
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-accelerated depolymerization catalyzed by Eosin Y†
Valentina
Bellotti
 ab,
Kostas
Parkatzidis
b,
Hyun Suk
Wang
b,
Nethmi
De Alwis Watuthanthrige
b,
Matteo
Orfano
a,
Angelo
Monguzzi
a,
Nghia P.
Truong
b,
Roberto
Simonutti
a and
Athina
Anastasaki
*b
ab,
Kostas
Parkatzidis
b,
Hyun Suk
Wang
b,
Nethmi
De Alwis Watuthanthrige
b,
Matteo
Orfano
a,
Angelo
Monguzzi
a,
Nghia P.
Truong
b,
Roberto
Simonutti
a and
Athina
Anastasaki
*b
aDepartment of Material Science, University of Milano-Bicocca, Via R. Cozzi, 55, 20125 Milan, Italy
bLaboratory of Polymeric Materials, Department of Materials, ETH Zurich, Vladimir-Prelog-Weg-5, Zurich, Switzerland. E-mail: athina.anastasaki@mat.ethz.ch
First published on 20th December 2022
Abstract
Retrieving the starting monomers from polymers synthesized by reversible deactivation radical polymerization has recently emerged as an efficient way to increase the recyclability of such materials and potentially enable their industrial implementation. To date, most methods have primarily focused on utilizing high temperatures (typically from 120 °C to 180 °C) to trigger an efficient depolymerization reaction. In this work, we show that, in the presence of Eosin Y under light irradiation, a much faster depolymerization of polymers made by reversible addition–fragmentation chain-transfer (RAFT) polymerization can be triggered even at a lower temperature (i.e. 100 °C). For instance, green light, in conjunction with ppm amounts of Eosin Y, resulted in the accelerated depolymerization of poly(methyl methacrylate) from 16% (thermal depolymerization at 100 °C) to 37% within 1 hour, and finally 80% depolymerization after 8 hours, as confirmed by both 1H-NMR and SEC analyses. The enhanced depolymerization rate was attributed to the activation of a macroCTA by Eosin Y, thus resulting in a faster macroradical generation. Notably, this method was found to be compatible with different wavelengths (e.g. blue, red and white light irradiation), solvents, and RAFT agents, thus highlighting the potential of light to significantly improve current depolymerization approaches.
Introduction
An attractive prospect for polymers synthesized by reversible deactivation radical polymerization (RDRP)1,2 is the possibility of reversing the polymerization and regenerating the monomers through a self-immolative approach.3 Previously, the vast majority of research focused on backbone degradation, which yields lower molecular weight products,3–7 and it is only recently that depolymerization back to monomers has attracted considerable attention and has been classified by IUPAC as one of the top 10 emerging technologies in chemistry.8 Regeneration of monomers can be particularly advantageous from a sustainability perspective as it allows the retrieved materials to be subsequently reused to either reconstruct the starting polymer or to create a new material.9–11 In order to achieve efficient unzipping of polymer chains, high end-group fidelity has to be first ensured during the respective polymerizations. This is shown by the fact that minimal depolymerization was observed when the end-groups were removed.12,13 As such, the vast majority of investigated depolymerizable polymers were synthesized by either reversible addition–fragmentation chain-transfer (RAFT) polymerization or atom transfer radical polymerization (ATRP),12–19 two of the most widely used RDRP techniques.20–23In seminal independent works by the groups of Gramlich18 and Raus,24 they showed that the depolymerization of bottlebrush polymers synthesized by either RAFT or ATRP was possible at relatively low temperatures (70 °C and 90 °C respectively). Although the perspective of these contributions was in favour of the polymerization of macromonomers by suppressing depolymerization, they set the foundation for subsequent depolymerization studies which primarily focused on the depolymerization of non-bulky polymers. For example, Ouchi and co-workers reported the depolymerization of poly(methyl methacrylate) (PMMA), previously synthesized by ATRP, in the presence of a ruthenium catalyst achieving up to 24% conversion at 120 °C.17 Matyjaszewski's group was able to significantly enhance the depolymerization yields (up to 67%) at 170 °C using copper catalysis.13,14 In the RAFT arena, our group recently reported the near-quantitative (up to 92%) depolymerization of a range of non-bulky, functional and cross-linked polymethacrylates at 120 °C.12,15 Furthermore, we were able to reuse the regenerated components either for resynthesizing the initial polymer or instead for creating a new material. It is noted that the regeneration of non-bulky monomers is typically triggered at increased temperatures (≥120 °C) when compared to those for the regeneration of macromonomers. In particular, PMMA yielded 86% of its monomer within 8 h at 120 °C. However, when the reaction was conducted at 100 °C, a much slower depolymerization rate was observed and the final yield reduced to 64%.12 We envisaged that we could improve not only the depolymerization rate but also the overall depolymerization conversion by leveraging the advantages of light catalysis, which have recently attracted much attention.25–33 Recent works by the groups of Boyer and others in the photoinduced electron/energy transfer (PET)-RAFT polymerization offer the possibility of utilizing photoredox catalysts as alternatives to activate a CTA under light irradiation.34–39 Homolytic cleavage of the C–S bond results in radical generation without the addition of exogenous radical sources,40,41 which could be advantageous for the depolymerization reaction. Among the variety of photoredox catalysts tested so far, Eosin Y is a well-known non-toxic organic metal-free catalyst employed for a variety of organic reactions42 and is the basis of our study. Herein, we report for the first time a light-accelerated depolymerization catalyzed by Eosin Y of PMMA with various solvents and CTAs under irradiation at various wavelengths.
Results and discussion
To initiate our study, we first repeated the thermal depolymerization of PMMA at 100 °C.12 It is noteworthy that the starting polymer was synthesized by RAFT polymerization using 2-cyano2-propyl dithiobenzoate as the chain transfer agent at 70 °C, resulting in a well-defined polymer with low dispersity (Đ = 1.14, Mn = 6300) and high end-group fidelity (Fig. S1 and S2†).43 The polymer was then rigorously purified by precipitation and no vinyl protons could be observed via1H-NMR, thus confirming the successful removal of any remaining monomers. The PMMA macroCTA was then subjected to our previously reported depolymerization conditions (100 °C, 5 mM)12 and in line with the original publication, a very slow depolymerization profile was observed with only 16% monomer generation achieved in the first 1 h, as can be seen in Fig. S3.† The total depolymerization conversion under thermal conditions did not surpass 66% at 8 h, after which no additional monomer regeneration was detected. Inspired by the previous works by Boyer and co-workers in PET-RAFT polymerization, we subsequently repeated the depolymerization under green light irradiation (λmax = 510 nm, Fig. S4†) in the presence of Eosin Y (Fig. 1a). To be able to simultaneously heat at 100 °C and irradiate, we constructed a home-made photothermal set-up, in which a strip of green LEDs was carefully positioned to circle around an oil bath, as illustrated in Fig. 1b. In the initial experiment, 12.5 ppm of Eosin Y with respect to the MMA repeating unit was employed, and there was significant acceleration of the depolymerization rate in the system. Within 1 h, a conversion of 26% was reached, a meaningful increase compared to the 16% reached with heat alone. However, the reaction conversion reached a plateau at 3 h (45% conversion) after which the conversion did not significantly increase. Although the final depolymerization conversion was similar compared to that of the thermal experiment, the presence of light and Eosin Y (12.5 ppm) considerably enhanced the depolymerization rate. Encouraged by these preliminary data, we then increased the Eosin Y concentration to 50 ppm. This resulted in not only a further increase in the depolymerization rate (37% conversion in 1 h), but also an increase in the final depolymerization conversion from 45% to 61% (Fig. 1c). In a similar way, when 100 ppm photocatalyst was employed, an even faster depolymerization rate was evident in the kinetics, accompanied by a final depolymerization conversion of 80% (Fig. 1e). Increasing the Eosin Y concentration to 150 ppm did not lead to further enhancement of the final depolymerization yield, although the rate of the reaction continued increasing (Fig. 1c). As the final depolymerization conversion was not improved at a higher Eosin Y loading, all subsequent experiments were conducted utilizing 100 ppm. It is worth mentioning that this is an uncontrolled depolymerization in which the polymer chains rapidly unzip back to the monomers, as can be seen in our SEC analysis (Fig. 1d), in a similar fashion to that in previous reports.12,13,15,21 Nevertheless, the rate of the depolymerization can be efficiently regulated by altering the photocatalyst concentration. Taken together, our current data unambiguously show that under irradiation of green light in the presence of Eosin Y, the depolymerization temperature can be lowered from 120 °C to 100 °C without compromising the final depolymerization conversion. Moreover, the photothermal system allowed significant acceleration of the depolymerization when compared with the purely thermal system (from 16% to 37% depolymerization within 1 h). For all the depolymerizations, we used a light intensity of 2.31 mW cm−2 (Table S1†), while when lower intensities were employed (i.e. 0.18 mW cm−2), slower depolymerization was observed, thus further highlighting the importance of light to accelerate the depolymerization. However, both experiments in the presence of light led to a significant acceleration of the rate when compared to the control experiment in the dark (Fig. S7†). PMMA with a higher degree of polymerization (DP) was also synthesized (DP = 260) and was subsequently subjected to our depolymerization conditions, reaching 62% conversion within 8 h (Fig. S8†). It is noteworthy that this is an improvement in the final depolymerization conversion when compared to our previously reported thermal approach.12 Moreover, under our optimized conditions, we depolymerized different polymers including poly benzyl methacrylate (PBzMA), poly dimethylaminoethyl methacrylate (PDMAEMA) and poly oligo (ethylene glycol) methyl ether methacrylate (POEGMA), resulting in very high conversions in all the cases (68–80%) (Fig. S9†).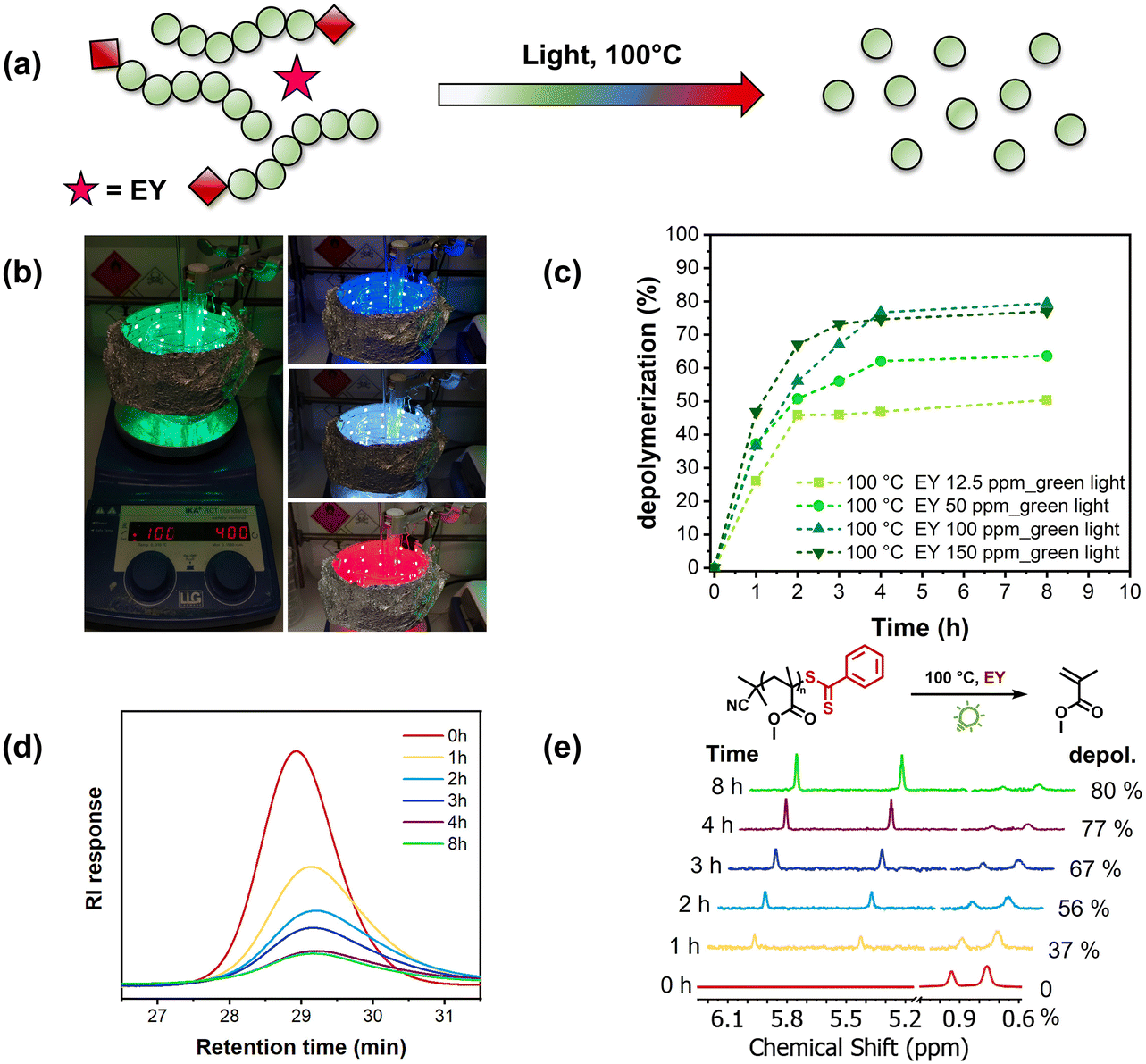 | ||
| Fig. 1 (a) Schematic illustration of reversing RAFT polymerization in the presence of Eosin Y under light irradiation. (b) Photothermal reactor set-up at different wavelengths. (c) Kinetics of light-accelerated depolymerization catalyzed by different concentrations of Eosin Y under green light. (d) Corresponding SEC traces of PMMA during depolymerization with 100 ppm Eosin Y. (e) 1H NMR spectra of the PMMA depolymerization, vinyl peak appearances over time confirming the monomer regeneration. Reactions were run in 1,4-dioxane at 5 mM and 100 °C in the presence of Eosin Y under green light. | ||
To expand the scope of this depolymerization study, we screened a number of different solvents under green light irradiation in the presence of 100 ppm Eosin Y. Reactions were conducted in dimethyl sulfoxide (DMSO), dimethyl formamide (DMF), toluene, and xylene (Fig. 2a, Fig. S11†). Xylene and toluene showed higher final depolymerization conversions (i.e. 52% and 50% respectively) when compared with DMF (i.e. 35%), in line with our previous report on thermal depolymerization.15 Interestingly, DMSO showed a final conversion of 82% within the same time frame, which is a very remarkable increase from the 35% conversion obtained by thermal depolymerization at 120 °C. This experiment suggests a much more efficient radical formation at the chain-end which is perhaps unsurprising considering that DMSO is the best solvent to conduct a PET-RAFT polymerization.44–46 In addition, DMSO facilitates proton extraction in the neutral form of Eosin Y to give the more catalytically active monoanionic and dianionic forms, increasing the photoredox performance.47 The possibility of conducting depolymerizations in different solvents with very high conversions expands the scope and applicability of the current approach.
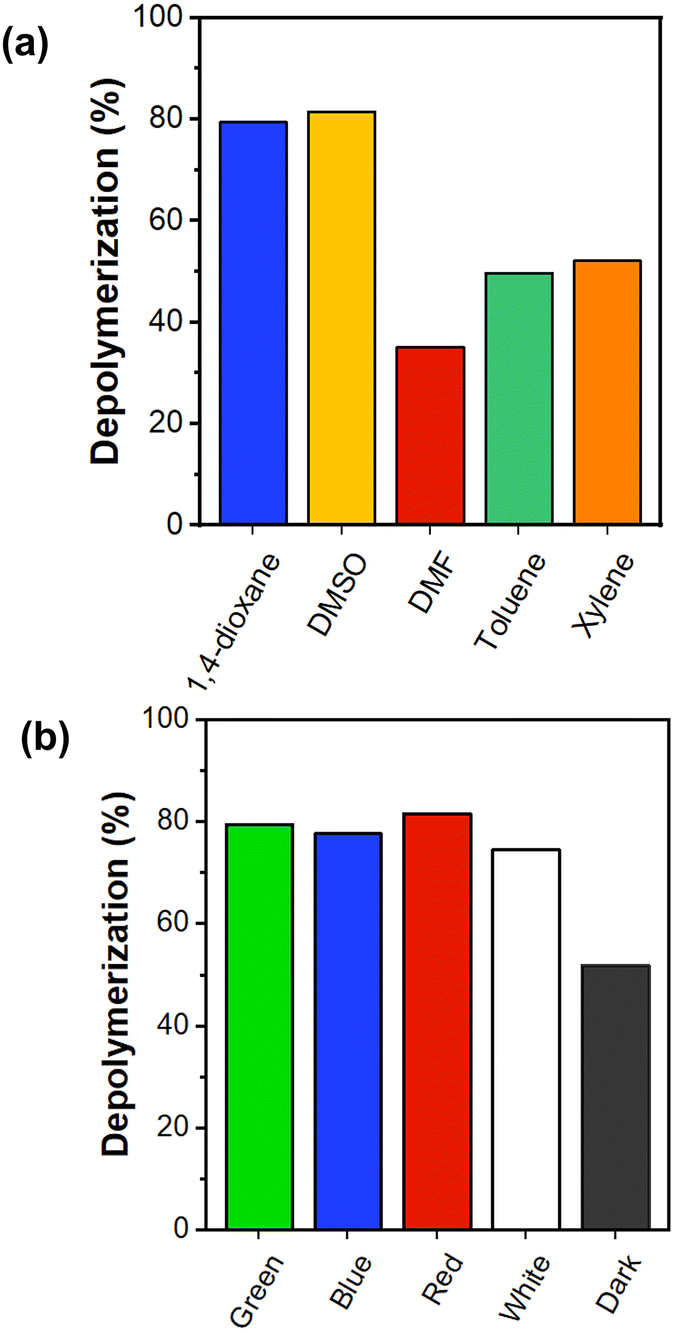 | ||
| Fig. 2 Depolymerization conversion after 8 hours of PMMA-DTB with 100 ppm Eosin Y at 5 mM and 100 °C using (a) different solvents under green irradiation, and (b) different wavelength irradiation in 1,4-dioxane. | ||
One of the particularly advantageous characteristics of PET-RAFT polymerization is its ability to be conducted under irradiation at a range of wavelengths.48 As such, we were interested in exploring the possibility of triggering an efficient depolymerization reaction by irradiating at different wavelengths. In particular, green light was compared with blue, red and white lights (Fig. S4 and S5†). As a control experiment, we performed the depolymerization in the dark under otherwise identical conditions. Fig. 2b shows that in the presence of 100 ppm Eosin Y under completely dark conditions at 100 °C, a maximum of 52% conversion could be reached within an 8 h timeframe. Pleasingly, all the different wavelengths employed resulted in a significantly high depolymerization conversion (Fig. S10†). For example, under blue light irradiation, a total conversion of 78% was detected after 8 h while white light resulted in slightly lower depolymerization conversions reaching 75%. Interestingly, the depolymerization proceeded in an efficient fashion even under red light irradiation, resulting in 82% conversion. This is rather surprising considering that the absorption of Eosin Y at this wavelength is incredibly low (Fig. S6†). In fact, usually other photocatalysts rather than Eosin Y are used under red light irradiation.49,50 However, it has been found that even though the best excitation wavelength is selected through the UV-visible spectrum, some catalysts might display effective photocatalysis at different wavelengths depending on the pathways available for intersystem crossing for their excited states (S1).51 Hence, it can be assumed that even this small absorption of red light irradiation appears adequate to excite Eosin Y to the singlet state which undergoes intersystem crossing, thus activating the C–S bond of the macroCTA and accelerating the depolymerization rates and yields. From these experiments, it can be concluded that regardless of the selected wavelength, any type of visible light irradiation investigated led to improved depolymerization conversions. These results significantly expand the application pool as different wavelengths can be used depending on the application. Since the RAFT chain-end was previously shown to undergo degradation under UV irradiation,52–54 the depolymerization experiments were only performed in the visible regime.
Last but not least, we studied the effect of the RAFT chain-end groups on the depolymerization rates and final depolymerization conversions. To do so, two more PMMAs were synthesized by RAFT polymerization by employing 2-cyano 2-propyl dodecyltrithiocarbonate (TTC) and 2-cyanobutan-2-yl 4-chloro-3,5-dimethyl-1H-pyrazole-1-carbodithioate (pyrazole-CD) as the RAFT agents (Fig. S12–S15†). Considering that comparable data were obtained for all the investigated wavelengths and that green light is the most commonly utilized light source for Eosin Y,42 we performed the subsequent experiments under green light irradiation. The TTC-macroCTA was first subjected to our previously optimized depolymerization conditions (i.e. 100 °C, green light, 100 ppm Eosin Y, 5 mM repeating unit concentration). Impressively, 50% monomer generation was detected in 1 h, which is a remarkable rate acceleration over the identical experiment performed exclusively under heat, which led to only 13% conversion (Fig. 3a). In addition, the thermal depolymerization led to a final conversion of 42% while the presence of green light/Eosin Y significantly enhanced the depolymerization conversion to 68%. Similar behaviour was also observed during the depolymerization of the pyrazoleCD-macroCTA (Fig. 3b). When only heat was applied, 14% depolymerization was detected in 1 h while this percentage doubled (31%) in the presence of green light/Eosin Y. Furthermore, while the thermal depolymerization resulted in a final conversion of 47%, the presence of green light/Eosin Y resulted in total depolymerization of 58% under comparable conditions. Our data highlight that the light/catalytic-depolymerization methodology can also be applied to various macroCTAs, thus expanding the potential of the current approach.
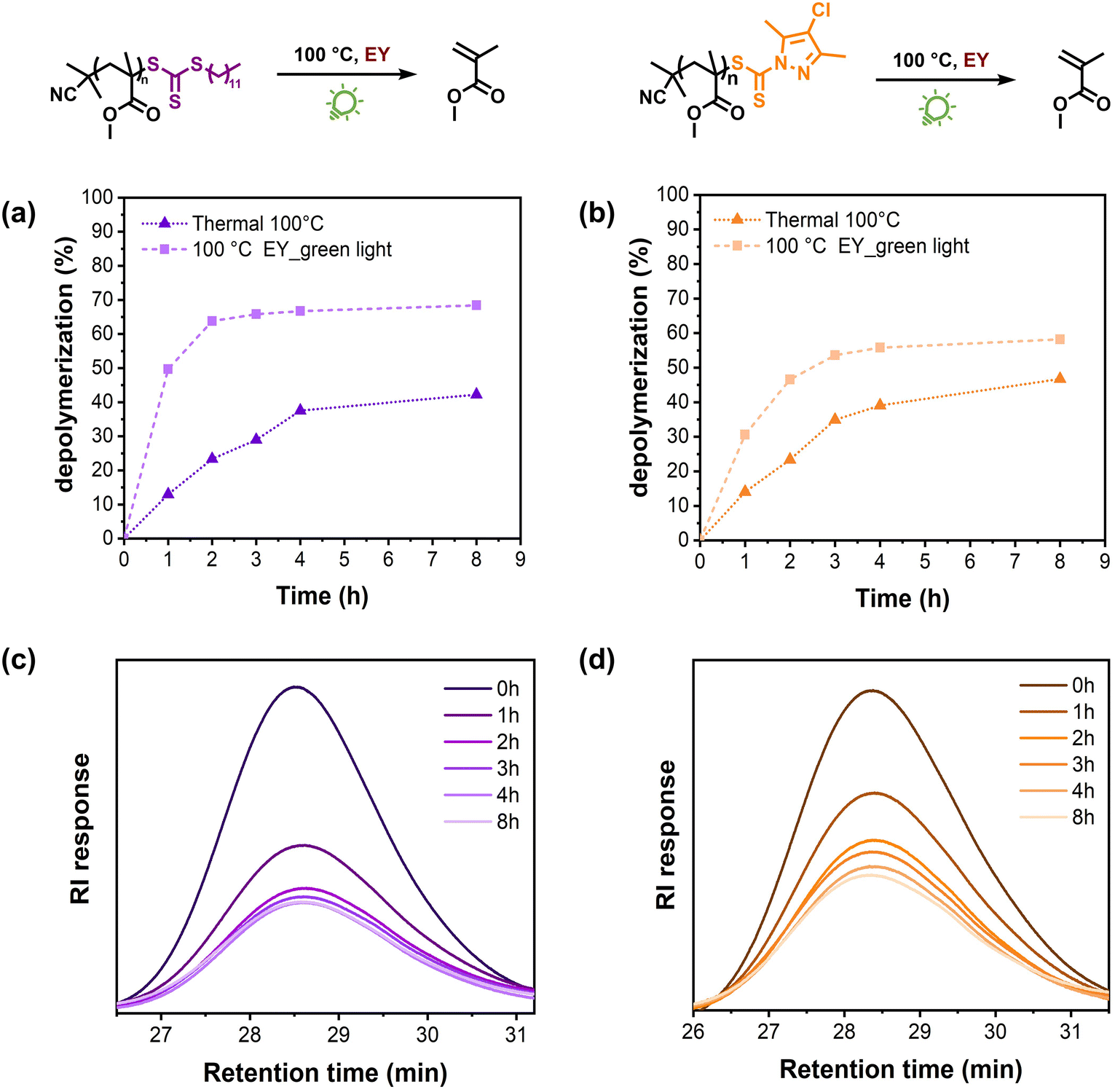 | ||
| Fig. 3 Kinetic study comparing green light-accelerated depolymerization of PMMA catalysed by Eosin Y at 100 °C versus thermal depolymerization at 100 °C using (a) TTC and (b) pyrazoleCD chain transfer agents, respectively. SEC traces at different depolymerization times (c) and (d). | ||
Conclusions
To summarize, in this work we report the first example of light-accelerated depolymerization in the presence of a photocatalyst. We show that under light irradiation and ppm concentrations of Eosin Y, faster depolymerization rates and higher overall depolymerization conversions can be achieved when compared to exclusively thermal depolymerization. Notably, our approach is applicable to different solvents with DMSO showing much higher depolymerization conversions (up to 82%) than those of current reports on thermal depolymerization at 120 °C, thus highlighting the potential of light to provide superior data over thermal depolymerizations. Notably, our approach operates at different wavelengths, ranging from blue, green, red, to white light irradiation, and can also be applied to different macroCTAs, further demonstrating the importance of light in efficient depolymerization strategies.Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. A. gratefully acknowledges the ETH Zurich for the financial support. KP thanks the Onassis Foundation as this scientific paper was partially supported by the Onassis Foundation – Scholarship ID: FZQ051-1/2020-2021. H. S. W. acknowledges the award of the Swiss Government Excellence Scholarship (ESKAS No. 2020.0324). N. D. A. W. acknowledges the financial support received from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 101030516. N. P. T. acknowledges the award of a DECRA Fellowship from the ARC (DE180100076). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (DEPO: Grant Agreement No. 949219).References
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef.
- A. Parkatzidis, K. Wang, H. S. Truong and N. P. Anastasaki, Chem, 2020, 6, 1575–1588 Search PubMed.
- M. R. Martinez and K. Matyjaszewski, CCS Chem., 2022, 4, 2176–2211 CrossRef.
- G. G. Hedir, C. A. Bell, N. S. Ieong, E. Chapman, I. R. Collins, R. K. O'Reilly and A. P. Dove, Macromolecules, 2014, 47, 2847–2852 CrossRef.
- N. De Alwis Watuthanthrige, J. A. Reeves, M. T. Dolan, S. Valloppilly, M. B. Zanjani, Z. Ye and D. Konkolewicz, Macromolecules, 2020, 53, 5199–5207 CrossRef CAS.
- J. A. Reeves, M. L. Allegrezza and D. Konkolewicz, Macromol. Rapid Commun., 2017, 38, 1–5 CrossRef PubMed.
- T. Nwoko, N. De Alwis Watuthanthrige, B. Parnitzke, K. Yehl and D. Konkolewicz, Polym. Chem., 2021, 12, 6761–6770 RSC.
- IUPAC Top Ten Emerging Technologies in Chemistry, https://iupac.org/what-we-do/top-ten/.
- C. Shi, L. T. Reilly, V. S. P. Kumar, M. W. Coile, S. R. Nicholson, L. J. Broadbelt, G. T. Beckham and E. Y. X. Chen, Chem, 2021, 7, 2896–2912 CAS.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed.
- H. S. Wang, N. P. Truong, Z. Pei, M. L. Coote and A. Anastasaki, J. Am. Chem. Soc., 2022, 144, 4678–4684 CrossRef CAS PubMed.
- M. R. Martinez, F. De Luca Bossa, M. Olszewski and K. Matyjaszewski, Macromolecules, 2022, 55, 78–87 CrossRef CAS.
- M. R. Martinez, S. Dadashi-Silab, F. Lorandi, Y. Zhao and K. Matyjaszewski, Macromolecules, 2021, 54, 5526–5538 CrossRef CAS.
- H. S. Wang, N. P. Truong, G. R. Jones and A. Anastasaki, ACS Macro Lett., 2022, 1212–1216 CrossRef CAS.
- D. J. Lloyd, V. Nikolaou, J. Collins, C. Waldron, A. Anastasaki, S. P. Bassett, S. M. Howdle, A. Blanazs, P. Wilson, K. Kempe and D. M. Haddleton, Chem. Commun., 2016, 52, 6533–6536 RSC.
- Y. Sano, T. Konishi, M. Sawamoto and M. Ouchi, Eur. Polym. J., 2019, 120, 109181 CrossRef CAS.
- M. J. Flanders and W. M. Gramlich, Polym. Chem., 2018, 9, 2328–2335 RSC.
- S. Huang, X. Su, Y. Wu, X.-G. Xiong and Y. Liu, Chem. Sci., 2022, 11352–11359 RSC.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- F. Lorandi, M. Fantin and K. Matyjaszewski, J. Am. Chem. Soc., 2022, 144, 15413–15430 CrossRef CAS.
- N. P. Truong, G. R. Jones, K. G. E. Bradford, D. Konkolewicz and A. Anastasaki, Nat. Rev. Chem., 2021, 5, 859–869 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- V. Raus, E. Čadová, L. Starovoytova and M. Janata, Macromolecules, 2014, 47, 7311–7320 CrossRef CAS.
- A. Theodorou, P. Mandriotis, A. Anastasaki and K. Velonia, Polym. Chem., 2021, 12, 2228–2235 RSC.
- A. Theodorou, E. Liarou, D. M. Haddleton, I. G. Stavrakaki, P. Skordalidis, R. Whitfield, A. Anastasaki and K. Velonia, Nat. Commun., 2020, 11, 1486 CrossRef CAS.
- G. Szczepaniak, J. Jeong, K. Kapil, S. Dadashi-Silab, S. S. Yerneni, P. Ratajczyk, S. Lathwal, D. J. Schild, S. R. Das and K. Matyjaszewski, Chem. Sci., 2022, 11540–11550 RSC.
- M. Rubens, P. Latsrisaeng and T. Junkers, Polym. Chem., 2017, 8, 6496–6505 RSC.
- K. Parkatzidis, M. Rolland, N. P. Truong and A. Anastasaki, Polym. Chem., 2021, 12, 5583–5588 RSC.
- K. Parkatzidis, S. Boner, H. S. Wang and A. Anastasaki, ACS Macro Lett., 2022, 11, 841–846 CrossRef PubMed.
- S. Dadashi-Silab, F. Lorandi, M. J. DiTucci, M. Sun, G. Szczepaniak, T. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2021, 143, 9630–9638 CrossRef PubMed.
- S. Dadashi-Silab, K. Kim, F. Lorandi, G. Szczepaniak, S. Kramer, L. Peteanu and K. Matyjaszewski, ACS Macro Lett., 2022, 11, 376–381 CrossRef.
- T. Junkers and J. Laun, Controlled reversible deactivation radical photopolymerization, in Photopolymerisation Initiating Systems, ed. J. Lalevée and J.-P. Fouassier, The Royal Society of Chemistry, Cambridge, 2018, pp. 244–273 Search PubMed.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef PubMed.
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef.
- J. Xu, K. Jung, N. A. Corrigan and C. Boyer, Chem. Sci., 2014, 5, 3568–3575 RSC.
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- K. Parkatzidis, N. P. Truong, M. N. Antonopoulou, R. Whitfield, D. Konkolewicz and A. Anastasaki, Polym. Chem., 2020, 11, 4968–4972 RSC.
- P. N. Kurek, A. J. Kloster, K. A. Weaver, R. Manahan, M. L. Allegrezza, N. De Alwis Watuthanthrige, C. Boyer, J. A. Reeves and D. Konkolewicz, Ind. Eng. Chem. Res., 2018, 57, 4203–4213 CrossRef.
- M. L. Allegrezza and D. Konkolewicz, ACS Macro Lett., 2021, 10, 433–446 CrossRef PubMed.
- M. L. Allegrezza, N. De Alwis Watuthanthrige, Y. Wang, G. A. Garcia, H. Ren and D. Konkolewicz, Polym. Chem., 2020, 11, 6129–6133 RSC.
- V. Srivastava and P. P. Singh, RSC Adv., 2017, 7, 31377–31392 RSC.
- R. Whitfield, K. Parkatzidis, N. P. Truong, T. Junkers and A. Anastasaki, Chem, 2020, 6, 1340–1352 Search PubMed.
- N. Corrigan, D. Rosli, J. W. J. Jones, J. Xu and C. Boyer, Macromolecules, 2016, 49, 6779–6789 CrossRef.
- S. Shanmugam, J. Xu and C. Boyer, Macromolecules, 2014, 47, 4930–4942 CrossRef.
- J. Yeow, R. Chapman, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 5012–5022 RSC.
- M. Majek, F. Filace and A. J. Von Wangelin, Beilstein J. Org. Chem., 2014, 10, 981–989 CrossRef PubMed.
- N. Corrigan, J. Yeow, P. Judzewitsch, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 5170–5189 CrossRef PubMed.
- H. Cao, G. Wang, Y. Xue, G. Yang, J. Tian, F. Liu and W. Zhang, ACS Macro Lett., 2019, 616–622 CrossRef PubMed.
- S. Allison-Logan, Q. Fu, Y. Sun, M. Liu, J. Xie, J. Tang and G. G. Qiao, Angew. Chem., Int. Ed., 2020, 59, 21392–21396 CrossRef.
- J. A. Reeves, N. De Alwis Watuthanthrige, C. Boyer and D. Konkolewicz, ChemPhotoChem, 2019, 3, 1171–1179 CrossRef.
- M. D. Thum, S. Wolf and D. E. Falvey, J. Phys. Chem. A, 2020, 124, 4211–4222 CrossRef.
- J. F. Quinn, E. Rizzardo, L. Barner, C. Barner-Kowollik and T. P. Davis, Am. Chem. Soc., Div. Polym. Chem., Prepr., 2002, 43, 319–320 Search PubMed.
- R. N. Carmean, C. A. Figg, G. M. Scheutz, T. Kubo and B. S. Sumerlin, ACS Macro Lett., 2017, 6, 185–189 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01383e |
| This journal is © The Royal Society of Chemistry 2023 |