
Lithium storage performance and mechanism of nano-sized Ti2InC MAX phase†
Xueqin
Xu
a,
Dawei
Sha
a,
Zhihua
Tian
a,
Fushuo
Wu
a,
Wei
Zheng
*b,
Li
Yang
a,
Shengyu
Xie
a,
Peigen
Zhang
 *a and
ZhengMing
Sun
a
*a and
ZhengMing
Sun
a
aSchool of Materials Science and Engineering, Southeast University, Nanjing 211189, P. R. China. E-mail: zhpeigen@seu.edu.cn; zmsun@seu.edu.cn
bSchool of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, P. R. China. E-mail: zhengwei22@seu.edu.cn
First published on 21st December 2022
Abstract
Fine powders of MAX phases (a family of layered carbides/nitrides) have been showing great promise in energy storage applications. A feasible method of obtaining nano-sized MAX phase particles is critical to realizing the practical application of the vast MAX phase family in more technologically important fields. Herein, ball milling, a commercial and feasible method, is employed to prepare nano-sized Ti2InC, which delivers a high specific capacity of 590 mA h g−1 after 500 cycles and maintains 574.4 mA h g−1 after 600 cycles at 0.1 A g−1 when used as a lithium storage anode. Compared with other methods (e.g., partial etching), decreasing the size of Ti2InC particles by ball milling can preserve the exfoliated indium (In) atoms, which have great volumetric and gravimetric capacities. In situ XRD analysis indicates that the capacity of the nano-sized Ti2InC primarily comes from the lithiation of elemental In exfoliated from Ti2InC, and in particular, the exfoliated In atoms by ball milling can increase the initial capacity. The lithiation/delithiation cycle can effectively activate and even exfoliate the Ti2InC grains, which accounts for the increasing capacity upon cycling.
New conceptsNano-sized MAX phases, a large family of layered compounds, have demonstrated promising energy storage performance due to their layer structure and A-site elements, which usually have a high specific capacity. In this paper, nano-sized Ti2InC MAX phase is obtained by ball milling, a feasible and commercially available route. While downsizing the Ti2InC grains, this method can reserve the exposed A-site atoms, which can increase the initial capacity. XPS and in situ XRD analysis indicates that the capacity of Ti2InC primarily comes from the lithiation of indium (In) atoms outside and in the lattice of Ti2InC. The charge/discharge cycles can effectively exfoliate the Ti2InC grains and shorten the diffusion path of lithium-ions to react with In atoms to contribute to the capacity via an alloying mechanism. |
1 Introduction
Recently, the energy storage application of MAX phases, a family of layered transition-metal carbides and/or nitrides,1,2 has received increasing attention.3 MAX phases have a generic formula Mn+1AXn (M: early transition metal; A: group A elements; X: C and/or N; normally, n = 1–3, referred to as 211, 312, or 413 phase, respectively).4 Owing to their layered structure, high theoretical capacity of A-site atoms, and good conductivity,5 MAX phases were once placed with high expectations to be used as anode materials;3,6–8 however, the performance cannot compare with the expectation.9,10 Just because of the early failure of the MAX phase in energy storage applications, researchers discovered MXenes as they tried to increase the interlayer spacing of the MAX phase by etching.11,12 Several papers published lately indicate that reducing the size of MAX phase materials can significantly augment their energy storage performance,13,14 which has renewed the interest of the MAX phase community in this topic. Micron-sized (or larger) MAX phase particles failed to deliver satisfactory electrochemical performance because their layered structure and the high specific capacity A-site elements cannot contribute.6,15 Reducing the particle size has been proven effective in leveraging the huge potential of the vast MAX phase family as anode materials from experimental results13,15,16 and theoretical calculations.7,9,17Several size-decreasing methods have been reported to unleash the energy storage potential of MAX phases, including sonic exfoliation,15,18 and partial etching.19 Ti2SnC nanosheets were obtained by Wu et al. via sonication in DMSO,18 and the small-sized Ti2SnC exhibited pleasant cycling stability and good specific capacity (735 mA h g−1 at 50 mA g−1) when used as anode materials in Li-ion batteries. Via sonication and annealing, the size of Ti2SC was significantly decreased to 100–200 nm, and delivered a specific capacity of 350 mA h g−1 at 400 mA g−1.20 Fan et al. found that partially etched Ti3AlC2 had much higher specific capacity (160 mA h g−1, 331.6 mA h cm−3 at 1C) when compared with the fully etched Ti3C2Tx (110 mA h g−1, 190.3 mA h cm−3 at 1C) and 99% capacity remained even after 1000 cycles, which was, at least in part, ascribed to the alloying of the residual Al in the unetched Ti3AlC2.19 Gogotsi et al. reported the similar performance profile of Nb2SnC to other MAX phases mentioned above; they investigated the interaction of Nb2SnC with Li ions, and found that alloying reaction between the Sn-atom layer in the MAX phase with Li ions can break down the MAX phase particles, and therefore led to an increased capacity from 87 to 150 mA h g−1 after 600 charge/discharge cycles.21
By exploring the energy storage performance and mechanism of various MAX phases, the prior endeavors have pointed out the right direction (size reduction) to functionalize the MAX phases. All the results show that MAX phases have unique advantages (e.g., good cycling stability) when used as battery anodes. And, compared with anodes made from their derivatives (MXenes, prepared by an expensive and hard-to-control etching and deintercalation technique that involves chemicals such as HF), MAX phases seem more feasible to be used as energy storage materials. In particular, the vast family of MAX phases (and their virtually unlimited solid solutions) provide composition space to fine-tune their performance. For example, the solid solution of Ti3Si0.75Al0.25C2 nanosheets was demonstrated to be a promising anode material for Li-ion batteries.15 Although the methods mentioned above can break down the particles of MAX phases, they are not ready for large-scale preparation. In particular, they cannot get rid of chemicals that are not friendly to the environment,15,18 and the exposed A-site atoms of the MAX phase cannot be fully conserved when they are treated in water-based media, which in turn sacrifices the contribution of the A-site elements (e.g., Sn, Al, In, etc., usually with high specific capacity).19 Considering the fact that A-site atoms account for 25% of the moles in the MAX phases (when n = 1), a large number of A-site atoms would be exposed as the MAX phase particle size decreases therefore, a size reduction method that can totally reserve the exposed A-site atoms would be necessary. Among many nano-material preparation processes, ball milling is an environmentally friendly method that is commercially available and widely used in the industry,22,23 and it is also feasible to achieve large-scale preparation of nano-sized MAX phases. Moreover, thanks to the relatively weak bonds between the MX and A layers in MAX phases, they are easy to break down during ball milling.24,25 Therefore, this paper aims to realize the nano-sized MAX phase by ball milling, taking Ti2InC as an example. And the energy storage performance of the nano-sized Ti2InC is investigated and its mechanism is discussed, and especially the role of A-site atoms is identified.
2 Experimental methods
2.1 Preparation of Ti2InC
Ti2InC was synthesized by pressureless sintering starting from Titanium (99.9% purity, 300 mesh, Sinopharm), Indium (99.90% purity, 700 mesh, Zhongxin new materials Co.), and graphite (99.85% purity, 500 mesh, Sinopharm). The three elemental powders (Ti/In/C) were mixed at a molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.95; then, the mixture was heated up to 1250 °C at 5 °C min−1 under the protection of argon (99.999%), and soaked for 1.5 h, following a previous work;26 finally, the product was washed in an HCl solution to remove the residual indium, filtered, and dried at 60 °C. The obtained Ti2InC was ball milled (stainless steel jar and milling balls) at 550 rpm for 300 min with a charge ratio of 10:1. The milled Ti2InC is named Ti2InC-300.
:1:0.95; then, the mixture was heated up to 1250 °C at 5 °C min−1 under the protection of argon (99.999%), and soaked for 1.5 h, following a previous work;26 finally, the product was washed in an HCl solution to remove the residual indium, filtered, and dried at 60 °C. The obtained Ti2InC was ball milled (stainless steel jar and milling balls) at 550 rpm for 300 min with a charge ratio of 10:1. The milled Ti2InC is named Ti2InC-300.
2.2 Material characterizations
An X-ray diffractometer (XRD, Haoyuan, DX-2700BH, China) with Cu Kα radiation (λ = 1.5406 Å) was employed to characterize the phase composition of the samples. The morphology and microstructure of the samples were characterized by scanning electron microscopy (SEM, Sirion 200, FEI) and transmission electron microscopy (TEM, TALOS F200X, FEI), respectively. The composition of the samples was analyzed by energy-dispersive X-ray spectroscopy (EDS). Moreover, X-ray photoelectron spectroscopy (XPS, ESCALAB 250 Xi) was used to analyze the valence state of the element In.2.3 Electrochemical tests
Electrochemical measurements were performed using CR2032 coin cells assembled in an argon-filled glove box to avoid moisture contamination. The working electrodes consisted of 80 wt% active material (Ti2InC or Ti2InC-300), 10 wt% carbon black, and 10 wt% polyvinylidene fluoride (PVDF). The three components were uniformly dispersed in 1-methyl-2-pyrrolidone (NMP), forming a slurry. The slurry was pasted onto a copper foil to form a working electrode and dried at 110 °C for 24 h under a vacuum to remove residual NMP. The areal density of the active material in the electrode was around 1.5 mg cm−2. Lithium foil was used as the counter and reference electrodes, and a polypropylene film (Celgard 2400) was used as a separator. The electrolyte employed was 1 M LiPF6 dissolved in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (1:1 v/v). The galvanic charge–discharge (GCD) tests were carried out by using a LANDCT2001C tester (Wuhan LANHE) in the voltage range of 0.01 to 3.0 V (vs. Li/Li+). Cyclic voltammetry (CV) was carried out using a CHI660E electrochemical workstation (Wuhan, China). CV curves were recorded at a scanning rate of 0.1, 0.2, 0.4, 0.6, 0.8, and 1.0 mV s−1. All electrochemical measurements were conducted at 25 °C. After 1000 charge/discharge cycles, the electrode materials (after being washed by DMC to remove the electrolyte) were characterized by XRD and TEM.
3 Results and discussion
3.1 Characterization of Ti2InC
XRD patterns in Fig. 1(a) indicate the pure phase of the as-prepared Ti2InC and Ti2InC-300. The experiment results coincide well with the Ti2InC phase (PDF#17-0435). The widening of the diffraction peaks of the sample Ti2InC-300 means that the size of the Ti2InC powder was reduced by ball milling without a phase transformation.6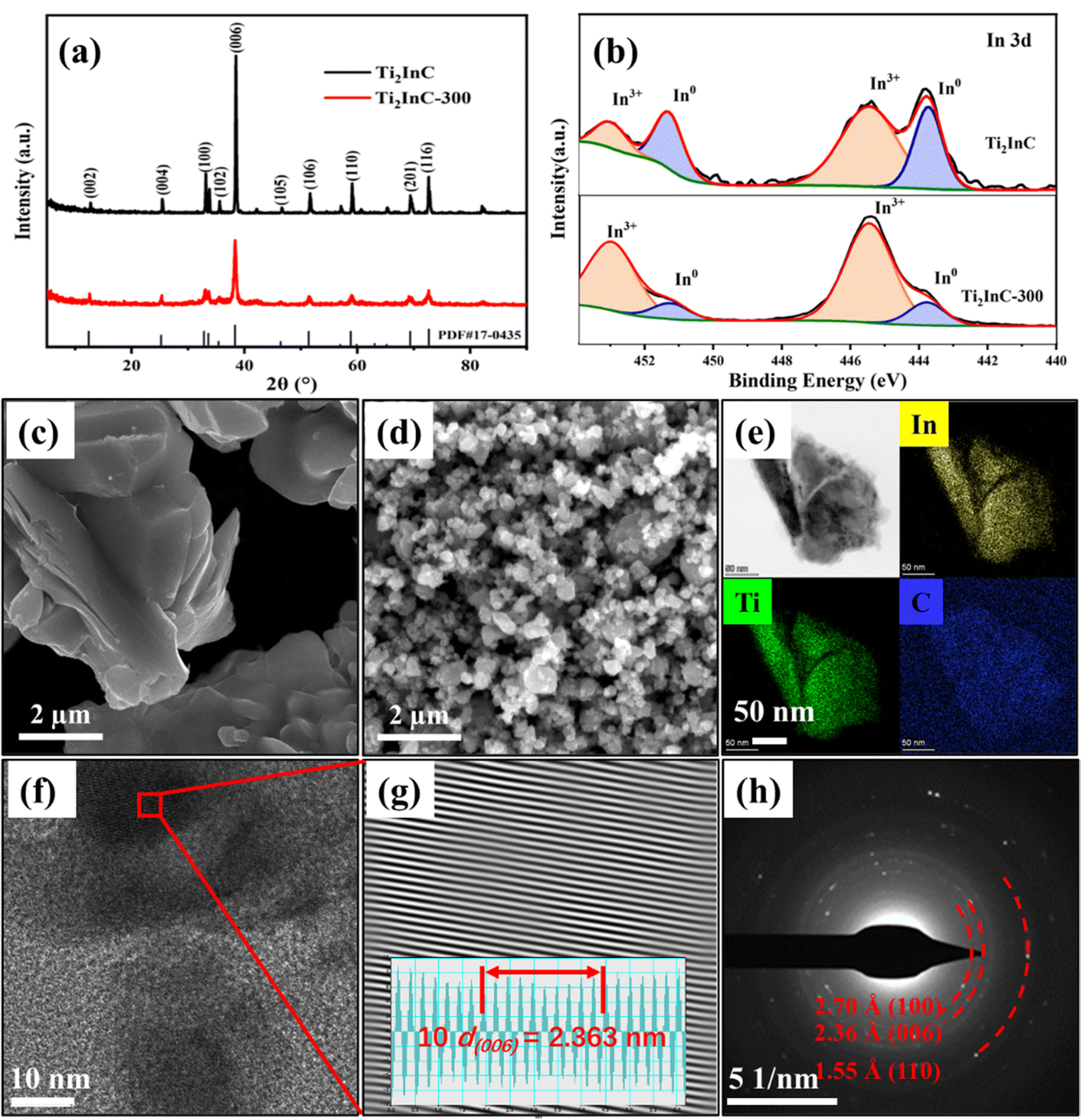 | ||
| Fig. 1 (a) XRD patterns of the Ti2InC and the Ti2InC-300. (b) In 3d XPS spectra of Ti2InC and Ti2InC-300. (c) and (d) SEM images for Ti2InC and Ti2InC-300, respectively. (e) TEM morphology and the distribution of the corresponding elements. (f) HRTEM morphology and (g) the corresponding IFFT image of the area marked with a red rectangle in (f). (h) The SAED pattern of the Ti2InC-300. | ||
Fig. 1(b) shows the high-resolution spectra of In 3d in the Ti2InC and the milled Ti2InC (Ti2InC-300). Briefly, peaks for indium located in the Ti2InC lattice (In0, 443.8 eV and 451.4 eV) and indium oxide (In3+, 445.5 eV and 453.1 eV) were observed for both Ti2InC and Ti2InC-300 samples.27,28 The surface In3+ of Ti2InC can show a relatively higher binding energy due to oxidation. Obviously, after ball milling, the proportion of surface In3+ increases significantly because of the increasing specific area and more In atoms are exposed and oxidized.
Fig. 1(c) and (d) are the SEM images of the Ti2InC and the ball-milled Ti2InC-300, which have a particle size of several micrometers and tens of nanometers, respectively. This means that ball milling can produce uniform nano-sized Ti2InC particles. The EDS elemental mappings of Ti2InC-300 in Fig. 1(e) indicate the homogeneous distribution of Ti, In, and C elements. Fig. 1(f) and (g) are the high-resolution TEM (HRTEM) image and the corresponding inverse fast Fourier transform (IFFT) image, which shows a clear lattice fringe (average interplanar spacing is 0.236 nm) corresponding to the (006) plane of Ti2InC. The SAED pattern of Ti2InC-300 particles, Fig. 1(h), indicates the existence of polycrystalline Ti2InC, and the interplanar spacings of (100), (006), and (110) conform to the lattice information of Ti2InC, suggesting that the size of Ti2InC-300 decreased after ball milling without phase transformation.
3.2 Electrochemical performance
Fig. 2(a) shows the typical cyclic voltammetry (CV) curves of a Ti2InC-300 electrode in the first three and the 1000th cycles at 0.1 mV s−1 between 0.01 and 3 V. During the first cathodic scan, the broad reduction peaks at 0.65 and 0.42 V are both ascribed to the reduction of In2O3 to produce amorphous In nanograins and a Li2O matrix, as shown by reaction (1).29 The two reduction peaks disappear in the following cycles, indicating the irreversibility of reaction (1) and the formation of the solid electrolyte interphase (SEI) layer during the 1st cycle,30 which is consistent with the first discharge–charge curve. The other two redox peaks at 0.55/0.68 and 0.44/0.46 V correspond to the multistep alloying and dealloying of Li/In, as shown by reaction (2), which occurs during the anodic scan and cathodic scan, respectively.31 After 1000 cycles, the specific capacity significantly increases. Due to the poor stability of the surface In, some original redox peaks disappear after 1000 cycles. At the same time, some new oxidation peaks appear at higher potential, which may be due to the opening of the lithium-ion diffusion channel between Ti2InC phase layers. This suggests that after cycling more lithium-ions can diffuse into the Ti2InC interlayer and more In atoms in the Ti2InC lattice come into play as active materials contributing to the capacity.| In2O3 + 6Li+ + 6e− ↔ 2In + 3Li2O | (1) |
| In + 4.33Li+ + 4.33e− ↔ Li4.33In | (2) |
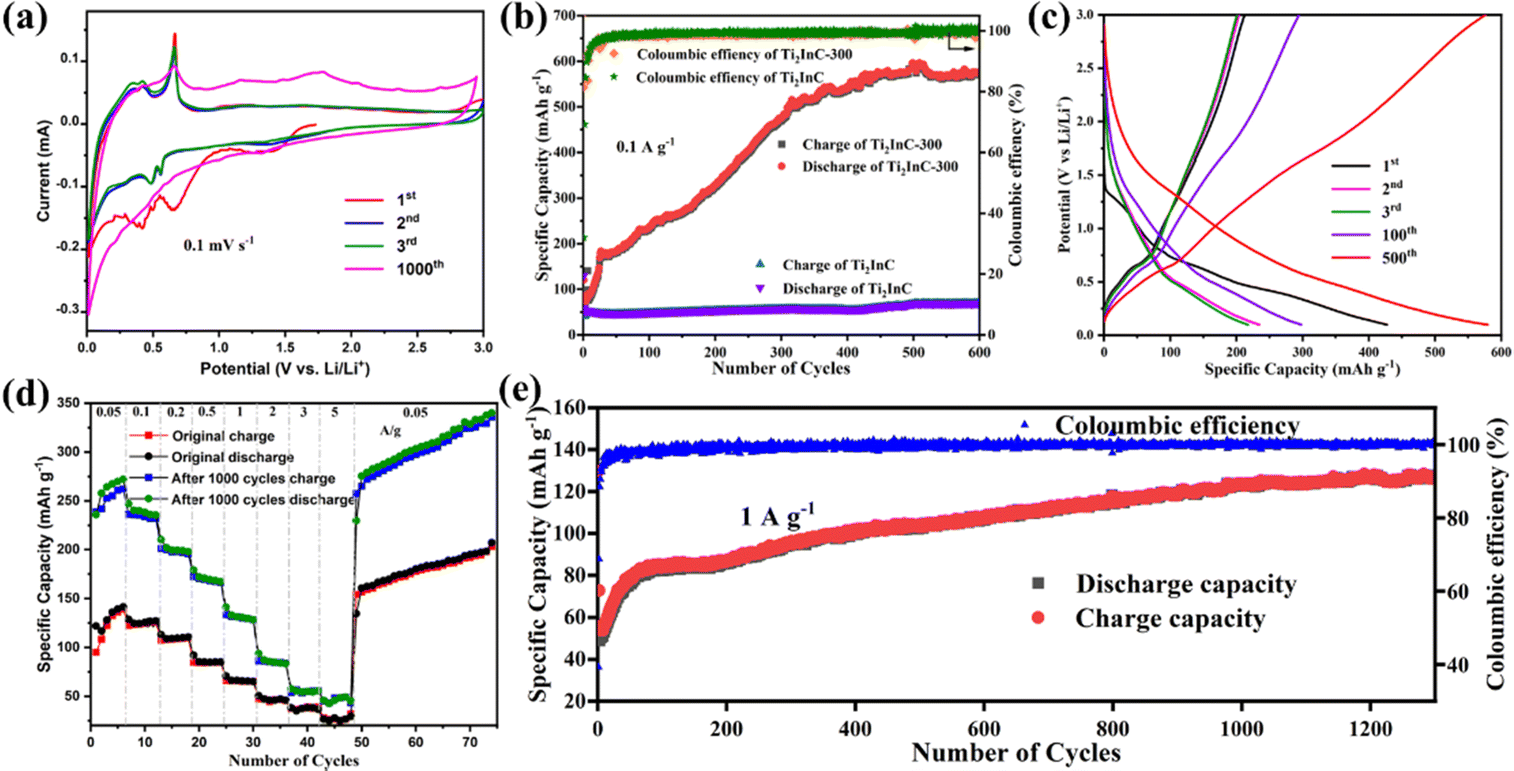 | ||
| Fig. 2 (a) CVs in the first three and the 1000th cycles at 0.1 mV s−1 between 0.01 and 3 V. (b) Cycling at 0.1 A g−1 for 600 cycles. (c) GCD profiles at 0.1 A g−1 within potentials from 0.01 to 3 V vs. Li/Li+. (d) Rate capabilities of the initial Ti2InC-300 electrode and Ti2InC-300 electrode after 1000 charge–discharge cycles at 1 A g−1. (e) Cycling at 1 A g−1 for 1300 cycles. | ||
Comparison of the cycling performances of Ti2InC and Ti2InC-300 at 0.1 A g−1 is shown in Fig. 2(b). Obviously, the nano-sized Ti2InC-300 electrode has a higher specific capacity. The first-cycle discharge specific capacity of Ti2InC-300 is 375 mA h g−1, which then decreases sharply for the second cycle due to the formation of the SEI layer and irreversible reactions.32 However, a significant increase in capacity is observed during the subsequent 500 cycles, and the capacity approaches 590 mA h g−1 after 500 cycles. This can be ascribed to the formation and continuous expansion of lithium-ion diffusion channels.14 It has been demonstrated that the interlayer spacing of nanometric MAX keeps increasing during reversible Li+ intercalation–deintercalation.33 A high reversible specific capacity of 574.4 mA h g−1 is delivered after 600 cycles, which indicates the excellent long-term cycling stability of Ti2InC-300 due to its unique structural features. It is worth noting that although the specific capacity of the micro-sized Ti2InC is low, an increase in its specific capacity can also be found after hundreds of cycles. But, after cycling, the capacity gap between nano-sized and micro-sized Ti2InC MAX gets wider, which suggests that lithium-ions can also diffuse into the interlayers of micro-sized Ti2InC slowly, alloying and de-alloying with In atoms, and the diffusion channels between the MX layer and A layer are gradually formed and expanded. It is just more challenging to form the lithium-ion diffusion channels in larger Ti2InC particles than the nano-sized particles, and as a result, the trend of rising capacity is relatively weak.
Fig. 2(c) shows galvanostatic charge–discharge curves of the Ti2InC-300 electrode at 0.1 A g−1 between 0.01 and 3.0 V. In the first discharge curve, there is only one discharge platform, which is due to the reduction of In2O3, producing amorphous In nanograins and a Li2O matrix, according to reaction (1). In the subsequent discharge curves, there is no obvious discharge platform. But in the charging curves, they plateau at 0.68 V, consistent with the CV curves.
The rate performance of Ti2InC-300 at current densities ranging from 0.05 to 3 A g−1 was tested before and after 1000 cycles at 1 A g−1. As presented in Fig. 2(d), when rated at current densities of 0.05, 0.1, 0.2, 0.5, 1, 2, 3, and 5 A g−1, the Ti2InC-300 electrode delivers initial reversible specific capacities of 141.6, 124.5, 110.0, 85.2, 65.5, 47.2, 37.0, and 28.5 mA h g−1, respectively. After 1000 cycles at 1 A g−1, the electrode delivers a reversible specific capacity of 281.4, 240.1, 201.3, 176.8, 136.7, 97.5, 68.3, and 46.7 mA h g−1, respectively. The lithium storage capacity of the electrode has significantly improved upon cycling, which is particularly obvious at smaller current densities. The increased specific capacity is mainly due to the fact that upon cycling the layer structure of Ti2InC is repeatedly expanded, and some diffusion channels generate, which makes more In atoms contribute to the capacity.21 But, at higher rates, the contribution of In atoms inside the Ti2InC grains is still limited due to the diffusion barriers. To further evaluate the cycling stability of the Ti2InC-300 electrode, it was cycled at 1 A g−1. As shown in Fig. 2(e), the Ti2InC-300 electrode shows an increasing specific capacity and reaches 130.7 mA h g−1 after 1300 cycles.
To further clarify the lithium-ion storage mechanism, in situ and ex situ XRD investigations of Ti2InC-300 electrodes during charge/discharge processes were performed. As shown in Fig. 3(a), in the first few cycles at 0.1 A g−1, the XRD peaks of the Ti2InC have no noticeable change. But after discharge, two more new diffraction peaks appear at 22.6° and 37.3°, highlighted in red belts in Fig. 3(a), corresponding to LixIn;29 the diffraction peaks disappear when charged to 0.68 V and appear again when discharged to 0.45 V, which is reversible. Therefore, it can be concluded that the battery capacity is partially provided by the lithiation of the surface In atoms in the initial cycle. In the original Ti2InC-300 electrode, the surface In atoms mainly exist in amorphous forms. As shown in Fig. 3(b), after one and two lithiation/delithiation cycles, In metal is detected. But after 1000 cycles, the diffraction peaks of In metal disappear, which might be ascribed to its amorphization caused by the poor cycling stability of In metal.34 Based on the XRD peaks of Ti2InC after cycling, the crystal structure of Ti2InC maintains well, which accounts for its good cycling stability. In Fig. S1 (ESI†), the full width at half maximum (FWHM) increases (from 0.0022 to 0.0035 in radian), meaning that Ti2InC can be exfoliated by lithiation/delithiation cycles, leading to size reduction, which is consistent with the TEM image shown in Fig. 3(c). The increased specific capacity and the widened oxidation peaks in the CV curves (Fig. 2(a)) imply that more In atoms located in the lattice points of Ti2InC-300 contribute to the specific capacity by Li–In alloying.
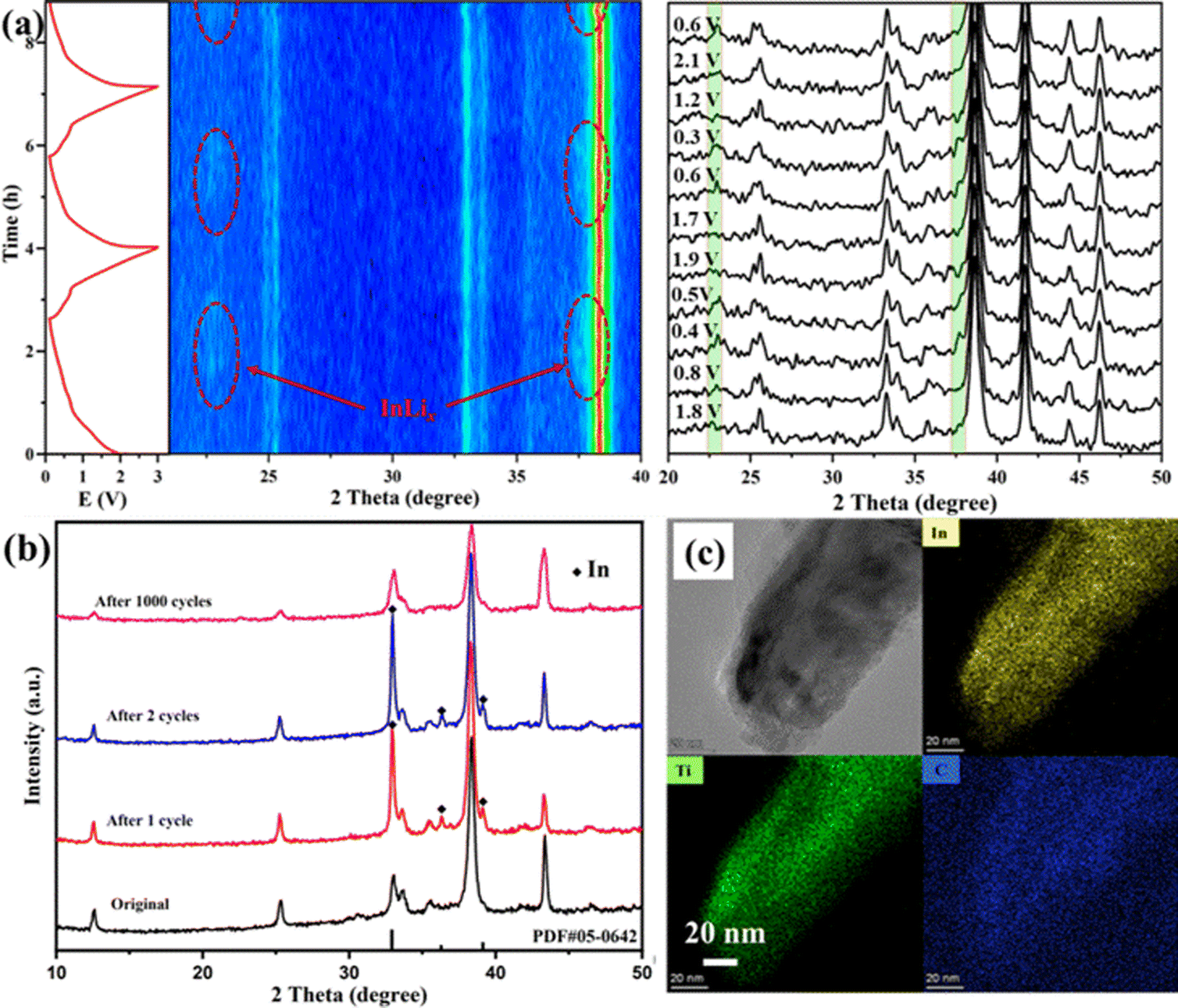 | ||
| Fig. 3 (a) In situ XRD patterns of the Ti2InC-300 electrode during the initial charge/discharge cycles at 0.1 A g−1 within potentials from 0.01 to 3 V vs. Li/Li+. (b) Ex situ XRD of the initial Ti2InC-300 electrode and electrode after being cycled. (c) TEM image and EDS elemental mappings of Ti2InC-300 after 1000 cycles at 1 A g−1. | ||
To reveal the reasons for the increasing capacity of the Ti2InC-300 electrode, CV tests under different scanning rates from 0.2 to 1 mV s−1 were performed to investigate its electrochemical kinetics before and after cycling. As shown in Fig. 4(a) and (b), the corresponding peak currents increase with increasing scanning rates, and the CV curves before and after cycling have obvious differences in shape and area. Generally, the peak current (i) in the CV profiles is dependent on the scanning rate (ν) according to the following equation:35
| i = aνb | (3) |
| i = k1v + k2v½ | (4) |
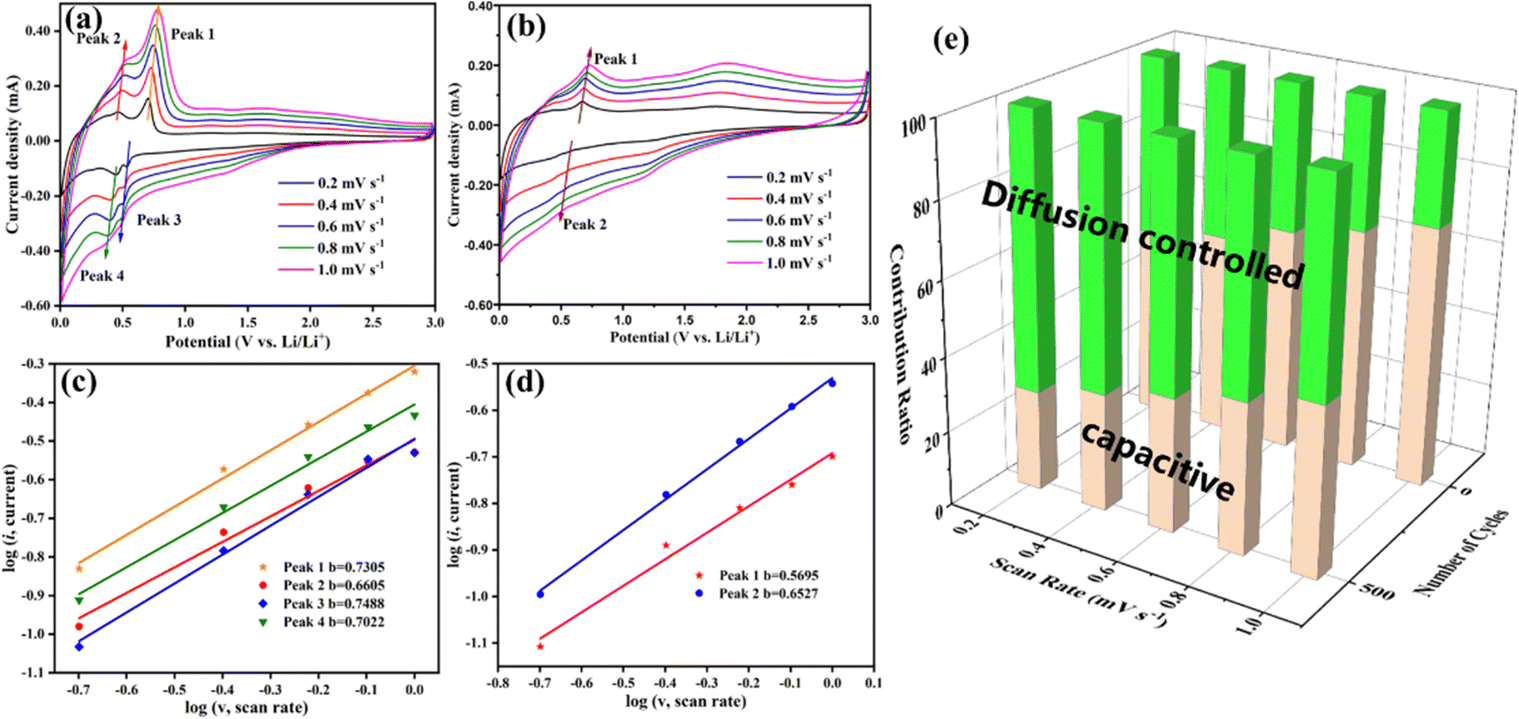 | ||
| Fig. 4 Electrochemical tests of the Ti2InC-300 electrodes before and after 500 cycles at 0.2 A g−1. (a) and (b) CV curves of the Ti2InC-300 before and after 500 cycles at different scanning rates. (c) and (d) Determination of the b-value of Ti2InC-300 before and after 500 cycles, respectively. (e) The capacitive and diffusion-controlled contribution ratios of the Ti2InC-300 electrode before and after cycling at various scanning rates (0.2 to 1.0 mV s−1). | ||
4 Conclusion
The nano-sized Ti2InC was realized by a feasible ball milling process, and it delivers a specific capacity of 590 mA h g−1 at 100 mA g−1. Compared with the unmilled Ti2InC, the exposed In atoms on the surface of the milled Ti2InC-300 can increase the initial lithium storage capacity. With the increasing number of cycles, the Ti2InC grains further break down, and therefore, the diffusion path for lithium-ions to react with In atoms located at the Ti2InC lattice points is shortened and more In atoms in Ti2InC can react with the lithium-ions and contribute to capacity via an alloying mechanism. This mechanism is verified by the result that diffusion-controlled battery behavior makes a dominant contribution to the capacity after cycling. The Ti2InC electrode demonstrates the combined advantages of layered materials and alloy-type materials, which has a favorable specific capacity and a longer cycle life. The results suggest that the large family of MAX phases are promising anode materials when they are downsized via a feasible method.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52171033) and the Natural Science Foundation of Jiangsu Province (BK20201283).References
- J. Zhou, Q. Tao, B. Ahmed, J. Palisaitis, I. Persson, J. Halim, M. W. Barsoum, P. O. Å. Persson and J. Rosen, Chem. Mater., 2022, 34, 2098–2106 CrossRef.
- Z. Tian, F. Wu, P. Hu, J. Ding, Y. Zhang, P. Zhang and Z. Sun, J. Alloys Compd., 2022, 894, 162429 CrossRef CAS.
- Y. Li, H. Shao, Z. Lin, J. Lu, L. Liu, B. Duployer, P. O. Persson, P. Eklund, L. Hultman and M. Li, Nat. Mater., 2020, 19, 894–899 CrossRef CAS.
- A. G. Zhou, Y. Liu, S. B. Li, X. H. Wang, G. B. Ying, Q. X. Xia and P. G. Zhang, J. Adv. Ceram., 2021, 10, 1194–1242 CrossRef CAS.
- J. Gonzalez-Julian, J. Am. Ceram. Soc., 2021, 104, 659–690 CrossRef CAS.
- J. Xu, M.-Q. Zhao, Y. Wang, W. Yao, C. Chen, B. Anasori, A. Sarycheva, C. E. Ren, T. Mathis, L. Gomes, L. Zhenghua and Y. Gogotsi, ACS Energy Lett., 2016, 1, 1094–1099 CrossRef CAS.
- J. Zhu, A. Chroneos, L. Wang, F. Rao and U. Schwingenschlögl, Appl. Mater. Today, 2017, 9, 192–195 CrossRef.
- C. Li, Z. Xue, J. Qin, M. Sawangphruk, S. Rajendran, X. Zhang and R. Liu, ChemistrySelect, 2019, 4, 7766–7772 CrossRef CAS.
- P. P. Filippatos, M. A. Hadi, S. G. Christopoulos, A. Kordatos, N. Kelaidis, M. E. Fitzpatrick, M. Vasilopoulou and A. Chroneos, Materials, 2019, 12, 4098 CrossRef CAS.
- S. Luan, J. Zhou, Y. Xi, M. Han, D. Wang, J. Gao, L. Hou and F. Gao, ChemistrySelect, 2019, 4, 5319–5321 CrossRef CAS.
- X. Xu, L. Yang, W. Zheng, H. Zhang, F. Wu, Z. Tian, P. Zhang and Z. Sun, Mater. Rep.: Energy, 2022, 2, 10080 Search PubMed.
- D. Sha, C. Lu, W. He, J. Ding, H. Zhang, Z. Bao, X. Cao, J. Fan, Y. Dou, L. Pan and Z. Sun, ACS Nano, 2022, 16, 2711–2720 CrossRef CAS.
- A. Sengupta, B. V. B. Rao, N. Sharma, S. Parmar, V. Chavan, S. K. Singh, S. Kale and S. Ogale, Nanoscale, 2020, 12, 8466–8476 RSC.
- J. G. Xu, M. Q. Zhao, Y. C. Wang, W. Yao, C. Chen, B. Anasori, A. Sarycheva, C. E. Ren, T. Mathis, L. Gomes, Z. H. Liang and Y. Gogotsi, ACS Energy Lett., 2016, 1, 1094–1099 CrossRef CAS.
- J. Xu, Q. Wang, B. Li, W. Yao and M. He, Nanomaterials, 2021, 11, 3449 CrossRef CAS.
- Y. Li, G. Ma, H. Shao, P. Xiao, J. Lu, J. Xu, J. Hou, K. Chen, X. Zhang, M. Li, P. O. A. Persson, L. Hultman, P. Eklund, S. Du, Z. Chai, Z. Huang, N. Jin, J. Ma, Y. Liu, Z. Lin and Q. Huang, Nano-Micro Lett., 2021, 13, 158 CrossRef CAS.
- P. Xiao, N. Jin and Z. Lin, Mater. Res. Lett., 2021, 9, 516–522 CrossRef CAS.
- H. Wu, J. Zhu, L. Liu, K. Cao, D. Yang, C. Gong, H. Lei, H. Hang, W. Yao and J. Xu, Nanoscale, 2021, 13, 7355–7361 RSC.
- X. Chen, Y. Zhu, X. Zhu, W. Peng, Y. Li, G. Zhang, F. Zhang and X. Fan, ChemSusChem, 2018, 11, 2677–2680 CrossRef CAS.
- J. Xu, H. Hang, C. Chen, B. Li, J. Zhu and W. Yao, Chin. Chem. Lett., 2022 DOI:10.1016/j.cclet.2022.05.014.
- S. S. Zhao, Y. Dall’Agnese, X. F. Chu, X. Zhao, Y. Gogotsi and Y. Gao, ACS Energy Lett., 2019, 4, 2452–2457 CrossRef CAS.
- A. J. Albaaji, E. G. Castle, M. J. Reece, J. P. Hall and S. L. Evans, Mater. Des., 2017, 122, 296–306 CrossRef CAS.
- M. Kumar, X. Xiong, Z. Wan, Y. Sun, D. C. Tsang, J. Gupta, B. Gao, X. Cao, J. Tang and Y. S. Ok, Bioresour. Technol., 2020, 312, 123613 CrossRef CAS PubMed.
- M. Sokol, V. Natu, S. Kota and M. W. Barsoum, Trends Chem., 2019, 1, 210–223 CrossRef CAS.
- Q. Zhang, Z. Tian, P. Zhang, Y. Zhang, Y. Liu, W. He, L. Pan, Y. Liu and Z. Sun, Mater. Today Commun., 2022, 31, 103466 CrossRef.
- S. Li, Y. S. Liu, Y. Zhang, L. X. Wang, P. G. Zhang, L. Pan, W. He, W. B. Tian and Z. M. Sun, J. Aust. Ceram. Soc., 2021, 57, 911–917 CrossRef.
- O. A. Balitskii and W. Jaegermann, Mater. Chem. Phys., 2006, 97, 98–101 CrossRef.
- Z. M. Detweiler, S. M. Wulfsberg, M. G. Frith, A. B. Bocarsly and S. L. Bernasek, Surf. Sci., 2016, 648, 188–195 CrossRef.
- C. Zhang, M. Liang, S.-H. Park, Z. Lin, A. Seral-Ascaso, L. Wang, A. Pakdel, C. Ó. Coileáin, J. Boland, O. Ronan, N. McEvoy, B. Lu, Y. Wang, Y. Xia, J. N. Coleman and V. Nicolosi, Energy Environ. Sci., 2020, 13, 2124–2133 RSC.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef.
- H. Yang, T. Song, S. Lee, H. Han, F. Xia, A. Devadoss, W. Sigmund and U. Paik, Electrochim. Acta, 2013, 91, 275–281 CrossRef CAS.
- F. Wang, X. Wang, Z. Chang, Y. Zhu, L. Fu, X. Liu and Y. Wu, Nanoscale Horiz., 2016, 1, 272–289 RSC.
- S. Zhao, Y. Dall’Agnese, X. Chu, X. Zhao, Y. Gogotsi and Y. Gao, ACS Energy Lett., 2019, 4, 2452–2457 CrossRef CAS.
- C. J. Zhang, M. Liang, S.-H. Park, Z. Lin, A. Seral-Ascaso, L. Wang, A. Pakdel, C. O. Coileain, J. Boland and O. Ronan, Energy Environ. Sci., 2020, 13, 2124–2133 RSC.
- S. S. Henrik Lindstrom, A. Solbrand, H. Rensmo, J. Hjelm and A. S.-E. L. A. Hagfeldt, J. Phys. Chem. B, 1997, 101, 7717–7722 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nh00489e |
| This journal is © The Royal Society of Chemistry 2023 |