
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stable organic electrochemical neurons based on p-type and n-type ladder polymers†
Han-Yan
Wu‡
a,
Jun-Da
Huang‡
ab,
Sang Young
Jeong
c,
Tiefeng
Liu
 a,
Ziang
Wu
c,
Tom
van der Pol
a,
Qingqing
Wang
a,
Marc-Antoine
Stoeckel
ab,
Qifan
Li
a,
Mats
Fahlman
a,
Deyu
Tu
a,
Han Young
Woo
c,
Chi-Yuan
Yang
ab and
Simone
Fabiano
*ab
a,
Ziang
Wu
c,
Tom
van der Pol
a,
Qingqing
Wang
a,
Marc-Antoine
Stoeckel
ab,
Qifan
Li
a,
Mats
Fahlman
a,
Deyu
Tu
a,
Han Young
Woo
c,
Chi-Yuan
Yang
ab and
Simone
Fabiano
*ab
aLaboratory of Organic Electronics, Department of Science and Technology, Linköping University, SE-60174 Norrköping, Sweden. E-mail: simone.fabiano@liu.se
bn-Ink AB, Bredgatan 33, SE-60221 Norrköping, Sweden
cDepartment of Chemistry, College of Science, Korea University, Seoul 136-713, Republic of Korea
First published on 15th July 2023
Abstract
Organic electrochemical transistors (OECTs) are a rapidly advancing technology that plays a crucial role in the development of next-generation bioelectronic devices. Recent advances in p-type/n-type organic mixed ionic-electronic conductors (OMIECs) have enabled power-efficient complementary OECT technologies for various applications, such as chemical/biological sensing, large-scale logic gates, and neuromorphic computing. However, ensuring long-term operational stability remains a significant challenge that hinders their widespread adoption. While p-type OMIECs are generally more stable than n-type OMIECs, they still face limitations, especially during prolonged operations. Here, we demonstrate that simple methylation of the pyrrole-benzothiazine-based (PBBT) ladder polymer backbone results in stable and high-performance p-type OECTs. The methylated PBBT (PBBT-Me) exhibits a 25-fold increase in OECT mobility and an impressive 36-fold increase in μC* (mobility × volumetric capacitance) compared to the non-methylated PBBT-H polymer. Combining the newly developed PBBT-Me with the ladder n-type poly(benzimidazobenzophenanthroline) (BBL), we developed complementary inverters with a record-high DC gain of 194 V V−1 and excellent stability. These state-of-the-art complementary inverters were used to demonstrate leaky integrate-and-fire type organic electrochemical neurons (LIF-OECNs) capable of biologically relevant firing frequencies of about 2 Hz and of operating continuously for up to 6.5 h. This achievement represents a significant improvement over previous results and holds great potential for developing stable bioelectronic circuits capable of in-sensor computing.
New conceptsElectronic devices capable of in-sensor computing have the potential to revolutionize the field of bioelectronics. Organic mixed ionic-electronic conductors (OMIECs) are well-suited as they efficiently couple ionic and electronic transport, thereby bridging electronics and biology. Recent advancements in the design and synthesis of p-type and n-type OMIECs have resulted in the development of power-efficient devices for various applications, including sensors, nervetronics, neural interfaces, and artificial synapses. Organic electrochemical neurons (OECNs) with ion-modulated spiking are the most recent addition to the bioelectronic toolbox, enabling the development of event-based sensors capable of local sensing, signal processing, and stimulation/actuation. However, current OECN technology suffers from poor stability due to degradation of the p-type OECT characteristics. Therefore, developing stable p-type OMIECs is crucial for achieving high-performance OECNs. Rigid ladder polymers can sustain a high level of electrochemical doping without experiencing conformational disorder. This leads to exceptional operational stability, high charge carrier mobility, and large volumetric capacitance. The use of both n-type and p-type ladder polymers, with carefully engineered backbone to ensure efficient charge transport, enables integrate-and-fire OECNs with biologically relevant firing frequencies and excellent stability, representing a significant improvement over previous results and opening for stable bioelectronic device operation. |
Introduction
Organic mixed ionic-electronic conductors (OMIECs) are versatile materials with widespread applications in various fields, such as bioelectronics, optoelectronics, and energy storage.1–17 One of the most notable features of OMIECs is their ability to efficiently transport both ionic and electronic charges, setting them apart from other organic semiconductors. This unique interaction allows for new approaches in signal transduction and charge transport.18 OMIECs are commonly utilized as channel materials in organic electrochemical transistors (OECTs),19 where ion-modulated doping and dedoping causes the channel conductance to change by orders of magnitude and allows for transistor switching. Due to their low operating voltage, ease of manufacturing, high transconductance, and biocompatibility, OECTs are ideal for chemical and biological sensing,20–22 large-area printed circuitry,23 and neuromorphic computing.24–40While the progress in OECT-based circuitry has primarily revolved around the exploration of the hole-transporting (p-type) polymer PEDOT:PSS,41–45 recent advances in the design and synthesis of p-type and n-type (electron-transporting) OMIECs have led to the development of power-efficient complementary OECT technologies.46–48 These advancements have not only opened new possibilities for achieving improved performance and energy efficiency in OECT-based amplifiers49,50 and digital logics46 but also facilitated the development of the first leaky-integrate-and-fire (LIF) type organic electrochemical neuron (OECN) with ion-modulated spiking.24 The breakthrough holds great promise for bioelectronic applications, enabling the development of event-based sensors capable of local sensing, signal processing, and stimulation of the nervous system. This requires the development of OECNs capable of operating stably with spike dynamics and voltage output on par with biology. However, the initial implementation of LIF-OECNs suffered from a low spiking frequency (20–200 mHz) and reduced voltage output during continuous operation for over 1 h. Although reducing the OECT channel dimensions could increase the spiking frequency, the instability of the output current in the p-type OECT remains a significant hurdle, leading to output voltage drop and poor operational stability. Therefore, developing stable p-type OMIECs is crucial for achieving high-performance OECNs and enabling their widespread implementation in bioelectronics.
Several p-type OMIECs have been reported for complementary OECT technologies, including the conjugated polyelectrolyte poly(3-carboxypentylthiophene) (P3CPT), as well as conjugated polymers bearing glycolated side chains such as p(g42T-T), p(gTDPPT), and gDPP-g2T.46,48,51–53 These p-type OMIECs exhibit excellent charge carrier mobility and large volumetric capacitance. However, several of them suffer from degradation during continuous operation in ambient. Strategies to improve the stability of p-type OMIECs include designing encapsulation layers54 or tuning the polymer's energetics55,56 to suppress electrochemical side reactions during OECT operation, incorporating high-boiling-point solvents and Lewis acids to alter the polymer chain arrangements and prevent oxidative degradation,57 and optimizing the polymer side chain distribution to regulate swelling and enhance stability.58,59 However, p-type OMIECs with improved stability often experience lower mobilities and volumetric capacitances, resulting in overall low complementary device performance.
Ladder-type polymers possess a rigid backbone structure consisting of double-strand chains linked by condensed π-conjugated units. This unique structure allows them to sustain high levels of chemical/electrochemical doping without experiencing conformational disorder.26,60,61 As a result, ladder-type polymers exhibit exceptional operational stability, high charge carrier mobility, and large volumetric capacitance.51 One prominent example of a ladder-type polymer is the n-type poly(benzimidazobenzophenanthroline) (BBL), which demonstrates outstanding operational stability in OECTs.48 Recently, Li et al.62 reported the ladder-type polymer poly[pyrrolo[3,2-b:4,5-b′]bis-[1,4]benzothiazine] (PBBT), which can function as a p-type OMIEC. While PBBT shows decent stability when used as the channel material of OECTs, it suffers from low mobility and low volumetric capacitance.63
Here, we present a practical approach to enhancing the mobility, volumetric capacitance, and stability of PBBT through backbone engineering. Our strategy involves methylating the PBBT polymer backbone to obtain the polymer poly[6-methyl-pyrrolo[3,2-b:4,5-b′]bis-[1,4]benzothiazine] (PBBT-Me, Fig. 1(a)). Notably, this subtle backbone modification leads to significant improvements in the material properties, including a remarkable 25-fold increase in hole mobility and an impressive 36-fold increase in μC* of PBBT-Me as compared to the non-methylated polymer PBBT-H, while maintaining similar stability. Using this newly developed polymer in combination with BBL, we successfully demonstrate complementary inverters with a record-high DC gain of 194 V V−1 and excellent stability. These state-of-the-art complementary inverters are then used to develop LIF-OECNs with firing frequencies of about 2 Hz and capable of continuous operation for up to 6.5 h. This achievement represents a substantial improvement compared to previous results and holds great potential for the development of stable bioelectronic circuits capable of in-sensor computing.
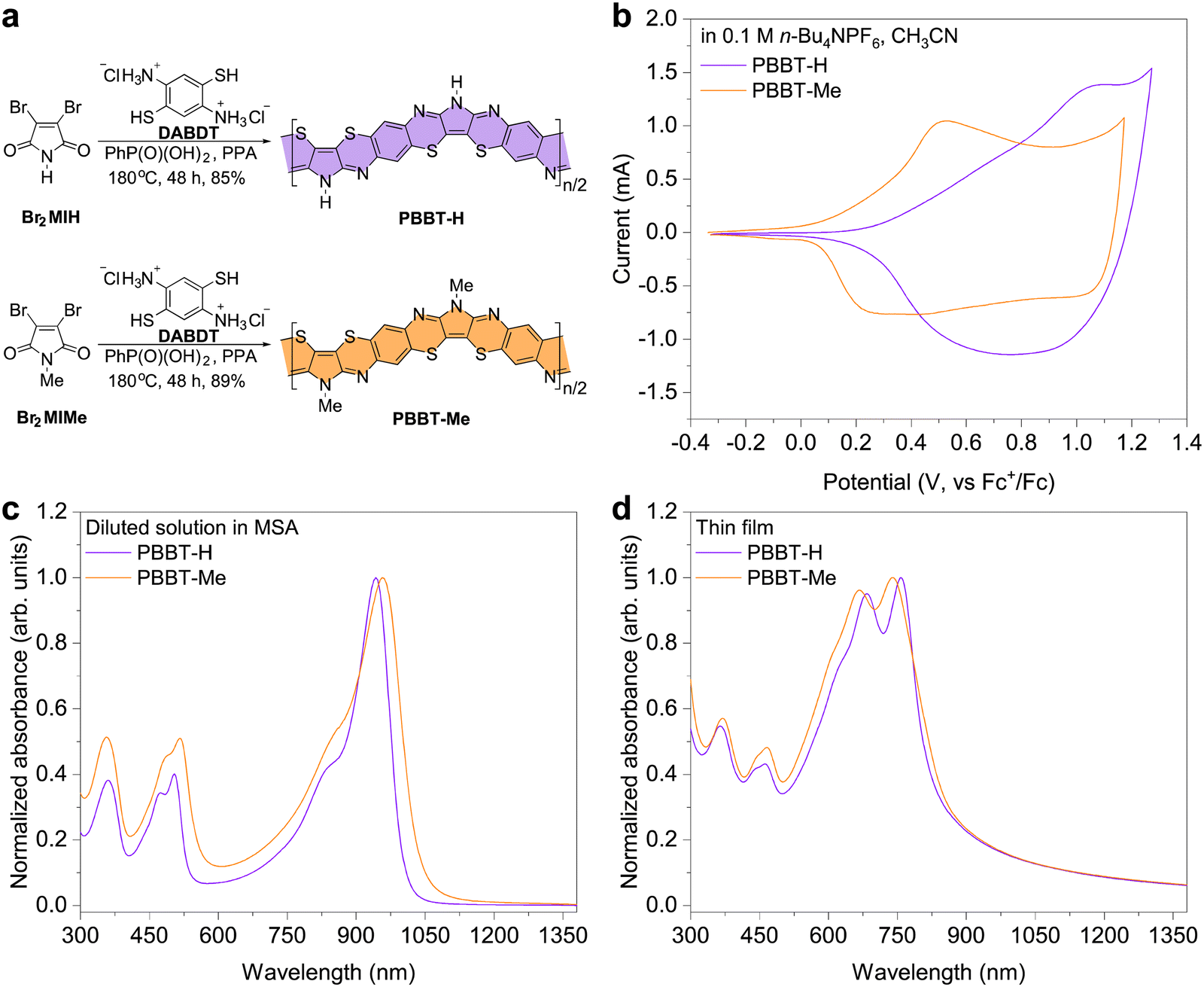 | ||
| Fig. 1 Synthesis and physicochemical properties of PBBT-H and PBBT-Me. (a) Synthetic route of PBBT-H and PBBT-Me. (b) CV of PBBT-H and PBBT-Me in 0.1 M n-Bu4NPF6 acetonitrile solution. (c) and (d) UV-vis-NIR absorption spectra of PBBT-H and PBBT-Me in diluted MSA solutions (c) and thin films (d). | ||
Results and discussion
PBBT-H and PBBT-Me were synthesized through a polycondensation reaction of 2,3-dibromomaleimide (Br2MIH) or 2,3-dibromomethylmaleimide (Br2MIMe) and 2,5-diaminobenzene-1,4-dithiol dihydrochloride (DABDT) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 polyphosphoric acid (PPA):phenylphosphonic acid64 (PhP(O)(OH)2) mixture at 180 °C (Fig. 1(a)). Fourier transform infrared spectroscopy and X-ray photoelectron spectroscopy confirmed successful methylation of the PBBT polymer backbone (Fig. S1 and S2, ESI†). Both ladder-type polymers have good solubility in methanesulfonic acid (MSA) at concentrations over 10 mg mL−1 and can be deposited in thin films by spin-coating their MSA solutions onto substrates at room temperature. The intrinsic viscosities of PBBT-H and PBBT-Me were measured to be 0.94 dL g−1 and 0.99 dL g−1 (in MSA, 25 °C), respectively, which indicates comparable molecular weights. Additionally, both polymers are thermally stable with decomposition temperatures over 540 °C, and no phase transitions were observed before decomposition (Fig. S3, ESI†).
:1 polyphosphoric acid (PPA):phenylphosphonic acid64 (PhP(O)(OH)2) mixture at 180 °C (Fig. 1(a)). Fourier transform infrared spectroscopy and X-ray photoelectron spectroscopy confirmed successful methylation of the PBBT polymer backbone (Fig. S1 and S2, ESI†). Both ladder-type polymers have good solubility in methanesulfonic acid (MSA) at concentrations over 10 mg mL−1 and can be deposited in thin films by spin-coating their MSA solutions onto substrates at room temperature. The intrinsic viscosities of PBBT-H and PBBT-Me were measured to be 0.94 dL g−1 and 0.99 dL g−1 (in MSA, 25 °C), respectively, which indicates comparable molecular weights. Additionally, both polymers are thermally stable with decomposition temperatures over 540 °C, and no phase transitions were observed before decomposition (Fig. S3, ESI†).
Cyclic voltammetry measurements show that PBBT-H and PBBT-Me have highest occupied molecular orbital (HOMO) energy levels of −5.06 eV and −4.87 eV, respectively. These measurements suggest that the electron-donating effect of the methyl group makes the HOMO level of PBBL-Me shallower by 0.2 eV. The UV-Vis-NIR absorption spectra of the two polymers in diluted MSA solutions show two absorption bands located at 300–600 nm and 800–1100 nm, respectively (Fig. 1(c)). The introduction of the methyl group in PBBT-Me compared to PBBT-H results in a redshift of the π–π transition in the range 800–1100 nm. This redshift indicates that the methyl group further enhances the electron-rich nature of the pyrrole unit, leading to a narrowing of the bandgap (Eg) in the protonated state in solution. The absorption spectra of the two polymers in the thin film state are also similar, with a slight redshifted absorption onset wavelength (λonset) for PBBT-Me (Fig. 1(d)). Note that the maximum absorption peaks in solutions are redshifted compared to those in thin films likely due to protonation of the polymer backbone in solution. With the λonset of PBBT-H being 829 nm and λonset of PBBT-Me being 868 nm, the optical Eg is calculated to be 1.50 eV for PBBT-H and 1.43 eV for PBBT-Me.
Next, we investigated PBBT-H and PBBT-Me thin film microstructure using grazing-incidence wide-angle X-ray scattering (GIWAXS) and atomic force microscopy (AFM). GIWAXS analysis (Fig. 2(a)–(c)) reveals that PBBT-H chains are arranged preferentially face-on on the substrate, while PBBT-Me chains are mostly oriented edge-on. Both PBBT-H and PBBT-Me display strong π–π stacking (010) peaks located at 1.82 Å−1, indicative of a similar π–π stacking distance dπ–π = 3.45 Å (Fig. 2(c) and Fig. S4, ESI†). However, PBBT-H exhibits a lamellar packing distance dlamellar = 8.73 Å [(100) peak at qxy = 0.72 Å−1], which is shorter than PBBT-Me's dlamellar = 9.82 Å [(100) peak at qz = 0.64 Å−1]. The observed difference in packing distance is consistent with the presence of the methyl group on the PBBT-Me backbone. Furthermore, PBBT-Me shows significantly enhanced π–π stacking crystallinity with smaller FWHM (0.1677 for PBBT-Me vs. 0.3256 for PBBT-H), longer coherence length (33.3 Å for PBBT-Me vs. 17.2 Å for PBBT-H), and smaller paracrystalline disorder (0.121 for PBBT-Me vs. 0.169 for PBBT-H) as shown in Fig. S4 and S5 of the ESI.† These results suggest that PBBT-Me may be more efficient in transporting charge carriers in the in-plane direction than PBBT-H. The AFM analysis shows that both PBBT-H and PBBT-Me thin films have a flat polycrystalline morphology (Fig. 2(d) and (e)). The surface roughness of PBBT-H was measured to be 7.89 nm, whereas PBBT-Me displayed a slightly smaller roughness of 6.15 nm. A summary of the solid-state microstructure characteristics can be found in Table 1.
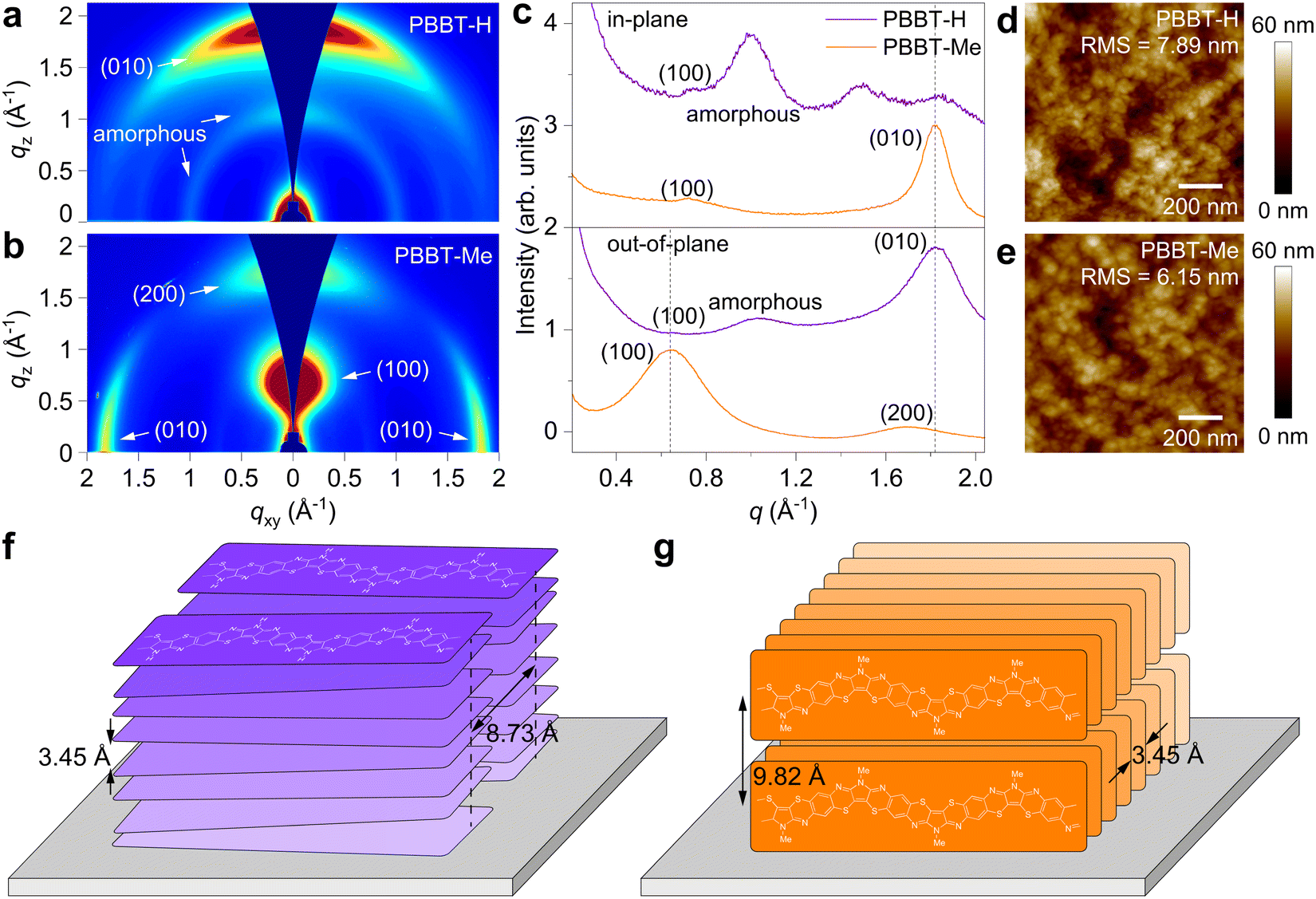 | ||
| Fig. 2 Thin-film microstructures. (a) and (b) 2D GIWAXS patterns of PBBT-H (a) and PBBT-Me (b). (c) In-plane and out-of-plane GIWAXS line cuts of PBBT-H and PBBT-Me thin films. (d) and (e) AFM height images of PBBT-H (d) and PBBT-Me (e). The root mean square (RMS) surface roughness is also reported. (f) and (g) Schematic of the PBBT-H and PBBT-Me chains packing. | ||
| Polymer | E g (eV) | HOMO (eV) | LUMO (eV) | Orientation | d lamellar (Å) | d π–π (Å) | L c,π–π (Å) |
|---|---|---|---|---|---|---|---|
| PBBT-H | 1.50 | −5.06 | −3.56 | Face-on | 8.73 | 3.45 | 17.2 |
| PBBT-Me | 1.43 | −4.87 | −3.44 | Edge-on | 9.82 | 3.45 | 33.3 |
Spectroelectrochemistry was performed to investigate the electronic structure of electrochemically doped PBBT-H and PBBT-Me thin films. The polymer thin films were coated on ITO substrates and subjected to a voltage bias applied to the Ag/AgCl electrode through a 0.1 M NaCl aqueous electrolyte (Fig. 3(a), (b) and Fig. S6 in the ESI†). Upon applying an oxidation voltage bias within the 0–1 V range, we observed that both PBBT-H and PBBT-Me undergo electrochemical doping with clear bleaching of the ground state (370–800 nm) and strong polaron absorption (>800 nm). The PBBT-H's polaronic species arise at an oxidation voltage of around 0.4 V, whereas those of PBBT-Me form at around 0.5 V (Fig. 3(c)). We attributed this difference to the introduction of the methyl group in PBBT-Me, which resulted in a slight increase in hydrophobicity (Fig. S14, ESI†), ultimately leading to a minor increase in the oxidation threshold.
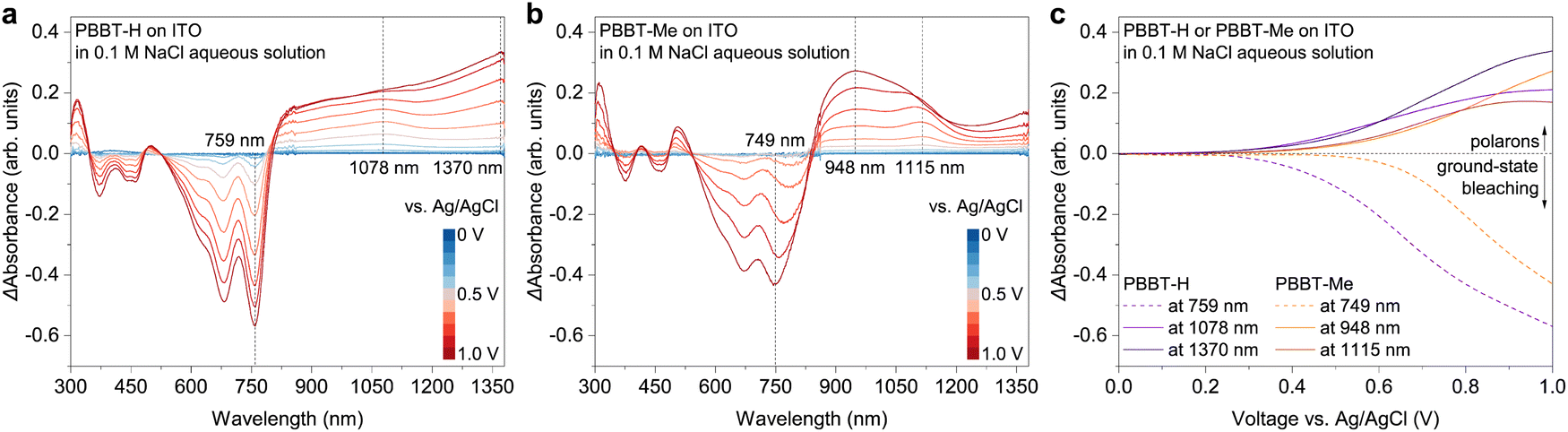 | ||
| Fig. 3 Differential spectroelectrochemistry. (a) and (b) Differential absorption spectra of PBBT-H (a) and PBBT-Me (b). (c) Differential absorption spectra of PBBT-H (c) and PBBT-Me (d) at select wavelengths. In all these spectroelectrochemistry measurements, 0.1 M NaCl aqueous solution was used as the electrolyte, Ag/AgCl electrode was grounded, and the voltage was applied to the polymer films scanning between 0 V and 1.0 V versus Ag/AgCl electrode. | ||
Next, we conducted a comprehensive characterization of the performance of PBBT-H and PBBT-Me in OECTs. OECTs were prepared by following the procedure reported in the Experimental Section, which involves using glass wafers with patterned metal and parylene layers. The channel materials were deposited onto the OECT substrates from their MSA solutions via spin-coating and then patterned using a sacrificial layer of parylene to produce the OECT channel (W/L = 400 μm/6 μm). The thickness of the polymer thin films (50 nm) was adjusted by controlling the concentration of the MSA solution. The OECT was then completed using 0.1 M NaCl aqueous solution as the electrolyte and an Ag/AgCl pellet as the gate electrode (Fig. S7, ESI†). Both PBBT-H and PBBT-Me exhibit typical p-type accumulation-mode characteristics, as evidenced by their output and transfer curves (Fig. 4(a)–(d)). The ON/OFF ratio of OECTs using PBBT-H and PBBT-Me as the channel materials are 4.5(± 0.5) × 103 and 2.5(± 0.3) × 106, respectively (Fig. 4(c) and (d)). The on-state current of PBBT-Me-based OECTs at VD = VG = −0.7 V is 511 ± 26 μA, which is approximately 10× higher than that of PBBT-H (50 ± 2 μA). The threshold voltage of PBBT-Me OECTs (0.52 ± 0.01 V) is slightly larger than that of PBBT-H (0.36 ± 0.01 V), and consistent with the spectroelectrochemistry experiments. The μC* values of PBBT-H and PBBT-Me were calculated to be 2.5 ± 0.1 F cm−1 V−1 s−1 and 92.3 ± 5.0 F cm−1 V−1 s−1, respectively (Fig. S8, ESI†). Electrochemical impedance spectroscopy (EIS) was used to evaluate the volumetric capacitance C* of the two polymers, resulting in values of 179 ± 5 F cm−3 for PBBT-H and 259 ± 6 F cm−3 for PBBT-Me (Fig. S9–S11, ESI†). Thus, the μOECT of PBBT-H and PBBT-Me was calculated to be (1.41 ± 0.04) × 10−2 cm2 V−1 s−1 for PBBT-H and 0.356 ± 0.028 cm2 V−1 s−1 for PBBT-Me. The higher mobility of PBBT-Me is consistent with the increased on-state current of PBBT-Me OECTs compared to PBBT-H OECTs and can be explained by the increased crystallinity, longer coherence length, and preferential edge-on orientation of PBBT-Me compared to PBBT-H, as revealed by the GIWAXS data (vide supra). This edge-on arrangement facilitates interchain hole transport, leading to more efficient charge transport in PBBT-Me. In addition, the max gm,norm of PBBT-Me is also increased by one order of magnitude compared to PBBT-H, from 1.11 ± 0.02 S cm−1 to 40.6 ± 1.5 S cm−1. Both PBBT-H and PBBT-Me OECTs exhibit remarkably high stability, with over 90% of the initial drain current retained after more than 4 hours of continuous operation (VD = VG = −0.6 V). This stability is among the highest reported for p-type OECTs (Fig. S12 and Table S1, ESI†).55–59 The PBBT-H OECTs have a transient response τON = 0.74 ± 0.02 ms, while that of PBBT-Me OECT is slightly slower at 3.19 ± 0.05 ms (Fig. S13, ESI†), likely due to the increased hydrophobicity of PBBT-Me layer (Fig. S14, ESI†). Table 2 summarizes the OECT characteristics of the two polymers.
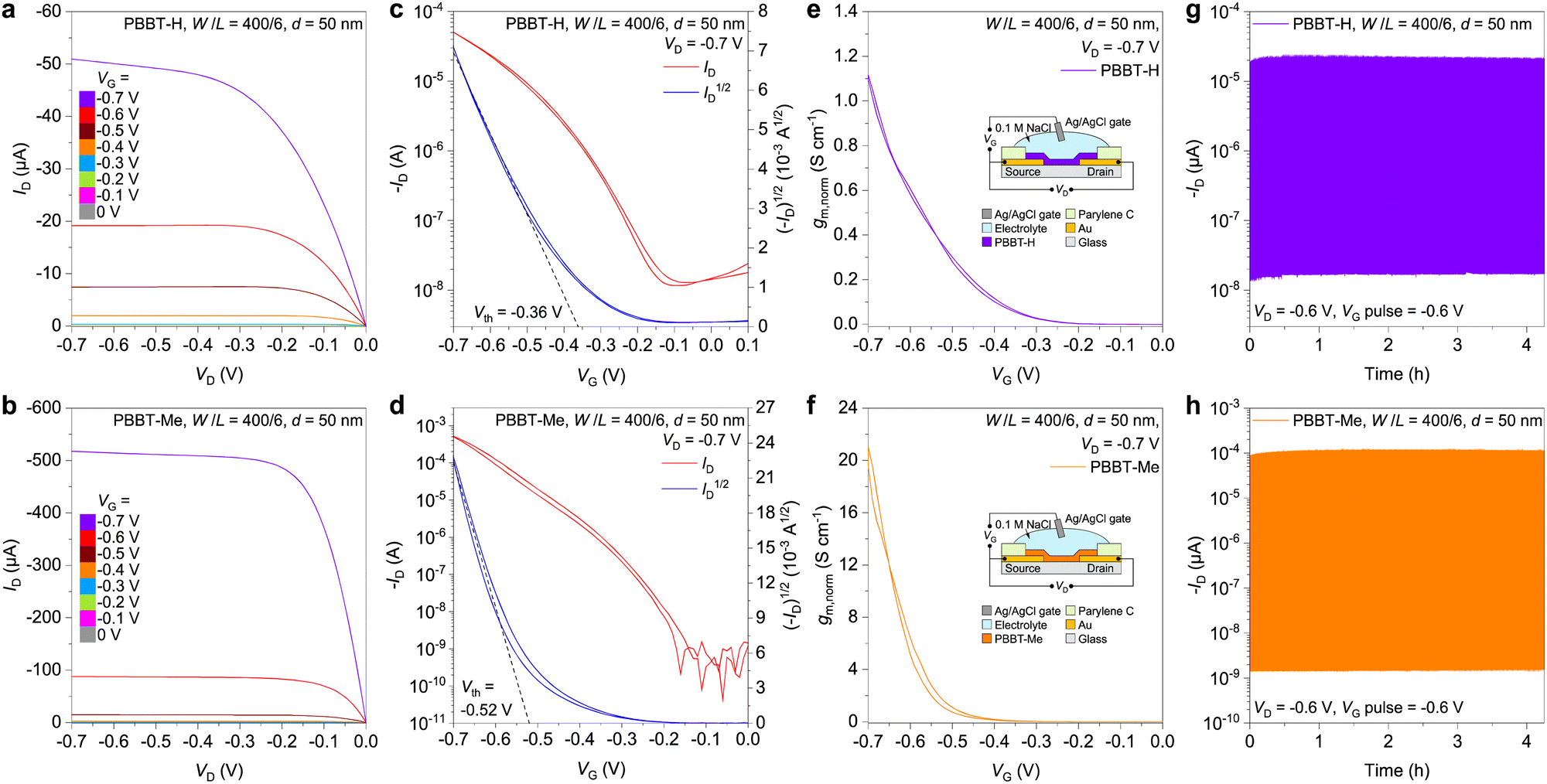 | ||
| Fig. 4 OECT performance. (a), (b) Output characteristics of OECTs based on PBBT-H (a) and PBBT-Me (b). (c), (d) Transfer characteristics of OECTs based on PBBT-H (c) and PBBT-Me (d). (e), (f) Geometry-normalized transconductance (gm,norm) of PBBT-H (e) and PBBT-Me (f). Inset: Schematic of the OECTs. (g) and (h) Stability of OECTs based on PBBT-H (g) and PBBT-Me (h). | ||
| Polymer | g m,norm (S cm−1) | V th (V) | I ON/IOFF | μC* (F cm−1 V−1 s−1) | C* (F cm−3) | μ (cm2 V−1 s−1) | τ ON, τOFF (ms) |
|---|---|---|---|---|---|---|---|
| PBBT-H | 1.11 ± 0.02 | −0.36 ± 0.01 | 4 × 103 | 2.52 ± 0.07 | 179 ± 5 | (1.41 ± 0.04) × 10−2 | 0.738 ± 0.018, 0.138 ± 0.011 |
| PBBT-Me | 40.6 ± 1.5 | −0.52 ± 0.02 | 2 × 106 | 92.3 ± 5.0 | 259 ± 6 | 0.356 ± 0.028 | 3.19 ± 0.02, 0.159 ± 0.014 |
To investigate their usage in circuits, we developed complementary electrochemical inverters utilizing PBBT-H or PBBT-Me as the hole-transporting materials and BBL as the electron-transporting material due to its low Vth = 0.15 V, high on/off ratio of 105, and high gm,norm = 11 S cm−1 (Fig. S15, ESI†). The OECT-based inverter schematic is presented in Fig. 5(a) and (b). The pull-up PBBT-H or PBBT-Me-based transistor in the OECT-based inverter switches on and off in response to a gradual change in gate voltage (Vin) between 0 V and Vin = VDD, while the pull-down BBL-based transistor works in the opposite manner. As a result, the inverter's output voltage (Vout) switches between approximately VDD and 0 V in response to Vin, as shown in Fig. 5(a) and (b). PBBT-H-based inverters operate at VDD as low as 0.2 V with voltage gains >8 V V−1 (Fig. 5(c)), while PBBT-Me-based inverters exhibit voltage gains >20 V V−1 at VDD = 0.3 V (Fig. 5(d), note that the threshold voltage of PBBT-Me based OECTs is 0.15 V higher than that of PBBT-H OECTs). At VDD = 0.7 V, PBBT-H-based inverters exhibit voltage gains of about 60 V V−1, while PBBT-Me-based inverters reach record high voltage gains of 194 V V−1, which is among the highest reported for complementary OECT inverters (Fig. S16 and Table S2, ESI†). Mixing PBBT-Me and BBL yields ambipolar mixtures with balanced hole and electron transport, resulting in complementary inverters with voltage gains larger than those recently reported for PBBT-L:BBL blends63 (Fig. S17, ESI†). This is consistent with the fact that PBBT-Me has significantly higher mobility and μC* than PBBT-H. Both PBBT-H and PBBT-Me-based inverters show exceptionally low static power consumption, ranging from 0.5 nW (VDD = 0.2 V) to 40 nW (VDD = 0.7 V), as well as peak power consumption during switching below 10 μW (Fig. S18, ESI†). Additionally, these inverters demonstrate swift transient switching with small τON and τOFF in the range of 1.5–4 ms (Fig. 5(e), (f) and Fig. S19, ESI†) and remarkable cycling stability (Fig. S20, ESI†). Considered altogether, these inverter characteristics are among the best reported for sub-1 V complementary inverters.46,49,52,63,65–69
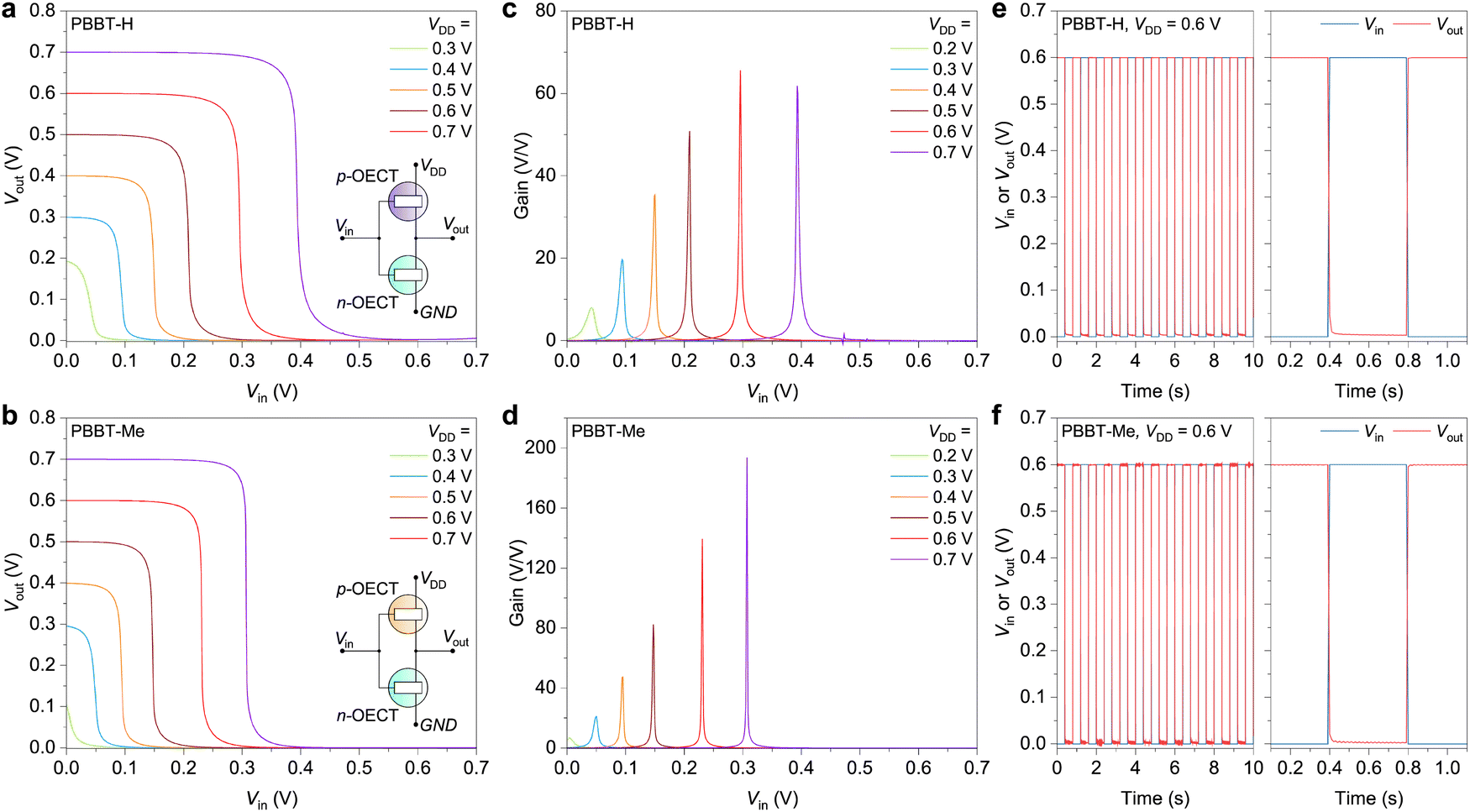 | ||
| Fig. 5 OECT-based inverters. (a) and (b) Typical voltage transfer characteristics (VTC) of the inverters at various supply voltages (from 0.2 to 0.7 V). (c) and (d) Voltage gains of the inverters at various supply voltages. (e) and (f) Input and output of the inverters showing “0” and “1” states. All the above inverters consisted of an n-type (BBL) and a p-type OECT (PBBT-H or PBBT-Me) OECT. | ||
Finally, we used the complementary electrochemical inverters described above to develop LIF type OECNs based on a modified version of the Axon-Hillock circuit (Fig. 6(a)).24 The OECNs comprising the p-type PBBT-Me and n-type BBL OECTs can fire at a frequency nearing 2 Hz and with voltage spike amplitude (Vfire,max) of around 0.4 V at input currents Iin > 2 μA (Fig. 6(b)–(d) and Fig. S21, S22 in the ESI†). OECNs made of the ambipolar PBBT-Me:BBL mixture show comparable spiking characteristics (Fig. S23 and S24, ESI†). This biologically plausible spiking frequency could mimic most neural firing rates and can be used to stimulate biological nerves.33 These OECNs can also be made solid state by using printable hydrogels46 based on polycationic polyquaternium-10 (PQ-10) for PBBT-Me OECTs and polyanionic poly(sodium-4-styrene sulfonate) (PSSNa) for BBL OECTs (Fig. S25 and S26 in the ESI†). Remarkably, these OECNs retain close to 90% of the initial spiking frequencies and voltage amplitudes for over 6.5 h of continuous spiking, outperforming similar LIF-OECNs comprising the p-type P(g42T-T) (Fig. S27 and S28, ESI†). This excellent operational stability is paramount for the development of stable event-based sensors.
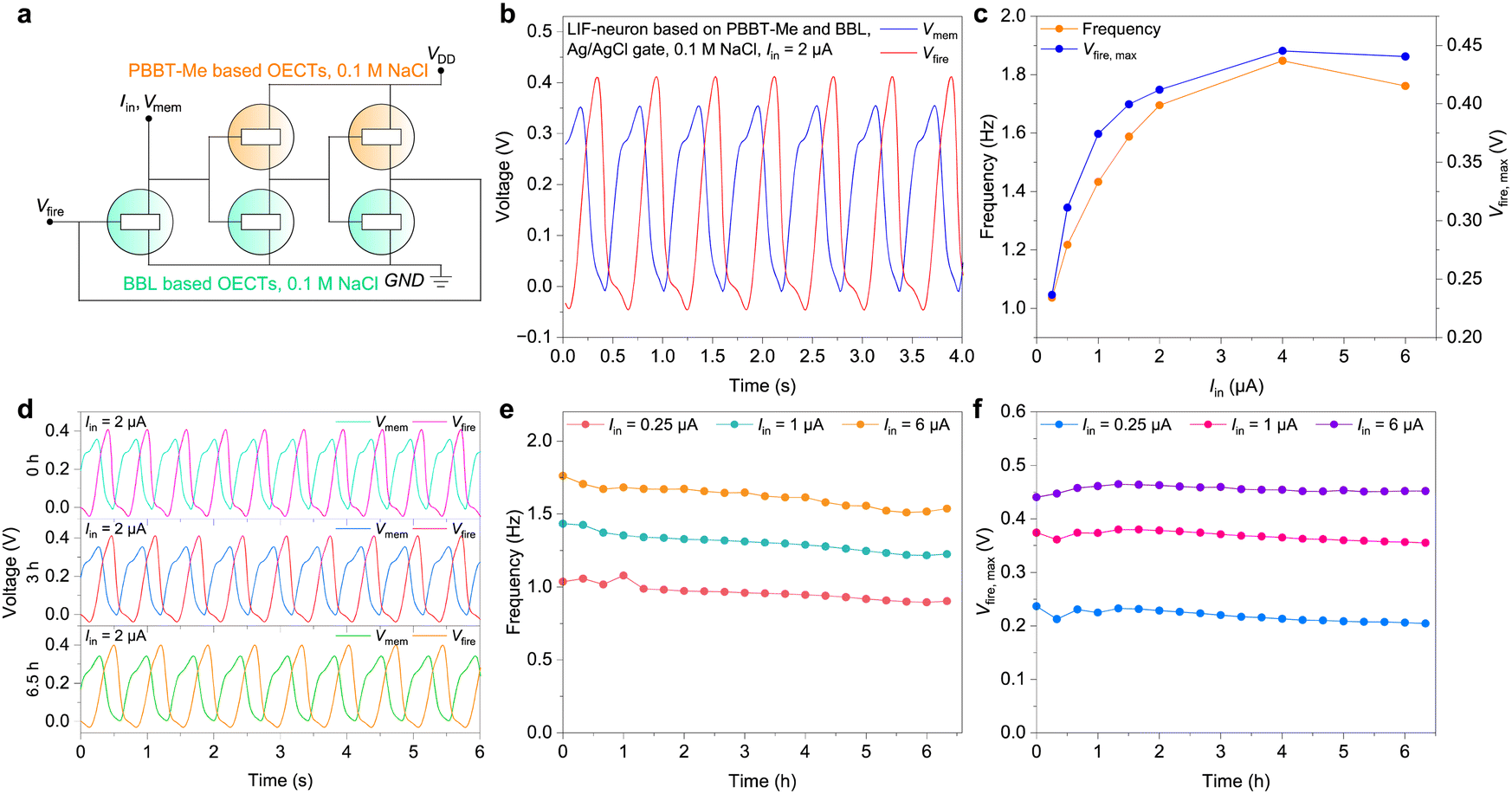 | ||
| Fig. 6 Organic electrochemical neurons based on PBBT-Me and BBL. (a) Schematic of the leaky-integrate-and-fire (LIF) type spiking neuron, comprising both p-type and n-type accumulation-mode OECTs. (b) Typical spiking behaviors of a LIF-neuron based on PBBT-Me and BBL, gated using Ag/AgCl and working at Iin = 2 μA in 0.1 M NaCl aqueous solution. (c) Spiking frequency and Vfire,max of the LIF-neuron at different Iin. (d) Spiking patterns at Iin = 2 μA for 0 h, 3 h, and 6.5 h. (e) Spiking frequencies of the LIF-neuron change over time at Iin = 0.25 μA, 1 μA, 6 μA during 6.5 h of continuous operation. (f) Evolution of the Vfire,max over time at Iin = 0.25 μA, 1 μA, 6 μA during 6.5 h of continuous operation. | ||
Conclusion
In conclusion, we demonstrated that simple methylation of PBBT-based ladder-type polymers yields a p-type OMIEC with notable OECT performance (i.e., gm,norm = 40.6 ± 1.5 S cm−1, μC* = 92.3 ± 5.0 F cm−1 V−1 s−1, and τON = 3.19 ± 0.02 ms). We attributed these remarkable characteristics to an enhanced intermolecular charge transport, which results from a rearrangement of the polymer chains induced by the introduction of the methyl group in the PBBT polymer backbone. In addition, the rigid ladder-type polymer backbone endows PBBT-Me with outstanding electrochemical stability, making it suitable for long-term operation. Complementary inverters based on PBBT-Me and BBL exhibit record-high voltage gains >190 V V−1, low static power consumption <40 nW, and excellent operational stability. This allowed us to build LIF-type OECNs with a stable operation for >6.5 h without significant degradation in terms of firing frequency (1–2 Hz) and voltage amplitude (0.4–0.5 V). These results open the path for developing high-performance complementary OECT-based amplifiers and artificial neurons for brain-inspired electronics.Experiment section
Synthesis
2,5-Diamino-1,4-benzenedithiol dihydrochloride (DABDT) was purchased from Tokyo Chemical Industry. 2,3-Dibromomaleimide (Br2MIH), polyphosphoric acid (PPA), phenylphosphonicacid (PhP(O)(OH)2), N-methylpyrrole, tetrahydrofuran (THF), N-bromosuccinimide (NBS) and methanesulfonic acid (MSA), n-hexane, nitric acid, ethyl acetate were purchased from Sigma-Aldrich.N-Methyl-3,4-dibromomaleimide (Br2MIMe), the polymerization precursor of PBBT-Me, was synthesized following a reported procedure.70N-Methylpyrrole (6.00 g, 73.97 mmol) was placed in a three-necked round-bottom flask, then dry THF (200 mL) was added and the system was kept under nitrogen atmosphere. After N-methylpyrrole was completely dissolved, the solution was cooled to −78 °C. NBS power (74.17 g, 416.73 mmol) was added in 10 portions during 20 minutes to keep the mixture at a low temperature. The mixture was stirred for 30 minutes at this temperature, then restored to room temperature and stirred for 12 hours. The solvent was removed by distillation under reduced pressure, and 20 mL of dichloromethane was added to the residual solid. The dichloromethane solution was isolated by filtration, concentrated to 10 mL, transferred to a 100 mL single-necked bottle and placed in ice bath. Concentrated nitric acid was added dropwise to make sure the temperature of the mixture wouldn’t increase. The reaction mixture was kept at 0 °C for 3 hours. The obtained solution was subsequently quenched with ice and water mixture and ethyl acetate (3 × 50 mL) was used to extract the soluble fraction. The organic layer was collected, successively washed with water, sodium bicarbonate, and brine, dried over anhydrous Na2SO4 and concentrated. The crude product was recrystallized from ethanol, and the final product Br2MIMe was obtained as colorless needles with 84% (16.77 g) yield. NMR: 1H (300 MHz, DMSO-d6): δ [ppm] 2.95 (s, 3H).
PBBT-Me was synthesized based on a procedure reported before.64 PPA (26 g) and PhP(O)(OH)2 (26 g) were added to a 250 mL three-necked flask fitted with an overhead stirrer and nitrogen inlet/outlet. The mixture of PPA and PhP(O)(OH)2 was deoxygenated by heating at 110°C for 2 hours with nitrogen bubbling through. The solution was cooled to room temperature and 2.00 g (8.16 mmol) of DABDT was added. The mixture was heated at 80°C for 12 hours to remove HCl, then Br2MIMe (2.19 g, 8.16mmol) was added. This solution was heated at 80 °C for another 12 hours, then heated at 150 °C for 24 hours. Next, the temperature was gradually increased to 180 °C and kept for 48 hours to complete the polymerization. The obtained black solution was cooled to room temperature and poured into 1 L of ethanol with the aid of a stirrer for better mixing. The obtained polymer precipitate was collected by filtration and washed with ethanol. It was then re-dissolved into methanesulfonic acid and re-precipitated in 1 L of ethanol. Again, the solid was collected by filtration, washed thoroughly with water and ethanol, and dried in vacuum to yield brown solid product PBBT-Me (1.97 g, 99.3% yield, η = 0.989 dL g−1 in MSA at 30 °C, Mv = 8.82 kDa). In estimation of Mv, the viscosity was measured with an Ubbelohde viscometer and calculated with48,71K = 5.11 × 10−6 g dL−1 and α = 1.34. Elemental anal. calcd for C11H5N3S2: C, 54.3; H, 2.1; N, 17.3; S, 26.4; found: C, 52.5; H, 2.6; N, 16.2; S, 28.5.
PBBT-H was synthesized following a reported procedure.64 The same setup was used as in PBBT-Me synthesis, and the synthesis procedure was similar, except that the reactant DBMI-Me was replaced with 2,3-dibromomaleimide. In brief, PPA (21 g) and PhP(O)(OH)2 (21 g) were added to a 250 mL three-necked flask fitted with an overhead stirrer and nitrogen inlet/outlet. The mixture of PPA and PhP(O)(OH)2 was heated at 110°C for 2 hours with nitrogen bubbling through. Then, the solution was cooled to room temperature, and 1.64 g (6.68 mmol) of DABDT was added. The reaction mixture was heated at 80°C for 12 hours to remove HCl, and then Br2MIH (1.70 g, 6.68mmol) was added. This mixture was kept at 80 °C for another 12 hours. Next, the solution was heated at 150 °C for 24 hours and then the temperature was gradually increased to 180 °C. The reaction mixture was kept at 180 °C for 48 hours to obtain PBBT-H polymer. The post-processing was the same as that of PBBT-Me. The final product PBBT-H was yield as brown solid product PBBT-H (1.27 g, 85% yield, η = 0.938 dL g−1 in MSA at 30 °C, Mv = 8.48 kDa). In estimation of Mv, the viscosity was measured with an Ubbelohde viscometer and calculated with71,72K = 5.11 × 10−6 g dL−1 and α = 1.34. Elemental anal. calcd for C10H3N3S2: C, 52.4; H, 1.3; N, 18.3; S, 28.0; found: C, 48.3; H, 2.3; N, 15.0; S, 25.6.
Thin-film casting
PBBT-H and PBBT-Me were dissolved in MSA at 60 °C for 2 hours and subsequently cooled to room temperature. The solutions were then spin-cast (1000 rpm, 60 s, acceleration 1000 rpm s−1) onto glass, ITO-glass or Si/SiO2 substrates to form thin films. The substrates covered with polymer films were immersed into deionized water to remove residual MSA and then blown dry with nitrogen. The concentration of the polymer solution was adjusted between 2–11 mg mL−1 to obtain thin films with thickness of 20–100 nm.Absorption spectra
Absorption spectra were performed with 7 μg mL−1 polymer-MSA solutions and 70 nm-thick polymer thin films on glass slides. Spectroelectrochemistry measurement was performed with indium tin oxide (ITO) glass substrates spin-cast on 70 nm-thick polymer thin films. The polymer films were grounded, and the substrates were immersed in 0.1 M NaCl solution, serving as the electrolyte. An Ag/AgCl pellet was used as the gate electrode applied with specific voltages through Keithley 4200A-SCS. Each voltage step was kept for 200 s before absorption spectra measuring, which was carried out with PerkinElmer Lambda 900 spectrometer at 25 °C.Electrochemistry
Electrochemical measurements were carried out with Potentiostat Biologic SP-200. In CV measurement, deoxygenized 0.1 M n-Bu4NPF6 anhydrous acetonitrile solution served as the electrolyte, saturated Ag/AgCl electrode worked as the reference electrode (RE), and a 10 mm × 10 mm platinum mesh was used as the counter electrode (CE). PBBT-H and PBBT-Me thin-films were spin-cast from 5 mg mL−1 MSA solutions onto chromium (5 nm)/gold (50 nm) deposited glass slides (5 mm × 5 mm) to make working electrode (WE). The scan rate used for CV measurement was 50 mV s−1. In electrochemical impedance spectroscopy (EIS) measurement, the electrolyte was 0.1 M NaCl aqueous solution, the RE was a saturated Ag/AgCl electrode, and the CE was a 10 mm × 10 mm platinum mesh. Cr/Au electrodes (on glass slides) were deposited with different amount of polymers through spin-coating to form films with various volumes. The obtained PBBT-H or PBBT-Me film with a volume of 7.5 × 10−7–5.1 × 10−6 cm3 serves as the WE. In each measurement of capacitance, WE was applied with bias of 0.6 V or 0.7 V vs. Ag/AgCl, and the frequency range was 0.1 Hz–10 kHz. A Randles circuit model Rs + Rp/CPE was used to fit the curves and to extract the capacitance. In this circuit, Rs is the resistance of active electrolyte, Rp represents the resistance in charge transfer, and CPE is a constant phase element.Grazing-incidence wide-angle X-ray scattering
GIWAXS experiments were performed following the reported procedure.48 All samples for GIWAXS measurements were deposited on cut silicon wafers and had a thickness of about 100 nm. They were measured at BeamLine 9A in the Pohang Accelerator Laboratory in South Korea. The X-ray energy was 11.08 eV and the incidence angle was 0.12°. Samples were measured in vacuum and the total exposure time was 10 s. The scattered X-rays were recorded by a charge-coupled device detector located 221.9351 mm from the sample.OECTs, complementary inverters, and LIF neurons
OECTs were fabricated following a procedure reported previously.26 Microscope glass slides were thoroughly cleaned via successive sonication in acetone, deionized water, and isopropanol, and then blown dry with nitrogen. Source/drain electrodes were thermally deposited with 5 nm Cr and 50 nm Au, and patterned with photolithography and wet etching. A first layer of parylene C (1 μm) was coated together with a drop of 3-(trimethoxysilyl) propyl methacrylate (A-174 Silane), to serve as an insulating layer between the metal electrode and the electrolyte. A dilution of industrial surfactant (2% Micro-90) was subsequently spin-coated before the depositing of a second layer (sacrificial layer) of parylene C (1 μm). To protect parylene C layers from a subsequent plasma reactive ion etching step (150 W, O2 = 500 sccm, CF4 = 1000 sccm, 380 s), a thick positive photoresist (5 μm, AZ 10XT 520CP) was spin-coated on top of the parylene C layers. A second photolithographic patterning step followed by an application of AZ developer was carried out to define the contact pads and the OECT channels. The subsequent plasma reactive ion etching step was applied to indiscriminately remove the organic layers including photoresist and parylene C, until the contact pads and the OECT channel area were exposed to the air, while the other part of the device was still covered with two layers of parylene C. The channel between source and drain was patterned to be W/L = 400 μm/6 μm. The obtained OECT device was spin-coated with PBBT-H, PBBT-Me or BBL MSA solutions to form polymer film of 50 nm. After removing the residual MSA by immersing the device in deionized water, the polymer film was dried with nitrogen flow. The sacrificial layer of parylene C was peeled off, removing the polymer film on top of it and leaving the polymer in the patterned area, which was the OECT channel and the electrode pads. Thus, the source and the drain electrode were connected by a micrometer-scale piece of p-type or n-type polymer. For all OECTs, 0.1 M NaCl aqueous solution was used as the electrolyte, and an Ag/AgCl pellet (purchased from VWR) was used as the gate electrode. The complementary inverters were assembled by connecting a p-type (PBBT-H or PBBT-Me) OECT and an n-type (BBL) OECT with silver paste. The LIF neurons were assembled by connecting two p-type (PBBT-Me) OECT and three n-type (BBL) OECTs as shown in Fig. 6(a). For processing convenience of the LIF neuron, the channel width to length ratio is W/L = 325/40 μm/μm for p-type OECTs and W/L = 1300/40 μm/μm for n-type OECTs. The LIF neurons based on P(g42T-T) and BBL were assembled in a similar way, and W/L was the same. The devices were characterized with Keilthley 4200A-SCS (with 4225-PMU Ultra Fast I-V Module and 4225-RPM Remote Amplifier/Switch Modules).Conflicts of interest
C.-Y. Y., M.-A. S., and S. F. are the co-founder of n-Ink AB. The other authors declare no competing interests.Acknowledgements
The authors thank Dr Zhixing Wu (Linköping U.) for help with TGA measurements. This work was financially supported by the Knut and Alice Wallenberg Foundation (2021.0058, 2022.0034, and WWSC), the Swedish Research Council (2020-03243, 2022-04053, and 2022-04553), the European Commission through the FET-OPEN project MITICS (GA-964677), and the Swedish Government Strategic Research Area in Materials Science on Functional Materials at Linköping University (Faculty Grant SFO-Mat-LiU 2009-00971). H. Y. Woo acknowledges the financial support from the National Research Foundation of Korea (2019R1A6A1A11044070, NRF2020M3H4A3081814).References
- I. Bargigia, L. R. Savagian, A. M. Osterholm, J. R. Reynolds and C. Silva, J. Am. Chem. Soc., 2021, 143, 294–308 CrossRef CAS PubMed.
- Z. Fan and J. Y. Ouyang, Adv. Electron. Mater., 2019, 5, 1800769 CrossRef CAS.
- K. R. Li, Y. L. Shao, H. P. Yan, Z. Lu, K. J. Griffith, J. H. Yan, G. Wang, H. W. Fan, J. Y. Lu, W. Huang, B. Bao, X. L. Liu, C. Y. Hou, Q. H. Zhang, Y. G. Li, J. S. Yu and H. Z. Wang, Nat. Commun., 2018, 9, 4798 CrossRef PubMed.
- P. Lundberg, Y. Tsuchiya, E. M. Lindh, S. Tang, C. Adachi and L. Edman, Nat. Commun., 2019, 10, 5307 CrossRef CAS PubMed.
- E. Macchia, R. A. Picca, K. Manoli, C. Di Franco, D. Blasi, L. Sarcina, N. Ditaranto, N. Cioffi, R. Osterbacka, G. Scamarcio, F. Torricelli and L. Torsi, Mater. Horiz., 2020, 7, 999–1013 RSC.
- L. Manjakkal, A. Pullanchiyodan, N. Yogeswaran, E. S. Hosseini and R. Dahiya, Adv. Mater., 2020, 32, 1907254 CrossRef CAS PubMed.
- D. Moia, A. Giovannitti, A. A. Szumska, I. P. Maria, E. Rezasoltani, M. Sachs, M. Schnurr, P. R. F. Barnes, I. McCulloch and J. Nelson, Energy Environ. Sci., 2019, 12, 1349–1357 RSC.
- B. D. Paulsen, K. Tybrandt, E. Stavrinidou and J. Rivnay, Nat. Mater., 2020, 19, 13–26 CrossRef CAS PubMed.
- R. A. Picca, K. Manoli, E. Macchia, L. Sarcina, C. Di Franco, N. Cioffi, D. Blasi, R. Osterbacka, F. Torricelli, G. Scamarcio and L. Torsi, Adv. Funct. Mater., 2020, 30, 1904513 CrossRef CAS.
- D. Scheunemann, V. Vijayakumar, H. Y. Zeng, P. Durand, N. Leclerc, M. Brinkmann and M. Kemerink, Adv. Electron. Mater., 2020, 6, 2000218 CrossRef CAS.
- S. Tang, A. Sandström, P. Lundberg, T. Lanz, C. Larsen, S. van Reenen, M. Kemerink and L. Edman, Nat. Commun., 2017, 8, 1190 CrossRef PubMed.
- S. van Reenen, R. A. J. Janssen and M. Kemerink, Adv. Funct. Mater., 2015, 25, 3066–3073 CrossRef CAS.
- A. V. Volkov, H. D. Sun, R. Kroon, T. P. Ruoko, C. Y. Che, J. Edberg, C. Muller, S. Fabiano and X. Crispin, ACS Appl. Energy Mater., 2019, 2, 5350–5355 CrossRef CAS.
- H. M. Wang, Y. F. Diao, Y. Lu, H. R. Yang, Q. J. Zhou, K. Chrulski and J. M. D'Arcy, Nat. Commun., 2020, 11, 3882 CrossRef CAS PubMed.
- C. C. Yu, C. Y. Wang, X. Liu, X. T. Jia, S. Naficy, K. W. Shu, M. Forsyth and G. G. Wallace, Adv. Mater., 2016, 28, 9349–9355 CrossRef CAS PubMed.
- E. Zeglio, A. L. Rutz, T. E. Winkler, G. G. Malliaras and A. Herland, Adv. Mater., 2019, 31, 1806712 CrossRef PubMed.
- X. N. Zhang, B. H. Wang, L. Z. Huang, W. Huang, Z. Wang, W. G. Zhu, Y. Chen, Y. L. Mao, A. Facchetti and T. J. Marks, Sci. Adv., 2020, 6, eaaz1042 CrossRef CAS PubMed.
- B. D. Paulsen, S. Fabiano and J. Rivnay, Annu. Rev. Mater. Res., 2021, 51, 73–99 CrossRef CAS.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, 17086 CrossRef CAS.
- D. Ohayon, G. Nikiforidis, A. Savva, A. Giugni, S. Wustoni, T. Palanisamy, X. X. Chen, I. P. Maria, E. Di Fabrizio, P. Costa, I. McCulloch and S. Inal, Nat. Mater., 2020, 19, 456–463 CrossRef CAS PubMed.
- J. Chen, W. Huang, D. Zheng, Z. Xie, X. Zhuang, D. Zhao, Y. Chen, N. Su, H. Chen, R. M. Pankow, Z. Gao, J. Yu, X. Guo, Y. Cheng, J. Strzalka, X. Yu, T. J. Marks and A. Facchetti, Nat. Mater., 2022, 21, 564–571 CrossRef CAS PubMed.
- C. Liao, C. Mak, M. Zhang, H. L. Chan and F. Yan, Adv. Mater., 2015, 27, 676–681 CrossRef CAS PubMed.
- P. Andersson Ersman, R. Lassnig, J. Strandberg, D. Tu, V. Keshmiri, R. Forchheimer, S. Fabiano, G. Gustafsson and M. Berggren, Nat. Commun., 2019, 10, 5053 CrossRef CAS PubMed.
- P. C. Harikesh, C.-Y. Yang, D. Tu, J. Y. Gerasimov, A. M. Dar, A. Armada-Moreira, M. Massetti, R. Kroon, D. Bliman, R. Olsson, E. Stavrinidou, M. Berggren and S. Fabiano, Nat. Commun., 2022, 13, 901 CrossRef CAS PubMed.
- T. Sarkar, K. Lieberth, A. Pavlou, T. Frank, V. Mailaender, I. McCulloch, P. W. M. Blom, F. Torricelli and P. Gkoupidenis, Nat. Electron., 2022, 5, 774–783 CrossRef.
- P. C. Harikesh, C.-Y. Yang, H.-Y. Wu, S. Zhang, M. J. Donahue, A. S. Caravaca, J.-D. Huang, P. S. Olofsson, M. Berggren, D. Tu and S. Fabiano, Nat. Mater., 2023, 22, 242–248 CrossRef CAS PubMed.
- J. Y. Gerasimov, R. Gabrielsson, R. Forchheimer, E. Stavrinidou, D. T. Simon, M. Berggren and S. Fabiano, Adv. Sci., 2019, 6, 1801339 CrossRef PubMed.
- J. Y. Gerasimov, D. Zhao, A. Sultana, T. Abrahamsson, S. Han, D. Bliman, D. Tu, D. T. Simon, R. Olsson, X. Crispin, M. Berggren and S. Fabiano, Adv. Electron. Mater., 2021, 7, 2001126 CrossRef CAS.
- S. K. Lee, Y. W. Cho, J. S. Lee, Y. R. Jung, S. H. Oh, J. Y. Sun, S. Kim and Y. C. Joo, Adv. Sci., 2021, 8, 2001544 CrossRef CAS PubMed.
- W. Wang, Y. Jiang, D. Zhong, Z. Zhang, S. Choudhury, J. C. Lai, H. Gong, S. Niu, X. Yan, Y. Zheng, C. C. Shih, R. Ning, Q. Lin, D. Li, Y. H. Kim, J. Kim, Y. X. Wang, C. Zhao, C. Xu, X. Ji, Y. Nishio, H. Lyu, J. B. H. Tok and Z. Bao, Science, 2023, 380, 735–742 CrossRef CAS PubMed.
- S. Wang, X. Chen, C. Zhao, Y. Kong, B. Lin, Y. Wu, Z. Bi, Z. Xuan, T. Li, Y. Li, W. Zhang, E. Ma, Z. Wang and W. Ma, Nat. Electron., 2023, 6, 281–291 CrossRef CAS.
- Y. Zhang, E. R. W. van Doremaele, G. Ye, T. Stevens, J. Song, R. C. Chiechi and Y. van de Burgt, Adv. Mater., 2022, 34, e2200393 CrossRef PubMed.
- J. Y. Gerasimov, D. Tu, V. Hitaishi, P. C. Harikesh, C. Y. Yang, T. Abrahamsson, M. Rad, M. J. Donahue, M. S. Ejneby, M. Berggren, R. Forchheimer and S. Fabiano, Adv. Sci., 2023, 10, e2207023 CrossRef PubMed.
- X. Ji, B. D. Paulsen, G. K. K. Chik, R. Wu, Y. Yin, P. K. L. Chan and J. Rivnay, Nat. Commun., 2021, 12, 2480 CrossRef CAS PubMed.
- S. Pecqueur, M. Mastropasqua Talamo, D. Guérin, P. Blanchard, J. Roncali, D. Vuillaume and F. Alibart, Adv. Electron. Mater., 2018, 4, 1800166 CrossRef.
- L. Liu, W. Xu, Y. Ni, Z. Xu, B. Cui, J. Liu, H. Wei and W. Xu, ACS Nano, 2022, 16, 2282–2291 CrossRef CAS PubMed.
- Y. Ni, H. Han, J. Liu, Y. Choi, L. Liu, Z. Xu, L. Yang, C. Jiang, W. Gao and W. Xu, Nano Energy, 2022, 104, 107898 CrossRef CAS.
- H. Wei, Y. Ni, L. Sun, H. Yu, J. Gong, Y. Du, M. Ma, H. Han and W. Xu, Nano Energy, 2021, 81, 105648 CrossRef CAS.
- S. Zhang, J. Guo, L. Liu, H. Ruan, C. Kong, X. Yuan, B. Zhang, G. Gu, P. Cui, G. Cheng and Z. Du, Nano Energy, 2022, 91, 106660 CrossRef CAS.
- Y. Peng, L. Gao, C. Liu, J. Deng, M. Xie, L. Bai, G. Wang, Y. Cheng, W. Huang and J. Yu, Nano Res., 2023 DOI:10.1007/s12274-023-5633-y.
- G. D. Spyropoulos, J. N. Gelinas and D. Khodagholy, Sci. Adv., 2019, 5, eaau7378 CrossRef CAS PubMed.
- J. Rivnay, P. Leleux, M. Sessolo, D. Khodagholy, T. Herve, M. Fiocchi and G. G. Malliaras, Adv. Mater., 2013, 25, 7010 CrossRef CAS PubMed.
- D. Khodagholy, J. Rivnay, M. Sessolo, M. Gurfinkel, P. Leleux, L. H. Jimison, E. Stavrinidou, T. Herve, S. Sanaur, R. M. Owens and G. G. Malliaras, Nat. Commun., 2013, 4, 2133 CrossRef PubMed.
- J. Rivnay, P. Leleux, M. Ferro, M. Sessolo, A. Williamson, D. A. Koutsouras, D. Khodagholy, M. Ramuz, X. Strakosas, R. M. Owens, C. Benar, J. M. Badier, C. Bernard and G. G. Malliaras, Sci. Adv., 2015, 1, e1400251 CrossRef PubMed.
- S. M. Kim, C. H. Kim, Y. Kim, N. Kim, W. J. Lee, E. H. Lee, D. Kim, S. Park, K. Lee, J. Rivnay and M. H. Yoon, Nat. Commun., 2018, 9, 3858 CrossRef PubMed.
- C. Y. Yang, D. Tu, T. P. Ruoko, J. Y. Gerasimov, H. Y. Wu, P. C. Harikesh, M. Massetti, M. A. Stoeckel, R. Kroon, C. Muller, M. Berggren and S. Fabiano, Adv. Electron. Mater., 2022, 8, 2100907 CrossRef CAS.
- Y. Yao, W. Huang, J. Chen, G. Wang, H. Chen, X. Zhuang, Y. Ying, J. Ping, T. J. Marks and A. Facchetti, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2111790118 CrossRef CAS PubMed.
- H. Y. Wu, C. Y. Yang, Q. F. Li, N. B. Kolhe, X. Strakosas, M. A. Stoeckel, Z. Wu, W. L. Jin, M. Savvakis, R. Kroon, D. Y. Tu, H. Y. Woo, M. Berggren, S. A. Jenekhe and S. Fabiano, Adv. Mater., 2022, 34, 2106235 CrossRef CAS PubMed.
- R. B. Rashid, W. Y. Du, S. Griggs, I. P. Maria, I. McCulloch and J. Rivnay, Sci. Adv., 2021, 7, eabh1055 CrossRef CAS PubMed.
- X. Ji, X. Lin and J. Rivnay, Nat. Commun., 2023, 14, 1665 CrossRef CAS PubMed.
- H. D. Sun, M. Vagin, S. H. Wang, X. Crispin, R. Forchheimer, M. Berggren and S. Fabiano, Adv. Mater., 2018, 30, 1704916 CrossRef PubMed.
- W. Huang, J. H. Chen, Y. Yao, D. Zheng, X. D. Ji, L. W. Feng, D. Moore, N. R. Glavin, M. Xie, Y. Chen, R. M. Pankow, A. Surendran, Z. Wang, Y. Xia, L. B. Bai, J. Rivnay, J. F. Ping, X. G. Guo, Y. H. Cheng, T. J. Marks and A. Facchetti, Nature, 2023, 613, 496 CrossRef CAS PubMed.
- P. Y. Li, J. W. Shi, Y. Q. Lei, Z. Huang and T. Lei, Nat. Commun., 2022, 13, 5970 CrossRef PubMed.
- S. Zhang, P. Ding, T. P. Ruoko, R. Wu, M. A. Stoeckel, M. Massetti, T. Liu, M. Vagin, D. Meli, R. Kroon, J. Rivnay and S. Fabiano, Adv. Funct. Mater., 2023 DOI:10.1002/adfm.202302249.
- X. Y. Luo, H. G. Shen, K. Perera, D. T. Tran, B. W. Boudouris and J. G. Mei, ACS Macro Lett., 2021, 10, 1061–1067 CrossRef CAS PubMed.
- A. Giovannitti, R. B. Rashid, Q. Thiburce, B. D. Paulsen, C. Cendra, K. Thorley, D. Moia, J. T. Mefford, D. Hanifi, W. Y. Du, M. Moser, A. Salleo, J. Nelson, I. McCulloch and J. Rivnay, Adv. Mater., 2020, 32, 1908047 CrossRef CAS PubMed.
- T. C. H. Castillo, M. Moser, C. Cendra, P. D. Nayak, A. Salleo, I. McCulloch and S. Inal, Chem. Mater., 2022, 34, 6723 CrossRef.
- A. Savva, R. Hallani, C. Cendra, J. Surgailis, T. C. Hidalgo, S. Wustoni, R. Sheelamanthula, X. X. Chen, M. Kirkus, A. Giovannitti, A. Salleo, I. McCulloch and S. Inal, Adv. Funct. Mater., 2020, 30, 1907657 CrossRef CAS.
- M. Moser, T. C. Hidalgo, J. Surgailis, J. Gladisch, S. Ghosh, R. Sheelamanthula, Q. Thiburce, A. Giovannitti, A. Salleo, N. Gasparini, A. Wadsworth, I. Zozoulenko, M. Berggren, E. Stavrinidou, S. Inal and I. McCulloch, Adv. Mater., 2020, 32, 2002748 CrossRef CAS PubMed.
- K. Xu, T. P. Ruoko, M. Shokrani, D. Scheunemann, H. Abdalla, H. Sun, C. Y. Yang, Y. Puttisong, N. B. Kolhe, J. S. M. Figueroa, J. O. Pedersen, T. Ederth, W. M. Chen, M. Berggren, S. A. Jenekhe, D. Fazzi, M. Kemerink and S. Fabiano, Adv. Funct. Mater., 2022, 32, 2112276 CrossRef CAS.
- S. Wang, H. Sun, U. Ail, M. Vagin, P. O. Persson, J. W. Andreasen, W. Thiel, M. Berggren, X. Crispin, D. Fazzi and S. Fabiano, Adv. Mater., 2016, 28, 10764–10771 CrossRef CAS PubMed.
- S. T. Wang, W. Hong, S. D. Ren, J. Li, M. Wang, X. K. Gao and H. X. Li, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4272–4276 CrossRef CAS.
- X. H. Wu, T. L. D. Tam, S. Chen, T. Salim, X. M. Zhao, Z. L. Zhou, M. Lin, J. W. Xu, Y. L. Loo and W. L. Leong, Adv. Mater., 2022, 34, 2206118 CrossRef CAS PubMed.
- T. L. D. Tam, M. Lin, S. W. Chien and J. W. Xu, ACS Macro Lett., 2022, 11, 110–115 CrossRef CAS PubMed.
- S. L. Zhang, M. Massetti, T. P. Ruoko, D. Y. Tu, C. Y. Yang, X. J. Liu, Z. A. Wu, Y. Lee, R. Kroon, P. O. A. Persson, H. Y. Woo, M. Berggren, C. Muller, M. Fahlman and S. Fabiano, Adv. Funct. Mater., 2022, 32, 2106447 CrossRef CAS.
- E. Stein, O. Nahor, M. Stolov, V. Freger, I. M. Petruta, I. McCulloch and G. L. Frey, Nat. Commun., 2022, 13, 5548 CrossRef CAS PubMed.
- J. J. Song, H. Liu, Z. Y. Zhao, X. Y. Guo, C. K. Liu, S. Griggs, A. Marks, Y. Zhu, H. K. W. Law, I. McCulloch and F. Yan, Sci. Adv., 2023, 9, eadd9627 CrossRef PubMed.
- J. J. Samuel, A. Garudapalli, A. A. Mohapatra, C. Gangadharappa, S. Patil and N. P. B. Aetukuri, Adv. Funct. Mater., 2021, 31, 2102903 CrossRef CAS.
- P. Romele, M. Ghittorelli, Z. M. Kovacs-Vajna and F. Torricelli, Nat. Commun., 2019, 10, 3044 CrossRef PubMed.
- E. Surmiak, A. Twarda-Clapa, K. M. Zak, B. Musielak, M. D. Tomala, K. Kubica, P. Grudnik, M. Madej, M. Jablonski, J. Potempa, J. Kalinowska-Tluscik, A. Domling, G. Dubin and T. A. Holak, ACS Chem. Biol., 2016, 11, 3310–3318 CrossRef CAS PubMed.
- C. P. Wong, H. Ohnuma and G. C. Berry, J. Polym. Sci., Part C: Polym. Symp., 1978, 65, 173–192 CAS.
- G. Berry and S. Yen, in Addition and Condensation Polymerization Processes, ed. N. A. J. Platzer, American Chemical Society, Washington, D.C., 1st edn, 1969, vol. 91, ch. 48, pp. 734–756 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh00858d |
| ‡ H.-Y. W. and J.-D. H. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |