Et3B for the CHO/CO2 copolymerization under various conditions. C3N3-Py-P3 is characterized by a relatively weak basicity (pKa = 26.5 in acetonitrile) and a significant size (1.3 nm). This compound activates the –OH species, but interactions with Et3B are relatively weak, as shown by NMR analyses, thus avoiding tight Lewis pair formation and concomitant deactivation of the catalytic setup. Just in contrast, at 80 °C and 1 MPa of CO2 pressure in presence of two equivalents Et3B per phosphazene molecule, a very well-defined product is received, showing neither ether linkages nor the formation of undesired cyclic carbonate (ĐM = 1.15–1.25). Under extreme conditions of 24000 equivalents of CHO relative to the phosphazene, still more than 50% of the epoxide are converted after 30 h, amounting to a TON of >12000. Interestingly, C3N3-Py-P3/Et3B also works under very mild conditions (25 °C, 0.1 MPa CO2), still achieving a TON of 320 (at higher Et3B loading) and a very well-defined polycarbonate product (>99% carbonate linkages), underlining that (co)catalyst design has tremendous potential for further optimizing performance. In spite of these benefits, the monomer scope is limited if the high standards of CHO copolymerization are applied. While sterically hindered PGE and SO are still polymerized with high selectivity, albeit much slower, for PO and BO a notable proportion of cyclic carbonate is observed alongside the polymer formation. Epichlorohydrin (ECH) and AGE are not polymerized at all, instead delivering exclusively the cyclic side product in high yield.
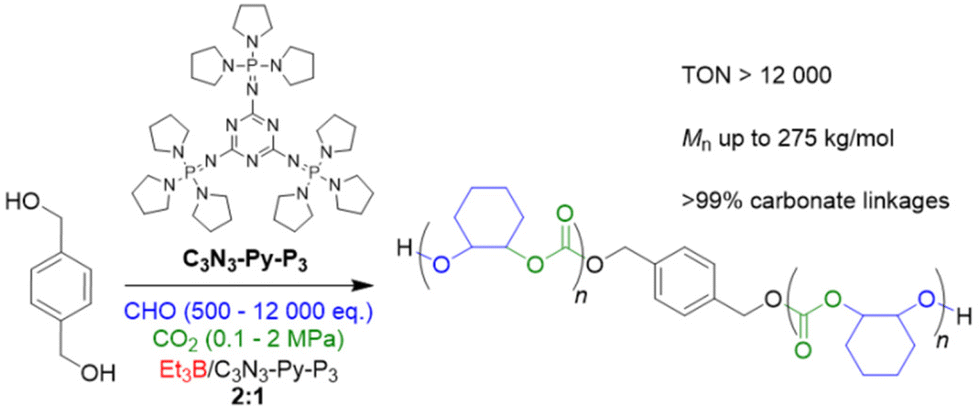 | ||
| Scheme 4 Polycarbonate formation from CHO/CO2, using a borane/phosphazene catalyst pair for polymerization under mild conditions. | ||
EO as epoxide component has received only limited attention in this context, but in a striking publication Feng, Ganou and co-workers have highlighted the potential of Et3B-mediated EO/CO2 copolymerization for producing a range of products with different and complementing properties.91 Thus, while PEG/PEO is water-soluble, the corresponding copolymer with high carbonate content is hydrophobic (with up to 50% of its weight deriving from CO2). On the other hand, a low carbonate content will generate a “PEG-like”, semicrystalline polymer which is water-soluble but degradable on account of the few remaining carbonate functionalities. Thus, using a combination of Et3B and onium salts under various reaction conditions (CO2 pressure, solvent, borane loading) an array of copolymers was prepared. In satisfying accordance with previous knowledge regarding borane-mediated epoxide copolymerization, the sterically unhindered and reactive EO requires a lowering of the Et3B-ratio to yield alternating comonomer incorporation: two equivalents of borane relative to onium salt result in a polymer with only 15% carbonate linkages (predominant formation of ether linkages). Lowering the Et3B proportion and increasing the CO2 pressure can push up the carbonate content accordingly. In hexane solvent, the dissociation of the intermediate ate-complexes is further disfavoured, suppressing EO homopolymerization (at the cost of somewhat slower polymerization kinetics). Under optimized conditions, high carbonate contents >90%, excellent selectivity for linear polymer (over cyclic carbonate side products) and molar masses up to 20 kg mol−1 were achieved (ĐM = 1.1–1.3). Using such hydrophobic polycarbonates as macroinitiators (including α,ω-dihydroxylated species), PEO blocks could be grown on them after releasing the carbon dioxide from the reactor and recharging with EO (Scheme 5).
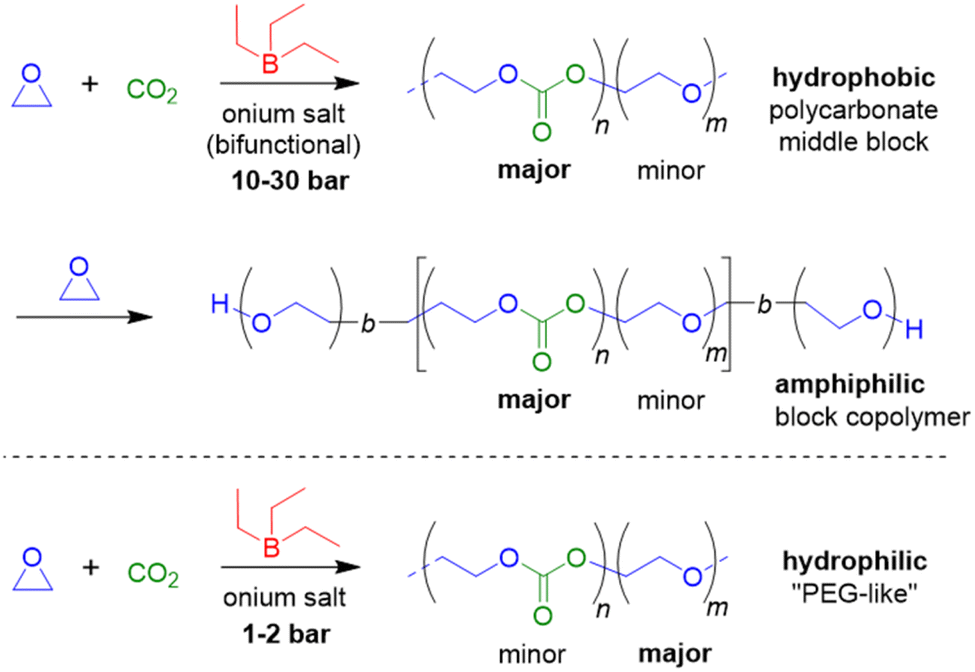 | ||
| Scheme 5 Top: formation of a CO2/EO-based polycarbonate, which can be used as a hydrophobic core to generate amphiphilic block copolymers after EO homopolymerization in a second step. Bottom: hydrophilic PEOEC, as resulting from the borane-mediated copolymerization under suitable conditions. | ||
The resulting, well-defined block copolymers are able to form micelles, in analogy to the well-known “Pluronic” non-ionic surfactants, however, with the added benefit of being sourced from CO2, being degradable and having a lower critical micelle concentration (cmc) than comparable Pluronics. In a final part of this study, poly(ethylene oxide-co-ethylene carbonate) (PEOEC) with particularly low carbonate content was targeted. Congruent with the observations discussed above, this could be achieved by an increase of the relative Et3B equivalents or a lowering of CO2 pressure (or a combination of both), giving access to polymers with tailored carbonate contents of 1.7–11.0%. Very high molar masses could be obtained this way (ĐM = 1.03–1.16). The polymers thus received were shown to be degradable as expected, displaying also a melting point close to that of PEG when the occurrence of carbonate functionalities was sufficiently low (<5%). Overall, this approach delivered colourless, metal-free polymers with adjustable polarity, with potential application as surfactants, for drug delivery or as polyol components.
Epoxide copolymerization using Et3B catalysis was also extended towards the heavier CO2 analogues, carbonyl sulfide (COS) and carbon disulfide (CS2). In 2017, Darensbourg, Zhang and co-workers demonstrated the exceptional performance of simple Et3B/Lewis base (amidine, guanidine or onium salt) catalyst pairs for the copolymerization of COS with PO and other epoxides (Scheme 6).92 Thus, transparent poly(monothiocarbonate)s were generated with high molar masses (Mn up to 92 kg mol−1) and completely alternating structure. Moreover, oxygen-sulfur exchange was fully suppressed (the polymer structure was not randomized to contain also thiocarbonate or carbonate moieties). In particular, the high head-to-tail content (>99%) and the colourlessness were considered advantageous compared to metal-based copolymerization catalysts. Mechanistically, it is interesting to note that amidine and guanidine functional groups are active for copolymerization. 1H NMR experiments suggested that addition of COS and PO to a DBU (1,8-diazabicyclo[5.4.0]undec-7-ene)/Et3B Lewis pair results in the formation of a DBU-PO-COS-Et3B species (the signal of the methine proton of PO shifts by 0.17 ppm), which can ring-open PO (Scheme 6). Initiation efficiency can be an issue under these conditions, especially if combined with swift propagation, engendering a loss of control over molar masses and broader molar mass distributions.
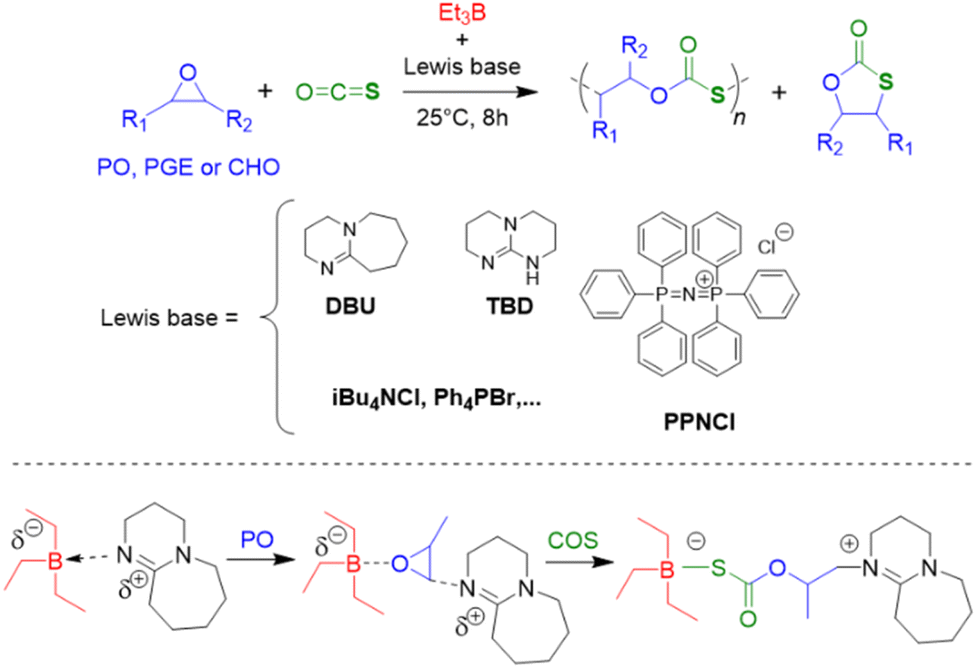 | ||
| Scheme 6 Top: general polymerization reaction for the preparation of well-defined poly(monothiocarbonate) via epoxide/COS copolymerization using Et3B in combination with several Lewis bases. Bottom: Et3B/DBU ion pair and its reactivity with the monomers. | ||
It should be noted that under the same conditions, no polymerization was observed when Et3B was exchanged for BCF or Ph3B. Also, an initial screening of frequently employed Lewis pairs (Ph3B/Ph3P, BCF/Ph3P or BCF/DBU) failed to produce the desired polymer, underlining the unique suitability of Et3B. Using the same polymerization strategy in the presence of mono- or dihydroxylated poly(ethylene glycol) (PEG) macroinitiators as chain-transfer agents (CTAs), well-defined AB or ABA block copolymers were constructed.93 Compared to the polymerization in the absence of CTAs, the excellent selectivity was retained while overall the catalytic reactivity even increased somewhat. Molar masses up to Mn = 50 kg mol−1 were achieved this way, overall displaying narrow mass distributions (ĐM < 1.3).
Interestingly, the catalytic activity for PO/COS copolymerization could be further improved by using N,N,N′,N′-tetramethylethylenediamine (TEED) in combination with Et3B.94 Indeed, a dramatic increase of turnover frequency (TOF) to 22500 h−1 was reported with this setup, presumably on account of a higher probability of monomer activation. The spacer length between nitrogen atoms was indeed found to be a key tuning parameter, with two methylene units, as present in TEED, being the optimum. The polymerization retained a high selectivity for strictly alternating copolymerization; moreover, once COS was consumed, the polymerization stopped even if excess of PO was still present. Further addition of COS then reactivated the polymerization. Several such “on–off” cycles were conducted and parallel GPC analysis proved the well-behaved, complete chain extension.
This behaviour markedly changes when more than one equivalent of Et3B per tertiary nitrogen moiety was present. Using a setup of PO/COS/Et3B/TEED of 500:200:2:0.5 (molar ratio), after rapid copolymerization, a block of PPO is slowly grown on the poly(monothiocarbonate), giving access to a one-pot, one-step process for preparing this interesting type of copolymer.
The terpolymerization of PO with both CO2 and COS has also been investigated recently.95 Using Et3B/amine pairs, sulfur-containing polycarbonates with high molar masses (Mn up to 59 kg mol−1) and moderate dispersity (ĐM < 1.56) could be obtained; by adjusting the COS feed, it was possible to adjust the thiocarbonate content in a range of 27–81%. Kinetic analyses showed that the polymer is characterized by a notable gradient (preferential incorporation of COS, Scheme 7 top). Interestingly, using the same reaction conditions with [PO]:[COS]:[Lewis base]:[Et3B] = 500:250:1:2 under 1.7 MPa of CO2 pressure, choice of the amine Lewis base had a significant impact on the polymer microstructure. For example, replacing N,N-dimethylcyclohexylamine by using N,N-diethylcyclohexylamine resulted in a loss of CO2 polymerizability. Use of a difunctional amine entailed a rise of polyether linkages. Similar to earlier studies, it was found that an increase in Et3B loading relative to Lewis base also entails a growing proportion of ether linkages while simultaneously the amount of cyclic side products decreased. The latter can most likely be explained by a more effective masking of the propagating chain end if more borane is present. This will inhibit the backbiting reaction necessary for forming the undesired cyclic product. Application of Bu3B resulted in similar behaviour while the arylborane dimesitylfluoroborane failed to show polymerization activity.
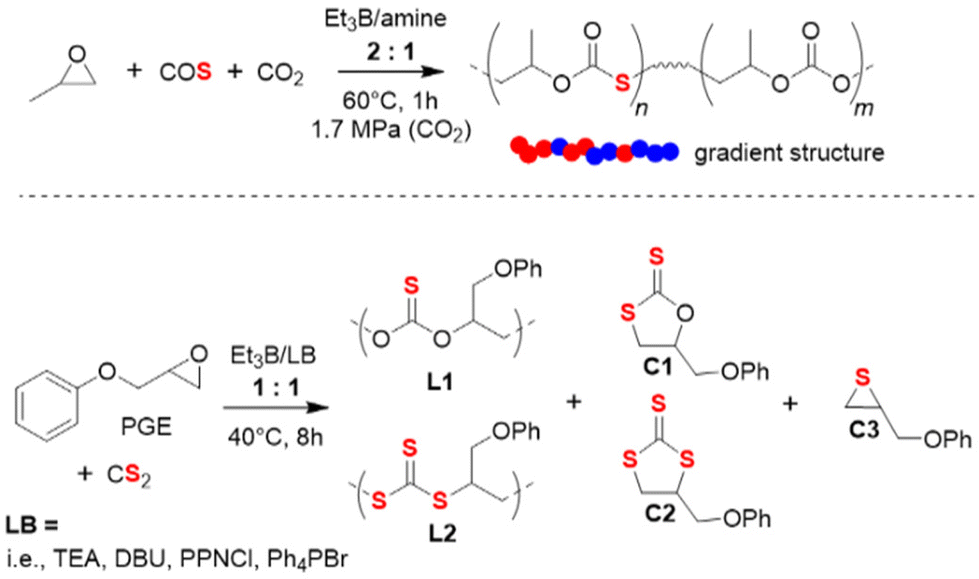 | ||
| Scheme 7 Top: terpolymerization of PO with CO2 and COS. Bottom: copolymerization of PGE with CS2, resulting in a polymer with two different types of linkage motifs and three cyclic side products. | ||
The Et3B-mediated copolymerization of CS2 with epoxides has received much less attention. In 2020, Zhang and co-workers have studied this, focusing on PGE as epoxide component.96 PGE was specifically selected because its reactivity with COS was found to be somewhat sluggish.92 This feature was hoped to simplify the array of different linkage motifs which can occur when copolymerizing cyclic ethers with CS2 under the excessive O/S scrambling (COS is an important intermediate). Indeed, when applied to PGE, catalyst setups using Et3B in combination with several Lewis bases resulted in just two different linkage types and three variants of cyclic by-products (Scheme 7, bottom). The relative proportion of L1 and L2 is decisive for the polymer properties, since the optical characteristics are largely dependent on the sulfur content. Indeed, this parameter could be impacted by tailoring the catalyst pair and the reaction conditions. Thus, using [PGE]:[CS2]:[Lewis base]:[Et3B] = 500:750:1:1 at 40 °C, an increase in the temperature entailed a strong increase in L2 over L1, meaning the polymer became richer in sulfur atoms. This, however, came at the cost of more undesired cyclic product and sharply decreasing molar masses. On the other hand, choice of the Lewis base also impacted this property: triethylamine (TEA) effected almost the double L2 content compared to Ph4PBr (32% vs. 17%). Curiously, when the well-known 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) was employed, no polymer at all was generated. Instead, almost exclusively C1 is formed. Application of 4-dimethylaminopyridine (DMAP) failed to yield any polymer at all. Under optimized conditions, Et3B/TEA delivered molar masses of up to 12 kg mol−1 (ĐM = 1.7, alongside 60% cyclic product).
Finally, it was also shown in an elegant study that the borane-mediated copolymerization can be extended to four-membered cyclic ethers (oxetane, (OX)), whereby choice of a suitable (pseudo-)halide initiator enabled a tailoring of the product spectrum from six-membered cyclic carbonate to polycarbonate, depending on the leaving group properties of the halide.97 The incorporation of oxetanes in such copolymers is usually challenging and success of the process is duly dependent on the involvement of Et3B in several transition sates and intermediates. While a high carbonate content was achieved, the molar mass distribution was somewhat broad (ĐM = 1.5–1.6) and molar masses limited to Mn < 10 kg mol−1.
Epoxide homopolymerization
As discussed above, the balancing of the borane equivalents relative to the number of propagating species is a key tuning site if copolymerization with carbon dioxide is desired. In general, an excess of Et3B will favour epoxide homopolymerization, whereby the specific number of borane equivalents is epoxide-dependent. This can be used to tailor polymer microstructures and also presents a powerful way of creating polyether structures in precisely defined ways.In 2018, the research groups of Zhao98 and Zhang99 independently described the Et3B-mediated homopolymerization of epoxides in the presence of organobases and alcohol-type initiators. These findings not only underlined high catalytic efficiency, but also provided valuable new insights in the polymerization mechanism. Thus, in his detailed work Zhao demonstrated that for EO homopolymerization, using tBu-P1/Et3B (1:3 molar ratio) under optimized conditions, a very high TOF of 6000 h−1 can be achieved, alongside low catalyst loadings (144 ppm by weight of base and borane combined) and well-defined PEO properties (ĐM < 1.3). Molar masses of well over 100 kg mol−1 were accessible this way in relatively short reaction times (few hours).
For PO, likewise impressive results were generated. Molar masses of up to Mn = 200 kg mol−1 were achieved in short reaction times (7 h, tBu-P2/Et3B = 1:3). As evidenced by the narrow molar mass distribution (ĐM < 1.1), MALDI-ToF MS and the linear development of molar mass with conversion, the notorious transfer-to-monomer side reaction is fully suppressed in this approach. It should be noted that this behaviour is vastly superior to KOH-mediated polymerization, but also outperforms previous organocatalysts such as N-heterocyclic carbenes (NHCs)58 or N-heterocyclic olefins (NHOs).59 Other epoxides such as BO, PGE, AGE, EHGE or tert-butyl glycidyl ether (TBGE) were also successfully converted to the corresponding homopolymers, albeit a significantly lower polymerization rate was observed.98,99 Interestingly, SO could not be converted, and also the polymerization of CHO was unsuccessful under these conditions.
Both Zhao and Zhang also found that phosphazene organobases are not strictly necessary to obtain polymerization of EO or PO; also the milder amidine or guanidine compounds DBU or MTBD (7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene) could be applied.98,99 Indeed, as shown later by Naumann and co-worker, even the rather weak nitrogen bases DABCO (1,4-diazabicyclo[2.2.2]octane) or DMAP are suitable.100 The polymerization rates clearly scale with the basicity of the corresponding organobase, resulting in rapid conversion for tBu-P2 and gentle monomer incorporation for DABCO or DMAP. Also NHCs have been applied jointly with Et3B to prepare polyethers.101
A closer inspection of polymerization kinetics revealed that a higher concentration of –OH functionalities ([hydroxyl], by adding a larger excess of alcohol initiator) entailed a significant slowdown of the polymerization rate. This is a crucial result, not only by having mechanistic implications, but also because epoxide polymerization in larger reactors is often started in the presence of a high amount of alcohol relative to epoxide (for safety reasons or to target oligomeric products). In the experiments conducted by Zhao an increase of [hydroxyl] from [tBu-P2]/[Et3B]/[hydroxyl]/[PO] = 1:3:2:1600 to 1:3:40:1600 halved the polymerization rate.98 An increase of the Et3B/organobase ratio entailed the expected increase of the polymerization rate, while the polymerizations became slower when less than two equivalents borane were present and often did not even start when less than one equivalent was used. Together with 1H and 11B NMR experiments, these findings served to come up with a more refined polymerization mechanism (Scheme 8). The two key species for monomer enchainment, activated monomer (AM) and activated hydroxyl (AH), are both delivered by Et3B-dependent equilibria ((1) and (2), respectively). It is important to note that the basicity of the organobase will regulate the activity of the AH; a stronger base can thus entail a faster propagation, in full accordance with above observations. In addition, the borane moiety will stabilize the negatively charged chain end, thereby acidifying the hydroxyl (rendering also comparatively weak organobases suitable for engendering polymerization). Simultaneously, this stabilization reduces the electron density at the alkoxides oxygen, likewise lowering its propensity for base-related side reactions. Transfer-to-monomer is thus effectively suppressed and very high molar masses are easily obtained.
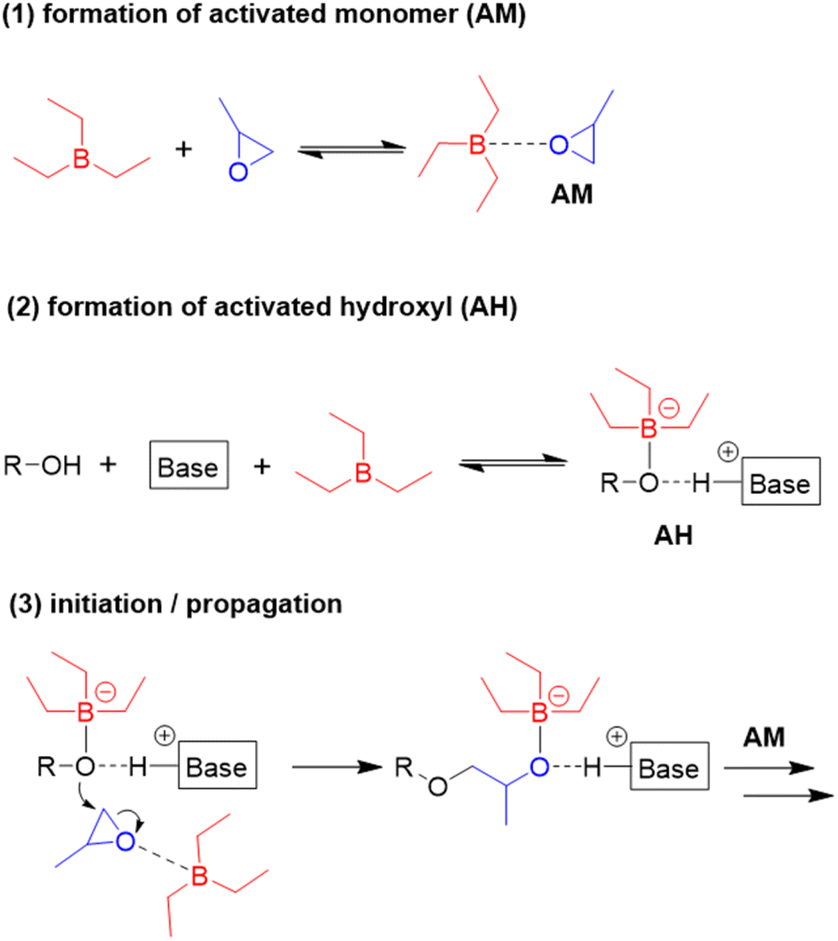 | ||
| Scheme 8 Polymerization mechanism for Et3B-mediated epoxide homopolymerization, as proposed by Zhao. | ||
Crucially, the AH itself is not able to achieve effective propagation. Uncomplexed Et3B, free to engage in the formation of AM, is necessary for this and consequently all factors that manipulate the concentration of AM will impact the polymerization rate. Competition for coordination to the borane Lewis acid is one such factor. One example for the latter is when larger amounts of hydroxyl species are present. Alcohols are slightly stronger bases than epoxides,102 which means they can effectively compete for interaction with Et3B even in bulk polymerization of, i.e., PO. Less of the borane is thus available for formation of AM, and the polymerization rate drops significantly. The addition of more equivalents of borane can partly compensate for this loss of activity. This behaviour also explains why functionalized monomers such as AGE are sometimes found difficult to polymerize; both the exocyclic oxygen as well as the double bond have been found to weakly interact with Et3B.86 Obviously, parameters such as temperature, polarity of solvent or steric effects will likewise impact the equilibria (1) and (2), albeit further research is necessary to clarify these effects. It should be noted that application of a weak organobase may lead to a situation where not exactly one equivalent of borane is consumed, since the less activated hydroxyl will bind the Et3B less effectively. Under these conditions a slow polymerization might be observed even when [Et3B] < [organobase]. Finally, it must be considered that the exchange between non-activated hydroxyls (dormant chain ends) and AH is much faster than propagation, which is typical for epoxide polymerizations with anionic character.1 As a result, all chains grow at the same speed and narrow molar mass distributions are obtained.
The ability to create such well-defined polyether moieties has been quickly taken up to realize improved material applications. Thus, Hadjichristidis, Sarathy and co-workers have tailored a range of all-polyether diblock or triblock architectures for use as lubricant additives, which is also of interest from an energy-saving point of view.103 While polyethers are generally known for beneficial properties in this regard (low pour point, high viscosity indices and flash points), they must be fine-tuned for best performance (thermal stability, rheological properties, antifriction properties). Such a screening was readily achieved by using tBu-P2/Et3B, creating polyethers such as PHO-b-PPO or POO-b-PPO-b-PSO via sequential monomer addition. Very well-defined block copolymers were received (ĐM = 1.04–1.12) and candidates with superior lubrication performance or improved thermal stability were identified.
Borane-mediated epoxide homopolymerization has also been used to create tailored variants98–100,104,105 of the “Pluronic”- or “Reverse Pluronic”-triblock copolyethers (Fig. 3). Here, the ability to finely tune the degree of polymerization, the hydrophilic to lipophilic balance (HLB) and the overall molar mass has been employed to establish libraries of polymers with a stepwise and systematic variation of properties. This is especially useful when applying these amphiphilic polymers for (soft-)templating processes in order to prepare, i.e., mesoporous materials. Mesopore diameter and the symmetry of pore arrangement are dependent on these parameters and very susceptible to small changes;15,106 the commercially available grades (i.e., F127, P123) are limited in this regard, in particular long PPO blocks are not provided. Thus, using NHO/Et3B, Naumann and Bruckner synthesized a polyether series of POn/2-EOm-POn/2 (m = 90–795, n/2 = 10–280) and applied the resulting block copolyethers as templates for ordered mesoporous carbon (OMC) materials104 or ordered mesoporous silica (OMS) materials.105 The resulting mesopores could be tuned continuously between ca. 5–20 nm, whereby a linear relationship between n/2 and mesopore diameter was observed (Fig. 3). It should be noted that success for these applications is dependent on very well defined polyethers and no limitations in designing m or n/2, underlining why the borane-mediated polymerization catalysis has already found application in this field of science.
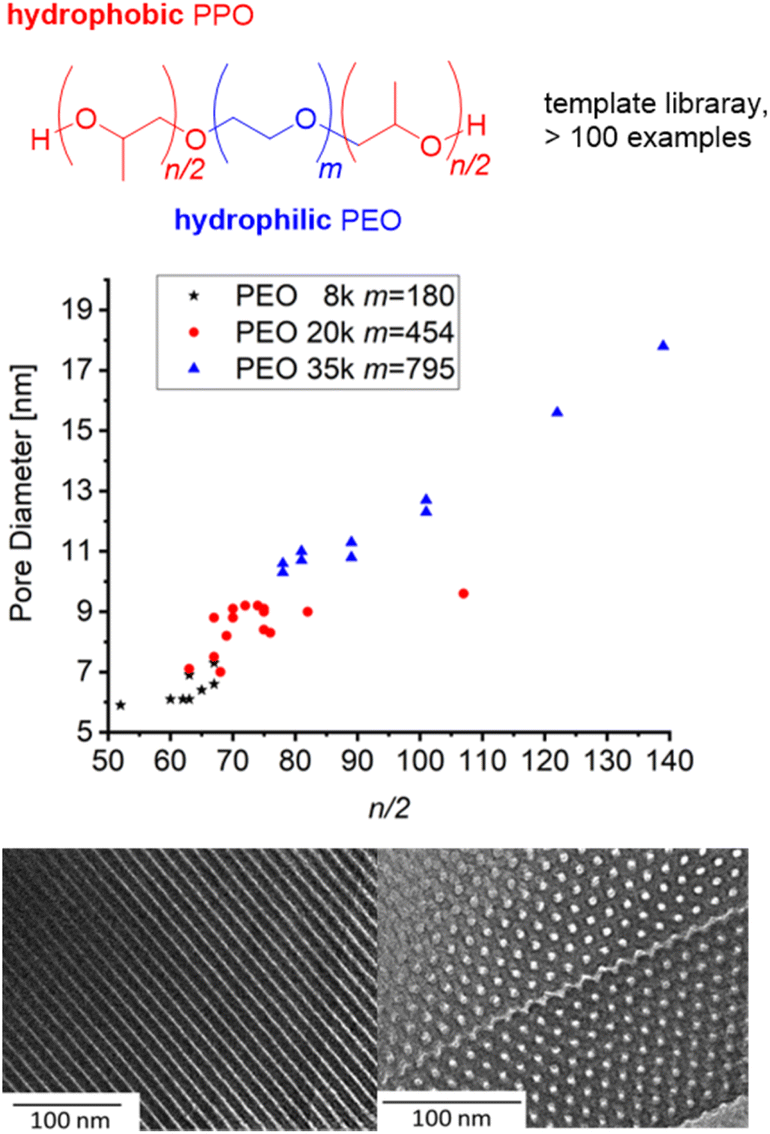 | ||
| Fig. 3 Amphiphilic block copolyether series based on PEO macroinitiators with different molar masses for OMC/OMS preparation. Middle: correlation of n/2 and pore diameter (OMC). Bottom: TEM of OMC based on PO82-EO454-PO82 (9 nm pore diameter). Reproduced from ref. 104 with permission from American Chemical Society, copyright 2021. | ||
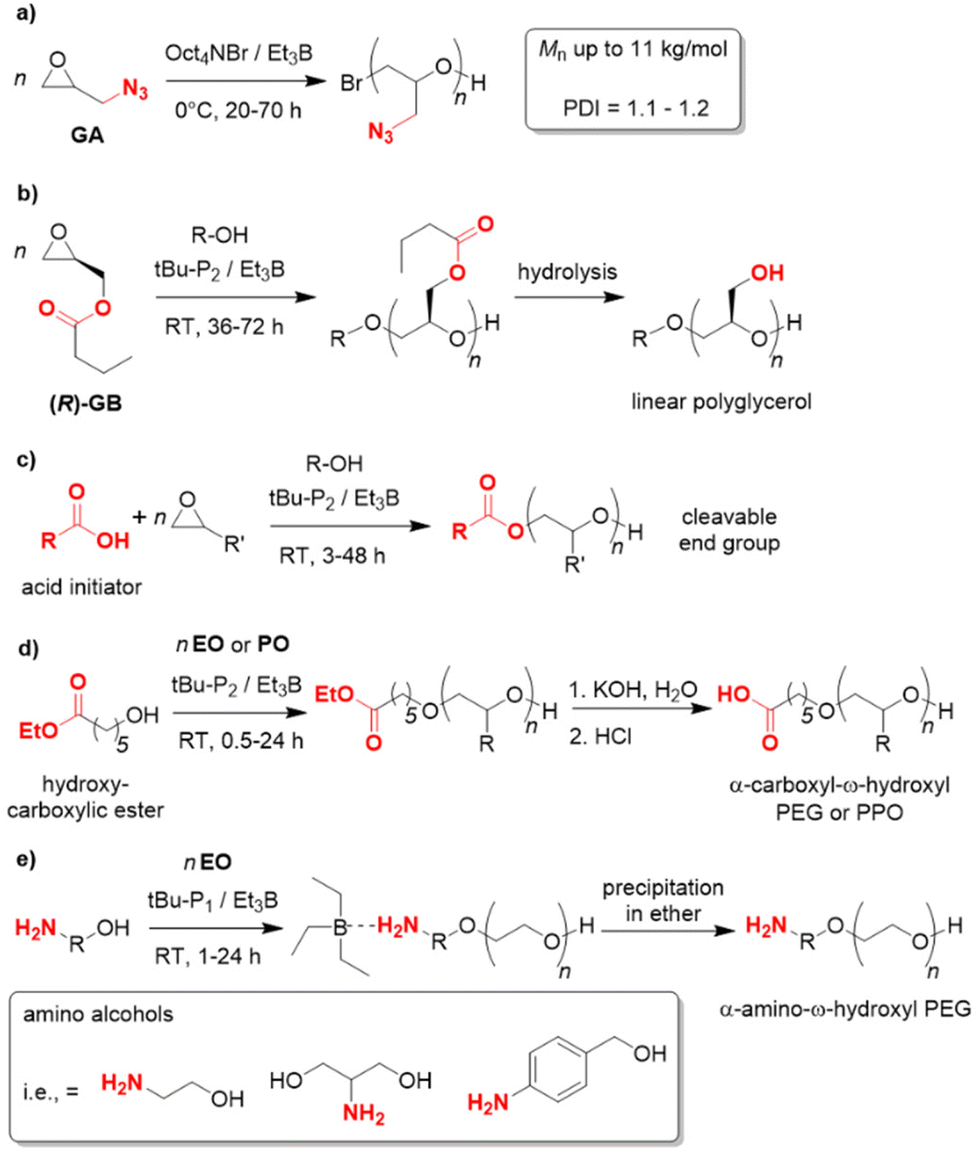
Since the polymerization conditions for the Et3B-catalyzed epoxide homopolymerization are usually gentle (0–50 °C) and, in particular, because the propagating chain end is efficiently masked to a reduced basicity, a notable functional group tolerance emerges in such polymerizations. In 2019, glyzidyl azide (GA) was successfully subjected to homopolymerization using Oct4NBr/Et3B at 0 °C (Scheme 9a).107 NMR investigations showed slight interaction with the oxygen atom in the monomer but also with the azide group. While transfer reactions could not be fully circumvented, still well-defined PGA resulted (Mn up to 11 kg mol−1, ĐM = 1.1–1.2). In view of the sensitive azide moiety, which is susceptible to nucleophiles and also acids, this feature is remarkable and the first report to polymerize this monomer. Copolymerization with carbon dioxide did also work out, as did postfunctionalization of the PGA (via “click”-chemistry). GA is used in the formulation of solid propellants, where also application of oligomeric/polymeric variants may be beneficial for future applications.
 | ||
| Scheme 9 Functional group tolerance as a distinctive feature of Et3B-mediated polymerization catalysis. | ||
Enantiopure glycidyl butyrates (GBs), compounds containing both an epoxide functionality and an ester group, were polymerized by Zhao and co-workers.108 Using Et3B/tBu-P2 (5:1, diol initiator), selective ROP exclusively via the epoxide moiety occurred. Indeed, DFT calculations suggested a preferred interaction of borane and epoxide, while coordination to the carbonyl oxygen is disfavoured, probably on account of steric reasons. This finding most likely explains why also a high loading of Et3B does not result in any kind of transesterification. The resulting polymers are well-defined (ĐM < 1.15) with molar masses (Mn) up to 40 kg mol−1. Interestingly, the ester side chains can be cleaved in a subsequent step via methanolysis, granting access to isotactic, linear polyglycerol (Scheme 9b). Such a very polar polyether is not accessible via direct polymerization of glycidol (GD, due to the inimer character of the latter), while a conventional anionic polymerization of GB would be detrimental for the ester functional groups. Borane catalysis thus provides a convenient route to these hydrophilic, linear polyglycerols. In the same work, block copolyethers with double hydrophilic or amphiphilic character were generated.
This potential for selective and functional group-tolerant polymerization was further illustrated by the group of Zhao,109–111 in particular for the preparation of end-functionalized polyethers such as PEG. Thus, carboxylic (di)acids were employed as initiators (tBu-P2/Et3B, 1:3) for the polymerization of EO, PO, BO or AGE.109 In all cases the desired polyethers were received (Scheme 9c). No transesterification was observed and narrow mass distributions were found (ĐM < 1.12). The thus functionalized and well-defined polymers are of interest for manifold applications, since non-alcoholic initiators allow for subsequent selective cleavage or modification of the polymer. This was illustrated in the same work by the construction of an enzymatically degradable polyurethane, starting from a diacid initiator and the resulting PPO macrodiol. Mechanistically, it is interesting to note that the polymerization displays a pronounced induction period (1–2 h) combined with a relatively swift ensuing epoxide conversion. 1H NMR observation reveals that the reaction between carboxylate and PO occurs during this induction time, then monomer consumption ensues (first-order kinetics regarding the epoxide). Clearly, the activated acid species is a weaker nucleophile than activated hydroxyl. Still, a very controlled polymer formation is guaranteed on account of the different acidity of –COOH and –OH. As long as unreacted acid initiator is present, the anionic (partial) charge will reside there and the newly formed hydroxyls will remain dormant. Only once a large majority of acid has been consumed (95% in these examples), the epoxide polymerization will kick in. Hence, slow initiation does not entail a broadening of molar mass distribution.
Similarly, the ability of the Et3B-catalyzed epoxide polymerization to conserve ester functionalities was employed to generate α-carboxyl-ω-hydroxyl PEG,110 which is a popular structure for bioconjugation. Usually, these compounds are prepared in more laborious ways, but using the beneficial features of borane catalysis a direct synthesis via hydroxyl-carboxylic esters as initiators is possible (Scheme 9d). NMR and MALDI-ToF MS analyses substantiated successful polymerization and subsequent hydrolysis of the ester moiety. At high Mn, a quantitative formation of the carboxylic acid end group is somewhat more difficult and longer reaction times must be considered. Instead of hydrolysis, also primary amines can be reacted with the polymer, delivering the corresponding α-amide-ω-hydroxyl PEG.
Likewise, α-amino-ω-hydroxy PEG is a sought-after motif for PEGylation since amino groups are much more capable in promoting conjugation reactions. Again the peculiarities of Et3B-catalysis allow for a reduction of synthetic effort. Thus, amino alcohols can be directly employed as initiators, using tBu-P1/Et3B, whereby propagation exclusively occurs via the –OH terminus (Scheme 9e).111 At first glance, this seems surprising given that the primary amine is the stronger nucleophile. However, it is exactly this feature which can be used against the amine when Et3B is present. The Lewis acid preferentially complexes the amine to such a degree that it is effectively masked and does not take part in the polymerization. The Et3B thus acts as a noncovalent protecting group. Indeed, control experiments show that in the absence of the borane, the amine will attack EO, while in the presence of one equivalent no reaction at all occurs (all the Et3B is consumed via amine complexation). If an excess of borane and a low phosphazene loading is applied, however, a smooth polymerization ensues, delivering well-defined amino-terminated polyether (Mn up to 10 kg mol−1, ĐM < 1.09).
In 2019, Zhao, Ling and co-workers took these efforts a step further by recognizing that the polymerization mechanism not only allows for tolerating (poly)esters but also for selectively polymerizing cyclic esters and epoxides from one-pot mixtures, notably in a manner where quantitative preference of the catalyst system for either ester or epoxide monomer can be switched back and forth several times.63 This process delivers remarkably well-defined poly(ester-ether) multiblock copolymers (up to pentadecablocks, see Scheme 10).
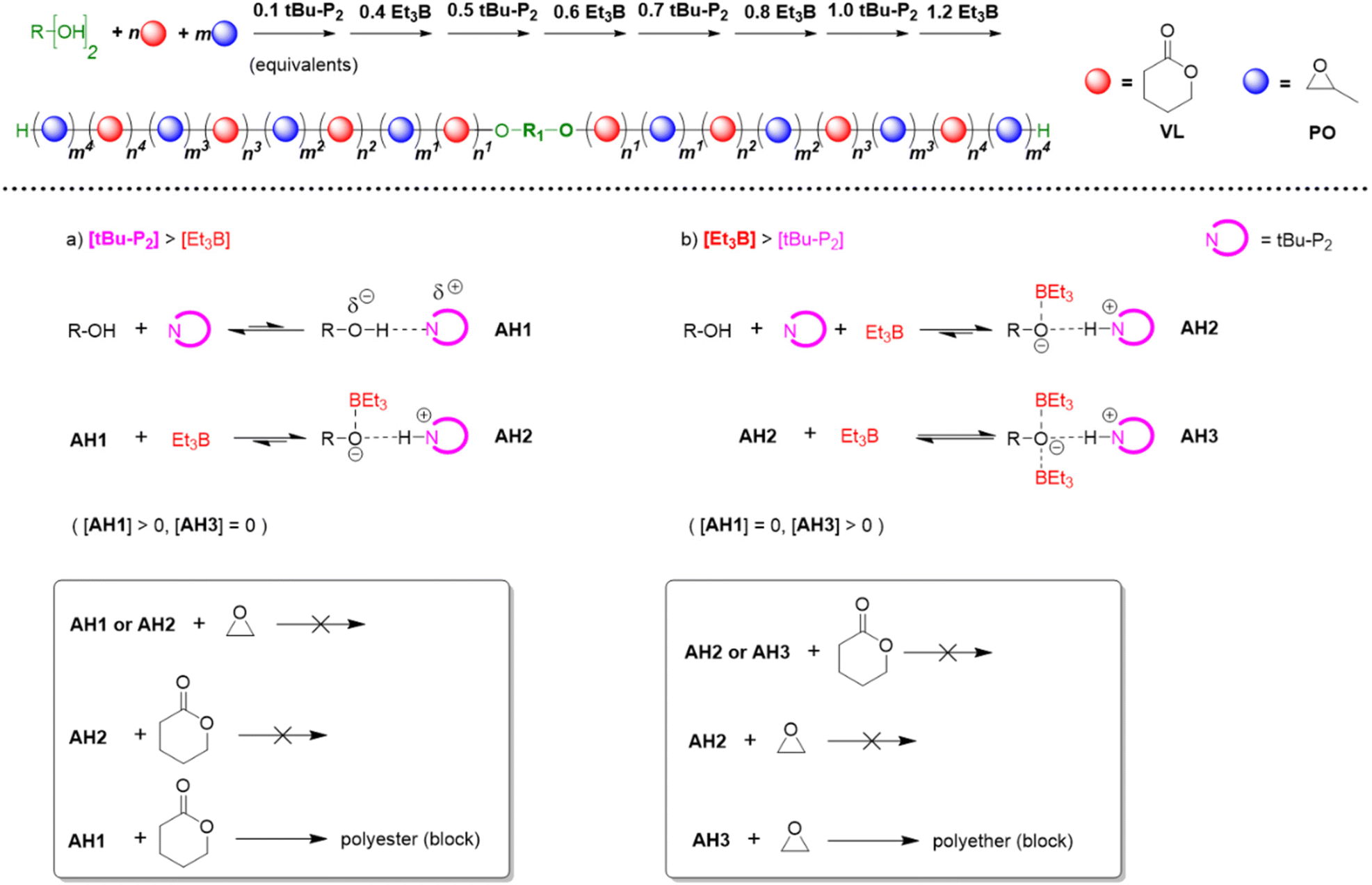 | ||
| Scheme 10 Top: using the Et3B/organobase stoichiometry to synthesize well-defined poly(ester-ether) multiblock copolymers. Bottom: different species of activated hydroxyl and respective selectivity. | ||
Key to this principle is judicious choice of the organobase/Et3B stoichiometry. In the absence of the borane Lewis acid or if the organobase is used in excess, cyclic esters such as ε-caprolactone (CL), δ-valerolactone (VL) or lactide (LA) are polymerized, while the epoxide remains untouched. In the reverse case, applying an excess of Et3B, only epoxide is enchained while transesterification does not occur. Note that the relative excess of either Lewis acid or organobase is slight, but is promptly translated in the aforementioned switch of selectivity, alternatingly turning the respective comonomer consumption on or off.
The block copolymers resulting from this strategy are interesting for a large number of prospective applications since it enables several polymer architectures which are usually not accessible (perhaps with the exception of laborious coupling approaches). Using conventional methods, a polyester block or polyester macroinitiator would suffer excessive transesterification during the reaction conditions necessary for epoxide enchainment. A seemingly simple ABA triblock copolymer with a central polyester block and two polyether terminal blocks would thus previously have posed a considerable obstacle, not to speak of multiblock motifs. Bio-related applications or the preparations of polymeric additives can be expected to profit from this advance.
The results also suggested that a modification of the assumed polymerization mechanism may be necessary. Based on NMR experiments and DFT calculations, Zhao and Ling proposed that overall three different activated hydroxyl species may be involved (Scheme 10).63 Here, AH1 represents the activated species resulting from interaction of the alcohol with the phosphazene base. This active chain end is able to polymerize lactones, but not the epoxide. If Et3B is added, it will be consumed by the oxyanionic chain end, forming AH2. This species obviously displays a reduced basicity and nucleophilicity; it is not able to enchain either lactone or epoxide, but as long as less than one equivalent of borane is present, a certain proportion of AH1 will persist and enable a controlled lactone polymerization. Crucially, if [Et3B] is increased further, all chain ends will be masked and the excess borane is suggested to be involved with the propagating chain-end, generating AH3. This species is now able to polymerize the epoxide (but not the lactone).
Thus, it is proposed that rather than direct monomer activation (AM, Scheme 8) – the shifts observed in NMR control experiments are described as indiscernible112 – an “insertion” between the alkoxide and boron atom takes place. DFT calculations were presented which support this mechanism, in particular it is shown that also the involvement of the second Et3B to form AH3 is favoured. The Gibbs free energy barrier lowers significantly from 29.7 kcal mol−1 to 10.6 kcal mol−1 when considering either AH2 or AH3 for epoxide opening, respectively. These findings are in accordance with the experimental results for poly(ester-ether) multiblock formation and also earlier research in that still two molecules of Et3B are required for the propagation step. In this context, a recent publication by Gnanou and Feng should be noted.113 It was found that setups inactive for lactone polymerization (one equivalent of Et3B per growing chain end, which means all oxyanionic species are capped by borane under formation of ate-complexes, analogous to AH2) can be activated for lactone conversion by addition of amines and (thio)ureas. As underpinned by detailed NMR investigations, the role of the amine thereby is to activate the monomer via H-bonding, while the urea derivative breaks up the ate-complex by trapping the borane species. This way, well-defined “polyester-first” block copolymers (PVL-b-PPO or PPO-b-PVL-b-PPO) were received. This technique is significant since it can avoid at all times the occurrence of free anionic species (AH1) while still lactone conversion is enabled, thus suppressing transesterification side reactions.
Copolymerization with anhydrides and lactones, terpolymerizations
As detailed above, Et3B-mediated epoxide (co)polymerization shows a pronounced tolerance for ester functionalities and also interacts well with carboxylates. In sum, this also suggests that copolymerization with anhydrides, delivering versatile polyester materials, is worth investigating. And indeed, in 2018, Zhang and co-workers described the copolymerization of several epoxides (PO, PGE, ECH) with a range of anhydrides (maleic anhydride (MA), phthalic anhydride (PA), succinic anhydride (SA) and diglycolic anhydride (DGA)).114 Impressively, under suitable conditions (1:1 ratio of Et3B and onium salts) perfectly alternating polymer microstructures were found, while undesired transesterification or polyether linkages were absent (Scheme 11, top). The strictly alternating nature of the polymer was also retained at 80 °C, where TOFs of 102 and 303 h−1 were achieved for the copolymerization of PO/MA and PO/PA, respectively. The well-behaved character of the copolymerization was attributed to the presence of the borane, which masks the propagating chain end, thereby reducing its basicity/nucleophilicity and thus its propensity for side reactions. This is fully in line with the considerations described earlier and in fact polymerizations with only the Lewis base present (i.e., PPNCl, R4NBr) result in rather regioirregular polyesters, slow polymerization, transesterification and, if stereocenters are present, pronounced epimerization.114,115
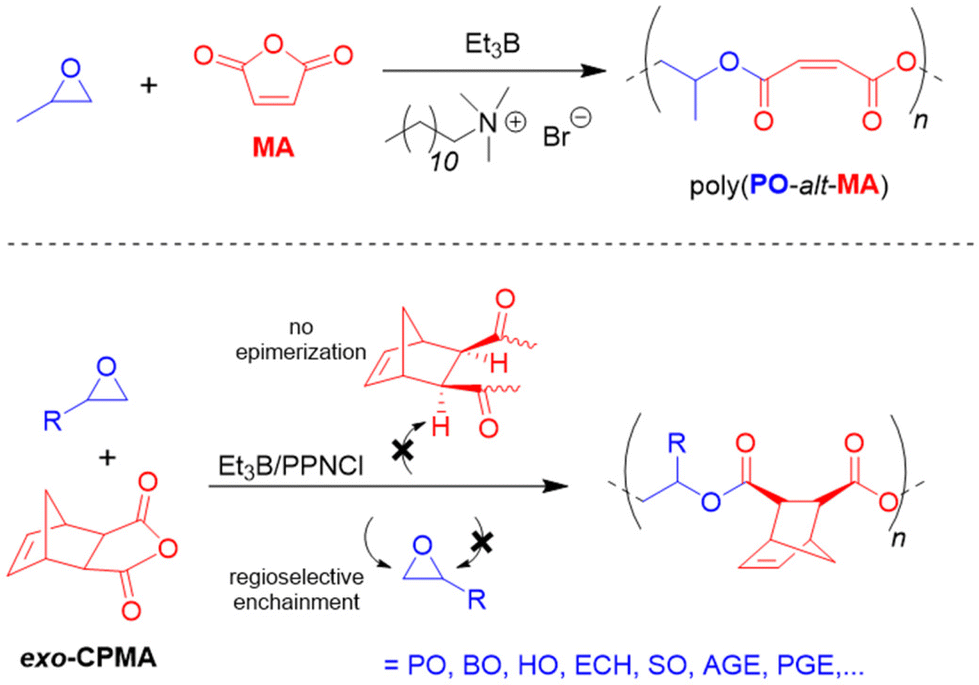 | ||
| Scheme 11 Selective copolymerization of epoxides and anhydrides (including tricyclic ones) using Et3B-cocatalysis. | ||
Albeit bimodal GPC results, a consequence of different competing initiating species (water, acid), can be challenging, this technique could still be used to construct well-defined block copolymers of the type poly[(PO-alt-MA)-b-(PO-alt-SA)]. A moderate regioselectivity of up to 77% head-to-tail arrangements was found. The same authors also investigated the impact of different Lewis acids/bases on polymerization rate and selectivity for the copolymerization of PO and biorenewable SA. It was found that the basicity of the organobase controls the speed of conversion (TEA (7 h−1) < DBU (9 h−1) < MTBD (11 h−1) < t-BuP1 (15 h−1) < t-BuP2 (20 h−1)), while selectivity for ester formation (in relation to polyether linkages) is only marginally influenced by this property.116 Li pointed out that also very low Et3B loadings (0.05 eq. relative to a phosphazene base) are sufficient for the copolymerization of PA with different epoxides.117
Wang, Li and co-workers described the highly regioselective and stereoregular copolmyerization of various epoxides with tricyclic anhydrides such as cis-5-norbornene-exo-2,3-dicarboxylic anhydride (exo-CPMA), which is the Diels–Alder product of cyclopentadiene and MA.115 Such copolymers can display a high Tg, but to achieve this feature, epimerization has to be suppressed and both the relative stereochemistry as well as the regioselectivity must be controlled. Faced with these requirements, the authors opted for borane cocatalysis, using Et3B/PPNCl (1:1 molar ratio). Also for this comonomer set, >99% selectivity for alternating enchainment is observed while for the anhydride components a zero-order kinetic dependence is found. Arrhenius analysis reveals a low activation energy of 50.4 kJ mol−1, which is less than a quarter of the value found for single PPNCl118 (214.7 kJ mol−1), underlining the key presence of Et3B. The regioregularity of poly(endo-CPMA-alt-PO) was very high (98% head-to-tail at 80 °C) and the borane-masked chain end was also found to avoid transesterification and epimerization in a quantitative manner. However, care must be taken to avoid any excess of Et3B, otherwise the expected epoxide homopolymerization will take over. An excess of PPNCl entails epimerization since not all of the propagating chain ends are masked by borane and a certain proportion of “free” oxyanionic species can abstract the α-H of the anhydride and scramble the stereoinformation accordingly. Overall, a broad range of epoxides was converted (Scheme 11, bottom). The Tg of the thus prepared materials could be modulated over a range of 33–95 °C, whereby especially the use of enantiopure epoxide and strict absence of anhydride epimerization led to the highest values.
Proper choice of the catalytic setup and the borane stoichiometry allows tuning this approach to yield polyester-polyether block copolymers in a one-step procedure. This only works if the epoxide homopolymerization kicks in after the anhydride has been consumed quantitatively and no tapering or undesired transesterification occurs. Both Zhao119 as well as Wang and Li120 demonstrated that this can be achieved in a straightforward manner, using mild phosphazene bases and Et3B as catalysts. Thus, using tBu-P1 and Et3B in a 3:1 or 2:1 molar ratio, well-defined blockcopolymers result, whereby the polyester block is generated by alternating anhydride/epoxide copolymerization, followed by epoxide homopolymerization. Albeit polymerization time can be 24 h or longer, this delivered molar masses up to 50 kg mol−1 and narrow distributions (ĐM = 1.03–1.04).119 At equimolar ratio, a tapered sequence distribution if found, as epoxide homopolymerization occurs prematurely. With an excess of borane, a gradient structure is formed. Interestingly, if EO is applied as epoxide component, amphiphilic block copolymers can be obtained this way in a single step,119 while an excess presence of Et3B can deliver statistical distribution of ester functionalities along the polyether chain (“degradable PEG”).121 If multifunctional alcohols are employed as initiators, also, i.e., star-shaped block copolymers can be synthesized.119 The well-defined character of the thus received copolymers is also reflected in macroscopic properties. PPO-b-P(PA-alt-PO)-b-PPO, for example, displays two distinct glass transition temperatures of Tg = −64.8 °C and 54.6 °C. PEO-b-P(PA-alt-EO)-b-PEO, on the other hand, is water-soluble and shows self-assembly behaviour. Using enantiopure (S)-PO, a high degree of isotacticity is found both in the polyester as well as in the polyether block of P((S)-PO)-b-P(PA-alt-(S)-PO)-b-P((S)-PO), indicating selective attack at the methylene moiety of the monomer (Scheme 12).120 Coherently, 13C NMR analysis also showed that head-to-head and tail-to-tail linkages were absent from the polyether blocks.
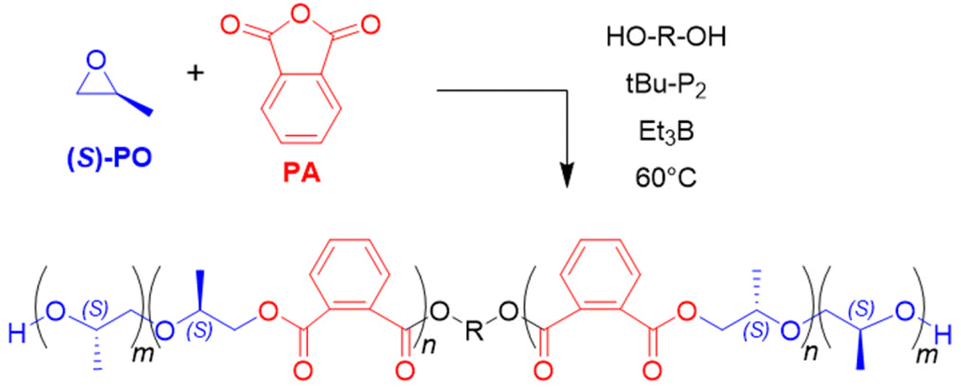 | ||
| Scheme 12 One-step preparation of P((S)-PO)-b-P(PA-alt-(S)-PO)-b-P((S)-PO) triblock copolymer. | ||
Xiao and Meng,122 Liu, Kang, and Li123 as well as Gnanou and Feng124 extended this principle to the one-pot, one-step terpolymerization of epoxide, anhydride and CO2, resulting in polyester-block-polycarbonate structures. According to the authors, this was the first time this feature was achieved in a metal-free manner. Thus, using a setup of CHO/PA/PPNCl/Et3B = 500:50:1:0.5 molar ratio under carbon dioxide pressure of 1 MPa (80 °C), it was found that in the initial phase of the copolymerization almost only polyester was formed.123 Once about 95% pf the PA was consumed, the polycarbonate formation ensued via the copolymerization of the epoxide with CO2 (Scheme 13). Consequently, the formed diblock copolymer showed only very little tapering and well-behaved characteristics (no polyether linkages, ĐM = 1.10). By adding the CO2 only after full PA consumption, perfect copolymers without any tapering were received.123,124
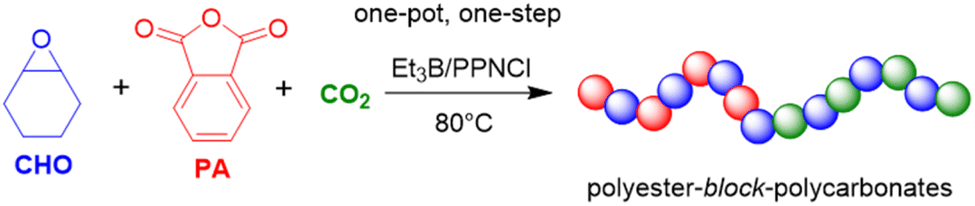 | ||
| Scheme 13 Et3B-cocatalyzed terpolymerization of epoxides, anhydrides and CO2. | ||
In full accordance with the previous discussion on epoxide/CO2 and epoxide/anhydride copolymerization above, also in this case a pronounced dependency on the type of epoxide and catalyst stoichiometry is found. Hence, under identical conditions, BO is only converted into the corresponding polyester and cyclic carbonate, while PO is still successfully used as comonomer to yield the diblock structure.123 A modified setup of CHO/PA/PPNCl/Et3B = 500:50:1:2 resulted in almost pure polycarbonate. Mechanistically, Et3B-activation of the epoxide is proposed, and it is noted that CO2 enchainment only becomes competitive once the carboxylate anion is not the dominant propagating species anymore (that is, in the (almost) absence of PA monomer). This is also supported by DFT calculations and frontier orbital analysis of the attack of the CHO-derived propagating oxyanion on PA and CO2, respectively. This characteristic can also be used to prepare random structures: if the feed contains very little anhydride component, a polycarbonate will form which contains distributed ester functionalities along the chain.124 A quaterpolymer using such an approach (CHO, PO, PA, CO2) delivered the corresponding polyester-block-polycarbonate, whereby the resulting material was found to have a notably high transparency.122
Given the chemoselective properties of the Et3B/organobase catalyst pair when cyclic esters are copolymerized with epoxides (see above, i.e.Scheme 10), also other types of terpolymerization are possible, resulting in intriguing polymer architectures from one-pot mixtures. In 2020, Wang and co-workers employed PO, PA and rac-lactide (rac-LA) to prepare triblock, pentablock or tapered pentablock terpolymers (Scheme 14).125 These copolymer structures were received from the same monomer mixtures in each case (PO/PA/rac-LA = 500:100:100, H2O as initiator) and selectivity was simply determined by the Et3B/DBU ratio. For the 1:1 ratio, the strictly alternating copolymerization of epoxide and anhydride component to yield polyester is followed by a second polyester block consisting of lactide repeat units while the excess PO remains untouched. If more borane is present (2:1 ratio) then an additional final PPO block will be formed. Thus, under these conditions the preference of the catalyst system is PO/PA copolymerization > rac-LA homopolymerization > PO homopolymerization. It should be noted that this example underlines the optimum borane loading for cyclic ester or epoxide selectivity is dependent on the chemical nature of both and potentially the pKa of the organobase (compare Scheme 10 for an example addressing VL and PO with tBu-P2 as organobase, respectively).63,126 If the excess of borane is further enhanced, then the expected increase of PO conversion and decrease of lactide conversion rate entails a tapering of the block structure (3:1 ratio). Similarly, the repeated addition of batches of such three-component monomer mixtures can lead to even larger multiblock copolymers, for example hepta- or undecablockcopolymers.126
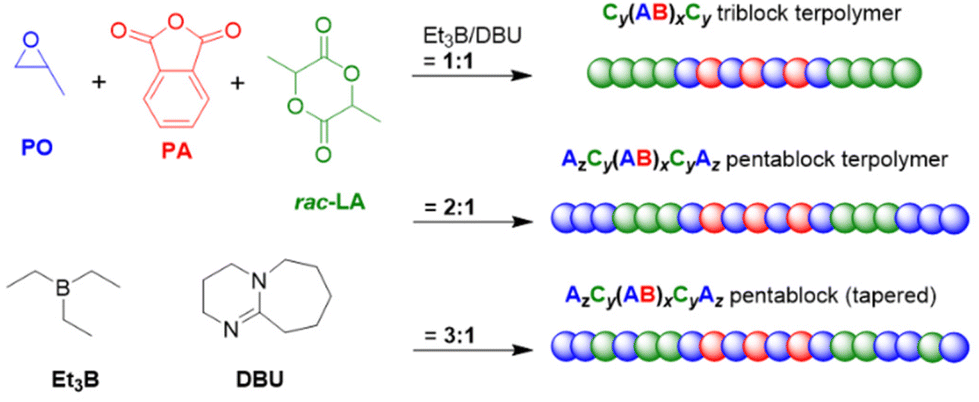 | ||
| Scheme 14 One-pot terpolymerization resulting in different polymer architectures depending on catalyst stoichiometry. | ||
Wang and co-workers also succeeded in the preparation of well-defined triblock copolymers (ĐM < 1.20) from mixtures of epoxides, anhydrides, rac-LA and vinyl monomers (such as styrene or methyl methacrylate).127 In this case Et3B/DBU was employed alongside a specific RAFT transfer agent (trithiocarbamate with a carboxylic group) so that the living radical polymerization of the vinyl monomer occurred alongside the ROP. The action of the borane was unperturbed by the presence of radical species (60 °C). The transfer agent thereby bridges the catalytic cycles and ensured the connection between the polyvinyl block and the two polyester blocks of the resulting P(vinyl)-b-P(anhydride-alt-epoxide)-b-P(rac-LA) polymer. Several different epoxides and anhydrides were successfully converted in this approach.
The direct copolymerization of L-LA and EO was described in 2019 by Gnanou and Feng.128 As discussed above, lactones such as CL or VL and various epoxides can be polymerized to yield exciting and well-defined multiblock structures, whereby the organobase/Et3B ratio controls which of both types of monomer is enchained.63 While block-like architecture is useful in many ways, in this work the statistical incorporation of the lactide in a PEO chain was achieved. Using five eq. of Et3B relative to the Lewis base and toluene as solvent, indeed the successful formation of copolymers was observed, as substantiated by careful NMR investigations. The copolymerization parameters were determined using an integrated, non-terminal approach. Using this excess of borane, the setup is clearly biased for epoxide polymerization and only relatively low degrees of ester content can be achieved (2–14%). Nonetheless, this method provides for a metal-free access to “degradable PEG”, including semi-crystallinity with a melting point depending on the ester content. It is currently not clear whether this strategy can be extended to other epoxides or cyclic esters. Recently, a statistical copolymer of VL and PO with high ester content was described by Feng and Gnanou, using Et3B/tBu-P4 (2:1 eq.) in the presence of ethylenediamine as additive (0.5 eq.).113 NMR investigations clearly demonstrated the statistical monomer incorporation of the well-defined polymer (ĐM = 1.1). Most likely the amine activates VL via H-bonding, thus partially compensating for the epoxide bias of the setup (PO is still incorporated somewhat faster).
Relatedly, Et3B has also been used to prepare “degradable PEG” via the polymerization of the cheap cyclic resource ethylene carbonate.129 Here, predominant decarboxylation during the propagation step entails the formation of a polyether main chain along which degradable carbonate functionalities are distributed.
Other monomers
Other types of comonomer have also been copolymerized with epoxides using Et3B-catalysis. In 2021, Zhao and co-workers described a new approach to generate main chain sulfur-containing polymers.130 This was achieved by copolymerizing epoxides with isothiocyanates in a strictly alternating manner, using a diol initiator and [tBu-P2] > [Et3B]. Interestingly, the resulting polythioimidocarbonates (Scheme 15) are free of ether linkages and also the formation of cyclic by-products (via backbiting) is suppressed, providing high molar masses and a narrow PDI (Mn > 50 kg mol−1, ĐM < 1.15). Crucially, once the catalyst stoichiometry is inverted ([Et3B] > [tBu-P2]), copolymerization ceases and epoxide homopolymerization is selectively switched on. DFT calculations suggest that an attack of the propagating oxyanion on the isocyanate is both kinetically and thermodynamically favoured by a low energy barrier and a considerable gain in Gibbs free energy, while the corresponding attack on the epoxide is accompanied by a significantly higher barrier. This is in agreement with the observed alternating copolymerization. While the protonated organobase is important in stabilizing further propagation, the role of Et3B is mainly to prohibit undesired backbiting, raising the energy barrier for this step to 38.5 kcal mol−1. If [Et3B] is then further increased, all alkoxides chain ends are borane-capped and cannot react anymore with the isocyanate. Epoxide homopolymerization then occurs, accordingly.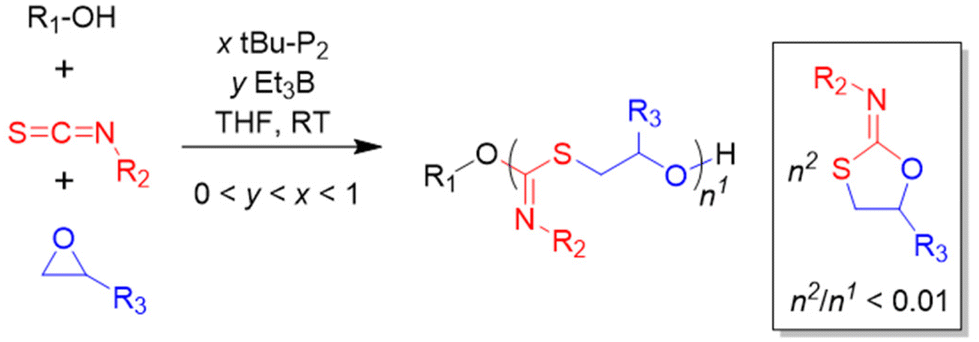 | ||
| Scheme 15 Alternating copolymerization of epoxides and isothiocyanates to yield polythioimidocarbonates. | ||
These desirable features were then used to construct block copolymers via in situ catalyst switches, i.e., PPO-b-P(PhITC-alt-PO)-b-PPO or P(PhITC-alt-PO-b-PPO-b-P(PhITC-alt-PO), whereby PhITC denotes phenyl isothiocyanate.130 Strikingly, in the presence of anhydride monomer (PA/PhITC/PO/-OH/tBu-P2/Et3B = 50:50:250:2:0.5:0.3), the one-step synthesis of well-defined block terpolymers succeeds. Starting from a diol, first the alternating copolymerization of anhydride and epoxide occurs, which is then followed by epoxide/isothiocyanate copolymerization, yielding P(PhITC-alt-PO)-b-P(PA-alt-PO)-b-P(PhITC-alt-PO) in 30 h reaction time.
In 2021, the copolymerization of several epoxides with an electron-poor isocyanate (p-tosyl isocyanate, TSI, Scheme 16 top) was described.131 With this specific monomer and under optimized conditions, it is possible to prepare the corresponding polyurethane, whereby neither allophanate nor isocyanurate is found as impurity. Also, the selectivity for linear polymer can be >99%, demonstrating that the formation of the cyclic oxazolidinone is suppressed, most likely because Et3B-capping of the propagating species disfavours backbiting. Further, selectivity for urethane formation of > 99% regarding the competing imidocarbonate motif can be achieved. Polyether linkages do not occur to a relevant degree, underlining the strictly alternating monomer incorporation, delivering well-defined polymer (1.1 < ĐM < 1.3). Depending on the concentration of Et3B, polymerization temperature and epoxide monomer, also higher (or even predominant) imidocarbonate content can be realized. Thus, importantly, the microstructure of the polymer can be tailored to some degree. A current limitation seems to be the strict requirement for a very electron-poor isocyanate. With hexyl isocyanate, only trimerization (and epoxide homopolymerization) is observed. This was attributed by the authors to the high reactivity of the isocyanate functional group, which means that Et3B-capping of the propagating species is not sufficient to prevent trimerization.
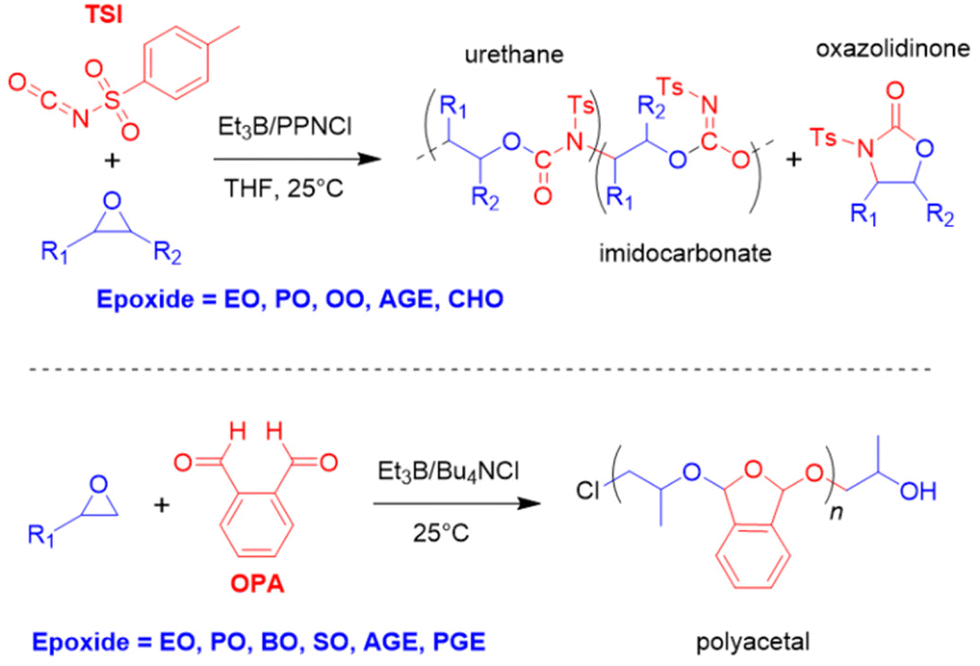 | ||
| Scheme 16 Top: borane-mediated copolymerization of epoxides and an electron-poor isocyanate and resulting motifs. Ts = tosyl. Bottom: alternating copolymerization of o-phthalaldehyde and epoxides at room temperature. | ||
In 2022, Feng, Gnanou and co-worker described the copolymerization of various epoxides with o-phthalaldehyde (OPA) to result in the corresponding polyacetals.132 Using Bu4NCl/Et3B (1:2 eq.), a relatively swift and well-controlled monomer conversion (ĐM = 1.02–1.17) was found, resulting in a microstructure with a high degree of alternating monomer incorporation (92–99%, Scheme 16 bottom). Strikingly, these reactions succeeded at room temperature, a notable difference to the typically low polymerization temperatures used for the homo- or copolymerization of this monomer (−78 °C/−36 °C). This can be most likely related to favourable polymerization kinetics, which quickly trap OPA repeat units by ring-opening of the activated epoxides. The range of suitable epoxides is broad, encompassing EO, PO, BO, OO, SO, AGE and PGE. These polymers are acid-degradable yet thermally stable up to 300 °C and display glass transition temperatures between 5.6 °C (poly(OPA-alt-OO)) and 65.7 °C (poly(OPA-alt-SO). Polymerization with an excess of PO delivered a tapered copolymer, where a segment with alternating microstructure was followed by ether-rich segments with randomly occurring acetal functionalities and a terminal pure polyether block. No polymerization was observed when iBu3B or Ph3B were employed as catalysts.
Modified boranes
Above examples underline the capabilities of Et3B to advance epoxide homo- and copolymerization, enabling fast reactions, well-defined and novel polymer architectures and a broad functional group tolerance. Nonetheless, Et3B is not without downsides. It has to be handled in solution, which in case of high borane loadings can introduce notable amounts of solvent into the polymerization reaction. The pyrophoric characteristics, its moderate toxicity and a certain degree of volatility (boiling point: 95 °C) means it has to be handled with care. Most importantly perhaps, it has been found that in the presence of a large excess of –OH species, epoxide homopolymerization is slowed down massively.98 This is important since such conditions are frequently employed on larger scale or in industrial alkoxylation reactors as a safety measure, where usually the monomer is gradually added to a mixture of catalyst and initiator (the latter almost always alcohols). Also, if oligoether products are targeted, similar unfavourable conditions for Et3B may apply. Finally, it should be noted that the usually invoked polymerization mechanism for epoxide homopolymerization requires an encounter of borane-activated epoxide and the borane-capped propagating chain end. Such a setup is vulnerable to dilution (i.e., very low catalyst loadings), in analogy to what is characteristic for Lewis pair polymerization (LPP).85 The corresponding zero order kinetics in epoxide monomer, indicating that only activated monomer reacts (the concentration of which is constant), has indeed been observed in some cases (see below).In sum, for the purpose of epoxide homo- and copolymerization, Et3B is a well available but not an optimized structure. The question how to modify it for further improved catalyst performance will be the subject of the following sections.
Trialkyl- or triaryl monoboranes
A first and obvious step to adapt the borane co-catalyst, regarding both its electron deficiency and steric demand, is an exchange of its ethyl moieties for other alkyl chains or aryl groups. Thereby, Lewis acidity of borane compounds is most often discussed by reporting the respective acceptor number (AN), determined via31P NMR (triethyl phosphine oxide as probe molecule, Gutmann–Beckett-method), which in the case of cationic epoxide polymerization scales qualitatively with the rate of conversion.133 For the polymerizations discussed in this review, which are more anionic in character, the situation is less clear-cut; a well-behaved correlation has not always been found in the studies which have specifically looked at this feature (see discussion below). This may be down to the notorious difference between Lewis acid strength and Lewis acid effect, which cannot be fully assessed by the Gutmann–Beckett approach alone.134 Nonetheless, excessively high or very low Lewis acidity has repeatedly been shown to quench polymerization activity, while moderate Lewis acids (comparable to Et3B) are usually the ones with superior performance. To put this into perspective, Table 1 lists AN values obtained for typical borane Lewis acids and some of the more complex borane catalysts described in detail further below.| Compound | AN | Solvent | Ref. |
|---|---|---|---|
| a See Scheme 22 for structure and full catalyst design. | |||
| BBr3 | 109.3 | Neat | 133 |
| BCl3 | 105.7 | Neat | 133 |
| BF3 | 88.5 | Neat | 133 |
| (C6F5)3B (BCF) | 76–81 | C6D6 | 72 |
| Ph3B | 66–73 | C6D6 | 72 |
| Ph3B3O3 (TPBX) | 49.1 | THF-d8 | 135 |
| Et3B | 24.1–30.3 | C6D6 | 136 and 137 |
| Borinanea | 23.4 | CD2Cl2 | 154 |
| BBNa | 21.7 | CD2Cl2 | 154 |
| Cy2Ba | 18.8 | CD2Cl2 | 154 |
| (OEt)3B | 17.1 | Neat | 133 |
| Bpina | 15.5 | CD2Cl2 | 154 |
As one of the closest Et3B-analogues, Bu3B, which is also commercially available, has been employed in some cases and generally shows a very comparable behaviour to Et3B;95,97 Zhang and co-workers noted that it may be beneficial on account of its increased size to further suppress side reactions when cyclic by-products can be formed via back-biting (i.e., copolymerization with CO2).89 Tri-sec-butylborane has been tested for the copolymerization of epoxides with TSI for polyurethane formation. This compound is a weaker Lewis acid than Et3B and its application resulted in a significantly reduced yield and selectivity.131
More interest was directed at derivatives with electron-withdrawing substituents. For example, in 2019 Kerton employed Ph3B and BCF to study the reaction of several epoxides with CO2.138 The authors found that under conditions where Et3B delivers strictly alternating polycarbonates (compare Schemes 1 and 2) both aryl boranes are more suitable to yield the cyclic carbonates. The latter reaction is achieved with high TON and TOF, clearly outperforming other organocatalysts. Conversion is somewhat slower for BCF, which was attributed to a stronger association between the borane and the nucleophile (chloride or DMAP). When the borane loading is increased relative to the nucleophile concentration, a mixture of cyclic product and polymer is received. Under optimized conditions, polycarbonate with >99% carbonate linkages is received when using CHO/CO2. No conversion (Ph3B) or exclusive polyether formation (BCF) is observed in the case of PO/CO2 copolymerization. This was later substantiated by Zhang and co-workers who also found that Ph3B does not produce polymer when applied to PO/CO2.89
The group of Kerton also succeeded in the preparation of block copolymers using a two-step process (Scheme 17).139 Thus, Ph3B/PPNCl (1:1) was applied to a mixture of epoxide and anhydride (5:1, CHO and PA, neat) and held at 100 °C for 30 min. After cooling to room temperature, 40 bar of CO2 pressure was used for 24 h (60 °C). The formation of a polyester-block-polycarbonate diblock structure was supported by 1H DOSY NMR analysis. Notably, in the first step, after full consumption of the anhydride component, no polyether formation is observed in spite of the excess of epoxide. This is in accordance with the inability of Ph3B to catalyse epoxide homopolymerization, both on its own75 as well as under conditions where Et3B delivers well-defined polyethers.98,99 However, most likely on account of its higher Lewis acidity compared to Et3B, the reaction has to be stopped timely after anhydride consumption, since otherwise severe transesterification will occur (which does not readily happen for Et3B). Both the polyester and the polycarbonate formation, under the conditions employed by Kerton and co-workers, cannot be catalysed by BCF. However, interestingly, the latter is able to selectively depolymerize the polycarbonate block (130 °C in CH2Cl2) of the polyester-block-polycarbonate structures whereby selectively cis-cyclohexene carbonate is produced while the polyester block remains remarkably untouched.139
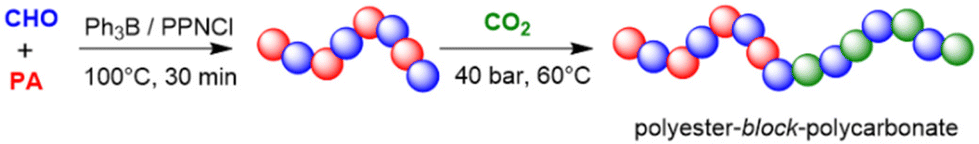 | ||
| Scheme 17 Ph3B as employed for block copolymer preparation in a two-step, one-pot strategy. | ||
Relatedly, Zhang and co-workers compared four different boranes (Et3B, diethylmethoxyborane (Et2(OMe)B, Bu3B and Ph3B) regarding the copolymerization of anhydride (SA) and epoxide (PO).116 Interestingly, in all cases the desired poly(propylene succinate) resulted, whereby the observed TOFs were 10 h−1, 9 h−1, 7 h−1 and 5 h−1, respectively, while the found ester selectivity (over ether formation) was 97%, 94%, 88% and 85%, respectively. Thus, both activity and selectivity are lower for the more Lewis acidic Ph3B compared to Et3B. Similar was reported by Li,117 Wang, Li and co-workers115 as well as Zhang.114
In sharp contrast, the aforementioned copolymerization of the electron-poor isocyanate TSI and PO to yield polyurethanes was rapid and strongly exothermic in the presence of Ph3B.131 Still, selectivity was retained to yield a strictly alternating monomer incorporation. TOFs of over 10000 h−1 and molar masses up to 225 kg mol−1 were achieved this way. The reaction was completed much faster than in the presence of Et3B.
BCF has been found to be able to homopolymerize PO, but the resulting material is typically regioirregular as discussed by Chen and co-workers in 2003 (showing undesired head-to-head and tail-to-tail linkages, entailing a mixture of primary and secondary hydroxyl end groups).75 The presence of an excess of alcohol initiator is necessary, otherwise isomerization of the monomer to propionaldehyde occurs, but such conditions simultaneously enforce relatively low molar masses. As a minor impurity, also cyclic PPO was observed; in later work by Barroso-Bujans the formation of polyether macrocycles, using glycidyl ether monomers, via application of BCF in the absence of protic initiators was further substantiated.140 BCF has also been used to increase the primary hydroxyl content of polyether polyols,141,142 a feature which is desirable for further reaction but not readily realized with i.e. DMC catalysis.
In general, epoxide polymerization catalysed by BCF is hard to control, which can be attributed to the polymerization mechanism: the strongly electrophilic borane will acidify involved R-OH functionalities, engendering a cationic polymerization mechanism, including elements of epoxide activation by coordination between the oxirane oxygen atom and the Lewis acid. The addition of Ti(OiPr)4143 or tributylamine/pyridine144 seems to improve control over the polymerization. BCF has also been employed in the copolymerization of vinyl ethers and isobutylene oxide (IBO).145 While not strictly a monoborane structure, the related triphenylboroxine (TPBX) has also successfully been applied to polymerize epoxides (TPBX/PPNCl = 2:1, 0–25 °C).146 The catalyst structure seems to be a challenging one as substitution reactions by the alkoxy anion with the phenyl moieties or even the B3O3 ring can occur. Accordingly, at lower temperatures the polymerization is more controlled, yet overall TPBX and versions thereof seem to be less robust and less active than Et3B.
Bifunctional mono-, di-, tri- and tetranuclear organoboron catalysts
A step-change in borane catalyst design for epoxide (co)polymerization occurred in 2020, when Wu and co-workers started to publish a series of papers describing novel, bifunctional borane catalysts (Fig. 4).147 A central idea to this design was the spatial proximity of the active centres, which can be guaranteed and tuned by linking them together via a suitable backbone, this way circumventing the unfavourable entropic situation with binary catalyst setups. This is of obvious benefit considering the polymerization mechanisms discussed above, where often two equivalents of borane are involved. Moreover, the introduction of an ammonium functionality in the backbone engenders bifunctionality and includes a nucleophilic counter ion (i.e., halide). Interestingly, this nucleophile is stabilized both by coulombic interaction with the positively charged nitrogen atom and by a (weak) dative coordination to the boron atom. This dynamic situation is even more pronounced if more than one boron functionality is part of the catalyst structure and it displays a specific profile in 11B NMR analyses, were broadened signals are found. Overall, this catalyst structure provides a multitude of tuning sites (Fig. 4, right). This includes (a) the number of boron centres and the electronic situation (degree of Lewis acidity) found for them, (b) steric hindrance around the ammonium motif, (c) type of nucleophilic anion and its interactions with ammonium and boron atom(s) and (d) linker length L (number of –CH2– units) between the nitrogen atom and the boron atom. This modular toolbox of catalysts can be further extended if, for example, nitrogen is exchanged for phosphorous (see below).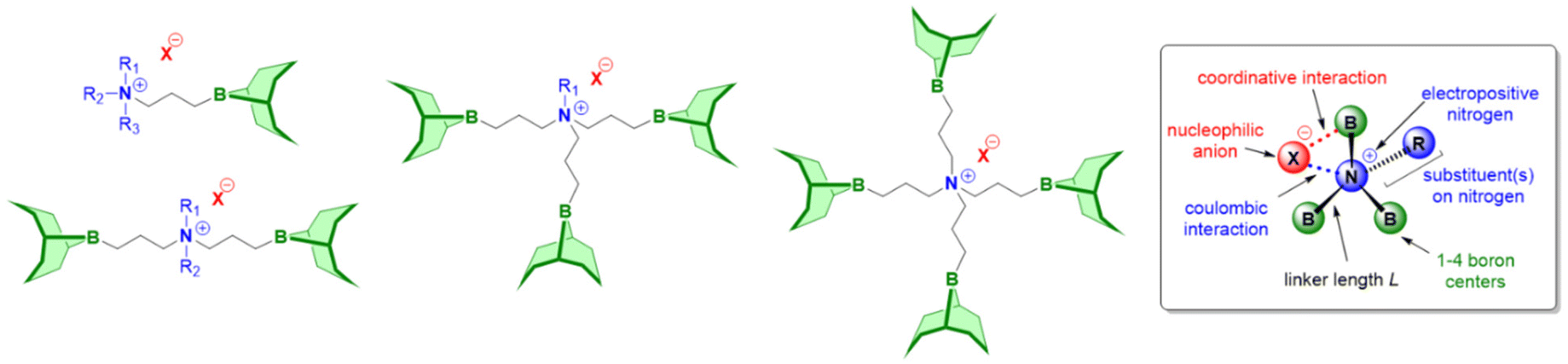 | ||
| Fig. 4 Modular and bifunctional mono-, di-, tri- and tetranuclear organoboron catalysts as reported by Wu and summary of tuning sites (insert). | ||
While these catalysts are obviously not commercially available, in contrast to the ones discussed previously, it should be stressed that their synthesis is easy and usually conducted in two high-yielding steps (reaction of a suitable amine with alkenyl halides to prepare the quaternary ammonium salt, followed by hydroboration using i.e. 9-BBN). Notably, some of the corresponding syntheses have been realized on the kg-scale.148 The catalyst class is described as white solids which can be stored for months under protective conditions. A certain sensitivity to oxygen is mentioned.147
A first focus of application for these catalysts was the reaction between epoxides and carbon dioxide.148,149 Here, a series of monoboron organocatalysts was applied to convert CHO and CO2 into the corresponding polycarbonate. This worked out very successfully, whereby >99% polymer selectivity (relative to the cyclic carbonate) and >99% carbonate linkages were found under relatively mild conditions (80 °C, 15 bar CO2). An odour- and colourless polymer was received via a single precipitation from ethanol.
Interestingly, a variation of L under otherwise identical structural features of the catalyst revealed that an increase from 3 up to 7 (heptamethylene, –(CH2)7–) proved beneficial, whereby a linker length of 5 displayed the highest TON (1550 compared to 330 for –(CH2)7–). Also, choice of the halide had a notable impact, as evident form the observed TOFs of 517 h−1 (bromide), 367 h−1 (chloride) and 317 h−1 (iodide). The three alkyl groups on the nitrogen had, in this case, only a very marginal influence on catalytic performance (comparing triethyl, tri-n-propyl and tri-n-butyl). An optimized structure was thus found in catalyst 1 (Scheme 18). A screening of reaction conditions showed that no relevant dependence on CO2 pressure exists, while the polymerization temperature has a massive impact. Thus, a series of experiments ranging from room temperature up to 150 °C showed that the TOF increases with higher temperature, to reach 4900 h−1 at 150 °C. This certainly underlines the thermal stability of 1, yet importantly, also the selectivity remained high (no cyclic by-product under these extreme conditions and still >99% carbonate linkages). Very low catalyst loadings could be applied (0.005 mol%), resulting in a TON of 13000 and catalytic efficiency of 5 kg polycarbonate per gram of 1. According to the authors this was a record achievement, unprecedented both regarding previous organocatalysts (including Et3B) but also metal-based setups.
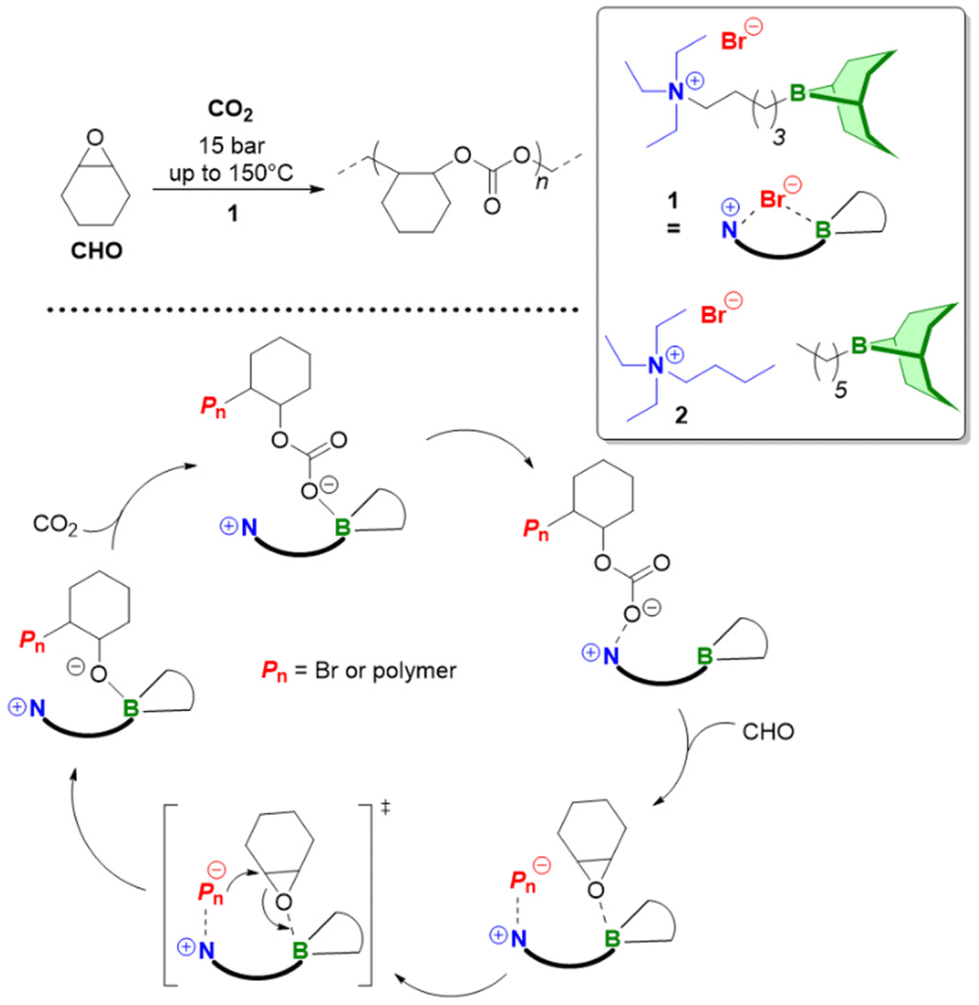 | ||
| Scheme 18 Polycarbonate formation from CO2 and CHO using a bifunctional monoborane as described by Wu. | ||
Perhaps especially interesting, also mechanistic investigations were conducted. Thus, kinetics revealed a first-order dependence in 1 as well as in CHO, while carbon dioxide consumption showed zero-order behaviour and was thus not considered the rate-determining step. Overall these results indicated the involvement of only one catalyst molecule in the rat-determining step, which was also confirmed by DFT calculations. Strikingly, control experiments with the binary analogue (2) of 1 suggested a strong synergistic effect between the Lewis acidic boron and the ammonium moiety: control experiments under identical conditions showed that 1 delivers a TOF that is hundredfold higher. Once the composition of 2 was adapted to 2 eq. of borane versus ammonium salt, performance went up (from 5 h−1 to 330 h−1) but still remained clearly inferior to 1 (4900 h−1). This was attributed to the enforced proximity of both functionalities in 1, which was also reflected in the respective 11B NMR analysis: while 2 showed a well-defined signal (including in the presence of ammonium species), 1 displayed a broad, weakly expressed signal. This was interpreted as supporting the intramolecular interaction of ammonium salt and boron atom (via the halide, Scheme 18). Based on these findings, a polymerization mechanism was proposed, whereby the synergistically confined Br− attacks CHO to form a stabilized alkoxides species, followed by CO2-insertion in the boron-alkoxide bond. The thus formed weakly nucleophilic carboxylate would dissociate from the boron and interact with the positively charged ammonium moiety, hence opening a coordination site for the epoxide. The catalytic cycle is closed by attack of the carboxylate on the activated CHO (Scheme 18).
As a limitation of the described catalyst, other epoxides like PO do not yield polycarbonate but the five-membered cyclic carbonate instead. However, this property was exploited to develop a modified catalyst with a very high activity for this kind of reaction.149 Under optimized conditions, TOFs of up to 11000 h−1 could be realized, whereby >99% selectivity for propylene carbonate was observed. A catalyst screening revealed that iodide was the best choice for the nucleophilic anion component, plausibly explained by its excellent leaving group properties, thus facilitating ring-closing (“back-biting”). Further, it was found that L = 3 delivered best results, in contrast to what was found for CHO/CO2 copolymerization. Interestingly, the 9-BBN derived catalyst proved superior to boron substituted with cyclohexyl groups or pinacol-based ligands. This methodology was extended to other epoxides (EO, BO, HO, SO, various glycidyl ethers), rendering the developed catalysts one of the most active and broadly applicable metal-free ones for cyclic carbonate preparation.
In 2021, the same group reported the selective and high-performing copolymerization of the electron-poor ECH epoxide monomer with CO2, using a borane co-catalyst bearing four flexible arms connected to a central ammonium motif (3, Scheme 19).150 With this catalyst, under mild conditions (25–40 °C, 25 bar CO2 pressure), a perfectly alternating polycarbonate was received, displaying >99% carbonate linkages and >99% selectivity for polymer formation over the cyclic product formation. The latter is very remarkable, since ring-closing is strongly favoured by the electron-withdrawing substituent on the epoxide. Extensive kinetic experiments revealed a zero-order dependence on CO2 and first-order kinetics in both 3 and ECH, suggesting that the rate-determining step is epoxide ring-opening and that one catalyst molecule is involved in the propagation step. Under optimized conditions, molar masses (Mn) of up to 36.5 kg mol−1 (ĐM = 1.22) were achieved, which according to the authors is the highest value obtained so far for the corresponding poly(chloropropylene carbonate). The resulting polymers offer exciting venues for i.e. postfunctionalizations.
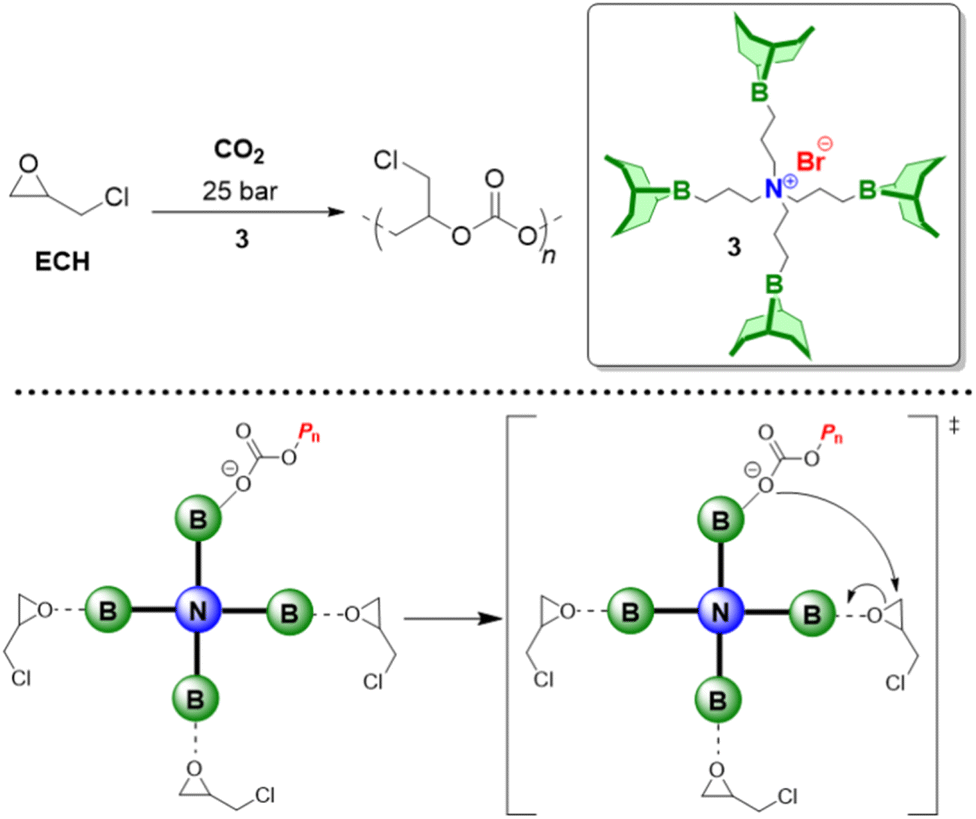 | ||
| Scheme 19 Copolymerization of ECH and CO2, using a tetranuclear organoboron catalyst. Bottom: detail of the polymerization mechanism. | ||
The catalyst structure was absolutely instrumental for achieving this performance. Control experiments using analogous tri-, di- and monoboranes as well as binary setups highlighted the superiority of 3. For example, using the analogue with three boron-bearing arms, only 76% polymer selectivity and 95% carbonate linkages were observed. Using the dinuclear compound, these values further shrank to 40% and 77%, respectively. Applying the monoboron catalyst, the activity was comparable to 3, yet as a product exclusively the cyclic, five-membered carbonate was received. The high density of Lewis acidic functionalities per nucleophile is accordingly crucial for maintaining selectivity and fast polymerization, most likely because the local density of activated epoxide in proximity to the boron-capped propagating chain-end is increased. The authors proposed an intriguing, cyclically sequential copolymerization mechanism, where essentially the propagating chain end, after CO2 insertion, can easily attack a neighbouring borane-activated epoxide monomer, thus “walking” along the four arms of the catalysts (Scheme 19).
In 2022, Feng, Gnanou and co-workers introduced the borinane motif for generating highly active catalysts for epoxide conversion. In addition to epoxide homopolymerization (see below), these are also powerful in catalysing the copolymerization of CO2 with various epoxides.151 Especially in combination with a bulky environment around the quaternary nitrogen atom and L = 5 (4, Scheme 20), well-defined polycarbonates are received from epoxides such as EO, PO, BO, OO or AGE and even oxetanes can be used as feedstock. For CHO, a monomer very well suited for this type of copolymerization, the productivity could be pushed up to 5.7 kg of polymer per gram of 4, a record value. While the same polymerization mechanism as proposed for, i.e., 1 (Scheme 18) is assumed, the borinane motif, where the boron atom is part of a six-membered cyclic ring, enhances reactivity by reducing the steric hindrance around the Lewis acid as compared to 9-BBN-derived catalysts. This has a subtle but crucial impact on activation energies, as suggested by DFT calculations.
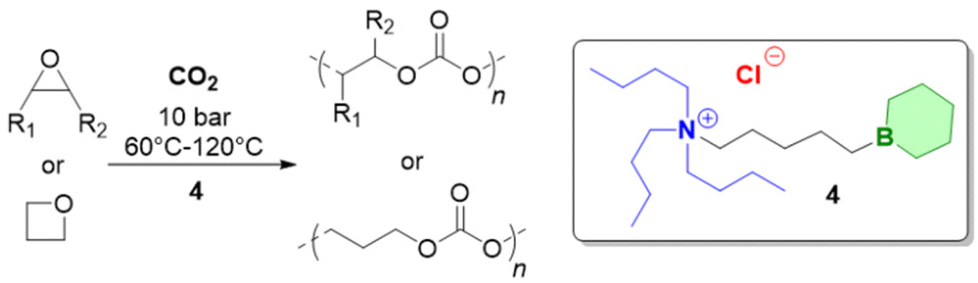 | ||
| Scheme 20 Borinane-type catalyst for polycarbonate production. | ||
The bifunctional, multinuclear borane catalysts were also applied as a powerful tool for the preparation of polyethers. Wu and co-workers employed a catalyst bearing two 9-BBN-derived borane functionalities (5) to realize an organocatalytic setup with unprecedented activity for epoxide homopolymerization.152 For example, 3000 eq. of PO are converted quantitatively in less than 6 h reaction time, whereby a narrow molar mass distribution is maintained. Using 30000 eq. of PO, monomer consumption is complete after 60 h, yielding PPO with a molar mass of Mn > 1000 kg mol−1. When using the more reactive EO, 10000 eq. can be converted in 20 minutes (TOF = 30000 h−1), rendering the reaction more than 25 times faster than with PO. Chain extension experiments, kinetic analyses and MALDI-ToF MS investigations underlined the living characteristics of this polymerization. Importantly, zero-order kinetics were found for PO dependence. Perhaps as a notable limitation of this catalyst, reactions had to be typically conducted at −20 °C to 0 °C, whereby higher temperatures quickly led to a loss of control. The exothermic ring-opening in combination with the high reaction rates must be considered.
Mechanistically, the success of 5 was attributed to the close proximity of two borane moieties (the flexible linkers are important in this regard), whereby one boron carries the propagating chain end which can easily attack the activated epoxide sitting on the other (Scheme 21). The quaternary nitrogen supports the propagation step by stabilizing the alkoxide chain-end and facilitating the transit to the second borane functionality. Indeed, control experiments using an analogous diborane with a neutral linker (where a pure methylene chain connects the two boranes) showed clearly inferior performance, thus underlining that also in this case a synergistic effect is operational. Interestingly, DFT calculations suggest that the nitrogen distributes much of its positive charge on the hydrogens of the methylene units in α and β position.
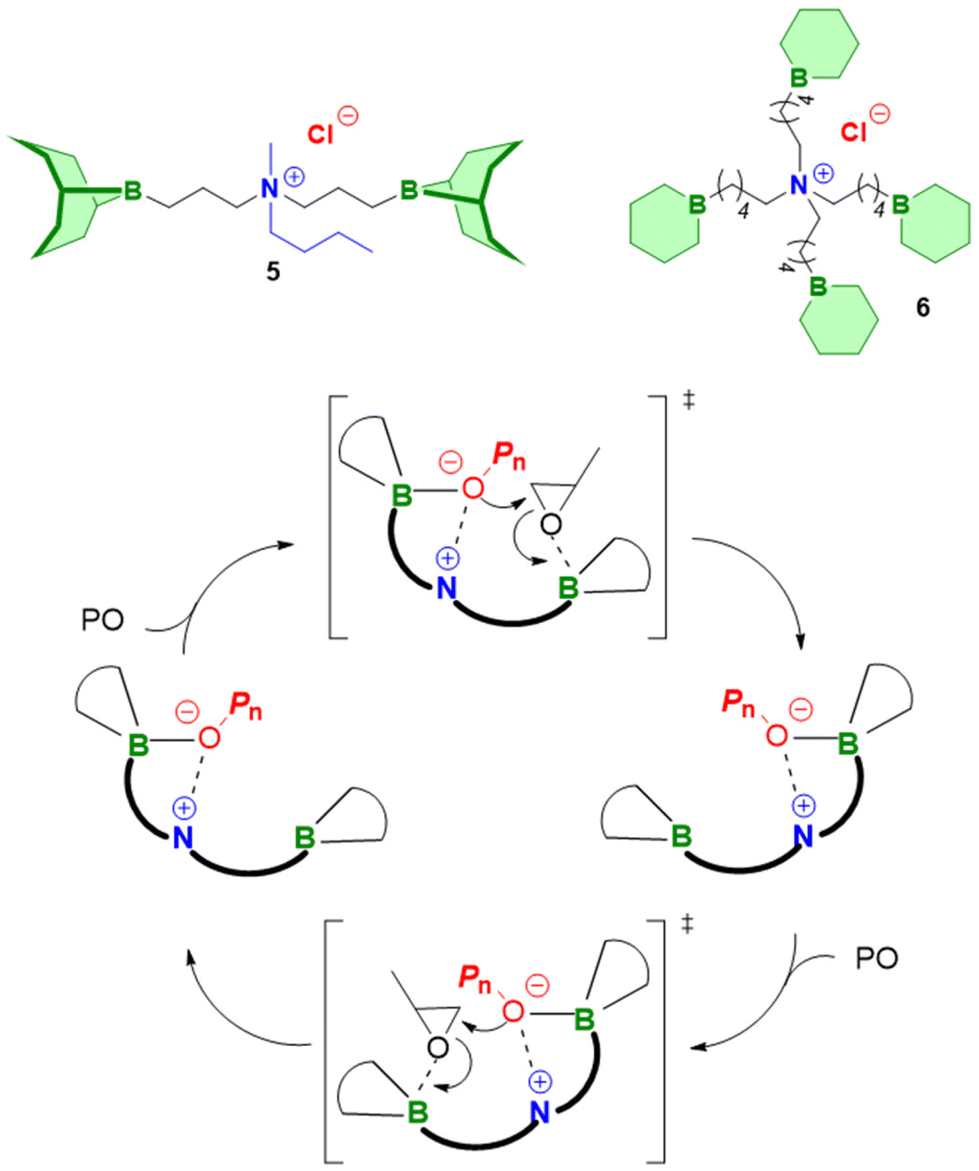 | ||
| Scheme 21 Bifunctional borane catalysts for epoxide homopolymerization and proposed mechanism. | ||
A reduction of steric hindrance around the Lewis acid by using the borinane motif and the application of a four-armed catalyst molecule (6) improved performance even further to achieve spectacular results.153 Thus, in the bulk polymerization of PO, 30000 eq. of the monomer were consumed within 10 minutes reaction time (0 °C). This value of 100% conversion went down respectively when analogous compounds to 6 were employed which possessed three (68%) or two (21%) borane functionalities. Obviously, the number of Lewis acidic moieties concentrated in the catalyst controls performance to a large degree. To put this into perspective, the very rapid PO consumption by application of 6 yields an exceedingly high TOF of 180000 h−1, which is again two orders of magnitude higher than found for 5. Crucially, with this setup very high molar masses can be constructed (Mn > 1000 kg mol−1, ĐM = 1.2) but also low molar mass target polymers or oligomers can be prepared. This is of importance for many polyether applications und usually means that either a high ratio of catalyst to monomer would be needed, or, more practically, that the polymerization is conducted in the presence of a significant excess of alcohol-type chain transfer agents (CTAs). As discussed above, such reaction conditions are problematic for Et3B, were only very slow monomer conversion results. Under application of 6, however, this limitation is removed and a range of alcohols (and water) can be used to precisely control the product degree of polymerization. This can be taken to extremes; for reactive EO, a catalyst loading in the ppb range (and 250000 eq. of CTA relative to the catalyst) can be applied. Within 48 h this leads to 90% conversion, which in turn translates into an average TOF of 187500 h−1 and a TON of 9 × 106. In sum, this means that 1 g of 6 can yield up to 600 kg of well-defined PEG (ĐM = 1.03), easily outperforming the current commercially employed setups. The monomer scope includes, apart from EO and PO, also BO, OO, AGE and PGE. For the glycidyl ethers somewhat reduced TONs and TOFs are observed, yet still the polymerization is very fast and allows for low catalyst loadings and well-defined product polyethers.
Yang, Wu and co-workers set themselves to the important task of systemizing the impact of the various catalyst tuning sites on performance in epoxide homopolymerization.154 They focused on four aspects, namely (a) electronic and steric situation of the borane functionality, (b) steric hindrance around the ammonium nitrogen atom, (c) the B–N distance and (d) the number of boranes in the catalyst molecule.
A first catalyst series targeted monoboron catalysts with boron centres of varying Lewis acidity, using their acceptor numbers as guideline (see also Table 1). A comparison of moieties with decreasing Lewis acid strength, specifically borinane (AN = 23.4) > BBN (21.7) > BCy2 (18.8) > Bpin (15.5), revealed that irrespective of the borane chosen, only negligible conversion of epoxide (PO) occurred. This nicely fits to the results obtained previously for Et3B, where the involvement of excess borane was crucial to achieve monomer activation, since one equivalent of borane (relative to organobase/nucleophile) is consumed for end-capping of the propagating chain. Since the monoboron bifunctional catalysts introduce exactly one boron per halide initiator, the absence of relevant polymerization is coherent.
Next, a similar series of diboron catalysts was investigated with a fixed L = 3 (Scheme 22) under identical reaction conditions (10000 eq. of PO, 6 h, −20 °C). Interestingly, under these conditions the BBN-bearing catalyst achieved 35% conversion and a TOF of 585 h−1. In contrast, the weaker Lewis acidic derivatives having BCy2 or Bpin groups did not show relevant conversion. The somewhat stronger borinane motif, on the other hand, delivered a conversion of 63% in only 2 h. Thus, moderately strong Lewis acidic boron centres seem favourable. It should be noted that the cited acceptor numbers are still low compared to BCF.72 It is expected that a too excessive Lewis acidity would lead to a loss of control over the polymerization, however, to date it is still an open question where the optimum boron Lewis acidity is located exactly. The current state of research suggests that AN = 20–40 may be preferable.
 | ||
| Scheme 22 Dinuclear organoboron catalysts as screened by Yang and Wu for epoxide homopolymerization. | ||
A systematic variation of L = 3, 4, 5 and 6 revealed a stepwise decrease of activity in this order, with an observed TOF of 585 h−1, 380 h−1, 325 h−1 and 113 h−1.154 A very comparable trend was also found for EO monomer (TOF = 24600 h−1, 16150 h−1, 12900 h−1 and 7750 h−1, respectively). Thus, gradually activity of the dinuclear catalysts decreases as the linker length increases. As both borane functionalities have to cooperate in the propagation step, the spatial arrangement is of obvious importance. This is also underlined when focusing on the steric pressure around the nitrogen atom. Thus, with the same borane moiety (BBN) and the same L (3), a dimethyl substitution pattern engenders of TOF of only 285 h−1, which increases to 690 h−1 for dibutyl and jumps to 1205 h−1 if a rigid pentamethylene dialkyl group is installed. Clearly, the steric encumbrance at the nitrogen reduces the available rotational conformations to a degree that is very favourable for the fitting spatial arrangement of the two boron centres. This was further substantiated by analysing the respective crystal structures. The Newman projection revealed that the less active catalysts (i.e., R1 = R2 = methyl) have the borane moieties in anti-positions, while the larger substituents, and especially the pentamethylene ring, enforce a gauche orientation. As the B–N+–B angle (θ) is reduced, the activity of the catalyst grows. This preorganization helps to achieve high catalysts activity. Coherently, computational studies have shown that the energy barrier for adopting an ideal conformation is lowered by a high steric demand around the nitrogen atom.154 Finally, in accordance with previous results, it was confirmed that catalyst activity multiplies with the number of boron groups, that is tri- and tetranuclear structures outperformed the dinuclear ones.
In the course of similar investigations, Wu's catalyst type was also applied with notable success to the copolymerization of epoxides and anhydrides.155 Using CHO and PA as model compounds, a screening of monoboron organocatalysts (L = 3, 4, 5, 7; halide = chloride, bromide, iodide) revealed L = 5 and Cl− (7, Scheme 23) as best suited for a fast reaction and high catalytic activity. Interestingly, the catalyst setup can tolerate temperatures up to 180 °C, which not only entails rapid monomer consumption but also renders the addition of solvent obsolete – the high viscosity at near complete conversion is less of an obstacle at these temperatures. The strictly alternating character of the copolymerization is also retained under such extreme reaction conditions. Molar masses of Mn up to >90 kg mol−1 and a TON of up to 9900 can be achieved under optimized conditions. At very low catalyst loading, at some point residual water and the hard to remove phthalic acid content limit the obtainable molar masses. Nonetheless, according to the authors the cited values are record data for CHO/PA copolymerization, outperforming competing metal-free and metal-based setups. Kinetic investigations find a zero-order dependence on PA, and DFT calculations support that ring-opening of the epoxide (by the carboxylate) is the rate-determining step. Interestingly, 11B NMR analyses confirm several of the key mechanistic steps, including CHO activation by the borane and the reversible coordination of boron and carboxylate.
 | ||
| Scheme 23 High-performing bifunctional boron catalyst for polyester formation from epoxide and anhydride. | ||
Also in 2022, several publications discussed the application of catalysts wherein the nitrogen atom was replaced by a phosphorus atom.156–158 Key rationale was the assumption that the larger phosphorous might offer a different set of sterically demanding substituents, which in turn could impact catalyst performance given the crucial role of the electropositive onium moieties in this type of bifunctional catalyst (see above). And indeed, the phosphonium analogues to Wu's catalysts were found to display even further improved performance, both for epoxide homopolymerization as well as for copolymerizations.
For polyether preparation, Wu157 as well as Zhong and Li156 successfully applied compound 8, which can be prepared in three steps (Scheme 24). At low reaction temperatures (−30 °C to −20 °C), rapid polymerizations ensued, delivering the corresponding α-bromide/ω-hydroxyl PPO, whereby chain extension experiments and narrow molar mass distributions underlined the controlled nature of the process. Under optimized conditions, TOFs of up to 3300 h−1 could be realized, which was about twice the value achieved by a comparable ammonium diborane catalyst. Interestingly, at very low catalyst loadings (PO/8 = 10000:1) molar masses of up to Mn = 490 kg mol−1 and 97% conversion in 6 h were observed.156 Catalyst 8 is also tolerant regarding the use of water as initiator.
 | ||
| Scheme 24 Phosphonium-type bifunctional diborane catalyst for epoxide homopolymerization. | ||
Kinetic experiments found a zero-order dependence with respect to PO concentration. Interestingly, as in the case of nitrogen-based catalysts, a synergistic effect was observed between phosphonium and borane functionalities: employment of a diborane with a neutral methylene linker and Ph4PBr (binary setup) entailed a significantly slower monomer conversion than use of 8. Conversion of BO and AGE also worked, however, monomer consumption was markedly slower in both cases.156
Various monoboron catalysts carrying phosphonium groups were employed for the copolymerization of CHO and PA, screening L = 3–6 and several alkyl- and aryl substituents on the phosphorous atom.157 At 120 °C, it was found that these catalysts delivered well-defined polyester with >99% ester linkages. Longer linker lengths delivered the best results (optimum: L = 5) as did soft alkyl phosphonium species. Regarding activity, an order of (nBu)3P > (nBu)2P(Ph) > (Ph)3P was found with the respective TOFs of 304 h−1, 226 h−1 and 164 h−1 (L = 3, PA/CHO/catalyst = 200:400:1, 120 °C). The latter was attributed to the high steric demand of the phenyl groups, which hamper the halide-phosphorous interaction, in turn strengthening the halide-boron interaction (compare Scheme 18). The catalyst can tolerate reaction temperatures up to 200 °C, whereby a high TOF of >2800 h−1 can be realized.
A direct comparison of P- and N-based monoboron catalysts (9 and 10, Fig. 5) shows that the better performance of the former is down to a shorter induction time followed by a faster propagation rate.15711B NMR analysis, crystal structure analysis and DFT calculations suggest that this improved behaviour can be related to a stronger coulombic interaction between halide and phosphonium, which in turn diminishes the halide-boron interaction, leaving the boranes more freedom to activate the epoxide monomer.
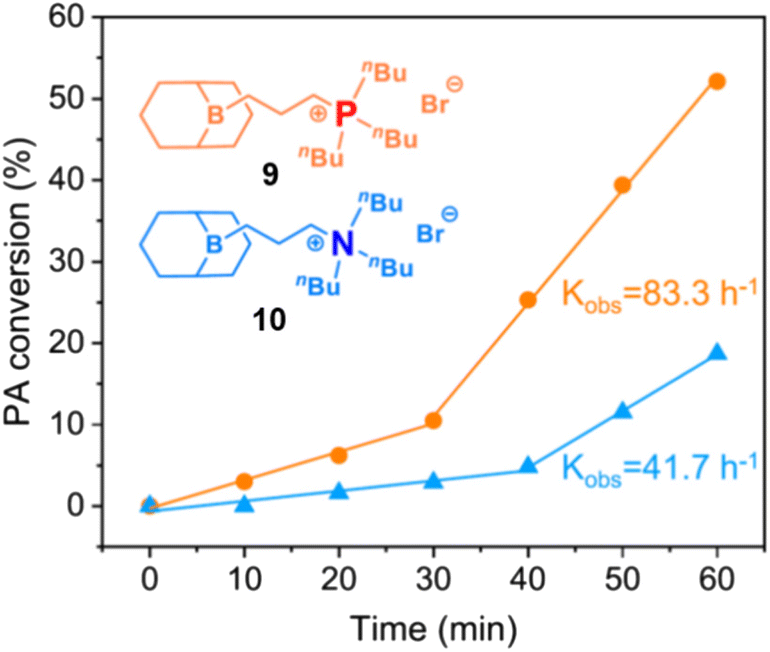 | ||
| Fig. 5 Direct comparison of P- and N-based monoboron catalyst for the copolymerization of CHO and PA (CHO/PA/9 or 10 = 1000:500:1, 120 °C). Adapted with permission from ref. 157. © 2022 American Chemical Society. | ||
Also in the copolymerization of epoxides with CO2 a superior performance of the phosphonium analogues was observed. Thus, with 9/CHO (80 °C, 1.5 MPa CO2 pressure) a TOF of 198 h−1 was found, double that of catalyst 10 under identical conditions.157 Catalyst optimization revealed L = 5 as optimum and it was demonstrated that temperatures up to 150 °C are easily possible, yielding rapid monomer consumption (5210 h−1). High molar masses (345 kg mol−1) and high TONs (up to 7800) were achieved.
Lin and co-workers (of ExxonMobile) deepened the understanding of this reaction by undertaking an extensive mechanistic investigation, screening 35 discrete catalysts (Fig. 6), including not only the quaternary phosphonium moieties, but also tertiary ones (which possess an acidic P–H group).158 Particular attention was paid to the phosphonium substituents (and their effect on the vacant σ*(P-R) orbital, which can interact with the halide), the borane substituents (and ensuing Lewis acidity), the linker length L and thermodynamic and kinetic properties of each catalyst for the copolymerization of CHO and CO2. The considerable amount of data necessary for this endeavour was harvested from in situ Raman spectroscopy. In using a non-isothermal temperature sweep technique, the authors were able to derive the entire kp-versus-temperature profile in one experiment for a given catalyst. The received amount of data was then subjected to Eyring fitting to yield ΔH‡ and ΔS‡. The polymerization rates at 80 °C (2.76 MPa carbon dioxide) were used to compare and rank the catalysts.
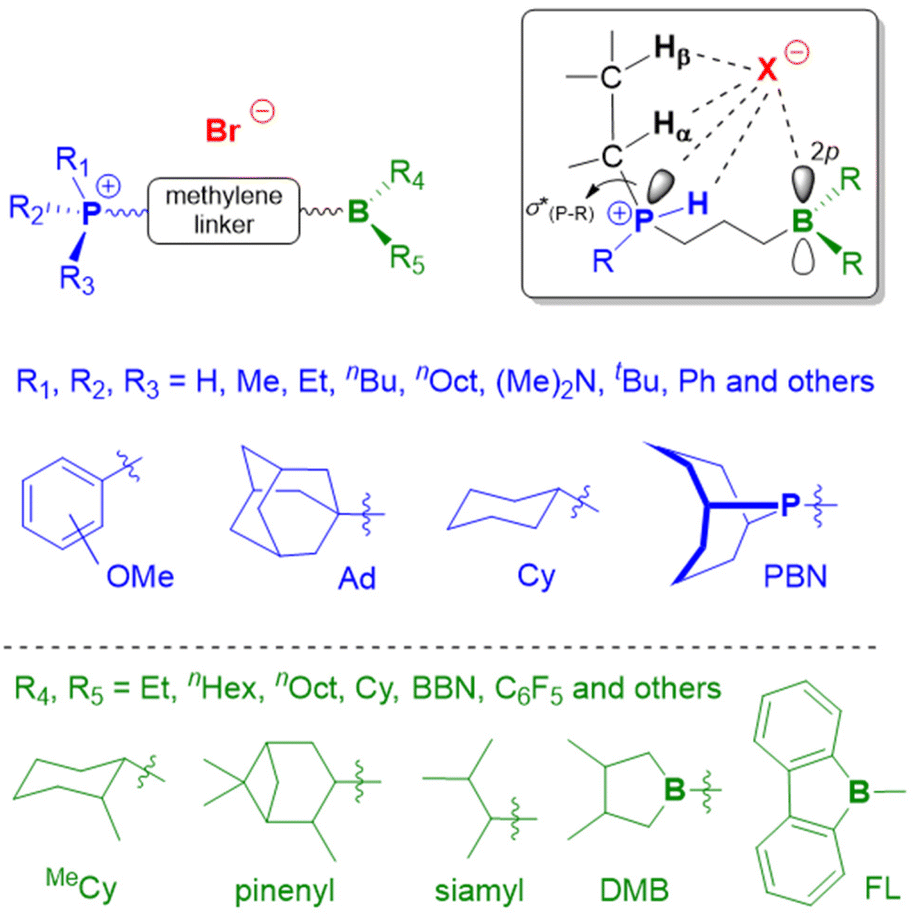 | ||
| Fig. 6 Tertiary and quaternary phosphonium-type monoborane catalysts as screened by Lin and co-workers. Insert: multiple interactions of halide and catalyst as tuning site. Note that Hα and Hβ act as H-bond donors. | ||
Regarding the quaternary phosphonium substituents, it was found that aryl groups slowed down the polymerization reaction (kobs = 0.7–0.9 × 10−4 s−1), while simple alkyl moieties (ethyl, butyl) delivered much faster monomer consumption (kobs = 1.9–2.1 × 10−4 s−1). This tendency is comparable to what was observed for epoxide/anhydride copolymerization (see above). Focusing on the nBu3P-motif and 9-BBN-derived borane groups, the linker length was also screened (L = 3, 4, 5, 6 and 10). The respective values for kobs (0.57 × 10−4 s−1, 1.4 × 10−4 s−1, 2.1 × 10−4 s−1, 1.6 × 10−4 s−1 and 0.74 × 10−4 s−1)158 revealed the pentamethylene linker as optimum, in perfect agreement with Wu's results.157 Measurement of borane Lewis acidity via the Gutmann-Beckett method, on the other hand, proved more difficult to interpret, most likely because the probe molecule (Et3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) not only interacts with the boron, but also with the phosphonium group (an effect which itself is dependent on L). Nonetheless, the broad variation of borane moieties revealed a very strong impact on performance. For example, the dimethylborolane (DMB) variant operates in a rather poor manner, compared to BBN (0.19 × 10−4 s−1vs. 2.1 × 10−4 s−1, respectively). This was attributed to the wider C–B–C angle, which aggravates sp2 to sp3 transition during the catalytic cycle. The use of much stronger Lewis acidic boranes (fluorenyl (FL) or C6F5-groups) resulted in a complete loss of activity. Likewise, the disiamylborane variant was fully inert, while the reduced steric demand exerted by octyl groups enabled moderate activity (kobs = 1.4 × 10−4 s−1). The fastest polymerization at 80 °C was displayed by the BCy2-motif (2.4 × 10−4 s−1), but also here slight further modifications (MeCy or pinenyl) shut down the polymerization activity completely. Temperature stability was also probed, revealing that the high polymerization temperature reported by Wu (150 °C)157 is not generally applicable to all of these phosphonium-based bifunctional catalysts. Irreversible catalyst decomposition seems to occur at T < 125 °C, for some compounds even at less than 100 °C.
O) not only interacts with the boron, but also with the phosphonium group (an effect which itself is dependent on L). Nonetheless, the broad variation of borane moieties revealed a very strong impact on performance. For example, the dimethylborolane (DMB) variant operates in a rather poor manner, compared to BBN (0.19 × 10−4 s−1vs. 2.1 × 10−4 s−1, respectively). This was attributed to the wider C–B–C angle, which aggravates sp2 to sp3 transition during the catalytic cycle. The use of much stronger Lewis acidic boranes (fluorenyl (FL) or C6F5-groups) resulted in a complete loss of activity. Likewise, the disiamylborane variant was fully inert, while the reduced steric demand exerted by octyl groups enabled moderate activity (kobs = 1.4 × 10−4 s−1). The fastest polymerization at 80 °C was displayed by the BCy2-motif (2.4 × 10−4 s−1), but also here slight further modifications (MeCy or pinenyl) shut down the polymerization activity completely. Temperature stability was also probed, revealing that the high polymerization temperature reported by Wu (150 °C)157 is not generally applicable to all of these phosphonium-based bifunctional catalysts. Irreversible catalyst decomposition seems to occur at T < 125 °C, for some compounds even at less than 100 °C.
Analysis of the enthalpic (ΔH‡) and entropic (ΔS‡) contributions to the Gibbs free energy of activation illuminated further aspects of the polymerization mechanism. For one, it was found that Lewis acidity and the enthalpy of activation are not correlated. Secondly, a trade-off between enthalpic and entropic contributions exists. For example, the more destabilized resting states (favourable enthalpic term) at higher L appear in conjunction with disadvantageous entropy – the longer B–P distance requires a “backfolding” for the cooperative propagation step. This may also explain why “sweet spots” are to be found with an optimal balancing of both thermodynamic contributions. As an example, for an investigated series of compounds, for L = 10, 5, and 3, ΔH‡ of 50.2 kJ mol−1, 67.7 kJ mol−1 and 56.7 kJ mol−1 is found. This is accompanied respectively by ΔS‡ = −145.2 J mol−1 K−1), −88.0 J mol−1 K−1) and −130.6 J mol−1 K−1).
Intriguingly, also the tertiary phosphonium-type catalysts were found to be very well-performing, especially at higher temperatures. Interesting perspectives for continuous solution polymerization with the ensuing lower viscosity at high temperatures can be envisioned for this type of catalyst, which is described for the first time in this paper. While not all mechanistic details are understood, it seems clear that the acidic proton is conducive for a rapid and well-controlled polymerization.
Diboranes with neutral (chiral) linkers
Also in 2022, the group of Naumann described organoboron catalysts for the stereoselective polymerization of rac-PO.66 Inspired by the polymerization mechanism for epoxide homopolymerization (see above and Scheme 25), involving two equivalents of Et3B, and by the work of Du on chiral diboranes for hydrogenation reactions,159 axially chiral backbones were used as linker structures. Starting from the well-available diol compounds (i.e., 1,1′-bi-2-naphthol, BINOL), a simple sequence of etherification followed by hydroboration yielded the corresponding diboranes 11 and 12. Interestingly, even for these non-optimized compounds, a moderate stereoselectivity (up to isotactic diad placement (m) = 88%) could indeed be achieved (−36 °C to 50 °C). While choice of the organobase (DBU, tBu-P2 or a specific NHO) had no impact on selectivity, the introduction of steric congestion (phenyl group, 12) was clearly favourable in this regard, albeit at the cost of a notably decreased polymerization rate. Moreover, dilution via suitable solvents (THF, toluene) proved beneficial. Most likely bimolecular interaction,159 which is thought to be non-selective, can be reduced this way. Molar masses of Mn up to 120 kg mol−1 could be realized, showing typically well-defined properties as found by GPC (ĐM = 1.05–1.25) and MALDI-ToF MS (end groups exclusively derived from initiator, BnOH). Kinetic experiments with enantiopure monomer revealed that (R)-11 is selective for (S)-PO (and (S)-BO), while (S)-11 shows the opposite preferences. In a typical experiment (2M in THF, RT, NHO/(R)-11/BnOH/enantiopure PO = 1:2:5:2000, molar ratio) a zero-order dependence on [PO] is found, with k(S-PO) 16 times faster than k(R-PO). Interestingly, using rac-11, the same order of selectivity is observed; tentatively this was attributed to a strong association of propagating chain end and diborane catalyst which would rule out a very frequent exchange of growing polyether chains between catalysts. If future research verifies this trend, catalyst syntheses could be simplified considerably.
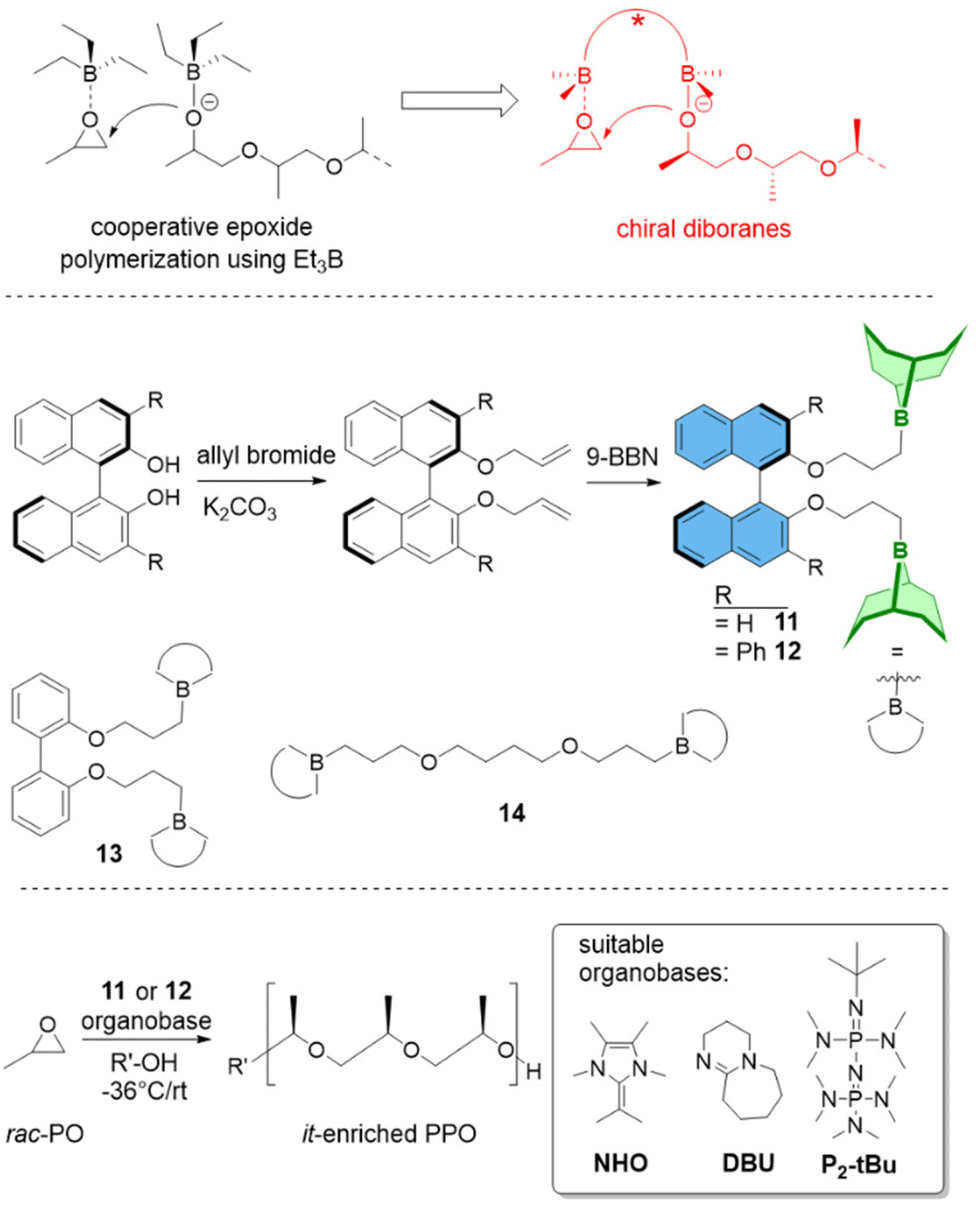 | ||
| Scheme 25 Research rationale (top), two-step synthesis (middle) and polymerization procedure (bottom) for the preparation of it-PPO. | ||
The isotactic-enriched (it-)PPO received under optimized conditions (m > 85%) proved to be semi-crystalline, which is obviously different from the fully amorphous PPO received from conventional anionic polymerization (KOH), from the action of Et3B or via any other borane catalysts described so far. Indeed, up this point the stereoselective polymerization of substituted epoxides had been an exclusive domain of metal-based catalysts.65 And while the most advanced of the latter are still clearly superior with regard to selectivity (with isotactic triad placement, mm, > 99% for PO)34 the functional group tolerance of the organoboron catalysts is a crucial difference of high practical importance. Indeed, using polycaprolactone or polylactide macroinitiators, it is possible to introduce blocks of it-PPO with high precision by the action of 11 or 12 (excess of borane functionalities relative to the organobase).66 Such “polyester-first” strategies and related approaches are considered to be of great benefit for the preparation of tacticity-tuned polymeric additives and, as outlined above, cannot be realized by conventional means.
It should be noted that 11 and 12 are no bifunctional catalysts in the sense of 1–10 as their linker setup is fully neutral. Structure–property relationships for this type of diborane catalysts are largely unknown to date. Control experiments using the non-chiral biphenyl analogue (13) and a linear variant (14) have nonetheless shed some light on promising catalyst structures for epoxide homopolymerization. While both 13 and 14 expectedly deliver practically atactic PPO (m < 57%), it is also found that under identical conditions the biphenyl catalyst consumes PO about fifty times faster than 14. Obviously, while in both cases the two borane groups, which need to act cooperatively in the propagation step, are separated by twelve atoms, the preorganization by the biphenyl backbone is clearly beneficial for rapid polymerization, perhaps in a similar way that has been described for the bifunctional catalysts according to Lin.158
Outlook and conclusion
The rise of Et3B and later generations of borane catalysts has provided spectacular advances for epoxide homo- and copolymerization. This is all the more impressive if it is considered how young the whole field of research actually is: more than 50% of the topic-related publications discussed herein have emerged in 2020 or later. Certainly the breadth of investigated catalyst structures is evolving rapidly and further breakthroughs can be expected in the near future.As illustrated by many of the examples discussed above, a unique combination of properties is responsible for this booming growth of interest. For one, the borane catalysis offers a metal-free approach to the corresponding polyethers and other copolymers. Thus, a potentially more sustainable route to typically achromatous product polymer is provided. This is further accentuated by the ultra-low catalyst loadings which can be applied in many cases, rendering catalyst removal redundant. The latter is especially true for the bifunctional/multiborane setups, which, on account of their much increased activity, can also be used in catalytic amounts relative to the CTA/initiator (alcohols). Overall, record values regarding TON, TOF and achievable molar masses have been realized using organoboron catalysts, including epoxide homopolymerization but also copolymerization with CO2 or anhydrides, not to speak of the corresponding terpolymerizations.
Apart from performance, also the functional group tolerance is a striking feature for borane catalysis. This is related to the moderate Lewis acidity of the employed borane species and the dual, cooperative role of the latter: a combination of monomer activation and chain-end capping (deactivation of the anionic, propagating species) allows for the polymerization of sensitive, highly functionalized monomers. Likewise, advanced or even novel polymer architectures have become possible, such as poly(ether-ester) multiblocks.
Also, borane catalysis has delivered the first example of organocatalytic, stereoselective polymerization of epoxides. Albeit in this case selectivity needs to be further improved and the complex structure–property correlations need to be understood further, this nicely complements the benefits for epoxide (co)polymerization achieved via borane catalysis.
Thus, the outlook is promising and this is perhaps also reflected by a first wave of patenting activities.160–162 Realization of borane-catalysed polymerization of O-heterocyclic monomers on technical scale will be a crucial step for advancing these technologies and might provide a further boost in research interest for this intriguing type of catalysis. Further, an extension to non-epoxide monomer feedstock can be observed. Lactone polymerization is an example,163,164 and very recently also acrylic monomers have been polymerized by a specific diborane catalyst.165 Likewise, the often underestimated possibility of structural or compositional changes of alkylboranes during the catalytic cycle may offer exciting venues for the development of novel (chemo)selectivities.166 Also, the conceptual closeness of (multi)borane catalyst design to Lewis pair polymerization85 might prove fruitful for the field as well. Very importantly, also classic organometallic chemistry may inspire further improved borane organocatalysts. An instructive example, Co(III)-Salen complexes published 2006–2010 combined the Lewis acidic metal centre with a Salen ligand carrying tertiary amine or ammonium functionalities (by which 2,4-dinitrophenolate counter ions were introduced).167 Thereby, two mechanistically important components were combined in one molecule, subsequently leading to much improved TONs, TOFs, selectivity and thermal stability for the copolymerization of epoxides and CO2. In a way, these can be understood as metal-based analogues to the bifunctional, multiborane catalysts discussed above.147 There may be more lessons to be learned from these or similar complexes, regarding for example catalyst recyclability or optimized catalyst geometry. Perhaps not coincidentally, a detailed study by Wu, Yang and co-workers recently found an ideal B–B distance of 769 pm for bifunctional diboron catalysts for epoxide homopolymerization (larger distances entailed a notable loss of activity).154 This is strongly reminiscent of Coates’ bimetallic Co(III) complexes, which likewise enable swift epoxide conversion and display Co–Co distances of 713 pm.65 Since in both cases the mechanism requires the cooperation of two Lewis acidic centres, similar geometrical constraints seem a plausible consequence. More rigid (multi)borane catalyst backbones seem especially recommended in this regard, perhaps opening exciting new venues for (i.e., stereoselective) epoxide (co)polymerization. Further overlaps of this kind may well exist and could potentially be fruitful for both metal-based and organocatalytic disciplines.
In sum, areas with particular high potential for the field may be identified as (a) a broadening of the monomer scope beyond O-heterocyclic monomers, (b) a more detailed understanding of borane deactivation or structural changes during catalysis, (c) a clarification of the role of Lewis acidity, monomer activation and polymerization mechanisms in general, (d) an improvement of stereocontrol, (e) immobilized and/or recyclable solutions and (f) realization of borane-catalyzed (co)polymerization on large scale.
This checklist, which is certainly not exhaustive, demonstrates that the disruptive impact of borane (co)catalysis on epoxide conversion and beyond can be well expected to continue in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. N. gratefully acknowledges financial support by the German Research Foundation (DFG, project NA 1206/3-1).References
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed.
- R. Klein and F. R. Wurm, Macromol. Rapid Commun., 2015, 36, 1147–1165 CrossRef CAS PubMed.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- H.-W. Engels, H.-G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CrossRef CAS PubMed.
- M. Greaves, E. Zaugg-Hoozemans, N. Khelidj, R. van Voorst and R. Meertens, Lubr. Sci., 2012, 24, 251–262 CrossRef CAS.
- F. Markus, J. R. Bruckner and S. Naumann, Macromol. Chem. Phys., 2020, 221, 1900437 CrossRef CAS.
- B. Zhang, X. Dong, J. Xuan and J. He, Color. Technol., 2013, 129, 377–384 CAS.
- T. Hargreaves, Chemical Formulation: An Overview of Surfactant-Based Preparations Used in Everyday Life, Royal Society of Chemistry, Cambridge, 2007 Search PubMed.
- C. Dingels, M. Schömer and H. Frey, Chem. Unserer Zeit, 2011, 45, 338–349 CrossRef CAS.
- S. Gupta, R. Tyagi, V. S. Parmar, S. K. Sharma and R. Haag, Polymer, 2012, 53, 3053–3078 CrossRef CAS.
- R. Greenwald, J. Controlled Release, 2001, 74, 159–171 CrossRef CAS PubMed.
- W. Wieczorek, Z. Florjanczyk and J. R. Stevens, Electrochim. Acta, 1995, 40, 2251–2258 CrossRef CAS.
- A. Nishimoto, K. Agehara, N. Furuya, T. Watanabe and M. Watanabe, Macromolecules, 1999, 32, 1541–1548 CrossRef CAS.
- H. Li, W. Tang, Y. Huang, W. Ruan and M. Zhang, Polym. Chem., 2019, 10, 2697–2705 RSC.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan, A. Stein and D. Zhao, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- A. Balint, M. Papendick, M. Clauss, C. Müller, F. Giesselmann and S. Naumann, Chem. Commun., 2018, 54, 2220–2223 RSC.
- J. A. Martens, J. Jammaer, S. Bajpe, A. Aerts, Y. Lorgouilloux and C. E. Kirschhock, Microporous Mesoporous Mater., 2011, 140, 2–8 CrossRef CAS.
- K. Shiraishi and M. Yokoyama, Sci. Technol. Adv. Mater., 2019, 20, 324–336 CrossRef CAS PubMed.
- A. Sunder, R. Mülhaupt, R. Haag and H. Frey, Adv. Mater., 2000, 12, 235–239 CrossRef CAS.
- Z. Geng, N. S. Schauser, J. Lee, R. P. Schmeller, S. M. Barbon, R. A. Segalman, N. A. Lynd and C. J. Hawker, Macromolecules, 2020, 53, 4960–4967 CrossRef CAS.
- A. Pitto-Barry and N. P. E. Barry, Polym. Chem., 2014, 5, 3291–3297 RSC.
- A. Z. Dookhith, N. A. Lynd, C. Creton and G. E. Sanoja, Macromolecules, 2022, 55, 5601–5609 CrossRef CAS.
- V. St-Onge, S. Rochon, J.-C. Daigle, A. Soldera and J. P. Claverie, Angew. Chem., Int. Ed., 2021, 60, 25897–25904 CrossRef CAS PubMed.
- T. Isono, H. Lee, K. Miyachi, Y. Satoh, T. Kakuchi, M. Ree and T. Satoh, Macromolecules, 2018, 51, 2939–2950 CrossRef CAS.
- P. Verkoyen and H. Frey, Polym. Chem., 2020, 11, 3940–3950 RSC.
- P. Dreier, A. Pipertzis, M. Spyridakou, R. Mathes, G. Floudas and H. Frey, Macromolecules, 2022, 55, 1342–1353 CrossRef CAS.
- R. Matthes and H. Frey, Biomacromolecules, 2022, 23, 2219–2235 CrossRef CAS PubMed.
- B. M. Lipinski, L. S. Morris, M. N. Silberstein and G. W. Coates, J. Am. Chem. Soc., 2020, 142, 6800–6806 CrossRef CAS PubMed.
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS.
- D. Darensbourg, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- X.-B. Lu, W.-M. Ren and G.-P. Wu, Acc. Chem. Res., 2012, 45, 1721–1735 CrossRef CAS PubMed.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- D. Ryzhakov, G. Printz, B. Jacques, S. Messaoudi, F. Dumas, S. Dagorne and F. Le Bideau, Polym. Chem., 2021, 12, 2932–2946 RSC.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS PubMed.
- Y. Xia and J. Zhao, Polymer, 2018, 143, 343–361 CrossRef CAS.
- H. J. Altmann, M. Clauss, S. König, E. Frick-Delaittre, C. Koopmans, A. Wolf, C. Guertler, S. Naumann and M. R. Buchmeiser, Macromolecules, 2019, 52, 487–494 CrossRef CAS.
- M. Jurrat, B. J. Pointer-Gleadhill, L. T. Ball, A. Chapman and L. Adriaenssens, J. Am. Chem. Soc., 2020, 142, 8136–8141 CrossRef CAS PubMed.
- R. Mohr, M. Wagner, S. Zarbakhsh and H. Frey, Macromol. Rapid Commun., 2021, 42, e2000542 CrossRef PubMed.
- W. T. Diment, W. Lindeboom, F. Fiorentini, A. C. Deacy and C. K. Williams, Acc. Chem. Res., 2022, 55, 1997–2010 CrossRef CAS PubMed.
- D. J. Darensbourg and W.-C. Chung, Polyhedron, 2013, 58, 139–143 CrossRef CAS.
- G. Odian and G. G. Odian, Principles of polymerization, Wiley-Interscience, Hoboken, NJ, 4th edn, 2004, Ch. 7.2 Search PubMed.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS PubMed.
- M. Fèvre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142–2172 RSC.
- D. Taton, Organic Catalysis for Polymerisation, ed. A. Dove, H. Sardon and S. Naumann, Royal Society of Chemistry, Cambridge, 2018, pp. 328–366 Search PubMed.
- S. Naumann and A. P. Dove, Polym. Int., 2016, 65, 16–27 CrossRef CAS.
- A.-L. Brocas, C. Mantzaridis, D. Tunc and S. Carlotti, Prog. Polym. Sci., 2013, 38, 845–873 CrossRef CAS.
- I. Kim, J.-T. Ahn, C. S. Ha, C. S. Yang and I. Park, Polymer, 2003, 44, 3417–3428 CrossRef CAS.
- N. J. Robertson, Z. Qin, G. C. Dallinger, E. B. Lobkovsky, S. Lee and G. W. Coates, Dalton Trans., 2006, 5390–5395 RSC.
- R.-J. Wei, X.-H. Zhang, B.-Y. Du, X.-K. Sun, Z.-Q. Fan and G.-R. Qi, Macromolecules, 2013, 46, 3693–3697 CrossRef CAS.
- E. J. Vandenberg, J. Polym. Sci., 1960, 47, 486–489 CrossRef CAS.
- E. J. Vandenberg, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 1423–1431 CrossRef CAS.
- R. C. Ferrier, S. Pakhira, S. E. Palmon, C. G. Rodriguez, D. J. Goldfeld, O. O. Iyiola, M. Chwatko, J. L. Mendoza-Cortes and N. A. Lynd, Macromolecules, 2018, 51, 1777–1786 CrossRef CAS.
- O. Rexin and R. Mülhaupt, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 864–873 CrossRef CAS.
- N. Takeda and S. Inoue, Makromol. Chem., 1978, 179, 1377–1381 CrossRef CAS.
- K. L. Peretti, H. Ajiro, C. T. Cohen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 11566–11567 CrossRef CAS PubMed.
- J. Raynaud, W. N. Ottou, Y. Gnanou and D. Taton, Chem. Commun., 2010, 46, 3203–3205 RSC.
- S. Naumann, A. W. Thomas and A. P. Dove, Angew. Chem., Int. Ed., 2015, 54, 9550–9554 CrossRef CAS PubMed.
- A. Labbé, S. Carlotti, C. Billouard, P. Desbois and A. Deffieux, Macromolecules, 2007, 40, 7842–7847 CrossRef.
- P. Walther, A. Krauß and S. Naumann, Angew. Chem., Int. Ed., 2019, 58, 10737–10741 CrossRef CAS PubMed.
- P. Walther, C. Vogler and S. Naumann, Synlett, 2020, 641–647 CAS.
- S. Liu, T. Bai, K. Ni, Y. Chen, J. Zhao, J. Ling, X. Ye and G. Zhang, Angew. Chem., Int. Ed., 2019, 58, 15478–15487 CrossRef CAS PubMed.
- J. Raynaud, C. Absalon, Y. Gnanou and D. Taton, J. Am. Chem. Soc., 2009, 131, 3201–3209 CrossRef CAS PubMed.
- M. I. Childers, J. M. Longo, N. J. van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed.
- A. Sirin-Sariaslan and S. Naumann, Chem. Sci., 2022, 13, 10939–10943 RSC.
- C. Zhang, X. Geng, X. Zhang, Y. Gnanou and X. Feng, Prog. Polym. Sci., 2023, 136, 101644 CrossRef CAS.
- N. V. Sutton and H. Schneider, Microchem. J., 1965, 9, 209–217 CrossRef CAS.
- https://www.sigmaaldrich.com/DE/de/product/aldrich/179701 , accessed 06/08/22.
- H. C. Brown and N. C. Hébert, J. Organomet. Chem., 1983, 255, 135–142 CrossRef CAS.
- E. Wiberg, N. Wiberg and G. Fischer, Lehrbuch der anorganischen Chemie, Walter de Gruyter, Berlin, New York, 102nd edn, 2007, p. 1127 Search PubMed.
- I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS.
- J. A. Plumley and J. D. Evanseck, J. Phys. Chem. A, 2009, 113, 5985–5992 CrossRef CAS PubMed.
- R. J. Mayer, N. Hampel and A. R. Ofial, Chem. – Eur. J., 2021, 27, 4070–4080 CrossRef CAS PubMed.
- See for example: D. Chakraborty, A. Rodriguez and E. Y.-X. Chen, Macromolecules, 2003, 36, 5470–5481 CrossRef CAS.
- D. P. Curran and T. R. McFadden, J. Am. Chem. Soc., 2016, 138, 7741–7752 CrossRef CAS PubMed.
- M. Kobayashi, T. Ishizone and S. Nakahama, Macromolecules, 2000, 33, 4411–4416 CrossRef CAS.
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- D. Zhang, H. Zhang, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2016, 49, 2484–2492 CrossRef CAS.
- D.-D. Zhang, X. Feng, Y. Gnanou and K.-W. Huang, Macromolecules, 2018, 51, 5600–5607 CrossRef CAS.
- N. G. Patil, S. K. Boopathi, P. Alagi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2019, 52, 2431–2438 CrossRef CAS.
- N. Patil, S. Bhoopathi, V. Chidara, N. Hadjichristidis, Y. Gnanou and X. Feng, ChemSusChem, 2020, 13, 5080–5087 CrossRef CAS PubMed.
- Z. Chen, J.-L. Yang, X.-Y. Lu, L.-F. Hu, X.-H. Cao, G.-P. Wu and X.-H. Zhang, Polym. Chem., 2019, 10, 3621–3628 RSC.
- M. Jia, N. Hadjichristidis, Y. Gnanou and X. Feng, ACS Macro Lett., 2019, 8, 1594–1598 CrossRef CAS PubMed.
- M. L. McGraw and E. Y.-X. Chen, Macromolecules, 2020, 53, 6102–6122 CrossRef CAS.
- M. Jia, D. Zhang, G. W. de Kort, C. H. R. M. Wilsens, S. Rastogi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2020, 53, 5297–5307 CrossRef CAS PubMed.
- D. Augustine, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2020, 53, 895–904 CrossRef CAS.
- P. Alagi, G. Zapsas, N. Hadjichristidis, S. C. Hong, Y. Gnanou and X. Feng, Macromolecules, 2021, 54, 6144–6152 CrossRef CAS.
- Y. Wang, J.-Y. Zhang, J.-L. Yang, H.-K. Zhang, J. Kiriratnikom, C.-J. Zhang, K.-L. Chen, X.-H. Cao, L.-F. Hu, X.-H. Zhang and B. Z. Tang, Macromolecules, 2021, 54, 2178–2186 CrossRef CAS.
- J. Zhang, L. Wang, S. Liu and Z. Li, Angew. Chem., Int. Ed., 2022, 61, e202111197 CAS.
- M. Jia, D. Zhang, Y. Gnanou and X. Feng, ACS Sustainable Chem. Eng., 2021, 9, 10370–10380 CrossRef CAS.
- J.-L. Yang, H.-L. Wu, Y. Li, X.-H. Zhang and D. J. Darensbourg, Angew. Chem., Int. Ed., 2017, 56, 5774–5779 CrossRef CAS PubMed.
- J.-L. Yang, X.-H. Cao, C.-J. Zhang, H.-L. Wu and X.-H. Zhang, Molecules, 2018, 23, 298 CrossRef PubMed.
- J.-L. Yang, H. Wang, L.-F. Hu, X. Hong and X.-H. Zhang, Polym. Chem., 2019, 10, 6555–6560 RSC.
- J. Kiriratnikom, J. Guo, X. Cao, M. U. Khan, C. Zhang and X. Zhang, J. Polym. Sci., 2022, 60, 3414–3419 CrossRef CAS.
- J.-L. Yang, L.-F. Hu, X.-H. Cao, Y. Wang and X.-H. Zhang, Chin. J. Chem., 2020, 38, 269–274 CrossRef CAS.
- C.-J. Zhang, S.-Q. Wu, S. Boopathi, X.-H. Zhang, X. Hong, Y. Gnanou and X.-S. Feng, ACS Sustainable Chem. Eng., 2020, 8, 13056–13063 CrossRef CAS.
- Y. Chen, J. Shen, S. Liu, J. Zhao, Y. Wang and G. Zhang, Macromolecules, 2018, 51, 8286–8297 CrossRef CAS.
- C.-J. Zhang, H.-Y. Duan, L.-F. Hu, C.-H. Zhang and X.-H. Zhang, ChemSusChem, 2018, 11, 4209–4213 CrossRef CAS PubMed.
- C. Vogler and S. Naumann, RSC Adv., 2020, 10, 43389–43393 RSC.
- Q. Song, J. Zhao, G. Zhang, D. Taton, F. Peruch and S. Carlotti, Eur. Polym. J., 2020, 134, 109839 CrossRef CAS.
- S. Penczek and J. Pretula, ACS Macro Lett., 2021, 10, 1377–1397 CrossRef CAS PubMed.
- F. T. Hong, V. Ladelta, R. Gautam, S. M. Sarathy and N. Hadjichristidis, ACS Appl. Polym. Mater., 2021, 3, 3811–3820 CrossRef CAS.
- F. Markus, C. Vogler, J. R. Bruckner and S. Naumann, ACS Appl. Nano Mater., 2021, 4, 3486–3492 CrossRef CAS.
- C. Vogler, S. Naumann and J. R. Bruckner, Mol. Syst. Des. Eng., 2022, 7, 1318–1326 RSC.
- S. Naumann, Org. Mater., 2021, 03, 283–294 CrossRef CAS.
- S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, Nat. Commun., 2019, 10, 293 CrossRef PubMed.
- S. Liu, L. Liu, Y. Zhou, Y. Chen and J. Zhao, Polym. Chem., 2022, 13, 3650–3659 RSC.
- Y. Chen, S. Liu, J. Zhao, D. Pahovnik, E. Žagar and G. Zhang, ACS Macro Lett., 2019, 8, 1582–1587 CrossRef CAS PubMed.
- S. Liu, L. Liu, Y. Chen and J. Zhao, Chem. Commun., 2020, 56, 12186–12189 RSC.
- Y. Chen, P. Zhang, S. Liu, D. Pahovnik, E. Žagar, J. Zhao and G. Zhang, ACS Macro Lett., 2021, 10, 737–743 CrossRef CAS PubMed.
- In this case, measurements were conducted in toluene-d8; in CDCl3, ref. 78 reports a shift of about 0.06 ppm (in both cases, 2 eq. of Et3B relative to PO were employed).
- J. Liu, Y. Gnanou and X. Feng, Macromolecules, 2022, 55, 1800–1810 CrossRef CAS.
- L.-F. Hu, C.-J. Zhang, H.-L. Wu, J.-L. Yang, B. Liu, H.-Y. Duan and X.-H. Zhang, Macromolecules, 2018, 51, 3126–3134 CrossRef CAS.
- H.-Y. Ji, X.-L. Chen, B. Wang, L. Pan and Y.-S. Li, Green Chem., 2018, 20, 3963–3973 RSC.
- L.-F. Hu, D.-J. Chen, J.-L. Yang and X.-H. Zhang, Molecules, 2020, 25, 253 CrossRef CAS PubMed.
- J. Zhang, L. Wang, S. Liu and Z. Li, J. Polym. Sci., 2020, 58, 803–810 CrossRef CAS.
- B. Han, L. Zhang, B. Liu, X. Dong, I. Kim, Z. Duan and P. Theato, Macromolecules, 2015, 48, 3431–3437 CrossRef CAS.
- H. Li, G. He, Y. Chen, J. Zhao and G. Zhang, ACS Macro Lett., 2019, 8, 973–978 CrossRef CAS PubMed.
- H.-Y. Ji, D.-P. Song, B. Wang, L. Pan and Y.-S. Li, Green Chem., 2019, 21, 6123–6132 RSC.
- V. K. Chidara, Y. Gnanou and X. Feng, Molecules, 2022, 27, 466 CrossRef CAS PubMed.
- S. Ye, W. Wang, J. Liang, S. Wang, M. Xiao and Y. Meng, ACS Sustainable Chem. Eng., 2020, 8, 17860–17867 CrossRef CAS.
- J. Zhang, L. Wang, S. Liu, X. Kang and Z. Li, Macromolecules, 2021, 54, 763–772 CrossRef CAS.
- V. K. Chidara, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2021, 54, 2711–2719 CrossRef CAS.
- S. Zhu, Y. Wang, W. Ding, X. Zhou, Y. Liao and X. Xie, Polym. Chem., 2020, 11, 1691–1695 RSC.
- G. He, H. Li and J. Zhao, Macromol. Chem. Phys., 2022, 223, 2100321 CrossRef CAS.
- S. Zhu, Y. Zhao, M. Ni, J. Xu, X. Zhou, Y. Liao, Y. Wang and X. Xie, ACS Macro Lett., 2020, 9, 204–209 CrossRef CAS PubMed.
- J. K. Varghese, N. Hadjichristidis, Y. Gnanou and X. Feng, Polym. Chem., 2019, 10, 3764–3771 RSC.
- N. von Seggern, T. Schindler and S. Naumann, Biomacromolecules, 2020, 21, 2661–2669 CrossRef CAS PubMed.
- T. Lai, P. Zhang, J. Zhao and G. Zhang, Macromolecules, 2021, 54, 11113–11125 CrossRef CAS.
- M. Jia, N. Hadjichristidis, Y. Gnanou and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 1593–1598 CrossRef CAS PubMed.
- N. Patil, Y. Gnanou and X. Feng, Macromolecules, 2022, 55, 7817–7826 CrossRef CAS.
- M. A. Beckett, G. C. Strickland, J. R. Holland and K. Sukumar Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS.
- P. Erdmann and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202114550 CrossRef CAS PubMed.
- M. A. Beckett, D. S. Brassington, P. Owen, M. B. Hursthouse, M. E. Light, K. Malik and K. Varma, J. Organomet. Chem., 1999, 585, 7–11 CrossRef CAS.
- R. C. Bauer, PhD Thesis, University of Alberta, Edmonton, 2009, p. 138.
- H. Li, A. J. A. Aquino, D. B. Cordes, F. Hung-Low, W. L. Hase and C. Krempner, J. Am. Chem. Soc., 2013, 135, 16066–16069 CrossRef CAS PubMed.
- K. A. Andrea and F. M. Kerton, ACS Catal., 2019, 9, 1799–1809 CrossRef CAS.
- K. A. Andrea, M. D. Wheeler and F. M. Kerton, Chem. Commun., 2021, 57, 7320–7322 RSC.
- I. Asenjo-Sanz, A. Veloso, J. I. Miranda, J. A. Pomposo and F. Barroso-Bujans, Polym. Chem., 2014, 5, 6905–6908 RSC.
- T. Miyajima, K. Nishiyama, M. Satake and T. Tsuji, Polym. J., 2015, 47, 771–778 CrossRef CAS.
- A. Raghuraman, D. Babb, M. Miller, M. Paradkar, B. Smith and A. Nguyen, Macromolecules, 2016, 49, 6790–6798 CrossRef CAS.
- A. Rodriguez-Delgado and E. Y.-X. Chen, Inorg. Chim. Acta, 2004, 357, 3911–3919 CrossRef CAS.
- S. E. Kim, Y.-R. Lee, M. Kim, E. Seo, H.-J. Paik, J. C. Kim, J.-E. Jeong, Y. I. Park, B.-S. Kim and S.-H. Lee, Polym. Chem., 2022, 13, 1243–1252 RSC.
- A. Kanazawa, S. Kanaoka and S. Aoshima, J. Am. Chem. Soc., 2013, 135, 9330–9333 CrossRef CAS PubMed.
- G.-G. Gu, L.-Y. Wang, R. Zhang, T.-J. Yue, B.-H. Ren and W.-M. Ren, Polym. Chem., 2021, 12, 6436–6443 RSC.
- G.-W. Yang, Y.-Y. Zhang and G.-P. Wu, Acc. Chem. Res., 2021, 54, 4434–4448 CrossRef CAS PubMed.
- G.-W. Yang, Y.-Y. Zhang, R. Xie and G.-P. Wu, J. Am. Chem. Soc., 2020, 142, 12245–12255 CrossRef CAS PubMed.
- Y.-Y. Zhang, G.-W. Yang, R. Xie, L. Yang, B. Li and G.-P. Wu, Angew. Chem., Int. Ed., 2020, 59, 23291–23298 CrossRef CAS PubMed.
- G.-W. Yang, C.-K. Xu, R. Xie, Y.-Y. Zhang, X.-F. Zhu and G.-P. Wu, J. Am. Chem. Soc., 2021, 143, 3455–3465 CrossRef CAS PubMed.
- C. Chen, Y. Gnanou and X. Feng, Polym. Chem., 2022, 13, 6312–6321 RSC.
- G.-W. Yang, Y.-Y. Zhang, R. Xie and G.-P. Wu, Angew. Chem., Int. Ed., 2020, 59, 16910–16917 CrossRef CAS PubMed.
- C. Chen, Y. Gnanou and X. Feng, Macromolecules, 2022, 55, 10662–10669 CrossRef CAS.
- H. Qi, R. Xie, G.-W. Yang, Y.-Y. Zhang, C.-K. Xu, Y. Wang and G.-P. Wu, Macromolecules, 2022, 55, 9081–9090 CrossRef CAS.
- R. Xie, Y.-Y. Zhang, G.-W. Yang, X.-F. Zhu, B. Li and G.-P. Wu, Angew. Chem., Int. Ed., 2021, 60, 19253–19261 CrossRef CAS PubMed.
- X. Wang, J. Hui, M. Shi, X. Kou, X. Li, R. Zhong and Z. Li, ACS Catal., 2022, 12, 8434–8443 CrossRef CAS.
- Y.-Y. Zhang, C. Lu, G.-W. Yang, R. Xie, Y.-B. Fang, Y. Wang and G.-P. Wu, Macromolecules, 2022, 55, 6443–6452 CrossRef CAS.
- J. Schaefer, H. Zhou, E. Lee, N. S. Lambic, G. Culcu, M. W. Holtcamp, F. C. Rix and T.-P. Lin, ACS Catal., 2022, 12, 11870–11885 CrossRef CAS.
- Y. Liu and H. Du, J. Am. Chem. Soc., 2013, 135, 6810–6813 CrossRef CAS PubMed.
- Y. Alzahrany, X. Feng, S. Boopathi, D. Zhang, N. Hadjichristidis and Y. Gnanou, Copolymerization of carbon dioxide and cyclic monomers to form polycarbonates, 2018, p. 2018/0051134A1 Search PubMed.
- G.-W. Yang, Y.-Y. Zhang and G.-P. Wu, Organic metal-free catalyst having both electrophilic and nucleophilic functions, preparation method therefor, and application thereof, 2020, 2020057356 Search PubMed.
- X. Feng, N. Patil, S. Boopathi, Y. Gnanou and N. Hadjichristidis, Polycarbonate polyols, 2020, 2020121262A3 Search PubMed.
- Y.-Y. Zhang, L. Yang, R. Xie, G.-W. Yang and G.-P. Wu, Macromolecules, 2021, 54, 9427–9436 CrossRef CAS.
- L. Yang, Y.-Y. Zhang, G.-W. Yang, R. Xie and G.-P. Wu, Macromolecules, 2021, 54, 5509–5517 CrossRef CAS.
- Y. Song, J. He, Y. Zhang, R. A. Gilsdorf and E. Y.-X. Chen, Nat. Chem., 2023, 15, 366–376 CrossRef CAS PubMed.
- H. Luo, Y. Zhou, Q. Li, B. Zhang, X. Cao, J. Zhao and G. Zhang, Macromolecules, 2023, 56, 1907–1920 CrossRef CAS.
- A reviewer is gratefully acknowledged for pointing out this conceptual parallel. See for example: (a) K. Nakano, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2006, 45, 7274–7277 CrossRef CAS PubMed; (b) E. K. Noh, S. J. Na, S. Sujith, S.-W. Kim and B. Y. Lee, J. Am. Chem. Soc., 2007, 129, 8082–8083 CrossRef CAS PubMed; (c) S. Sujith, J. K. Min, J. E. Seong, S. J. Na and B. Y. Lee, Angew. Chem., Int. Ed., 2008, 47, 7306–7309 CrossRef CAS PubMed; (d) W.-M. Ren, X. Zhang, Y. Liu, J.-F. Li, H. Wang and X.-B. Lu, Macromolecules, 2010, 43, 1396–1402 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |