
Stabilizing Cu0–Cu+ sites by Pb-doping for highly efficient CO2 electroreduction to C2 products†
Xiaodong
Ma
ab,
Xinning
Song
 ab,
Libing
Zhang
ab,
Limin
Wu
ab,
Jiaqi
Feng
ab,
Shunhan
Jia
ab,
Xingxing
Tan
ab,
Liang
Xu
ab,
Xiaofu
Sun
*ab and
Buxing
Han
*abc
ab,
Libing
Zhang
ab,
Limin
Wu
ab,
Jiaqi
Feng
ab,
Shunhan
Jia
ab,
Xingxing
Tan
ab,
Liang
Xu
ab,
Xiaofu
Sun
*ab and
Buxing
Han
*abc
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Colloid and Interface and Thermodynamics, Center for Carbon Neutral Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: sunxiaofu@iccas.ac.cn; hanbx@iccas.ac.cn
bSchool of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
cShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, P. R. China
First published on 15th August 2023
Abstract
The electrochemical CO2 reduction reaction (CO2RR) can convert CO2 to C2 hydrocarbons and oxygenate over Cu-based catalysts, and has great potential to store renewable energy and close the carbon cycle. Developing a facile method to modify the local electronic structure of Cu is a useful way to design efficient catalysts. Herein, we design a Pb-doping Cu2O catalyst with controllable Cu0–Cu+ sites. The catalyst generated a high C2 faradaic efficiency (FE) of 83.9% with a current density of 203.8 mA cm−2 at −1.1 V vs. RHE in a flow cell. In situ X-ray absorption spectroscopy and Raman spectroscopy revealed that the Pb doping in Cu2O could stabilize the Cu0–Cu+ structure and enhance the CO adsorption and C–C coupling, leading to high activity for C2 product formation. Theoretical calculations also show that Pb doping could reduce the energy barrier for both CO2 activation and C–C coupling processes.
Introduction
The electrochemical CO2 reduction reaction (CO2RR) can convert CO2 into valuable fuels or chemicals, which have great potential to mitigate the greenhouse effect, alleviate the energy crisis, and store intermittent electrical energy.1–4 C2 products including ethylene, ethanol, and acetic acid have higher energy density and economic value per unit mass compared with their C1 counterparts.5–9 Current research has paid increasing attention to the enhancement of conversion and selectivity for CO2-to-C2 products. Cu-based catalysts have been widely studied owing to their excellent performance in the electrochemical CO2RR to C2+ products.10–15 Previous studies show that the Cu0–Cu+ sites in Cu-based catalysts are the key active sites for producing the C2+ products, which are favorable for CO2 activation and C–C coupling.16–20 Therefore, it is of interest to modify the local electronic structure of Cu to achieve high activity and selectivity for CO2-to-C2 products.Unfortunately, the surface Cu+ is usually unstable, and it is prone to be reduced to Cu0 at the high applied reducing potentials during the CO2RR, leading to the loss of the performance of the CO2RR to give C2 products.16,21–25 Hence, it is extremely important to stabilize Cu+ in the catalysts.26,27 Some strategies such as surface modification,17 electropolishing,28 and doping29 can stabilize the surface Cu+ species. Among them, introducing a p-block metal dopant into Cu has been shown to be a promising strategy. It has been reported that the introduction of a p-block metal can effectively stabilize Cu+ in Cu-based catalysts.30–33 Not only that, some p-block metal elements such as In, Sn, Bi, and Pb have O affinity and can reduce the reaction energy barrier of the CO2RR, as well as show high overpotentials for the H2 evolution reaction (HER).34–37 For example, Xie et al. screened Cu-based bimetallic catalysts for the CO2RR to form C2+ products and found that Sn, In, Pb, and Bi in Cu-based bimetallic catalysts exhibited performances for inhibiting the HER.35 Bai et al. used first-principles calculations to find that Sn doping into Cu-based catalysts can effectively suppress the HER without altering the activity toward CO2 reduction, thereby improving the FE of ethanol.36 Li et al. found that introducing Sn can enhance the surface oxophilicity of Cu–Sn alloy catalysts, which plays an important role in guiding the protonation of the key oxygenic intermediate and transforming CO2 into ethanol.34 Wang et al. developed Cu–Bi bimetallic aerogels as catalysts, which successfully improved the faradaic efficiency (FE) of ethylene in the CO2RR.37 Therefore, developing facile methods to construct p-block metal doped Cu catalysts with controllable Cu0–Cu+ sites is an effective way to realize the CO2-to-C2 products with high efficiency.
In this work, we have designed a Pb-doped Cu2O electrocatalyst for highly efficient CO2 reduction to C2 products. It was discovered that the FE of C2 products could reach 83.9% with good stability. Experimental and density functional theory (DFT) studies indicated that the doping of Pb in the catalyst can stabilize the Cu0–Cu+ structure, which could reduce the energy barrier of CO2 activation and C–C coupling. As a result, it can improve the performance of the CO2RR to form C2 products.
Results and discussion
The detailed fabrication procedures of the electrocatalysts are discussed in the Methods section. In brief, as shown in Fig. 1A, 1 mmol of copper sulfate, 5 mL of ethanol, and 2 mL of oleic acid were added into 15 mL of deionized water and heated to 80 °C. Then, 5 mL of 1 M NaOH solution and 5 mL of 2 M glucose solution were added into the flask. The obtained reddish-brown precipitate was Cu2O.38 Subsequently, the Cu2O powder was evenly dispersed in the lead acetate solution and stirred for a long time without light and air. The mass fraction of Pb doped into Cu2O can be controlled with different stirring times. After stirring, we can obtain a series of Pb/Cu2O-x catalysts, where x is the mass fraction of Pb in the catalysts measured by inductively coupled plasma optical emission spectroscopy (ICP-OES). The measured mass fraction of Pb was 1.2%, 2.1%, 3.4%, and 6.2%, respectively. Pb/Cu2O-x was taken and uniformly loaded on a gas diffusion electrode (GDE) and electrochemically activated in a 3 M KOH aqueous solution. Finally, the e-Pb/Cu2O-x GDE was obtained, where e stands for “after electrochemical activation”.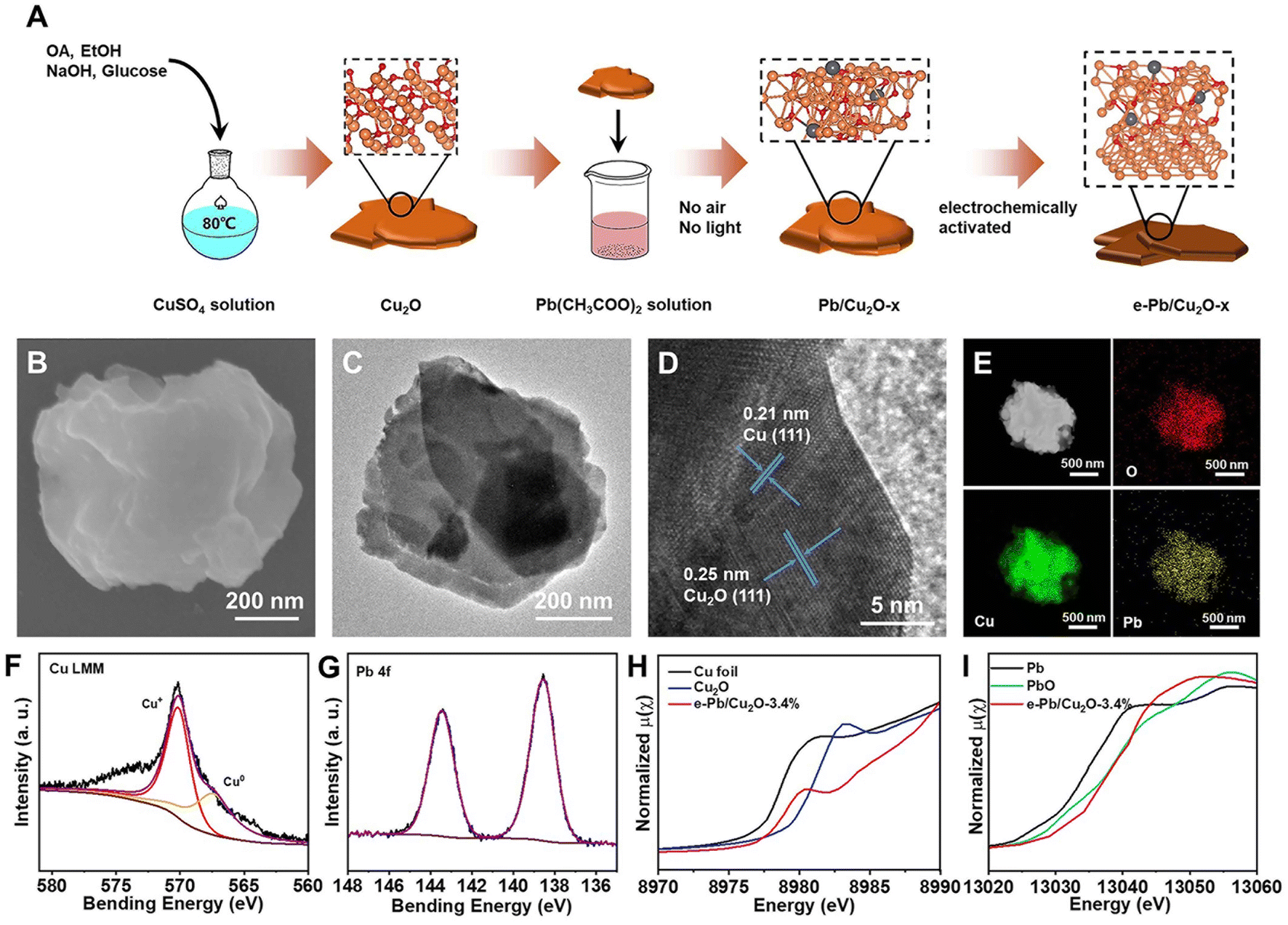 | ||
| Fig. 1 (A) Schematic illustration of the preparation of e-Pb/Cu2O-x. (B) SEM image of e-Pb/Cu2O-3.4%. (C and D) TEM and HRTEM images of e-Pb/Cu2O-3.4%. (E) Elemental mappings of e-Pb/Cu2O-3.4%. (F) Cu LMM XPS spectra of e-Pb/Cu2O-3.4%. (G) Pb 4f XPS spectra of e-Pb/Cu2O-3.4%. (H) Cu K-edge XANES spectra of e-Pb/Cu2O-3.4%. (I) Pb L-edge XANES spectra of e-Pb/Cu2O-3.4%. | ||
The structure of e-Pb/Cu2O-x was confirmed through powder X-ray diffraction (XRD) characterization, which showed the diffraction peaks of Cu, Cu2O, and PbO (Fig. S1†). This result preliminarily indicated the co-existence of Cu0 and Cu+ in the catalysts. The scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of e-Pb/Cu2O-3.4% are shown in Fig. 1B and C. We can see that e-Pb/Cu2O-3.4% has a sheet-like morphology with an average diameter ranging from 400 to 600 nm. Other e-Pb/Cu2O-x had similar morphological characteristics (Fig. S2†). The high-resolution TEM (HRTEM) images in Fig. 1D and Fig. S3† show that the spacings of the lattice fringe for all of the e-Pb/Cu2O-x were 0.25 nm and 0.21 nm, which can be assigned to the (111) plane of Cu2O and (111) plane of Cu, respectively.39,40 Elemental distribution mappings illustrated a uniform distribution of Cu, O, and Pb over the entire architecture (Fig. 1E).
X-ray photoelectron spectroscopy (XPS) analysis was performed to study the composition and chemical nature of the catalysts. The spectra of Cu 2p for e-Pb/Cu2O-3.4% (Fig. S4†) displayed the fitting peaks, which were attributed to Cu+ or Cu0 (932.1 eV and 951.9 eV),41,42 and the Auger LMM spectra of Cu further clarified two fitting peaks at 569.9 eV and 567.9 eV that belonged to Cu+ and Cu0 (Fig. 1F).42,43 The spectra of Pb 4f (Fig. 1G) displayed the two fitting peaks of Pb at 138.5 and 143.2 eV, indicating that the valence of Pb in e-Pb/Cu2O-3.4% was +2.44 The same conclusions can be obtained from the XPS spectra of other e-Pb/Cu2O-x samples (Fig. S5–S7†). Furthermore, X-ray absorption spectroscopy (XAS) was used to explore the detailed electronic structures of e-Pb/Cu2O-3.4%. The X-ray absorption near-edge structure (XANES) spectra showed that the near-edge absorption energy (E0) of Cu K-edge for e-Pb/Cu2O-3.4% was between those of Cu and Cu2O (Fig. 1H). It can be concluded that the average valence of Cu in e-Pb/Cu2O-3.4% was between 0 and +1.45–47 The coordination structures for Cu can be confirmed by the extended X-ray absorption fine structure (EXAFS) spectra (Fig. S8A and S9A†). The Cu–O coordination of Cu2O and e-Pb/Cu2O-3.4% were at the same peak (1.52 Å). The peak of the Cu–Cu coordination (2.61 Å) of e-Pb/Cu2O-3.4% was between Cu and Cu2O. These results further indicated the coexistence of Cu2O and Cu structures in e-Pb/Cu2O-3.4%.
Similarly, the E0 of Pb L-edge was almost the same as that of PbO (Fig. 1I), demonstrating that the valence of Pb in e-Pb/Cu2O-3.4% was +2.48 The EXAFS spectra of Pb (Fig. S8B and S9B†) also showed that the Pb–O and Pb–Pb coordination was similar to those of PbO. These results are consistent with the data of XRD, XPS, and HRTEM.
The CO2RR electrocatalytic activity of the as-prepared catalysts was carried out in a flow cell. In a typical experiment, the 3 M KOH aqueous solution was used as the electrolyte. 1H nuclear magnetic resonance (NMR) spectroscopy and gas chromatography (GC) were adopted to determine and quantify the liquid and gas products from the CO2RR. We can detect ethanol, acetate, formate, and trace methanol in liquid products, and H2, CO, C2H4, and trace CH4 in gas products. It can be found that the total FE of C2 products with e-Pb/Cu2O-x showed volcano-shaped dependence on the applied potentials (Fig. 2A). Significantly, e-Pb/Cu2O-3.4% had the best activity with the highest C2 product FE. The maximum C2 product FE could reach 83.9% with a current density of 203.8 mA cm−2 at −1.1 V vs. RHE (Fig. 2B and C), which is comparable to those of many reported systems (Table S1†). The FEs of C2H4, ethanol, and acetate were 32.8%, 42.5%, and 8.6%, respectively. Different Pb contents in the catalysts can affect the CO2-to-C2 product performance obviously (Fig. S10†). The FE of C2 products increased with the increase of Pb. However, when the mass fraction of Pb increased to 6.2%, the yield of CO increased, and the FE of the C2 product was only 54.0%. In addition, different Pb contents in the catalysts can also affect the HER in the CO2RR (Fig. S11†). The FE of H2 was reduced until the Pb increased to 3.4%. Then the H2 FE increased when the mass fraction of Pb increased to 6.2%. We then performed electrochemical impedance spectroscopy (EIS) to investigate the electrode/electrolyte interface properties. The Nyquist plots were obtained by running the experiments at an open circuit potential (Fig. 2D). e-Pb/Cu2O-3.4% had lower interfacial charge transfer resistance, which ensures a faster electron transfer during the reaction. These results indicated that appropriate Pb doping in e-Pb/Cu2O-x could improve the C2 product selectivity.
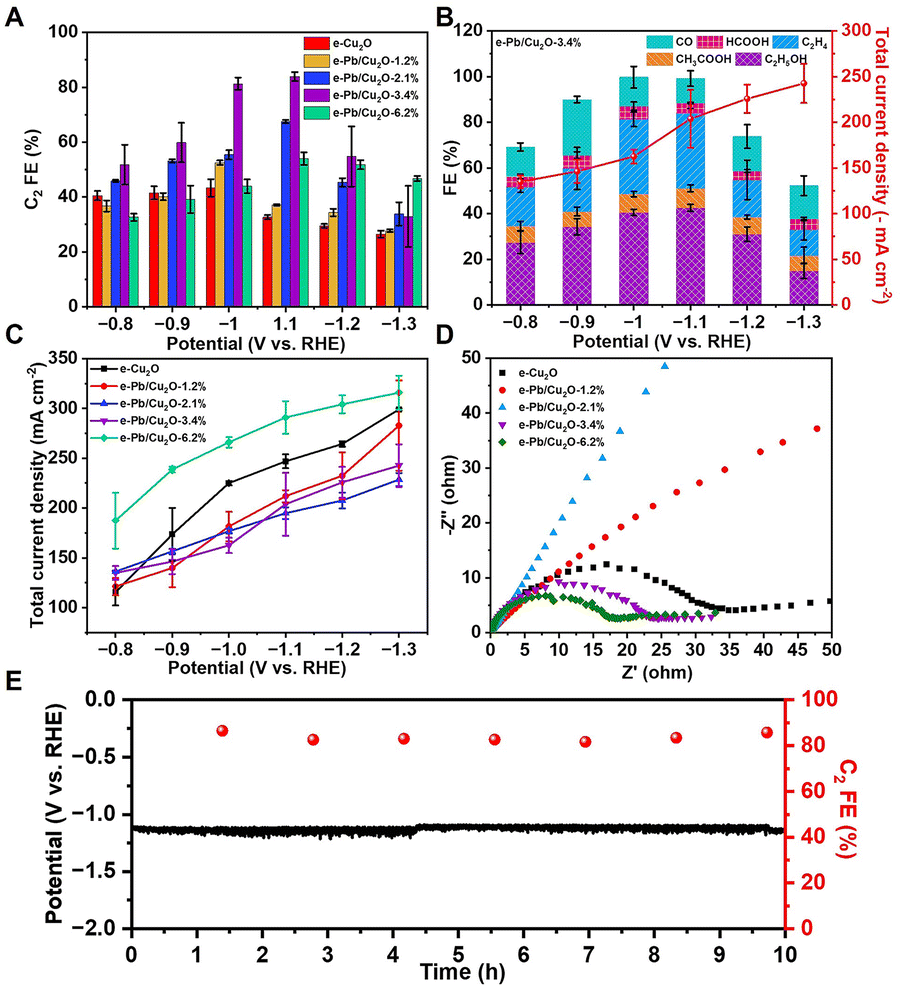 | ||
| Fig. 2 (A) The FE for C2 production over various catalysts at different applied potentials. (B) The FE of different products and total current density for e-Pb/Cu2O-3.4% at different applied potentials. (C) The total current density for e-Cu2O, e-Pb/Cu2O-1.2%, e-Pb/Cu2O-2.1%, e-Pb/Cu2O-3.4% and e-Pb/Cu2O-6.2% at different applied potentials. (D) Electrochemical impedance spectra for e-Cu2O, e-Pb/Cu2O-1.2%, e-Pb/Cu2O-2.1%, e-Pb/Cu2O-3.4%, and e-Pb/Cu2O-6.2% at the open circuit potential (OCP) shown as a Nyquist plot. (E) Stability test for e-Pb/Cu2O-3.4% at a constant current density of 200 mA cm−2. | ||
The stability of e-Pb/Cu2O-3.4% in the flow cell system at a constant current density of 200 mA cm−2 is shown in Fig. 2E. There was no obvious decay in the potential and FE of C2 products during the 10 h test. e-Pb/Cu2O-3.4% after the reaction, denoted as e-Pb/Cu2O-3.4%-R, was examined by TEM and XRD. The morphology and structure of the catalysts did not change notably, further indicating its excellent stability (Fig. S12 and S13†). The fine structure was analyzed by XAS (Fig. S14†). The valence and structure of Cu and Pb in e-Pb/Cu2O-3.4%-R were almost the same as those in e-Pb/Cu2O-3.4%. These results suggest that the structure of the catalyst can remain stable during the CO2RR.
To explore the behavior of e-Pb/Cu2O-3.4% in the CO2RR, we carried out detailed experimental analysis. In situ XAS was first performed to monitor the valence change and coordination environment of Cu under different potentials. In Fig. 3A, the Cu K-edge E0 of e-Pb/Cu2O-3.4% reflects the change in the oxidation state. Compared with the data of the Cu K-edge E0 of Cu foil and Cu2O, we can determine the valence of Cu in e-Pb/Cu2O-3.4% at each applied potential (Fig. 3B). It can be seen that the average valence of Cu in e-Pb/Cu2O-3.4% decreased from +0.95 at OCP to +0.8 at −0.9 V vs. RHE, and finally it would be stable around +0.4 under a higher potential. This indicated that the local electronic structure of Cu with positive valence sites could be retained during the CO2RR.45–47 From the EXAFS spectra in Fig. 3C, it can be seen that the Cu–Cu and Cu–O coordination is close to those in Cu2O at −0.9 V vs. RHE, suggesting that e-Pb/Cu2O-3.4% could still maintain the Cu2O structure, but a small amount of Cu0 appeared. When the applied potential increased to a value higher than −1.0 V vs. RHE, the Cu–Cu coordination of e-Pb/Cu2O-3.4% was close to that of Cu foil, and the Cu–O coordination of e-Pb/Cu2O-3.4% was close to that of Cu2O. The complete Cu2O structure could not be maintained in the catalysts, and a part of Cu+ was reduced to Cu0. These results prove that the Cu0–Cu+ structure exists in e-Pb/Cu2O-3.4% during the electrochemical CO2RR.
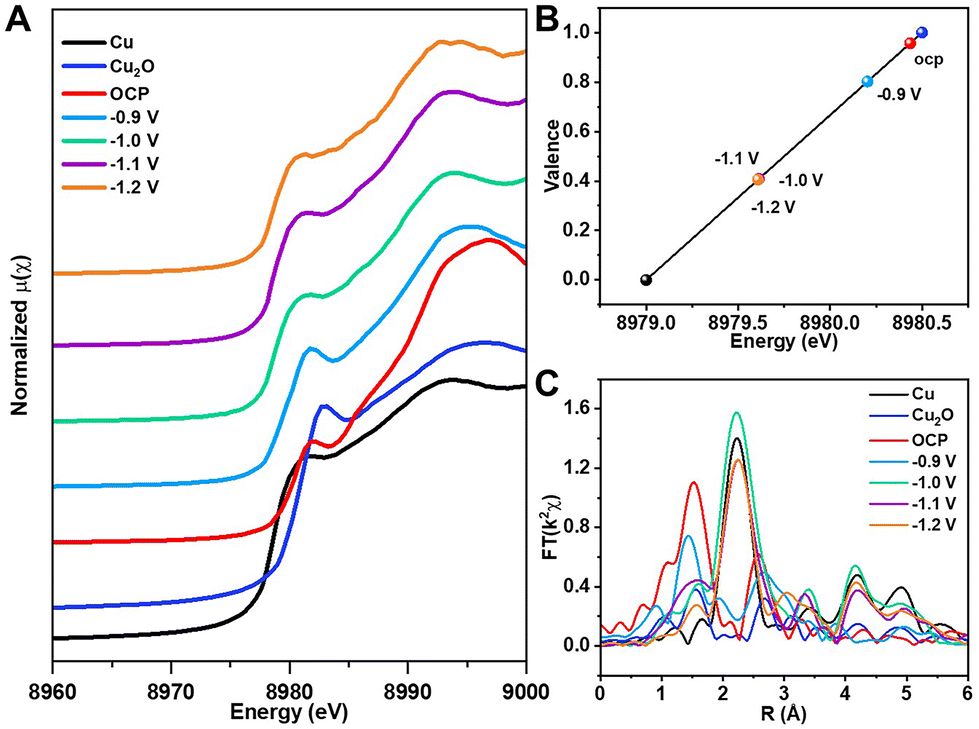 | ||
| Fig. 3 (A) In situ XANES spectra at Cu K-edge for e-Pb/Cu2O-3.4% at different applied potentials. (B) The average oxidation state of Cu in e-Pb/Cu2O-3.4% at different applied potentials from Cu K-edge XANES. (C) R spatial data of Cu K-edge EXAFS spectra for e-Pb/Cu2O-3.4% at different applied potentials. | ||
In situ Raman spectroscopy measurements were conducted to identify the possible intermediates. As shown in Fig. 4A, the Raman peaks around 2060 cm−1, 1556 cm−1, and 1436 cm−1 can be attributed to *CO, *COO−, and *HCOO−, respectively.15,49–53 These important reaction intermediates were adsorbed on the surface of e-Pb/Cu2O-3.4% during the CO2RR, which could promote the occurrence of C–C coupling and increase the FE of C2 products. The peak at 603 cm−1 was attributed to the Cu2O structure.50,53,54 However, it would disappear when the applied potential was higher than −1.1 V vs. RHE. Moreover, the new characteristic peak appeared at 525 cm−1, which can be attributed to CuOx.50,53,54 It suggested the existence of the Cu0–Cu+ structure in the catalysts. The signal of Cu–CO was also detected at 361 cm−1, indicating that the generated CO was adsorbed on the catalyst.
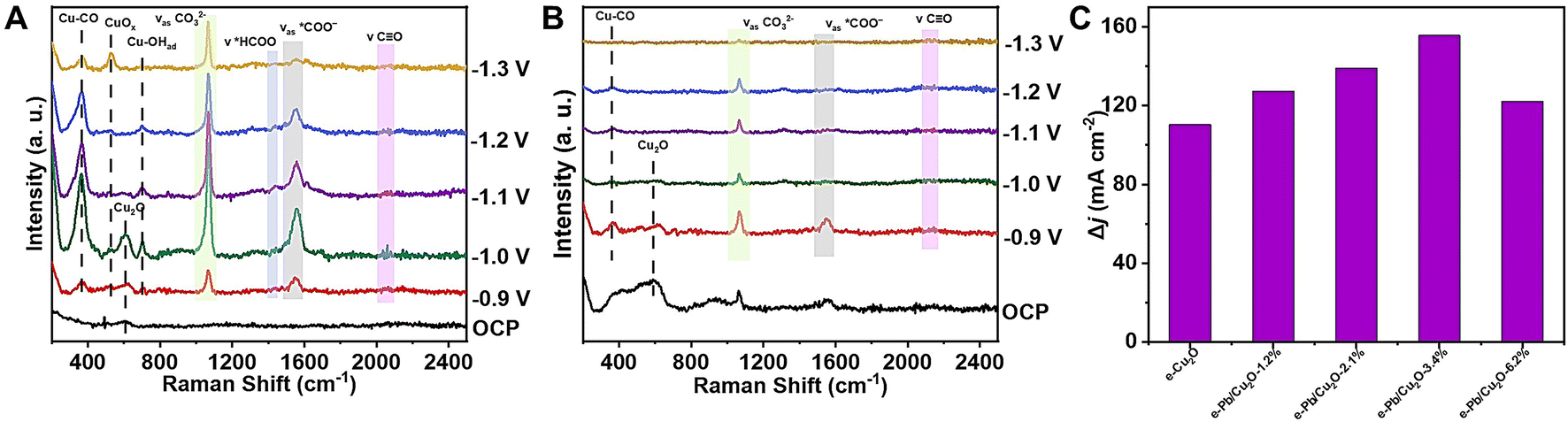 | ||
| Fig. 4 (A) In situ Raman spectra at different applied potentials for e-Pb/Cu2O-3.4%. (B) In situ Raman spectra at different applied potentials for e-Cu2O. (C) The results of CO adsorption responses for the catalysts. | ||
For comparison, we also obtained the in situ Raman spectra over e-Cu2O (Fig. 4B). At a potential of −1.1 V vs. RHE, we cannot observe the CuOx characteristic peak at 525 cm−1, indicating that Cu2O was reduced to Cu during the reaction.55 Compared with the Cu0–Cu+ structure, Cu0 had a poor performance for C–C coupling. Therefore, the doping of Pb in the catalyst could stabilize the Cu0–Cu+ structure and improve the catalytic performance for CO2-to-C2 products.
According to Raman data, CO is an important intermediate in CO2-to-C2 products. The increase of CO adsorption on the catalysts can increase the C2 product FE. The detailed experimental procedures of the gas electroresponse experiments are discussed in the Methods section. The gas electroresponse experiments clearly showed the capacity for the adsorption of CO on e-Pb/Cu2O-x and e-Cu2O (Fig. 4C).56 Compared with other materials, e-Pb/Cu2O-3.4% showed the best capacity of adsorbing CO. This indicated that moderate Pb doping can improve the adsorption of a *CO intermediate, leading to high catalytic performance for C2 product formation.
To better understand the reaction mechanism, we performed correlative theoretical calculations through DFT. All the simulated data and detailed procedures are shown in the ESI.† Based on the materials characterization, we have established the structural model of e-Pb/Cu2O-3.4%. For e-Pb/Cu2O-3.4%, a Cu(111) and Cu2O(111) facet heterostructure was used as a model and a part of Cu in Cu2O was replaced with Pb (Fig. S15†). We named it Cu(111)–Cu2O(111)–Pb. Previous studies showed that the adsorption of *CO on Cu+ is stronger than that on Cu0.28,57 The Cu2O(111) facet and Cu(111) were used to compare with Cu(111)–Cu2O(111)–Pb to explore the effect of Pb doping.
The catalytic pathway of CO2 activation is illustrated in Fig. 5A. It can be seen that the formation of *COOH from *CO2 on the three models was highly endergonic and acted as the rate-determining step. The Gibbs free energy for *CO2 to *COOH on Cu(111)–Cu2O(111)–Pb was only about 0.33 eV, which was much lower than that on Cu2O(111) (0.73 eV) and Cu(111) (0.85 eV). This indicated that the doping of Pb greatly reduced the reaction energy barrier of *CO2 to *COOH, which contributed to easier CO2 activation. The C–C coupling process was studied subsequently (Fig. 5B). We have compared different coupling pathways (*CO–*CO, *CO–*COH, *CO–*CHO, and *CHO–*CHO) and found that the *CO–*COH coupling had a lower Gibbs free energy. The Gibbs free energy of this coupling process on Cu(111)–Cu2O(111)–Pb was −0.31 eV, while it would be highly endergonic on Cu2O(111) (0.69 eV) and Cu(111) (2.26 eV).
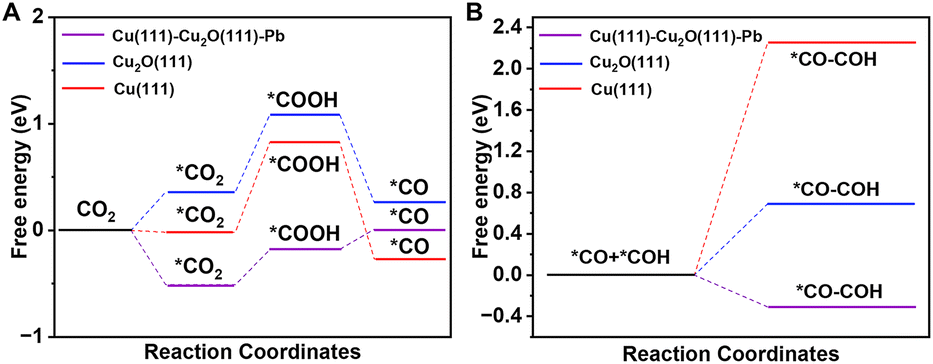 | ||
| Fig. 5 (A) Gibbs free-energy diagrams for CO2 activation on different simulated models. (B) Gibbs free-energy diagrams for C–C coupling on different simulated models. | ||
Therefore, by combining the above simulation data, it is found that Pb doping can be beneficial not only for the activation of CO2 but also the improvement of the C–C coupling, leading to the enhancement of the CO2RR to form C2 products.
Conclusions
In summary, a series of Pb-doped Cu2O catalysts have been successfully designed and synthesized for the efficient electrochemical CO2RR to form C2 products. e-Pb/Cu2O-3.4% exhibited the highest performance with a C2 product FE of 83.9% in the flow cell. The catalyst also showed good stability in 10 h. In situ characterization revealed that Pb doping could stabilize the Cu0–Cu+ structure in e-Pb/Cu2O-3.4% during the electrochemical CO2RR, which had strong adsorption ability for the *CO intermediate. DFT calculations suggested that the doping of Pb could reduce the reaction energy barrier of *CO2 to *COOH and C–C coupling processes simultaneously. This work provides a facile strategy for the design of Cu-based catalysts to improve the production of C2 products from the CO2RR. We believe that it may inspire new exploration of electrocatalyst design in the future.Author contributions
X.D.M., X.F.S., and B.X.H. proposed the project, designed the experiments, and wrote the manuscript; M.X.D. performed the whole experiments; X.N.S., L.B.Z., L.M.W., J.Q.F., S.H.J., X.X.T., and L.X. performed the analysis of experimental data; X.F.S. and B.X.H. supervised the whole project.Data availability
All experimental data are available in the ESI.†Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The work was supported by the National Key Research and Development Program of China (2020YFA0710203), National Natural Science Foundation of China (22002172, 22203099, 21890761, and 22121002), Beijing Natural Science Foundation (J210020), China Postdoctoral Science Foundation (2022M713200), and Photon Science Center for Carbon Neutrality. The X-ray absorption spectroscopy measurements were performed at Beamline 1W1B and 4B9A at Beijing Synchrotron Radiation Facility (BSRF).References
- I. Sullivan, A. Goryachev, I. A. Digdaya, X. Li, H. Atwater, D. A. Vermaas and C. Xiang, Nat. Catal., 2021, 4, 952–958 CrossRef CAS.
- Y. Zhai, P. Han, Q. Yun, Y. Ge, X. Zhang, Y. Chen and H. Zhang, eScience, 2022, 2, 467–485 CrossRef.
- L. Zhang, J. Feng, S. Liu, X. Tan, L. Wu, S. Jia, L. Xu, X. Ma, X. Song, J. Ma, X. Sun and B. Han, Adv. Mater., 2023, 35, 2209590 CrossRef CAS PubMed.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, Z. Zhang and B. Han, Angew. Chem., Int. Ed., 2016, 55, 6771–6775 CrossRef CAS PubMed.
- P. Li, J. Bi, J. Liu, Y. Wang, X. Kang, X. Sun, J. Zhang, Z. Liu, Q. Zhu and B. Han, J. Am. Chem. Soc., 2023, 145, 4675–4682 CrossRef CAS PubMed.
- W. Nie, G. P. Heim, N. B. Watkins, T. Agapie and J. C. Peters, Angew. Chem., Int. Ed., 2023, 62, e202216102 CrossRef CAS PubMed.
- D. Wang, J. Mao, C. Zhang, J. Zhang, J. Li, Y. Zhang and Y. Zhu, eScience, 2023, 3, 100119 CrossRef.
- R. De, S. Gonglach, S. Paul, M. Haas, S. S. Sreejith, P. Gerschel, U. P. Apfel, T. H. Vuong, J. Rabeah, S. Roy and W. Schofberger, Angew. Chem., Int. Ed., 2020, 59, 10527–10534 CrossRef CAS PubMed.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- L. Xu, X. Ma, L. Wu, X. Tan, X. Song, Q. Zhu, C. Chen, Q. Qian, Z. Liu, X. Sun, S. Liu and B. Han, Angew. Chem., Int. Ed., 2022, 61, e202210375 CrossRef CAS PubMed.
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. F. Guzman, J. Feijoo, P. C. Chen, H. Wang, C. J. Pollock, X. Huang, Y. T. Shao, C. Wang, D. A. Muller, H. D. Abruna and P. Yang, Nature, 2023, 614, 262–269 CrossRef CAS PubMed.
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Joule, 2021, 5, 429–440 CrossRef CAS.
- Y. Liang, J. Zhao, Y. Yang, S. Hung, J. Li, S. Zhang, Y. Zhao, A. Zhang, C. Wang, D. Appadoo, L. Zhang, Z. Geng, F. Li and J. Zeng, Nat. Commun., 2023, 14, 474 CrossRef CAS PubMed.
- Y. Yang, A. He, H. Li, Q. Zou, Z. Liu, C. Tao and J. Du, ACS Catal., 2022, 12, 12942–12953 CrossRef CAS.
- X. Yan, C. Chen, Y. Wu, Y. Chen, J. Zhang, R. Feng, J. Zhang and B. Han, Green Chem., 2022, 24, 1989–1994 RSC.
- X. Yuan, S. Chen, D. Cheng, L. Li, W. Zhu, D. Zhong, Z. J. Zhao, J. Li, T. Wang and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 15344–15347 CrossRef CAS PubMed.
- C. Lim, M. Yilmaz, J. M. Arce-Ramos, A. D. Handoko, W. J. Teh, Y. Zheng, Z. Khoo, M. Lin, M. Isaacs, T. Tam, Y. Bai, C. K. Ng, B. S. Yeo, G. Sankar, I. Parkin, K. Hippalgaonkar, M. Sullivan, J. Zhang and Y. Lim, Nat. Commun., 2023, 14, 335 CrossRef CAS PubMed.
- M. Li, Y. Ma, J. Chen, R. Lawrence, W. Luo, M. Sacchi, W. Jiang and J. Yang, Angew. Chem., Int. Ed., 2021, 60, 11487–11493 CrossRef CAS PubMed.
- P. M. Krzywda, A. P. Rodríguez, N. E. Benes, B. T. Mei and G. Mul, Appl. Catal., B, 2022, 316, 121512 CrossRef CAS.
- C. Liu, X. Zhang, J. Huang, M. Guan, M. Xu and Z. Gu, ACS Catal., 2022, 12, 15230–15240 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- H. Li, T. Liu, P. Wei, L. Lin, D. Gao, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2021, 60, 14329–14333 CrossRef CAS PubMed.
- J. Sang, P. Wei, T. Liu, H. Lv, X. Ni, D. Gao, J. Zhang, H. Li, Y. Zang, F. Yang, Z. Liu, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2022, 61, e202114238 CrossRef CAS PubMed.
- Y. Zang, T. Liu, P. Wei, H. Li, Q. Wang, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2022, 61, e202209629 CrossRef CAS PubMed.
- J. Feng, L. Wu, S. Liu, L. Xu, X. Song, L. Zhang, Q. Zhu, X. Kang, X. Sun and B. Han, J. Am. Chem. Soc., 2023, 145, 9857–9866 CrossRef CAS PubMed.
- J. Liu, L. Cheng, Y. Wang, R. Chen, C. Xiao, X. Zhou, Y. Zhu, Y. Li and C. Li, J. Mater. Chem. A, 2022, 10, 8459–8465 RSC.
- S. Mu, H. Lu, Q. Wu, L. Li, R. Zhao, C. Long and C. Cui, Nat. Commun., 2022, 13, 3694 CrossRef CAS PubMed.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- Y. Wang, L. Cheng, Y. Zhu, J. Liu, C Xiao, R. Chen, L. Zhang, Y. Li and C. Li, Appl. Catal., B, 2022, 317, 121650 CrossRef CAS.
- R. Imania, Z. Qiu, R. Younesi, M. Pazokib, D. Fernandes, P. Mitev, T. Edvinsson and H. Tian, Nano Energy, 2018, 49, 40–50 CrossRef.
- W. Zhang, P. He, C. Wang, T. Ding, T Chen, X. Liu, L Cao, T. Huang, X. Shen, O. Usoltsev, A. Bugaev, Y. Lin and T. Yao, J. Mater. Chem. A, 2020, 8, 25970 RSC.
- W. Guo, S. Liu, X. Tan, R. Wu, X. Yan, C. Chen, Q. Zhu, L. Zheng, J. Ma, J. Zhang, Y. Huang, X. Sun and B. Han, Angew. Chem., Int. Ed., 2021, 60, 21979–21987 CrossRef CAS PubMed.
- M. Li, N. Song, W. Luo, J. Chen, W. Jiang and J. Yang, Adv. Sci., 2023, 10, 2204579 CrossRef CAS PubMed.
- M. Xie, Y. Shen, W. Ma, D. Wei, B. Zhang, Z. Wang, Y. Wang, Q. Zhang, S. Xie, C. Wang and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202213423 CrossRef CAS PubMed.
- X. Bai, L. Shi, Q. Li, C. Ling, Y. Ouyang, S. Wang and J. Wang, Energy Environ. Mater., 2022, 5, 892–898 CrossRef CAS.
- Y. Wang, L. Cheng, Y. Zhu, J. Liu, C. Xiao, R. Chen, L. Zhang, Y. Li and C. Li, Appl. Catal., B, 2022, 317, 121650 CrossRef CAS.
- C. Zhan, Q. Wang, L. Zhou, X. Han, Y. Wanyan, J. Chen, Y. Zheng, Y. Wang, G. Fu, Z. Xie and Z. Tian, J. Am. Chem. Soc., 2020, 142, 14134–14141 CrossRef CAS PubMed.
- P. Wang, H. Yang, Y. Xu, X. Huang, J. Wang, M. Zhong, T. Cheng and Q. Shao, ACS Nano, 2021, 15, 1039–1047 CrossRef CAS PubMed.
- Y. Jiang, T. Xia, L. Shen, J. Ma, H. Ma, T. Sun, F. Lv and N. Zhu, ACS Catal., 2021, 11, 2949–2955 CrossRef CAS.
- X. Ma, L. Xu, S. Liu, L. Zhang, X. Tan, L. Wu, J. Feng, Z. Liu, X. Sun and B. Han, Chem. Catal., 2022, 2, 3207–3224 CrossRef CAS.
- S. Lee, H. Jung, N. Kim, H. Oh, B. Min and Y. Hwang, J. Am. Chem. Soc., 2018, 140, 8681–8689 CrossRef CAS PubMed.
- X. Chang, T. Wang, Z. Zhao, P. Yang, J. Greeley, R. Mu, G. Zhang, Z. Gong, Z. Luo, J. Chen, Y. Cui, G. Ozin and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 15415–15419 CrossRef CAS PubMed.
- T. Ng, C. Chan, M. Lo, Z. Guan and C. Lee, J. Mater. Chem. A, 2015, 3, 9081–9085 RSC.
- S. F. Hung, A. Xu, X. Wang, F. Li, S. H. Hsu, Y. Li, J. Wicks, E. G. Cervantes, A. S. Rasouli, Y. C. Li, M. Luo, D. H. Nam, N. Wang, T. Peng, Y. Yan, G. Lee and E. H. Sargent, Nat. Commun., 2022, 13, 819 CrossRef CAS PubMed.
- X. Wang, P. Ou, A. Ozden, S.-F. Hung, J. Tam, C. M. Gabardo, J. Y. Howe, J. Sisler, K. Bertens, F. P. G. de Arquer, R. K. Miao, C. P. O’Brien, Z. Wang, J. Abed, A. S. Rasouli, M. Sun, A. H. Ip, D. Sinton and E. H. Sargent, Nat. Energy, 2022, 7, 170–176 CrossRef CAS.
- J. Li, A. Ozden, M. Wan, Y. Hu, F. Li, Y. Wang, R. R. Zamani, D. Ren, Z. Wang, Y. Xu, D. H. Nam, J. Wicks, B. Chen, X. Wang, M. Luo, M. Graetzel, F. Che, E. H. Sargent and D. Sinton, Nat. Commun., 2021, 12, 2808 CrossRef CAS PubMed.
- C. Li, Z. Li, J. Wang, W. Xiong, H. Yan, Y. Bai, D. O'Hare and Y. Zhao, Chem. Eng. J., 2023, 462, 141926 CrossRef CAS.
- G. Shi, Y. Xie, L. Du, X. Fu, X. Chen, W. Xie, T. Lu, M. Yuan and M. Wang, Angew. Chem., Int. Ed., 2022, 61, e202203569 CrossRef CAS PubMed.
- H. Li, P. Wei, D. Gao and G. Wang, Curr. Opin. Green Sustainable Chem., 2022, 34, 100589 CrossRef CAS.
- Z. Z. Niu, F. Y. Gao, X. L. Zhang, P. P. Yang, R. Liu, L. P. Chi, Z. Z. Wu, S. Qin, X. Yu and M. R. Gao, J. Am. Chem. Soc., 2021, 143, 8011–8021 CrossRef CAS PubMed.
- Z. Pan, K. Wang, K. Ye, Y. Wang, H.-Y. Su, B. Hu, J. Xiao, T. Yu, Y. Wang and S. Song, ACS Catal., 2020, 10, 3871–3880 CrossRef CAS.
- X. Chen, D. A. Henckel, U. O. Nwabara, Y. Li, A. I. Frenkel, T. T. Fister, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2019, 10, 672–682 CrossRef.
- Y. Zhao, X. Zu, R. Chen, X. Li, Y. Jiang, Z. Wang, S. Wang, Y. Wu, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2022, 144, 10446–10454 CrossRef CAS PubMed.
- M. F. Saleem, Y. A. Haleem, W. Sun, L. Ma and D. Wang, J. Raman Spectrosc., 2020, 51, 1286–1294 CrossRef CAS.
- B. Yang, K. Liu, H. Li, C. Liu, J. Fu, H. Li, J. E. Huang, P. Ou, T. Alkayyali, C. Cai, Y. Duan, H. Liu, P. An, N. Zhang, W. Li, X. Qiu, C. Jia, J. Hu, L. Chai, Z. Lin, Y. Gao, M. Miyauchi, E. Cortes, S. A. Maier and M. Liu, J. Am. Chem. Soc., 2022, 144, 3039–3049 CrossRef CAS PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, Fig. S1–S16 and Table S1. See DOI: https://doi.org/10.1039/d3gc01506h |
| This journal is © The Royal Society of Chemistry 2023 |