
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
InP/ZnS quantum dot photoluminescence modulation via in situ H2S interface engineering†
Xiang-Bing
Fan
 a,
Dong-Wook
Shin
a,
Sanghyo
Lee
a,
Junzhi
Ye
b,
Shan
Yu
c,
David J.
Morgan
d,
Adrees
Arbab
a,
Jiajie
Yang
a,
Jeong-Wan
Jo
a,
Yoonwoo
Kim
a,
Sung-Min
Jung
a,
Philip R.
Davies
d,
Akshay
Rao
b,
Bo
Hou
e and
Jong Min
Kim
*a
a,
Dong-Wook
Shin
a,
Sanghyo
Lee
a,
Junzhi
Ye
b,
Shan
Yu
c,
David J.
Morgan
d,
Adrees
Arbab
a,
Jiajie
Yang
a,
Jeong-Wan
Jo
a,
Yoonwoo
Kim
a,
Sung-Min
Jung
a,
Philip R.
Davies
d,
Akshay
Rao
b,
Bo
Hou
e and
Jong Min
Kim
*a
aDepartment of Engineering, University of Cambridge, Cambridge, CB3 0FA, UK. E-mail: jmk71@cam.ac.uk
bThe Cavendish Laboratory, Department of Physics, University of Cambridge, Cambridge, CB3 0HE, UK
cSchool of New Energy and Materials, Southwest Petroleum University, Chengdu, 610500, China
dSchool of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK
eSchool of Physics and Astronomy, Cardiff University, Cardiff, CF24 3AA, UK
First published on 18th January 2023
Abstract
InP quantum dots (QDs) are attracting significant interest as a potentially less toxic alternative to Cd-based QDs in many research areas. Although InP-based core/shell QDs with excellent photoluminescence properties have been reported so far, sophisticated interface treatment to eliminate defects is often necessary. Herein, using aminophosphine as a seeding source of phosphorus, we find that H2S can be efficiently generated from the reaction between a thiol and an alkylamine at high temperatures. Apart from general comprehension that H2S acts as a S precursor, it is revealed that with core etching by H2S, the interface between InP and ZnS can be reconstructed with S2− incorporation. Such a transition layer can reduce inherent defects at the interface, resulting in significant photoluminescence (PL) enhancement. Meanwhile, the size of the InP core could be further controlled by H2S etching, which offers a feasible process to obtain wide band gap InP-based QDs with blue emission.
New conceptsWe report a new straightforward interface engineering concept by in situ H2S generation from a thiol and an alkylamine at high temperatures for highly photoluminescent InP/ZnS QD synthesis. Apart from previous reports that use other recipes to generate H2S as a S resource, we further find that the large amount of highly reactive H2S can etch the InP core and reconstruct the interface between InP and ZnS by S2− incorporation. Such a transition layer can reduce the inherent defects at the interface, resulting in significant PLQY enhancement. In addition, we find that the phosphorus is further oxidized after the interface treatment, indicating that the extent of oxidized phosphorous at the interface has no direct correlation with the PLQY, which differs from the recent reports that claim an oxide-free phosphorus interface is beneficial for higher PLQY of InP-based QDs. Furthermore, we show that this in situ H2S interface engineering can also offer an additional size control of the InP core, which reveals the mechanism for blue emission InP-based QD synthesis. We believe that our methodology provides new concepts for interface science, which shall have a significant impact on the synthesis of high-quality InP-based QDs and other nanocrystals. |
Introduction
With unique optoelectronic properties stemming from nanoscale dimensions,1 quantum dots (QDs) are emerging semiconductor materials that underpin the development of future photonics and electronics applications including lighting,2,3 displays,4,5 photovoltaics,6 photosynthesis,7 and biosensing.8 Environment-friendly indium phosphide (InP)-based QDs (Cd/Pb-free), with a direct band gap in the visible light region, tuneable emission bandwidth, and practicable material stability,9,10 have become strong candidates to achieve breakthroughs in the aforementioned fields. For example, InP-based QDs have been recently used as photoluminescent materials in the liquid crystal display market.11 The latest advances in InP QD light-emitting diodes (LEDs) are pushing this research field towards a next-generation self-emissive QD-LED display.12–16Nevertheless, because of the difficulties such as covalent In-P bond formation, high reactivity to be oxidized, and lack of green precursors,9 the development of InP QD synthesis has lagged behind that of related II–VI materials such as CdSe QDs. It is generally agreed that InP QDs with bright PL could be realized by the protection of the external ZnSe and ZnS shells. However, the photoluminescence quantum yield (PLQY) varies from 20% to near 100% for the InP core/shell QDs depending on different core and shell growth methods.9,13,17–20 It has been reported that the defects from oxidative species and the lattice mismatch at the interface are probably the reasons for the poor PLQY of the InP core/shell QDs.13,17,21,22 Consequently, additional interface treatments such as HF etching of the InP core are used to achieve high photoluminescence InP-based QDs.13,23 However, the harsh and complicated processing may hinder large-scale QD production for future optoelectronic applications.
Here we propose a straightforward synthetic route to grow highly photoluminescent InP/ZnS QDs based on a facile interface engineering by in situ generated H2S from a thiol and an alkylamine at high temperatures. It is revealed that an adequate amount of highly reactive H2S can etch the InP core and create a transition layer at the interface, effectively eliminating the inherent interface defects and relaxing the lattice strain during the shell growth, which leads to PLQY enhancement. Furthermore, the in situ H2S treatment also offers additional size controllability of the InP core, which opens more possibilities for the emission wavelength of InP-based QDs.
Results and discussion
Synthesis and optical characterization
Two growth steps were implemented to synthesize InP/ZnS QDs: (i) InP core QDs were prepared by the aminophosphine route with different ratios of ZnCl2 and ZnI2 to tune the InP core size;24,25 (ii) a ZnS shell was grown on the InP core by direct injection of zinc stearate (ZnSt2) as a Zn precursor and 1-dodecanethiol (DDT) as a S precursor, followed by raising the temperature to 300 °C and maintaining for 60 min (see details in the ESI†). The absorption and PL spectra of InP and InP/ZnS QDs are shown in Fig. 1. The absorption increase in the ultraviolet region confirmed ZnS shell formation (Fig. S1A, ESI†).25 Interestingly, we find that after shell growth on the InP core, the absorption peak of the InP/ZnS QDs was largely blue shifted from 505 nm to 470 nm (Fig. 1A), which shows an abnormal behaviour compared to the optical properties of previously reported InP/ZnS QDs where the PL peaks are nearly unchanged or a little red shifted.24–26 This blueshift was also observed in the PL spectrum, with a peak at 521 nm of the InP/ZnS QDs for green emission (Fig. 1B). Compared to the poor PL from the InP QDs (PLQY ∼1%), the PLQY increased to 85% after ZnS shell coating (Fig. S1B, ESI†), which is among the best results for InP/ZnS QDs synthesized from either tris(trimethylsilyl)-phosphine ((TMS)3P) or aminophosphine.18,19,25,27 During the temporal shell growth process, the increase of PLQY was correlated with the blueshift of the PL peak at high temperature (Fig. S1C and D, ESI†). | ||
| Fig. 1 Optical properties of InP and InP/ZnS QDs synthesized under different conditions. The absorption (A) and PL (B) spectra of InP and InP/ZnS QDs; the inset is a photograph of the InP/ZnS QD solution under UV light. The absorption (C) and PL (D) spectra of InP/ZnS QDs synthesized under different shell growth conditions. | ||
Next, we investigated various shell growth parameters by changing the precursors and reaction temperature (Fig. 1C and D). Initially, sulfur powder dissolved in trioctylphosphine as a common S precursor (TOPS) was employed for ZnS shell growth.18,24,25 The PLQY of InP/ZnS QDs synthesized with TOPS was only 56% (Fig. 1D). Compared to the InP QDs, a peak blueshift of the InP/ZnS QDs with TOPS was also observed (absorption peak 484 nm, PL peak 541 nm), but much weaker than those with DDT reacted at 300 °C. Considering that the lower PLQY may be caused by the different roles and reactivities between TOPS and DDT, the synthesis parameters when using TOPS were further modified. However, as shown in Fig. S2A and S2C (ESI†), changing the moles of the TOPS precursor or prolonging the shell growth time could hardly obtain a comparable PLQY for InP/ZnS QDs.
Subsequently, to control the reactivity of the precursors with DDT and ZnSt2, the shell growth temperature was varied from 210 to 300 °C in the presence of DDT (Fig. S2B, ESI†). Even though DDT can decompose and form the ZnS shell between 200 °C and 240 °C according to the previous literature for InP QD synthesis,28,29 such a temperature range did not show high PLQY in our case (PLQY 39%, PL peak 537 nm). With a longer reaction time, the PLQY of InP/ZnS QDs synthesized with DDT at 240 °C still could not exceed 50% (Fig. S2C, ESI†). As the lower temperature with a longer reaction time cannot achieve a desired PLQY, we believe that the process at a high temperature of 300 °C is significantly essential for obtaining a high PLQY. Furthermore, by introducing DDT after the TOPS reaction at 300 °C, the PLQY of the InP/ZnS QDs was around 67% (Fig. S2D, ESI†). However, it is still lower than that of the directly synthesized InP/ZnS QDs with DDT. Based on these experiments, we exclude the key role of DDT as an additional surface ligand for achieving a higher PLQY.
Surprisingly, we find that the value of PLQY was correlated with the extent of PL peak blueshift. When using TOPS or DDT at a lower temperature, a lower PLQY was always associated with less peak movement compared to InP/ZnS-DDT/300 QDs. It is therefore important to understand how this peak shift happens. Accordingly, DDT and ZnSt2, which existed in InP/ZnS-DDT/300 QD synthesis, were introduced individually. The results show that DDT was critical for the blueshift of the PL peak with increased PLQY, while the addition of ZnSt2 alone yielded a small redshift for the PL peak with limited PLQY improvement (Fig. S3A, ESI†). Such results exclude the possibility that the Zn carboxylic precursor causes the blueshift with acid corrosion or water formation on the InP core.30,31 Moreover, a moderately excessive amount of DDT to ZnSt2 in the system benefited the PLQY (Fig. S3B, ESI†) and other thiols and zinc carboxylic compounds with different aliphatic chains provided similar PL properties compared to the typical InP/ZnS-DDT/300 QD synthesis (Fig. S3C, ESI†).
Finally, we repeated the synthesis of InP/ZnS-DDT/300 QDs to confirm the reproducibility of the proposed method, resulting in a PLQY of 84% ± 3 (highest 87%) for different batches with negligible peak wavelength variation (Fig. S4A, ESI†). The reproducibility is due to the simple and convenient synthesis procedure, since it avoids elaborate precursor preparation, complicated interface treatment like HF etching, and multiple core or shell growths. The obtained InP/ZnS QDs had good stability with the PLQY maintained over one year (Fig. S4B, ESI†), demonstrating excellent protection of the InP core by the ZnS shell.
Structure and component characterization
In general, the blueshift of absorption and PL spectra of QDs can be caused by the formation of new core compounds,32 Zn doping in the core,33 and a size decrease or morphology change with increased quantum confinement effect.34 The X-ray diffraction patterns (XRD; Fig. 2A) reveal that the InP QDs have a zinc blende structure. Upon shell growth, the peaks shift to higher angles from InP to ZnS.25 The TEM images prove that the morphologies for both InP and InP/ZnS QDs were quasi-spherical (Fig. S5, ESI†). The HRTEM image for the InP/ZnS-DDT/300 QDs (Fig. S6, ESI†) shows the interplanar spacing as 0.319 nm, which is between the (111) facets of zinc blende InP (0.339 nm) and ZnS (0.312 nm). Based on the XRD and TEM results, no other side products can be identified. The size of the as-prepared InP core was about 2.5 nm (Fig. 2B and Fig. S7, ESI†), which is close to the literature data.24,35 For the InP/ZnS-DDT/300 QDs, the size increased to 4.6 nm, larger than InP/ZnS-TOPS/300 QDs (3.7 nm) and InP/ZnS-DDT/240 QDs (3.3 nm). Elemental analysis of the QDs by inductively coupled plasma optical emission spectrometry (ICP-OES; Fig. 2C and Table S1, ESI†) and scanning electron microscopy energy dispersive X-ray spectroscopy (SEM-EDS; Table S2, ESI†) presented similar ratios of P to In (0.8–0.9) for all the QDs. The similar P to In ratios in the InP/ZnS QDs suggest that the blueshift of the peak wavelength does not result from a large amount of Zn replacing In and alloying into the InP core.33 | ||
| Fig. 2 Structure and component characterization of InP and InP/ZnS QDs synthesized under different conditions. (A) The XRD patterns of InP and InP/ZnS QDs. (B) The TEM size distributions of InP and InP/ZnS QDs. (C) Elemental composition results by ICP-OES of InP and InP/ZnS QDs. | ||
Alternatively, a size decrease of the InP core might happen during shell growth, especially for the reaction with DDT at 300 °C. We notice that H2S can be generated in situ by DDT and oleylamine (OLA) at high temperatures.36 Thus we focused on the appearance of H2S in our shell growth processes. To verify H2S gas generation, a Pb2+ aqueous solution was used (Fig. S8A, ESI†) and resulted in black PbS precipitates.37 It is noted that the black precipitate started appearing after the temperature was raised above 250 °C and a large amount of precipitate was observed at the end of the reaction (Fig. 3A and B). EDS and XRD further confirmed the PbS black precipitate (Fig. S9, ESI†), indicating the formation of H2S during the shell growth by DDT at 300 °C. H2S is an acidic gas and can etch the InP structure,38 resulting in a size decrease of the InP core, which contributed to the blueshift of the absorption and PL spectra. In other shell growth processes such as TOPS at 300 °C or at lower temperature with DDT at 240 °C, the Pb2+ solution became slightly darker but remained transparent (Fig. 3C and D), indicating a small amount of H2S release under these conditions. Besides DDT and high temperature, we find that OLA is also necessary for efficient H2S generation. If OLA is removed or replaced by a carboxylic acid such as oleic acid (OA), the generation of H2S is also minimized (Fig. S8B and C, ESI†).
 | ||
| Fig. 3 (A–D) Detection of H2S gas generation by Pb2+ aqueous solution. (A) Pb2+ solution before reaction. (B–D) Pb2+ solution after absorption under different reaction conditions: (B) OLA/DDT at 300 °C, (C) OLA/TOPS at 300 °C, and (D) OLA/DDT at 240 °C. (E–G) Detection of H2S gas generation by GC-FPD. (E) Comparison between the gas extracted from the flask, H2S standard gas (H2S in Ar) and blank (Ar). (F) Time monitoring of H2S gas generation from DDT and OLA during heating at 300 °C. The time before 0 min represents the heating-up period. (G) Comparison of H2S generation under different reaction conditions. | ||
To further confirm the in situ generation of H2S, gas chromatography with a flame photometric detector (GC-FPD) was used to monitor the possible gas generation during the reaction.39 The gas was extracted from the reaction flask, injected into the GC-FPD, and then compared with standard H2S gas. It can be clearly seen that the reaction flask contains H2S gas with retention time (RT) = 1.5 min (Fig. 3E), while some other S-containing gases, such as carbonyl sulfide (COS, RT ∼ 1.7 min), methanethiol (CH3SH, RT ∼ 2.3 min), ethanethiol (C2H5SH, RT ∼ 3.7 min), and carbon disulfide (CS2, RT ∼ 4.3 min), were not detected in the reaction system. We further monitored the H2S generation during the shell growth process with different heating times (Fig. 3F). This confirms that H2S was largely generated when the temperature reached 300 °C and the amount of H2S was gradually increased as the heating time went on. Besides, we also compared H2S generation using GC-FPD under different shell growth conditions (Fig. 3G), which shows that H2S generation with DDT at 300 °C is much higher than those under other conditions including DDT at 240 °C, TOPS at 300 °C and TOPS at 240 °C. Such results accord well with the findings in H2S detection with the Pb2+ solution above.
Although in situ generation of H2S by some other recipes in QD synthesis has been reported, most works focus on H2S as a S resource.37,40–42 The significant role of H2S over other S precursors for higher PLQY is still unclear. To understand the chemical change in the InP surface composition, X-ray photoelectron spectroscopy (XPS) was employed. From the In 3d spectra, we find a peak shift for the InP/ZnS-DDT/300 QDs compared to the others (Fig. 4A), indicating a different chemical environment of In after exposure to a large amount of H2S. Considering the high reactivity of H2S with InP, the peak shift to higher binding energy probably occurred from the contribution of newly formed In-S bonds at the interface.38,41 A further peak deconvolution for the In 3d5/2 spectrum shows that two peaks with 444.6 eV and 445.2 eV are well fitted to the spectrum (Fig. S10A, ESI†), where the former 444.6 eV comes from In–P bonds, and the latter 445.2 eV matches the binding energy of P–In–S bonds in the literature.43 For other QDs, there is still an In 3d peak shift compared to the In–P value (Fig. S10A, ESI†). This smaller peak shift can be assigned to In–O bonds or fewer In–S bonds.31,44,45 For the InP/ZnS QDs synthesized in the absence of H2S (InP/ZnS-TOPS/300 and InP/ZnS-DDT/240 QDs), the interface treatment also occurred, mainly from the reaction between the InP core and H2O generated by the side reaction of carboxylic acid during the shell growth process.17,31 However, the H2O generation is much slower and insufficient under these conditions, resulting in an inadequate interface layer.
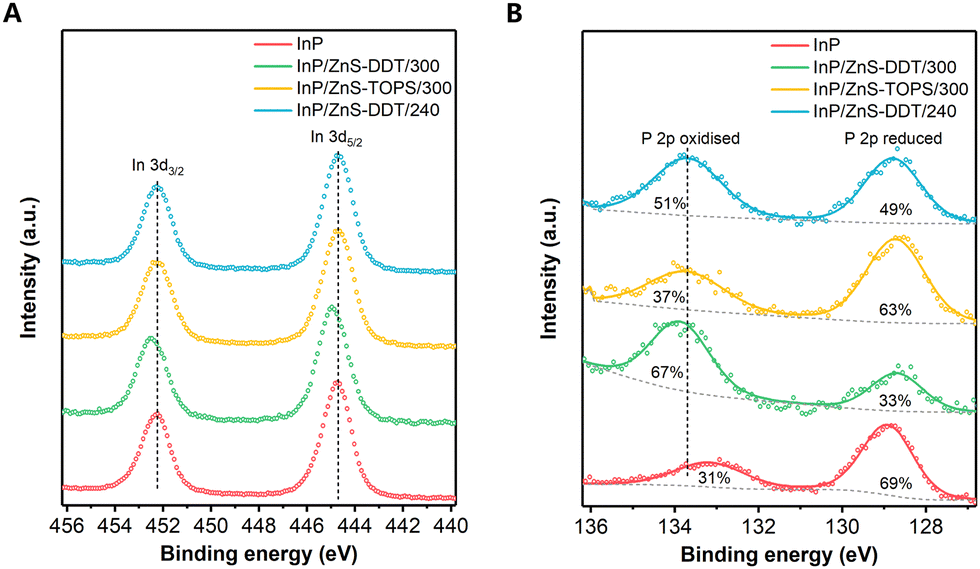 | ||
| Fig. 4 The X-ray photoelectron spectroscopy profiles of InP and InP/ZnS QDs synthesized under different conditions. (A) The In 3d spectra of InP and InP/ZnS QDs. (B) The P 2p spectra of InP and InP/ZnS QDs; the values inside the peaks are the percentages of oxidized and reduced P. | ||
The XPS P 2p spectra were also measured and show two chemical environments for P for the QDs (Fig. 4B): (i) the peak at around 129 eV is characteristic of reduced P (P−3) in InP; (ii) the peak at 133–134 eV is indicative of a positive oxidation state as either P3+ or P5+. For the InP core QDs without shell protection, the surface of the InP core was easily oxidized by O2 and H2O during QD synthesis or purification, resulting in P3+/P5+ compounds.17,46 For InP/ZnS QDs, we find that the P3+/P5+ in InP/ZnS-DDT/300 QDs (67% ratio of P3+/P5+ to all P) is much higher than that under other shell growth conditions (37–51%). Considering the highest PLQY observed for the InP/ZnS-DDT/300 QDs, our finding indicates that the content of oxidized phosphorous at the interface has no direct correlation with the PLQY of InP/ZnS QDs, which differs from a recent report that claims an oxide-free phosphorus interface is beneficial for the higher PLQY of InP QDs.13 As shown in Fig. S10B (ESI†), fitting of the P 2p3/2 spectra at 133–134 eV shows the peak for InP/ZnS-DDT/300 QDs at a binding energy of 133.7 eV, which is higher than those of InP/ZnS-TOPS/300 and InP/ZnS-DDT/240 QDs (133.4 eV). Based on our observation and the literature reports,38,41,47 the increased oxidization of P (P3+/P5+) in InP/ZnS-DDT/300 QDs was contributed from the efficient H2S reaction by core etching and interface reconstruction, probably with some P–S bonds.38,47,48 Besides, the XPS spectra of Zn 3d for all QDs were nearly the same (Fig. S11A, ESI†), showing that this interface engineering with H2S barely influenced the ZnS shell. Furthermore, we do not observe any sulfur oxidation around 168–169 eV in the S 2p spectra (Fig. S11B, ESI†). The slight peak shift of S binding energy for QDs with DDT was mainly attributed to the different S resources, where thiols have higher binding energies than the inorganic sulfide.42
Photophysical study
Next, photophysical studies of InP and InP/ZnS QDs were conducted to understand the internal relationship between interface states and photoluminescence. Generally, dynamic decays in a very short time (<100 ps) are relevant to inherent defects, while longer decay processes (∼ns) are derived from surface traps.49,50 The time-resolved photoluminescence (TRPL) spectra by time-correlated single-photon counting (TCSPC) firstly show the dynamic decay of PL in the nanosecond range (Fig. 5A). The longer PL lifetime of the InP/ZnS QDs compared to InP QDs is well understood, as excitons can be effectively isolated from the core surface after shell coating. Therefore, the fast decay process by nonradiative recombination of charge carriers occurring at the surface of the InP core was largely suppressed and the PLQY was improved with slower dynamic decay.49,51 However, no significant difference in the TRPL curves was observed among these InP/ZnS QDs, with similar average lifetimes around 75–80 ns (Fig. 5A and Table S3, ESI†), implying that surface trapping is not the only factor for the different PLQYs of the QDs. This is also consistent with the fact that some InP-based QDs with very thick shells and slow TRPL decay in the nanosecond range have a modest PLQY (below 50%),17,35,52 even though the thick shell can keep wave functions of excitons away from the surface of the QDs.15,18 | ||
| Fig. 5 Photophysical study of InP and InP/ZnS QDs synthesized under different conditions. (A) The time-resolved photoluminescence spectra of InP and InP/ZnS QDs. (B) The TA kinetics spectra for excitation bleaching signal (average from peaks ± 10 nm) of InP and InP/ZnS QDs. (C-F) TA mapping of InP and InP/ZnS QDs in different wavelengths and delay times. | ||
Furthermore, the QDs were characterized by the ultrafast transient absorption (TA) spectra in the picosecond range. All the TA spectra of InP/ZnS QDs (Fig. 5C–F) display two prominent features, typical excitation photo-bleaching and featureless broad photoinduced absorption. Unlike the similarity of the decays in the nanosecond range, InP/ZnS QDs with TOPS/300 and DDT/240 show a faster recovery of excitation bleaching than InP/ZnS-DDT/300 QDs (Fig. 5B), indicating an ultrafast recombination process in the picosecond time range. Fast exciton decays are normally relevant to inherent defects,49,50 and therefore the ultrafast recombination in QDs with TOPS/300 and DDT/240 can be attributed to inherent defects in the QDs, which are mainly at the interface or inside the InP core. We believe that the superior high PLQY of InP/ZnS-DDT/300 QDs could be attributed to their fewer inherent defects.
Due to the large lattice mismatch between InP and ZnS, inherent defects will be formed at the interface by the growing ZnS shell on the InP core, especially without any transition layer that leads to a very low PLQY.21 For general InP/ZnS QD synthesis, the interface treatment by side reactions is not efficient enough to remove the interface defects. Unlike the external surface traps that can be isolated by increasing shell thickness, the inherent defects at the interface still exist after shell growth, which leads to moderate PLQY of QDs.17,31 Differently, the in situ generated H2S can provide abundant highly reactive H2S to the surface of the InP core in a short time. Therefore, the pristine InP surface structure can be etched and reconstructed with S2− incorporation, accompanied by the decrease of the InP core size. Followed by the growth of the ZnS shell, this transition layer could reduce the interface defects, relax the lattice strain and lead to extended conformal ZnS shell growth.53 This hypothesis can also explain the thicker shell and more regular shape observed in TEM for the InP/ZnS-DDT/300 QDs.
The above analysis may decipher the apparent contradiction that some InP-based QD synthesis with high PLQY turns out to contain more P in a positive oxidation state,21,23,54 despite the supposition that the P3+/P5+-free InP core benefits a better PLQY.9,13 From our results, the H2S interface engineering with more oxidized P can also reduce the interface defects, resulting in improvement of the PLQY. Therefore, we assume that rather than oxidation states, introducing a highly reactive chemical is more crucial to reconstructing the interface with fewer inherent defects for high PLQY QD synthesis, which includes previously reported HF,13,23,55 ZnF2,20 (NH4)2S,56 or H2S in this work.
Extended synthesis for QD emission at different wavelengths
The developed facile InP/ZnS QD synthetic method based on H2S interface engineering was well utilized to synthesize QDs with emission that covers the most range of visible light. Firstly, the PL peak wavelength of the InP/ZnS QDs could be precisely controlled by changing the reaction temperature during the InP core formation of different core sizes, varying from 500 to 550 nm with most PLQY over 80% (Fig. 6A). As the InP/ZnS QDs synthesized with H2S generation can get higher PLQY with similar emission wavelengths for InP/ZnS-TOPS/300 and InP/ZnS-DDT/240 QDs, it further shows that the main contribution from H2S is interface engineering to increase the PLQY rather than just tuning the emission wavelength. | ||
| Fig. 6 Extended synthesis of InP/ZnS QDs for different wavelength emissions. (A) The PL spectra of green emission InP/ZnS QDs synthesized under different core formation temperatures. All PLQYs of the InP/ZnS QDs are over 80% except for the InP core formation at 220 °C, for which the PLQY is 75%. (B) The PL spectra of InP/ZnS QDs with different Zn halide precursors used for the InP core formation. The red line is for aminophosphines injected twice. (C) The PL spectra of blue emission InP/ZnS QDs synthesized under different shell growth conditions. (D) The PL spectrum of InP/ZnS QDs synthesized with ZnSt2 and sulfur powder (8 mmol) for shell formation. The shell growth temperature is 300 °C. | ||
Secondly, significant InP size change and emission modulation to red, orange or blue could be realized by tuning different halogen precursors used at the beginning of the synthesis (see details in the ESI†).24,25 Orange and red emissions of InP/ZnS QDs were obtained from ZnCl2 precursor (Fig. 6B) and blue emission of InP/ZnS QDs was synthesized from ZnI2 precursor (Fig. 6C). In particular, the InP/ZnS QDs at 468 nm with a 54% PLQY (Fig. 6C, S12A, S12B, ESI†) provide a promising and feasible blue emission InP-based QD synthesis approach, which shall be vital for QD lighting and display applications. Further studies show that a blueshift in the PL spectrum was also observed during the shell growth (Fig. S12C and D, ESI†), which agrees with the green emission InP/ZnS QD synthesis. Control experiments show that, without efficient generation of H2S, the PL peaks of InP/ZnS QDs were located at the wavelength region for cyan emission (482–506 nm) with a lower PLQY (Fig. 6C). This further confirms the contribution of H2S by interface engineering to increase the PLQY and tuning of the emission wavelength. Moreover, introducing more DDT and ZnSt2 for a second shell growth allowed the PLQY to increase to 66% (Fig. 6C). Although there have been several reports for InP-based QD synthesis with blue emissions,45,57,58 the mechanism for obtaining short PL wavelength of InP-based QDs with a high PLQY is still confused. Generally, the realization of a smaller InP core for blue emission conflicts with the effective elimination of defects. Due to the rapid nucleation process, a small size of InP QDs should be obtained with a quick termination of the core formation, easily leaving a lot of trap states on the surface of the InP core.9 Furthermore, it is more difficult to achieve controllable epitaxial growth of shells for the smaller size of core QDs.9 Our findings show that fine control of the smaller InP size can be realized by getting a large core first, then reducing the core size by H2S etching, resulting in a shorter emission wavelength (blue region) with a high PLQY.
The success of blue colour emission through H2S generation inspired us to promote the PL of InP QDs to deep blue emission. However, it did not work well by further changing the In and Zn precursors. Therefore, we developed another procedure to accelerate the reaction between H2S and the InP core by increasing the H2S generation rate. Instead of DDT/OLA or TOPS, we found that a combination of sulfur powder with OLA (S/OLA) can result in a much faster H2S generation,40,59,60 which enlarges the etching extent of the InP core. As shown in Fig. 6D, a larger blueshift of PL to deep blue emission at 435 nm with a PLQY of around 18% is observed, which over-ranges the typical emission wavelength of the reported InP-based QDs.10 Elemental analysis confirms that the structure of InP was partially maintained after the shell growth (Table S4, ESI†). Although further optimization is needed, this as-proposed photoluminescence modulation via in situ H2S interface engineering has offered strong promise for deep blue emission InP-based QD synthesis.
Conclusions
In summary, we propose an easy strategy for photoluminescence modulation of InP-based QDs by in situ H2S interface engineering. Comparison with different shell growth conditions confirms that H2S can be generated at an appropriate rate from DDT and OLA at high temperatures. The efficient H2S etching with S2− incorporation into the InP core causes interface reconstruction, reduces the inherent defects and relaxes the lattice strain at the interface during shell growth, leading to a higher PLQY of QDs. Furthermore, the in situ H2S interface engineering can also offer an additional size control of the InP core, which brings more possibilities for blue emission InP QD synthesis. We believe that the as-proposed in situ H2S generation approach can provide a convenient interface treatment strategy and enable an additional PL wavelength tunability, which shall have a significant impact on the synthesis of high-quality InP-based QDs and other nanocrystals.Author contributions
X. B. F., B. H., and J. M. K. conceived the idea. X. B. F., D. W. S., and S. L. designed the experiments. X. B. F., D. W. S., S. L., and J. J. Y. performed the QD synthesis and characterization. X. B. F. and S. Y. performed the GC measurements. J. Z. Y. and A. R. performed the femtosecond transient absorption measurements. D. J. M. and P. R. D. performed the XPS measurements. X. B. F., D. W. S., and S. Y. drafted the manuscript. All the authors analyzed the data and commented on the manuscript. B. H. and J. M. K. supervised the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from European Commission Horizon 2020 (685758), Engineering and Physical Sciences Research Council (EPSRC, EP/P027628/1) and International Exchanges 2021 Cost Share award from Royal Society and Natural Science Foundation of China (G114171 and NSFC22211530070). The XPS data collection was performed at the EPSRC National Facility for XPS (HarwellXPS), operated by Cardiff University and UCL, under contract No. PR16195. We would like to thank Dr Yuqi Chen (University of Cambridge), Dr Xue-Wang Gao (Technical Institute of Physics and Chemistry, CAS), and Mr Yijiang Chen (Southwest Petroleum University, China) for their assistance in the sample characterization.References
- F. P. García de Arquer, D. V. Talapin, V. I. Klimov, Y. Arakawa, M. Bayer and E. H. Sargent, Science, 2021, 373, 8541 CrossRef PubMed.
- Z. Yang, M. Gao, W. Wu, X. Yang, X. W. Sun, J. Zhang, H.-C. Wang, R.-S. Liu, C.-Y. Han, H. Yang and W. Li, Mater. Today, 2019, 24, 69–93 CrossRef CAS.
- A. R. C. Osypiw, S. Lee, S.-M. Jung, S. Leoni, P. M. Smowton, B. Hou, J. M. Kim and G. A. J. Amaratunga, Mater. Adv., 2022, 3, 6773–6790 RSC.
- Y. Shu, X. Lin, H. Qin, Z. Hu, Y. Jin and X. Peng, Angew. Chem., Int. Ed., 2020, 59, 22312–22323 CrossRef CAS PubMed.
- T. Meng, Y. Zheng, D. Zhao, H. Hu, Y. Zhu, Z. Xu, S. Ju, J. Jing, X. Chen, H. Gao, K. Yang, T. Guo, F. Li, J. Fan and L. Qian, Nat. Photonics, 2022, 16, 297–303 CrossRef CAS.
- M. Yuan, M. Liu and E. H. Sargent, Nat. Energy, 2016, 1, 16016 CrossRef CAS.
- X.-B. Li, C.-H. Tung and L.-Z. Wu, Nat. Rev. Chem., 2018, 2, 160–173 CrossRef CAS.
- K. D. Wegner and N. Hildebrandt, Chem. Soc. Rev., 2015, 44, 4792–4834 RSC.
- S. Tamang, C. Lincheneau, Y. Hermans, S. Jeong and P. Reiss, Chem. Mater., 2016, 28, 2491–2506 CrossRef CAS.
- P. Reiss, M. Carriere, C. Lincheneau, L. Vaure and S. Tamang, Chem. Rev., 2016, 116, 10731–10819 CrossRef CAS PubMed.
- A. K. Srivastava, W. Zhang, J. Schneider, J. E. Halpert and A. L. Rogach, Adv. Sci., 2019, 6, 1901345 CrossRef CAS PubMed.
- J. Lim, M. Park, W. K. Bae, D. Lee, S. Lee, C. Lee and K. Char, ACS Nano, 2013, 7, 9019–9026 CrossRef CAS PubMed.
- Y.-H. Won, O. Cho, T. Kim, D.-Y. Chung, T. Kim, H. Chung, H. Jang, J. Lee, D. Kim and E. Jang, Nature, 2019, 575, 634–638 CrossRef CAS PubMed.
- Y. Li, X. Hou, X. Dai, Z. Yao, L. Lv, Y. Jin and X. Peng, J. Am. Chem. Soc., 2019, 141, 6448–6452 CrossRef CAS PubMed.
- E. Jang, Y. Kim, Y.-H. Won, H. Jang and S.-M. Choi, ACS Energy Lett., 2020, 5, 1316–1327 CrossRef CAS.
- Z. Wu, P. Liu, W. Zhang, K. Wang and X. W. Sun, ACS Energy Lett., 2020, 5, 1095–1106 CrossRef CAS.
- J. L. Stein, W. M. Holden, A. Venkatesh, M. E. Mundy, A. J. Rossini, G. T. Seidler and B. M. Cossairt, Chem. Mater., 2018, 30, 6377–6388 CrossRef CAS.
- D. Hahm, J. H. Chang, B. G. Jeong, P. Park, J. Kim, S. Lee, J. Choi, W. D. Kim, S. Rhee, J. Lim, D. C. Lee, C. Lee, K. Char and W. K. Bae, Chem. Mater., 2019, 31, 3476–3484 CrossRef CAS.
- B. Chen, D. Li and F. Wang, Small, 2020, 16, 2002454 CrossRef CAS PubMed.
- H. Li, W. Zhang, Y. Bian, T. K. Ahn, H. Shen and B. Ji, Nano Lett., 2022, 22, 4067–4073 CrossRef CAS PubMed.
- M. D. Tessier, E. A. Baquero, D. Dupont, V. Grigel, E. Bladt, S. Bals, Y. Coppel, Z. Hens, C. Nayral and F. Delpech, Chem. Mater., 2018, 30, 6877–6883 CrossRef CAS.
- F. W. Eagle, N. Park, M. Cash and B. M. Cossairt, ACS Energy Lett., 2021, 6, 977–984 CrossRef CAS.
- Y.-C. Pu, H.-C. Fan, J.-C. Chang, Y.-H. Chen and S.-W. Tseng, J. Phys. Chem. Lett., 2021, 12, 7194–7200 CrossRef CAS PubMed.
- S. Yu, X.-B. Fan, X. Wang, J. Li, Q. Zhang, A. Xia, S. Wei, L.-Z. Wu, Y. Zhou and G. R. Patzke, Nat. Commun., 2018, 9, 4009 CrossRef PubMed.
- M. D. Tessier, D. Dupont, K. De Nolf, J. De Roo and Z. Hens, Chem. Mater., 2015, 27, 4893–4898 CrossRef CAS.
- L. Li and P. Reiss, J. Am. Chem. Soc., 2008, 130, 11588–11589 CrossRef CAS PubMed.
- K. Nemoto, J. Watanabe, H.-T. Sun and N. Shirahata, Nanoscale, 2022, 14, 9900–9909 RSC.
- T. Kim, S. W. Kim, M. Kang and S.-W. Kim, J. Phys. Chem. Lett., 2012, 3, 214–218 CrossRef CAS.
- W.-S. Song, H.-S. Lee, J. C. Lee, D. S. Jang, Y. Choi, M. Choi and H. Yang, J. Nanopart. Res., 2013, 15, 1750 CrossRef.
- E. Ryu, S. Kim, E. Jang, S. Jun, H. Jang, B. Kim and S.-W. Kim, Chem. Mater., 2009, 21, 573–575 CrossRef CAS.
- H. Virieux, M. Le Troedec, A. Cros-Gagneux, W.-S. Ojo, F. Delpech, C. Nayral, H. Martinez and B. Chaudret, J. Am. Chem. Soc., 2012, 134, 19701–19708 CrossRef CAS PubMed.
- V. Srivastava, V. Kamysbayev, L. Hong, E. Dunietz, R. F. Klie and D. V. Talapin, J. Am. Chem. Soc., 2018, 140, 12144–12151 CrossRef CAS PubMed.
- F. Pietra, L. De Trizio, A. W. Hoekstra, N. Renaud, M. Prato, F. C. Grozema, P. J. Baesjou, R. Koole, L. Manna and A. J. Houtepen, ACS Nano, 2016, 10, 4754–4762 CrossRef CAS PubMed.
- A. M. Smith and S. Nie, Acc. Chem. Res., 2010, 43, 190–200 CrossRef CAS PubMed.
- K. Kim, D. Yoo, H. Choi, S. Tamang, J.-H. Ko, S. Kim, Y.-H. Kim and S. Jeong, Angew. Chem., Int. Ed., 2016, 55, 3714–3718 CrossRef CAS PubMed.
- D. Yoo, M. Kim, S. Jeong, J. Han and J. Cheon, J. Am. Chem. Soc., 2014, 136, 14670–14673 CrossRef CAS PubMed.
- Z. Li, Y. Ji, R. Xie, S. Y. Grisham and X. Peng, J. Am. Chem. Soc., 2011, 133, 17248–17256 CrossRef CAS PubMed.
- A. J. Nelson, S. Frigo and R. Rosenberg, J. Appl. Phys., 1992, 71, 6086–6089 CrossRef CAS.
- W. Wardencki and B. Zygmunt, Anal. Chim. Acta, 1991, 255, 1–13 CrossRef CAS.
- J. W. Thomson, K. Nagashima, P. M. Macdonald and G. A. Ozin, J. Am. Chem. Soc., 2011, 133, 5036–5041 CrossRef CAS PubMed.
- L. Xi, D.-Y. Cho, M. Duchamp, C. B. Boothroyd, J. Y. Lek, A. Besmehn, R. Waser, Y. M. Lam and B. Kardynal, ACS Appl. Mater. Interfaces, 2014, 6, 18233–18242 CrossRef CAS PubMed.
- A. C. Berends, W. van der Stam, J. P. Hofmann, E. Bladt, J. D. Meeldijk, S. Bals and C. de Mello Donega, Chem. Mater., 2018, 30, 2400–2413 CrossRef CAS PubMed.
- K. Huang, R. Demadrille, M. G. Silly, F. Sirotti, P. Reiss and O. Renault, ACS Nano, 2010, 4, 4799–4805 CrossRef CAS PubMed.
- H. Xu, Y. Wang, X. Dong, N. Zheng, H. Ma and X. Zhang, Appl. Catal., B, 2019, 257, 117932 CrossRef CAS.
- Z. Cui, S. Mei, Z. Wen, D. Yang, S. Qin, Z. Xiong, B. Yang, H. He, R. Bao, Y. Qiu, Y. Chen, W. Zhang, F. Xie, G. Xing and R. Guo, Small, 2022, 18, 2108120 CrossRef CAS PubMed.
- A. Buffard, S. Dreyfuss, B. Nadal, H. Heuclin, X. Xu, G. Patriarche, N. Mézailles and B. Dubertret, Chem. Mater., 2016, 28, 5925–5934 CrossRef CAS.
- K. Z. Liu, Y. Suzuki and Y. Fukuda, Appl. Surf. Sci., 2004, 237, 623–626 CrossRef.
- M. Pelavin, D. N. Hendrickson, J. M. Hollander and W. L. Jolly, J. Phys. Chem., 1970, 74, 1116–1121 CrossRef CAS.
- W. Yang, Y. Yang, A. L. Kaledin, S. He, T. Jin, J. R. McBride and T. Lian, Chem. Sci., 2020, 11, 5779–5789 RSC.
- K. E. Knowles, E. A. McArthur and E. A. Weiss, ACS Nano, 2011, 5, 2026–2035 CrossRef CAS PubMed.
- K. Wu, N. Song, Z. Liu, H. Zhu, W. Rodríguez-Córdoba and T. Lian, J. Phys. Chem. A, 2013, 117, 7561–7570 CrossRef CAS PubMed.
- K. R. Reid, J. R. McBride, N. J. Freymeyer, L. B. Thal and S. J. Rosenthal, Nano Lett., 2018, 18, 709–716 CrossRef CAS PubMed.
- M. Rafipoor, D. Dupont, H. Tornatzky, M. D. Tessier, J. Maultzsch, Z. Hens and H. Lange, Chem. Mater., 2018, 30, 4393–4400 CrossRef CAS.
- K. Kim, Y.-H. Suh, D. Kim, Y. Choi, E. Bang, B. H. Kim and J. Park, Chem. Mater., 2020, 32, 2795–2802 CrossRef CAS.
- T. Kim, K.-H. Kim, S. Kim, S.-M. Choi, H. Jang, H.-K. Seo, H. Lee, D.-Y. Chung and E. Jang, Nature, 2020, 586, 385–389 CrossRef CAS PubMed.
- V. Sayevich, Z. L. Robinson, Y. Kim, O. V. Kozlov, H. Jung, T. Nakotte, Y.-S. Park and V. I. Klimov, Nat. Nanotechnol., 2021, 16, 673–679 CrossRef CAS PubMed.
- W. Shen, H. Tang, X. Yang, Z. Cao, T. Cheng, X. Wang, Z. Tan, J. You and Z. Deng, J. Mater. Chem. C, 2017, 5, 8243–8249 RSC.
- W. Zhang, S. Ding, W. Zhuang, D. Wu, P. Liu, X. Qu, H. Liu, H. Yang, Z. Wu, K. Wang and X. W. Sun, Adv. Funct. Mater., 2020, 30, 2005303 CrossRef CAS.
- B. Hou, D. Benito-Alifonso, R. Webster, D. Cherns, M. C. Galan and D. J. Fermín, J. Mater. Chem. A, 2014, 2, 6879–6886 RSC.
- S.-Y. Yoon, J.-H. Kim, E.-P. Jang, S.-H. Lee, D.-Y. Jo, Y. Kim, Y. R. Do and H. Yang, Chem. Mater., 2019, 31, 2627–2634 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nh00436d |
| This journal is © The Royal Society of Chemistry 2023 |