
A CoOxHy/β-NiOOH electrocatalyst for robust ammonia oxidation to nitrite and nitrate†
Sam
Cohen
 ab,
Sam
Johnston
a,
Cuong K.
Nguyen
a,
Tam D.
Nguyen
a,
Dijon A.
Hoogeveen
a,
Daniel
Van Zeil
a,
Sarbjit
Giddey
b,
Alexandr N.
Simonov
*a and
Douglas R.
MacFarlane
*a
ab,
Sam
Johnston
a,
Cuong K.
Nguyen
a,
Tam D.
Nguyen
a,
Dijon A.
Hoogeveen
a,
Daniel
Van Zeil
a,
Sarbjit
Giddey
b,
Alexandr N.
Simonov
*a and
Douglas R.
MacFarlane
*a
aSchool of Chemistry, Monash University, Clayton, VIC 3800, Australia. E-mail: alexandr.simonov@monash.edu; douglas.macfarlane@monash.edu
bCSIRO Energy, Private Bag 10, Clayton South, Victoria 3169, Australia
First published on 4th August 2023
Abstract
As the global demand for fertilisers and other nitrogenous products increases, so does the demand for robust, cost-effective and sustainable alternatives to the Ostwald process for the oxidation of ammonia to NOx compounds. Attention has turned to the electrochemical ammonia oxidation to nitrite and nitrate ([NO2/3]−) – a process that could enable distributed production of these important commodity chemicals. Studies of the ammonia oxidation reaction (AOR) for the synthesis of [NO2/3]− are now trending towards more selective and cheaper catalytic materials, rather than the optimisation of Pt and other known noble metal-based catalysts that are subject to poisoning and/or corrosion. Towards this goal, we describe a composite of CoOxHy and β-NiOOH on a Ni foam substrate as an electrocatalyst for the AOR that enables generation of [NO2/3]− under alkaline conditions over extended periods of operation. Specifically, the average [NO2/3]− yield rate of 1.5 ± 0.5 nmol s−1 cm−2 with a faradaic efficiency of 79% ± 10% is demonstrated over 4 days of continuous operation. These results represent a step forward in the development of more robust, corrosion-resistant, and industrially practical materials for the sustainable production of nitrates and nitrites.
Introduction
In the scope of the increasing global impetus for a transition of the chemical industry towards sustainable methods, one important emerging area of research and development is focused on the production of fertilisers from green feedstocks.1–6 Primarily, nitrate is utilised in fertilisers to enhance plant flower and fruit growth, particularly as ammonium nitrate, but also in the form of calcium, potassium and magnesium salts.7–9 This makes nitrate a key component in the large-scale production of fertilisers. Nitrates and nitrites are also utilised in many other industries, including in food preservation, pharmaceuticals, and in the production of explosives.10–13Currently, the primary industrial-scale method for converting NH3 into nitrates is the Ostwald process whereby ammonia is converted into a solution of nitric acid in a multistage oxidation reaction typically catalysed by a Pt/Rh gauze (eqn (1)), before being converted into ammonium or potassium nitrate.2,3,14,15
| NH3(g) + 2O2(g) ⇌ HNO3(aq) + H2O(g) | (1) |
However, the Ostwald process suffers from a 2%, or more, loss of N, where ammonia is converted into nitrous oxide (N2O) or dinitrogen gas via side reactions (2) and (3).14
| 2NH3(g) + 2O2(g) ⇌ N2O(g) + 3H2O(g) | (2) |
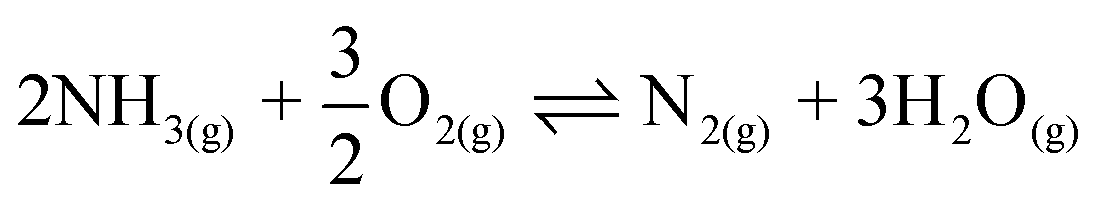 | (3) |
As N2O is a highly potent greenhouse gas (global warming potential rating of 298 relative to CO2 which has a rating of 1), its generation is also a significant environmental issue.16 While a number of techniques exist for the mitigation of N2O produced during the Ostwald process, they are either inefficient or expensive to implement.14,17–20
Nitrite is widely used in a variety of applications including the production of nitroso and isonitroso compounds, synthetic caffeine, pharmaceuticals and herbicides as well as an anti-corrosion agent for the treatment of metal surfaces.13,21 The production of nitrite on an industrial scale is carried out in a variation of the Ostwald process to generate nitrous fumes, which are a combination of NO and NO2 (eqn (4)). The fumes are then passed through a solution of sodium carbonate and sodium hydroxide to generate sodium nitrite and carbon dioxide gas.13
| 2NO(g) + 2NO2(g) + 2NaOH(aq) + Na2CO3(aq) ⇌ 4NaNO2(aq) + CO2(g) + H2O(l) | (4) |
When the number and scale of applications for nitrites and nitrates ([NO2/3]−), as well as the issues associated with their production, are considered, the need for sustainable, environmentally benign methods of their synthesis becomes clear. Among different options, an electrochemical conversion of ammonia to [NO2/3]− presents a prominent possibility to create a sustainable process at various scales with minimal environmental impact. Ideally, this electrochemical route to nitrite and nitrate would employ low-cost catalysts based on abundant elements, coupled with the well-known oxygen reduction reaction.
The electrochemical ammonia oxidation reaction (AOR) has been studied for over a century, with research in the field focused on applications such as fuel cells, wastewater treatment, chemical sensing, and others.22–29 In aqueous alkaline solutions, the major AOR products of interest are N2 (eqn (5); standard potential E0 = −0.772 V vs. standard hydrogen electrode (SHE), pH = 14; hereinafter at 25 °C), nitrite (eqn (6); E0 = −0.174 V vs. SHE, pH = 14) or nitrate (eqn (7); E0 = −0.132 V vs. SHE, pH = 14), depending on the intended application (standard potentials were calculated using tabulated thermodynamic data30,31).
| NH3(aq) + 3OH−(aq) ⇌ ½N2(g) + 3H2O(l) + 3e− | (5) |
| NH3(aq) + 7OH−(aq) ⇌ NO2−(aq) + 5H2O(l) + 6e− | (6) |
| NH3(aq) + 9OH−(aq) ⇌ NO3−(aq) + 6H2O(l) + 8e− | (7) |
While much of this research revolves around the fundamentals of the reaction, the bulk has focused on platinum-based catalysts. As such, the AOR on platinum has become a benchmark in the field and is well understood, though the majority of these studies target the oxidation of ammonia to N2.32,33 However, issues with high catalyst cost and severe issues with adsorptive poisoning as well as interest in the nitrite/nitrate products have motivated researchers to consider more practical, non-noble-metal electrocatalysts.34–37 The major challenge with these materials is in the relatively low faradaic efficiencies of the AOR (FE, the fraction of charge passed that is consumed in producing the desired product, as opposed to undesirable side reactions). They also typically require more positive potentials than platinum to sustain high rates of the NH3 oxidation to the desired nitrogenous product.34,36,38,39
According to recent studies, copper, cobalt, iron, and nickel oxides lead the way as prime candidates for cost-effective AOR electrocatalysis.35,37,40–43 Nickel oxides in particular have shown much promise for the oxidation of ammonia to N2.37,44–46 To this end, Choueiri et al. have demonstrated the mechanism and ability of β-NiOOH as an electrocatalyst for the oxidation of ammonia, computationally identifying the (0001) facet as providing the lowest energy pathway to dinitrogen.31 Further work by Medvedev et al. used similar nickel hydroxide electrodes for the electrooxidation of ammonia to nitrate at 72% faradaic efficiency.4 However, there was a noted decrease in catalyst activity after 52 h of operation, and in further investigations at a positive potential of ca. 2.1 V vs. reversible hydrogen electrode (RHE) significant degradation of the catalyst was visually observed.4 While some electrocatalysts have shown promise in resisting the effects of poisoning, there are still issues with catalyst longevity due to corrosion.47 Formation of surface β-NiOOH has been shown to assist in stabilising Ni electrodes in this context.48
Herein, we describe an alternative approach to improve the longevity of noble-metal-free AOR catalysts. Specifically, we demonstrate that the formation of β-phase oxyhydroxides of nickel in combination with cobalt (oxy)hydroxides provides high robustness to the AOR electrodes and prevents their dissolution during operation on a timescale of days.
Results and discussion
The aim of the present study was to utilise cobalt oxide's high electrocatalytic activity towards the AOR to form nitrite and nitrate while mitigating its poisoning and dissolution due to corrosion and complexation.A nickel foam electrode, which had been subjected to prior voltammetric cycling in 0.1 M KOH to form a β-NiOOH surface layer (Fig. S1†), was used as the electrode substrate. The electrochemical formation of β-NiOOH in KOH is a well understood process, having been examined by Alsabet et al., whereby a characteristic α-NiOOH reversible formation peak and a β-NiOOH irreversible formation peak can be identified in cyclic voltammetry.48,49 Herein, the β-NiOOH-modified nickel electrode was then functionalised with cobalt oxides by galvanostatic electrooxidation of a cobalt acetate solution at a potential of 1.0 V vs. SHE for 1 min (Fig. S2†). To facilitate the formation of the CoOxHy species, 1 M NH4OH was injected into the solution 30 s after the electrodeposition was commenced.50 Finally, hydrothermal treatment was used to stabilise the CoOxHy layer (full experimental details are provided in the Experimental section).51
Scanning electron microscopy (SEM) coupled to energy dispersive X-ray spectroscopic (EDS) elemental mapping of the obtained surface shows a coating of cobalt-based compounds, with cracks displaying β-NiOOH/Ni underneath (Fig. 1A–C). Transmission electron microscopic (TEM) analysis of the material exfoliated by ultrasonication of the electrode suggests that the deposited cobalt-based layer is comprised of nanocrystals with a typical size of 6.8 ± 1.4 nm (n = 100) (Fig. 1D). The observed lattice fringes of 0.24 nm are consistent with the (101) plane of the β-CoOOH phase (PDF 9009885).
 | ||
| Fig. 1 (A) Scanning electron micrograph, (B–C) corresponding energy-dispersive X-ray spectroscopic mapping for (B) Ni and (C) Co of an as-prepared CoOxHy/β-NiOOH/Ni composite electrode. (D) Lower and higher (inset) transmission electron micrographs (lines and arrows in the inset highlight the interlayer distance of ca. 0.24 nm). (E) X-ray diffractogram of the electrodeposited material removed from the CoOxHy/β-NiOOH/Ni composite electrode (blue) compared to the simulated background (black) and diffractogram (red); vertical lines show positions and relative intensities of the reflections for Co(OH)2 (PDF 1548811) and CoOOH (PDF 9009885). | ||
The presence of the latter was additionally confirmed by X-ray diffraction analysis, which also showed a significant contribution of Co(OH)2 to the material produced on the nickel foam surface (Fig. 1E). Of the surface layer, the Co(OH)2 and CoOOH phases have mean crystallite sizes of ca. 20 and 15 nm, respectively, and are present in a ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, as derived from the fitting of the XRD data (Fig. 1E).
:1 molar ratio, as derived from the fitting of the XRD data (Fig. 1E).
Overall, the physical characterisation confirms that the employed fabrication method produces a dense coating of cobalt hydroxide/oxyhydroxide particles over the pre-oxidised nickel foam.
Electrocatalytic performance
Aiming to examine the electrocatalytic activity of the catalysts, cyclic voltammetry and chronoamperometry were the primary electrochemical tools utilised in this study. All electrochemical measurements were conducted in a standard three-electrode cell with a peristaltically recirculated headspace to ensure no loss of ammonia that may evaporate from the electrolyte solution. Since ammonia solutions have relatively low conductivities (on the order of mS cm−1), a 0.1 M KnHmPO4 (pH 7.4) electrolyte was added. Since both potassium and phosphate are commonly used fertiliser components, this choice of additive was consistent with our goal of providing an outcome that could be used directly, for example in fertigation or hydroponics. Measurements were undertaken without (pH 7.4) and with (pH 11.3) the addition of 0.5 M NH3. At the latter pH, the reversible potential for reaction (5) is ca. −0.6 V vs. SHE.In the absence of NH3, cyclic voltammograms recorded for the pre-oxidised nickel foam, with and without cobalt hydroxide/oxyhydroxide deposited, exhibited redox processes corresponding to the Ni and Co redox transformations respectively (Fig. 2), as expected from the physical characterisation data (Fig. 1). In the presence of NH3, both unmodified and pre-oxidised nickel foam electrodes exhibited significantly enhanced oxidative currents at potentials more positive than ca. 0.8 V vs. SHE, prior to the onset of the oxygen evolution reaction, which can be attributed to the AOR (Fig. 2A and B).48,49 The pre-oxidised electrode exhibited notably higher activity in the cyclic voltammograms, possibly due to the higher electrochemically active surface area induced by voltammetric pre-treatment (Fig. S1†). Modification of such electrodes with the cobalt hydroxide/oxyhydroxide layer resulted in less notable differences between the cyclic voltammograms recorded with and without ammonia present (Fig. 2C). At the same time, more relevant to the true electrocatalytic activity potentiostatic experiments produced a different trend, as discussed below.
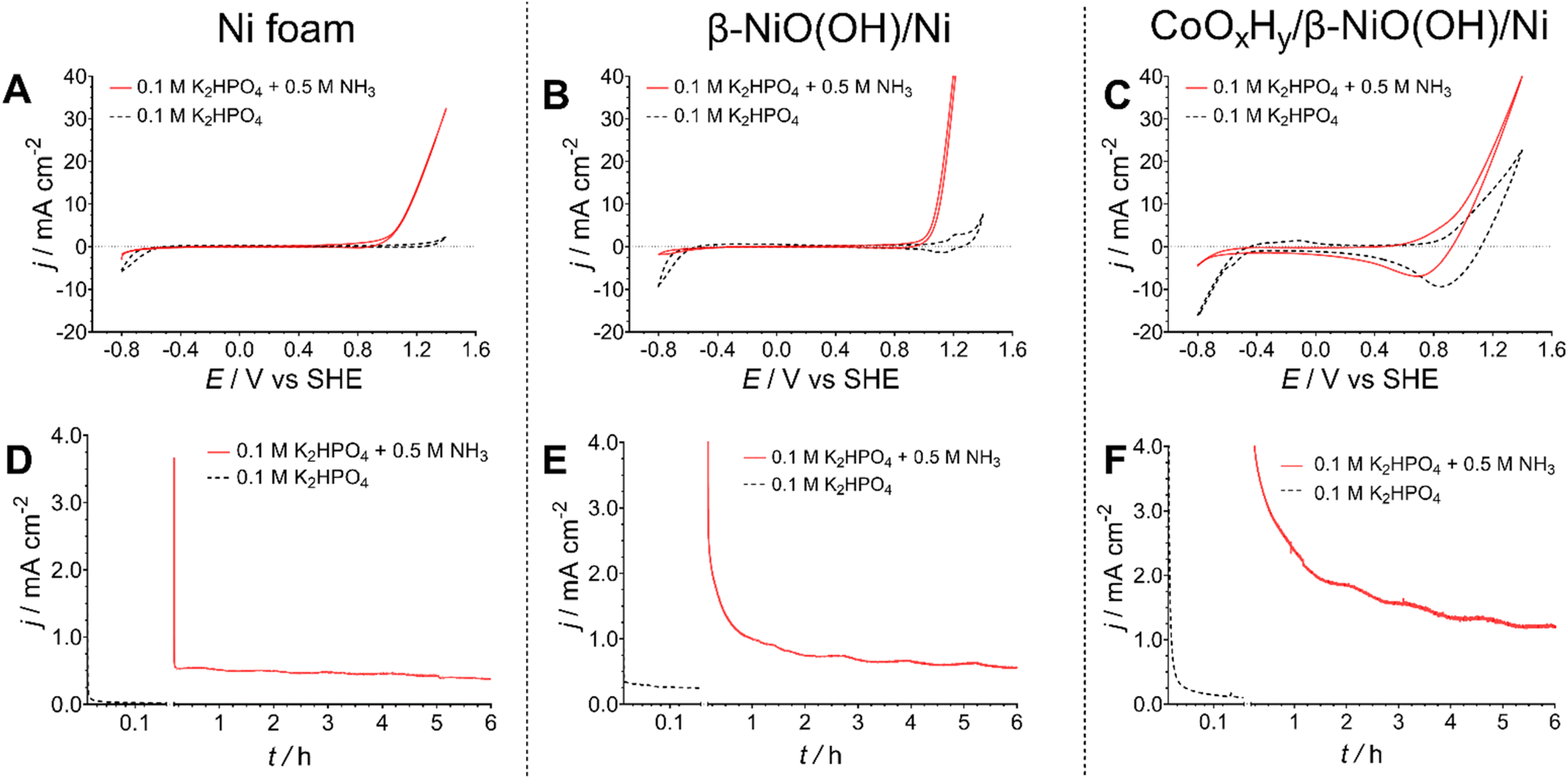 | ||
| Fig. 2 (A–C) Cyclic voltammograms (v = 0.050 V s−1; 3rd cycle shown), and (D–F) chronoamperograms (E = 1.000 ± 0.003 V vs. SHE) for (A and D) nickel foam, (B and E) β-NiOOH/Ni and (C and F) CoOxHy/β-NiOOH/Ni electrodes recorded in 0.1 M K2HPO4 without (dotted black) and with (solid red) 0.5 M NH3 present. Currents are normalised to the geometric surface area of the electrodes (1 cm2); data are presented as average derived from tests of n = 3 independent electrodes of each type. Fig. S3† compares these data for different electrodes under the same testing conditions. | ||
First, a series of short 10 min chronoamperometric tests were performed at different potentials within the range of 0.8–1.3 V vs. SHE to determine the potential enabling the highest faradaic efficiency for the conversion of ammonia to nitrate and nitrite (Table S1†). These experiments showed that the best results are consistently achieved at 1.0 V vs. SHE. As such, this potential was used for all subsequent chronoamperometric experiments.
Under the potentiostatic conditions, the highest AOR current densities were provided by the CoOxHy-modified electrodes (1.2 ± 0.2 mA cm−2), while the cobalt-free pre-oxidised and bare Ni foams demonstrated comparable activity after ca. 6 h of tests (0.73 ± 0.15 and 0.48 ± 0.02 mA cm−2, respectively) (Fig. 2D–F). Equally importantly, the ion chromatographic analysis of the electrolyte solutions after these experiments demonstrated that untreated and pre-oxidised Ni foam electrodes with no deposited Co materials produce low amounts of nitrite and nitrate with an overall faradaic efficiency towards [NO2/3]− of 19 ± 2% and 29 ± 8%, respectively (Fig. 3A and Table S4†). This can be compared to the highly reproducible faradaic efficiency of 79 ± 10% provided by the CoOxHy-modified electrodes. Correspondingly, the bimetallic cobalt–nickel system also provided the highest cumulative nitrate and nitrite yield rates of 1.5 ± 0.5 nmol s−1 cm−2, notably excelling the untreated (0.2 ± 0.02 nmol s−1 cm−2) and electrochemically pre-oxidised (0.4 ± 0.1 nmol s−1 cm−2) Ni foam electrodes (Fig. 3B and Table S4†). The AOR selectivities towards nitrate and nitrate were approximately similar to that of the best performing CoOxHy/β-NiOOH/Ni catalyst. Another major product formed during the electrooxidation experiments is most likely N2 and potentially NOx(g) species, since the OER rates are very low at the potential of 1.0 V vs. SHE used in these experiments (Fig. 2D–F).
 | ||
| Fig. 3 (A) Faradaic efficiency of the AOR towards [NO2/3]−, (B) yield rate of [NO2/3]− and (C) metal corrosion rate during 6 hours chronoamperometric oxidation at 1.0 V vs. SHE of aqueous 0.1 M K2HPO4 + 0.5 M NH3 solutions using 1 cm2 nickel foam electrodes that were unmodified, electrochemically oxidised (β-NiOOH/Ni), and electrochemically oxidised and modified with cobalt hydroxide/oxyhydroxide (CoOxHy/β-NiOOH/Ni). Data are shown as average ± standard deviation for n = 3 independent samples of each type of the electrode. Extended experimental data are provided in Table S4.† | ||
Among possible gaseous NOx by-products, the most undesirable is nitrous oxide. To quantify the amount of N2O generated during the AOR catalysed by CoOxHy/β-NiOOH/Ni, an extended 24 h potentiostatic electrolysis experiment was undertaken using a sealed cell containing 20 mL of 0.1 M K2HPO4 + 0.5 M NH3 with a 20 mL air headspace, which was sampled before commencement (t = 0) and after experiment completion (t = 24 h). A N2O concentration of 0.5 ± 0.1 ppm was recorded in the ambient air, as well as in the t = 0 and t = 24 samples. This important result confirms that negligible, if any, amounts of N2O are generated during ammonia electrooxidation with the CoOxHy/β-NiOOH/Ni electrodes.
We have additionally tested the catalytic activity of the CoOxHy powder exfoliated from the electrode for the chemical oxidation of NH3 by dissolved O2 in air-saturated electrolyte solutions. These control experiments showed that the cobalt hydroxide/oxyhydroxide species might indeed be catalytically active for this reaction with the predominant formation of NO3−, but the rate of nitrate generation was approximately 5-fold lower than those achieved in the electrochemical experiments (Table S3†). Nevertheless, although the major [NO2/3]− products are generated via the AOR, direct catalytic oxidation of ammonia might also make a measurable contribution to the production of nitrate.
Furthermore, the CoOxHy/β-NiOOH/Ni electrode was found to be highly stable against corrosion during the AOR, which is a well-known limitation of many, especially non-noble-metal-based electrocatalysts. The stability was assessed through quantification of the rates of metal dissolution into the electrolyte solutions during the 6 h chronoamperometric experiments by inductively coupled plasma mass spectrometry (ICP-MS). The corrosion rates of the unmodified and pre-oxidised nickel foam electrodes were approximately similar at 4.4 ± 0.5 and 5.5 ± 0.5 nmol s−1 cm−2, respectively. In contrast, the CoOxHy/β-NiOOH/Ni system lost only very small and very similar amounts of Co and Ni at a cumulative rate of only 0.3 ± 0.2 nmol s−1 cm−2 under the same conditions (Fig. 3C and Table S4†).
We have additionally investigated nickel foam electrodes that were not electrochemically pre-oxidised prior to modification with the cobalt hydroxide/oxyhydroxide layer. Such electrodes were losing CoOxHy very rapidly even when put in contact with an ammonia-containing electrolyte solution without any potential applied (observed with a naked eye, Fig. S4†) and produced highly inconsistent electrochemical and AOR data.
SEM analysis of the electrodes before and after 6 hours of the AOR at 1.0 V vs. SHE revealed notable changes to the surface morphology of all examined electrodes (Fig. 4). The surface of the untreated nickel foam developed a large number of cracks, which are attributed to the metal corrosion (Fig. 4A and B). Qualitatively similar surface fissures were found for the β-NiOOH/Ni electrode, but with even higher frequency (Fig. 4C and D). These morphological changes might be associated with the expansion and contraction of the material due to the nickel phase transformations induced electrochemically and through redox reactions with dissolved ammonia. The presence of the latter also facilitates corrosion of the material.
 | ||
| Fig. 4 SEM images of the (A and B) unmodified Ni foam, (C and D) electrochemically pre-oxidised Ni foam (β-NiOOH/Ni), (E and F) electrochemically deposited CoOxHy on unmodified nickel foam, and (G and H) β-NiOOH/Ni functionalised with cobalt hydroxide/oxyhydroxide layer (CoOxHy/β-NiOOH/Ni). Images were taken (A, C, E and G) before and (B, D, F and H) after 6 hours of chronoamperometric tests at 1.000 ± 0.003 V vs. SHE in 0.1 M K2HPO4 + 0.5 M NH3. | ||
Nickel foam electrodes, that have not been subjected to electrochemical pre-oxidation prior to coating with CoOxHy, produced distinct clusters of the cobalt-based flakes on the surface (Fig. 4E), which were essentially completely removed after the AOR (Fig. 4F). This is consistent with the visually observed CoOxHy detachment mentioned above (Fig. S4†), as well as colouration of the electrolyte solution during the electrochemical tests. EDS confirmed the loss of CoOxHy from the electrode surface that was not pre-treated to produce a β-NiOOH layer (Fig. S5†)
The CoOxHy/β-NiOOH/Ni electrodes also underwent changes after the AOR tests (Fig. 4G and H). However, the thick CoOxHy layer was still present on the surface of the underlying nickel-based support, consistent with the improved corrosion resistance of these electrodes (Fig. 3C).
Collectively, the electrochemical activity, metal corrosion and microscopic data suggest that the CoOxHy/β-NiOOH interface is critical for the efficient and stable operation of the electrode during the AOR. This improved performance might be associated with the favourable effect of the β-NiOOH interlayer on the homogeneity of electrodeposition of relatively thick and uniform CoOxHy coatings. These cobalt-based coatings exhibit higher selectivity towards the AOR, while protecting the underlying nickel-based support from corrosion (Fig. 3C and 4H).
To further probe the durability of the composite CoOxHy/β-NiOOH/Ni electrodes, an extended AOR test spanning over 4 days was undertaken at 1.0 V vs. SHE (Fig. 5A). After a slight drop over the initial ca. 6 h, the performance was seen to stabilise during the subsequent 18 hours of the experiment. Further, the electrooxidation rate deteriorated and fluctuated to some extent until starting to steadily increase over the final 12 h of tests. Critically, the faradaic efficiency towards [NO2/3]− was as high as ca. 77% with nitrite and nitrate contributing ca. 27 and 50%, respectively.
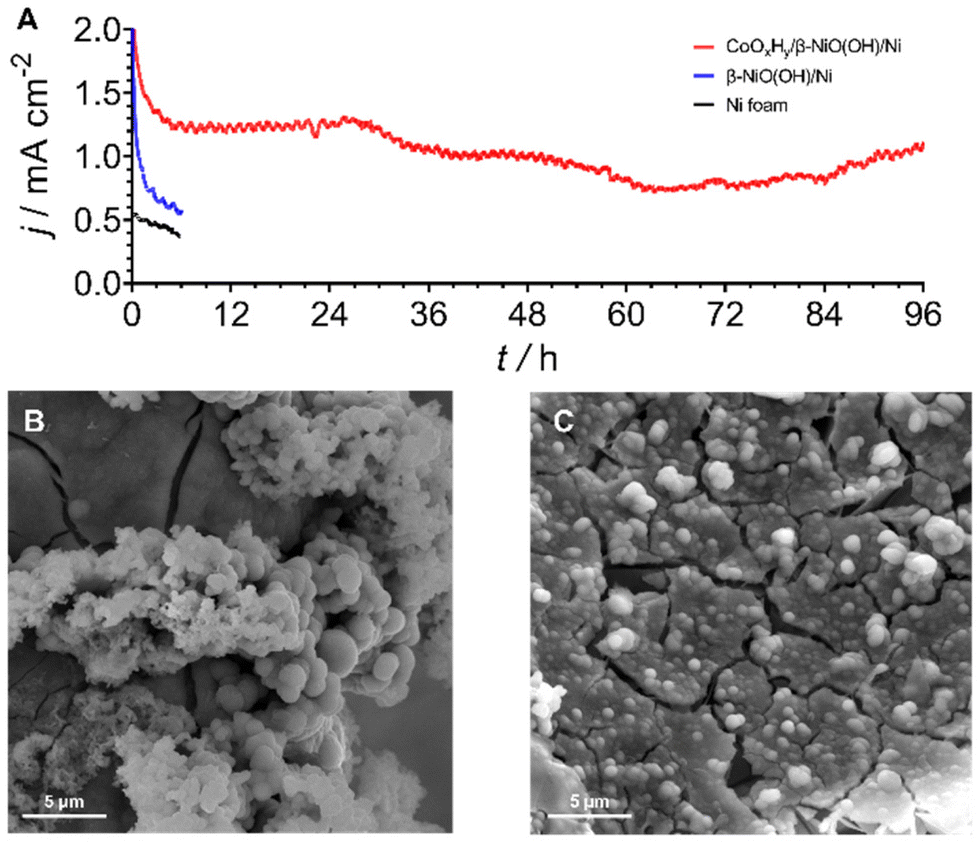 | ||
| Fig. 5 (A) Long-term chronoamperometric (E = 1.000 ± 0.003 V vs. SHE) AOR test of CoOxHy/β-NiOOH/Ni (red) in 0.1 M K2HPO4 + 0.5 M NH3. Short-term (6 h) data for β-NiOOH/Ni (blue) and Ni foam (black) are shown for comparison. Currents are normalised to the geometric surface area of the electrodes (1 cm2). (B and C) SEM images of the CoOxHy/β-NiOOH/Ni electrode (B) as-prepared and (C) after the test shown in panel A. | ||
This level of performance is similar to that found in the short-term experiments (Fig. 3). SEM examination showed that CoOxHy was still present on the electrode surface following the 4 days of chronoamperometric AOR testing (Fig. 5B and C). ICP-MS analysis of the electrolyte solution after this experiment showed that the amount of Ni and Co dissolved from the electrode surface was 8.3 ± 0.7 and 28.5 ± 1.2 μmol cm−2, respectively (Table S5†). Comparisons of these values to the corrosion data recorded after 6 h tests indicate that the loss of the catalytically active cobalt and especially underlying nickel species decelerates with time, further highlighting the robustness of the electrode (Fig. 3B and Table S5†). Overall, the CoOxHy/β-NiOOH/Ni system presents one of the most stable catalysts for the ammonia oxidation to [NO2/3]− reported to date.4
Conclusion
The key result of this study is the outstanding longevity of the CoOxHy/β-NiOOH/Ni electrodes during ammonia electrooxidation to nitrite and nitrate at a relatively high faradaic efficiency. While the electrocatalyst does not feature the highest yield rate reported to date,4,37 its high selectivity and resistance to corrosion and poisoning provide a strong platform for further investigations and performance optimisation. Improvements in the areal current densities may arise from nanostructuring of the CoOxHy catalyst layer, for example through optimisations of the electrodeposition conditions.52,53The results presented herein specifically highlight the critical importance of the catalyst|electrode interface to the stability of the AOR anodes. This knowledge might aid future design of the conceptually similar non-noble-metal catalytic systems for the sustainable and N2O-emission-free electrosynthesis of nitrite and nitrate.
Experimental
Materials
Electrolyte solutions were prepared using Na2SO4 (Merck, ≥99%, ACS Reagent), KOH (Merck, ≥85%, acidimetric), NH4OH (Sigma-Aldrich, 28% NH3 in H2O, ≥99.99% trace metals basis), K2HPO4 (Merck, ≥99%, ACS Reagent), KH2PO4 (Merck, ≥99%, ACS Reagent), and deionised H2O, which was produced by a Satorius™ Arium® Comfort II water purification system and had a resistivity of 18.2 MΩ cm at 23 ± 2 °C. Water of the same quality was used for all operations requiring H2O. Hygroscopic reagents were stored under vacuum.Catalyst precursor used to produce the electro deposition solutions was (CH3COO)2Co·4H2O (Sigma-Aldrich, ≥98%). Electrodes were produced using a Ni foam (Goodfellow, thickness 1.6 mm, bulk density 0.45 g cm−3, porosity 95%).
For the ion chromatography calibration, NaNO2 (Sigma-Aldrich, ReagentPlus®, ≥99%) and NaNO3 (Sigma-Aldrich, ACS reagent, ≥99.0%) were used as received.
Electrochemical methods
All electrochemical measurements were conducted using Gamry Instruments Interface 1000-E potentiostats in a three-electrode configuration at ambient temperature (23 ± 2 °C). A 3 cm × 1 cm platinised titanium mesh auxiliary electrode was used for all electrochemical experiments.As ammonia is known to penetrate several kinds of frits and membranes, a custom-designed reference system37 was employed to ensure that the electrode potentials remained stable throughout the course of testing (Fig. S6†). This was achieved by using a narrow PTFE tube filled with agar and 0.1 M K2SO4 which served as a salt bridge, connecting the electrolyte solution to a standard Ag|AgCl|KClsat. CHI instruments reference electrode. The design proved effective, with an average potential of ca. 0.191 ± 0.003 V vs. SHE at 23 ± 2 °C. Reference electrode potential was regularly measured against a saturated calomel reference electrode (SCE) prior to and after electrochemical experiments. Measured potentials were converted to the SHE scale assuming ESCE = 0.246 V vs. SHE.
Preparation of working electrodes
Nickel foam was cut into 2 cm × 1 cm segments and kept in a 3:1:1 vol. mixture of H2O:acetone:iso-propanol for 12 hours under ambient conditions to ensure any residual surface organics were removed. Following this treatment, electrodes were washed via 3 repeated cycles of sonication (Branson B5500R-dth bath with an operating power of 175 W) in water. Finally, the electrodes were washed with water and allowed to dry in a desiccator filled with silica gel beads under vacuum over ca. 16 hours.
Electrochemical pre-oxidation of the nickel foam to produce β-NiOOH/Ni was undertaken in aqueous 0.1 M KOH without stirring. First, the electrode was kept at a constant potential of −0.4 V vs. SHE for 200 s and then at −0.2 V vs. SHE for 400 s to reduce the nickel surface.48 Further, cyclic voltammetry (100 cycles) between −1.0 and 1.2 V vs. SHE at a scan rate of 0.050 V s−1 was performed (Fig. S1†). Following electrochemical treatment, the electrodes were washed by sonication in water (Branson B5500R-dth bath with an operating power of 175 W) for 30 seconds and rinsed with water. Afterwards, the electrodes were allowed to dry in a small benchtop vacuum desiccator with silica gel beads over ca. 16 hours.
For the CoOxHy deposition, an aqueous electrolyte solution (20 mL) containing 0.5 mM of (CH3COO)2Co·4H2O and 0.1 M Na2SO4 was used. Electrodeposition was performed at a constant potential 1.0 V vs. SHE for 60 seconds under stirring provided by a 1 mm x 5 mm cylindrical Teflon stirring bar. After 30 seconds, 1 M NH4OH was added swiftly to the electrolyte solution. After 60 seconds, the reference and counter electrodes were removed from the solution leaving the working electrode immersed. The deposition vessel was immediately sleeved and sealed in a stainless-steel autoclave (30 mL internal volume) and placed in an oven preheated to 120 °C for 12 hours. Upon cooling down of the autoclave to ambient temperature (ca. 12 h), the electrode was washed and sonicated in water (1 min) and stored in a desiccator under vacuum. The electrodes were used within 1 h after preparation.
In order to avoid any catalytic contributions from the electrode connector, titanium electrode holders were used, as described previously.37 Notwithstanding the fact that titanium has unmeasurably low electrooxidation catalytic activity under the conditions examined herein,37 titanium holders were kept above the electrolyte solution during testing. The geometric surface area of the working electrodes was 1 cm2.
Electrocatalytic tests
Experiments were conducted in a hermetically sealed glass cell equipped with a fritted (P4 sintered glass) compartment for the auxiliary electrode and rubber septum for sampling. A 0.1 M KnHmPO4 electrolyte solution (pH 7.4) was made from 80.2 mL of 0.1 M K2HPO4 and 19.8 mL of 0.1 M KH2PO4. 48.7 mL of the 0.1 M KnHmPO4 electrolyte solution was used for all measurements, with NH4OH (14.8 M stock) added in a 1.3 mL aliquot to produce an NH3 concentration of 0.5 M following preliminary electrochemical measurements in the absence of ammonia.The NH3-containing electrolyte solution within the cell was continuously agitated by air bubbles from the headspace using a peristaltic pump. This approach was applied to avoid the loss of NH3 from the setup while maintaining an equilibrium between ammonia dissolved in the electrolyte solution and partially evaporated to the headspace.
Electrolyte solution samples of 0.1 mL were taken using a syringe with a stainless needle for analysis prior to the commencement of cyclic voltammetry and chronoamperometry in the presence of NH3, and immediately after the electrochemical experiment was concluded. pH was measured using a smartCHEM multiparameter benchtop pH meter equipped with an Ionode IJ series pH probe.
Ion chromatography
Concentrations of nitrite and nitrate in the electrolyte solutions were analysed and quantified by ion chromatography (IC) on a DIONEX Integrion high pressure ion chromatograph from ThermoFisher Scientific. The ion chromatograph was equipped with a Dionex ADRS 600 2 mm RFIC™ ionic conductivity detector, an electrolytically regenerated conductivity suppressor, and a combination of Dionex IonPac™ AS1-HC-4 μm high-capacity analytical column and Dionex IonPac™ AG11-HC-4 μm high-capacity guard column. Analysis of chromatograms was done using the Chromeleon™ 7.2.10 ES ThermoFisher Scientific software suite.Sample preparation was performed by diluting the electrolyte solution aliquots (0.1 mL) by a factor of 10 with water to achieve a similar electrolyte concentration and pH as in the IC eluent. This dilution was performed to avoid damage to the column, as well as ensuring elution times were reliable.
Ion chromatographic analysis consisted of a 20 minutes run at a flow rate of 0.3 mL min−1 and a data collection rate of 10 Hz. A custom eluent gradient curve was developed specifically for the task of effectively separating and quantifying NO2− and NO3− in 0.1 M K2HPO4 + 0.5 M NH3 solutions, as well as providing adequate separation of other anions which may be present in solution. The KOH eluent concentration was kept constant at 8 mM from 0 to 11 min, and then increased to 40 mM at a ramp rate of 20 mM min−1. The ramp was then reversed at the same rate, returning to a value of 8 mM at the 15 min mark, continuing through at that level until the measurement was concluded at the 20 min. All key components, including the conductivity detection cell, conductivity suppressor, column and eluent lines were maintained at a temperature of 30 °C during the measurements.
Gas chromatography
At each measurement, an airtight syringe was used to collect 12 mL gas sample from the headspace both before the commencement of electrocatalytic experiments, and after 24 hours of electrocatalytic operation. 12 ml was taken at a t = 0 to quantify the ambient N2O in the headspace after ammonia had been added to the solution. The headspace was allowed to refill, and the cell was sealed again. Then the experiment was started. The collected gas sample was immediately transferred into an airtight 12 mL glass Exetainer® vial with grey silicon septa (Labco, UK) which was pre-evacuated and flushed with argon and then re-evacuated. The N2O concentration in the gas samples was measured using a gas chromatograph (Agilent 7890A) fitted with a Gerstel MPS autosampler.The samples were injected with a volume of 2500 μL via an autosampler. CO2 was measured using a thermal conductivity detector (TCD) with a detection limit of 400 pg tridecane mL−1. CH4 was measured using a flame ionisation detector (FID) with a detection limit of 1.5 pg C s−1 of tridecane. A micro electron capture detector (ECD) was used to measure N2O with a detection limit of 5.5 fg lindane mL−1. The retention times and peak heights were recorded and interpreted using Agilent Technologies OpenLab Software Suite. The N2O concentration was determined with the help of a calibration curve prepared from the reference gas with a known concentration of 1 ppm. The CH4 concentration was determined with a calibration curve prepared from the reference gas with a known concentration of 5.5 ppm. The CO2 concentration was determined with a calibration curve prepared from the reference gas with a known concentration of 650 ppm. Linear interpolation of the gas concentrations was used to calculate the concentration and cumulative N2O, CH4 and CO2 following the calculation and procedure outlined by van Zwieten et al.54
Scanning electron microscopy
A scanning electron microscope FEI Quanta 3D FIBSEM equipped with a TEAM X-ray analysis system as well as a 10 mm2 SDD with ultra-thin window was used. EDS analysis was conducted using a JEOL 7001F SEM instrument equipped with an Oxford Instruments X-Max 80 silicon drift type detector. EDS data were collected in mapping mode with high count rates (>10 kcps) with a probe current of 14 nA and an accelerating voltage of 15 kV. Samples were mounted onto SEM sample holders by double-sided sticky carbon tape without any additional coatings. All images are taken at 5 mA beam current and 10 mm working distance.Transmission electron microscopy
TEM analysis was conducted using a FEI Tecnai F20 FEGTEM instrument. The samples were scratched off the Ni foam and suspended in ethanol by ca. 10 minutes ultrasonication. The sample was then drop-cast onto a lacey carbon Cu grid, followed by air-drying at room temperature before TEM imaging.X-ray diffraction
XRD analysis was undertaken using a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation (40 mA and 40 kV). Diffractograms were collected in a 10–80° range at a 0.02° step and 0.5 s step−1 sampling rate.Author contributions
S. C. conceived and did the electrode preparation, ion chromatography, and co-wrote the manuscript. S. J. assisted with the ion chromatography and electrochemical experiments, and co-wrote the manuscript. C. K. N. assisted with the ion chromatography, gas chromatography, and collected SEM images. T. D. N. collected TEM images. D. A. H. recorded and analysed ICP-MS data. D. V. Z. collected and analysed XRD, SEM and EDS data. S. G., A. N. S. and D. R. M. conceived and directed the project. A. N. S. and D. R. M. co-wrote the manuscript.Conflicts of interest
The authors state that there are no conflicts to declare.Acknowledgements
Parts of this work were undertaken using the facilities provided by the Monash Centre for Electron Microscopy and Monash X-ray platform. This study was supported by the Australian Research Council (Discovery Project, DP200101491; Future Fellowship to ANS, FT200100317) and the CSIRO Hydrogen Energy Future Science Platform (FSP).References
- M. Nagpal and R. Kakkar, Int. J. Hydrogen Energy, 2018, 43, 12168–12188 CrossRef CAS.
- L. Green, Int. J. Hydrogen Energy, 1982, 7, 355–359 CrossRef CAS.
- E. Gülzow and M. Schulze, Mater. Fuel Cells, 2008, 1, 64–100 Search PubMed.
- J. J. Medvedev, Y. Tobolovskaya, X. V. Medvedeva, S. W. Tatarchuk, F. Li and A. Klinkova, Green Chem., 2022, 24, 1578–1589 RSC.
- G. Strickland, Int. J. Hydrogen Energy, 1984, 9, 759–766 CrossRef CAS.
- O. Oenema, H. P. Witzke, Z. Klimont, J. P. Lesschen and G. L. Velthof, Agric., Ecosyst. Environ., 2009, 133, 280–288 CrossRef CAS.
- J. F. Herencia, P. A. García-Galavís, J. A. R. Dorado and C. Maqueda, Sci. Hortic., 2011, 129, 882–888 CrossRef.
- S. Y. Chung, J. S. Kim, M. Kim, M. K. Hong, J. O. Lee, C. M. Kim and I. S. Song, Food Addit. Contam., 2003, 20, 621–628 CrossRef CAS PubMed.
- C. W. Liu, Y. Sung, B. C. Chen and H. Y. Lai, Int. J. Environ. Res. Public Health, 2014, 11, 4427 CrossRef CAS PubMed.
- A. P. Akolzin, Y. Y. Kharitonov and S. G. Kovalenko, Mater. Corros., 1987, 38, 417–421 CrossRef CAS.
- F. Z. Dörwald, Lead Optimization for Medicinal Chemists: Pharmacokinetic Properties of Functional Groups and Organic Compounds, Wiley, 2012, pp. 66–67 Search PubMed.
- M. Bohnet, Ullmann's encyclopedia of industrial chemistry, 6th edn, 2003 Search PubMed.
- W. Laue, M. Thiemann, E. Scheibler and K. W. Wiegand, Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, Ltd, 2000 Search PubMed.
- Z. Kirova-Yordanova, Energy, 2011, 36, 3733–3744 CrossRef CAS.
- V. Smill and R. A. Streatfeild, Enriching the earth: Fritz Haber, Carl Bosch, and the transformation of world food production, MIT press, 2002 Search PubMed.
- D. Reay, C. Sabine, P. Smith and G. Hymus, Intergovernmental Panel on Climate Change. Fourth Assessment Report. Geneva, Switzerland: Inter-gov- ernmental Panel on Climate Change, Cambridge University Press, Cambridge, UK, 2007. Available from: https://www.ipcc.ch, Intergovernmental Panel on Climate Change, 2007.
- K. Siddharth, Y. Chan, L. Wang and M. Shao, Curr. Opin. Electrochem., 2018, 9, 151–157 CrossRef CAS.
- V. Jevtovic-Todorovic, J. Beals, N. Benshoff and J. W. Olney, Neuroscience, 2003, 122, 609–616 CrossRef CAS PubMed.
- L. K. Schneider, A. Wüst, A. Pomowski, L. Zhang and O. Einsle, Met. Ions Life Sci., 2014, 14–113, 177–210 Search PubMed.
- M. Weiss, J. Haufe, M. Carus, M. Brandão, S. Bringezu, B. Hermann and M. K. Patel, J. Ind. Ecol., 2012, 16, S169–S181 CrossRef CAS.
- A. P. Akolzin, Y. Y. Kharitonov and S. G. Kovalenko, Mater. Corros. und Korrosion, 1987, 38, 417–421 CrossRef CAS.
- B. A. López de Mishima, D. Lescano, T. Molina Holgado and H. T. Mishima, Electrochim. Acta, 1998, 43, 395–404 CrossRef.
- X. Ji, C. E. Banks and R. G. Compton, Analyst, 2005, 130, 1345–1347 RSC.
- L. Marinčić and F. B. Leitz, J. Appl. Electrochem., 1978, 8, 333–345 CrossRef.
- E. P. Bonnin, E. J. Biddinger and G. G. Botte, J. Power Sources, 2008, 182, 284–290 CrossRef CAS.
- Z. F. Li, Y. Wang and G. G. Botte, Electrochim. Acta, 2017, 228, 351–360 CrossRef CAS.
- H. Zöllig, C. Fritzsche, E. Morgenroth and K. M. Udert, Water Res., 2015, 69, 284–294 CrossRef PubMed.
- F. Vitse, M. Cooper and G. G. Botte, J. Power Sources, 2005, 142, 18–26 CrossRef CAS.
- S. P. S. Badwal, A. P. Kulkarni, H. Ju and S. Giddey, Electrochemical Science for a Sustainable Society: A Tribute to John O'M Bockris, Springer International Publishing, 2017, pp. 223–259 Search PubMed.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, 84th edn, 2003, CRC Press Search PubMed.
- R. M. Choueiri, S. W. Tatarchuk, A. Klinkova and L. D. Chen, Electrochem. Sci. Adv., 2022, 2, e2100142 CAS.
- H. Gerischer and A. Mauerer, J. Electroanal. Chem., 1970, 25, 421–433 CrossRef CAS.
- H. G. Oswin and M. Salomon, Can. J. Chem., 1963, 41, 1686–1694 CrossRef CAS.
- W. Xu, D. Du, R. Lan, J. Humphreys, D. N. Miller, M. Walker, Z. Wu, J. T. S. Irvine and S. Tao, Appl. Catal., B, 2018, 237, 1101–1109 CrossRef CAS.
- S. Johnston, L. Kemp, B. Turay, A. N. Simonov, B. H. R. Suryanto and D. R. MacFarlane, ChemSusChem, 2021, 14, 4793–4801 CrossRef CAS PubMed.
- R. Lan and S. Tao, Electrochem. Solid-State Lett., 2010, 13, B83–B86 CrossRef CAS.
- S. Johnston, S. Cohen, C. K. Nguyen, K. N. Dinh, T. D. Nguyen, S. Giddey, C. Munnings, A. N. Simonov and D. R. MacFarlane, ChemSusChem, 2022, 15, e202200614 CrossRef CAS PubMed.
- H. Kim, M. W. Chung and C. H. Choi, Electrochem. Commun., 2018, 94, 31–35 CrossRef CAS.
- S. Le Vot, L. Roué and D. Bélanger, J. Power Sources, 2013, 223, 221–231 CrossRef CAS.
- M. Jabłońska and R. Palkovits, Appl. Catal., B, 2016, 181, 332–351 CrossRef.
- M. Cooper and G. G. Botte, J. Electrochem. Soc., 2006, 153, A1894 CrossRef CAS.
- D. J. Little, D. O. Edwards, M. R. Smith and T. W. Hamann, ACS Appl. Mater. Interfaces, 2017, 9, 16488–16494 CrossRef PubMed.
- K. Schmidt-Szałowski, K. Krawczyk and J. Petryk, Appl. Catal., A, 1998, 175, 147–157 CrossRef.
- F. Almomani, R. Bhosale, M. Khraisheh, A. Kumar and M. Tawalbeh, Int. J. Hydrogen Energy, 2020, 45, 10398–10408 CrossRef CAS.
- Y. J. Shih, Y. H. Huang and C. P. Huang, Electrochim. Acta, 2018, 263, 261–271 CrossRef CAS.
- A. Kapałka, A. Cally, S. Neodo, C. Comninellis, M. Wächter and K. M. Udert, Electrochem. Commun., 2010, 12, 18–21 CrossRef.
- N. M. Adli, H. Zhang, S. Mukherjee and G. Wu, J. Electrochem. Soc., 2018, 165, J3130–J3147 CrossRef CAS.
- M. Alsabet, M. Grden and G. Jerkiewicz, Electrocatalysis, 2014, 5, 136–147 CrossRef CAS.
- M. Alsabet, M. Grden and G. Jerkiewicz, Electrocatalysis, 2011, 2, 317–330 CrossRef CAS.
- A. D. Jagadale, D. P. Dubal and C. D. Lokhande, Mater. Res. Bull., 2012, 47, 672–676 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- Z. Zhou, C. Chen, M. Gao, B. Xia and J. Zhang, Green Chem., 2019, 21, 6699–6706 RSC.
- B. Zhu, C. Chen, L. Huai, Z. Zhou, L. Wang and J. Zhang, Appl. Catal., B, 2021, 297, 120396 CrossRef CAS.
- L. van Zwieten, S. Kimber, S. Morris, A. Downie, E. Berger, J. Rust and C. Scheer, Aust. J. Soil Res., 2010, 48, 555–568 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01835k |
| This journal is © The Royal Society of Chemistry 2023 |