
Injectable thermoresponsive hydrogels based on (Me)PEG–poly(menthide) amphiphilic block copolymers from bioderived lactone†
Mehmet Onur
Arıcan
 a,
Tuğba
Koldankaya
a,
Serap
Mert
abc,
Handan
Çoban
d,
Sezgi
Erdoğan
a and
Olcay
Mert
*ad
a,
Tuğba
Koldankaya
a,
Serap
Mert
abc,
Handan
Çoban
d,
Sezgi
Erdoğan
a and
Olcay
Mert
*ad
aDepartment of Polymer Sci. and Technol., Kocaeli University, 41001, Kocaeli, Turkey. E-mail: olcay.mert@kocaeli.edu.tr; Tel: +902623032018
bCenter for Stem Cell and Gene Therapies Res. and Pract., Kocaeli University, Turkey
cDepartment of Chemistry and Chemical Processing Technol., Kocaeli University, Turkey
dDepartment of Chemistry, Kocaeli University, Turkey
First published on 13th February 2023
Abstract
In conjunction with the rise in cancer incidence-mortality and handicaps of conventional poly(ethylene glycol)-based polylactide, poly(lactide-co-glycolide), or poly(ε-caprolactone) (PEG-based PLA, PLGA, or PCL) injectable thermoresponsive hydrogel platforms, formulating novel biomaterials exploiting sustainable resources for local drug release purposes has currently become critical. From this point of view, we synthesized MePEG–poly(menthide) (MePEG–PM) diblock and poly(menthide)–PEG–poly(menthide) (PM–PEG–PM) triblock copolymers through ring-opening polymerization of (−)-menthide (70%), acquired from (−)-menthone, a readily accessible ketone derivative of the natural product (−)-menthol, using MePEG and PEG as initiators and Sn(Oct)2 as a catalyst with high conversions (>97%), narrow molecular weight distributions (1.12–1.22), and monomodal GPC traces. The molecular weights of MePEG–PM diblock and PM–PEG–PM triblock copolymers evaluated by GPC and calculated from 1H NMR were close to the theoretical values and increased linearly with increasing monomer-to-initiator ratios. Structural determination of the copolymers was performed by comprehensive analyses via two-dimensional 1H–1H COSY and 1H–13C HMQC techniques. The critical point in the thermoresponsive phase transition behavior was found to be the length of the PM component, which was meticulously tuned during the synthesis of MePEG–PM and PM–PEG–PM. Specifically, injectable thermoresponsive hydrogels based on these diblock and triblock copolymers prepared with lower (Me)PEG/PM ratios were found to be suitable copolymer formulations for local therapy applications as they showed fluid characteristics (sol form) at around 40–44 °C and turned into a gel form after cooling to body temperature. Moreover, the hydrolytic degradation of block copolymers in PBS at two different pH values (6.5 and 7.4) at 37 °C resulted in very high degradations (>50% at 30 days), indicating quite impressive results considering the copolymers to be used in local drug delivery systems.
1. Introduction
Cancer can be characterized by the growth of abnormal cells beyond normal limits, which can then invade adjacent environments of the body and/or spread to other organs because of genetic factors as well as external agents such as physical, chemical, and biological carcinogens. Millions of people are diagnosed with cancer each year and almost 10 million deaths or roughly one in six deaths occurred worldwide in 2020 due to cancer.1,2 Systemic chemotherapy is the most common therapeutic method in cancer treatment. In this method, drugs are taken in maximum tolerable doses and transferred directly into the systemic circulation through the vein, which results in serious toxicities in healthy tissues ranging from neutropenia to cardiomyopathy. Besides, only a fraction of the administered dose can reach tumor cells, which limits therapeutic efficacy and increases toxicity in healthy tissues.3 In this regard, drug delivery/release systems have been developed to increase the effectiveness of chemotherapy and reduce its systemic side effects, and therefore have shown particular promises in the past decades. Among them, especially local drug delivery systems have aroused special interest in cancer treatment. This method increases the effectiveness of therapy by preventing chemotherapeutic drugs in systemic circulation, reducing toxicity in normal tissues, and providing local, constant, and controlled release.3–7Injectable polymeric hydrogels are in the form of a fluid aqueous solution prior to administration, but when injected, they show rapid gel formation under physiological conditions.8–11 There are two main categories of injectable hydrogels, physical and chemical, depending on the type of cross-linking. Chemically cross-linked hydrogels are formed using Schiff bases, enzymes, Michael-addition reactions, and photopolymerization. However, the applications of chemically cross-linked hydrogels are restricted by the necessity for enzymes, crosslinking chemicals, photoinitiators, and/or organic solvents during the fabrication process despite their high mechanical characteristics. On the other hand, physically cross-linked ones can be fabricated in response to environmental conditions including the pH, temperature, glucose, electric field, magnetic field, or combinations of these by using amphiphilic block copolymers. They do not involve any chemical reaction, thus providing a mild environment during the preparation of hydrogels. Moreover, the biocompatibility problems caused by monomer or initiator residues observed in some chemical cross-linked hydrogels are not encountered in physically cross-linked hydrogels.8,11,12 In particular, temperature-responsive hydrogels have received a lot of attention as injectable materials owing to their self-gelling characteristics exploiting body temperature without requiring any extra chemical treatment among stimulus-responsive physically cross-linked hydrogels. These in situ hydrogel systems used in drug release offer easy production with low cost, as well as fast, painless, and easy application thanks to small needle sizes. Before the administration, the carrier is in a flowable form with a low viscosity, making the process easier and not too painful for the patient, and gelation occurs at the tumor site just after being administered in the fluid form.8,11,13
Temperature-dependent reversible gel to sol transitions of the block copolymers consisting of PEG and biodegradable polyester were drastically influenced by the copolymer concentration in an aqueous phase. Accordingly, the concentrated copolymer solutions form a gel at a lower temperature and a sol at a higher temperature.14–16 At high concentration levels, the hydrophobic segments of block copolymer chains associate with each other through hydrophobic–hydrophobic interactions, that is, by packing of the hydrophobic polyester blocks, resulting in copolymer gelation. However, the chain packing structure of the gel could be disrupted by the partial dehydration and shrinkage of the PEG chains at elevated temperatures, resulting in a decrease in micelle volume. This reduces the attractive forces between micelles and allows the gel to flow. The second important point in the fluidization of the gel is that chains in the polyester–PEG block copolymer are prone to be more mobile and diffusible at high temperatures.16
The first study on temperature-responsive hydrogel platforms was carried out using poly(N-isopropyl acrylamide) (PNIPAAm) in 1967, and the reversible phase transition behavior of PNIPAAm was investigated.17,18 PNIPAAm and its copolymers are one of the most employed polymers for utilization in the area of drug delivery systems.9 However, it should be considered that PNIPAAm-based hydrogels are not biodegradable, and thus may accumulate in the body and cause toxic effects.12,17 Pluronic or poloxamer triblock copolymers, poly(ethylene glycol)/poly(propylene glycol) (PEG/PPG), as potential drug carriers are also quite widely studied because they have excellent biocompatibility and temperature responsive characteristics. Nonetheless, PEG/PPG-based copolymers suffer from their non-biodegradability, high permeability, short retention times of a few days in the body (i.e., they show rapid gel-erosion properties, thus not suitable for long-term drug release applications), and poor mechanical properties.17,19,20 Therefore, extensive efforts have been made to incorporate biodegradable fractions into PEG blocks to formulate biodegradable injectable systems.
Jeong et al. first synthesized PEG–PLLA diblock and PEG–PLLA–PEG triblock copolymers having sol–gel transition with decreasing temperature in 1997.14 This pioneering work of Jeong et al. started a new trend for injectable biomaterials, and subsequently many biodegradable injectable systems have been prepared with various macromolecular structures such as diblock, triblock, multiblock, and graft architectures.11 In a study carried out by our group, temperature-responsive PLLA–mPEG (polylactide–methoxy poly(ethylene glycol)) was employed to prevent the conversion of a biologically active lactone form of camptothecin (CPT) and topotecan (TPT) anticancer drugs into their toxic carboxylate form in aqueous medium at pH 7.4.21 Nevertheless, PLA based copolymers were not convenient for applications requiring short-term drug release due to the semicrystalline nature of PLA units.22 Hydrogels produced from PEG-based poly(lactide-co-glycolide) (PLGA) copolymers are another popular class of temperature-responsive biodegradable injectable systems. Zentner et al. compared the release behavior of a paclitaxel anti-cancer drug from poloxamer F-127 and PLGA–PEG–PLGA (23 wt%, OncoGel™: Regel®/paclitaxel) and found that paclitaxel is released from pluronic F-127 and PLGA–PEG–PLGA in one day and approximately 50 days, respectively.23 This result indicates that the PLGA–PEG–PLGA hydrogel provides a better and more controlled release of paclitaxel. In another study, the local simultaneous release of OncoGel™ and temozolomide dramatically prolongs the survival of a rodent 9L gliosarcoma model without any systemic or neurological toxicity.24 However, transferring or weighing PLGA-based temperature-responsive copolymers is often problematic because of their sticky paste morphology. Additionally, the requirement of several hours for PLGA–PEG–PLGA triblock copolymers to dissolve in water and an extremely slow redissolution/reconstitution process limit their biomedical applications.25,26
Although plenty of PLA- and/or PLGA-based temperature-responsive hydrogels have been reported as injectable hydrogels so far, these kinds of systems still have some limitations as aforementioned. For this reason, new kinds of hydrogel platforms, poly(substituted glycolide)-based (PSG) biomaterials, were prepared by our group to tailor the characteristics of PLA- and/or PLGA-hydrogels in a controlled manner and investigate their biological functions via leveraging the chemical strategies.22,27,28 In the first study, the temperature-responsive features of poly(diisopropyl glycolide)–PEG (PDIPG–PEG) di- and tri-block copolymers were examined for localized drug delivery purposes. However, 57% of paclitaxel was released in two months from the prepared paclitaxel-loaded PDIPG–PEG hydrogels owing to the semicrystalline structure of PDIPG blocks.28 In another study conducted by our group, amine-functionalized PLA–PEG block copolymers were designed to address the problems caused by non-functionalized PLA and PLGA copolymers such as not enough binding sites for biological molecules except for end groups, slow biodegradation, and a slow drug release profile due to high hydrophobicity. When amine-functional PLA–PEG hydrogels were compared with traditional PLA–PEG hydrogels, more effective drug release behavior (up to 95% drug release was achieved at the end of 20 days while this rate is 29% in PLA–PEG hydrogels) and faster degradation in the hydrolytic medium (at the end of 1 month, 47% degradation of amine-functional PLA–PEG copolymers vs. 13.8% degradation of PLA–PEG copolymers) were achieved.27 However, the biggest challenge of these biomaterials is the necessity of the protection of the amine group before the polymerization because unprotected monomers undoubtedly impede the polymerization due to the free amine groups.27 Recently, we engineered temperature-responsive hydrogel-based systems comprising (Me)PEG–PIBL block copolymers using an isobutyl lactide (or isobutyl-methyl glycolide, IBL) monomer with similar strategies as in previous studies.27,28 These systems were found to exhibit a quite good gel–sol transition behavior when heated to around physiological temperature for use in stimulus-responsive local site drug delivery targets. In addition, a more effective drug release profile was achieved with these PIBL-based hydrogels compared to PLA-based systems (5.7% versus 57% at two weeks) because of the intrinsic features of PIBL blocks (i.e., lower glass transition temperature and amorphous structure). Furthermore, there was no cell damage or cell morphological changes when a triblock copolymer was employed in in vitro cell viability assays for testing human primary dermal fibroblasts and L929.22 On the other hand, the degradation of PLA, PLGA, and PSGs (i.e., PDIPG, P(Z)NEtMG (amine-functional PSG), and PIBL) to lactic acid or glycolic acid derivatives resulted in the accumulation of these acids which sometimes leads to the denaturation of biomacromolecules.20,29
To address the problems arising from the above-mentioned PEG–PLA, PEG–PLGA, and PSG copolymers, another important strategy is the production of temperature-responsive hydrogels using hydrophilic PEG and poly(ε-caprolactone) (PCL), an FDA-approved polymer widely studied in biomedical applications. PEG-based PCL hydrogels offer a wide gelation range and are softer. Therefore, one of the advantages of this system over the PLA/PLGA system is the ease of injection without needle clogging.20,26,29 On the other hand, the most important disadvantage of PCL is that it is obtained from petroleum-based sources. Therefore, new chemical-based products are being manufactured utilizing feedstocks from plants as alternative sustainable resources in response to the negative environmental impacts of oil-based materials.
Our primary initial focus was to design and fabricate injectable thermoresponsive hydrogel platforms from sustainable resources for local drug delivery purposes. To this end, research interest in the preparation of polymeric materials with equivalent or better features exploiting renewable monomers is expanding. Terpenes and terpenoids are valuable substances in this setting due to their variety and abundance.30 (−)-Menthol, a natural material valued for its cooling properties when inhaled or administered to the skin, is one example of such a resource. Every year, large quantities of (−)-menthol are extracted from the Mentha arvensis plant for use in the medicinal, flavor and fragrance, and confectionery sectors. Numerous biosynthetic derivatives of (−)-menthol are accessible, and its chemical conversions and derivatives are quite common. One particular example is that (−)-menthone, a commercially available ketone derivative of (−)-menthol, can easily be converted into a seven-membered lactone (−)-menthide via the straightforward Baeyer–Villiger oxidation.31 Hillmyer and coworkers obtained a poly(menthide) (PM) homopolymer with an Mn value up to 91.000 g mol−1 in a controlled manner using zinc-alkoxide (ZnEt2) as the catalyst.31 In addition, PLA–b-PM–b-PLA triblock copolymers were synthesized from D-, L-, and D,L-lactide, respectively, taking into account the stereochemistry effect on biorenewable self-assembled thermoplastic elastomer applications in the presence of dihydroxy PM, obtained from the ring-opening polymerization (ROP) reaction of (−)-menthide with diethylene glycol.32,33 This versatile triblock copolymer has also been reported to be valuable as hydrolytically degradable pressure-sensitive adhesives.34 Thermoplastic elastomers or pressure-sensitive adhesives were also prepared from PM with tulipalin A, a natural material found in tulip Tulipa gesneriana L., or with γ-methyl-α-methylene-γ-butyrolactone.35,36 Polyurethane film formulations were prepared with three armed PMs for use in bio-based thermoset materials for flexible or rigid foams.30
Early studies with biodegradable poly(menthide) polymers obtained from renewable resources have generally focused on the thermoplastic elastomeric behavior of these polymers in the literature.32–36 Considering the importance of cancer treatment and sustainable polymeric materials, herein, we report, for the very first time, temperature-responsive poly(menthide)–PEG block copolymers synthesized from the ROP of (−)-menthide with a hydrophilic and biocompatible poly(ethylene glycol) macro-initiator to evaluate their potential utility as injectable hydrogels at the local site. Thanks to PEG, these temperature-responsive hydrogels showed injectable fluid characteristics at around 40–44 °C and formed a gel at body temperature, indicating that these systems could be potential candidates for local drug delivery systems.
2. Materials and methods
2.1. Materials
(−)-Menthone (Sigma-Aldrich, 90%) and 3-chloroperbenzoic acid (meta-chloroperbenzoic acid, m-CPBA) (Sigma-Aldrich, 77%) were used as received without any purification in the synthesis of the (−)-menthide monomer. Sodium metabisulfite (sodium disulfite, Na2S2O5) (Merck), sodium carbonate (Na2CO3) (Merck), sodium chloride (NaCl) (Sigma-Aldrich, 99%), and sodium sulfate (Na2SO4) (Sigma-Aldrich, 99%) were used for the purification of (−)-menthide. Celite (Sigma-Aldrich) was utilized to remove the salts formed in the reaction medium during the (−)-menthide synthesis. 4-Methoxybenzaldehyde (p-anisaldehyde, p-methoxybenzaldehyde) (Sigma-Aldrich, 98%) was used in the preparation of p-anisaldehyde stain. Methoxy poly(ethylene glycol) (MePEG-2000) (Fluka) and poly(ethylene glycol) (PEG-2000) (Sigma) homopolymers were used as macro-initiators and stannous octoate (Sn(Oct)2) (Aldrich, 95%) was used as a catalyst. Paclitaxel (Alfa Aesar, 99.5%), chosen as an anticancer drug, was used for drug release studies. Tween 80 (Merck) was used to increase the solubility of paclitaxel in drug release. Ethyl acetate (Sigma-Aldrich, 99.5%) and hexane (Sigma-Aldrich, 95%) were used as mobile phases for column chromatography during monomer synthesis. Tetrahydrofuran (Sigma, 99.9%) was used for sample preparation in GPC analyses. Acetonitrile (Sigma, 99.9%) was filtered and used as a mobile phase for HPLC analysis.2.2. Characterization
ATR-FTIR analyses were performed using an ATR Bruker-Tensor 27 model spectrometer with 30 scans in the range of 600–4000 cm−1 and a resolution of 4 cm−1. NMR analyses used for the structural determination of (−)-menthide and copolymers were performed using a Bruker Avance III 400 MHz NMR spectrometer. The molecular weights and molecular weight distributions (PDI) of copolymers were evaluated using gel permeation chromatography with a Viscotek system equipped with a column furnace with a pump, a RI detector (VE 3580 RI Detector), and 3 columns (two columns 300 × 8 mm Low Org Viscotec LT4000L, and a front column 10 × 4.6 mm Viscotek TGuard, Org Guard Col). It was carried out at 35 °C using a GPCmax Autosampler system. The sample concentration was determined as 5 mg mL−1 in THF and the injection volume was determined as 100 μL. The calibration curve was made with polystyrene standards in the range of 1.2–400 kDa. Data were acquired from the OmniSEC 5.12 program. The thermal characteristics of copolymers were investigated using a Mettler Toledo DSC1 Star System device under a N2 atmosphere in two heating steps at a heating/cooling rate of 10 °C min−1 in a range of −50–140 °C. The thermal degradation behaviors of copolymers were determined by thermogravimetric analyses using a TGA 1 STAR System device under a nitrogen atmosphere at a flow rate of 30 mL min−1. In the analysis, the samples were heated from 25 °C to 600 °C at a rate of 10 °C min−1. The aggregation behavior and hydrodynamic sizes of the (Me)PEG–PM thermoresponsive system were determined with the dynamic light scattering (DLS) technique using a ZetaSizer Nano ZS90 (Malvern Instruments) at two different temperatures. Before the measurements, the prepared hydrogels were filtered through a 0.45 μm pore diameter filter and maintained at relevant temperatures for at least 10 min for equilibration. Rheological measurements were conducted using an Anton Paar modular compact rheometer (MCR302). The solid-like (Me)PEG–PM hydrogels were placed on a precooled plate (∼4 °C). The viscosity (η) and shear stress (τ) of the hydrogels were recorded upon temperature sweeping from 4 °C to 60 °C at a heating rate of 5 °C min−1, a 0.4 mm gap between parallel plates, and at a constant shear rate (1 s−1). Release studies of paclitaxel from the prepared copolymer hydrogels were performed using a VWR incubator microplate shaker at 37 °C and 150 rpm as the shaking speed. Quantitative determinations of paclitaxel loading and released paclitaxel from the hydrogels were performed using a 1260 Infinity Agilent HPLC device comprising of a 4.6 × 150 mm ZORBAX SB-C18 column, a 3.5 μm column and a UV detector. The obtained data were calculated using the Agilent ChemStation software program.2.3. Synthesis of the (−)-menthide monomer (2)
(−)-Menthone (1, 6.13 mL, 31.92 mmol) was added to a solution of 3-chloroperbenzoic acid (mCPBA) (8.63 g, 38.52 mmol) in anhydrous dichloromethane (100 mL) while stirring in an ice bath under an argon atmosphere. The mixture was brought to room temperature, and the reaction was continued at a stirring rate of 1000 rpm for 24 hours. The white precipitates (salts) formed during the reaction were removed using a Celite pad with dichloromethane as an eluent. The filtrate was washed with sodium metabisulfite (10%) (Na2S2O5, 1 × 120 mL), saturated sodium carbonate (Na2CO3, 1 × 120 mL), distilled water (2 × 120 mL), and saturated sodium chloride (NaCl, 1 × 120 mL), respectively. Then, the organic phase was dried with sodium sulfate (Na2SO4) before it was removed with an evaporator under reduced pressure. The yellowish crude product was purified by the column chromatography method using an ethyl acetate/hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) mobile phase (Rf: 0.6). A TLC plate was stained with p-anisaldehyde stain and maintained at 60 °C for about 2 minutes to determine the spots since (−)-menthone and (−)-menthide were not UV active. A colorless liquid product was obtained with 86% yield after column chromatography, and crystallization with ethyl acetate/hexane at −22 °C overnight was performed. Finally, sublimation was performed (70%) to overcome the bimodal molecular distribution problems in the ring-opening polymerization.31,35,371H NMR (400 MHz, CDCl3) δ: 0.91 (2 × d, 2 × CH3); 0.98 (d, CH3); 1.25 (m, CH); 1.55 (m, CH); 1.83 (m, 4 × CH); 2.46 (m, CH2); 4.01 (dd, CH). 13C NMR (100 MHz, CDCl3) δ: 17.15, 18.44, 24.00, 30.47, 30.99, 33.37, 37.49, 42.62, 84.74, 174.93. ATR-FTIR (νmax/cm−1): 2962, 2937, 2916, 2874 (CH); 1713 (C
:5) mobile phase (Rf: 0.6). A TLC plate was stained with p-anisaldehyde stain and maintained at 60 °C for about 2 minutes to determine the spots since (−)-menthone and (−)-menthide were not UV active. A colorless liquid product was obtained with 86% yield after column chromatography, and crystallization with ethyl acetate/hexane at −22 °C overnight was performed. Finally, sublimation was performed (70%) to overcome the bimodal molecular distribution problems in the ring-opening polymerization.31,35,371H NMR (400 MHz, CDCl3) δ: 0.91 (2 × d, 2 × CH3); 0.98 (d, CH3); 1.25 (m, CH); 1.55 (m, CH); 1.83 (m, 4 × CH); 2.46 (m, CH2); 4.01 (dd, CH). 13C NMR (100 MHz, CDCl3) δ: 17.15, 18.44, 24.00, 30.47, 30.99, 33.37, 37.49, 42.62, 84.74, 174.93. ATR-FTIR (νmax/cm−1): 2962, 2937, 2916, 2874 (CH); 1713 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
O).
2.4. Preparation of MePEG–PM diblock and PM–PEG–PM triblock copolymers
Briefly, Sn(Oct)2 (21.3 mg, 0.05 mmol), MePEG-2000 (3, 240 mg, 0.12 mmol), and (−)-menthide (2, 851.3 mg, 5.00 mmol) were added into the polymerization tube to synthesize MePEG–PM diblock copolymer 8. The reaction was carried out in a solvent-free medium under an argon atmosphere at 140 °C with stirring for four days. The synthesized diblock copolymer was dissolved in a trace amount of dichloromethane (1 mL) and precipitated with excess cold methanol (10 mL) by keeping it at −22 °C overnight. Afterward, it was centrifuged at 12000 rpm −20 °C for 5 minutes, the precipitate was separated by decantation and dried under vacuum. MePEG–PM diblock copolymers 5–7 were also synthesized by the same method as mentioned above. 1H NMR (400 MHz, CDCl3) δ: 0.86 (d, 6H, 2 × CH3); 0.92 (d, 3H, CH3); 1.15 (m, 1H, CH); 1.29 (m, 1H, CH); 1.50 (m, 2H, CH2); 1.80 (m, 1H, CH); 1.92 (m, 1H, CH); 2.05 (m, 1H, CH); 2.29 (m, 1H, CH); 3.36 (s, 3H, –OCH3); 3.63 (s, 4H, 2 × CH2); 4.70 (m, 1H, CH). 13C NMR (100 MHz, CDCl3) δ:17.62, 18.73, 19.82, 28.51, 30.41, 31.20, 32.66, 42.02, 70.61, 78.38, 173.04. ATR-FTIR (νmax/cm−1): 2958, 2880 (CH); 1727 (CO).
PM–PEG–PM triblock copolymers 9–12 having various molecular weights were also obtained by the above-mentioned protocol, except that PEG was used instead of MePEG. 1H NMR (400 MHz, CDCl3) δ: 0.87 (d, 6H, 2 × CH3); 0.92 (d, 3H, CH3); 1.14 (m, 1H, CH); 1.30 (m, 1H, CH); 1.50 (m, 2H, CH2); 1.80 (m, 1H, CH); 1.91 (m, 1H, CH); 2.07 (m, 1H, CH); 2.30 (m, 1H, CH); 3.63 (s, 4H, 2 × CH2); 4.70 (m, 1H, CH). 13C NMR (100 MHz, CDCl3) δ:17.63, 18.74, 19.84, 28.53, 30.42, 31.22, 32.64, 42.07, 70.69, 78.37, 173.00. ATR-FTIR (νmax/cm−1): 2958, 2876 (CH); 1727 (CO).
2.5. Investigation of the thermoresponsive characteristics of MePEG–PM and PM–PEG–PM copolymers
The thermoresponsive features of copolymers were assessed after determining their ability to form hydrogels. For this purpose, suspensions of various concentrations of copolymers in distilled water with different ratios of the (Me)PEG/PM content were tested whether a homogeneous mixture was formed or not. The samples were stored in a refrigerator at 4 °C for an hour to equilibrate before investigating the thermoresponsive properties of (Me)PEG–PM copolymers having suitable compositions and concentrations. Then the prepared hydrogels were examined by the method of inverting the vial in a temperature-controlled water bath by increasing two degrees in the range of 4–60 °C. The critical gel–sol transition temperature of each sample was found when the gel turned into a sol form by inverting the vial containing the copolymers.2.6. Preparation of paclitaxel-loaded hydrogels and drug release studies
The paclitaxel anticancer drug was effectively loaded into the MePEG–PM diblock 6 and PM–PEG–PM triblock 10 hydrogels; both hydrogels show ∼40 °C gel–sol transition temperatures, at a rate of 1 wt%. Preparation of drug-loaded hydrogels was carried out briefly as follows: 1.50 mg of paclitaxel, 150 mg of MePEG–PM diblock copolymer 6, and 350 μL of distilled water were mixed in a 1.5 mL vial with the help of a vortex and a micro-spatula at room temperature until a homogeneous hydrogel was formed (at least 15 minutes). The drug-loaded triblock hydrogel was prepared with 1.1 mg of paclitaxel, 110 mg of PM–PEG–PM triblock copolymer 10, and 390 μL of distilled water. Finally, the drug-loaded hydrogels were maintained at 4 °C for 60 minutes to equilibrate. Then, 500 μL of phosphate-buffered saline containing 1 wt% Tween 80 (PBS/Tween 80, 99/1, w/w) at different pH values (7.4 and 6.5) was added on top of the drug-loaded hydrogels. Drug release studies were carried out by placing these samples in an incubator with a temperature of 37 °C and a stirring speed of 150 rpm. PBS/Tween 80 (99/1, w/w) phases on the hydrogels were collected at the 1st and 24th hours and every 24 hours in separate eppendorfs, and fresh PBS/Tween 80 phases were added onto the hydrogels for 11 days. The collected supernatants were stored at −22 °C until analysis by HPLC. In order to obtain reproducible results in drug release studies, three replicates were studied for each hydrogel.21,22,27,28 The release amounts of the paclitaxel anticancer drug were determined by HPLC analyses using acetonitrile/water (60/40) as the mobile phase at 227 nm. The stock solution of paclitaxel was prepared by weighing 1.5 mg of paclitaxel into a vial and diluting it with 1.5 mL of acetonitrile to get 1000 μg mL−1. The calibration curve was prepared by using 5 different standard solutions in the range of 31.25–500 μg mL−1 by the serial dilution method from the stock solution. Chromatographic conditions: the flow rate was determined to be 1 mL min−1, the injection volume was 20 μL, the retention time was 6 minutes, and the curve equation was found to be “y = 36.44x + 45.13 (R2: 0.99990)”. After preparing the calibration curve, the samples with and without the drug were diluted 1/5 with acetonitrile and analyzed with HPLC. As a result of the analysis, the retention time of the paclitaxel anticancer drug was determined to be 3.7 minutes (Fig. S1†). The data were obtained by using the Agilent ChemStation package program analysis.2.7. Hydrolytic degradation study
The hydrolytic degradation study of MePEG–PM diblock copolymer 6 and PM–PEG–PM triblock copolymer 10 was performed in phosphate-buffered saline (PBS) buffer at pH: 6.5 and 7.4 at 37 °C. Incubation was performed by taking 15 mg of polymer and 4 mL of PBS into a 10 mL test tube. At specified time intervals (10 and 30 days), the copolymers were removed from the incubator and properly washed with distilled water to get rid of salt residues; however, the copolymer was dissolved in the aqueous phase; therefore, all the aqueous phase was lyophilized overnight. They were dissolved in THF, filtered with a 0.45 μm pore-diameter filter, and analyzed with GPC to determine their degradation rates.272.8. Statistical analysis
All data were expressed with the means ± standard deviation (SD) (n = 3) and analyzed using one-way analysis of variance (ANOVA) followed by Tukey's post hoc test with IBM SPSS® Statistics software. The statistical significance was reported as *p < 0.05, **p < 0.01, and ***p < 0.001.3. Results and discussion
3.1. Preparation of block copolymers
Synthesis of (−)-menthide monomer 2 from (−)-menthone 1 was carried out by a simple Baeyer–Villiger reaction, widely used for the oxidation of ketones to esters or lactones in organic synthesis, in the presence of 3-chloroperbenzoic acid (mCPBA) under an inert atmosphere in anhydrous dichloromethane (Scheme 1).31 Structural characterization of compound 2 was previously performed using spectroscopic techniques in the literature (Fig. 2).31,37,38 The data provided for (−)-menthide monomer 2 in these studies include 1H- and 13C-NMR analyses. However, since there are many overlapping CH and CH2e, d, g, and f peaks at the same location in the monomer, detailed mapping for each peak via COSY and HMQC 2D NMR spectroscopy was reported for the first time in this work (Fig. S20 and S21†). The cross-peaks of horizontal and vertical axes in the COSY 2D NMR spectra indicate the interactions of neighboring protons, confirming the junction points. Briefly, the off-diagonal peaks at points 1–6 represent the coupling of protons of “a and b” with “g”, “c” with “f”, “d” with “e”, “e” with “d”, “e” with “i”, and “h” with “f” in the COSY 2D NMR spectrum of (−)-menthide 2 (Fig. S20†). Points 1–5 correspond to the direct proton-carbon shift correlation of g, e, f, d, and h coded signals in (−) menthide 2 in the HMQC 2D NMR spectrum, respectively (Fig. S21†).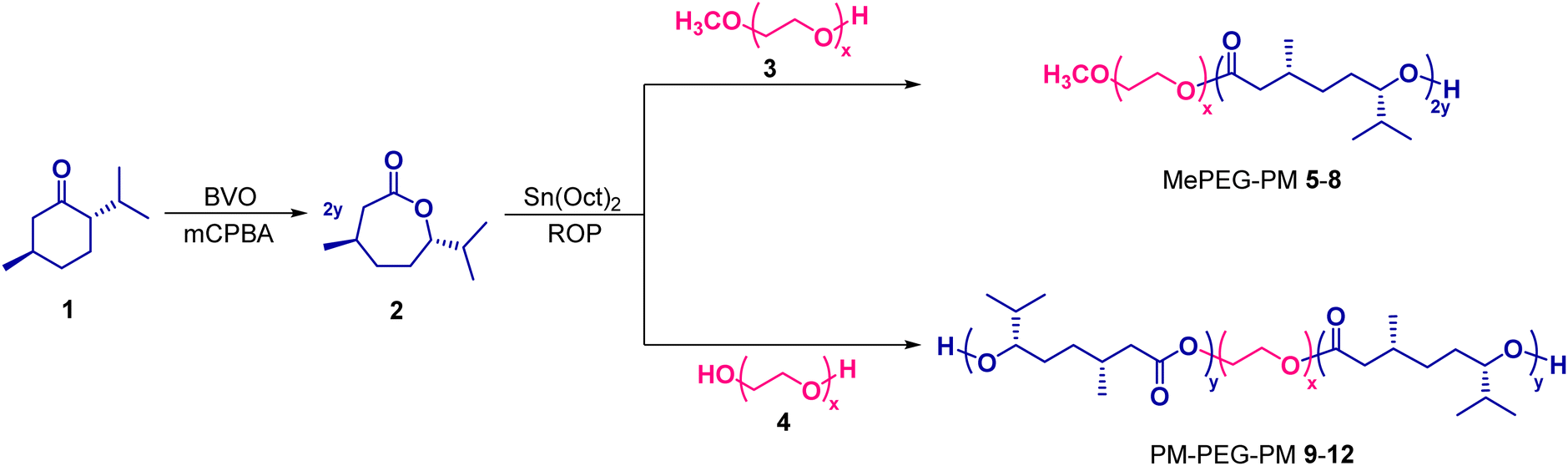 | ||
| Scheme 1 ROP of (−)-menthide catalyzed by stannous octoate in the presence of PEG-based macroinitiators. | ||
MePEG–PM diblock 5–8 and PM–PEG–PM triblock 9–12 copolymers were prepared through the ROP of (−)-menthide 2 using MePEG or PEG as macro-initiators and stannous octoate as the catalyst under an argon atmosphere in a solvent-free medium (Scheme 1 and Table 1). The length of each component ((Me)PEG/PM) during copolymer synthesis was adjusted carefully. The molecular weights of MePEG and PEG macroinitiators used in the synthesis were particularly preferred as 2000 Da. Because high molecular weight PEGs (over ∼10000 Da) were not suitable for filtration through the human kidney membrane due to the large hydrodynamic radius of PEG in an aqueous phase.14,22,28 Furthermore, the molecular weights of (Me)PEG/PM copolymers were maintained in a specific range (usually less than 10000 Da) to make them a homogeneous suspension for the gel–sol transition by varying the mole ratio of (−)-menthide while maintaining a constant mole ratio of the initiator, as shown in Table 1. The thermoresponsive phase transition behavior of the copolymers varies depending on the ratio of hydrophobic and hydrophilic blocks in the chain. If the PM repeating units were high in the copolymer, a non-homogeneous suspension of the copolymer in the aqueous medium was obtained due to the overwhelming hydrophobicity. On the other hand, if the PEG segment was too long in the copolymer, the gel–sol transition temperature increased highly and became higher than the physiological temperature, or the copolymer might lose the sol–gel transition due to its high hydrophilic character.22
| Polymera | [M0]:[I0]:[C] |
Time (day) |
M
n,GPCb (Da) |
M
n,NMRc (Da) |
M
n, thd (Da) |
Đ
Mb |
RUc | RUd |
|---|---|---|---|---|---|---|---|---|
| a All conversions calculated from the 1H NMR spectra of the copolymers using the methine protons of the unreacted monomer (δ = 4.01 ppm) and copolymer (δ = 4.70 ppm) are >97%. b Molecular weight and distribution were determined by GPC with a RI detector, calibrated with linear polystyrene standards using THF as a mobile phase. c Determined from the 1H NMR spectra of the copolymers using the signals of the (Me)PEG block and PM block. d Calculated from conversion using the feed ratio and molecular weight of the monomer and (Me)PEG. RU: repeating unit. | ||||||||
| MePEG–PM 5 | 1/0.12/0.05 | 1 | 3430 | 2850 | 3420 | 1.12 | 5.0 | 8.3 |
| MePEG–PM 6 | 2/0.12/0.05 | 2 | 4650 | 4280 | 4790 | 1.13 | 13.4 | 16.4 |
| MePEG–PM 7 | 3/0.12/0.05 | 3 | 5770 | 5980 | 6230 | 1.22 | 23.4 | 24.9 |
| MePEG–PM 8 | 5/0.12/0.05 | 4 | 8550 | 8230 | 8900 | 1.21 | 36.6 | 40.5 |
| PM–PEG–PM 9 | 1/0.12/0.05 | 1 | 3950 | 3160 | 3420 | 1.08 | 6.8 | 8.3 |
| PM–PEG–PM 10 | 2/0.12/0.05 | 2 | 5110 | 4090 | 4790 | 1.13 | 12.3 | 16.4 |
| PM–PEG–PM 11 | 3/0.12/0.05 | 3 | 6200 | 5320 | 6240 | 1.12 | 19.5 | 24.9 |
| PM–PEG–PM 12 | 5/0.12/0.05 | 4 | 10930 |
9030 | 8920 | 1.15 | 41.3 | 40.7 |
MePEG–PM diblock 5–8 and PM–PEG–PM triblock copolymers 9–12 were obtained with high conversion (>97%) and narrow molecular weight distributions (ĐM: 1.08–1.22), and the molecular weight of the block copolymers acquired from the GPC and NMR analyses were found to be quite compatible with the theoretical values. Moreover, the molecular weight of the copolymers increased with an increase in the mole of (−)-menthide, indicating the well-controlled polymerization although longer times are required at higher mole ratios (Table 1). The shift of the retention volume of copolymers to the left in the GPC chromatograms was accurately correlated with increasing molecular weights (Fig. 1). It is noteworthy that there was an absence of unreacted (Me)PEG even before the purification steps of all copolymers (data not shown), proving that the homopolymerization of (−)-menthide 2 did not occur under the selected polymerization conditions.
 | ||
| Fig. 1 Chromatographic characterization. Overall GPC chromatograms of diblock copolymers 5–8 (A) and triblock copolymers 9–12 (B). | ||
Di- and triblock copolymers 8 and 12 showed strong bands related to both the PEG and PM blocks in the ATR-FTIR spectrum (Fig. 2B). The –C–H vibrations of PM and the –CH2– vibrations of the (Me)PEG parts of the copolymers overlapped at around 2900 cm−1. The characteristic carbonyl (CO) stretching vibration shifted from 1713 cm−1 in the (−)-menthide monomer to 1727 cm−1 in both copolymers (Fig. 2B). Similar peak assignments were also obtained in MePEG–PM diblock copolymers 5–7 and PM–PEG–PM triblock copolymers 9–11 in the ATR-FTIR spectra, as shown in the ESI (Fig. S2–S7†).
 | ||
| Fig. 2 Spectroscopic characterization-1. Schematic representation for the location of NMR signals in compounds 2, 8, and 12 (A). Overall ATR-FTIR (B), 1H NMR (C), and 13C NMR spectra (D) of compounds 2, 8, and 12. | ||
The resonances in 4.70 ppm (–CH– (i)) and 0.9–2.3 ppm ranges (–CH3 (a, b, and c), –CH2 (d, e, and h), and –CH– (f and g) signals) belong to PM blocks while the signals at 3.36 (k) and 3.63 ppm (l) are characteristic methylene protons (–OCH2CH2O) within the PEG block and methoxy protons (–OCH3) at the end of the PEG block (if MePEG used), respectively, when the 1H NMR spectra of MePEG–PM diblock copolymer 8 and PM–PEG–PM triblock copolymer 12 were examined (Fig. 2C). The shift of the signal at 4.01 ppm belonging to the methine proton (i) in the (−)-menthide monomer to 4.70 ppm in the copolymers indicates the successful ring-opening polymerization in the presence of (Me)PEG. The multiplet peak (m) in the range of 4.15–4.30 ppm represented the α-methylene protons of PM-connecting PEG units (PM-COO-CH2-CH2–) and the protons of the hydroxyl end groups (–OCOCH2CH(CH3)CH2CH2CH(iPr)OH).22,27,28 Various carbon resonances related to PM blocks in copolymers 8 and 12 appeared at 78 ppm (–CH– (i)) in the range of 17–42 ppm (–CH3– (a, b, and c), –CH2– (d, e, and h), and –CH– (f and g) signals), and at 173 ppm (–CO (j)), respectively, while methylene carbon in the (Me)PEG units resonated at 71 ppm in the 13C NMR spectrum. Particularly, the peaks of methine (i) and carbonyl carbons (j) appearing at 85 ppm and 175 ppm in the monomer (2) shifted to 78 ppm and 173 ppm in copolymers 8 and 12, respectively (Fig. 2D). All of these findings confirmed the successful synthesis of diblock 8 and triblock 12 copolymers. Similar observations were obtained in the 1H and 13C NMR spectra of MePEG–PM diblock 5–7 and PM–PEG–PM triblock copolymers 9–11, as can be seen in the ESI (Fig. S8–S19†).
The resonance order of the –CH and –CH2 groups in the monomer differed in the polymers because of the relieving ring strain as a result of ring opening in the presence of PEG. In order to confirm the above assignments, di- and triblock copolymers were also analyzed by two-dimensional NMR techniques: COSY and HMQC. The off-diagonal peaks at points 1–5 indicate the coupling of neighboring protons of “a and b” with “g”, “c” with “f”, “e” with “i”, “g” with “i”, and “h” with “f” in the 1H–1H COSY 2D NMR spectrum of di- and triblock copolymers (Fig. 3A and S22†). Direct proton-carbon shift correlation of e, f, g, d, and h coded signals indicated as points 1–5, respectively, confirms skeleton connectivities and assignments as mentioned above for di- and triblock copolymers when the HMQC 2D NMR spectrum was examined (Fig. 3B and S23†).
 | ||
| Fig. 3 Spectroscopic characterization-2. COSY 2D NMR (A) and HMQC 2D NMR (B) spectra of MePEG–PM diblock copolymer 7. | ||
The thermal features of MePEG–PM diblock and PM–PEG–PM triblock copolymers were investigated by DSC analyses to examine the effect of each block length on Tg and Tm (Fig. 4A and B). PM is an amorphous polymer with a glass transition temperature at −25 (°C).33 On the other hand, MePEG 3 and PEG 4 are semicrystalline polymers that exhibit melting endotherms at 53.2 °C and 54.3 °C, respectively (Fig. 4A and B). It was observed that these copolymers exhibited the characteristic features of both PM and MePEG blocks in the DSC thermograms (Table 2). With the introduction of PM into the PEG chain, the Tm of the PEG unit decreased from 53.2 °C to 52.0 °C in copolymer 5. As the ratio of [M0]:[I0] increased, the Tm of the PEG block decreased from 52.0 °C in copolymer 5 to 50.4 °C and then 44.7 °C in copolymers 6 and 8, respectively. Similar behaviors were observed in the triblock copolymers as well. In other words, as the molecular weight of the PM chain in the block copolymer increases, the Tm of the PEG unit decreases from 54.3 °C to 44.4 °C in copolymer 9, 42.4 °C in copolymer 10, and finally 30.6 °C in copolymer 12. The longer the PM block length, the lower the Tm and the degree of crystallinity of (Me)PEG as reflected by the melting enthalpy values (ΔHm). The fact that the presence of PM blocks attached to PEG blocks lowers the melting temperature of PEG units, indicating that the crystallization of the component in copolymers is significantly affected by the presence of the other moiety.22,27,28 The glass transition of copolymers having the lowest PM block length (i.e., MePEG–PM diblock copolymer 5 and PM–PEG–PM triblock copolymer 9) couldn't be observed in the DSC thermograms. However, it was also noted that the Tg values of both diblock and triblock copolymers, which were quite close to that of the PM homopolymer, increased as the PM block length in the copolymer increased.
 | ||
| Fig. 4 Thermal characterization. Overall DSC thermograms of MePEG 3, MePEG–PM diblock copolymers 5, 6, and 8 (A), PEG 4, and PM–PEG–PM triblock copolymers 9, 10, and 12 (B), and overall TGA thermograms of compounds 3–6, 8–10, and 12 (C). | ||
| Polymer |
T
ga (°C) |
T
ma (°C) |
ΔHa (J g−1) | PEG | PM |
Y
cb (%) |
PEG/PMb (%) | PEG/PMc (%) |
M
nc |
||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
T
onsetb (°C) |
T
maxb (°C) |
T
onsetb (°C) |
T
maxb (°C) |
||||||||
| NO: not observed.a Measured by DSC analysis after the second heating run.b Determined by the TGA analysis. Yc: char yield after 600 °C heating. Tonset: initial decomposition temperature. Tmax: temperature at the maximum mass loss.c Calculated from the 1H NMR spectra of the copolymers. | |||||||||||
| MePEG 3 | NO | 53.2 | 176.5 | 380 | 401 | — | — | 1.5 | 100–0 | 100–0 | 2000–0 |
| MePEG–PM 5 | NO | 52.0 | 96.5 | 373 | 406 | 240 | 283 | 5.9 | 68–32 | 70–30 | 2000–850 |
| MePEG–PM 6 | −27.1 | 50.4 | 73.0 | 381 | 407 | 297 | 315 | 4.7 | 53–47 | 47–53 | 2000–2280 |
| MePEG–PM 8 | −23.5 | 44.7 | 36.8 | 386 | 401 | 299 | 347 | 3.3 | 26–74 | 24–76 | 2000–6230 |
| PEG 4 | NO | 54.3 | 197.9 | 378 | 398 | — | — | 0.2 | 100–0 | 100–0 | 0–2000–0 |
| PM–PEG–PM 9 | NO | 44.4 | 70.2 | 370 | 404 | 224 | 300 | 6.7 | 64–36 | 63–37 | 580–2000–580 |
| PM–PEG–PM 10 | −31.1 | 42.4 | 60.0 | 381 | 404 | 305 | 329 | 6.8 | 57–43 | 49–51 | 1045–2000–1045 |
| PM–PEG–PM 12 | −19.4 | 30.6 | 23.0 | 381 | 400 | 287 | 347 | 3.5 | 24–76 | 22–78 | 3515–2000–3515 |
When the thermal stability behaviors of MePEG–PM diblock and PM–PEG–PM triblock copolymers were investigated by TGA analyses, two-stage degradation behavior was noted (Fig. 4C). The decomposition of the PM block resulting from the breakdown of ester bonds was observed at ∼220–350 °C in the first stage while the degradation of ether bonds in the PEG chains was detected at ∼370–400 °C in the second stage. In addition, the mass% loss values acquired from TGA analyses were consistent with the data obtained from the 1H NMR spectrum (Table 2). Another result revealed from the TGA analyses was that the stability of copolymers increased with the increase of the chain length of the PM block. For instance, while the Tmax value of diblock copolymer 5 (Mn,NMR: 2850 Da) was 283 °C, the Tmax values of diblock copolymers 6 (Mn,NMR: 4280 Da) and 8 (Mn,NMR: 8230 Da) were found to be 315 °C and 347 °C, respectively. Similarly, the Tmax values increased from 300 °C in triblock copolymer 9 (Mn,NMR: 3160 Da) to 329 °C in triblock copolymer 10 (Mn,NMR: 4090 Da), and finally 347 °C in triblock copolymer 12 (Mn,NMR: 9030 Da).
3.2. Determination of the gel–sol transition temperatures of hydrogels
We aim to determine suitable hydrogels that exhibit sol behavior for injection in the range of 40–44 °C and can quickly form gels when cooled to body temperature. Thus, various concentrations of MePEG–PM diblock and PM–PEG–PM triblock copolymers were tested to determine gel–sol transition temperatures using the test tube inverting method. To illustrate this, the representative gel-to-sol transition images of MePEG–PM diblock hydrogel 6 at a concentration of 28.5% with increasing temperatures are given in Fig. 5A. Diblock hydrogel 6 showed a gel form between 25 °C and 38 °C (1), started to liquefy at 40 °C (2), and lastly turned into a sol form at 44 °C (3) as a result of the decreasing interaction between the hydrophobic poly(menthide) chains (Fig. 5A). It was observed that if the hydrophobic unit (poly(menthide)) in the copolymer chain is considerably higher than the hydrophilic unit ((Me)PEG), a homogeneous mixture cannot be obtained even at very low concentrations for MePEG–PM diblock 7 and 8 and PM–PEG–PM triblock 11 and 12 copolymers.21,22,27,28 All in all, MePEG–PM diblock hydrogels 5 and 6 and PM–PEG–PM triblock hydrogels 9 and 10 showed the desired gel–sol phase transition at around 40–44 °C at concentrations of 33.5%, 28.5%, 44%, and 19.5%, respectively. Different trends in the transition curves compared to our previously published work27 were obtained with the (Me)PEG–PM hydrogels, in other words, much steeper slopes were observed in this work. These results are attributed to the less water uptake tendency of (Me)PEG–PM hydrogels in comparison with amine-functional PLLA-based hydrogels having more hydrophilic structures because of the amine groups on the polymer backbone.15 As can be seen in Fig. 5B, the aqueous solutions of MePEG–PM diblock hydrogels 5 and 6 and PM–PEG–PM triblock hydrogels 9 and 10 were in the sol phase at low concentration levels and high temperatures and were in the gel phase at high concentration levels and low temperatures. It was also concluded that an increase in the PM block length in copolymers considerably affects the copolymer concentrations for the same gel–sol transition temperature, as expected. For example, a suitable gel–sol transition temperature (40 °C) was obtained at 33.4% for MePEG–PM diblock hydrogel 5 while it was at 28.3% for MePEG–PM diblock hydrogel 6 (Fig. 5B). | ||
| Fig. 5 Representative gel-to-sol transition images of diblock hydrogel 6 at the temperature ranging from 25 °C to 45 °C (A). Gel–sol transition temperatures of diblock hydrogels 5 and 6 and triblock hydrogels 9 and 10 at different concentrations (B). Overall release profiles of diblock hydrogel 6 and triblock hydrogel 10 at different pH values during 11 days (C). Viscosity and shear stress of diblock hydrogel 6 at 28.5% concentration (D) and triblock hydrogel 10 at 19.5% concentration as a function of temperature (E). DLS curves of diblock hydrogel 6 (1.0 wt%) (F) and triblock hydrogel 10 in water (1.0 wt%) at different temperatures (G). | ||
Furthermore, oscillatory rheological measurements of MePEG–PM diblock hydrogel 6 and PM–PEG–PM triblock hydrogel 10 were examined at the determined concentrations to evaluate the mechanical properties (i.e., viscosity and shear stress). As shown in Fig. 5D and E, diblock hydrogel 6 showed higher viscosity and shear stress compared to triblock hydrogel 10, which could be ascribed to the higher concentration of the diblock hydrogel (870 Pa s/Pa at 28.5% vs. 70 Pa s/Pa at 19.5% concentration levels).39 The viscosity and shear stress values of diblock hydrogel 6 gradually decreased from ∼870 Pa s/Pa to 18 Pa s/Pa as the temperature increased until 50 °C indicating that the intermolecular interactions (i.e., hydrogen bonding and dipole–dipole interactions) between the hydrophobic PM blocks decreased, and then a slight increase in the values was observed between 50 °C and 60 °C but this increase was still not enough to turn into a gel form considering the values of initial gel states (Fig. 5D). In contrast, triblock hydrogel 10 exhibited a different behavior from diblock hydrogel 6. In other words, temperature-dependent increases were observed in the values of viscosity and shear stress up to 50 °C, and then a sharp decrease occurred in the range of 50 °C–60 °C. Even though this type of behavior is obtained in systems showing a sol-to-gel transition,39 the curve seen here only refers that the amount of aggregation in the system increased as the temperature increased and then decreased pronouncedly over 50 °C, which proves a gel-to-sol-transition, because our sample was in the gel state at the beginning of the analysis at 4 °C. Of note, both hydrogels exhibited slightly higher gel-to-sol transition temperatures during the rheology analyses as compared to the results in the test tube inverting method (i.e., 50 °C vs. 44 °C). This could be explained by the heating rate or ageing time of the samples prior to analysis by both methods.40,41
It has been widely acknowledged that these kinds of amphiphilic block copolymers self-assemble into micelles consisting of a hydrophobic core (i.e., PM) and a hydrophilic shell (i.e., PEG) in an aqueous solution.42 Accordingly, DLS analyses of the diluted solutions of (Me)PEG–PM block copolymers in water (1.0 wt%) as a function of temperature were performed to understand the microstructure of the thermoresponsive system (Fig. 5F and G). Based on the findings, the size of micellar aggregates increased as the PM unit increased in the block copolymer because of the stronger attraction between hydrophobic PM units, as expected (Table S1†).43 In accordance with the gel to sol transition mechanism, the hydrodynamic sizes of diblock hydrogels 5 and 6 and triblock hydrogel 9 slightly decreased with an increase in the temperature from 25 °C to 50 °C except for somehow slight increase in triblock hydrogel 10 (Table S1,†Fig. 5F, G, and S24†), which was also compatible with the results obtained from rheology measurements (Fig. 5D and E). A temperature higher than 50 °C was also examined for DLS analyses; however, the hydrogels were fully dehydrated at 60 °C within 10 min and precipitated because of the fully compact packing of hydrophobic chains and the removal of absorbed water,16 which also explains why we observe increasing modulus between 50 °C and 60 °C while examining diblock hydrogel 6 (Fig. 5D). In summary, (Me)PEG–PM thermoresponsive core–shell structures form a gel due to the chain packing characteristics of hydrophobic PM and undergo a sol transition upon heating due to the partial dehydration and shrinkage of the PEG chains, leading to a decrease in the micelle volume, reducing the attractive forces between micelles, and finally allows the gel to flow.15,16
3.3. Release studies of the paclitaxel anticancer drug from hydrogels
Fig. 5C shows the release profile of paclitaxel from MePEG–PM diblock hydrogel 6 and PM–PEG–PM triblock hydrogel 10. MePEG–PM diblock hydrogel 6 and PM–PEG–PM triblock hydrogel 10 showed 45.3% and 38.2% of the drug release by the end of 10 days at pH 7.4, respectively, without any significant burst effect. The drug release from MePEG–PM diblock hydrogel 6 was faster in the first days and then the release rate slowed down, whereas PM–PEG–PM triblock hydrogel 10 showed a controlled release of paclitaxel in a close ratio every day from the beginning to end (Fig. 5C). The rapid release of diblock hydrogel 6 compared to triblock hydrogel 10 could be attributed to the much faster 10-day degradation data of diblock hydrogel 6 described below. Therefore, paclitaxel may have been released more rapidly by breaking the ester bonds in PM in diblock copolymer 6. Overall, both the copolymer hydrogels (i.e., 6 and 10) maintained their integrity during the 10 days of paclitaxel release but the hydrogel dimensions started to decrease after 10 days. On the other hand, it was found that the hydrogel size decreased faster when copolymers 5 and 9 having lower molecular weights were tested for drug release studies (data not shown). This situation might be related to the amorphous nature of the copolymers and the high content of the hydrophilic PEG moiety compared to the PM unit in the block chains.22 Furthermore, the effect of pH of the release medium on drug release was evaluated, and no major changes were observed except for small differences due to not being their pH-responsive characteristics. Namely, a bit more drug was released from diblock hydrogel 6 at pH 6.5 in the first five days while no significant difference was observed between days 6 and 11. In contrast, for triblock hydrogel 10, a slightly larger amount of the drug was released at acidic pH during the period of days 6–10, whereas there was no significant difference in drug release in the first five days and the last day. Statistical significance data are shown in the ESI (Fig. S25†).3.4. Investigation of the decomposition behaviors of copolymers in hydrolytic media
The degradation behaviors of MePEG–PM diblock copolymer 6 and PM–PEG–PM triblock copolymer 10, tested in drug release studies, were investigated in PBS at pH values 6.5 and 7.4 at 37 °C and at two different time intervals (10 and 30 days). (Table 3). The degradation rate and polydispersity indexes (ĐM, Mw/Mn) of the copolymers increased with time elapsed. The hydrolytic degradation of the poly(menthide) homopolymer was quite slow and it also retained 96.5% of its original molecular weight at the end of 45 weeks based on the SEC measurements.44 With the incorporation of PLA units to both ends, the polylactide–poly(menthide)–polylactide copolymer (7.6-33-7.6) retained 90% of its original molecular weight at the end of 12 weeks and degraded by almost 50% at the end of 45 weeks.44 Notably, the hydrophilic (Me)PEG initiator showed a significant effect on the degradation of MePEG–PM diblock copolymer 6 and PM–PEG–PM triblock copolymer 10 in the current work (Fig. 6 and Table 3). Fig. 6A, B, D and E show the GPC chromatograms of MePEG–PM diblock copolymer 6, PM–PEG–PM triblock copolymer 10, and their 10 days and 30 days of degradation. The retention volumes of the diblock and triblock copolymers shifted higher values from 15.78 and 15.62 to 15.87 and 15.65 in 10 days and 15.92 and 15.83 in 30 days at pH 6.5. A similar degradation behavior was observed at pH 7.4. Thus, the number average molecular weight of copolymers 6 and 10 decreased as degradation time increased at pH 6.5 and pH 7.4 (Table 3 and Fig. 6C, F). Furthermore, the GPC traces of the block copolymers showed that the distribution curves had a tail toward lower molecular weight, as shown in Fig. 6A, B, D and E. Both the copolymers showed very high degradation rates in all pH media, reaching 54–57% molecular weight loss while the molecular weight distribution (ĐM) increased from 1.13 to 1.20–1.25 in 4 weeks (Table 3 and Fig. 6C, F), which indicates quite impressive results considering the copolymers as drug delivery vehicles. In addition, MePEG–PM diblock copolymer 6 degraded significantly more than PM–PEG–PM triblock copolymer 10 at the end of day 10 but to a similar degree at day 30. The reason for this can be explained as follows: the PEG moiety of MePEG–PM diblock copolymer 6 is only attached at one end to the PM blocks, and thus, it has more freedom of movement and is more prone to swelling.45 | ||
| Fig. 6 Decomposition profiles of MePEG–PM diblock copolymer 6 at pH 6.5 (A) and at pH 7.4 (B), decomposition profiles of PM–PEG–PM triblock copolymer 10 at pH 6.5 (D) and at pH 7.4 (E), and Mn and ĐM values of MePEG–PM diblock copolymer 6 (C) and PM–PEG–PM triblock copolymer 10 (F) during 30 days of hydrolytic degradation in PBS at different pH values at 37 °C (*pH: 7.4). | ||
| Polymer | Temperature (°C) | Time (day) | pH |
M
n,GPCa (Da) |
Đ
Ma |
Loss%a |
Retention volumea (mL) |
|---|---|---|---|---|---|---|---|
| a Determined by GPC analysis. Equation of “[(Man,0 − I) − (Man,t − I)]/(Man,0 − I) × 100” was used for the calculation of Mn loss% of the copolymers. | |||||||
| MePEG–PM 6 | 37 | 0 | 4650 | 1.13 | — | 15.78 | |
| 10 | 6.5 | 4090 | 1.16 | 25.81 | 15.87 | ||
| 30 | 3410 | 1.25 | 57.14 | 15.92 | |||
| 10 | 7.4 | 4120 | 1.15 | 24.42 | 15.87 | ||
| 30 | 3460 | 1.22 | 54.84 | 15.93 | |||
| PM–PEG–PM 10 | 37 | 0 | 5110 | 1.13 | — | 15.62 | |
| 10 | 6.5 | 4730 | 1.18 | 14.45 | 15.65 | ||
| 30 | 3670 | 1.20 | 54.75 | 15.83 | |||
| 10 | 7.4 | 4800 | 1.15 | 11.79 | 15.67 | ||
| 30 | 3700 | 1.19 | 53.61 | 15.85 | |||
4. Conclusion
Cancer cases and death rates continue to increase day by day despite significant advances and efforts in cancer research to reduce cancer risks. The low solubility and the systemic side effects of currently used anticancer agents in cancer treatment have pushed scientists towards better alternatives with new solutions. Local drug delivery systems developed for this objective have a substantial place in cancer treatment since 85% of all cancers are solid tumors. More effective treatment is provided by protecting healthy tissues with localized drug release systems and controlled drug release in the target tumor area. With the intention of advancing to this profession, (Me)PEG–PM diblock and triblock copolymers with various compositions were synthesized with high conversions (>97%) and narrow molecular weight distributions (1.12–1.22) under inert conditions in a solvent-free medium by the ROP of (−)-menthide utilizing a Sn(Oct)2 catalyst in the presence of biocompatible MePEG and PEG macroinitiators. According to the findings, the formation of homogeneous hydrogels was highly dependent on the length of hydrophobic poly(menthide) units in the copolymer composition. MePEG–PM diblock copolymers 5 and 6 and PM–PEG–PM triblock copolymers 9 and 10 were homogenously suspended while hydrogel formation could not be achieved with MePEG–PM diblock 7 and 8 and PM–PEG–PM triblock 11 and 12 copolymers. It was also revealed that the gel–sol transition temperatures shifted to lower concentrations as the PM composition in the copolymer increased. 40% and 45% of the total paclitaxel were released from the diblock and triblock hydrogels in 10 days. Finally, the existence of MePEG/PEG units in MePEG–PM and PM–PEG–PM improved the hydrophilicity of the copolymers and facilitated rapid degradation based on the degradation studies. The pH of the medium also exerted a slight effect on the release and degradation behaviors of the block copolymers. The degradation rates of copolymers from the GPC analysis, 12–26% at the end of 10 days and 54–57% at the end of 30 days, are quite important because they demonstrate how quickly hydrogels will degrade if employed inside the body. The fact that the novel thermoresponsive hydrogels fabricated within the scope of this study, which are ready to be injectable sol forms at around 40–44 °C, solidified by cooling to body temperature, exhibit sustained drug release, and degrade rapidly in the hydrolytic environment, suggests that these materials could serve as promising drug carriers in the treatment of local solid tumors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Scientific and Technological Research Council of Turkey (TUBITAK) (project number: 119Z137) and by Kocaeli University (BAP 2016/073 HD). We thank Prof. Dr. Mehmet Kodal from Kocaeli University for the DSC, TGA, and rheometer access; Rumeysa Yıldırım and Gizem Urtekin from Kocaeli University for help with the DSC and TGA analyses; and Tuğba Erol from Anton Paar for help with the rheology analyses.References
- X. Ma and H. Yu, Yale J. Biol. Med., 2006, 79, 85–94 Search PubMed.
- World, Health Organization “Cancer, Key Facts; What causes cancer?”. https://www.who.int/news-room/fact-sheets/detail/cancer. Last access date: 29 Ağustos 2022.
- R. De Souza, P. Zahedi, C. J. Allen and M. Piquette-Miller, Drug Delivery, 2010, 17, 365–375 CrossRef CAS PubMed.
- G. Chang, T. Ci, L. Yu and J. Ding, J. Controlled Release, 2011, 156, 21–27 CrossRef CAS PubMed.
- T. Ci, L. Chen, L. Yu and J. Ding, Sci. Rep., 2014, 4, 5473 CrossRef CAS PubMed.
- A. Fakhari and J. Subramony, J. Controlled Release, 2015, 220, 465–475 CrossRef CAS PubMed.
- M. Norouzi, B. Nazari and D. W. Miller, Drug Discovery Today, 2016, 21, 1835–1849 CrossRef CAS PubMed.
- C. T. Huynh, M. K. Nguyen and D. S. Lee, Macromolecules, 2011, 44, 6629–6636 CrossRef CAS.
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2002, 54, 37–51 CrossRef CAS PubMed.
- E. Ruel-Gariepy and J. C. Leroux, Eur. J. Pharm. Biopharm., 2004, 58, 409–426 CrossRef CAS PubMed.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- C. He, S. W. Kim and D. S. Lee, J. Controlled Release, 2008, 127, 189–207 CrossRef CAS PubMed.
- K. Krukiewicz and J. K. Zak, Mater. Sci. Eng., C, 2016, 62, 927–942 CrossRef CAS PubMed.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CrossRef CAS PubMed.
- B. Jeong, D. S. Lee, J.-I. shon, Y. H. Bae and S. W. Kim, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 751–760 CrossRef CAS.
- H. Mao, G. Shan, Y. Bao, Z. L. Wu and P. Pan, Soft Matter, 2016, 12, 4628–4637 RSC.
- S. S. Liow, A. A. Karim and X. J. Loh, MRS Bull., 2016, 41, 557–566 CrossRef CAS.
- J. S. Scarpa, D. D. Mueller and I. M. Klotz, J. Am. Chem. Soc., 1967, 89(24), 6024–6030 CrossRef CAS.
- C. Gong, S. Shi, P. Dong, B. Kan, M. Gou, X. Wang, X. Li, F. Luo, X. Zhao, Y. Wei and Z. Qian, Int. J. Pharm., 2009, 365, 89–99 CrossRef CAS PubMed.
- H. Hyun, Y. H. Kim, I. B. Song, J. W. Lee, M. S. Kim, G. Khang, K. Park and H. B. Lee, Biomacromolecules, 2007, 8, 1093–1100 CrossRef CAS PubMed.
- O. Mert, G. Esendağlı, A. L. Doğan and A. S. Demir, RSC Adv., 2012, 2, 176–185 RSC.
- D. Çetin, M. O. Arıcan, H. Kenar, S. Mert and O. Mert, Macromolecules, 2021, 54, 272–290 CrossRef.
- G. M. Zentner, R. Rathi, C. Shih, R. C. Mc, M.-H. Seo, H. Oh, B. G. Rhee, J. Mestecky, Z. Moldoveanu, M. Morgan and S. Weitman, J. Controlled Release, 2001, 72, 203–215 CrossRef CAS PubMed.
- A. K. Vellimana, V. R. Recinos, L. Hwang, K. D. Fowers, K. W. Li, Y. Zhang, S. Okonma, C. G. Eberhart, H. Brem and B. M. Tyler, J. Neurooncol., 2013, 111, 229–236 CrossRef CAS PubMed.
- M. J. Hwang, J. M. Suh, Y. H. Bae, S. W. Kim and B. Jeong, Biomacromolecules, 2005, 6, 885–890 CrossRef CAS PubMed.
- G. Ma, B. Miao and C. Song, J. Appl. Polym. Sci., 2010, 116, 1985–1993 CrossRef CAS.
- M. O. Arıcan, S. Erdoğan and O. Mert, Macromolecules, 2018, 51, 2817–2830 CrossRef.
- M. O. Arıcan and O. Mert, RSC Adv., 2015, 5, 71519–71528 RSC.
- A. Basu, K. R. Kunduru, S. Doppalapudi, A. J. Domb and W. Khan, Adv. Drug Delivery Rev., 2016, 107, 192–205 CrossRef CAS PubMed.
- S. A. Gurusamy-Thangavelu, S. J. Emond, A. Kulshrestha, M. A. Hillmyer, C. W. Macosko, W. B. Tolman and T. R. Hoye, Polym. Chem., 2012, 3, 2941–2948 RSC.
- D. Zhang, M. A. Hillmyer and W. B. Tolman, Biomacromolecules, 2005, 6, 2091–2095 CrossRef CAS PubMed.
- C. L. Wanamaker, M. J. Bluemle, L. M. Pitet, L. E. O'Leary, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2009, 10, 2904–2911 CrossRef CAS PubMed.
- C. L. Wanamaker, L. E. O'Leary, N. A. Lynd, M. A. Hillmyer and W. B. Tolman, Biomacromolecules, 2007, 8, 3634–3640 CrossRef CAS PubMed.
- J. Shin, M. T. Martello, M. Shrestha, J. E. Wissinger, W. B. Tolman and M. A. Hillmyer, Macromolecules, 2011, 44, 87–94 CrossRef CAS.
- K. Ding, A. John, J. Shin, Y. Lee, T. Quinn, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2015, 16, 2537–2539 CrossRef CAS PubMed.
- J. Shin, Y. Lee, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2012, 13, 3833–3840 CrossRef CAS PubMed.
- J. A. Wilson, S. A. Hopkins, P. M. Wright and A. P. Dove, Biomacromolecules, 2015, 16, 3191–3200 CrossRef CAS PubMed.
- V. Alphand and R. Furstoss, Tetrahedron: Asymmetry, 1992, 3, 379–382 CrossRef CAS.
- H. F. Darge, A. T. Andrgie, E. Y. Hanurry, Y. S. Birhan, T. W. Mekonnen, H. Y. Chou, W. H. Hsu, J. Y. Lai, S. Y. Lin and H. C. Tsai, Int. J. Pharm., 2019, 572, 118799 CrossRef CAS PubMed.
- C. Gong, Z. Qian, C. Liu, M. Huang, Y. Gu, Y. Wen, B. Kan, K. Wang, M. Dai, X. Li, M. Gou, M. Tu and Y. Wei, Smart Mater. Struct., 2007, 16, 927–933 CrossRef CAS.
- C. B. Liu, C. Y. Gong, M. J. Huang, J. W. Wang, Y. F. Pan, Y. D. Zhang, G. Z. Li, M. L. Gou, K. Wang, M. J. Tu, Y. Q. Wei and Z. Y. Qian, J. Biomed. Mater. Res., Part B, 2008, 84, 165–175 CrossRef PubMed.
- D. S. Lee, M. S. Shim, S. W. Kim, H. Lee, I. Park and T. Chang, Macromol. Rapid Commun., 2001, 22, 587–592 CrossRef CAS.
- C. Chassenieux, T. Nicolai and L. Benyahia, Curr. Opin. Colloid Interface Sci., 2011, 16, 18–26 CrossRef CAS.
- C. L. Wanamaker, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2009, 10, 443–448 CrossRef CAS PubMed.
- S. Li, H. Garreau, B. Pauvert, J. McGrath, A. Toniolo and M. Vert, Biomacromolecules, 2002, 3, 525–530 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01452a |
| This journal is © The Royal Society of Chemistry 2023 |