
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetically localized and wash-free fluorescence immunoassay (MLFIA): proof of concept and clinical applications†
S.
Delshadi‡
 *abc,
M.
Fratzl
acd,
O.
Ramel
a,
P.
Bigotte
a,
P.
Kauffmann
a,
D.
Kirk
a,
V.
Masse
a,
M. P.
Brenier-Pinchart
be,
H.
Fricker-Hidalgo
be,
H.
Pelloux
be,
F.
Bruckert
f,
C.
Charrat
b,
O.
Cugat
c,
N. M.
Dempsey
d,
T.
Devillers
d,
P.
Halfon
g,
A.
Leroy
b,
M.
Weidenhaupt
f and
P. N.
Marche
b
*abc,
M.
Fratzl
acd,
O.
Ramel
a,
P.
Bigotte
a,
P.
Kauffmann
a,
D.
Kirk
a,
V.
Masse
a,
M. P.
Brenier-Pinchart
be,
H.
Fricker-Hidalgo
be,
H.
Pelloux
be,
F.
Bruckert
f,
C.
Charrat
b,
O.
Cugat
c,
N. M.
Dempsey
d,
T.
Devillers
d,
P.
Halfon
g,
A.
Leroy
b,
M.
Weidenhaupt
f and
P. N.
Marche
b
aMagIA diagnostics, 15 rue Maréchal Leclerc, 38130 Échirolles, France. E-mail: sarah.delshadi@magia-diagnostics.com
bUniv. Grenoble Alpes, Inserm U1209, CNRS UMR 5309, IAB, 38000 Grenoble, France
cUniv. Grenoble Alpes, CNRS, Grenoble INP, G2Elab, 21 Av. des Martyrs, 38000 Grenoble, France
dUniv. Grenoble Alpes, CNRS, Grenoble INP, Institut Néel, 25 Av. des Martyrs, 38042 Grenoble, France
eService de Parasitologie-Mycologie, CHU Grenoble Alpes, 38000 Grenoble, France
fUniv. Grenoble Alpes, CNRS, Grenoble INP, LMGP, 38000 Grenoble, France
gHopital Europeen, Laboratoire Alphabio-Biogroup, 13003 Marseille, France
First published on 26th January 2023
Abstract
Immunoassays are used for many applications in various markets, from clinical diagnostics to the food industry, generally relying on gold-standard ELISAs that are sensitive, robust, and cheap but also time-consuming and labour intensive. As an alternative, we propose here the magnetically localized and wash-free fluorescence immunoassay (MLFIA): a no-wash assay to directly measure a biomolecule concentration, without mixing nor washing steps. To do so, a fluorescence no-wash measurement is performed to generate a detectable signal. It consists of a differential measurement between the fluorescence of fluorophores bound to magnetic nanoparticles specifically captured by micro-magnets against the residual background fluorescence of unbound fluorophores. Targeted biomolecules (antibodies or antigens) are locally concentrated on micro-magnet lines, with the number of captured biomolecules quantitatively measured without any washing step. The performance of the MLFIA platform is assessed and its use is demonstrated with several biological models as well as clinical blood samples for HIV, HCV and HBV detection, with benchmarking to standard analyzers of healthcare laboratories. Thus, we demonstrated for the first time the versatility of the innovative MLFIA platform. We highlighted promising performances with the successful quantitative detection of various targets (antigens and antibodies), in different biological samples (serum and plasma), for different clinical tests (HCV, HBV, HIV).
Introduction
Immunoassays use antibodies to specifically capture, detect and quantify molecules of interest.1 Immunoassays are used for many applications in clinical diagnostics, pharmacology, food industry and even environmental monitoring.1–3 The power of immunoassays resides essentially in the specificity of antibodies, able to discriminate discrete targets (antigens) in a complex mixture of molecules. The enzyme linked immunosorbent assay (ELISA) is considered as the gold standard of immunoassays and is routinely performed in specialized laboratories for biological research, in vitro diagnostics, and biotechnology.4–6ELISAs can be performed to quantify analytes (sandwich: antigen detection) or an immune response (serology: antibody detection). The technique is sensitive, generic, robust, and cheap and is commonly used to detect infectious agents, to evaluate the humoral immune response (e.g. to detect hepatitis viruses or human immunodeficiency virus; HIV), or to quantify biomarkers (e.g. CRP and TSH) for diagnosing specific diseases (e.g., inflammation and thyroid abnormality).7–11 Nevertheless, the standard process is lengthy as analytes and detection antibodies are added successively to the functionalized surface of multi-well plates.1,12 Binding molecules of interest is diffusion-limited since the capture occurs on a surface (microwell surface), whereas the molecule of interest is distributed homogenously in a volume (sample in a microwell). Furthermore, multiple washes are needed to remove unbound molecules in the successive immunoassay steps (Fig. S1†). Finally, washing steps generate liquid biological waste that must be discarded in specific containers with a local procedure (e.g. liquids contaminated with HIV, SARS-CoV-2, etc.).
Several technological improvements have been introduced to ease and speed up agitation and washing. (i) 96 well plate based automated commercial equipment has first considerably accelerated the entire procedure, but at the expense of instrument size, hardware complexity and high purchase price. Consequently, automated ELISA has been dedicated to high-volume processing of samples per day, restricting its use in large hospitals or medical analysis facilities employing trained specialists, with an incompressible time to results of a few hours. This led to the development of (ii) faster, higher throughput, versatile, and fully automated systems such as the Architect from Abbott or the COBAS from Roche, routinely used for the detection of hundreds of immune parameters such as cardiac or infectious disease markers, and recently adapted to COVID-19 detection.13,14 They are based on the use of micrometric size magnetic particles as a functionalized surface, which replace the bottom of the well as a solid substrate (Fig. S1†). As they provide more coating surface with a higher surface-to-volume ratio (compared to coated microwells), the automated systems achieve faster detection times, and can process many more tests. Yet, they still require heavy and costly equipment and are thus limited to a clinical environment.
Several wash-free immunoassays have been reported already.15–21 In the work by Kim et al., a nanomachine that transduces a protein signal to a nucleic acid output could achieve a real-time and fast detection in whole blood and plasma.15 Akama et al. and Byrnes et al. proposed a digital ELISA for sensitive detection, relying either on nanoreactors,17 or fluorescence proximity-based digital droplet immunoassay,18 while Dixon et al. presented a peptide-based complementation system.16 Overall, these methods – relying on DNA amplification, additional PCR steps, or peptide synthesis – are good and promising early-stage approaches that, however, necessitate further development, with the challenge of having specific steps that might be difficult to automate and/or to integrate. Some homogeneous no-wash tests are already commercialized (AlphaLISA and Cisbio), respectively measuring singlet oxygen transfer and fluorescence resonance energy transfer.19–21 In both cases, the principle is based on the use of donor and acceptor beads, functionalized with antigens or antibodies. When they are at a distance, the beads do not interact. In the presence of the molecule to be detected, the beads are colocalized by the immunological complex. The proximity of the beads causes the transfer of molecules from the donor bead to the receiving bead. Relying on cumbersome equipment, these commercial no-wash tests may be more adapted for high-throughput life-science experiments, rather than for diagnostic tests.
This is why there has been a growing demand for simplified and fast immunoassays, with reduced hands-on time, that could be used in less stringent environments, particularly for low-income settings.22–24 Such a demand has been exacerbated by the pandemic and the growing need for fast assays to be performed off-lab, for timely prevention and control of the COVID spread.25 So far, two approaches dominate the immunodiagnostic point-of-care (POC) market.
(i) Lateral flow tests, also known as strip tests or lateral immunochromatographic assays, are simple paper-based tools for performing immunoassays without the need for specialized and costly equipment.26 They are easy to use, cost effective, and compatible with complex samples such as blood or urine, but are generally only qualitative with a subjective readout.24,27 (ii) To overcome these issues, microfluidic lab-on-a-chip (LOC) immunoassays for medical diagnostics have been developed, reducing the size of the biochemical device down to a single portable and disposable chip. In these systems, pipetting, reagent mixing, washing and result reading are considered as distinct steps needing specific solutions, all of which must fit on a tiny device.28 To connect all these steps, microfluidic channels transport analytes and reagents from one chamber to the next.29,30 Such systems can thus offer higher sensitivity31 but the complexity and cost of the pumps required to drive the fluids limit their widespread use to high value tests (such as emergency testing, oncology, etc.).32 For these reasons, LOCs are seldom used for massive testing of infectious diseases.
Here, we propose to simplify the biochemical protocol instead of miniaturizing existing lab-grade immunoassays, with an ELISA approach that can be used at a point of need in lab antennas for a limited number of samples. The magnetically localized and wash-free fluorescence immuno-assay (MLFIA) was developed as a generic immunoassay that eliminates most pipetting, and all mixing and washing steps.33,34 This no-wash assay allows the direct measurement of protein concentration, without requiring the separation of the target molecule from unbound molecules by washing steps. MLFIA detection principle is based on the differential measurement between the fluorescence of detection antibodies bound to magnetic nanoparticles (MNPs), specifically captured by the micro-magnets, against the residual background fluorescence of unbound detection antibodies in the sample. We obtain an immunoassay that locally concentrates targeted biomolecules (antibodies or antigens) on the micro-magnet lines, with the number of captured biomolecules quantitatively measured without any washing step. The MLFIA platform reported here is assessed for performance and demonstrated with several biological models as well as clinical samples.
Presentation of the MLFIA technology
MLFIA works as illustrated in Fig. 1. The sample (analyte) is added to the MLFIA reagent mixture, consisting of functionalized nanoparticles with antigens or antibodies to capture the target molecule and fluorescent detection antibodies. The mixture is incubated to let the antibody–antigen binding occur (Fig. 1A-1). Due to the fast diffusion and high concentration of nanoparticles and reagents, immune complexes form on the surface of the MNPs in minutes (Fig. 1A-2). 5 μL of the reaction volume is then charged into a cartridge chamber situated above a micro-magnet array consisting of stripes of alternating magnetization (100 μm large and 10 mm long) (Fig. 1A-3). This short distance leads to high magnetic field gradients acting on the MNPs. The size of the magnetic nanoparticles and the high magnetic field gradients at the junction between adjacent stripes lead to a discriminative detection of free and bound detection antibodies within minutes (Fig. 1A-4). MLFIA quantification measures the fluorescence specifically associated with the micro-magnet junctions, hence the name magnetically localized fluorescence immuno-assay or MLFIA. As described in Fig. 1B, the baseline (background signal) corresponds to the amount of free detection antibodies not adsorbed by the magnetic surface. The peak heights of the specific signal are proportional to the concentration of antibodies/antigens present in the analyte. The overall process lasts less than 15 min and does not require any washing, while giving quantitative and high sensitivity results. The different steps are further described below.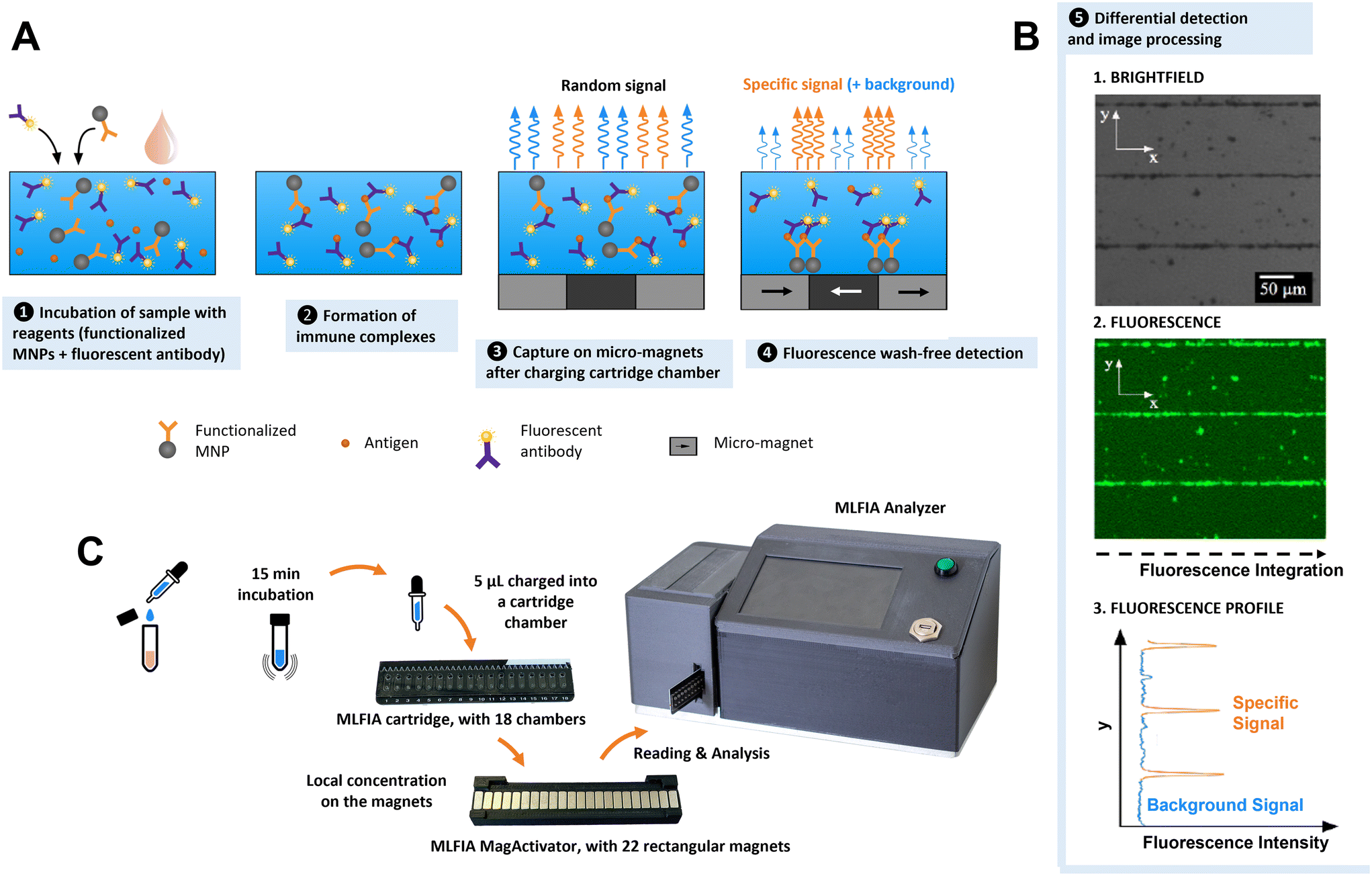 | ||
| Fig. 1 MLFIA technology and protocol. (A) MNPs, with the immune complexes bound on their surface, are captured by the micro-magnets after activation by the MagActivator. Due to the strip-shaped micro-magnet layout, the magnetically localized immune complexes can form fluorescent lines. (B) Integrating those lines yields a signal proportional to the immune complexes formed. (C) Workflow and photos of the MLFIA cartridges, dedicated array of mm sized magnets (MagActivator), and analyzer. | ||
1. Immune complexes with nanoscale magnetic particles
Compared to microscale magnetic particles, nanoscale particles have a higher diffusivity and better surface-to-volume ratio.33,34 Combined with the local capture on micro-magnet arrays through two-way coupling, simple immunologic assays are possible without washing steps.33 Such a magnetically localized immunoassay advantageously reduces the sample volume and enables a wash-free detection and a quantitative measurement within minutes (Fig. S2†).2. MLFIA 18-chamber microfluidic cartridge
To facilitate manipulation and measurement, we developed a microfluidic MLFIA cartridge with 18 chambers (Fig. 1C and S3†). The cartridge consists of 3 elements: a micro-magnet surface, a channel layer and a rigid capping layer, with more details in the Experimental section.In order to activate the micro-magnets incorporated below the chambers, the cartridge must be placed on an external device, the “MagActivator”. The MagActivator consists of an array of 22 mm-sized magnets increasing the reach of the micro-magnet stray field while magnetically saturating MNPs in the chamber (Fig. S4†). This allows to trigger the MNP capture at the junctions of the micro-magnets (Fig. 1A-4). Thus, the fluorescence signal associated with the MNPs is concentrated at the junctions between magnetic domains, where the magnetic field norm is the highest (Fig. 1B).
3. Fluorescence based detection
Immunoassays are often quantified using detection antibodies coupled to enzymes, which catalyze the generation of a detectable signal.1,35–37 In MLFIA, signals emitted by the detection antibody must remain localized to allow accurate quantification. Therefore, colorimetric enzymes are not appropriate for quantification in MLFIA, since their soluble products diffuse. Alternatively, electrochemical signals are local, but their detection requires the deposition of micro-electrodes on the micro-magnets, which is technologically challenging.38,39 Radioactive signals remain localized and detectable, but their usage generates waste and safety issues.35 Thus, fluorescence was identified as the ideal detection solution for POC integration. Fluorescence is easy to detect via an optical sensor. There is a large panel of possible fluorochromes, and a fluorescent label does not diffuse once associated with detection antibodies, allowing precise localization of the signals. We selected allophycocyanin (APC) fluorochromes due to their brightness and their emission spectrum in red, making them compatible with biological samples such as blood, and suitable for applications that require high sensitivity.4. Signal processing
In order to quantify MLFIA, the cartridge is inserted in a dedicated MLFIA analyzer (Fig. 1C and S5†). The analyzer allows the imaging of the fluorescence with an adapted optical arrangement. An algorithm, described in the Experimental section, then integrates each pixel value along the x direction (Fig. 1B). A specific fluorescence signal is obtained by integrating the surface below the curve and removing the baseline (unbound fluorescence signal). Some signals can be generated by autofluorescence of the nanoparticles in the absence of specific binding on the MNP surface.Results
Magnetic nanoparticle selection and functionalization
In the magnetic particle based MLFIA immunoassay, the immune complexes do not form on a plane surface as in classical ELISAs, but on the surface of functionalized magnetic nanoparticles. Therefore, a careful MNP selection is crucial for the assay performance. The MNPs used in MLFIA shall respect certain dimension criteria: their size must be small enough and they shall be monodispersed, to avoid sedimentation and to offer a maximised coating surface, while remaining large enough to be easily and rapidly captured within seconds by micro-magnets (Fig. S6A†). Our previous experiments showed that the particles with sizes between 100 nm and 200 nm are easily capturable.33To perform immunoassays, the particles must be functionalized. Three particle coatings commonly used for surface functionalization were considered (Fig. S6B†): protein A or protein G, streptavidin and carboxylic acids (COOH). Protein A or protein G coatings are only compatible with antibody coatings, and thus not usable for serologies. Streptavidin coatings need an extra biotinylization step before antibody or antigen functionalization, thus making the overall process lengthier. Finally, COOH coatings covalently bind to amine functions and are thus directly compatible with antibody and antigen functionalization. To develop a generic protocol adaptable to any kind of protein, we favored a particle functionalization protocol that could be molecule independent. Therefore, particle carboxylation was chosen as the functionalization method.
To facilitate the particle functionalization protocol (which includes magnetic particle separation steps), particles need to be easily dispersible, implying that no residual magnetization should remain after removing the magnetic field (i.e., they should show superparamagnetic behavior). To avoid chemical interactions, the magnetic particles should be covered with bio-compatible materials. We selected two comparable carboxylic acid-coated superparamagnetic 200 nm nanoparticles fitting the required criteria: Merck Estapor MNPs and Chemicell MNPs. Both particle types were carboxylated by the supplier, facilitating the surface functionalization process. Magnetic characterization confirmed their superparamagnetic behavior with no residual macroscopic magnetization (Fig. S7†). Characterization by Scanning Electron Microscopy (SEM) (Fig. 2A) indicated that the size definition and the monodispersity of the Merck particles were superior to those of the Chemicell ones. Therefore, we selected the 200 nm Estapor MNPs from Merck to pursue our development.
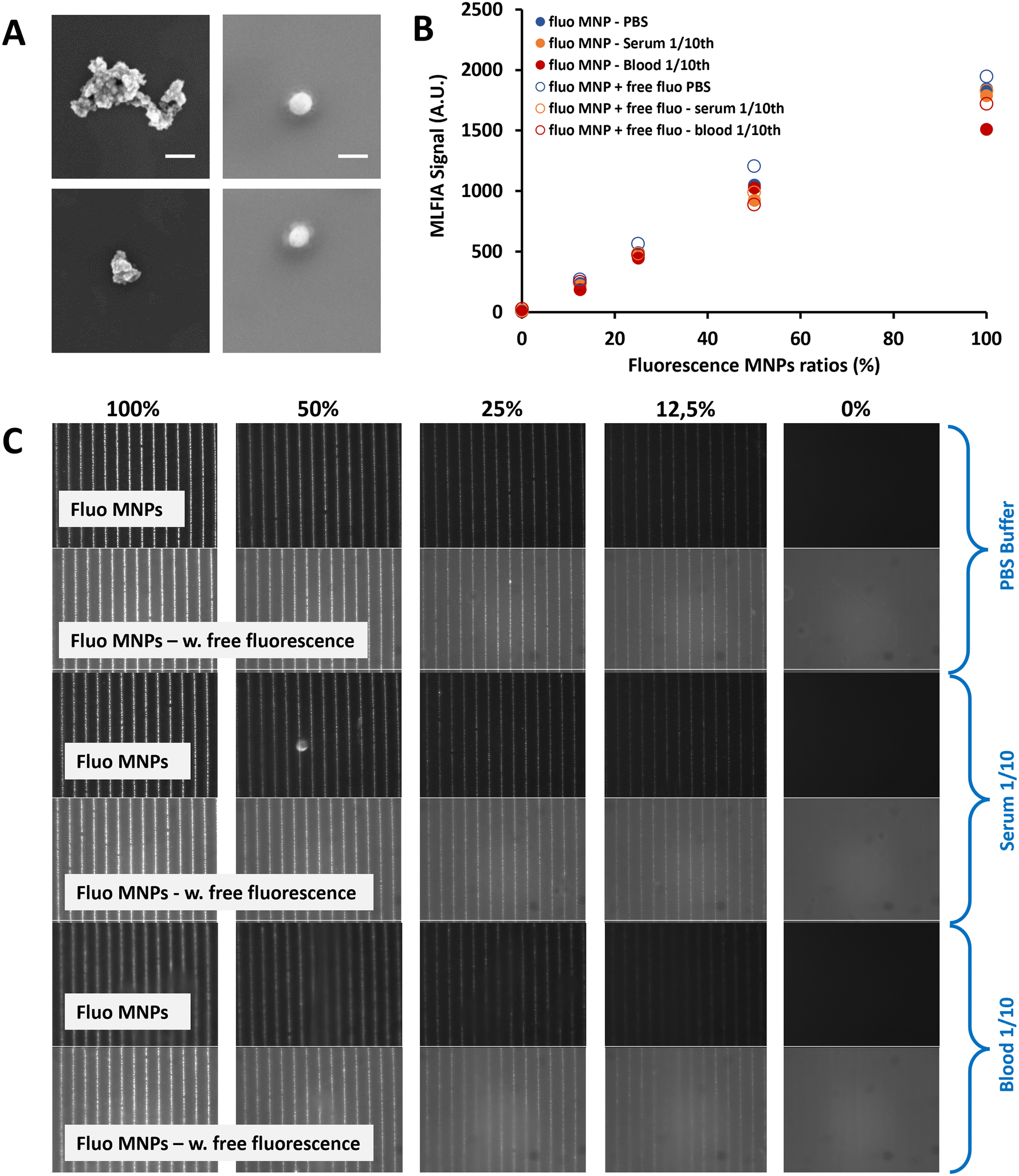 | ||
| Fig. 2 (A) SEM images from Chemicell fluidMAG 200 nm (left) and Merck Estapor 200 nm MNPs (right). Scale bar = 200 nm (2 images per MNP reference). (B) MLFIA detection for different fluorescent MNP ratios, at a constant number of MNPs, in a blank background (full circles) and fluorescent background (empty circles), respectively in PBS (blue markers), serum (orange markers) and whole blood (red markers). Experiment realized in duplicate (n = 2). (C) Typical images obtained using the MLFIA microscope at different ratios of fluorescent MNPs, without (top) or with (bottom) a fluorescent background, for PBS, serum and blood samples. | ||
MLFIA concept validation using the detection of fluorescent MNPs
To validate the MLFIA concept, we analyzed the proportionality of the MLFIA signal using the MNPs covalently coupled to fluorescent antibody anti-mouse immunoglobulin G APC (anti-mouse IgG APC). Therefore, the fluorescent MNP solution was progressively diluted (from 0 to 100%) in phosphate buffered saline (PBS) buffer and analyzed using the MLFIA system. The results are shown in Fig. 2C. It is worth noting that, in order to keep a constant amount of total MNPs, the solution was diluted in PBS buffer containing non-fluorescent MNPs coupled to ovalbumin (OVA). They do not influence the fluorescence signal. However, this allowed us to exclude any unwanted magnetic interference effects due to the reduced amount of MNPs. To allow for the MNP capture according to the MLFIA protocol, the cartridge with chambers loaded with the mentioned dilution was placed in contact with the MagActivator. The relative fluorescence intensity along the micro-magnetic stripe lines increased with the quantity of loaded fluorescent MNPs (Fig. 2C). This confirmed that the particle functionalization enabled an efficient binding of fluorescent probes (here; antibodies), and that the MNPs could be locally captured on micro-magnets. After image processing, Fig. 2B shows that the fluorescence intensity on the lines was increasing linearly (R2 = 0.99) with the amount of fluorescent MNPs in the sample (Fig. 2B, blue full circles), validating our detection method in the absence of a fluorescent supernatant, as well as its proportionality.To push this assessment forward, the same fluorescent antibody (anti-mouse IgG APC) was added to the MNP suspension (10 μg mL−1), with again a fluorescent/non fluorescent MNP ratio varying from 0 to 100%. The differential measurement of the fluorescence intensity for the MNPs localized on magnetic junctions and the non-specific signal measured away from these junctions allowed the quantification of the signal. The detection signal remained proportional (R2 = 0.99) to the quantity of the fluorescent MNPs, despite the increase of the fluorescence background (Fig. 2B, blue empty circles). Furthermore, the fluorescence signals remained similar at a 100% fluorescence ratio, with or without fluorescence in the background (1949 A.U versus 1846 A.U respectively). This validates the wash-free detection concept and demonstrates that the presence of fluorescence in the supernatant does not affect the quantification of the specific fluorescence. Further details are provided in Fig. S8.†
To validate the MLFIA concept in a complex matrix, we repeated this experiment with fluorescent MNPs in serum (orange) and veinous blood 10× diluted (red), with/without fluorescence in the matrix. This experiment is described in Fig. 2B and C and confirms the possibility to apply this assay directly to blood, with, for example, a signal of 1051 A.U. for PBS (CV of 1%), 925 A.U. for serum (CV of 7%) and 1027 A.U for blood (CV of 4%) at a 50% fluorescence ratio. More interestingly, the detection signals remain proportional to the quantity of fluorescent MNPs, despite the increase of the fluorescence background, and despite the matrix considered: R2 = 0.98, 0.99, and 0.99 in PBS, blood, and serum, respectively, with 50% background fluorescence. Relying on these positive results for fluorescent MNPs, we are strongly confident that MLFIA is compatible with complex matrices including sera and whole blood.
MLFIA validation using the detection of specific antibodies
As the signal proportionality with fluorescent MNPs was assessed, the next step was to evaluate the immunoassay concept. In the following experiment, MNPs are supposed to become fluorescent only after they form immune complexes in the presence of a target molecule.As a first biological proof of concept, we assessed MLFIA for the detection of an anti-ovalbumin monoclonal antibody produced in mouse (anti-OVA mAb). Therefore, we suspended (1) MNPs coated with ovalbumin, (2) fluorescent detection antibodies (anti-mouse IgG APC) and (3) anti-OVA mAb in a PBS buffer supplemented with bovine serum albumin (BSA) (1 mg mL−1) (Fig. 3A). The MLFIA reagent amount (see the Experimental section) was chosen according to the optimizations performed for a previously described one-wash magnetic ELISA combining MNPs and micro-magnets.34
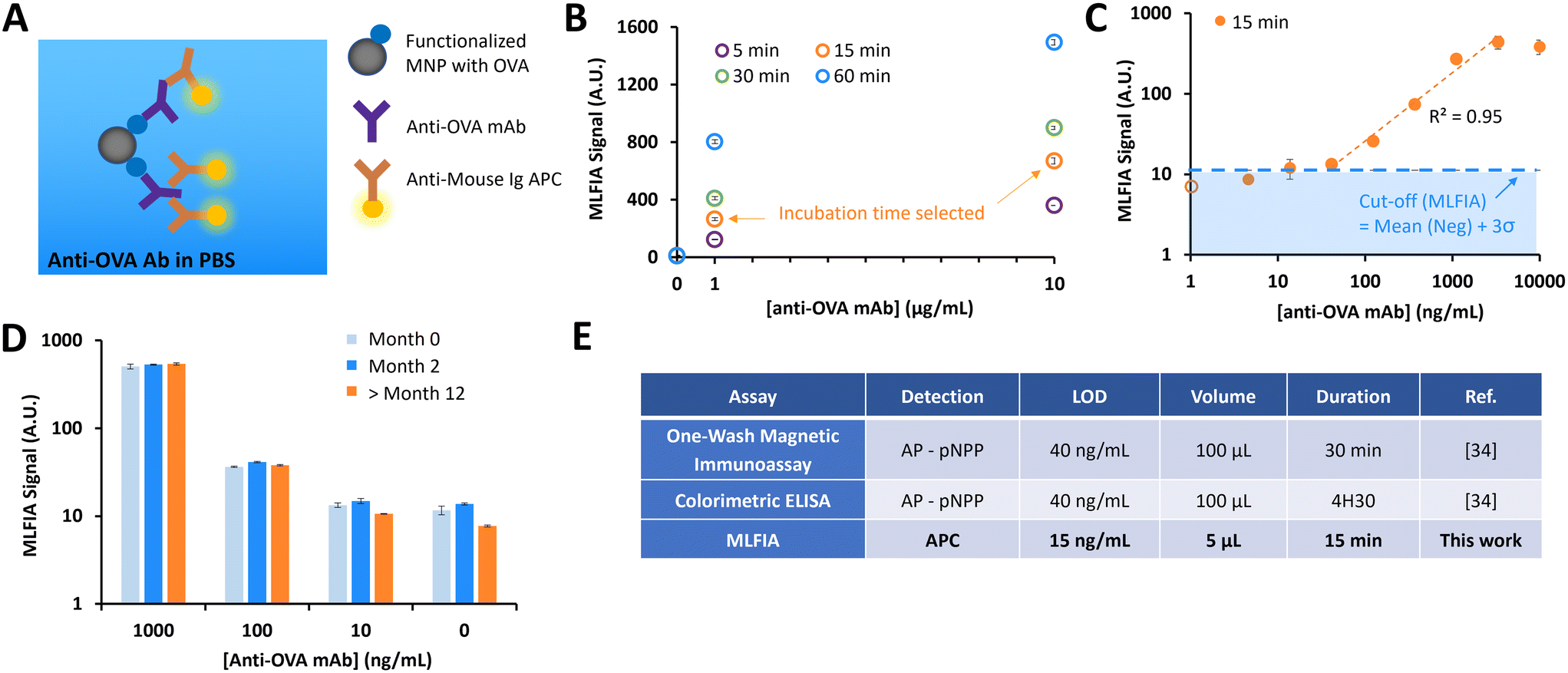 | ||
| Fig. 3 First validation of our technology on a simple biological target (anti-OVA mAb), in a PBS buffer. (A) Schematic of the reaction. (B) MLFIA signals for different incubation times; 5, 15, 30 and 60 min for the detection of 1 and 10 μg ml−1 of anti-OVA mAb. (C) MLFIA signals, linear range of detection and LOD. The empty circle represents the value at 0 ng ml−1. (D) Stability of functionalized OVA-MNPs over time, with MLFIA signals for different concentrations of anti-OVA Ab, at month 0, month 2, and beyond month 12 following the MNP functionalization. (E) MLFIA performance versus two different immunoassays under similar experimental conditions, for the detection of anti-OVA mAb in PBS. B–D experiments were realized in duplicate (n = 2). | ||
We first compared the detection signal for incubations of 5 min, 15 min, 30 min and 60 min (Fig. 3B). The detection kinetics were rapid; even an incubation of only 5 min was enough to detect signals from the solution of 10 μg mL−1 anti-OVA mAb, but also from the 1 μg mL−1 solution. For practical reasons, we set the incubation time at 15 min.
We then evaluated the analytical sensitivity of the detection, for a concentration of anti-OVA mAb in a buffer varying from 1 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ng mL−1 (Fig. 3C). The linear range of the detection was between 40 ng mL−1 and 3300 ng mL−1. To assess the limit of detection (LOD), we processed blank buffer samples (N = 3) and used the mean value + 3σ as a cut-off to define a sample as positive. Here, the LOD in this experimental configuration was 15 ng mL−1.
000 ng mL−1 (Fig. 3C). The linear range of the detection was between 40 ng mL−1 and 3300 ng mL−1. To assess the limit of detection (LOD), we processed blank buffer samples (N = 3) and used the mean value + 3σ as a cut-off to define a sample as positive. Here, the LOD in this experimental configuration was 15 ng mL−1.
Finally, we assessed the stability of the MNP coating with OVA over time. We compared the detection signals for OVA functionalized MNPs stored at 4 °C for 1 week, 2 months and more than a year (Fig. 3D). Comparable results were obtained, independent of storage time, emphasizing the long-term robustness of our grafting method.
MLFIA comparison with previously developed immunoassays
We compared the MLFIA with “standard” immunoassays previously developed in-house, using the same reagents (Fig. 3E). The performance indicators (LOD and dynamic range) of MLFIA were compared to those of a colorimetric ELISA and a one-wash magnetic immunoassay.34 All assays were performed using identical concentrations of OVA coating (10 μg mL−1) for the functionalization of multi-well plates or MNPs.With an incubation time of 15 min, MLFIA gave an LOD of 15 ng mL−1 for anti-OVA mAb in PBS. In comparison, we obtained an LOD of 40 ng mL−1 with the one-wash magnetic immunoassay as well as with the colorimetric ELISA. Thus, we improved the LOD while reducing the sample volume, the quantity of reagents, and the duration. Furthermore, the protocol is greatly simplified in the absence of washing steps.
MLFIA for HCV antibody detection in blood samples
Previous experiments considered simple biological models in PBS buffer.34 In this experiment, we used the MLFIA protocol obtained from the ovalbumin model to the detection of HCV antibodies. To do so, we functionalized the MNPs with a recombinant HCV fusion protein (NS3/core/NS4/NS5) and used as fluorescent detection antibodies, anti-human immunoglobulin coupled to APC (anti-human IgG APC) (Fig. 4A).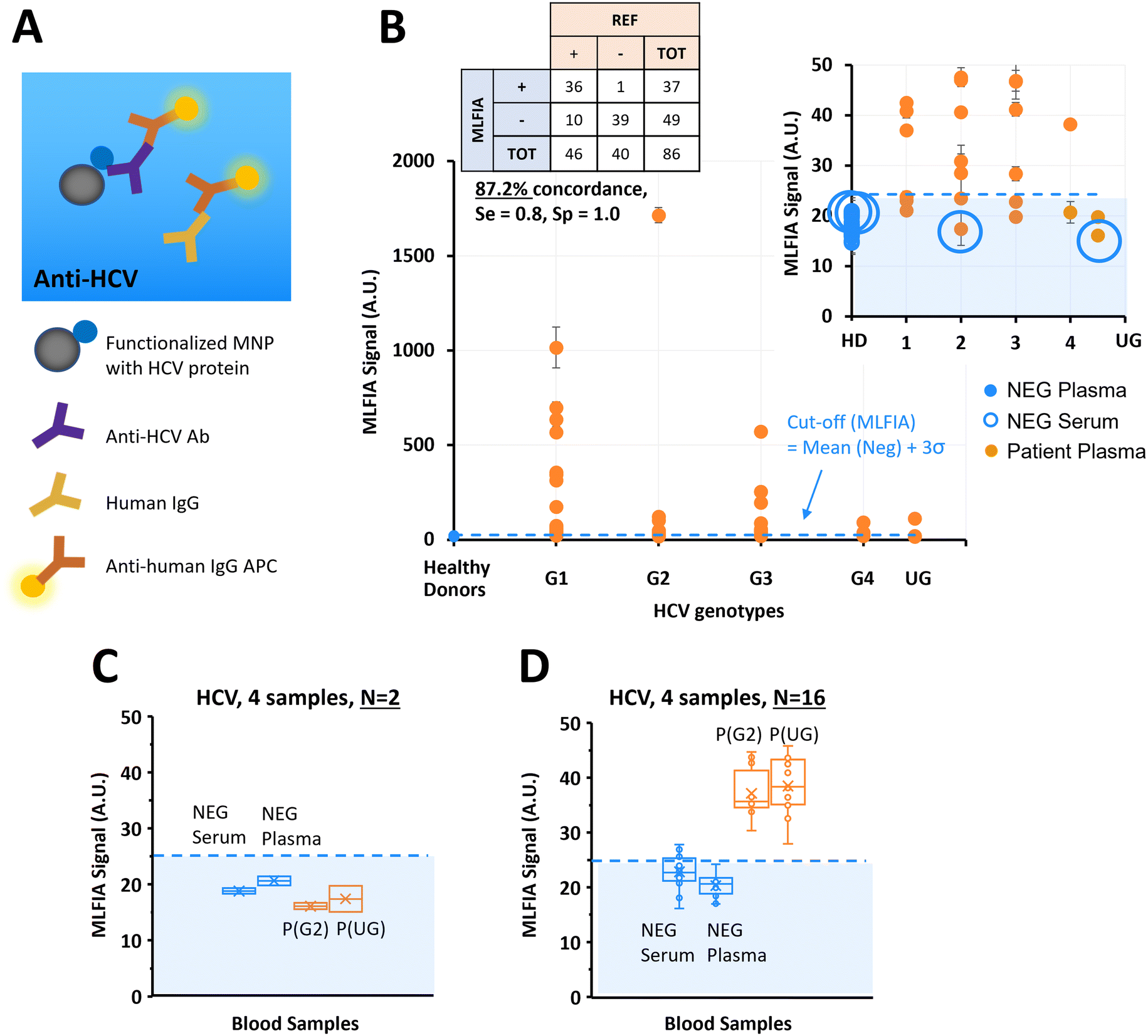 | ||
| Fig. 4 Validation of our technology on a clinical target; HCV. (A) Schematic of the biological reaction model. (B) Benchmarking of MLFIA versus blood laboratory test for HCV antibody detection in plasma and serum samples, with 40 negative control samples (blue markers) and 46 samples from patients (orange markers) of different HCV genotypes indicated by G1, G2, G3, G4 and UG for HCV genotypes 1, 2, 3, 4 and undetermined genotype. Experiment realized in duplicate (n = 2). (C and D) To illustrate variability, 4 samples were selected for additional processing in replicate (n = 16) and compared with the values obtained in experiment B with n = 2; 1 NEG plasma, 1 NEG serum, and 2 samples from patients. For all benchmarking experiments, the MLFIA cut-off is defined as mean (NEG controls) + 3σ, and indicated with a blue dotted line. | ||
To demonstrate MLFIA applicability to the detection of a clinical target in a real sample, we processed 46 plasma samples of HCV patients, and 40 negative control samples from 20 healthy donors (1 serum and 1 plasma for each of the 20 healthy donors), with HCV genotypes provided and characterized by a blood bank (Fig. 4B).
All 40 negative samples were identified as negative by MLFIA, except one plasma sample which had a much higher MLFIA signal. Because the serum or plasma for this same blood drawing was however clearly negative, we assumed a user or machine error, and put this sample value aside (as false positive) in our cut-off calculation.
For the infected patient samples, 36 out of the 46 positive samples were detected as positive by MLFIA, using a threshold defined by the mean MLFIA signal of negative samples + 3σ. Interestingly, all HCV genotypes were similarly detected. This confirmed that MLFIA could detect antibodies in blood samples, despite the presence of other antibodies without interfering with the specific detection. When compared with blood laboratory testing, MLFIA achieves an overall concordance of 87.2%, a diagnostic sensitivity of 0.8, and a specificity of 1.0.
To understand further the discordance between the MLFIA results and the results provided by the laboratory, we re-processed 4 samples, which are indicated with large blue circles in Fig. 4B; the 2 control samples with the highest MLFIA signal (1 serum and 1 plasma) and the 2 patient samples with the lowest MLFIA signal. In the first experimental batch, we processed all the samples at once, with 15 min incubation, all within 10 cartridges for N = 2 replicate. Fig. 4C presents the values initially obtained for these 4 samples; the 2 patient samples are below the MLFIA cut-off, and thus are considered as negative for the presence of anti-HCV antibodies. However, when re-processing these 4 samples within 4 different cartridges and for N = 16 replicate, the 2 patient samples are above the MLFIA cut-off (Fig. 4D) and become positive to anti-HCV antibodies. Fig. 4D however shows a large spread of data points for a single sample (CV ranging from 9.44% to 13.65%, with in particular one negative serum varying from 16.11 AU to 27.83 AU, and a positive plasma varying from 27.94 AU to 45.77 AU), highlighting a reproducibility issue between cartridges, that is due to the early stage of technological development.
MLFIA for HBV HBsAg detection in blood samples
After demonstrating the applicability of MLFIA to antibody detection, we demonstrated its applicability to antigen detection. In this experiment, MLFIA is used for the detection of hepatitis B surface antigen (HBsAg) in blood samples. To do so, we functionalized the MNPs with an anti-HBsAg antibody and used as fluorescent detection antibodies, a second anti-HBsAg coupled to APC (Fig. 5A).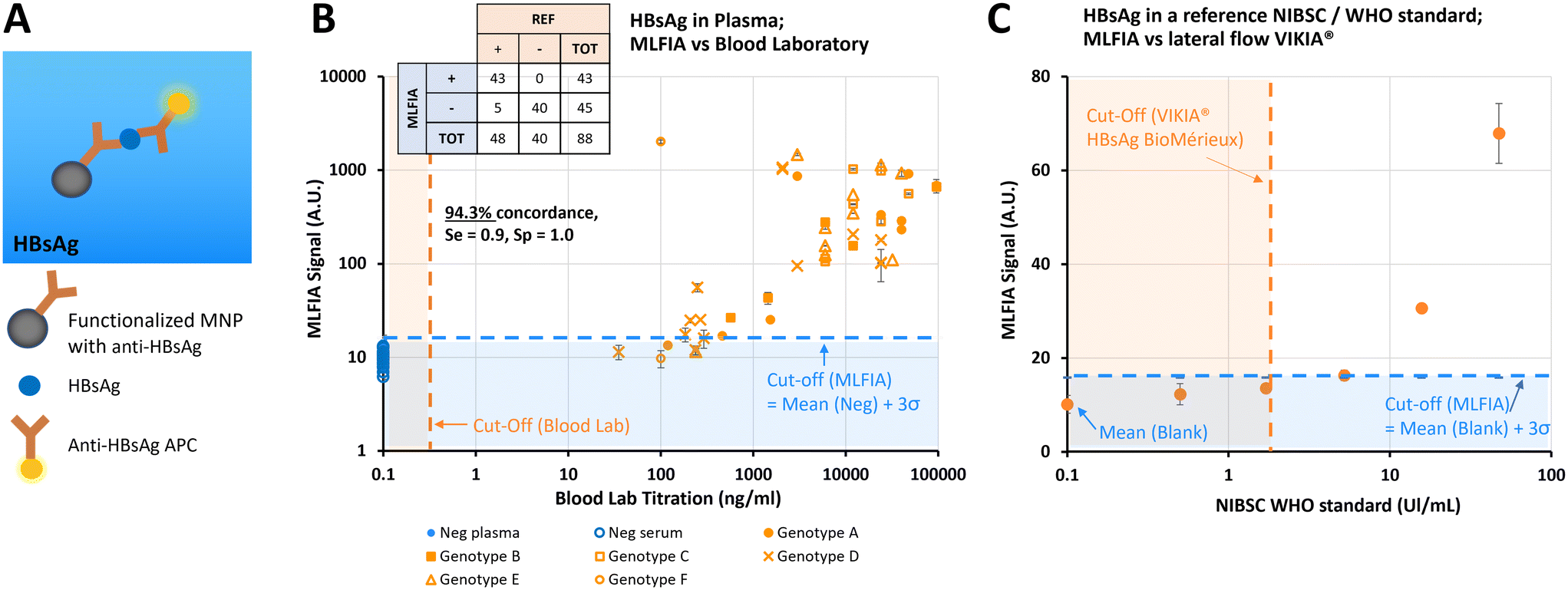 | ||
| Fig. 5 Validation of our technology on a clinical target; HBsAg. (A) Schematic of the biological reaction model. (B) Benchmarking of MLFIA versus blood lab test for HBsAg detection in plasma and serum samples, with 40 negative control samples (blue markers) and 48 samples from patients (orange markers). Blood lab cut-off is indicated with an orange dotted line. Experiment realized in duplicate (n = 2). (C) Benchmarking of MLFIA versus the commercial lateral flow VIKIA test, from BioMérieux. The VIKIA cut-off is indicated with an orange dotted line. A blue dotted line represents the blank value for MLFIA. For both benchmarking experiments, the MLFIA cut-off is defined as mean (NEG controls) + 3σ, and indicated with a blue dotted line. Experiment realized in triplicate (n = 3). | ||
We processed 48 plasma samples of HBV infected patients, and 40 negative control samples from 20 healthy donors (20 serum and 20 plasmas, from each of the 20 donors), all provided and characterized by a blood bank (Fig. 5B).
All negative samples were identified as negative by MLFIA, while 43 out of the 48 positive samples were detected as positive by MLFIA, using a threshold defined with the negative samples. This corresponds to an overall concordance of 94.3%, a diagnostic sensitivity of 0.9, and a specificity of 1.0.
However, the signal was not entirely proportional to the blood laboratory titration (Fig. 5B), which was expected as the sample matrix differs from one donor to another, with different subtypes and genotypes of HBsAg. This confirmed however that MLFIA can detect all the clinical isoforms of HBsAg successfully.
Finally, in Fig. 5C, we compared the MLFIA HBsAg analytical sensitivity with a recognized commercial lateral flow test (VIKIA from BioMérieux), using an HBsAg reference sample accredited by the World Health Organization (WHO). The reference sample was diluted from 0.5 to 46 UI mL−1. Using the cut-off defined with the samples from the healthy donors, MLFIA can detect HBsAg from a concentration as low as 5 UI mL−1. The dots at 0.5 and 1.7 UI mL−1 are below the defined cut-off, but well above the blank value. In comparison, the cut-off of the VIKIA is 2 UI mL−1.
MLFIA for HIV antibody detection in blood samples
After demonstrating the applicability of MLFIA to antibody and antigen detection, we performed another antibody detection. In this experiment, MLFIA is used for the detection of HIV antibodies in blood samples. To do so, we functionalized the MNPs with an HIV recombinant gp41 protein and used as fluorescent detection antibodies, anti-human IgG APC (Fig. 6A).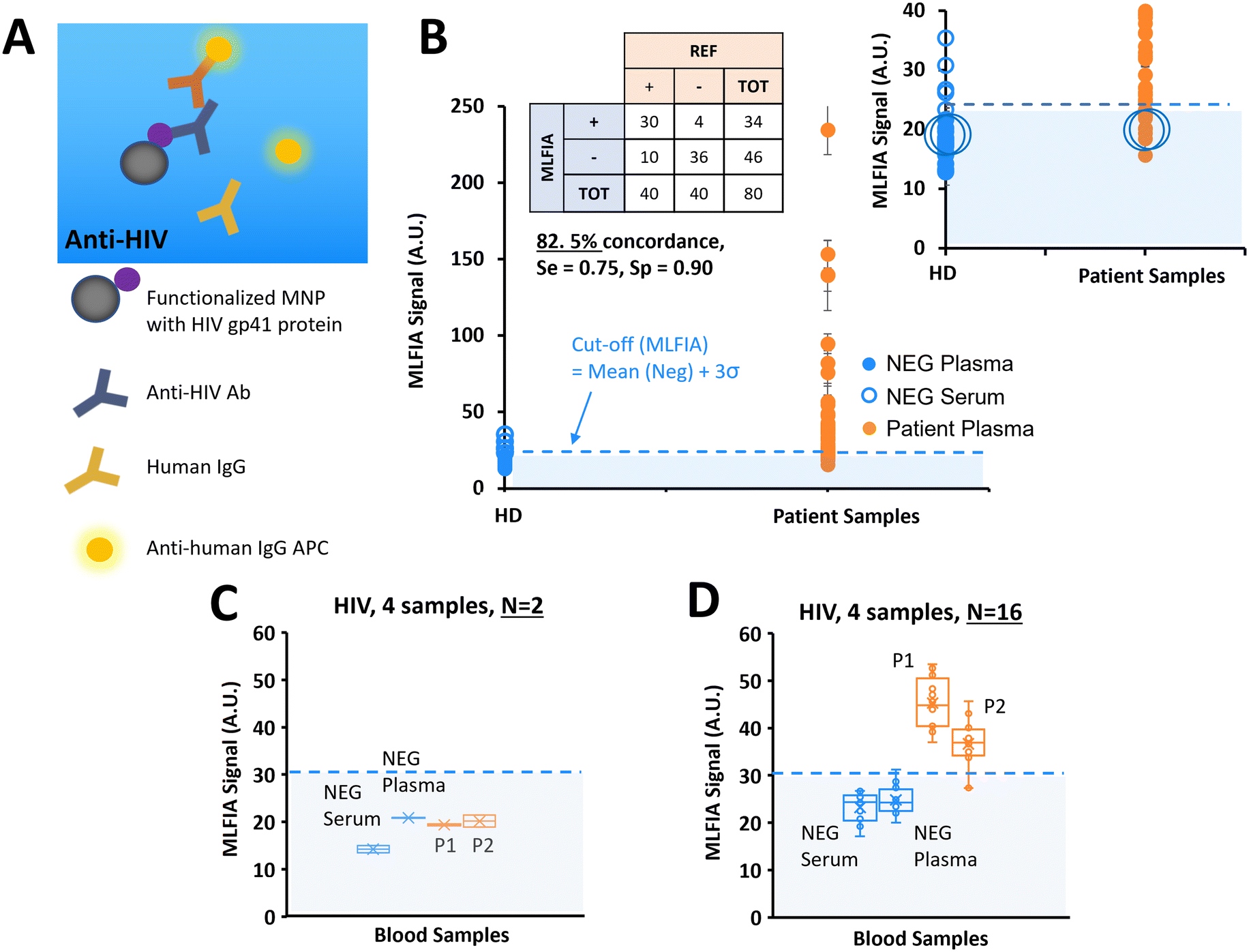 | ||
| Fig. 6 Validation of our technology on a clinical target; HIV. (A) Schematic of the biological reaction model. (B) Benchmarking of MLFIA versus blood lab test for HIV detection in plasma and serum samples, with 40 negative control samples (blue markers, 20 plasma, 20 serums) and 40 plasma samples from patients (orange markers). Experiment realized in duplicate (n = 2). (C and D) To illustrate cartridge variability, 4 samples were selected for additional processing in replicate (n = 16) and compared with the values obtained in experiment B with n = 2; 1 NEG plasma, 1 NEG serum, and 2 samples from patients. For all benchmarking experiments, the MLFIA cut-off is defined as mean (NEG controls) + 3σ, and indicated with a blue dotted line. | ||
We analysed 40 plasma samples from HIV infected patients, and 40 negative control samples from 20 healthy donors (1 serum and 1 plasma from each of the 20 donors) with HIV genotypes provided and characterized by a blood bank (Fig. 6B).
36 negative samples were identified as negative by MLFIA, and 4 were considered false positive. The 2 highest values correspond to the plasma and serum samples of the same patient. Because they appeared as outliers when plotting negative samples with a boxplot, we put these 4 sample values aside in our cut-off calculation. For the samples from the patients, 30 out of the 40 positive samples were detected as positive by MLFIA. When compared with blood laboratory tests, we have an overall concordance of 82.5%, a diagnostic sensitivity of 0.75, and a specificity of 0.90.
As before, for the detection of HCV antibodies, we re-processed 4 samples, labelled with large blue circles in Fig. 6B; 2 control (NEG) samples (1 serum and 1 plasma) and 2 samples from the patients (P1 and P2) were identified as negative by MLFIA. Fig. 6C presents the values initially obtained for these 4 samples in duplicate; the 2 samples from the patients are below the MLFIA cut-off. However, when re-processing these 4 samples within 4 different cartridges and for N = 16 replicate, the 2 samples from the patients are above the MLFIA cut-off (Fig. 6D) and become positive, as for the HCV experiment.
Discussion
We developed the magnetically localized and wash-free fluorescence immunoassay (MLFIA), using functionalized MNPs, micro-magnets and a localized fluorescence detection. We validated the MLFIA concept, first (i) by detecting and quantifying fluorescent MNPs, then (ii) by detecting and quantifying anti-OVA mAb. We then successfully compared the detection of anti-OVA mAb in PBS with immunoassays using the same reagents: MLFIA presents a better analytical sensitivity with an LOD of 15 ng mL−1 in PBS, while using a lower volume of the sample and reagents, and also being faster, enabling detection in 15 min. We then demonstrated the versatility of the MLFIA platform with the detection of various targets (antigens and antibodies), in different environments (serum and plasma from blood samples), for 3 major viral infections in humans: HCV, HBV, and HIV. Benchmarking showed a correlation with standard analyzers of healthcare labs (Abbott Prism or Architect) with the following concordances: 87.2% for HCV, 94.3% for HBsAg, and 82.5% for HIV. Thus, we demonstrated for the first time the successful detection of antigens or antibodies, without washing steps, even in a medium such as blood plasma and serum. MLFIA provides a user-friendly simplified process and exhibited a very promising diagnostic sensitivity and specificity, with some reproducibility issues that can be easily addressed in the future.The MLFIA technology relies on the use of highly diffusive functionalized superparamagnetic nanoparticles as a support for surface reactions, highly effective striped micro-magnet traps and the elimination of all washing steps. Instead of a simple supernatant measurement as shown by Kim et al.40 and Wosnitza et al.,41 MLFIA allows for a more robust differential measurement of specific and background signals, performed on small sample volumes and without agitation. MLFIA is faster than non-magnetic homogenous immunoassays,19 and simpler than microfluidic immunoassays.42 The absence of washing steps makes the overall protocol simple, while enabling a simpler fluidic design for the cartridge, without the need for a wash buffer to be pumped through the chip. It also enables the processing of very small volumes of patient samples for a less invasive assay, of reagents for a lower reagent consumption and lower cost per assay. Besides, we use a generic protocol with a simple grafting – that can be easily adapted for different assays (sandwich and serology) and even matrices (PBS, serum, plasma) – and a generic reading and analysis method, easily and quickly applicable in the future to the development of new PoC assays.
When compared to commercialized homogeneous no-wash tests (AlphaLisa, Cisbio),19–21 the bead size (250–350 nm diameter) and reaction volume (5 μl) are comparable. The MLFIA assay duration, however, is faster (>1–2 h with the incubation included for these commercial assays, versus 30 min for MLFIA). Their analyzer (i.e. a multimode microplate reader) is more cumbersome, both in terms of overall size and weight (around 10 kg, versus 3 kg for MLFIA platform) and probably more expensive. Overall, these commercial no-wash tests seem to be more adapted for high-throughput life-science experiments, rather than for diagnostic tests. The MLFIA platform, on the other hand, being more compact, easily transportable, and probably cheaper, would be more adequate for small labs which do not require high throughputs and cannot purchase such big equipment.
The absence of washing steps reduces greatly liquid waste and therefore attracts great interest to analyse blood samples of infected people. Thus, we addressed the capacity of MLFIA to detect 3 major viral infections, namely HIV and hepatitis virus B and C. For both HIV and HCV infections, the specific antibodies were measured, whereas for HBV infections, the HBsAg was quantified accordingly to the current assays. We determined a variability limitation for this first platform, which was confirmed by processing the same samples on more cartridges. Several causes for such variability have been identified and classified and are illustrated in Table S1;† the presence of artefacts on the image (bubbles, dusts, fluorophore aggregates) causes undesired fluorescence heterogeneity, abnormal capture phenomena, cartridge optical misalignment, and suboptimal optical focusing. Different approaches can be used in parallel to address this variability issue. (i) We first need to stabilize the consumable design, to set-up an automated and robust fabrication process, and to perform its assembly in a clean-room environment. (ii) The cartridge interfacing with the analyzer is crucial as well, with the need for a tighter alignment with the optical module, from a cartridge to the other. This may be implemented with an x–y–z autofocus module. (iii) Adding an integrated mixing step would be beneficial as well to avoid detection antibodies aggregation leading to fluorophore aggregates. (iv) We could also increase the incubation time for a higher signal gain and for experimental convenience. Indeed, in this study, we processed 86 clinical samples in a single session, with a 15 min incubation. However, such a short incubation time made it difficult to handle well such a number of samples from patients, inducing a time effect on the results, between the first and last samples. This would be obviously different in a clinical scenario where not that many samples from patients would be analysed in one session with only one analyzer. (v) The capture of the nanoparticles is not entirely reproducible from one chamber to another. We are working on micro-magnet optimization to make the capture more localized and more reproducible, to ultimately improve robustness and thus analytical and diagnostic sensitivity.
The level of diagnostic sensitivity was variable from one test to the other, with 0.8 for HCV, 0.9 for HBsAg, and 0.75 for HIV, respectively. For HBsAg, we detect an antigen. This is the easiest scenario, with no interfering antibodies, as the antibodies used are only specific to HBsAg, thus providing images with a strong contrast. For the serological cases of HIV and HCV, however, there is a competition between the target antibody in the samples from patients (anti-HIV or HCV) and all the other antibodies of the patient's plasma/serum for the binding of the labelled detection antibody (anti-human IgG APC). An alternative would be to use an antigen for the detection, tagged with APC, rather than an anti-human antibody. As a result, we capture fewer specific antibodies against HIV or HCV, resulting in images that have a lower contrast, the most challenging situation for our image analysis algorithm. To increase the detection signal, we could also add other antigenic targets on the MNPs, for example to capture other antibodies produced by the patient in response to an HIV infection (gp120, P24).43,44 For HCV, we already used a fusion protein that contains several proteins of VHC (NS3/core/NS4/NS5), i.e., different epitopes to enhance the probability of recognition by different antibodies.45,46
Ultimately, all the characteristics of MLFIA make it an ideal candidate for POC technology. From the earliest phases of the MLFIA technology design, we kept in mind the attributes defined by the World Health Organization (WHO) for disease control by point-of-care testing, namely the ASSURED criteria: affordable, sensitive, specific, user-friendly, rapid and robust, equipment free, and deliverable to end-users.47 MLFIA is a versatile, inexpensive, and easy-to-handle technology that could be operated by untrained personnel. The sample droplet could be deposited on a surface or in a capillary channel and immediately analysed, with no further mixing, nor pumping, allowing for cheap and disposable assay. It has potential for numerous applications and different surroundings, from in-the-field infectious disease testing in developing countries, emergency diagnostics, and routine home-based monitoring, to food industry routine tests or even biodefense applications.
The analyzer
The currently used analyzer is a prototype requiring external computation and a power source. Moving MLFIA towards POC application, we could consider a smartphone-based detection approach since smartphones are ubiquitous even in remote areas, usually include high-quality image sensors, are easy to program and use, and come with standard communication ports. Another possibility, more suitable than “volatile” smartphone technology, could be a dedicated portable MLFIA analyzer, designed specifically to be fully automated, autonomous, robust, and reliable. Pricing-wise, the current analyzer being simply composed of epifluorescence optics, a mechanical conveyer and embedded electronics, its cost structure will remain competitive compared to existing 96 well plate analyzers, and thus targeted to POC applications.The consumable
Another strategic step towards a POC device would be the re-design of the MLFIA cartridge. While this multi-chamber cartridge performs well, it is not suitable for POC diagnostic purposes as it still requires a centrifugation step to extract plasma from whole blood, micro-pipettes, and a laboratory facility, with equipment and skilled technicians, while enabling only one type of assay. A cartridge enabling for multiple single assays could be a more robust, reliable, and industrially feasible approach compared to developing multiplex assays. Instead of measuring multiple parameters in a single chamber, each parameter could be read in its own chamber with lyophilized probes, with several chambers per system and a single filling port to directly bring in the sample. The design of the cartridge could be simple enough to envision a low fabrication and assembly cost, which is facilitated also by the possibility to use common manufacturing plastic materials (such as PMMA). The pricing structure “per assay” must consider the reagent consumption as well, that is significantly reduced. As an illustration, the quantity of the reagent needed for MLFIA during OVA assay was more than 15 times less than that for ELISA. Applying the MLFIA to capillary blood analysis would be a crucial step as well, with most probably a dilution step to reduce interference of the numerous blood cells with the optical measurement and facilitate the flow of the sample into the chip. The possibility to process small volumes of blood samples also opens the possibility to apply such an assay to rare samples, including but not limited to new-born testing.48Experimental
Particles, reagents, and blood samples
200 nm polystyrene MNPs, containing iron oxide superparamagnetic nanoparticles and coated with carboxylic acid, were purchased from Merck GmbH (M1-020/50). 200 nm MNPs, covered with a hydrophilic polymer containing iron oxide and coated with carboxylic acid, were purchased from Chemicell GmbH (4115).Bovine serum albumin (BSA) (A4503), ovalbumin (A5503), mouse monoclonal anti-ovalbumin (A6075), mouse serum (M5905), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) (E6383), N-hydroxysuccinimide (NHS) (130672), MES (M3671), PBS (P4417), and Tween (P1379) were all purchased from Sigma Aldrich. Fluorescent anti-mouse immunoglobulins G (115-136-072) and anti-human immunoglobulins G antibodies (109-136-127) were purchased from Jackson Immuno. HBsAg antibodies (CK1374) were purchased from Aalto Bio Reagents, HCV NS3/core/NS4/NS5 recombinant protein from Tebu-bio (R01600) and HIV recombinant Gp41 (IBAG48) protein from Infinity Biomarkers. International HBV HBsAg standard was provided by NIBSC (12/226).
Blood donors were recruited, and informed consent was obtained according to the appropriate clinical protocols from the National French Blood Bank (EFS Rhônes Alpes for the healthy donors, and EFS Tour for the non-healthy donors). Blood samples were drawn from the volunteers with no known illness or fever at the time of blood draw. For each donor, peripheral blood was collected and decanted in dry tubes to obtain sera and in EDTA-coated tubes to obtain plasma. The blood samples were analyzed in central blood labs using chemiluminescence immunoassays (Abbott Prism or Architect) and the results were used as a benchmark for MLFIA.
MNP functionalization and characterization
Cartridges
The cartridges used for the MLFIA approach were made of four different layers (Fig. S3†): (1) a 1 mm thick polymer layer, forming a rigid base, (2) a 12 μm thick magnetic layer, consisting of 1 μm anisotropic hard ferrite particles embedded in a polymer matrix with a volume fraction of about 50%. The hard ferrite particles were oriented in-plane prior to the reticulation of the polymer matrix, and subsequently magnetized in-plane to form stripe-like magnetic domains with a width of 100 μm. (3) A 250 μm thick polymer layer with 18 die-cut 7 × 2.4 mm chambers. (4) The 18 chambers covered by a 1.5 mm thick transparent PMMA layer, with 18 filling ports and 18 venting ports (all laser cut), allowing the chambers to be filled with a pipette. One length of the PMMA is cut with gears in order to move the cartridge around in the analyzer with the motor. All components were assembled using acrylic adhesive, and being entirely inert, they could be stored indefinitely under normal storage conditions.MagActivator
The MagActivator consisted of 22 rectangular magnets, 10 × 4 × 1 mm, made of neodymium (NdFeB) and purchased from Supermagnete (Q-10-04-01-N). The 22 magnets were embedded in a dedicated black plastic support, 3D printed using a STREAM 30 Pro MK2 (Volumic). The magnets were placed head-to-tail to guarantee a homogeneous magnetic field, similar between the extremities and centre of the cartridge (Fig. S4†).Localised and wash-free immunoassay
Functionalised magnetic nanoparticles (10 μg mL−1 of coated protein) and detection antibodies of interest (2 μg mL−1) were mixed with 20 μL of sample in a microtube. After a 15 min incubation at room temperature, 5 μL of the mixture was injected in one well of an 18-well cartridge, with repetition of this operation as needed for duplicate. Once full, the entire cartridge was deposited on the MagActivator for 1 min to activate MNP capture (Fig. 1). The cartridge was then inserted into the MLFIA laboratory analyzer prototype.MLFIA analyzer
The MLFIA analyzer consists of a miniaturized epifluorescence microscope, adapted for far-red fluorescence imaging to be compatible with the spectrum required by APC. It is composed of an optical module, a mechanical module, an electronic module and a 5-inch touchscreen (Fig. S5†). The optical module is composed of a cube (CM1-DCH/M, from Thorlabs) including a fluorescence filter set (39007, from Chroma) and a dichroic mirror (AT655dc, from Chroma), an illumination module composed of a condenser lens (ACL2520U-A, from Thorlabs) and a red LED (SP-01-R5 LED, 637 nm, from Luxeon), an objective (Ricoh FP-RR27, from Stemmer) and a camera (Mako G125, from Edmund Optics). This optical module enables an image magnification of ×3.5. The mechanical module is composed of a 3D printed rail and a motor to allow the automatic displacement of the cartridge under the optical module thanks to the gear of the cartridge. One after the other, the 18 chambers are positioned under the optical axis and 3 pictures are taken per chamber, at 3 different exposure times (600, 700 and 800 ms), so that the mean of the 3 data can define the final signal. The field of view of the images acquired by the analyzer is 1.1 × 1.5 mm. The different modules are positioned in 3D printed structures, all printed with the STREAM 30 Pro MK2 (Volumic), with a stage to position the optical module, and a casing including a dedicated spot for the touchscreen.Image processing
Pictures are processed using an algorithm based on OpenCV 4.5 and MKL 2021. This processing includes several steps: first, the filtering of any stains, bubbles, or artifacts on the picture with traditional techniques. Then, the location and orientation of the magnetic lines were determined by convolution with a Dirac comb. Orientation was then corrected, if necessary, to ascertain that the lines were vertical in the processed picture. Lastly, this picture was integrated along the vertical axis into a 1D profile, on which the specific signal could be discriminated from the background using the positions previously identified (Fig. S8†).Author contributions
SD, MF, PK, FB, OC, NMD, PNM, TD, MW, CC and AL contributed to the design of the technology. Specifically, MF, PK, NMD, OC, and TD designed the micro-magnetic sources and MagActivator, MF, OC, SD, CC and AL contributed to the cartridge design, PK, SD, OC, DK, and FB contributed to the design of the analyzer and image processing, and SD, PNM, FB, and MW contributed to the design of the immunological MagIA protocol. PB, OR, VM and SD developed the OVA, HIV, HCV, and HBsAg immunoassays and performed the biological experiments. MPBP, HFH, PH and HP provided the biological samples and assisted SD in the clinical interpretation of the results, to assess the versatility of the technology and its first application fields (HIV, HBV, HCV). SD wrote the manuscript, with the support of all the authors. All the authors reviewed the manuscript.Conflicts of interest
The authors disclosed the following conflicts of interest: some of the authors (SD, PK, MF, FB, OC, PH, DK, PB, OR, VM) have financial interests in MagIA, the company spun off from the MLFIA technology, and in the intellectual property described herein.Acknowledgements
We thank our clinical advisors, Dr Sylvie Larrat and Dr Patrice Faure, the nurses, and all blood donors for their contributions toward blood and tissue sample collection, for clinical samples provided by the EFS Tour and EFS Grenoble. In terms of funding, the LANEF funded SD's PhD. The French Ministry of Research and Education funded MF's PhD. The SATT Linksium, BPI France and Région Auvergne-Rhône-Alpes funded the research presented in the project.Notes and references
- D. Wild, The Immunoassay Handbook. Theory and Applications of Ligand Binding, ELISA and Related Techniques, Newnes, 2013 Search PubMed.
- W. Scheller, U. Wollenberger, A. Warsinke and F. Lisdat, Curr. Opin. Biotechnol., 2001, 35–40 CrossRef PubMed.
- Y. Song, Y.-Y. Huang, X. Liu, X. Zhang, M. Ferrari and L. Qin, Trends Biotechnol., 2014, 132–139 CrossRef CAS PubMed.
- R. M. Lequin, Clin. Chem., 2005, 51(12), 2415–2418 CrossRef CAS PubMed.
- S. Aydin, Peptides, 2015, 72, 4–15 CrossRef CAS PubMed.
- M. Alhajj and A. Farhana, StatPearls, 2022 Search PubMed.
- S. Hosseini, P. Vázquez-Villegas, M. Rito-Palomares and S. O. Martinez-Chapa, Enzyme-Linked Immunosorbent Assay (ELISA), Springer, 2018, pp. 67–115 Search PubMed.
- D. Desai, G. Wu and M. H. Zaman, Lab Chip, 2011, 11(2), 194–211 RSC.
- S. K. Vashist and J. H. Luong, Handbook of immunoassay technologies: approaches, performances, and applications, Academic Press, 2018 Search PubMed.
- T. S. Hnasko and R. M. Hnasko, Methods Mol. Biol., 2015, 1318, 87–96 CrossRef PubMed.
- R. O'Kennedy and C. Murphy, Immunoassays: development, applications and future trends, CRC Press, 2017 Search PubMed.
- J. Dong and H. Ueda, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9(5), e1457 Search PubMed.
- S. Meschi, F. Colavita, L. Bordi, G. Matusali, D. Lapa, A. Amendola and C. Castilletti, J. Clin. Virol., 2020, 129, 104539 CrossRef CAS PubMed.
- C. S. Lau, S. P. Hoo, Y. L. Liang, S. K. Phua and T. C. Aw, Pract. Lab. Med., 2021, 25, e00230 CrossRef PubMed.
- D. Kim, O. B. Garner, A. Ozcan and D. Di Carlo, ACS Nano, 2016, 10, 7467–7475 CrossRef CAS PubMed.
- A. S. Dixon, S. J. Kim, B. K. Baumgartner, S. Krippner and S. C. Owen, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- K. Akama, N. Iwanaga, K. Yamawaki, M. Okuda, K. Jain, H. Ueno, N. Soga, Y. Minagawa and H. Noji, ACS Nano, 2019, 13, 13116–13126 CrossRef CAS PubMed.
- S. A. Byrnes, T. Huynh, T. C. Chang, C. E. Anderson, J. J. McDermott, C. I. Oncina, B. H. Weigl and K. P. Nichols, Anal. Chem., 2020, 92, 3535–3543 CrossRef CAS PubMed.
- L. Beaudet, R. Rodriguez-Suarez, M. H. Venne, M. Caron, J. Bédard, V. Brechler, P. Parent and M. Bielefeld-Sévigny, Nat. Methods, 2008, 5, 10–11 CrossRef.
- M. Bielefeld-Sevigny, Assay Drug Dev. Technol., 2009, 7, 90–92 CrossRef CAS PubMed.
- F. Degorce, A. Card, S. Soh, E. Trinquet, G. P. Knapik and B. Xie, Curr. Chem. Genomics, 2009, 3, 1 CrossRef PubMed.
- G. Wu and M. H. Zaman, Bull. W. H. O., 2012, 90, 914–920 CrossRef PubMed.
- C. Carrell, A. Kava, M. Nguyen, R. Menger, Z. Munshi, Z. Call and C. Henry, Microelectron. Eng., 2019, 206, 45–54 CrossRef CAS.
- S. Chevaliez and J. M. Pawlotsky, J. Hepatol., 2018, 69(4), 916–926 CrossRef PubMed.
- C. Wang, M. Liu, Z. Wang, S. Li, Y. Deng and N. He, Nano Today, 2021, 37, 101092 CrossRef CAS PubMed.
- P. Yager, T. Edwards, E. Fu, K. Helton, K. Nelson, M. R. Tam and B. H. Weigl, Nature, 2006, 412–418 CrossRef CAS PubMed.
- G. A. Posthuma-Trumpie, J. Korf and A. van Amerongen, Anal. Bioanal. Chem., 2009, 393(2), 569–582 CrossRef CAS PubMed.
- S. Haeberle and R. Zengerle, Lab Chip, 2007, 1094–1110 RSC.
- L. R. Volpatti and A. K. Yetisen, Trends Biotechnol., 2014, 32(7), 347–350 CrossRef CAS PubMed.
- W. Su, X. Gao, L. Jiang and J. Qin, J. Chromatogr. A, 2015, 13–26 CrossRef CAS PubMed.
- H. Jiang, X. Weng and D. Li, Microfluid. Nanofluid., 2011, 10(5), 941–964 CrossRef CAS.
- A. K. Au, H. Lai, B. R. Utela and A. Folch, Micromachines, 2011, 2(2), 179–220 CrossRef.
- M. Fratzl, S. Delshadi, T. Devillers, F. Bruckert, O. Cugat, N. M. Dempsey and G. Blaire, Soft Matter, 2018, 14(14), 2671–2681 RSC.
- S. Delshadi, G. Blaire, P. Kauffmann, M. Fratzl, T. Devillers, D. Delabouglise and P. N. Marche, Bioanalysis, 2017, 9(6), 517–526 CrossRef CAS PubMed.
- E. Engvall, Clin. Chem., 2010, 56(2), 319–320 CrossRef CAS PubMed.
- E. Engvall and P. Perlmann, Immunochemistry, 1971, 8(9), 871–874 CrossRef CAS PubMed.
- K. Shah and P. Maghsoudlou, Br. J. Hosp. Med., 2016, 77(7), C98–C101 CrossRef PubMed.
- M. Thangamuthu, C. Santschi and O. J. F. Martin, Biosensors, 2018, 8(2), 34 CrossRef PubMed.
- D. Evans, K. I. Papadimitriou, N. Vasilakis, P. Pantelidis, P. Kelleher, H. Morgan and T. Prodromakis, Sensors, 2018, 18(11), 4011 CrossRef PubMed.
- D. Kim, H. J. Kwon and K. Shin, et al. , ACS Nano, 2017, 11(8), 8448–8455 CrossRef CAS PubMed.
- C. I. Woznitza, K. Lechner and S. Giehring, BMC Proc., 2015, 9(9), 62 CrossRef.
- H. Jiang, X. Weng and D. Li, Microfluid. Nanofluid., 2011, 10(5), 941–964 CrossRef CAS.
- T. D. Ly, L. Martin, D. Daghfal, A. Sandridge, D. West, R. Bristow and S. G. Devare, J. Clin. Microbiol., 2001, 39(9), 3122–3128 CrossRef CAS PubMed.
- S. K. Barik, K. K. Mohanty, D. Bisht, B. Joshi, S. Jena and S. P. Tripathy, J. AIDS Clin. Res., 2018, 9(3), 1–5 Search PubMed.
- S. Kamili, J. Drobeniuc, A. C. Araujo and T. M. Hayden, Clin. Infect. Dis., 2012, 55, S43–S48 CrossRef PubMed.
- B. D. Smith, J. Drobeniuc, A. Jewett, B. M. Branson, R. S. Garfein, E. Teshale and C. M. Weinbaum, J. Infect. Dis., 2011, 204(6), 825–831 CrossRef CAS PubMed.
- N. P. Pai, C. Vadnais, C. Denkinger, N. Engel and M. Pai, PLoS Med., 2012, e1001306 CrossRef PubMed.
- S. Wilson, M. K. Bohn and K. Adeli, eJIFCC, 2021, 32(2), 145 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2lc00926a |
| ‡ Institute of Engineering Univ. Grenoble Alpes. |
| This journal is © The Royal Society of Chemistry 2023 |