
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermoresponsive block copolymers of increasing architecture complexity: a review on structure–property relationships
Anna P.
Constantinou
 ,
Lezhi
Wang†
,
Shaobai
Wang†
and
Theoni K.
Georgiou
*
,
Lezhi
Wang†
,
Shaobai
Wang†
and
Theoni K.
Georgiou
*
Department of Materials, Imperial College London, South Kensington Campus, SW7 2AZ, UK. E-mail: t.georgiou@imperial.ac.uk
First published on 23rd November 2022
Abstract
Thermogels are an exciting class of stimuli responsive materials with many promising applications ranging from the medical field to additive manufacturing. This review focuses on the structure–property relationship of thermoresponsive block copolymers, with emphasis on the effect of architecture. Polymers based on Pluronic®, N-isopropylacrylamide, oligo(ethylene glycol) (meth)acrylate units, and 2-oxazoline units, which are amongst the most studied thermoresponsive units, are discussed. The effect of the polymer's architecture is crucial for controlling the thermoresponsive properties, such as cloud point and gelation temperature.
 Anna P. Constantinou | Dr Anna Constantinou obtained a BSc degree in Chemistry from the University of Cyprus in 2014, and an MSc degree in Advanced Materials from the Department of Materials at Imperial College London (ICL) in 2015. For her MSc thesis Dr Constantinou was granted the James S. Walker Award by the IOM3 (Institute of Materials, Minerals and Mining). Consequently, she obtained her PhD in Polymer Chemistry from ICL (awarded 2019). She has been working on well-defined polymers, specialising on thermoresponsive gels for biomedical applications. After her PhD studies, she was awarded an EPSRC Doctoral Prize fellowship (2018–2019) to work on in-depth physicochemical characterisation and drug delivery applicability of thermogels. She then worked as a Research Associate exploring the use of well-defined polymers in antibacterial and emulsion applications in Prof. Theoni K. Georgiou's group until April 2022. She is now a Research Associate for Professor Molly Stevens, working on polymers for biomedical applications. |
 Lezhi Wang | Ms Lezhi Wang obtained an MEng degree in Materials Science and Engineering from the Department of Materials at Imperial College London in 2020. She undertook her final year project on “Thermoresponsive gels” in Professor Theoni K. Georgiou's group at ICL. In 2021, she started her PhD studies in the same group and her main research interest involves the synthesis and characterisation of well-defined thermoresponsive polymers. |
 Shaobai Wang | Mr Shaobai Wang obtained his BEng degree in Polymer Materials and Engineering from South China University of Technology in 2016, and an MSc degree in Advanced Materials Science and Engineering from Imperial College London in 2021. In 2022 he started his PhD studies under the supervision of Professor Theoni K. Georgiou. His PhD thesis focuses on stimuli responsive polymeric materials for biomedical applications. |
 Theoni K. Georgiou | Professor Theoni K. Georgiou obtained a PhD in Polymer Chemistry under the supervision of Professor Costas Patrickios at the University of Cyprus in 2006. She then worked as a Postdoctoral Fellow under the supervision of Professor Antonios Mikos at Rice University, USA. In 2007 she moved to the University of Hull as a Research Councils of UK (RCUK) Academic Fellow. In 2014 she joined the Department of Materials at Imperial College London as a Lecturer where she is now a Professor in Polymer Chemistry. She is an expert in the synthesis and characterisation of well-defined polymers of various architectures with a special focus in group transfer polymerisation (GTP). She has special interest in thermoresponsive polymers and specifically thermogels and how these can be tailored by varying the polymer chemistry, molar mass and polymer architecture. In 2017 she was awarded the 2016 Macro Group UK Young Researchers Medal for “contributions to polymer science which show outstanding promise for the future”. She is an active member of the polymer community. Specifically, she has served as the Chair of Macrogroup UK (2019–2022), is a member of the editorial board for several polymer journals and is an Associate Editor for European Polymer Journal. |
Introduction
Thermoresponsive polymers, also called temperature-responsive polymers, are polymers which exhibit a drastic change in their solubility as a response to temperature. When reduction in solubility at elevated temperatures is detected, this behaviour is characterised as a lower critical solution temperature (LCST) behaviour, while an upper critical solution temperature (UCST) is presented when the solubility is enhanced upon heating. This change in solubility can be manifested by means of (i) cloud point (CP), i.e., the solution turns cloudy/opaque, (ii) precipitation, i.e., complete phase separation to a solid (polymer) and a liquid (solvent), and (ii) gelation, i.e., formation of a 3-D polymer network, Fig. 1, for LCST-type systems. The evolution of the CP with the concentration is depicted as a parabolic curve with the minimum corresponding to the LCST, Fig. 1(a), even though the terms CP and LCST are often used interchangeably in the literature. Similarly, a parabolic curve with a maximum is representative of the UCST-type behaviour. An appropriate combination of the polymer structure and the solvent triggers a reversible transition from solution to gel, which is favoured at higher concentrations, Fig. 1(b). These systems are widely known by the terms thermoresponsive gels, thermoreversible gels, thermogelators, and thermogels. Further heating of the gel might lead to destabilisation, such as gel syneresis, i.e., exclusion of the solvent from the gel matrix due to internal stresses, and precipitation.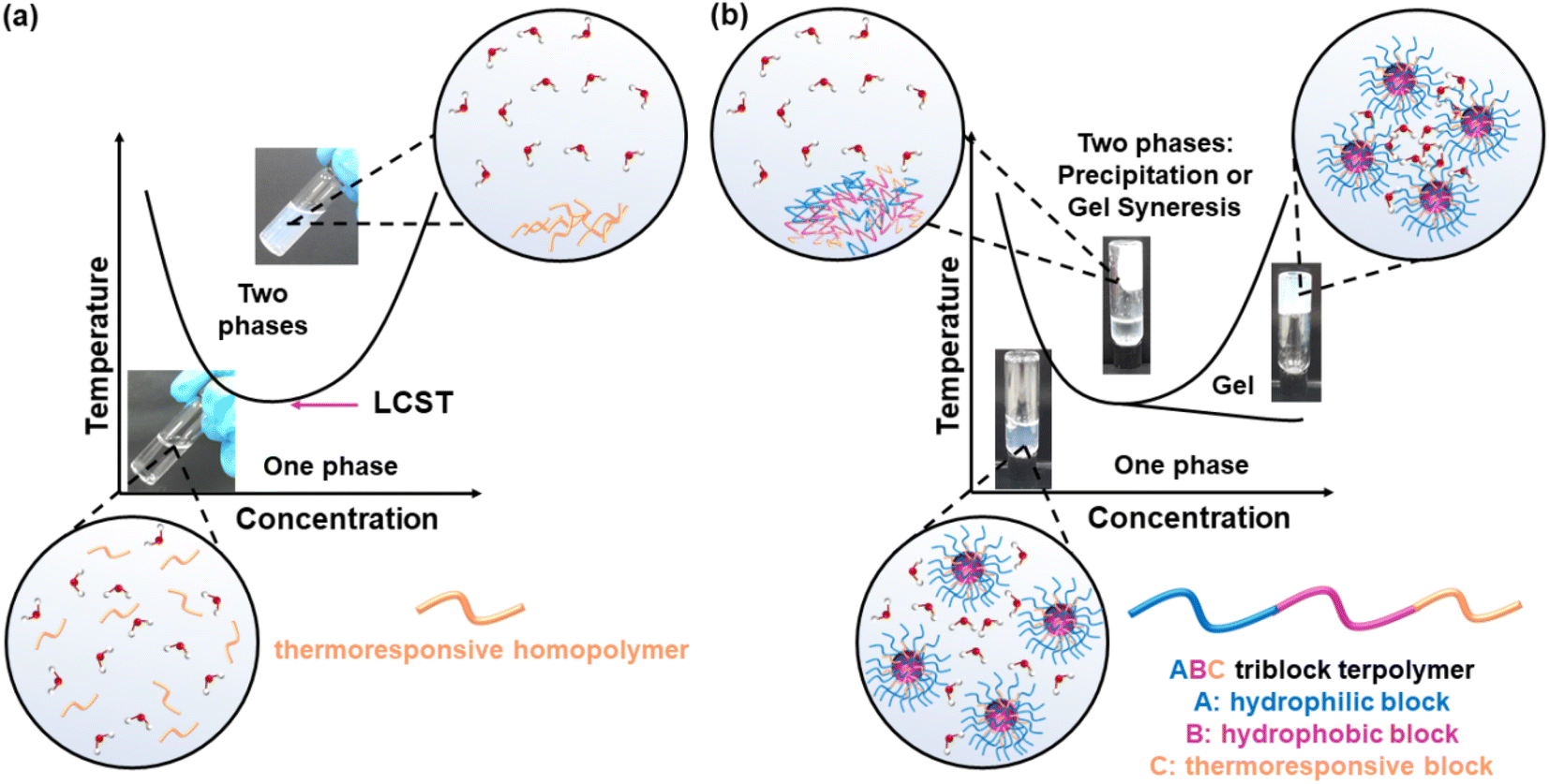 | ||
| Fig. 1 Phase diagrams of (a) an aqueous solution of a thermoresponsive homopolymer presenting LCST behaviour and (b) an aqueous solution of a thermoresponsive ABC triblock terpolymer with LCST type thermogelling behaviour; A, B, and C blocks are shown in blue, purple, and orange, respectively. | ||
The LCST behaviour is attributed to the “hydrophobic effect”.1,2 Thermodynamically speaking, an increased change in the entropy of the system (ΔS) takes place upon heating, which contributes to lowering the Gibbs free energy (ΔG): ΔG = ΔH − TΔS. In more detail, when the temperature of the thermoresponsive polymer solution increases, the interactions, such as hydrogen bonding (if any), between the polymer and the water molecules are weakened, and the entropic contribution of the water increases.1,2 Therefore, driven by the change in the entropy of the system, a thermoresponse takes place, during which the separation between the polymer chains and the water molecules occurs, Fig. 2.
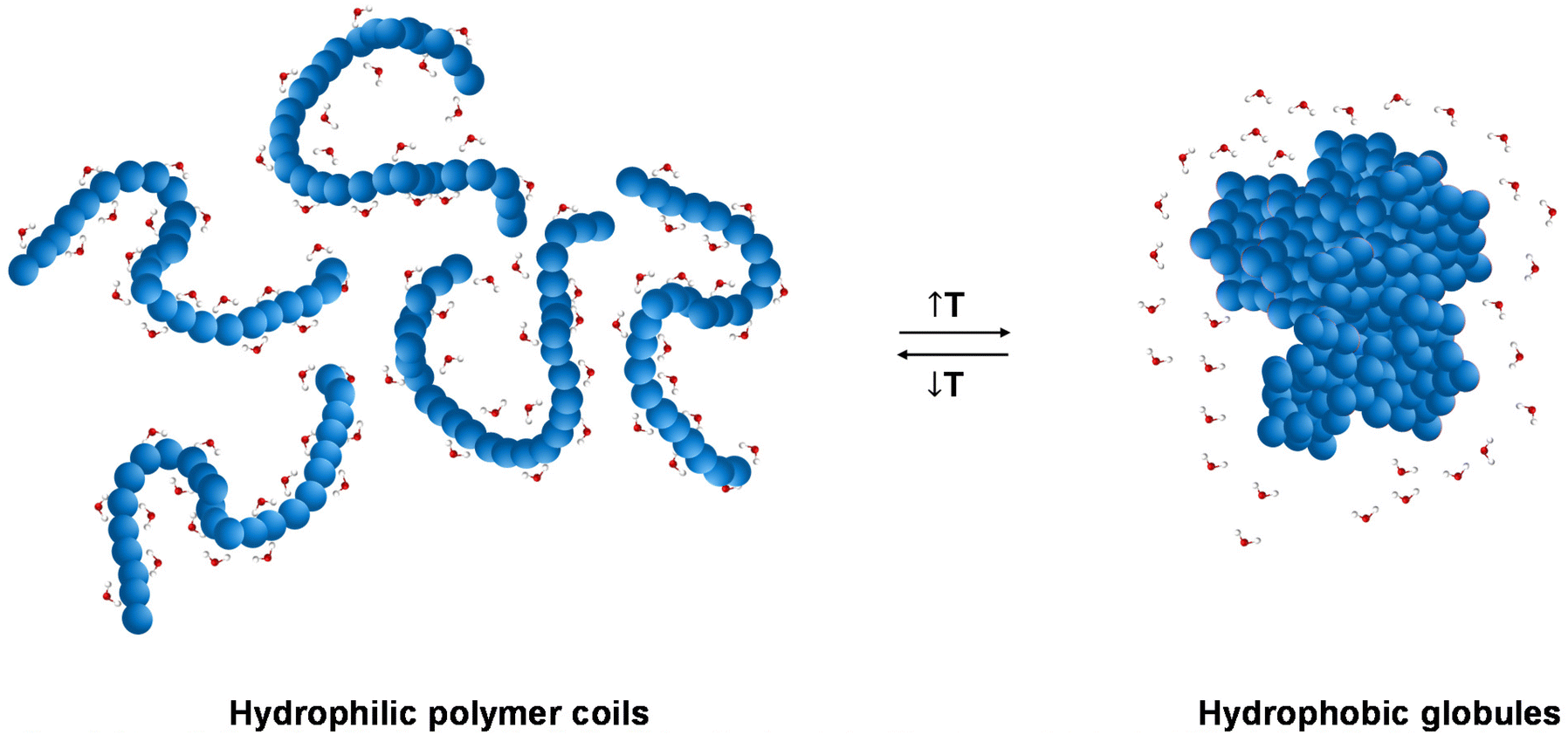 | ||
| Fig. 2 Schematic illustration of the “hydrophobic effect”, with a transition from hydrophilic polymer coils below the LCST to hydrophobic globules above the LCST. | ||
Thermoresponsive polymers have a wide range of applications, from biomedical applications, like drug delivery and tissue engineering,3–6 to catalysis,7–9 purification,10,11 and additive manufacturing (3D printing) and fabrication of 3-D structures and objects,12,13Fig. 3. In the healthcare sector, the scientific interest has been focused on LCST-type systems, as the structural integrity of biologicals, such as proteins, genes, drugs, and cells, is preserved throughout the temperature window of the application. Tissue engineering and drug delivery are amongst the most popular applications of thermoresponsive gels with the LCST, which are used as depots for tissue regeneration or sustained and topical delivery, respectively.5 Injectability is a key feature which makes thermoresponsive gels popular candidates in biomedical engineering as they offer a minimally invasive alternative to traditional implantation.14 In addition to this, the high content of water and the soft tissue-resembling properties support their suitability in the tissue engineering field.15 Thus, this literature review focuses on summarising and discussing LCST-type polymers.
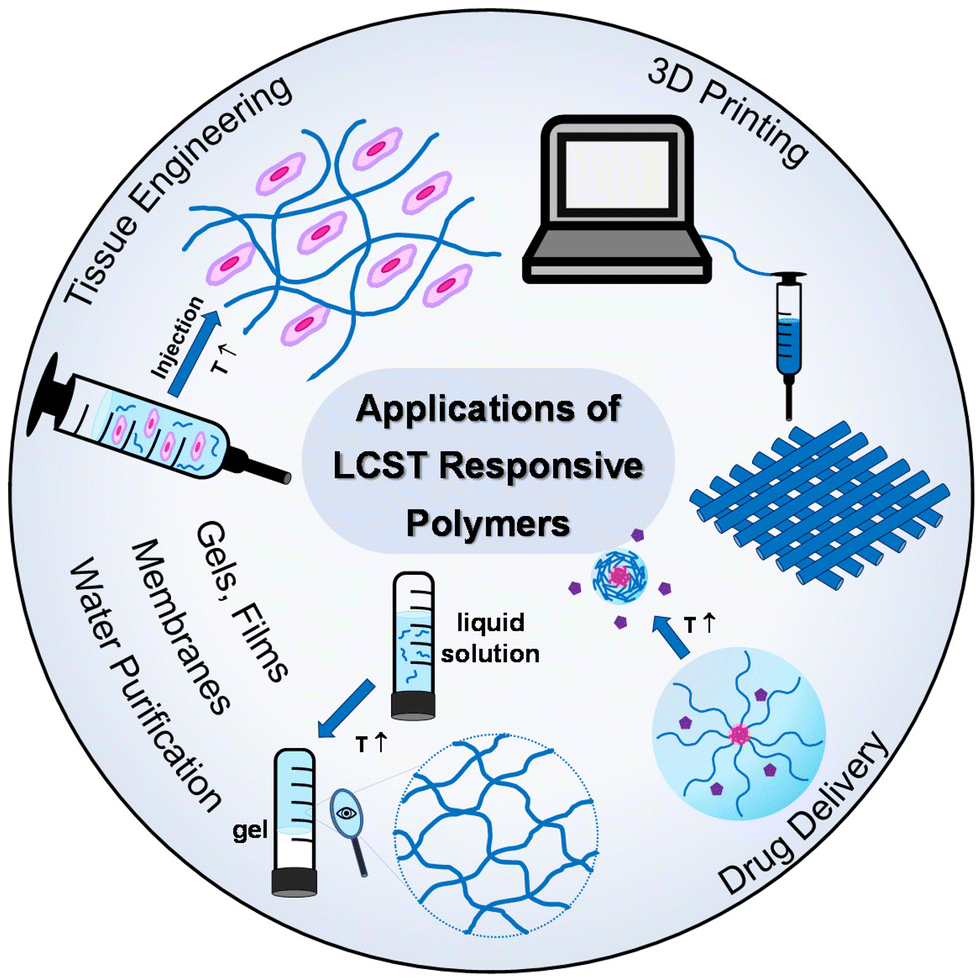 | ||
| Fig. 3 Applications of thermoresponsive polymers with a lower critical solution temperature (LCST). | ||
LCST-type thermoresponsive units
Several LCST-type thermoresponsive units, whose homo- or co-polymerisation produces thermoresponsive polymers have been well established in the literature. Amongst the most popular ones are N-isopropylacrylamide (NIPAAm) and ethylene glycol (EG), often combined with propylene glycol (PG), e.g., Pluronic® polymers.16 Other units presenting LCST-type thermoresponse are 2-(dimethylamino) ethyl methacrylate (DMAEMA),17 EG-based (meth)acrylate units,17,18 and N-vinylcaprolactam (VCL).19 2-Oxazoline-20 and peptide-based thermoresponsive polymers have also been reported. The chemical structures, names and abbreviations of these units are shown in Fig. 4.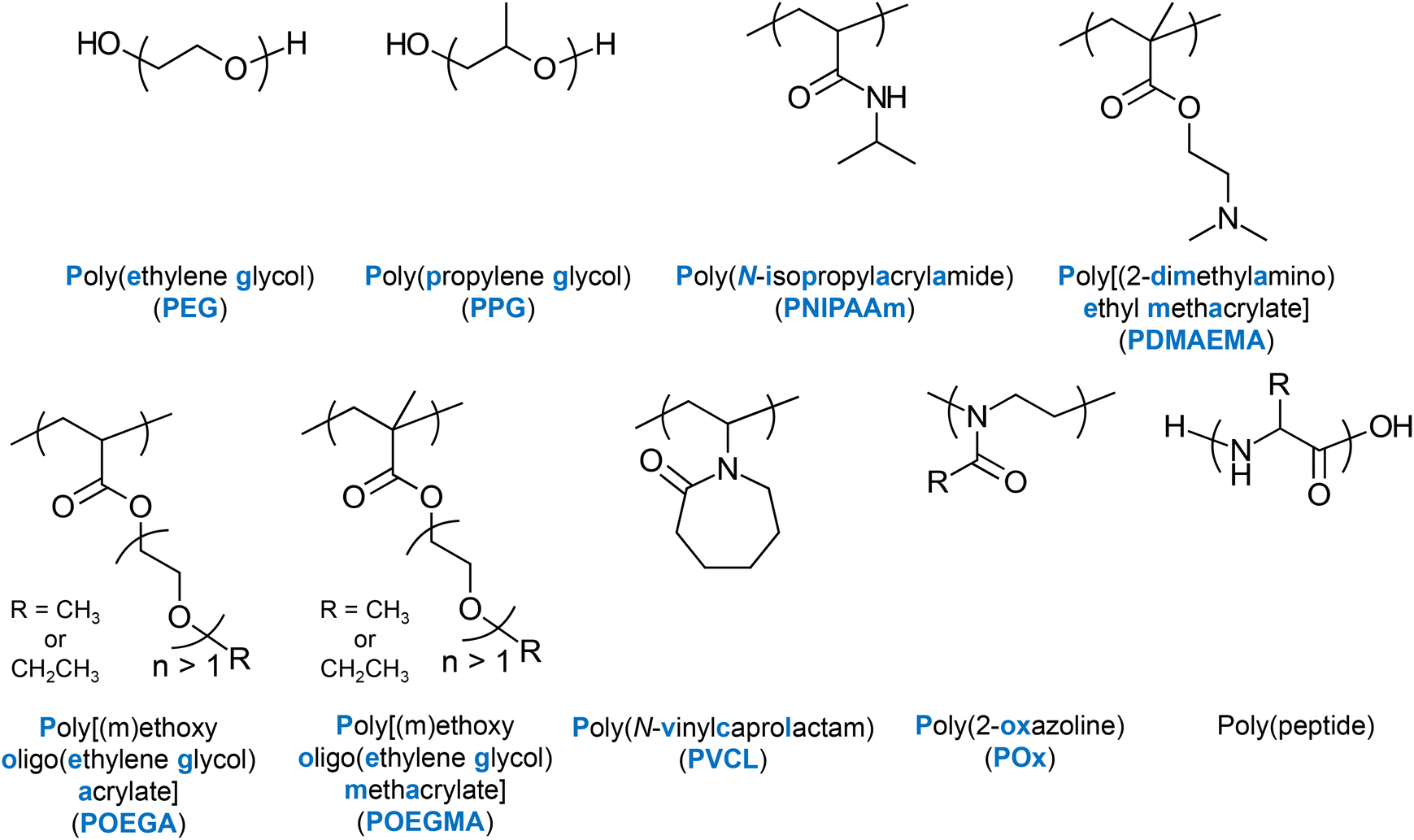 | ||
| Fig. 4 Chemical structures, names, and abbreviations (if any) of the main thermoresponsive polymers reported in the literature. | ||
Block copolymers
It is widely accepted that block copolymers, defined as “polymers composed of block macromolecules”,21 have gained popularity over statistical copolymers, despite the ease in the synthesis of the latter. It is well known that the architecture governs the final properties, and several advantages of the block copolymers have been well established. Amphiphilic block copolymers, i.e., polymers with both hydrophilic and hydrophobic blocks, self-assemble into well-ordered structures when dissolved in water, such as micelles of various shapes, including spherical core–shell micelles, flower-like micelles, and polymersomes, Fig. 5. The size of the self-assembled structures depends on the polymer characteristics, such as molar mass and composition,22 while the type and shape, such as spherical versus worm-like,23 or micelles versus polymersomes/polymer vesicles,24 can be controlled by varying the comonomer composition (hydrophilic/hydrophobic ratio) and the preparation method. Polymersome formation can also be temperature-driven, as previously reported for ABC triblock copolymers based on EG (A block), a random copolymer of EG and butylene glycol (B block), and isoprene (C block), respectively.25 In these self-assembled structures, the hydrophobic blocks interact with each other, thus avoiding interactions with water, while the hydrophilic ones interact with water, either via hydrogen bonding, ion–dipole, or dipole–dipole interactions. Adopting such configurations enhances the water solubility, in contrast to the statistical copolymers, which do not self-assemble into well-ordered structures, due to the lack of distinct hydrophobic and hydrophilic blocks. In addition, hydrophobic drugs can be solubilised in the hydrophobic part of these self-assembled structures, thus enabling these structures to act as drug delivery vehicles. Block copolymers also favour the formation of physical gels via interconnection of the micelles, with their statistical counterparts being unable to form gels or the gels formed are less stable.26,27 Physical gels have been extensively studied by researchers in the tissue engineering and drug delivery fields for curing numerous diseases, such as cancer and diabetes, thus highlighting the significance of studying block copolymers.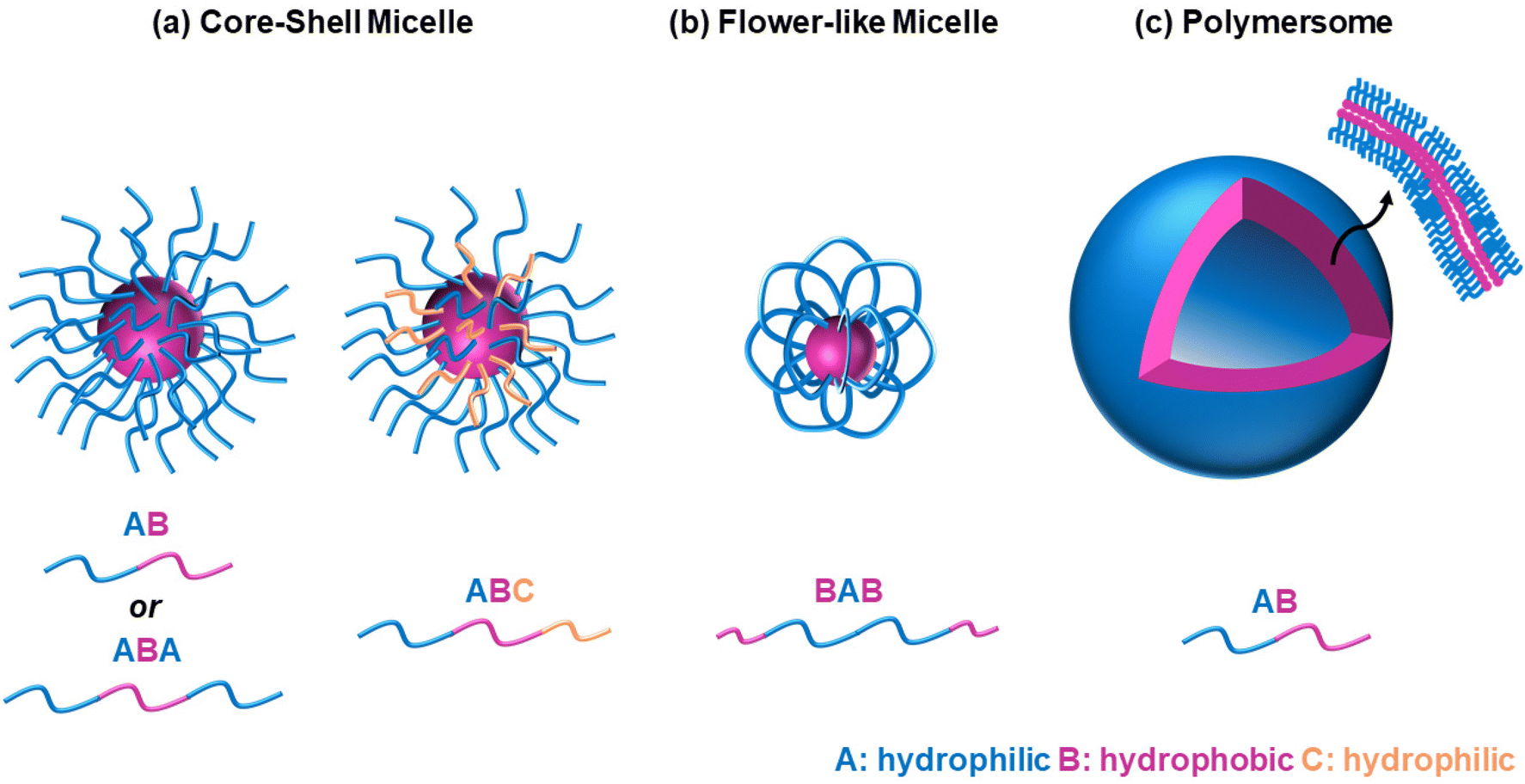 | ||
| Fig. 5 Self-assembled structures adopted by amphiphilic block copolymers: (a) core–shell micelles by the AB, ABA, and ABC architectures, (b) flower-like micelles by the BAB architecture, and (c) polymersomes by the AB architecture. Blue and purple represent the hydrophilic and hydrophobic blocks, respectively, while when a second hydrophilic block is present, this is shown in orange. In the self-assembled structures, the compact hydrophobic part is represented by purple spheres. | ||
Block copolymers can be categorised by the number of blocks, with thermoresponsive diblock copolymers being the most commonly reported because they are the easiest to make. Thermoresponsive triblock bipolymers, i.e., polymers with three blocks and two different repeating units, namely ABA, have been extensively studied, with the thermoresponsive gels applied in clinical trials falling into this category. These are Pluronic® F127 (A = EG and B = PG) and ReGel® (A = poly(L-lactide-co-glycolide, PLGA, and B = EG). Thermoresponsive triblock terpolymers, i.e., polymers with three blocks, each consisting of a different repeating unit, have also been studied, but fewer studies have been reported, due to their more difficult synthesis, as discussed in the following section. Increasing the complexity of the architecture to tetrablock (four blocks), pentablock (five blocks), hexablock (six blocks), heptablock (seven blocks), etc. opens new possibilities for developing novel thermoresponsive polymers with exciting self-assembling properties. Therefore, this review focuses on summarising and discussing studies on thermoresponsive multiblock copolymers, i.e., block copolymers with more than two blocks, and the effect of temperature on their self-assembly and gelation properties. Each section summarises studies based on the same thermoresponsive unit, and the studies are discussed based on the increasing complexity of architecture, as can be seen in Table 1. The effect of architecture on the properties is identified and conclusions are drawn, whenever possible.
| Main category of thermoresponsive polymers | Architecture | Comonomer(s) | Ref. | |
|---|---|---|---|---|
| Abbreviations (in the order they appear in the Table): EG, ethylene glycol; PG, propylene glycol, DEAEMA, 2-(diethylamino)ethyl methacrylate; tBAEMA, 2-(t-butylamino)ethyl methacrylate; DMAEMA, 2-(dimethylamino)ethyl methacrylate; DiPAEMA, 2-(diisopropylamino)ethyl methacrylate NIPAAm, N-isopropylacrylamide; LA, lactic acid; GA, glycolic acid; CL, ε-caprolactone; St, styrene; tBuSt, 4-t-butylstyrene; BrBzA, 3,5-dibromobenzyl acrylate; EtHexA, 2-ethylhexyl acrylate; OcDecA, octadecyl acrylate; AA, acrylic acid; Adac, adamantane acrylate; VP, N-vinylpyrrolidone; TEMPO, 2,2,6,6-tetramethylpiperidine-1-oxyl; HTPB, hydroxyterminated polybutadiene; HEMA, 2-hydroxyethyl methacrylate; EtA, ethyl acrylate; MA-POSS, 3-methacryloxypropylheptaphenyl polyhedral oligomeric silsesquioxanes; mPEGV, poly(ethylene glycol) methyl ether vinylphenyl; MAGlc, poly(3-O-methacryloyl-α,β-D-glucopyranose; BuA, n-butyl acrylate; NAM, 4-acryloylmorpholine; NBOCAEMA, 2-((((2-nitrobenzyl)oxy)carbonyl)amino) ethyl methacrylate; DEAEA, 2-(dimethylamino)ethyl acrylate; mOEGA480, methoxy oligo(ethylene glycol) acrylate with an average Mn of 480 g mol−1; DMAAm, N,N-dimethylacrylamide; DEAAm, N,N-diethylacrylamide; PEGMA-co-PPGMA, poly(ethylene glycol) methacrylate-co-poly(propylene glycol) methacrylate; DiBuAAm, N,N-dibutylacrylamide; BuMA, n-butyl methacrylate, mDEGMA, methoxy di(ethylene glycol) methacrylate; TEGMA, tri(ethylene glycol) methyl ether methacrylate; mOEGMA300, methoxy oligo(ethylene glycol) methacrylate with an average Mn of 300 g mol−1; mOEGMA500, methoxy oligo(ethylene glycol) methacrylate with an average Mn of 500 g mol−1; MMA, methyl methacrylate; EtMA, ethyl methacrylate; HexMA, n-hexyl methacrylate, VB, 4-vinyl benzyl; mTEGMA, methoxy tri(ethylene glycol) methacrylate; mMEGA, methoxy mono(ethylene glycol) acrylate; mDEGA, methoxy di(ethylene glycol) acrylate; eDEGA, ethoxy di(ethylene glycol) acrylate; MPC, 2-methacryloyloxyethyl phosphorylcholine; mPEG, monomethoxy poly(ethylene glycol); HMAAm, N-hydroxymethyl acrylamide; eDEGMA, ethoxy di(ethylene glycol) methacrylate; MAEtMAm, 2-(methacryloyloxy)ethyltrimethylammonium iodide; RhoMA, rhodamine-containing methacrylate; HEA, 2-hydroxyethyl acrylate; VCL, N-vinylcaprolactam; tBA, t-butyl acrylate; EtOx, 2-ethyl-2-oxazoline; nProOx, 2-n-propyl-2-oxazoline; iProOx, 2-isopropyl-2-oxazoline; MeOx, 2-methyl-2-oxazoline; BuOx, 2-butyl-2-oxazoline; iBuOx, 2-isobutyl-2-oxazoline. | ||||
| Pluronic® (EG/A and PG/B) | Triblock (ABA/BAB) | — | 32–38 | |
| Pentablock | DEAEMA, tBAEMA, DMAEMA, DiPAEMA, NIPAAm | 39–49 | ||
| Multiblock | — | 50 | ||
| EG-containing degradable | Triblock (ABA/BAB) | LA, GA, CL & derivatives, depsipeptides | 51–71 | |
| Pentablock | CL, LA, L-lysine, L-histidine, γ-benzyl-L-glutamate | 72–74 | ||
| Hyperbranched | PG, CL | 75 | ||
| Multiblock | PG, 1,4-butylene adipate, betulin | 76, 77 | ||
| Alternating block | Hydrophobic polyesters | 78 | ||
| Acrylamide | NIPAAm | Triblock (ABA/BAB) | St, tBuSt, BrBzA, EtHexA, OcDecA, AA & derivatives, DMAAm, Adac, ionic liquid, EG, VP, betaine ester, TEMPO, HTPB, HEMA, EtA | 79–94 |
| Triblock (ABC) | CL, MA-POSS, mPEGV, St, MAGlc, HEMA, EG, LA, ethylene, propylene, BuA, NAM, NBOCAEMA, DEAEA, mOEGA480 | 95–107 | ||
| Tetrablock | DMAAm, EG, St, DMAEMA | 108, 109 | ||
| Pentablock | DEAEMA, EG, DEAAm, PG, AA, tBA | 110–114 | ||
| Multiblock | EG, DEAAm | 115 | ||
| Star | EG, [R]-3-hydroxybutylate, PEGMA-co-PPGMA | 116 and 117 | ||
| DEAAm | Triblock (ABA) | EG | 118 | |
| Triblock (ABC) | EG, DiBuAAm | 119 | ||
| Amine - DMAEMA | Triblock (ABA/BAB) | 6-O-methacryloyl-D-galactopyranose, BuMA | 120–122 | |
| Triblock (ABC) | mDEGMA, TEGMA, mOEGMA300, mOEGMA500, MMA, EtMA, BuMA, HexMA | 26 and 123–126 | ||
| Tetrablock | OEGMA300, BuMA | 127 | ||
| Pentablock | BuMA | 128 | ||
| Heptablock | ||||
| Nonablock | ||||
| Graft | VB, CL | 129 | ||
| EG-based (meth)acrylate | mDEGMA | Triblock (ABA/BAB) | mOEGA480, HEMA, MPC | 130–132 |
| Triblock (ABC) | mPEG, HMAAm, DEAEMA, eDEGMA, MAEtMAm, RhoMA, BuMA, mOEGMA300 | 133–136 | ||
| Tetrablock | mOEGMA300, BuMA | 137 | ||
| Star | mOEGA480, mOEGMA500, CL | 130 and 138 | ||
| mTEGMA | Pentablock | EG, DMAEMA, BuMA | 139 | |
| mOEGMA300 | Triblock (ABC) | BuMA, DEAEMA | 140 | |
| mOEGMA500 | Triblock (ABA/BAB) | CL | 141 | |
| mMEGA | Triblock (ABA/BAB) | HEA | 142 | |
| mDEGA | Triblock (ABA/BAB) | St | 143–145 | |
| Star | ||||
| eDEGA | Triblock (ABA/BAB) | mOEGA480 | 146 | |
| Other | VCL | Triblock (ABA/BAB) | VP, tBA, AA | 147–149 |
| Polypeptides | Tetrablock | Amino acids | 150 | |
| EtOx | Triblock (ABA/BAB) | LA, CL | 151 | |
| nProOx | Triblock (ABA/BAB) | EtOx, MeOx | 152 and 153 | |
| iProOx | Triblock (ABA/BAB) | MeOx, BuOx | 154 | |
| MeOx & iBuOx | Triblock (ABA/BAB) | — | 155 | |
Da Silva et al. reported the synthesis and investigation of a library of ABA triblock copolymers consisting of PEG as the B central block and two thermoresponsive outer A blocks, varying from NIPAAm, to DMAEMA, to methoxy di(ethylene glycol) methacrylate (mDEGMA).28 Extensive SANS measurements revealed that the self-assembly at the nanoscale driving the thermogelling properties was highly depended on the MM of the blocks and the chemical nature of the thermoresponsive unit. Gels with a sharp sol–gel transition were formed by the mDEGMA- and NIPAAM-based analogues when the MM of the blocks was targeted to 10 kDa (B block) and either 10 or 20 kDa (A block). These copolymers adopted spherical or ellipsoid micellar structures, and the network formation was attributed to the bridging of the flower-like micelles by unimers.28
Synthetic approaches
The synthesis of block copolymers has been more challenging compared to statistical copolymers, as it requires multiple and controlled synthetic steps, and thus it has only been achieved with the discovery of living and controlled polymerisation techniques. These techniques include, but are not limited to, anionic polymerisation, ring opening polymerisation (ROP), group transfer polymerisation (GTP), reversible addition–fragmentation chain-transfer (RAFT) polymerisation, and atom transfer radical polymerisation (ATRP). The synthesis of block copolymers via these techniques is carried out in a controlled manner, with sequential addition of monomers leading to chain extension, which is possible due to the living/controlled nature of these polymerisation techniques. In 2016, Gody et al. published a report on the experimental requirements of chain growth polymerisation techniques that are needed to precisely control the polymer structure.29 They reported that a minimum degree of polymerisation of 6 is required for producing icosablock copolymers with 95% fidelity, i.e., to ensure that each polymer chain contains at least one repeating unit for the targeted block.29The difficulty of the synthesis relies on several factors, such as the number of blocks and the symmetry of the structure. While diblock copolymers are easily synthesised via a two-step synthetic procedure, the synthesis of multiblock copolymers is more complicated, as has been highlighted by a recent review by Becer's group,30 because the polymer chain should be extended without termination. In reality, termination might be detected, but this should be kept at minimum to ensure that the molar mass distribution of the final polymer is monomodal, i.e., polymer chains from different synthetic steps do not co-exist, thus ensuring reproducibility of the properties. The conventional way of synthesising block copolymers is sequential polymerisation by using a monofunctional initiator, thus resulting in unidirectional growth of the polymer chain. Triblock terpolymers (ABC), tetrablock ter- and quater-polymers, etc. are inevitably synthesised using a monofunctional initiator, thus increasing the complexity of their synthesis, evident from the limited studies published. On the other hand, a bifunctional initiator can be used for the synthesis of symmetric polymers, such as ABA triblock bipolymers and ABCBA pentablock terpolymers, to complete the synthesis in two and three steps, respectively, instead of the three and five steps needed, respectively, when using a monofunctional initiator. Another approach that has been followed to synthesise ABCBA pentablock terpolymers is the conversion of a commercially available triblock bipolymer to a bifunctional initiator, followed by chain growth and consequent formation of a pentablock terpolymer in only one step. A combination of polymerisation techniques might be adopted to obtain the desired polymer structure. For example, for synthesising ABA triblock copolymers based on NIPAAm and poly(2-ethyl-2-oxazoline), Sahn et al. synthesised the middle oxazoline-based block via living cationic ring-opening polymerisation, while RAFT polymerisation facilitated the synthesis of the outer NIPAAm-based blocks.31 These synthetic routes are illustrated schematically in Fig. 6.
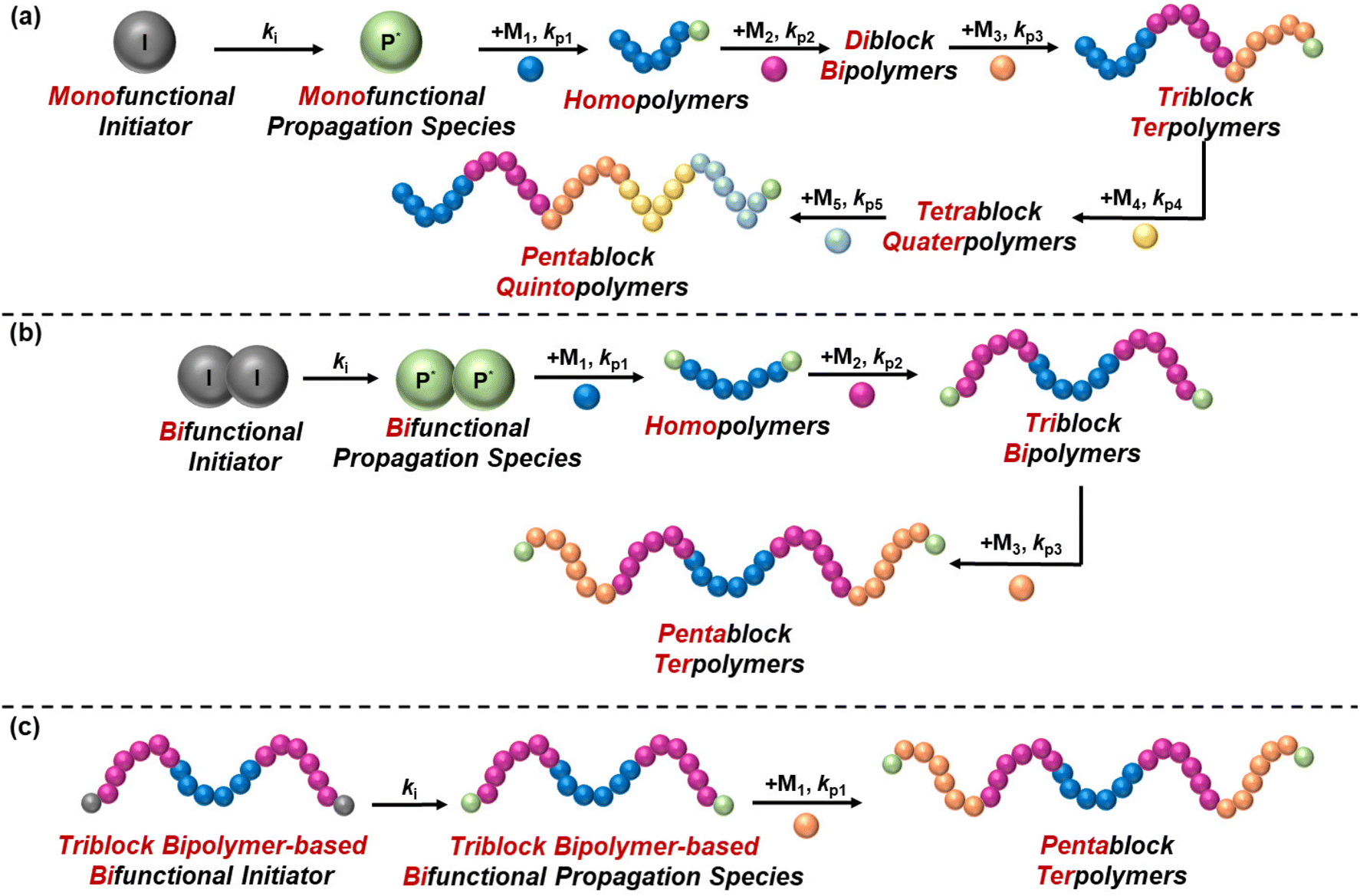 | ||
| Fig. 6 Common synthetic approaches followed for the synthesis of thermoresponsive block copolymers: (a) synthesis of block copolymers using a monofunctional initiator, with unidirectional chain extension taking place upon addition of a different monomer. The chain growth from a homopolymer to a pentablock quintopolymer is given as an example. (b) Synthesis of block copolymers using a bifunctional initiator, with bidirectional chain growth taking place upon addition of a different monomer. The chain growth from a homopolymer to a symmetrical pentablock terpolymer is shown. (c) One-step synthesis of pentablock terpolymers using a triblock bipolymer-based bifunctional initiator, obtained by modification of a commercially available triblock bipolymer, with bidirectional chain growth. | ||
Characterisation methods
Monitoring the polymer synthesis and characterising the polymer structure are necessary, with gel permeation chromatography (GPC) and nuclear magnetic resonance spectroscopy (NMR) being the most popular techniques amongst polymer chemists. Thus, GPC and NMR are normally used to monitor and confirm the molar mass and composition of the polymers, respectively. The following paragraphs will focus on how the thermoresponsiveness of the polymers is studied.The thermoresponse of the polymers in solution can be monitored via a plethora of techniques, Fig. 7. Temperature-driven cloudiness/turbidity is detected visually or by ultraviolet-visible (UV-Vis) spectroscopy; the CP is determined by UV-Vis as the temperature at which the transmittance drops to 50%. A complementary technique for determining the CP is differential scanning calorimetry (DSC), including microDSC, with the phase separation presented as an endothermic peak on the DSC thermogram.47,48,156,157 The effect of temperature on the self-assembly of thermoresponsive polymers has also received much attention, with dynamic light scattering (DLS) being used as an indication of the presence and size of the unimers (free polymer chains in solution), micelles or bigger aggregates, and static light scattering (SLS) being used to determine the aggregation number. DLS temperature ramps can also be performed to determine the CP as the temperature at which aggregation occurs at 50%. However, one should keep in mind that the theory of DLS assumes spheres; thus more powerful techniques, such as small-angle neutron scattering (SANS) and small-angle X-ray scattering (SAXS), could be used to determine more accurately the shape and size of the self-assembled structures. Gel formation as a response to temperature can be easily determined by the tube inversion method, as the temperature at which the sample does not flow when the vial is inverted, indicating a solid-like material. Rheological measurements can also detect the gelation temperature (Tgel), which is defined as the temperature at which the shear elastic modulus (also called the storage modulus, G′) overcomes the shear viscous modulus (also called the loss modulus, G′′), thus indicating a temperature-driven transition from a free-flowing solution to an elastic gel. In addition to the Tgel, rheology provides further insight into the rheological properties of the sample, such as the strength of the gel, gel destabilisation, and changes in the viscosity of the sample as a response to temperature when no gelation is detected. Both techniques determining the gelation are complementary and should be applied for a complete study.
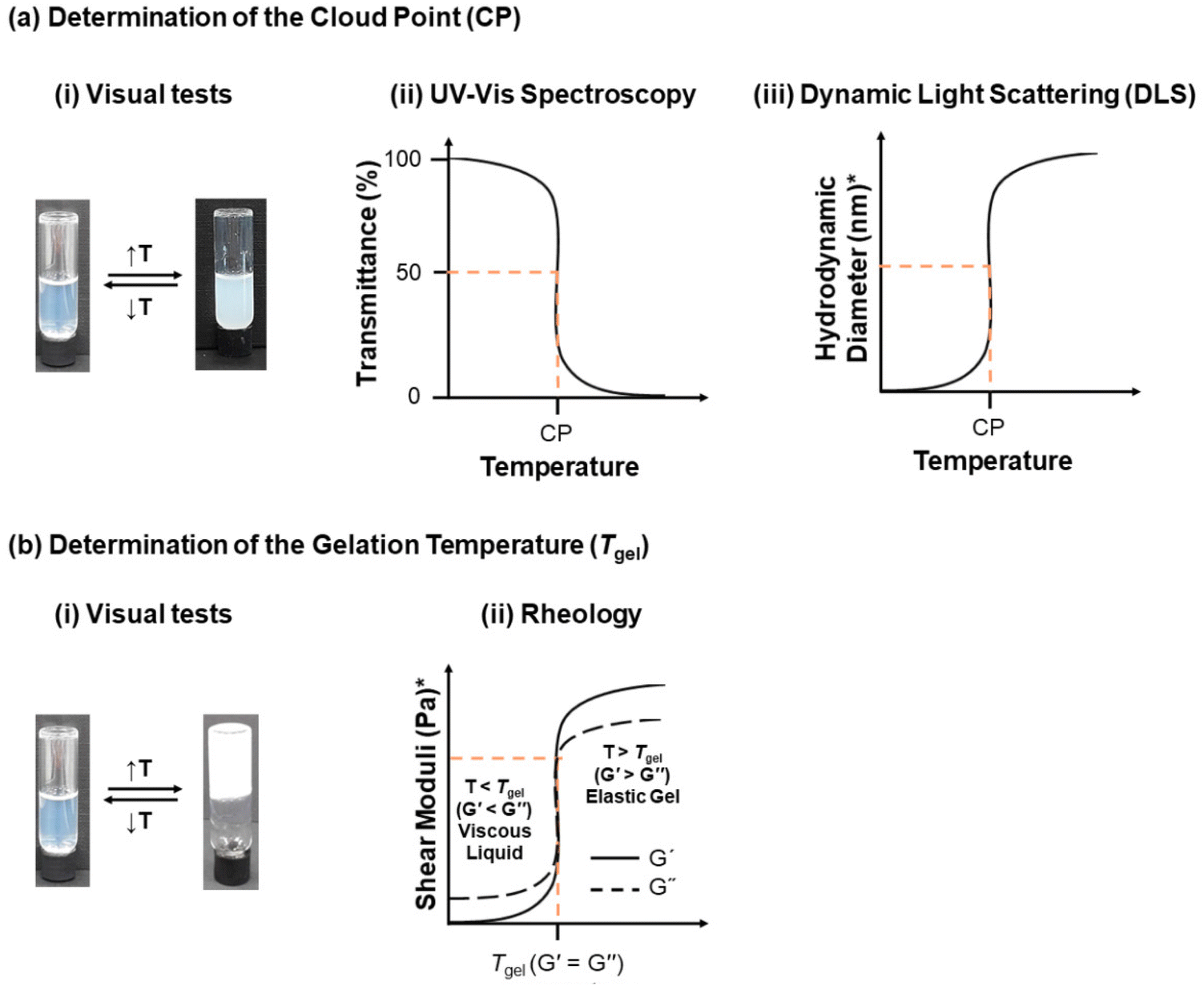 | ||
| Fig. 7 (a) Determination of the cloud point (CP) by (i) visual tests, as the temperature at which the solution turns cloudy, (ii) UV-Vis spectroscopy, as the temperature at which the transmittance drops to 50%, and (iii) dynamic light scattering (DLS), as the temperature at which 50% aggregation occurs. (b) Determination of the gelation temperature (Tgel) by (i) visual tests, as the temperature at which the sample does not flow upon tube inversion, and (ii) rheology, as the temperature at which the shear storage (shear elastic) modulus, G′, overcomes the shear loss (shear viscous), G′′, modulus. * The y axis indicated should be plotted in a logarithmic scale. | ||
Pluronic®-based polymers
Pluronic® polymers are commercially available thermoresponsive polymers with temperature driven micellisation, aggregation, and gelation. As previously mentioned, these polymers are ABA triblock copolymers with A and B being based on EG and PG, respectively. It has been previously reported that the critical gelation concentration (CGC) and Tgel of these polymers decrease with the increase in MM,32,33 as expected, since the viscosity of a polymer solution increases with its MM (Mark–Houwink's equation21). On the other hand, a decrease in the CP of Pluronic® solutions is observed with a decrease in MM.34 When comparing Pluronic® polymers (ABA) with their reverse counterparts (BAB), known as reverse Pluronic® polymers, it is generally concluded that the CP of the ABA-type polymers increases with increasing polymer concentration, while the opposite trend is reported for the BAB-type polymer.35 For example, while the CP of Pluronic® L44 (EG10-b-PG23-b-EG10) increases from 62 °C (at 2 mM) to 73 °C (at 50 mM), the CP of reverse Pluronic® 10R5 (PG8-b-EG22-b-PG8) decreases from 74 °C (at 2 mM) to 58 °C (at 50 mM).35 Similar observations were made for Pluronic® L64 (EG13-b-PG30-b-EG13), with the CP increasing from 52 °C (at 1 w/v%) to 58 °C (25 w/v%).36 In a study by Zhou and Chu,37 Pluronic® L64 was compared to reverse Pluronic® 17R4 (PG14-b-EG24-b-PG14), and it was found that the ABA triblock bipolymer forms micelles in a wider range of temperatures and concentrations, while its BAB counterpart presents only a narrow window of micelle formation under the same conditions.37 In addition to this, the CPs of the BAB are lower than the ones of the ABA polymer.37 These observations indicate an architecture effect on the micellisation and CP, with the ABA triblock bipolymer forming more stable micelles in wider temperature and concentration ranges compared to the BAB one, presumably due to the entropically more favourable self-assembly into core–shell micelles (ABA), as opposed to the flower-like micelles (BAB).Well-defined reverse Pluronic® polymers have been synthesised by Markus et al. via organocatalytic polymerisation; the Đ values varied from 1.02 to 1.07.38 The MM of the middle hydrophilic block ranged from 4 to 20 kg mol−1, while the DP of the outer hydrophobic blocks varied from 12 to 128. Copolymers with 20 kg mol−1 middle block and the DP of the outer blocks varying between 77 and 128 formed gels with an exceptionally high storage modulus, which is tenfold relative to Pluronic® F127. In addition, gel formation was detected at three times lower gelation concentration than Pluronic® F127.38 Even though the authors have attributed these superior thermogelling properties to both the increased MM and BAB architecture, for clear trends to be established, a comparison should be made between copolymers differing in only one structural parameter.
Mallapragada and co-workers have previously reported the synthesis and investigation of thermoresponsive pentablock terpolymers with Pluronic® F127 (EG99-b-PG66-b-EG99, MM ≈ 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1) as a central triblock terpolymer.39–46 The synthesis of the pentablock terpolymer was carried out by bidirectional chain-extension of Pluronic® F127, via ATRP39,40,42–46 or oxyanionic polymerisation,41,44 with outer blocks consisting of an amine-containing methacrylate monomer, which provides pH-response. In most of the studies, 2-(diethylamino)ethyl methacrylate (DEAEMA) was used,39,41–46 while in one of the studies the hydrophobicity of the amine-containing monomer was varied from 2-(t-butylamino)ethyl methacrylate (tBAEMA), to DMAEMA, to DEAEMA, to 2-(diisopropylamino)ethyl methacrylate (DiPAEMA).40 Copolymers with different MMs of the outer blocks were studied, thus simultaneously altering the total MM and composition of the copolymers. More specifically, pentablock terpolymers, with the total MM ranging from 12800 to 22000 g mol−1, were reported, which preserved thermogelation. The Tgel could be tuned by changing the pH, with the lower pH increasing the Tgel.40 Thus, these copolymers were studied as pH-responsive release depots, as gel dissolution, and thus release of the compound of interest, was promoted by decreasing the pH. Nile blue chloride,41 lysozyme,42 plasmid DNA,44 and siRNA combined with gold nanoparticles46 were tested, and it was found that the lower the pH, the faster the release.41,42 The cytotoxic nature of these copolymers was highly associated with the content of DEAEMA, with the higher content increasing the cytotoxicity,44 similarly to DMAEMA-based systems,158–161 as the positive charges destabilise the negatively charged cellular membrane. Interestingly, the thermogelation could be tuned by changing the hydrophilicity of the amine-containing outer blocks.40 While the tBAEMA-based polymers were too hydrophilic, thus preventing gelation, the DiPAEMA-based ones were too hydrophobic, thus making the polymers insoluble. Both DMAEMA- and DEAEMA-based polymers formed gels, with the latter lowering the Tgel and increasing the gel rigidity, presumably due to the increased hydrophobicity, thus enhancing the “hydrophobic effect”. A comparison between a DEAEMA-containing pentablock terpolymer with Pluronic® F127 revealed the formation of more rigid gels by the latter, thus indicating a disturbance of the gelation by the addition of the outer blocks, attributed to the change in the micelle packing,40 as revealed by SANS and nuclear magnetic resonance spectroscopy.43 Interestingly, one of the DEAEMA-based pentablock copolymers has recently been used for membrane fabrication, and its pH- and thermo-response were found to control the permeability of the membrane, which increased by increasing the pH and temperature.39
400 g mol−1) as a central triblock terpolymer.39–46 The synthesis of the pentablock terpolymer was carried out by bidirectional chain-extension of Pluronic® F127, via ATRP39,40,42–46 or oxyanionic polymerisation,41,44 with outer blocks consisting of an amine-containing methacrylate monomer, which provides pH-response. In most of the studies, 2-(diethylamino)ethyl methacrylate (DEAEMA) was used,39,41–46 while in one of the studies the hydrophobicity of the amine-containing monomer was varied from 2-(t-butylamino)ethyl methacrylate (tBAEMA), to DMAEMA, to DEAEMA, to 2-(diisopropylamino)ethyl methacrylate (DiPAEMA).40 Copolymers with different MMs of the outer blocks were studied, thus simultaneously altering the total MM and composition of the copolymers. More specifically, pentablock terpolymers, with the total MM ranging from 12800 to 22000 g mol−1, were reported, which preserved thermogelation. The Tgel could be tuned by changing the pH, with the lower pH increasing the Tgel.40 Thus, these copolymers were studied as pH-responsive release depots, as gel dissolution, and thus release of the compound of interest, was promoted by decreasing the pH. Nile blue chloride,41 lysozyme,42 plasmid DNA,44 and siRNA combined with gold nanoparticles46 were tested, and it was found that the lower the pH, the faster the release.41,42 The cytotoxic nature of these copolymers was highly associated with the content of DEAEMA, with the higher content increasing the cytotoxicity,44 similarly to DMAEMA-based systems,158–161 as the positive charges destabilise the negatively charged cellular membrane. Interestingly, the thermogelation could be tuned by changing the hydrophilicity of the amine-containing outer blocks.40 While the tBAEMA-based polymers were too hydrophilic, thus preventing gelation, the DiPAEMA-based ones were too hydrophobic, thus making the polymers insoluble. Both DMAEMA- and DEAEMA-based polymers formed gels, with the latter lowering the Tgel and increasing the gel rigidity, presumably due to the increased hydrophobicity, thus enhancing the “hydrophobic effect”. A comparison between a DEAEMA-containing pentablock terpolymer with Pluronic® F127 revealed the formation of more rigid gels by the latter, thus indicating a disturbance of the gelation by the addition of the outer blocks, attributed to the change in the micelle packing,40 as revealed by SANS and nuclear magnetic resonance spectroscopy.43 Interestingly, one of the DEAEMA-based pentablock copolymers has recently been used for membrane fabrication, and its pH- and thermo-response were found to control the permeability of the membrane, which increased by increasing the pH and temperature.39
Pentablock terpolymers with outer NIPAAm-based blocks, instead of amino-containing methacrylate units, were also investigated.47–49 In one of the studies, three different Pluronic® polymers were used, namely Pluronic® F108, F68, and F127.48 By increasing the DP of the hydrophobic PG-based block, and by increasing the length of the NIPAAm-based outer blocks while keeping the Pluronic® constant, a decrease in the LCST and a higher aggregation number were recorded.48 A similar pentablock terpolymer consisting of Pluronic® F127 was investigated via micro-differential scanning calorimetry (micro-DSC), SANS and NOESY experiments, which revealed two phase transitions corresponding to the thermoresponse of PG and NIPAAm, respectively.47,48 It was confirmed that the thermoresponse of NIPAAm units triggered the change from classical core–shell micelles to flower-like micelles.47,48
In another study, Pluronic® F127 multiblock copolymers were produced by: (i) coupling of Pluronic® F127, i.e., four times and (ii) random coupling of EG- and PG-based blocks, with the final copolymers having a similar content of EG but a different MM of Pluronic® F127.50 In both cases, the viscosity of the final product exceeded that of Pluronic® F127 by fifteen and six times, respectively. Prolonged anti-restenosis drug release was observed for the four-times coupled Pluronic® F127 (forty days) compared to Pluronic® F127 (seven days).50 However, the favourable gelation characteristics of the synthesised polymers cannot be purely attributed to the multiblock architecture, as the increased MM does play a strong role in the viscosity of the solution, and consequently the gelation, as previously mentioned. Therefore, for clear architecture effects to be drawn, systematic studies need to be carried out, in which the feature of interest should be varied while keeping all the other parameters constant.
EG-containing degradable polymers
Thermoresponsive ABA and BAB triblock copolymers based on EG (A unit) and PLGA (B unit) have been reported in the literature, with the BAB counterparts of a specific MM and composition being trademarked as ReGel®.51 Specifically, ReGel® polymers’ MM varies from 3100 to 4500 g mol−1, while the composition of the degradable component, i.e., PLGA, varies from 51 to 83 w/w%, in which the glycolate (GA) content varies from 15 to 35 w/w%.51 Both structures have been patented by Macromed Inc.,52–54 with the work to be strongly associated with Prof Sung Wan Kim (1940–2020, co-founder of Macromed Inc.).162The composition and the MM of these copolymers strongly control their gelation properties. When the MM of the central EG-based block was kept constant (BAB-type), the gelation temperature was lower for polymers with a higher MM of the PLGA block and a higher content of D,L-lactate (D,L-LA).55–58 The latter is expected as LA is more hydrophobic than GA, owing to the extra methyl group in its structure, thus enhancing the “hydrophobic effect”, responsible for thermoresponse. Similarly, the longer the EG-based block, the higher the gelation temperature.57,59 Similar observations were made for the ABA type polymers, as the lower the D,L-LA content, the higher the CGC.60 On the other hand, when L-LA was used to fabricate BAB-type triblock copolymers, the Tgel decreased as the content in L-LA decreased, indicating that the effect of stereochemistry, i.e., the increase in crystallinity, overcomes the effect of hydrophobicity.61
The content and block length of these BAB triblock copolymers affect their properties at the nanoscale, as revealed by SANS. While an ellipsoid structure is adopted by the BAB copolymer with a shorter EG-based block in diluted solutions, core–shell spherical micelles are detected for the one with a longer EG-based block under the same conditions.59 On the other hand, at higher concentrations, both copolymers adopt the core–shell spherical micelle configuration, and their thermally induced gelation is attributed to the packing of the micelles into cylinders.59
When the ABA and BAB copolymers are compared, the critical gelation concentration (CGC) and the Tgel of the latter are lower than the ones of the former, a feature attributed to the bridging of adjacent flower-like micelles by the PLGA blocks.56,60,62 Monte-Carlo simulations in combination with experimental work revealed a lower CGC, Tgel, and precipitation temperature of the BAB-systems compared to those of the corresponding ABA and AB polymers.63 The physical crosslinking of the core–shell micelles formed by the ABA and AB polymers was attributed to the formation of hydrophobic channels, throughout the gelation window. On the other hand, the thermogelation of the flower-like micelles adopted by the BAB polymer chains takes place in two steps: (i) gelation with a dominant crosslinking factor with the formation of hydrophilic bridges at lower temperatures, and (ii) gelation with a dominant crosslinking factor with the formation of hydrophobic channels at higher temperatures. These are shown schematically in Fig. 8.
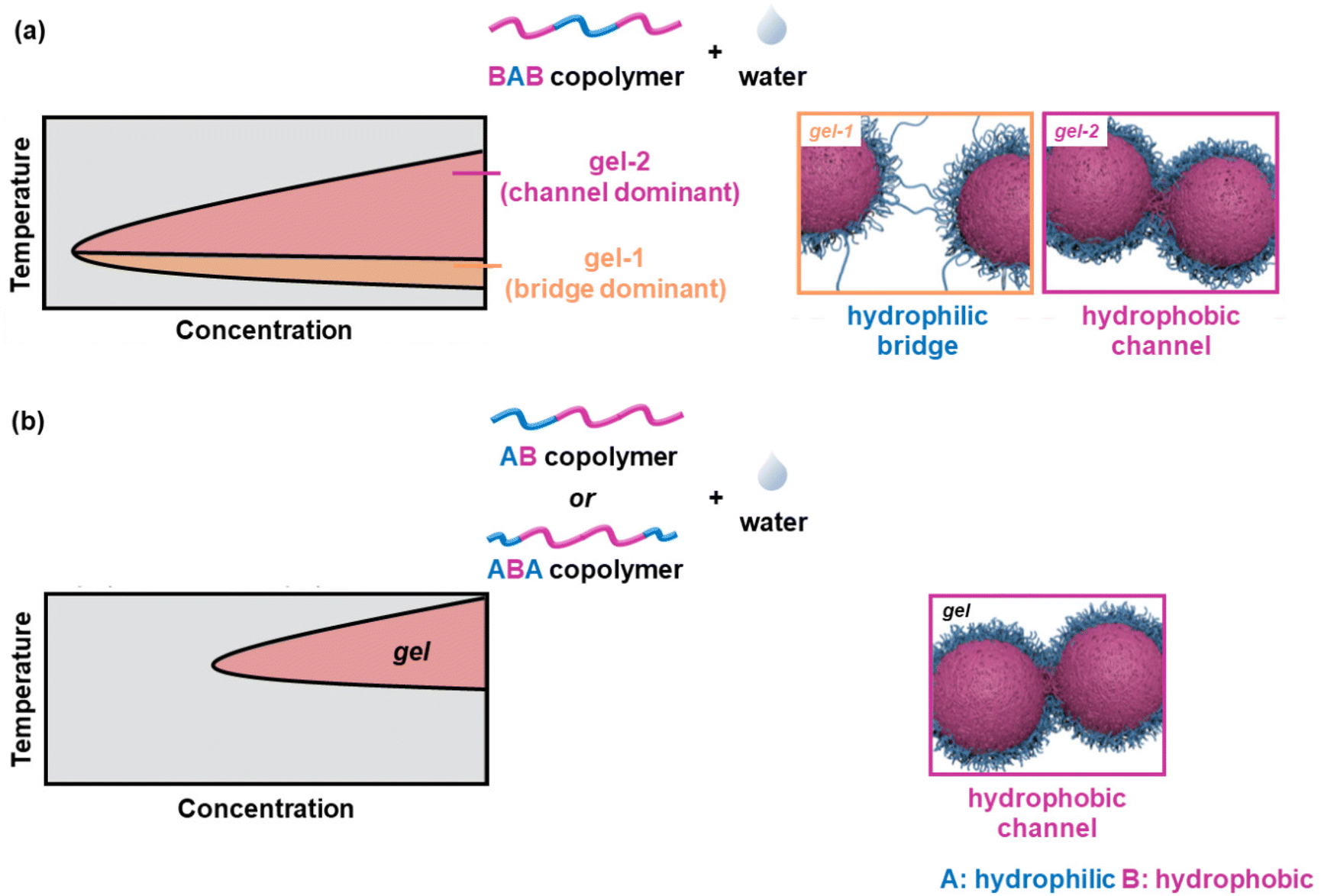 | ||
| Fig. 8 (a) Schematic of the phase diagram of a PLGA-PEG-PLGA triblock copolymer (left) and the suggested gelation mechanism (right), and (b) schematic of the phase diagram of a PEG-PLGA diblock copolymer and a PEG-PLGA-PEG triblock copolymer (left) and the suggested gelation mechanism (right). A and B correspond to the hydrophilic PEG block (in blue) and the hydrophobic PLGA block (in purple), respectively. PEG and PLGA stand for poly(ethylene glycol) and poly(D,L-lactide-co-glycolide), respectively. Adapted with permission from S. Cui, L. Yu, and J. Ding, Macromolecules, 2019, 52, 3697–3715. Copyright 2019 American Chemical Society. | ||
ε-Caprolactone (CL) was also used as a repeating unit in combination with EG for the synthesis of degradable ABA and BAB triblock copolymers. It should be reminded that the hydrophobicity increases from GA to LA to CL, thus offering opportunities for modulating the gelation properties of the polymers and their degradability rate.
Studies on BAB triblock copolymers based on EG (A unit) and a random copolymer (B block) of CL with either LA64,65 or GA66 were reported. One of the studies on LA-containing polymers reported that the higher the MM of the middle block, the lower the gelation concentration.65 On the other hand, the second study reported a lower Tgel, a wider gelation region, and a higher storage modulus, but a higher gelation concentration for the polymer with a longer EG-based middle block.64 However, direct comparison should be avoided due to the difference in both MM and composition. SANS studies revealed the transition from core–shell micelles to rod-like objects for the polymer with a longer middle block, while the one with a shorter middle block existed as cylinders, regardless of the temperature. This was attributed to the shorter middle block being unable to form a loop, thus promoting intermicellar bridging.64 As far as GA-containing copolymers are concerned, it was observed that the higher the CL/GA ratio, the lower the Tgel, despite the increase in the hydrophobicity of the structure, indicating that other factors, such as crystallisation, play an important role in the gelation properties.66
In some of the studies, BAB triblock copolymers based on EG (A unit) and CL's derivatives (B unit) were investigated.67,68 In particular, the CL unit consisted of a benzyl carboxylate group at the α-position, which was hydrolysed to different extents to give carboxylic acid groups, which are pH-responsive. Increasing the content of the hydrophobic benzyl groups decreased the CP and the Tgel and increased the strength of the gels. As far as the carboxyl-containing copolymers are concerned, their micellisation was controlled by the pH, with the neutrally charged polymers self-assembling into micelles at lower concentrations.67,68 However, depending on the polymerisation conditions, i.e., bulk versus solution, and polymerisation time, branching occurred, thus gelation or water-insolubility were promoted, depending on the level of branching.163 This indicates the importance of synthesising well-defined polymers, thus ensuring reproducibility in the synthesis and the final properties.
Degradable BAB and ABA copolymers consisting of EG (A block) and either CL or depsipeptides (B block) were studied,69,70 revealing that the architecture has an important role in the final properties. Concerning the study on CL-based copolymers, a lower Tgel, a wider gelation area and a higher storage modulus of the gels were obtained when the BAB-type copolymers were used, a feature attributed to the bridging of the flower-like micelles.69 This is in agreement with the observations made on triblock copolymers based on PLGA instead of CL. In contrast, the ABA-type depsipeptide-containing polymers, which form classical core–shell micelles, showed temperature-driven gelation, while the BAB-type ones only presented a CP upon heating, which decreased by increasing the hydrophobicity of the depsipeptide.70 However, it should be noted that in the latter study both the composition and the MM varied, thus illustrating the importance of performing systematic studies for clear conclusions to be drawn.
In an interesting study by Mohammadi et al., several degradable triblock copolymers were studied.71 In more detail, systems containing BAB-type copolymers with the A block consisting of EG and the B block consisting of GA/LA, LA and LA/CL were investigated. An injectable system which gels at body temperature was formed by the LA-containing copolymer. On the other hand, GA/LA copolymers of different structures were mixed to achieve the same result, similarly to the LA/CL based copolymers. These thermogelling systems were studied as drug release depots, with the LA/CL containing systems prolonging the release,71 presumably due to the increased hydrophobicity. This illustrates the potential of designing optimal thermogelling systems by blending of thermoresponsive polymers.
Degradable pentablock copolymers based on EG, CL and/or LA were also reported in the literature. In one of the studies, ABCBA pentablock terpolymers consisting of EG (A block), CL (B block) and LA (C block) of different hydrophobic contents showed that the higher the content of CL, the lower the Tgel, and the higher the rigidity of the gel.72 In another study, ABCBA pentablock terpolymers with various ratios of D-LA (A block), L-LA (B block) and EG (C block) showed UCST-type gelation, attributed to the intermicellar aggregation and bridging, revealed by SAXS.73 This observation indicates that slight variations in the position and chemical nature of the polymers can result in important differences in their thermogelling properties. Interestingly, copolymers with an equimolar content of enantiomers showed a wider gelation area and a higher storage modulus, as the gel destabilised at higher temperatures. When the pentablock copolymers were compared with their corresponding triblock copolymers, the gels destabilised at lower temperatures from ABCBA, to the ACA/BCB enantiomeric mixture, to the isotactic BCB, to the atactic copolymer based on D,L-LA instead. This indicates that the position of the blocks and the degree of crystallinity are of great importance for the gelation.73
Hybrid ABCBA pentablock copolymers consisting of the hydrophilic peptide L-lysine (A unit), a hydrophobic statistical copolymer of poly(L-histidine)-co-poly(γ-benzyl-L-glutamate) as the B block, and hydrophilic and thermoresponsive EG (C unit) have been recently reported by Skoulas et al.; L-histidine is pH-responsive.74 Physical hydrogels were formed at room temperature, which are temperature-insensitive when L-histidine is neutrally charged. However, when L-histidine is protonated by lowering the pH from 7.4 to 6.5, weaker gels with temperature-tuneable viscoelastic properties are observed.74
Degradable thermoresponsive copolymers with hyperbranched/multiblock architectures were also reported in the literature. In one of the studies, triblock and hyperbranched copolymers based on EG, PG and CL were compared with the hyperbranched architecture lowering the CP, the critical gelation temperature (CGT) and the critical gelation concentration.75 Interestingly, the CGC values reported were as low as 4.3 wt%, depending on the composition. These superior gelation properties of the hyperbranched architecture were attributed to the enhanced chain interactions through hydrogen bonding between the branches.75 Multiblock terpolymers based on 1,4-butylene adipate, EG and PG, and multiblock bipolymers based only on EG and PG were also reported, with the BA content enhancing the gelation characteristics.76 A thermoresponsive multiblock bioconjugate was reported, namely (EG23-b-betulin-b-EG23)6, which showed UCST-type behaviour with a transition from a turbid gel to a transparent solution to precipitation upon heating.77
An interesting study on thermoresponsive amphiphilic block copolymers with alternating hydrophilic and hydrophobic blocks has been recently published by Kostyurina et al.78 This library of thermoresponsive polymers consisted of polyester hydrophobic blocks and PEG hydrophilic blocks of varying lengths. A linear relationship between the CP and the length of the hydrophobic block was revealed, while a logarithmic relationship between the CP and the length of the hydrophilic block was established instead. Amphiphilic alternating copolymers with short PEG blocks favour the formation of highly ordered polymer networks characterised by liquid crystalline cubic phases, as revealed by SAXS studies.78
Acrylamide thermoresponsive units
Several (meth)acrylamide homopolymers have been reported to present thermoresponse. In 1975, Taylor and Cerankowski reported the CP of several (meth)acrylamide homopolymer solutions at 1% in water, such as N-ethylacrylamide (CP = 74 °C), NIPAAm (CP = 32 °C),164N,N-diethylacrylamide (DEAAm, CP = 25 °C), and N-cyclopropylacrylamide (CP = 57 °C).165 The structures and properties of these units are shown in Fig. 9. Amongst these, NIPAAm has been undoubtedly the most popular thermoresponsive unit, and it has been extensively studied for its thermoresponsive properties, including thermogelation. The relevant studies are discussed in the following paragraphs, with the major interest being focused on the effect of architecture on the thermoresponsive properties. | ||
| Fig. 9 Chemical structures and properties of thermoresponsive acrylamide units. | ||
In several studies, NIPAAm (A unit) was combined with styrene (St, B unit) to form BAB triblock copolymers, which adopt flower-like configuration when dissolved in aqueous media.79–82 SANS studies on copolymers containing deuterated St revealed the formation of a St-based hydrophobic core, and a NIPAAm-based hydrophilic corona below the CP. These micelles collapsed and formed clusters once the CP was reached. When a higher content of St was used, micelles, aggregates, and clusters formed both below and above the CP, as revealed by SANS and light scattering.79–82 When varying the hydrophobic monomer from St to 4-t-butyl St (tBuSt), 3,5-dibromobenzyl acrylate (BrBzA), 2-ethylhexyl acrylate (EtHexA), to octadecyl acrylate (OcDecA), it was observed that the CGC decreases, and the gels were stronger, as the glass transition temperature of the hydrophobic monomer decreased.83 On the other hand, no effect on the CP was observed, presumably due to the hydrophobic block being collapsed in the core of the micelle.83
Several other units were used to form BAB triblock copolymers with NIPAAm forming the central hydrophilic and thermoresponsive block. Acrylic acid (AA) was incorporated into the structure to exhibit pH-responsive properties in addition to the thermoresponse; deprotonation of the carboxylic acid units disrupted thermoresponse, as expected.84 Post-polymerisation modification of these copolymers with iron oxide introduced magnetic-responsive properties,84 while phosphorylation of the AA units decreased the Tgel, with NIPAAm117-b-AA(PEA)303-b-NIPAAm117 forming hydrogels under physiological conditions at only 2 wt%; PEA stands for O-phosphoethanolamine.85 When the outer blocks consisted of a random copolymer of N,N-dimethylacrylamide (DMAAm) and adamantane acrylate (Adac), complexation with cyclodextrin was achieved, thus forming thermoresponsive supramolecular glycopolymers.86 Incorporation of an ionic liquid, namely 3-methyl-1-(4-vinylbenzyl)-1-imidazolium chloride (A block) into the structure of BAB NIPAAm based triblock copolymers formed thermogels.87
ABA triblock copolymers, with NIPAAm forming the outer blocks, were also synthesised, and investigated by several groups. Increasing the length of the hydrophilic EG-based block resulted in an increase in the CP, as the “hydrophobic effect” was disrupted,88 while increasing the length of the thermoresponsive NIPAAm block in copolymers containing N-vinylpyrrolidone (VP) decreased both the CGC and the Tgel of the system.89 A positively charged betaine ester was incorporated into the central block to electrostatically attract the negatively charged antimicrobial drug, salicylate, thus forming biocompatible antimicrobial drug release depots based on thermogelling wound dressing.90 Redox active polymers were also investigated by incorporating 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) units (B unit) into the structure, and the redox activity was affected by NIPAAm's thermoresponse.91 In one of the studies, hydrophobic hydroxyterminated polybutadiene (HTPB) was used to form the central block, and its epoxidation increased the CP due to the introduction of more water molecules.92
Two studies have been reported in the literature in which ABA and BAB NIPAAm-based triblock copolymers were compared, and the effect of the architecture was discussed. In the first study, 2-hydroxyethyl methacrylate (HEMA) was used to fabricate the B block, and the ABA-type copolymers presented a lower Tgel than the BAB one.93 In the second study, ethyl acrylate (EtA) was used as the hydrophobic B unit instead.94 While the critical micellisation concentration (CMC) value of the ABA architecture was lower than its BAB counterpart, the CP of the former was higher than the latter.94 These observations can be explained upon consideration of the self-assembled structures these copolymers adopt. The ABA architecture, with the hydrophobic block in the middle, forms traditional core–shell micelles, which are easier to form (lower CMC) and remain stable up to higher temperatures (higher CP). On the other hand, the BAB architecture forms flower-like micelles, whose formation requires folding of the polymer chain into a loop, thus their self-assembly is less favourable (higher CMC) and these copolymers destabilise more easily upon heating (lower CP), due to the folding of the polymer chain.
Several studies reported the synthesis of triblock terpolymers based on NIPAAm and investigation of their thermoresponsive properties in terms of the CP. When PEG-PCL-PNIPAAM copolymers (ABC structure) were studied, the CP decreased upon increasing the DP of the hydrophobic and thermoresponsive blocks.95 When their ACB counterparts were tested, the CP decreased as a function of the polymer concentration.96 When the middle block in the ABC structure was changed to 3-methacryloxypropylheptaphenyl polyhedral oligomeric silsesquioxanes (MA-POSS), similar observations were recorded, i.e., the CP decreased when the DP of NIPAAm increased.97 Double thermoresponsive ACB triblock terpolymers based on poly(ethylene glycol) methyl ether vinylphenyl (mPEGV, A block), NIPAAm (B block) and St (C block) presented two CP values, and the higher the DP of NIPAAm, the more pronounced the transmittance change.98 When the change in the size as a function of temperature was monitored, collapse of the NIPAAm-based block on the hydrophobic core occurred upon NIPAAm's thermoresponse, leading to the shrinkage of the micelles above the CP.95,96
Two studies on triblock quaterpolymers, i.e., polymer with three different blocks, with one block consisting of repeating units with different chemical nature, were reported in the literature. In one of the studies, thermoresponsive glycopolymers were studied; these polymers were poly(3-O-methacryloyl-α,β-D-glucopyranose (MAGlc)-poly(HEMA-g-PCL))-PNIPAAm.99 An increase in the CP was recorded as the DP of both the hydrophilic MAGlc and the thermoresponsive NIPAAm blocks increased, due to the increased hydrophilicity in their structure.99 When PEG-poly(HEMA-g-LA)-PNIPAAm was studied, collapse of the NIPAAm block on the hydrophobic central block, and thus shrinkage of the micelles occurred above the CP, similarly to the previous observations.100 It is worth noting that the highly hydrophilic PEG corona prevented aggregation above the CP.100
Lodge and co-workers have previously reported several studies on thermogels from ABC triblock terpolymers, with A, B, and C blocks being based on poly(ethylene-alt-propylene), EG, and NIPAAm, respectively.101–103 In the first study on this subject, the ABC triblock terpolymers out-performed their CBC counterparts, by favouring gelation at lower concentrations. Detailed analysis on the ABC triblock terpolymers via cryogenic transmission electron microscopy, cryogenic scanning electron microscopy, and SANS revealed the formation of two-compartment polymer networks with distinct poly(ethylene-alt-propylene) and PNIPAAm cores, which are connected via hydrophilic PEG bridges. When dissolved in the ionic liquid 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide, NIPAAm presents a UCST-behaviour instead, with two-compartment ion gels being formed above the UCST.101–103
In an intriguing study by Onoda et al., precise control of the sol–gel transition by blending two ABC triblock terpolymers was reported.104 More specifically, the two ABC triblock terpolymers consisted of DMAAm as the central block and a random copolymer of NIPAAm and n-butyl acrylate (BuA) as the A block, but they differed in the third block; this was based either on a random copolymer of NIPAAm and BuA, but with a different composition from the first block, or a NIPAAm homopolymer. The Tgel of the former was lower than that of the latter, due to the incorporation of more hydrophobic BuA units, as expected. Interestingly, the Tgel of the polymer blends varied linearly with the content in the two copolymers, while keeping the strength of the gel constant.104
Temperature- and photo-responsive hydrogels were fabricated using ABC triblock terpolymers, where A, B and C correspond to NIPAAm, 4-acryloylmorpholine (NAM), and 2-((((2-nitrobenzyl)oxy)carbonyl)amino) ethyl methacrylate (NBOCAEMA), respectively.105 Self-assembly into core–shell micelles with the hydrophobic NBOCAEMA and the hydrophilic NIPAAm and NAM was recorded below the LCST of NIPAAm, and temperature-induced gelation occurred upon heating above the LCST. An increase in the Tgel upon UV-crosslinking of the micellar core was observed, thus offering a second trigger for the gel dissolution, in addition to temperature. These novel hydrogels were tested for co-delivery of the hydrophobic drug doxorubicin and the hydrophilic drug gemcitabine.103
Two studies reported the synthesis and investigation of thermoresponsive tetrablock terpolymers. In one of the studies, ABAB multiblock copolymers based on DMAAm and NIPAAm were investigated.108 When comparing two tetrablock copolymers with similar compositions and different MMs, gelation was observed only for the higher MM copolymer, due to the hydrophilic blocks being long enough to act as bridges. When compared to the corresponding BAB triblock copolymer, the Tgel decreased and the strength of the gel increased, as the complexity of the architecture increased.108 The self-assembly of EG-b-St-b-NIPAAm-b-DMAEMA tetrablock quaterpolymers was tuned by changing the temperature and the pH, thus employing the thermoresponsive properties of NIPAAm and the pH-responsive properties of DMAEMA.109 In addition, by changing the pH of the solution from acidic to alkaline, the CP of the copolymers was decreased, due to the increase in the hydrophobicity of their structure.109
Two studies investigated ABCBA pentablock terpolymers based on the pH responsive DEAEMA (A block), the thermoresponsive NIPAAm (B block) and the hydrophilic EG (C block).110,111 Both studies investigated the effect of DEAEMA's pH response on the self-assembly and the CP. In an acidic environment, the amine groups are protonated, thus temperature-driven micellisation due to NIPAAm's thermoresponse was observed. On the other hand, at alkaline pH, DEAEMA is hydrophobic, thus self-assembly at room temperature was observed. An increase in the pH lowers the CP, as expected, due to favouring the “hydrophobic effect”.110,111
In a recent study by Lv et al., double stimuli responsive ABCBA pentablock quaterpolymers have been shown to form different types of self-assembled structures depending on the pH and temperature.114 More specifically, these copolymers consisted of thermosensitive NIPAAm (A unit), PG (C unit), and a pH-responsive block based on a random copolymer of AA and t-butyl acrylate (tBA), but they differed in the length of the NIPAAm block and the ratio of AA:tBA. Interestingly, the degree of deprotonation of the AA units could determine the reversibility of the thermosensitive process, and tune the types of self-assembled nanostructures, with cylinders and spheres being identified.114
Degradable NIPAAm-containing pentablock terpolymers have also been reported in the literature. The ABCBA pentablock terpolymers were based on NIPAAm (A unit) and EG (C unit), while the B unit was based on a degradable hydrophobic monomer, either CL112 or LA.113 In both studies, longer outer NIPAAm chains favoured thermogelation. In the study by Abandansari et al., the best-performing polymer (CL-based) presented the Tgel at 32 °C and the storage modulus (G′) at 25000 Pa, when the concentration was 20 wt%. Most importantly, the storage modulus drastically increased to 60000 Pa when the concentration increased to 30 wt%.112 Concerning the LA-containing pentablock terpolymer, gelation in a physiological medium was visually detected at 10 w/w% and 30 °C, which was not the case for its triblock ABC analogue, whose aqueous solutions were unable to form gels.113
Interestingly, a study on well-defined NIPAAm-based multiblock copolymers has been reported by Simula et al.115 In this study, thermoresponsive triblock and pentablock copolymers were synthesised via aqueous Cu(0) mediated living radical polymerisation (SET-LRP), with excellent control over the molecular mass distribution (Đ < 1.19), quantitative conversions, and time-efficiency. Increasing the DP of NIPAAm (B unit) in BAB triblock copolymers with the PEG central block from 10 to 160 decreases the CP from 71.3 °C to 41.2 °C. Chain extension with a second thermoresponsive unit, namely DEAAm (C unit), to form a CBABC pentablock terpolymer reduced the CP from 71.3 °C to 52.5 °C. Substituting the thermoresponsive DEAAm unit with the hydrophilic DMAAm unit disrupted the thermoresponse, as no CP was detected up to 90 °C.115
Two studies discussed the thermoresponse of NIPAAm-based star copolymers and their comparison with the corresponding linear analogues.116,117 When A2BA2 copolymers (A: NIPAAm, B: EG) were compared with ABA triblock copolymers with the same MM and composition, a similar thermogelling behaviour was recorded. On the other hand, the gelation characteristics were favoured as the complexity of the architecture increased from ABC, ABCBA, and (ABC)4 (A: [R]-3-hydroxybutyrate, B: NIPAAm, C: poly(ethylene glycol) methacrylate-co-poly(propylene glycol) methacrylate), PEGMA-co-PPGMA. Specifically, the higher the complexity of the architecture the stronger the gels, and the lower the Tgel, with both ABCBA and (ABC)4 forming gels at room temperature; the strength of the latter increased upon NIPAAm's thermoresponse.116,117
As previously mentioned, DEAAm is a less famous thermoresponsive unit, whose homopolymers have been reported to show a CP at 25 °C by Taylor and Cerankowski,165 and 33 °C by Idziak et al.166 The CP being close to the physiological temperature makes this unit a suitable candidate for designing thermoresponsive polymers. In a study by Haddow et al., ABA triblock copolymers based on DEAAm (A unit) and EG (B unit) were synthesised, and the MM of the blocks was varied. Copolymers with a longer PEG middle block favoured gelation, due to the effective bridging of the micelles and the formation of spherical and elliptical aggregates upon heating, thus leading to gel formation.118 Grubbs and co-workers reported the synthesis and investigation of two ABC triblock copolymers with A, B and C being based on EG, DEAAm, and N,N-dibutylacrylamide (DiBuAAm), respectively.119 When the thermoresponsive middle block was short, gelation driven by a sphere to worm transition was recorded, while an increase in its length disrupted thermogelation due to difficulties in the re-arrangement of the self-assembled structures.119
Amine-based methacrylate thermoresponsive units
The DMAEMA unit has been extensively reported in the synthesis of polymers for various applications. Even though DMAEMA has been mostly incorporated into the polymer structure to exhibit pH-responsive properties, reports on its thermoresponse have also been published. The latter are discussed in the following paragraphs with an increased architecture complexity.In one of the studies, AB diblock and ABA and BAB triblock copolymers consisting of hydrophilic 6-O-methacryloyl-D-galactopyranose (A unit) and pH- and temperature-responsive DMAEMA (B unit) were synthesised and investigated in terms of their thermoresponse.120 At alkaline pH both units are water-soluble and thus they exist as unimers in solution below the LCST of DMAEMA. Upon heating, temperature-driven micellisation was induced, leading to the formation of traditional core–shell micelles by the AB and ABA copolymers, and flower-like micelles by the BAB ones, with their CP being detected between 35 and 55 °C, depending on their MM and composition.120 Direct comparison between the different architectures cannot be made due to the variations in the MM and composition, which also affect the CP.
BAB triblock copolymers with DMAEMA units forming the A block and a random copolymer of DMAEMA and n-butyl methacrylate (BuMA) forming the B block were reported by Lauber et al.121 These copolymers formed hydrogels at room temperature when the degree of ionisation of DMAEMA was kept at low values. However, the viscosity of the hydrogels decreased, and eventually homogeneous runny solutions were formed when protonating the DMAEMA units and thus increasing the hydrophilicity of the structure. Interestingly, upon heating of the solutions at high degrees of the protonation of DMAEMA, irreversible hydrogels were formed.121
A study on the same repeating units has been reported by our group, with symmetric and asymmetric BAB triblock copolymers being investigated.122 While keeping both the MM and composition constant, within the same family, the Tgel of the copolymers increased by increasing the asymmetry from BAB to B′AB′2, to B′′AB′′4. This trend was attributed to the insufficient bridging between the neighbouring flower-like micelles, thus interrupting the gelation.122
To the best of our knowledge, only our group has reported the synthesis and investigation of triblock terpolymers consisting of DMAEMA as the thermoresponsive unit (C unit).26,123–126 In all the studies, the A block was based on a methoxy EG-based methacrylate unit, e.g., methoxy oligo(ethylene glycol) methacrylate with an average Mn of 300 g mol−1 (mOEGMA300), which is hydrophilic and thermoresponsive at a temperature higher than 70 °C.17,167 Note that the thermoresponse of the OEGMA monomer depends on the number of EG groups and the chemistry of the terminal group, as will be discussed in more detail in the relevant section. The B block consisted of a hydrophobic unit with a linear alkyl side chain, e.g., BuMA. When investigating the MM and composition, it was observed that the gelation was favoured when the MM, the hydrophobic content, and the length of the hydrophobic side chain were increased, and when the length of the hydrophilic EG-based unit was decreased. The complexity of the architecture was varied by changing the position of the blocks in the structure, from ABC to ACB to BAC.26,124,126 From our studies, it was concluded that the ABC architecture, i.e., the one with the hydrophobic block in the middle, shows the most favourable characteristics for injectable systems, as it favours self-assembly to smaller micelles and thus better dissolution at room temperature, while it presents a clear solution–gel transition upon heating.26,124,126
The only study on tetrablock terpolymers with DMAEMA as a thermoresponsive unit has been reported by our group in 2018.127 In this study, the copolymers consisted of mOEGMA300, BuMA and DMAEMA as A, B, and C units respectively. The MM and composition were kept constant within the same family and the position of the blocks was varied as follows: ABCA, ABAC, ACAB, BACB, BABC, ABCB, CABC, CACB, and ACB. The position of the blocks strongly controlled the solubility, self-assembly and gelation properties of their aqueous solutions, with the BABC and ACBC architectures showing the widest gelation areas.127
Patrickios and co-workers have investigated multiblock bipolymers composed of DMAEMA and BuMA as the A and B blocks, respectively.128 BAB, ABABA, BABABAB and ABABABABA copolymers with the DPA and DPB being kept the same in different copolymers, but simultaneously changing the total MM and composition, were investigated, and gelation was reported for the heptablock bipolymer. When the composition and total MM were kept constant in BAB, BABABAB and ABABABABA copolymers, only the triblock and heptablock copolymers formed gels. It was concluded that the gelation was favoured when the BuMA block is located at the end of the polymer chain, with the hydrophilic and thermoresponsive DMAEMA block being long enough to facilitate micelle bridging. When the DMAEMA block was incorporated at the end of the polymer chain, electrostatic repulsion between the positively charged amine groups interrupted gelation.128
DMAEMA-containing thermoresponsive multiblock brush type graft terpolymers were reported.129 CLxCH2CH2[O(CLy)]CH2-b-VBz-g-DMAEMAw copolymers, with VB standing for 4-vinyl benzyl and x, y, z, and w denoting the DP of the corresponding units, showed a CP which was strongly controlled by the content of DMAEMA.129
EG-based (meth)acrylate thermoresponsive units
(Meth)acrylate units with EG on the side chain have received much scientific interest as alternative thermoresponsive units, due to the combination of their thermoresponsive ability and PEG's biocompatible nature.168 It is well-documented that depending on the number of EG groups on the side chain, the hydrophobicity/hydrophilicity and thermoresponse of these (meth)acrylate polymers can be varied.17,18,167 As an example, methoxy mono(ethylene glycol) methacrylate (MEGMA) homopolymers are water-insoluble, while methoxy oligo (ethylene glycol) methacrylate with an average Mn of 500 g mol−1 (mOEGMA500) shows no thermoresponse up to 95 °C.17 In addition, changing the end group on the side chain from hydroxyl to methyl to ethyl, the CP decreases as the hydrophobicity increases.17,18 The chemical nature of the backbone, i.e., acrylate versus methacrylate, also affects the CP, with the thermoresponse of the acrylate units being affected by the length and the end group of the side chain, similarly to their methacrylate analogues.18 The chemical structures and properties of EG-based (meth)acrylate units are summarised in Fig. 10.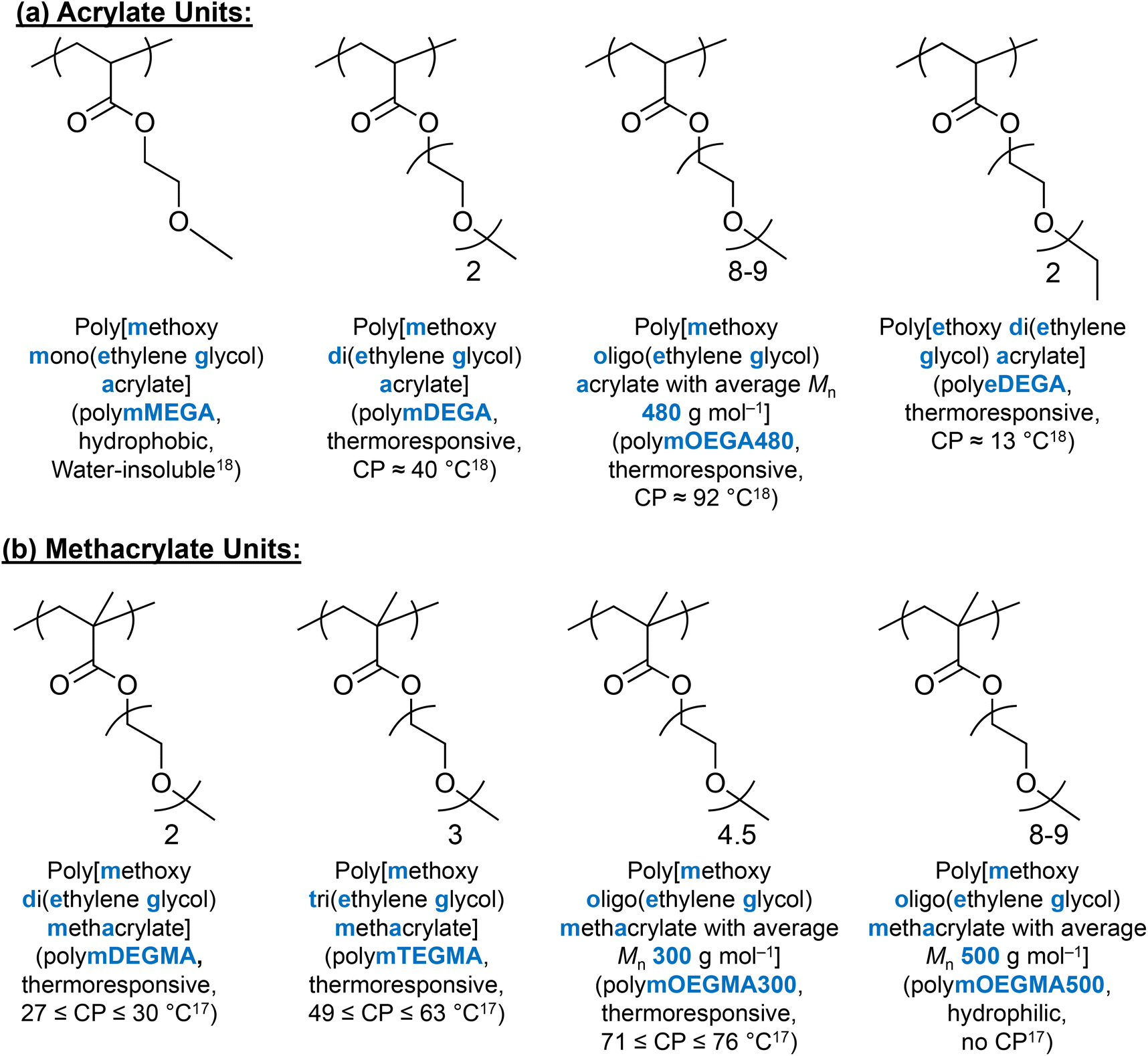 | ||
| Fig. 10 Chemical structures and properties of (a) acrylate units and (b) methacrylate units discussed in this review. | ||
Thermoresponsive ABA and BAB triblock copolymers consisting only of EG-based (meth)acrylate units were reported by Xiang et al.130 In this study, the triblock copolymers were based on methoxy oligo(ethylene glycol) acrylate with an average Mn of 480 g mol−1 (mOEGA480) and mDEGMA and were compared with their corresponding diblock and star copolymers [both (AB)3 and (BA)3], with the attention being focused on the effect of matching the polarity of the end group (phenyl versus carboxyl) with the polarity of the adjacent block. Generally, mismatching the polarity disturbs the self-assembly and sufficient hydration, thus leading to aggregation.130
Negru et al. reported a study on BAB triblock copolymers consisting of EG (A unit) and a random copolymer of ethoxy di(ethylene glycol) acrylate (eDEGA) and mOEGA480.146 It was observed that increasing the DP of either block resulted in a decrease in the Tgel, while when the composition was systematically varied, the Tgel decreased as the hydrophilic mOEGA480 content decreased.146
In three of the studies, hydrophilic and thermoresponsive methoxy di(ethylene glycol) acrylate (mDEGA, A unit) was combined with hydrophobic styrene (St, B unit) to synthesise amphiphilic BAB triblock copolymers.143–145 Miasnikova et al. investigated copolymers with a fixed DP of St, and a varied DP of the mDEGA block.144 They observed that the copolymer with the shortest DEGA block was insoluble in water, while solubility was enhanced when longer mDEGA blocks were incorporated into the structure, which also increased the CP of these copolymers. While the composition effect dominated the thermoresponse in diluted solutions, the MM effect controlled the gelation in concentrated solutions, with the CGC decreasing with the increasing length of the mDEGA block, due to the easier bridging.144 In another study by the same group, the BAB triblock copolymers were compared with their corresponding AB diblock copolymers and (BA)3 star copolymers.143 Interestingly, increasing the complexity of the architecture from diblock to triblock to star copolymers favours the thermoresponse by reducing the CP.143 In addition to the reports on micellisation and gelation, thermoresponsive films based on these triblock copolymers were also investigated.145 By increasing the temperature of the films, water was excluded from the structure and shrinkage was observed, driven by DEGA's thermoresponse.145
Studies in which triblock copolymers consisted of a hydrophilic and thermoresponsive EG-based (meth)acrylate unit and 2-hydroxyethyl (meth)acrylate were reported. In one of the studies, two ABA triblock copolymers based on mDEGMA (A block of constant length) and HEMA (B block of varied length) were compared, and it was revealed that the CP, the CGC and the Tgel decreased, while the strength of the formed gels increased by increasing the hydrophobic HEMA content.131 BAB triblock copolymers with the B block being based on 2-hydroxyethyl acrylate (HEA) and the A block being based either only on methoxy mono(ethylene glycol) acrylate (mMEGA) or a gradient copolymer of HEA and mMEGA were reported.142 It was observed that the BAB triblock copolymers presented lower CPs than their corresponding triblock-gradient copolymers, thus illustrating an architecture effect on the thermoresponse.142
In two of the studies, ATRP facilitated the synthesis of ABA triblock copolymers consisting of an EG-based methacrylate unit and either PCL141 or 2-methacryloyloxyethyl phosphorylcholine (MPC).132 When increasing the length of the mOEGMA500 block in mOEGMA500-b-PCL-b-mOEGMA500 polymers, the CMC and the CP decreased, thus indicating that the MM effect governs the composition effect.141 Biocompatible mDEGMA113-b-MPC243-b-mDEGMA113 copolymers formed physical gels, with the Tgel decreasing from 32 to 19 when the concentration increased from 5 to 25 wt%.132
Synthesising triblock terpolymers offers the possibility of incorporating different units into the structure, which may provide additional properties, e.g., pH-response. To the best of our knowledge, no study has investigated the effect of architecture of EG-based (meth)acrylate triblock terpolymers, i.e., the position of the blocks, and how it affects the thermoresponsive properties. Thus, this paragraph discusses studies in which the architecture was kept constant. In one of the studies, Liu et al. investigated thermo- and pH-responsive ABC triblock copolymers based on monomethoxy poly(ethylene glycol) (mPEG, A block), mDEGMA-co-N-hydroxymethyl acrylamide (mDEGMA-co-HMAAm, B block) and DEAEMA (C unit).133 These copolymers presented temperature-driven micellisation at acidic pH, due to the thermoresponse of mDEGMA, and their CP decreased by increasing the content of HMAAm.133 Double thermoresponsive ABC triblock copolymers were reported by Hu et al.134 These polymers consisted of ethoxy di(ethylene glycol) methacrylate (eDEGMA, A block) and 2-(methacryloyloxy)ethyltrimethylammonium iodide (MAEtMAm, B block), while the C block was based on a random copolymer of mDEGMA with rhodamine-B containing methacrylate (RhoMA). Heating the samples above the CP of eDEGMA resulted in micellisation, while in concentrated solutions, physical hydrogels were formed when the system was heated above the CP of mDEGMA. Interestingly, incorporation of thermoresponsive silica nanoparticles resulted in the formation of a hybrid gel with improved mechanical strength, due to the enhanced intermicellar bridging.134 Our group has previously reported a study on mOEGMA300-b-BuMA-b-DEAEMA copolymers, where BuMA stands for n-butyl methacrylate, and the aqueous solutions were investigated at an initial pH value, at which the DEAEMA copolymers are not protonated, and thus they are hydrophobic.140 It was found that gelation was promoted when BuMA and DEAEMA contents increased due to the enhanced hydrophobicity.140 When replacing the DEAEMA block with the thermoresponsive mDEGMA, and when the composition is targeted appropriately, a promising biocompatible thermogelling system is fabricated, which has a low gelation concentration (2 w/w%) and shows great potential in injectable gel applications.135,136
Only our group has previously published a study on EG-based (meth)acrylate tetrablock terpolymers.137 The composition and MM of the copolymers were kept constant, while the architecture was varied from ABCA, ABAC, ACAB, BACB, BABC, BABC, ABCB, CABC, CACB and ACBC; A, B, and C blocks consisted of mOEGMA300, BuMA, and mDEGMA, respectively. When comparing these architectures, it was concluded that higher CPs are detected for copolymers with the hydrophobic BuMA block being adjacent to the thermoresponsive mDEGMA one, as upon thermoresponse of mDEGMA, self-assembled structures with a BuMA- and mDEGMA-based core and a hydrated OEGMA300 corona may be formed. Similarly, self-assembled structures which can preserve a hydrophilic OEGMA300 corona upon the thermoresponse of mDEGMA favour gelation, with the ABCA architecture forming physical gels at 5 w/w% at physiological temperature.137
A study on an (A-co-B)-b-C-b-D-b-C-b-(A-co-B) pentablock quaterpolymer has been published by Papadakis and co-workers.139 The central D block consisted of EG, while the C block was based on DMAEMA. The outer blocks consisted of a random copolymer of BuMA and methoxy tri(ethylene glycol) methacrylate (mTEGMA). It should be noted that this study was performed in a pH range at which DMAEMA is positively charged, thus the thermoresponse is provided only by mTEGMA. A thorough physicochemical investigation has been performed using a combination of DLS, SLS, SANS and SAXS, which revealed significant hydration of the hydrophobic core and the highly swollen hydrophilic corona, with the number of loops and tangling chains being highly dependent on the temperature and pH.139
Thermoresponsive four-arm star copolymers with the arms being formed by a triblock terpolymer have been reported in the literature.138 The architecture of the arms varied from ABC to ACB, where A, B, and C correspond to CL, mOEGMA500, and mDEGMA, respectively. In either case, the length of the outer block was varied, which resulted in two opposite trends: (i) the longer the mDEGMA block in (ABC)4 star copolymers, the more favoured the thermoresponse was (both CP and gelation), and (ii) the longer the mOEGMA500 block in (ACB)4 star copolymers, the more interrupted the gelation was, due to the increase in hydrophilicity. A 10 wt% solution of an (ABC)4 copolymer formed gels at a physiologically applicable temperature.138 This study indicates an architecture effect, according to which the position of the block is crucial for promoting gelation.
Polymers based on other thermoresponsive units
Several studies have investigated the thermoresponsive properties of polymers based on VCL, peptides, and oxazolines. Due to the limited number of studies in which the effect of architecture has been investigated, the following paragraphs discuss studies in which generally the effects of the polymer structure, i.e., architecture, composition, and MM, on the thermoresponse have been reported.Three studies on VCL-based copolymers have been published and the effect of composition and MM on the thermoresponsive properties has been discussed.147–149 Two of the studies have been carried out by Debuigne's group, in which BAB double thermoresponsive copolymers were studied; the B block consisted of VCL, while the A block was a random copolymer of VCL and N-vinylpyrrolidone (VP).147,148 It should be pointed out that VP, which consists of a five-membered ring, is more hydrophilic when compared to VCL (seven-membered ring). Two CP values were detected, which correspond to the thermoresponse of VCL and VP. It was observed that the longer the VCL-based outer blocks, the lower the CP and Tgel and the higher the strength of the gels. Addition of salts lowered the thermoresponsive point, due to the ionic strength effect.147,148 The third study investigated the CP of BAB triblock copolymers based on VCL (A block) and a random copolymer of tBA and AA (B block).149 In this study, the Turbiscan Lab instrument was used for the first time to determine the CP of these copolymers, with the CP increasing with the content of AA.149
To the best of our knowledge, only one study reported thermoresponsive LCST type polypeptides.150 An elastin-like (AB)2 “tetrablock” copolymer with MESLLP{[(VPGVG)2-(VPGEG)-(VPGVG)2]10-b-[VGIPG]60}2V was biosynthesised by Martín et al.; the amino acids used are methionine (M), glutamic acid (E), serine (S), leucine (L), proline (P), valine (V), glycine (G), and isoleucine (I). This polypeptide was double-responsive, with the first block being pH-responsive and the second block showing thermoresponse. Temperature-triggered gelation was observed in concentrated samples (5 to 15 wt%), with the storage modulus increasing with the concentration. Interestingly, patterns with different shapes, such as hexagonal, circular, square pits, grooves, and cylindrical pillars, were created via replica molding, thus indicating the potential applicability of these polypeptides in the bioengineering field.150
Poly(2-oxazoline)s are a special class of polymers with the tertiary amide bond participating in the backbone of the polymers, Fig. 3.20 By substituting the side chain with hydrophobic moieties, the hydrophilicity of the structure changes. For example, polymers consisting of 2-methyl-2-oxazoline (MeOx) are very hydrophilic, thus showing no thermoresponse, while polymers based on 2-butyl-2-oxazoline (BuOx) are very hydrophobic, and are thus water-insoluble. Poly(2-oxazoline)s bearing alkyl chains with intermediate lengths are thermoresponsive, with the transition temperature being controlled by the chemical nature of the side chain.20 For example, high MM polymers of 2-ethyl-2-oxazoline (EtOx) present a CP at around 61 to 69 °C,169 and polymers of 2-isopropyl-2-oxazoline (iProOx) present a CP at around 26 to 34 °C,170,171 while their analogues with linear side chains, namely 2-n-propyl-2-oxazoline (nProOx), show a CP at 25 °C.172 The structure of poly(2-oxazoline)s and their respective LCST values (if any) reported in the literature are summarised in Fig. 11.
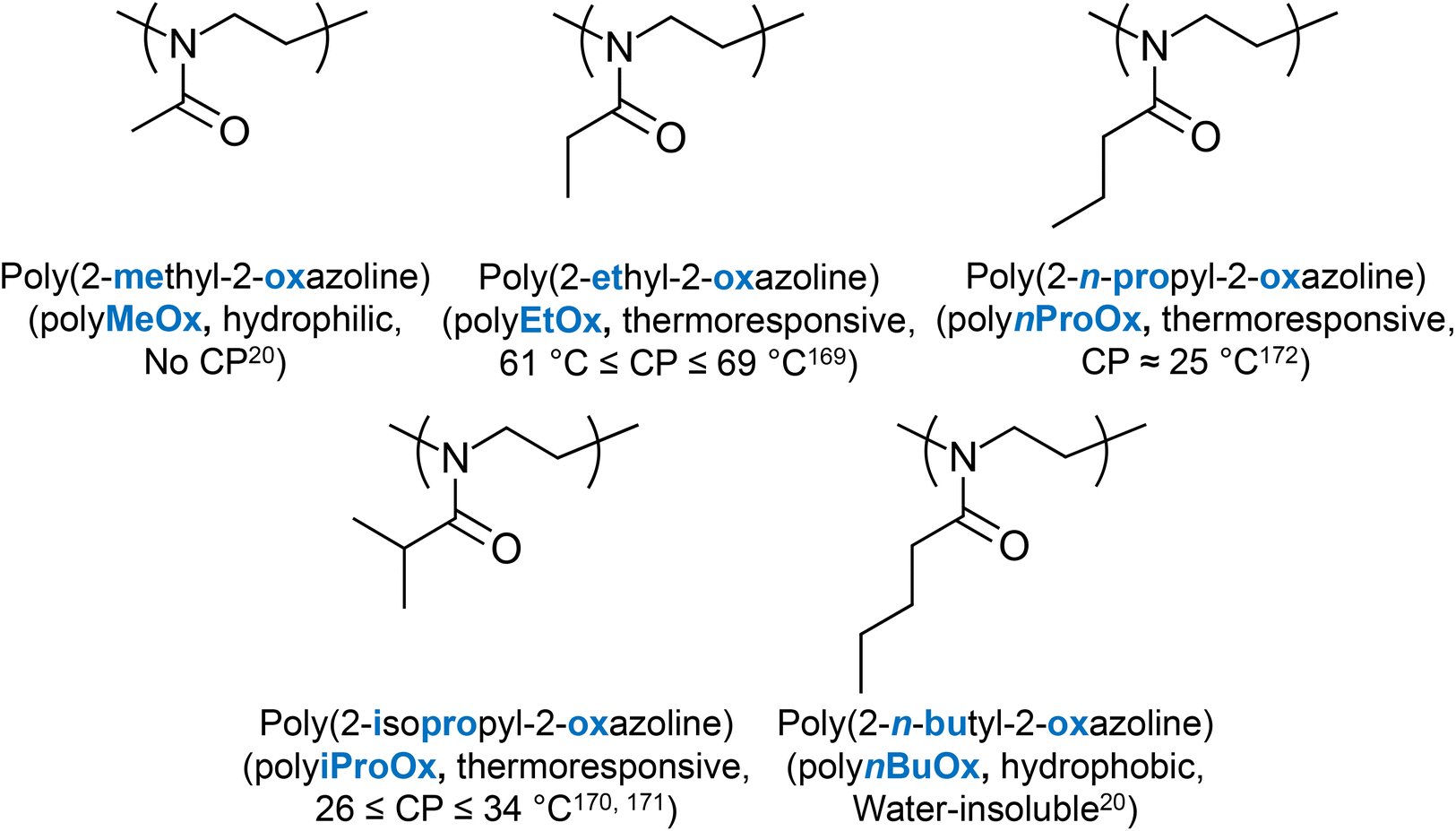 | ||
| Fig. 11 Chemical structures and properties of poly(2-oxazoline)s. | ||
A few studies have been reported on the effect of the polymer structure of poly(2-oxazoline)s on their properties. In one of the studies, ABA copolymers based on hydrophilic MeOx (A block) and iProOx-co-BuOx (B block, thermoresponsive) were investigated, and it was found that the higher the content of hydrophobic BuOx, the lower the CP,154 as expected. Luxenhofer's group reported several studies on oxazoline-based thermogelling triblock copolymers.152,155,173,174 In three of the studies, the ABA triblock copolymers consisted of hydrophilic MeOx (A unit) and a hydrophobic block based on either 2-isobutyl-2-oxazoline (iBuOx), 2-phenethyl-2-oxazoline (PhEtOx) or 2-benzhydryl oxazine (BzHOx) as the B unit.155,173,174 While none of these units are thermoresponsive, the combination resulted in the formation of thermoresponsive gels, and by changing the B block from alkyl to aromatic, the thermogelling behaviour changed from the LCST to UCST, when concentrated solutions (20 wt%) were tested.155,173,174 Intriguingly, the UCST-type thermogel based on PhEtOx was used to fabricate bioink for printing.173 Interestingly, the same group has investigated the effect of the block position on the thermoresponsive properties of triblock bipolymers, i.e., ABA versus BAB.152 These copolymers consisted of MeOx (A unit) and 2-n-propyl-2-oxazoline (nProOx, B unit, thermoresponsive), and they presented temperature-driven self-assembly, but no gelation. While the BAB architecture showed aggregation, presumably due to the flower-like structure, the ABA one formed smaller self-assembled structures.152 In a similar study on flower-like micelles adopted by the BAB architecture, where the A and B units are EtOx and nProOx, respectively, it was observed that the content and length of the hydrophilic middle block were crucial in gel formation, with the aqueous solution of a copolymer with 78 wt% of EtOx forming a hydrogel at 20 wt% at physiological temperature.153
EtOx (A unit) was combined with either L-LA175 or CL151 (B unit) to form degradable BAB triblock copolymers. It was found that the CP of the L-LA-based copolymers increased with the MM,175 similarly to what has been previously observed for Pluronic® polymers, while the gelation was favoured by increasing the content of the hydrophobic CL,151 as expected.
Effect of architecture – synopsis
In summary, the architecture has a key role in the thermoresponsive properties of the polymers. However, the effect of architecture depends on the chemical nature of the thermoresponsive units and the repeating units they are copolymerised with. The trends reported in the literature are summarised in the following paragraphs and the general trends identified are listed in Table 2.| Architecture | Effect |
|---|---|
| Abbreviations: CP, cloud point; CMC, critical micellisation concentration; CGC, critical gelation concentration; Tgel, gelation temperature; G′, storage modulus. Hydrophilic, hydrophobic, and thermoresponsive blocks are shown in blue, purple, and orange, respectively. In the case of Pluronic® polymers and their reverse counterparts, the B block, namely PPG, is also thermoresponsive at lower temperatures, and its thermoresponse promotes the self-assembly. | |

|
CPBAB < CPABA37,94 |
| CMCABA < CMCBAB94 | |
| CGCBAB < CGCABA56,60,62,63 | |
| T gel, BAB < Tgel, ABA56,60,62,63,69 | |
| G′BAB >G′ABA69 | |

|
ABC: clear sol–gel transition26,124,126 |
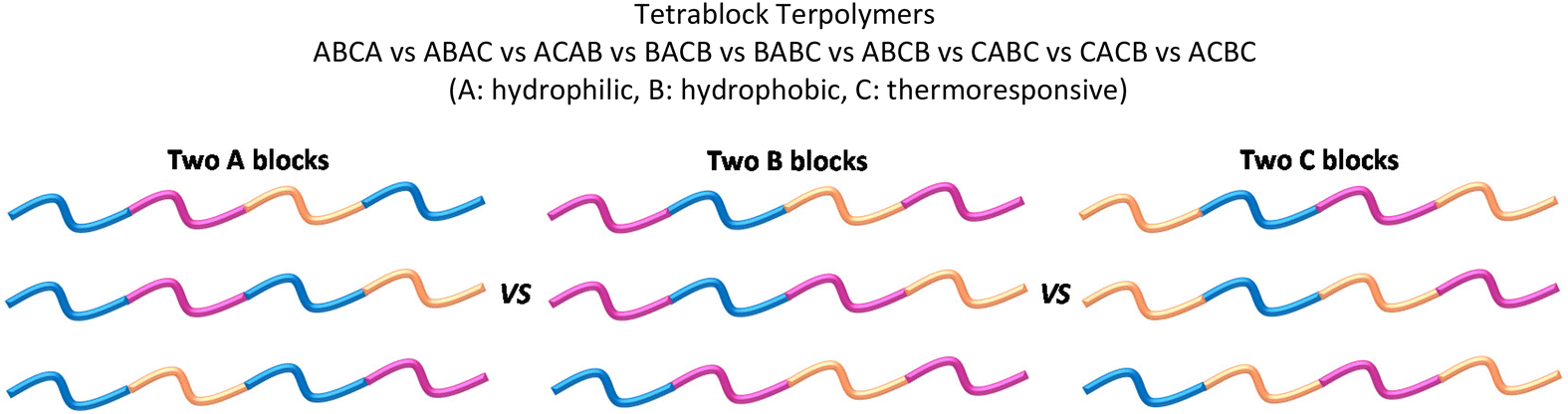
|
When C is next to B → ↑CP & ↓Tgel & ↓CGC127,137 |
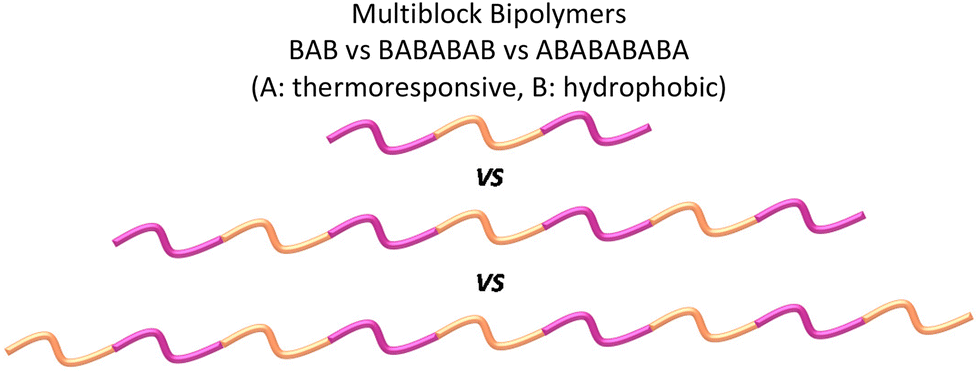
|
When B is an outer block → Gelation128 |
|
CPstar < CPlinear143 |
| T gel, star < Tgel, linear116 | |
The effect of the position of the blocks within the polymer chain on the thermoresponse is highlighted by the triblock bipolymers, i.e., ABA- and BAB-type architecture, Table 2. For example, Pluronic® polymers (ABA) present a CP which increases with the polymer concentration, while their reverse counterparts (BAB) show the opposite trend at the specific concentrations tested that ranged from 5 to 50 mM.35 The micellisation of these copolymers is also affected by the architecture,37 with the micellisation of the ABA architecture being favoured compared to the BAB one, presumably due to its self-assembled structure, i.e., core–shell micelles (ABA) versus flower-like micelles (BAB); the latter show a lower CP.37 As far as ABA and BAB triblock copolymers based on PEG (A block) and PLGA (B block) are concerned, the CGC and Tgel of the BAB architecture are lower than those of the ABA one, due to the two PLGA outer blocks joining two different adjacent micelles, thus resulting in bridging.56,60,62,63 Similar trends were observed when substituting the PLGA block with PCL, with the BAB counterpart showing a lower Tgel, a wider gelation area and a higher storage modulus69 In NIPAAm-based polymers, when HEMA was used as a comonomer, the ABA-type copolymers presented a lower Tgel than the BAB ones,93 but when EtA was used instead, the CMC value of the ABA architecture was lower, and the CP was higher than the BAB one.94 In poly(2-oxazoline)s, aggregation was observed for the BAB copolymers based on MeOx (A unit) and nProOx (B unit), with better-defined self-assembled structures being adopted by the ABA one.152 Generally, it can be concluded that the formation of well-defined micelles by the ABA architecture is favoured due to the formation of traditional core–shell micelles, as the adaptation of flower-like micelles might be less favourable entropically. As far as gelation is concerned, the BAB architecture with the B unit being hydrophobic favours the formation of the network by bridging of adjacent flower-like micelles.
Moving to triblock terpolymers, to the best of our knowledge, only our group has reported the effect of architecture within the triblock terpolymer structure.26,124,126 It has been concluded that the ABC architecture outperforms the ACB and BAC architectures by presenting a clear sol–gel transition, i.e., a transition from a solution to gel without solubility issues, upon heating; the A, B and C blocks were based on an EG-based methacrylate unit, an alkyl methacrylate unit, and DMAEMA, respectively.26,124,126
Concerning tetrablock copolymers, our group has systematically investigated the effect of the block position within the tetrablock terpolymer architecture in both DMAEMA-127 and DEGMA-containing137 thermoresponsive polymers. In both studies the hydrophilic and hydrophobic blocks were based on mOEGMA300 (A block) and BuMA (B block), respectively, while DMAEMA or DEGMA were the C block. It has been generally observed that architectures with the thermoresponsive block being adjacent to the hydrophobic one show a higher CP, presumably due to the re-arrangement of the core–shell micelles upon heating, with the thermoresponsive block collapsing in the hydrophobic block upon thermoresponse. Interestingly, these structures favour thermogelation, as this re-arrangement might lead to the formation of well-hydrated mOEGMA300 bridges, thus leading to network formation. Due to the different chemical nature of these two families, i.e., ionic versus non-ionic, the best architectures were ACBC and ABCA for the DMAEMA- and DEGMA-containing polymers, respectively.127,137
When multiblock bipolymers consisting of DMAEMA (A unit) and BuMA (B unit) were systematically compared, only the BAB triblock and BABABAB heptablock copolymers showed gelation, while the corresponding ABABABABA nonablock copolymer did not.128 This was attributed to the hydrophobic outer blocks facilitating bridging, in contrast to the positively charged DMAEMA outer blocks preventing gelation.128
Increasing the transition to a more complex topology, specifically from a linear block to a star structure generally favours thermoresponse. For example, in NIPAAm-containing polymers, lower Tgel and stronger gels were formed by the (ABC)4 architecture compared to the corresponding ABC one.116 Similar findings were reported when the ABCBA pentablock terpolymer and the ABC triblock terpolymer were compared.116 Changing the architecture from AB to BAB to (BA)3, in mDEGA-containing polymers, reduced the CP.143
Conclusions
Thermoresponsive polymers are polymers whose solubility changes as a response to temperature. Amongst them, LCST-type polymers are of major importance in biomedical applications. In this review, we summarised the studies in which thermoresponsive block copolymers of increasing architecture complexity have been reported. We have identified four main categories of polymers, depending on the general chemical structures, such as Pluronic®-, EG-containing degradable, acrylamide-, and EG (meth)acrylate-based polymers. Other thermoresponsive families, such as poly(2-oxazoline)s, are also discussed. The number of blocks and the position of the blocks within the polymer chain are of major importance, with BAB triblock bipolymers favouring gelation over their ABA counterparts, due to efficient micelle bridging; A and B are thermoresponsive and hydrophobic, respectively. In triblock and tetrablock terpolymers, architectures with (i) the hydrophilic and/or thermoresponsive blocks at the end of the polymer chain, surrounding the hydrophobic one, and (ii) the thermoresponsive block being next to the hydrophobic block (in tetrablock terpolymers), favour thermogelation. This may be attributed to the easier rearrangement of the micelles upon thermoresponse, while preserving well-hydrated bridges. Moving from linear to star architectures, thermoresponse is favoured as the complexity of architecture increases. It should be kept in mind that although in this review we aimed to compare how the repeating unit/block position affects the thermoresponse, accurate comparisons can only be made when the composition and the overall MM are kept constant, as both parameters affect the thermoresponsive properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
EPSRC is acknowledged for APC's funding (EPSRC IAA, EP/R511547/1).References
- N. T. Southall, K. A. Dill and A. D. J. Haymet, J. Phys. Chem. B, 2002, 106, 521–533 CrossRef CAS.
- J. C. Foster, I. Akar, M. C. Grocott, A. K. Pearce, R. T. Mathers and R. K. O'Reilly, ACS Macro Lett., 2020, 1700–1707 CrossRef CAS.
- V. Nele, J. P. Wojciechowski, J. P. K. Armstrong and M. M. Stevens, Adv. Funct. Mater., 2020, 30, 2002759 CrossRef CAS.
- M. T. Cook, P. Haddow, S. B. Kirton and W. J. McAuley, Adv. Funct. Mater., 2021, 31, 2008123 CrossRef CAS.
- A. P. Constantinou and T. K. Georgiou, Polym. Int., 2021, 70, 1433–1448 CrossRef CAS.
- C. De Las Heras Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- S. Ikegami and H. Hamamoto, Chem. Rev., 2009, 109, 583–593 CrossRef CAS PubMed.
- H. Hamamoto, Y. Suzuki, Y. M. A. Yamada, H. Tabata, H. Takahashi and S. Ikegami, Angew. Chem., Int. Ed., 2005, 44, 4536–4538 CrossRef CAS PubMed.
- D. E. Bergbreiter, B. L. Case, Y. Liu and J. W. Caraway, Macromolecules, 1998, 31, 6053–6062 CrossRef CAS.
- I. Lokuge, X. Wang and P. W. Bohn, Langmuir, 2007, 23, 305–311 CrossRef CAS.
- K. Nishimori, M. Maruyama, Y. Shimazaki, M. Ouchi and H. Yoshida, ACS Appl. Polym. Mater., 2019, 1, 1925–1929 CrossRef CAS.
- M. Zhang, A. Vora, W. Han, R. J. Wojtecki, H. Maune, A. B. A. Le, L. E. Thompson, G. M. McClelland, F. Ribet, A. C. Engler and A. Nelson, Macromolecules, 2015, 48, 6482–6488 CrossRef CAS.
- V. G. Rocha, E. García-Tuñón, C. Botas, F. Markoulidis, E. Feilden, E. D'Elia, N. Ni, M. Shaffer and E. Saiz, ACS Appl. Mater. Interfaces, 2017, 9, 37136–37145 CrossRef CAS.
- V. Pertici, T. Trimaille and D. Gigmes, Macromolecules, 2020, 53, 682–692 CrossRef CAS.
- B. V. K. J. Schmidt, Macromol. Rapid Commun., 2022, 2100895 CrossRef CAS.
- S. Lanzalaco and E. Armelin, Gels, 2017, 3, 36 CrossRef.
- Q. Li, A. P. Constantinou and T. K. Georgiou, J. Polym. Sci., 2021, 59, 230–239 CrossRef CAS.
- G. Vancoillie, D. Frank and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS.
- J. Liu, A. Debuigne, C. Detrembleur and C. Jérôme, Adv. Healthcare Mater., 2014, 3, 1941–1968 CrossRef CAS PubMed.
- R. Hoogenboom and H. Schlaad, Polym. Chem., 2017, 8, 24–40 RSC.
- A. D. McNaught and A. Wilkinson, https://goldbook.iupac.org/terms/view/B00685, (accessed 2020).
- A. P. Constantinou, G. Patias, B. Somuncuoğlu, T. Brock, D. W. Lester, D. M. Haddleton and T. K. Georgiou, Polym. Chem., 2021, 12, 3522–3532 RSC.
- N. Ghasdian, D. M. A. Buzza, P. D. I. Fletcher and T. K. Georgiou, Macromol. Rapid Commun., 2015, 36, 528–532 CrossRef CAS.
- X. Zhang, C. Contini, A. P. Constantinou, J. J. Doutch and T. K. Georgiou, J. Polym. Sci., 2021, 59, 1724–1731 CrossRef CAS.
- Y. Cai, K. B. Aubrecht and R. B. Grubbs, J. Am. Chem. Soc., 2011, 133, 1058–1065 CrossRef CAS PubMed.
- M. A. Ward and T. K. Georgiou, Polym. Chem., 2013, 4, 1893–1902 RSC.
- S. Sugihara, K. Hashimoto, S. Okabe, M. Shibayama, S. Kanaoka and S. Aoshima, Macromolecules, 2004, 37, 336–343 CrossRef CAS.
- M. A. da Silva, P. Haddow, S. B. Kirton, W. J. McAuley, L. Porcar, C. A. Dreiss and M. T. Cook, Adv. Funct. Mater., 2021, 2109010 Search PubMed.
- G. Gody, P. B. Zetterlund, S. Perrier and S. Harrisson, Nat. Commun., 2016, 7, 10514 CrossRef CAS.
- V. P. Beyer, J. Kim and C. R. Becer, Polym. Chem., 2020, 11, 1271–1291 RSC.
- M. Sahn, T. Yildirim, M. Dirauf, C. Weber, P. Sungur, S. Hoeppener and U. S. Schubert, Macromolecules, 2016, 49, 7257–7267 CrossRef CAS.
- M. Vadnere, G. Amidon, S. Lindenbaum and J. L. Haslam, Int. J. Pharm., 1984, 22, 207–218 CrossRef CAS.
- K. H. Sun, Y. S. Sohn and B. Jeong, Biomacromolecules, 2006, 7, 2871–2877 CrossRef CAS.
- M. Almeida, M. Magalhães, F. Veiga and A. Figueiras, J. Polym. Res., 2017, 25, 31 CrossRef.
- B. Naskar, S. Ghosh and S. P. Moulik, Langmuir, 2012, 28, 7134–7146 CrossRef CAS PubMed.
- J. P. Mata, P. R. Majhi, C. Guo, H. Z. Liu and P. Bahadur, J. Colloid Interface Sci., 2005, 292, 548–556 CrossRef CAS.
- Z. Zhou and B. Chu, Macromolecules, 1994, 27, 2025–2033 CrossRef CAS.
- F. Markus, J. R. Bruckner and S. Naumann, Macromol. Chem. Phys., 2020, 221, 1900437 CrossRef CAS.
- C. Bar, N. Çağlar, M. Uz, S. K. Mallapragada and S. A. Altinkaya, ACS Appl. Mater. Interfaces, 2019, 11, 31367–31377 CrossRef CAS.
- M. D. Determan, J. P. Cox, S. Seifert, P. Thiyagarajan and S. K. Mallapragada, Polymer, 2005, 46, 6933–6946 CrossRef CAS.
- B. C. Anderson, S. M. Cox, P. D. Bloom, V. V. Sheares and S. K. Mallapragada, Macromolecules, 2003, 36, 1670–1676 CrossRef CAS.
- M. D. Determan, J. P. Cox and S. K. Mallapragada, J. Biomed. Mater. Res., 2007, 81A, 326–333 CrossRef CAS.
- M. D. Determan, L. Guo, C. Lo, P. Thiyagarajan and S. K. Mallapragada, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 78, 021802 CrossRef.
- A. Agarwal, R. Unfer and S. K. Mallapragada, J. Controlled Release, 2005, 103, 245–258 CrossRef CAS.
- S. Peleshanko, K. D. Anderson, M. Goodman, M. D. Determan, S. K. Mallapragada and V. V. Tsukruk, Langmuir, 2007, 23, 25–30 CrossRef CAS PubMed.
- M. Uz, S. K. Mallapragada and S. A. Altinkaya, RSC Adv., 2015, 5, 43515–43527 RSC.
- A. Mei, X. Guo, Y. Ding, X. Zhang, J. Xu, Z. Fan and B. Du, Macromolecules, 2010, 43, 7312–7320 CrossRef CAS.
- P. Parekh, S. Ohno, S. Yusa, C. Lv, B. Du, D. Ray, V. K. Aswal and P. Bahadur, Polym. Int., 2020, 69, 1113–1121 CrossRef CAS.
- Y. Wu, X. Liu, Y. Wang, Z. Guo and Y. Feng, Macromol. Chem. Phys., 2012, 213, 1489–1498 CrossRef CAS.
- D. Cohn, A. Sosnik and A. Levy, Biomaterials, 2003, 24, 3707–3714 CrossRef CAS.
- G. M. Zentner, R. Rathi, C. Shih, J. C. McRea, M. Seo, H. Oh, B. G. Rhee, J. Mestecky, Z. Moldoveanu, M. Morgan and S. Weitman, J. Controlled Release, 2001, 72, 203–215 CrossRef CAS.
- R. C. Rathi, G. M. Zentner and B. Jeong, MacoMed Inc.US6201072B1, 2001.
- R. C. Rathi, G. M. Zentner and B. Jeong, MacroMed Inc.US6117947, 2000.
- R. C. Rathi and G. M. Zentner, MacroMed Inc.US6004573, 1999.
- D. Lee, M. Shim, S. Kim, H. Lee, I. Park and T. Chang, Macromol. Rapid Commun., 2001, 22, 587–592 CrossRef CAS.
- M. S. Shim, H. T. Lee, W. S. Shim, I. Park, H. Lee, T. Chang, S. W. Kim and D. S. Lee, J. Biomed. Mater. Res., 2002, 61, 188–196 CrossRef CAS.
- N. Y. Steinman, M. Haim-Zada, I. A. Goldstein, A. H. Goldberg, T. Haber, J. M. Berlin and A. J. Domb, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 35–39 CrossRef CAS.
- L. L. Osorno, D. E. Maldonado, R. J. Whitener, A. N. Brandley, A. Yiantsos, J. D. R. Medina and M. E. Byrne, J. Appl. Polym. Sci., 2020, 137, 48678 CrossRef CAS.
- N. K. Khorshid, K. Zhu, K. D. Knudsen, S. Bekhradnia, S. A. Sande and B. Nyström, Macromol. Biosci., 2016, 16, 1838–1852 CrossRef CAS PubMed.
- B. Jeong, Y. H. Bae and S. W. Kim, Macromolecules, 1999, 32, 7064–7069 CrossRef CAS.
- C. Chen, L. Chen, L. Cao, W. Shen, L. Yu and J. Ding, RSC Adv., 2014, 4, 8789–8798 RSC.
- Y. J. Kim and S. W. Kim, Controlled Drug Delivery from Injectable Biodegradable Triblock Copolymer, Polymer Gels, ACS symposium series, 2002, ch. 20, pp. 300–311 Search PubMed.
- S. Cui, L. Yu and J. Ding, Macromolecules, 2019, 52, 3697–3715 CrossRef CAS.
- J. E. Nielsen, K. Zhu, S. A. Sande, L. Kováčik, D. Cmarko, K. D. Knudsen and B. Nyström, J. Phys. Chem. B, 2017, 121, 4885–4899 CrossRef CAS.
- D. A. Prince, I. J. Villamagna, C. C. Hopkins, J. R. de Bruyn and E. R. Gillies, Polym. Int., 2019, 68, 1074–1083 CrossRef CAS.
- L. Yu, W. Sheng, D. Yang and J. Ding, Macromol. Res., 2013, 21, 207–215 CrossRef CAS.
- N. Safaei Nikouei and A. Lavasanifar, Acta Biomater., 2011, 7, 3708–3718 CrossRef CAS PubMed.
- N. S. Nikouei, M. R. Vakili, M. S. Bahniuk, L. Unsworth, A. Akbari, J. Wu and A. Lavasanifar, Acta Biomater., 2015, 12, 81–92 CrossRef CAS.
- S. J. Bae, J. M. Suh, Y. S. Sohn, Y. H. Bae, S. W. Kim and B. Jeong, Macromolecules, 2005, 38, 5260–5265 CrossRef CAS.
- Y. Ohya, H. Yamamoto, K. Nagahama and T. Ouchi, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3892–3903 CrossRef CAS.
- M. Mohammadi, K. Patel, S. P. Alaie, R. B. Shmueli, C. G. Besirli, R. G. Larson and J. J. Green, Acta Biomater., 2018, 73, 90–102 CrossRef CAS.
- S. Bobbala, V. Tamboli, A. McDowell, A. K. Mitra and S. Hook, AAPS J., 2016, 18, 261–269 CrossRef CAS.
- H. Mao, G. Shan, Y. Bao, Z. L. Wu and P. Pan, Soft Matter, 2016, 12, 4628–4637 RSC.
- D. Skoulas, G. Mangiapia, D. Parisi, M. Kasimatis, E. Glynos, E. Stratikos, D. Vlassopoulos, H. Frielinghaus and H. Iatrou, Macromolecules, 2021, 54, 10786–10800 CrossRef CAS.
- Z. Li, Z. Zhang, K. L. Liu, X. Ni and J. Li, Biomacromolecules, 2012, 13, 3977–3989 CrossRef CAS PubMed.
- C. Liu, Z. Zhang, K. L. Liu, X. Ni and J. Li, Soft Matter, 2013, 9, 787–794 RSC.
- J. Zhao, J. Jeromenok, J. Weber and H. Schlaad, Macromol. Biosci., 2012, 12, 1272–1278 CrossRef CAS PubMed.
- E. Kostyurina, J. U. De Mel, A. Vasilyeva, M. Kruteva, H. Frielinghaus, M. Dulle, L. Barnsley, S. Förster, G. J. Schneider, R. Biehl and J. Allgaier, Macromolecules, 2022, 55(5), 1552–1565 CrossRef CAS.
- A. Papagiannopoulos, J. Zhao, G. Zhang, S. Pispas and A. Radulescu, Eur. Polym. J., 2014, 56, 59–68 CrossRef CAS.
- J. Adelsberger, A. Kulkami, A. Jain, W. Wang, A. M. Bivigou-Koumba, P. Busch, V. Pipich, O. Holderer, T. Hellweg, A. Laschewsky, P. Müller-Buschbaum and C. M. Papadakis, Macromolecules, 2010, 43, 2490–2501 CrossRef CAS.
- J. Adelsberger, E. Metwalli, A. Diethert, I. Grillo, A. M. Bivigou-Koumba, A. Laschewsky, P. Müller-Buschbaum and C. M. Papadakis, Macromol. Rapid Commun., 2012, 33, 254–259 CrossRef CAS.
- J. Adelsberger, I. Grillo, A. Kulkarni, M. Sharp, A. M. Bivigou-Koumba, A. Laschewsky, P. Müller-Buschbaum and C. M. Papadakis, Soft Matter, 2013, 9, 1685–1699 RSC.
- A. M. Bivigou-Koumba, E. Görnitz, A. Laschewsky, P. Müller-Buschbaum and C. M. Papadakis, Colloid Polym. Sci., 2010, 288, 499–517 CrossRef CAS.
- C. Kuo, T. Liu, A. Hardiansyah, C. Lee, M. Wang and W. Chiu, Nanoscale Res. Lett., 2014, 9, 520 CrossRef PubMed.
- Z. Lin, S. Cao, X. Chen, W. Wu and J. Li, Biomacromolecules, 2013, 14, 2206–2214 CrossRef CAS PubMed.
- N. Cakir, G. Hizal and C. R. Becer, Polym. Chem., 2015, 6, 6623–6631 RSC.
- E. Karjalainen, V. Khlebnikov, A. Korpi, S. Hirvonen, S. Hietala, V. Aseyev and H. Tenhu, Polymer, 2015, 58, 180–188 CrossRef CAS.
- A. Maleki, K. Zhu, R. Pamies, R. R. Schmidt, A. Kjøniksen, G. Karlsson, J. G. Hernández Cifre, J. García De La Torre and B. Nyström, Soft Matter, 2011, 7, 8111–8119 RSC.
- H. Cong, J. Li, L. Li and S. Zheng, Eur. Polym. J., 2014, 61, 23–32 CrossRef CAS.
- L. Mi, H. Xue, Y. Li and S. Jiang, Adv. Funct. Mater., 2011, 21, 4028–4034 CrossRef CAS.
- T. Uemukai, T. Hioki and M. Ishifune, Int. J. Polym. Sci., 2013, 2013, 196145 Search PubMed.
- Y. Luo, J. Zhang, F. Han, F. Xu, Y. Chen and R. Liu, J. Appl. Polym. Sci., 2015, 132, 41877 Search PubMed.
- X. Zhao, W. Liu, D. Chen, X. Lin and W. W. Lu, Macromol. Chem. Phys., 2007, 208, 1773–1781 CrossRef CAS.
- Y. Yu, D. Hong, Z. Liu, F. Jia, Y. Zhou and C. Leng, J. Polym. Res., 2013, 20, 235 CrossRef.
- X. Cao, Y. Chen, W. Chai, W. Zhang, Y. Wang and P. Fu, J. Appl. Polym. Sci., 2015, 132, 41361 Search PubMed.
- J. Chen, M. Liu, H. Gong, Y. Huang and C. Chen, J. Phys. Chem. B, 2011, 115, 14947–14955 CrossRef CAS.
- Y. Zheng, L. Wang, R. Yu and S. Zheng, Macromol. Chem. Phys., 2012, 213, 458–469 CrossRef CAS.
- Q. Li, F. Huo, Y. Cui, C. Gao, S. Li and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2266–2278 CrossRef CAS.
- Y. Luo, L. Liu, X. Wang, H. Shi, W. Lv and J. Li, Soft Matter, 2012, 8, 1634–1642 RSC.
- C. Wu, A. Ying and S. Ren, Colloid Polym. Sci., 2013, 291, 827–834 CrossRef CAS.
- C. Zhou, M. A. Hillmyer and T. P. Lodge, J. Am. Chem. Soc., 2012, 134, 10365–10368 CrossRef CAS PubMed.
- C. C. Hall, C. Zhou, S. P. O. Danielsen and T. P. Lodge, Macromolecules, 2016, 49, 2298–2306 CrossRef CAS.
- C. Zhou, G. E. S. Toombes, M. J. Wasbrough, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2015, 48, 5934–5943 CrossRef CAS.
- M. Onoda, T. Ueki, R. Tamate, A. M. Akimoto, C. C. Hall, T. P. Lodge and R. Yoshida, ACS Macro Lett., 2018, 7, 950–955 CrossRef CAS.
- C. Wang, G. Zhang, G. Liu, J. Hu and S. Liu, J. Controlled Release, 2017, 259, 149–159 CrossRef CAS.
- A. Skandalis and S. Pispas, Polymer, 2018, 157, 9–18 CrossRef CAS.
- D. Giaouzi and S. Pispas, Polymers, 2020, 12, 1382 CrossRef CAS PubMed.
- Z. Ge, Y. Zhou, Z. Tong and S. Liu, Langmuir, 2011, 27, 1143–1151 CrossRef CAS.
- J. Chen, M. Liu, H. Gong, G. Cui, S. Lü, C. Gao, F. Huang, T. Chen, X. Zhang and Z. Liu, Polym. Chem., 2013, 4, 1815–1825 RSC.
- X. Li, R. Fan, Y. Wang, M. Wu, A. Tong, J. Shi, M. Xiang, L. Zhou and G. Guo, RSC Adv., 2015, 5, 101494–101506 RSC.
- G. Isapour, R. Lund, K. Zhu, Z. Quan, K. D. Knudsen and B. Nyström, Eur. Polym. J., 2015, 70, 79–93 CrossRef CAS.
- H. S. Abandansari, E. Aghaghafari, M. R. Nabid and H. Niknejad, Polymer, 2013, 54, 1329–1340 CrossRef CAS.
- V. Pertici, C. Pin-Barre, C. Rivera, C. Pellegrino, J. Laurin, D. Gigmes and T. Trimaille, Biomacromolecules, 2019, 20, 149–163 CrossRef CAS PubMed.
- C. Lv, J. Gao, K. An, J. Nie, J. Xu and B. Du, Macromolecules, 2021, 54, 6489–6501 CrossRef CAS.
- A. Simula, V. Nikolaou, A. Anastasaki, F. Alsubaie, G. Nurumbetov, P. Wilson, K. Kempe and D. M. Haddleton, Polym. Chem., 2015, 6, 2226–2233 RSC.
- G. Barouti, S. S. Liow, Q. Dou, H. Ye, C. Orione, S. M. Guillaume and X. J. Loh, Chem. – Eur. J., 2016, 22, 10501–10512 CrossRef CAS.
- M. Teodorescu, I. Negru, P. O. Stanescu, C. Draghici, A. Lungu and A. Sârbu, J. Macromol. Sci., Pure Appl. Chem., 2011, 48, 177–185 CrossRef CAS.
- P. J. Haddow, M. A. da Silva, D. B. Kaldybekov, C. A. Dreiss, E. Hoffman, V. Hutter, V. V. Khutoryanskiy, S. B. Kirton, N. Mahmoudi, W. J. McAuley and M. T. Cook, Macromol. Biosci., 2021, 2100432 Search PubMed.
- Z. Sun, Y. Tian, W. L. Hom, O. Gang, S. R. Bhatia and R. B. Grubbs, Angew. Chem., Int. Ed., 2017, 56, 1491–1494 CrossRef CAS PubMed.
- H. Arslan, O. Zırtıl and V. Bütün, Eur. Polym. J., 2013, 49, 4118–4129 CrossRef CAS.
- L. Lauber, J. Santarelli, O. Boyron, C. Chassenieux, O. Colombani and T. Nicolai, Macromolecules, 2017, 50, 416–423 CrossRef CAS.
- M. A. Ward and T. K. Georgiou, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2850–2859 CrossRef CAS.
- M. A. Ward and T. K. Georgiou, Soft Matter, 2012, 8, 2737–2745 RSC.
- M. A. Ward and T. K. Georgiou, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 775–783 CrossRef CAS.
- A. P. Constantinou and T. K. Georgiou, Polym. Chem., 2016, 7, 2045–2056 RSC.
- A. P. Constantinou, H. Zhao, C. M. McGilvery, A. E. Porter and T. K. Georgiou, Polymers, 2017, 9, 31 CrossRef PubMed.
- A. P. Constantinou, N. Sam-Soon, D. R. Carroll and T. K. Georgiou, Macromolecules, 2018, 51, 7019–7031 CrossRef CAS.
- M. Popescu, I. Athanasoulias, C. Tsitsilianis, N. A. Hadjiantoniou and C. S. Patrickios, Soft Matter, 2010, 6, 5417–5424 RSC.
- T. Sanal, O. Oruç, T. Öztürk and B. Hazer, J. Polym. Res., 2015, 22, 3 CrossRef.
- X. Xiang, X. Ding, N. Chen, B. Zhang and P. A. Heiden, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2838–2848 CrossRef CAS.
- Y. Zhang and W. Zhao, Iran. Polym. J., 2015, 24, 481–490 CrossRef CAS.
- M. Nagao, J. Sengupta, D. Diaz-Dussan, M. Adam, M. Wu, J. Acker, R. Ben, K. Ishihara, H. Zeng, Y. Miura and R. Narain, ACS Appl. Bio Mater., 2018, 1, 356–366 CrossRef CAS.
- S. Liu, L. Tian, H. Mao, W. Ning, P. Shang, J. Wu and X. Shi, J. Polym. Res., 2018, 25, 264 CrossRef.
- B. Hu, W. Fu and B. Zhao, Macromolecules, 2016, 49, 5502–5513 CrossRef CAS.
- A. P. Constantinou, B. Zhan and T. K. Georgiou, Macromolecules, 2021, 54, 1943–1960 CrossRef CAS.
- A. P. Constantinou, V. Nele, J. J. Doutch, J. S. Correia, R. V. Moiseev, M. Cihova, D. C. A. Gaboriau, J. Krell, V. V. Khutoryanskiy, M. M. Stevens and T. K. Georgiou, Macromolecules, 2022, 55(5), 1783–1799 CrossRef CAS.
- A. P. Constantinou, K. Zhang, B. Somuncuoğlu, B. Feng and T. K. Georgiou, Macromolecules, 2021, 54(13), 6511–6524 CrossRef CAS.
- W. Zhu, A. Nese and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1942–1952 CrossRef CAS.
- F. A. Jung, P. A. Panteli, C. Ko, J. Kang, L. C. Barnsley, C. Tsitsilianis, C. S. Patrickios and C. M. Papadakis, Macromolecules, 2019, 52, 9746–9758 CrossRef CAS.
- A. P. Constantinou, T. Lan, D. R. Carroll and T. K. Georgiou, Eur. Polym. J., 2020, 130, 109655 CrossRef CAS.
- M. Luzón, T. Corrales, F. Catalina, V. San Miguel, C. Ballesteros and C. Peinado, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4909–4921 CrossRef.
- W. Steinhauer, R. Hoogenboom, H. Keul and M. Moeller, Macromolecules, 2013, 46, 1447–1460 CrossRef CAS.
- A. Miasnikova and A. Laschewsky, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3313–3323 CrossRef CAS.
- A. Miasnikova, A. Laschewsky, G. De Paoli, C. M. Papadakis, P. Müller-Buschbaum and S. S. Funari, Langmuir, 2012, 28, 4479–4490 CrossRef CAS PubMed.
- Q. Zhong, E. Metwalli, M. Rawolle, G. Kaune, A. M. Bivigou-Koumba, A. Laschewsky, C. M. Papadakis, R. Cubitt and P. Müller-Buschbaum, Macromolecules, 2013, 46, 4069–4080 CrossRef CAS.
- I. Negru, M. Teodorescu, P. O. Stanescu, C. Draghici, A. Lungu and A. Sârbu, Soft Mater., 2013, 11, 149–156 CrossRef CAS.
- A. Kermagoret, K. Mathieu, J. Thomassin, C. Fustin, R. Duchêne, C. Jérôme, C. Detrembleur and A. Debuigne, Polym. Chem., 2014, 5, 6534–6544 RSC.
- J. Thomassin, K. Mathieu, A. Kermagoret, C. Fustin, C. Jérôme and A. Debuigne, Polym. Chem., 2015, 6, 1856–1864 RSC.
- M. L. Tebaldi, L. A. Fiel, A. M. Santos, S. S. Guterres and A. R. Pohlmann, J. Macromol. Sci., Part A: Pure Appl. Chem., 2013, 50, 581–587 CrossRef CAS.
- L. Martín, F. J. Arias, M. Alonso, C. García-Arévalo and J. C. Rodríguez-Cabello, Soft Matter, 2010, 6, 1121–1124 RSC.
- C. Kim, S. C. Lee, S. W. Kang, I. C. Kwon and S. Y. Jeong, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 2400–2408 CrossRef CAS.
- A. Zahoranová, M. Mrlík, K. Tomanová, J. Kronek and R. Luxenhofer, Macromol. Chem. Phys., 2017, 218, 1700031 CrossRef.
- B. D. Monnery and R. Hoogenboom, Polym. Chem., 2019, 10, 3480–3487 RSC.
- M. Hruby, S. K. Filippov, J. Panek, M. Novakova, H. Mackova, J. Kucka, D. Vetvicka and K. Ulbrich, Macromol. Biosci., 2010, 10, 916–924 CrossRef CAS.
- M. M. Lübtow, M. Mrlik, L. Hahn, A. Altmann, M. Beudert, T. Lühmann and R. Luxenhofer, J. Funct. Biomater., 2019, 10, 36 CrossRef.
- M. Wagner, C. Pietsch, A. Kerth, A. Traeger and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 924–935 CrossRef CAS.
- P. Alexandridis and J. F. Holzwarth, Langmuir, 1997, 13, 6074–6082 CrossRef CAS.
- T. K. Georgiou, M. Vamvakaki, C. S. Patrickios, E. N. Yamasaki and L. A. Phylactou, Biomacromolecules, 2004, 5, 2221–2229 CrossRef CAS.
- P. van de Wetering, J. Cherng, H. Talsma and W. E. Hennink, J. Controlled Release, 1997, 49, 59–69 CrossRef CAS.
- P. van de Wetering, E. E. Moret, N. Schuurmans-Nieuwenbroek, M. J. van Steenbergen and W. E. Hennink, Bioconjugate Chem., 1999, 10, 589–597 CrossRef CAS PubMed.
- C. V. Synatschke, A. Schallon, V. Jérôme, R. Freitag and A. H. E. Müller, Biomacromolecules, 2011, 12, 4247–4255 CrossRef CAS PubMed.
- T. Okano and K. Kim, Regener. Ther., 2020, 14, 330–331 CrossRef.
- N. Ghasemi, M. R. Vakili and A. Lavasanifar, ACS Appl. Polym. Mater., 2021, 3, 2608–2617 CrossRef CAS.
- A. Narumi, S. Sato, X. Shen and T. Kakuchi, Polym. Chem., 2022, 13, 1293–1319 RSC.
- L. D. Taylor and L. D. Cerankowski, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 2551–2570 CrossRef CAS.
- I. Idziak, D. Avoce, D. Lessard, D. Gravel and X. X. Zhu, Macromolecules, 1999, 32, 1260–1263 CrossRef CAS.
- J. Lutz, A. Hoth and K. Schade, Des. Monomers Polym., 2009, 12, 343–353 CrossRef CAS.
- J. Lutz, Adv. Mater., 2011, 23, 2237–2243 CrossRef CAS.
- P. Lin, C. Clash, E. M. Pearce, T. K. Kwei and M. A. Aponte, J. Polym. Sci., Part B: Polym. Phys., 1988, 26, 603–619 CrossRef CAS.
- J. Zhao, R. Hoogenboom, G. Van Assche and B. Van Mele, Macromolecules, 2010, 43, 6853–6860 CrossRef CAS.
- U. Hiroshi and K. Shiro, Chem. Lett., 1992, 21, 1643–1646 CrossRef.
- J. Park and K. Kataoka, Macromolecules, 2007, 40, 3599–3609 CrossRef CAS.
- L. Hahn, E. Karakaya, T. Zorn, B. Sochor, M. Maier, P. Stahlhut, S. Forster, K. Fischer, S. Seiffert, A. Pöppler, R. Detsch and R. Luxenhofer, Biomacromolecules, 2021, 22, 3017–3027 CrossRef CAS PubMed.
- L. Hahn, L. Keßler, L. Polzin, L. Fritze, S. Forster, H. Helten and R. Luxenhofer, Macromol. Chem. Phys., 2021, 222, 2100114 CrossRef CAS.
- C. Wang and G. Hsiue, Biomacromolecules, 2003, 4, 1487–1490 CrossRef CAS PubMed.
Footnote |
| † These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |