
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A measurement and modelling investigation of the indoor air chemistry following cooking activities†
Helen L.
Davies
 a,
Catherine
O'Leary
b,
Terry
Dillon
b,
David R.
Shaw
a,
Marvin
Shaw
b,
Archit
Mehra
c,
Gavin
Phillips‡
c and
Nicola
Carslaw
*a
a,
Catherine
O'Leary
b,
Terry
Dillon
b,
David R.
Shaw
a,
Marvin
Shaw
b,
Archit
Mehra
c,
Gavin
Phillips‡
c and
Nicola
Carslaw
*a
aDepartment of Environment and Geography, University of York, Heslington, York, UK. E-mail: nicola.carslaw@york.ac.uk
bWolfson Atmospheric Chemistry Laboratories, Department of Chemistry, University of York, Heslington, York, UK
cDepartment of Physical, Mathematical and Engineering Sciences, University of Chester, Chester, UK
First published on 14th August 2023
Abstract
Domestic cooking is a source of indoor air pollutants, including volatile organic compounds (VOCs), which can impact on indoor air quality. However, the real-time VOC emissions from cooking are not well characterised, and similarly, the resulting secondary chemistry is poorly understood. Here, selected-ion flow-tube mass spectrometry (SIFT-MS) was used to monitor the real-time VOC emissions during the cooking of a scripted chicken and vegetable stir-fry meal, in a room scale, semi-realistic environment. The VOC emissions were dominated by alcohols (70% of total emission), but also contained a range of aldehydes (14%) and terpenes (5%), largely attributable to the heating of oil and the preparation and heating of spices, respectively. The direct cooking-related VOC emissions were then simulated using the Indoor Chemical Model in Python (INCHEM-Py), to investigate the resulting secondary chemistry. Modelling revealed that VOC concentrations were dominated by direct emissions, with only a small contribution from secondary products, though the secondary species were longer lived than the directly emitted species. Following cooking, hydroxyl radical concentrations reduced by 86%, while organic peroxy radical levels increased by over 700%, later forming secondary organic nitrates, peroxyacylnitrates (PANs) and formaldehyde. Monoterpene emissions were shown to drive the formation of secondary formaldehyde, albeit to produce relatively modest concentrations (average of 60 ppt). Sensitivity analysis of the simulation conditions revealed that increasing the outdoor concentrations of ozone and NOx species (2.9× and 9×, respectively) resulted in the greatest increase in secondary product formation indoors (≈400%, 200% and 600% increase in organic nitrates, PANs and formaldehyde production, respectively). Given the fact that climate change is likely to result in increased ozone concentrations in the future, and that increased window-opening in response to rising temperatures is also likely, higher concentrations of indoor oxidants are likely in homes in the future. This work, therefore, suggests that cooking could be a more important source of secondary pollutants indoors in the future.
Environmental significance statementDomestic cooking emits a range of volatile organic compounds (VOCs), which subsequently react with oxidants (O3/OH/NO3) in ambient air, to form a plethora of secondary products. Some directly emitted and secondary compounds are associated with adverse health effects, therefore, understanding factors that affect their emission and formation is vital. Here, scripted cooking experiments are performed to obtain real-time VOC mixing ratios and emission rates, which are subsequently used in an indoor air chemistry model to investigate factors affecting secondary product formation. This work reveals strong links between secondary product formation and indoor oxidant levels and ventilation, both of which may be affected by future climate change. Our results will provide pointers to influence human behaviour change around cooking. |
1 Introduction
On average, people in developed countries spend approximately 90% of their time indoors,1 and the majority of their exposure to inhalable pollutants occurs in indoor environments. Therefore, the sources and quantities of potentially harmful pollutants are important considerations in the field of indoor air quality (IAQ) research. There are a wide variety of sources of one of the most common groups of indoor air pollutants, known as volatile organic compounds (VOCs). These sources include building materials (composite-woods, plastics, adhesives, floorings, etc.2), personal care products (shampoos, soaps, skin care products, etc.3), appliances (stoves, photocopiers, fires, air fresheners etc.2,4), domestic activities, such as cooking and cleaning,5,6 and the people inhabiting the space.7 Some of the species that are emitted or formed indoors are associated with adverse health effects. For example, small aldehydes, such as formaldehyde, acetaldehyde and acrolein, produced during cooking and combustion,8,9 have been linked to the induction and exacerbation of asthma,10 and formaldehyde is accepted to be a sensory irritant and human carcinogen.11 As such, workplace exposure limits for some chemicals such as formaldehyde exist,12 however, the toxicological effects of many VOCs produced indoors, are poorly understood.Indoors, VOCs can react with oxidants and radicals, such as ozone (O3), and the hydroxyl (OH) and nitrate (NO3) radicals, and form a wide range of secondary species, some of which are more harmful to health than the parent VOC.13 As an example, monoterpenes, such as limonene, are present in many cleaning and personal care products, due to their pleasant aromas.14 However, monoterpenes can be highly reactive with OH, O3 and NO3 (ref. 15 and 16) and, through multiple oxidation steps, can form a wide range of secondary products, such as organic nitrates, carbonyls (including formaldehyde and glyoxal), peroxyacylnitrates (PANs) and particulate matter (PM), among others.17 Some of these secondary products have been associated with detrimental health effects,18,19 though the toxicology of these, and many other secondary species, remains poorly characterised.
Domestic cooking is known to be a major source of VOCs. The types and quantities of VOCs produced depends strongly on the types of foods being cooked, the cooking method (e.g. boiling, frying, etc), the oil type and the type of stove.20 To date, the impact of cooking on IAQ has largely been limited to heating single ingredients in chamber-scale experiments, to determine the emissions in a highly controlled environment. For example, chamber experiments have revealed that the heating of cooking oils yields aldehydes, and the type of oil affects which aldehydes are emitted.21 The heating of herbs and pepper in another study, resulted in emission of terpene species.22
Aside from the chamber experiments, the House Observations of Microbial and Environmental Chemistry (HOMEChem) campaign was a large-scale project which investigated the effects of cooking and cleaning on IAQ in a test house.23 For this campaign, the researchers cooked various different meals, carried out cleaning with different products, and conducted a number of ‘layered’ days which involved both cooking and cleaning. This yielded large amounts of data relating to the emissions of VOCs and particles from the different activities. Alongside the determination of real-time particle concentrations and particle number size distributions as a result of cooking,24 the study also resulted in a ranking of the most-produced VOCs in the house, both during cooking, and when the house was unoccupied.5 However, the real-time concentrations of VOCs during cooking was restricted to a short time period around the cooking events.
When VOCs are emitted, they react readily with OH, and if they contain a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond (e.g. monoterpenes and sesquiterpenes), with O3. These oxidation reactions lead to the formation of numerous species, including peroxy radicals (RO2) and hydroperoxy radicals (HO2). After formation, RO2 species predominantly react with nitric oxide (NO), to form mainly alkoxy radicals (RO), but also organic nitrates (RNO3), through a minor pathway.
C double bond (e.g. monoterpenes and sesquiterpenes), with O3. These oxidation reactions lead to the formation of numerous species, including peroxy radicals (RO2) and hydroperoxy radicals (HO2). After formation, RO2 species predominantly react with nitric oxide (NO), to form mainly alkoxy radicals (RO), but also organic nitrates (RNO3), through a minor pathway.
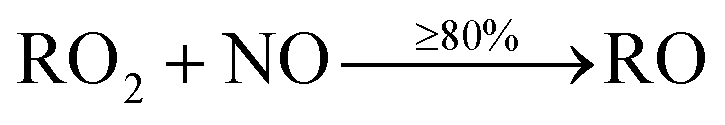 | (1) |
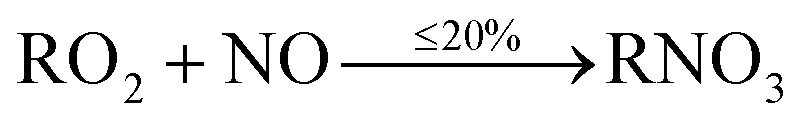 | (2) |
Depending on the specific species, at least 80% of the reaction between RO2 and NO proceeds via(1), while up to 20% is via(2). Production of organic nitrates is important to consider as they have the potential for causing a reduction in lung function following inhalation, and formation of nitric acid in lung fluid.25,26
The formation of RO species in (1) enables the subsequent formation of species such as aldehydes, via reaction with ambient O2.
| RO + O2 → HO2 + RCHO | (3) |
For instance, formaldehyde can be formed as a secondary product when a methoxy radical (CH3O), formed through (1), reacts with ambient O2via(3). The carcinogenicity and mutagenicity of formaldehyde is well documented, with the World Health Organization giving a guideline maximum exposure of 80 ppb for 30 min, which is considered to be low enough to prevent cancerous, and non-cancerous toxic effects.11,27,28
A subset of peroxy radicals, known as peroxyacetyl radicals (RCO3) can react with NO2, and form peroxyacyl nitrate (RCO3NO2, PAN) species as follows:
| RCO3 + NO2 → RCO3NO2. | (4) |
PANs are of interest as an indoor pollutant as they are known to be lachrymators, mutagens and phytotoxins.19
While significant work has been conducted on the effects of cooking on IAQ, understanding of the evolution of the chemistry before, during, and after cooking is absent. One of the aims of the IMPeCCABLE (IMPacts of Cooking and Cleaning on indoor Air quality: towards healthy BuiLdings for the futurE) project is to understand the emissions of a wide range of VOCs during cooking, and the evolution of the resulting secondary chemistry. Here, the first campaign of the IMPeCCABLE project is reported, where experiments were carried out in the DOMEstic Systems and Technology InCubator (DOMESTIC) facility at the University of Chester. The DOMESTIC facility was used to allow cooking (and cleaning) activities to be carried out in a simplified, room-scale environment, with a multitude of diagnostic instruments. A suite of online and offline diagnostic equipment was used to monitor the real-time concentrations of 43 VOCs, PM, O3 and NOx (NO, NO2) species, both indoors and outdoors. The experimental campaign yielded emission rates for VOCs emitted during the cooking experiments, and these were then used to model the experiments using a new, open-source, indoor chemistry box-model, the Indoor Chemical Model in Python (INCHEM-Py),29 to provide further insight into the secondary chemistry.
2 Methods
2.1 The DOMESTIC facility and diagnostic equipment
A one-month long experimental campaign was carried out during May 2021, at the Thornton Science Park, University of Chester, to investigate the effects of cooking and cleaning on IAQ. Experiments were carried out in the DOMESTIC facility, which consists of two 6.1 m shipping containers. Cooking experiments took place in one of the containers, which was fitted with an electric stove. The space was split into the main area (≈4.3 × 2.2 × 2.3 m), and a small bathroom at one end (≈1.5 × 2.2 × 2.3 m), as shown in Fig. S1.† The door to the bathroom was kept shut to reduce the influence of the extra volume on the results. The second container (≈5.8 × 2.2 × 2.3 m) contained an array of instrumentation, and was linked to the first via a duct, which housed the sample lines. In addition to the instrumentation container, diagnostic equipment was also housed in the Wolfson Atmospheric Chemistry Laboratory (WACL) Air Sampling Platform (WASP) van, which provides a mobile laboratory.30A Voice200 selected-ion flow-tube mass spectrometer (SIFT-MS, Syft Technologies, Christchurch, New Zealand) was deployed to measure targeted VOC mixing ratios both inside and outside the container. The SIFT-MS principles of operation are discussed in detail elsewhere,30–32 with only operating conditions listed here. The SIFT-MS was operated with a flow tube temperature of 120 °C, pressure of 460 mTorr, a flowtube voltage of 25 V, a sample flowrate of 100 sccm and a nitrogen (N2, Research grade, BOC) carrier gas flow of 1 Torr per L per s which was maintained throughout the measurement period. The microwave ion source current was operated at 40 mW and 300 mTorr pressure.
The SIFT-MS was used to measure 43 different VOCs and inorganic gases using selective ion monitoring mode (SIM), with a dwell time of 0.1 seconds per mass/charge ratio (m/z). This resulted in a total measurement cycle of 8.2 s. The compounds measured by SIFT-MS and the corresponding reagent ions, molecular masses and product ion molecular formulae are shown in Table S1.† The SIFT-MS was used to measure both indoor and outdoor concentrations via multi-port switching, using polytetrafluoroethylene (PTFE)-internally-coated solenoid valves (12VDC, GEMS30). A sequential cycle of 30 minutes indoor/5 minutes outdoor sampling was used for the duration of the measurement periods. The exception was during the cooking experiments, when only indoor measurements were taken for at least 20 minutes before and after the cooking start time (t0).
The instrument background was assessed daily (for 3 minutes at a time) by sampling zero air from an in-house heated palladium alumina-based zero air generator in SIM mode.30 The 3 minutes average background mixing ratios were subtracted from both indoor and outdoor ambient mixing ratios. Sensitivities for the compounds detected by SIFT-MS were determined daily from automated calibrations performed using a gas dilution unit that was developed in-house.30 Calibrations were performed using two standards: a mixed gas standard and a limonene-only standard. The mixed gas standard was a 1 ppm gravimetrically prepared standard (National Physical Laboratories, NPL) containing methanol, ethanol, acetone, isoprene, acetonitrile, octane, nonane, decane, benzene, toluene, m-xylene and 1,2,4-trimethylbenzene, in ultrahigh purity N2 (NPL). The limonene standard was produced in house by injecting a controlled amount of liquid standard (Sigma Aldrich, 99.8% purity) into an evacuated aluminium Experis cylinder and pressurised with research grade N2 (N6, BOC). The resulting gas mixture was allowed to equilibrate for 7 days at room temperature before the limonene concentration was determined via gas chromatography with a flame ionisation detector (GC-FID), which itself was calibrated using a 1 ppm limonene in N2 mixture as its primary standard (NPL). Daily multipoint calibrations were performed by diluting each standard with ambient humidity zero air for a concentration range of 1 to 10 ppbv (mixed standard) and 1.8 to 18 ppbv (limonene standard). Compounds which were not in the calibration standards were quantified based on literature values of branching ratios and reaction rate constants for reagent ions with the specific compounds. The branching ratios and rate constants used are given in Table S1.† Table S2† shows the species that were measured by the SIFT during the DOMESTIC campaign, their limits of detection (LOD), and whether or not the species are calibrated against a gas standard. The uncertainties in the SIFT measurements are quantified and discussed in the ESI.†
In addition to the online SIFT-MS monitoring, canister samples were taken during the campaign for offline analysis after the campaign. Samples were analysed using simultaneous gas chromatography time-of-flight mass spectrometry (GC-ToF-MS) and GC-FID. The GC system used was an Agilent 7890A GC System, with an Agilent CP9223 VF-WAXms (60 m × 250 μm × 250 μm) column coupled to the ToF-MS detector, and an Agilent CP7565 CP-Al2O3/Na2SO4 (50 m × 320 μm × 5 μm) column coupled to the FID detector.
The canister samples were first diluted with lab supply zero air to a pressure of 1 bar above atmospheric. Before firing onto the column, 500 mL of sample was flowed onto traps held at −30 °C. Injection of sample to column was performed in splitless mode. The oven temperature was programmed to start at 40 °C, hold for 3 min, then increase at 2.5 °C min−1 to 80 °C, and then at 10 °C min−1 to 250 °C. The column flow rates were 1.5 mL min−1 (CP9223 VF-WAXms) and 3 mL min−1 (CP-Al2O3/Na2SO4), and the carrier gas was N2. Peaks were assigned using GC-ToF-MS data aided by comparison with the National Institute of Standards and Technology (NIST) 2011 mass spectral library. The ratio of FID peak areas for identified monoterpenes was used to split the SIFT-MS monoterpene response.
2.2 Ventilation
All tests were conducted under natural ventilation conditions only. The air change rate (ACR) was measured using acetonitrile tracer releases on 6 days. 0.2% acetonitrile in N2 was released into the space after cooking, then the concentration decay monitored using SIFT-MS. The ACR was estimated through log-linear regression of the background subtracted concentrations over 2 hours following the release using eqn (5), as follows:| ln(C) = −λt + ln(C0) | (5) |
2.3 Cooking protocol
A chicken and vegetable stir-fry (Table 1) was used as the model meal, and was based on a published recipe.33 All ingredients were obtained from a well-known UK supermarket. The chef entered the container, cooked the meal, then left. To ensure that the measured VOC emissions derived only from the actual cooking (rather than the period after), the pan containing the finished meal was sealed with a lid at the end of cooking and removed from the container when the chef exited the room. All diagnostics, including the SIFT-MS, continued to monitor the evolution of VOC concentrations for the hours following the cooking.| Time (s) | Step | Notes |
|---|---|---|
| a Vegetable mix contained pre-chopped beansprouts, carrot, green cabbage, chinese leaf, bamboo shoot, water chestnuts, red pepper, mange tout, and white onion. b Sweet chilli stir-fry sauce contained water, sugar, rice wine vinegar, tomato purée, cornflour, garlic, ginger, red chilli, salt. | ||
| ≈−500 | Spice preparation | 10 g each of fresh garlic, ginger, and chilli peeled and chopped in food processor |
| ≈−250 | Pan preheating started | Back right ring turned on |
| 0 (t0) | Oil heating 1 | 10 mL rapeseed oil added to preheated pan (at 180 °C) |
| 60 | Fry chicken | 200 g of diced chicken added to pan |
| 270 | Chicken set aside | Fried chicken removed from pan and set aside |
| 300 | Oil heating 2 | 10 mL of fresh rapeseed oil added to pan |
| 360 | Add spices | Prepared spices added to pan |
| 380 | Add mixed vegetables | Pre-chopped vegetable mixture added to pana |
| 540 | Add noodles | 155 ± 5 g of noodles added to pan |
| 600 | Add fried chicken | Fried chicken added back to pan |
| 660 | Add sauce | 192 ± 10 g of sauce added to panb |
| 720 | End | Pan covered and removed from room |
Every stir-fry was cooked on the rear-right ring of an electric ceramic stove (KDC5422A, Beko) in a 24 cm stainless steel frying pan (Morphy Richards). Throughout cooking, the pan was heated on setting 4, where the maximum setting 6 corresponds to 1.7 kW heating power. During the pan preheat period, the pan surface temperature was measured with a surface temperature thermocouple (RS Pro) until it reached 180 °C, at which point, rapeseed oil was added to the pan and the cooking process detailed in Table 1 began.
2.4 Experimental reproducibility
During the campaign, multiple experiments were carried out, which focused on the effects of cooking a chicken and vegetable stir-fry, the effects of cleaning using various products, and the effects of the two activities combined. In order to assess how applicable the results (for example, calculated VOC emission rates) of this study are to a wider range of scenarios, the reproducibility of identical experiments was assessed. As this was the first attempt to carry out an experimental campaign at this scale, only two cooking experiments were exact repeats. The same stir-fry was also cooked on two further days, however, these were ‘layered days’, when other activities also took place in the container before and after. The timings of the cooking steps (as shown in Table 1) were kept consistent when the stir-fry was cooked on a cooking-only or a layered day, therefore, the cooking experiment data can still be compared from all 4 repeats. In general, data are reproducible in timing of emission peaks, while there is some variation in the concentrations at the peaks on different days. Fig. S3† shows the mixing ratios of three representative species that are emitted during each of the stir-fry repeats, measured using SIFT-MS. For the following analyses, the data from the two cooking-only days are averaged to understand VOC emissions from an average stir-fry, given there are no other activities on those days which may have influenced the measurements.2.5 INCHEM-Py
INCHEM-Py is a new, open source, indoor air chemistry box model, with v1.2 used for this work.29,34 INCHEM-Py creates and solves a series of ordinary differential equations to calculate indoor species concentrations over time. The model uses the near-explicit Master Chemical Mechanism (MCM), which details the reaction pathways for over 140 different VOCs,15,35–38 and includes further reaction mechanisms unique to the indoor environment, totalling over 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 different reactions and over 6000 species. For example, although not in the MCM, a reaction scheme for camphene is included in the model (as per Carslaw, 2007 (ref. 16)), where initial oxidation steps with OH, O3 and NO3 are unique for camphene. Further degradation then proceeds via the same oxidation pathways as β-pinene, due to camphene also having a CC double bond outside the carbon ring.16
000 different reactions and over 6000 species. For example, although not in the MCM, a reaction scheme for camphene is included in the model (as per Carslaw, 2007 (ref. 16)), where initial oxidation steps with OH, O3 and NO3 are unique for camphene. Further degradation then proceeds via the same oxidation pathways as β-pinene, due to camphene also having a CC double bond outside the carbon ring.16
INCHEM-Py creates and solves the following general equation for the concentration, C, of each species i:
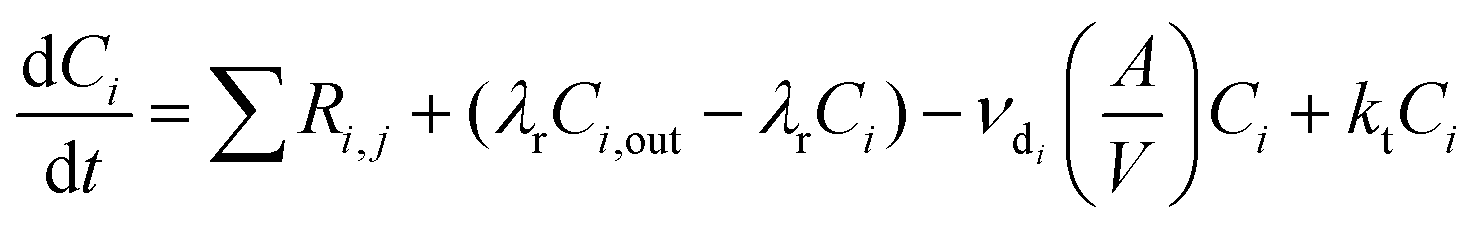 | (6) |
For surface deposition, species each have a deposition velocity which, combined with the SAV of the room, gives a rate of irreversible loss of species to the surface, regardless of the surface material. However, for O3 and hydrogen peroxide (H2O2), the deposition velocity is dependent on the surface material, with specific SAVs given for metal, glass, wood, plastic, paint, paper, concrete, soft furnishing and skin.34,39 Following deposition of O3 and/or H2O2, there is surface-dependent emission of aldehydes, based on Carter et al., 2023.39
The concentrations of gases outdoors are split into species with diurnally varying concentrations (O3, NO and NO2) and 110 VOCs with static outdoor concentrations.39 Data from the European Air Quality Database, from the ‘GB0586A, suburban London, 0.070766 51.45258’ monitoring station were used, from which average hourly concentrations were calculated from measurements taken over a 3 month period (July, August and September) for O3, NO and NO2.41
The indoor background VOC concentrations are determined by the outdoor concentrations and the ACR. A review by Nazaroff, 2021,42 suggested that the mean ACR for residential dwellings is 0.5 h−1. Therefore, for the simulations in this study, an ACR of 0.5 h−1 was used. The combination of the outdoor concentrations and the ACR resulted in simulated indoor concentrations for O3, OH and NO3 of ≈3.5 ppb, 6 × 105 molecules per cm3 and 2.5 ppt, respectively, at midday.
To determine the light levels in the simulated kitchen, a latitude of 51.45°N was used (the same as the suburban London monitoring station), and the window was assumed to be glazed with low emissivity glass, which allows the transmission of light in the wavelength range of 330–800 nm.43 Each simulated cooking activity took place at 1 pm, in June. Indoors, incandescent lighting was assumed to be on between the hours of 07:00 and 19:00.
To account for people in the simulations, it was assumed that one person was in the room at all times, therefore, the additional skin surface is considered (as described above), as well as breath emissions. Breath emissions include acetone, ethanol, methanol, isopropanol and isoprene, with emission rates for adults and children described in.44,45 These rates were used instead of the isoprene emissions measured in DOMESTIC, as the SIFT-MS isoprene measurements also included a measurement of furan, so were not entirely due to the person's breath.
To simulate cooking experiments, timed VOC emissions were determined from the experimental SIFT-MS data by calculating the rate of increase in species concentrations during cooking. Many species displayed multiple emission peaks and each of these separate peaks were noted and used as an input into INCHEM-Py (term 4 of eqn (6)). Emissions were included for the following species: ethanol, octane, nonane, acetaldehyde, propanal, acrolein, hexanal, heptanal, octanal, nonanal, limonene, camphene, α-pinene, β-caryophyllene and 1,2,4-trimethylbenzene. Examples of fitted emissions are shown in Fig. S4.†
3 Results and discussion
3.1 Total species groups emissions
We identified 27 VOC species that were emitted during the stir-fry cooking experiment. The total measured concentrations of different homologous groups – the alkanes, aldehydes, terpenes and alcohols – over time are shown in Fig. 1, where the different colours show the fraction of the total concentration that is accounted for by individual species. The cooking period is shown by the grey shaded area, starting at t0 (when oil is first added to the heated pan), and extending to the time point when cooking ended (720 s).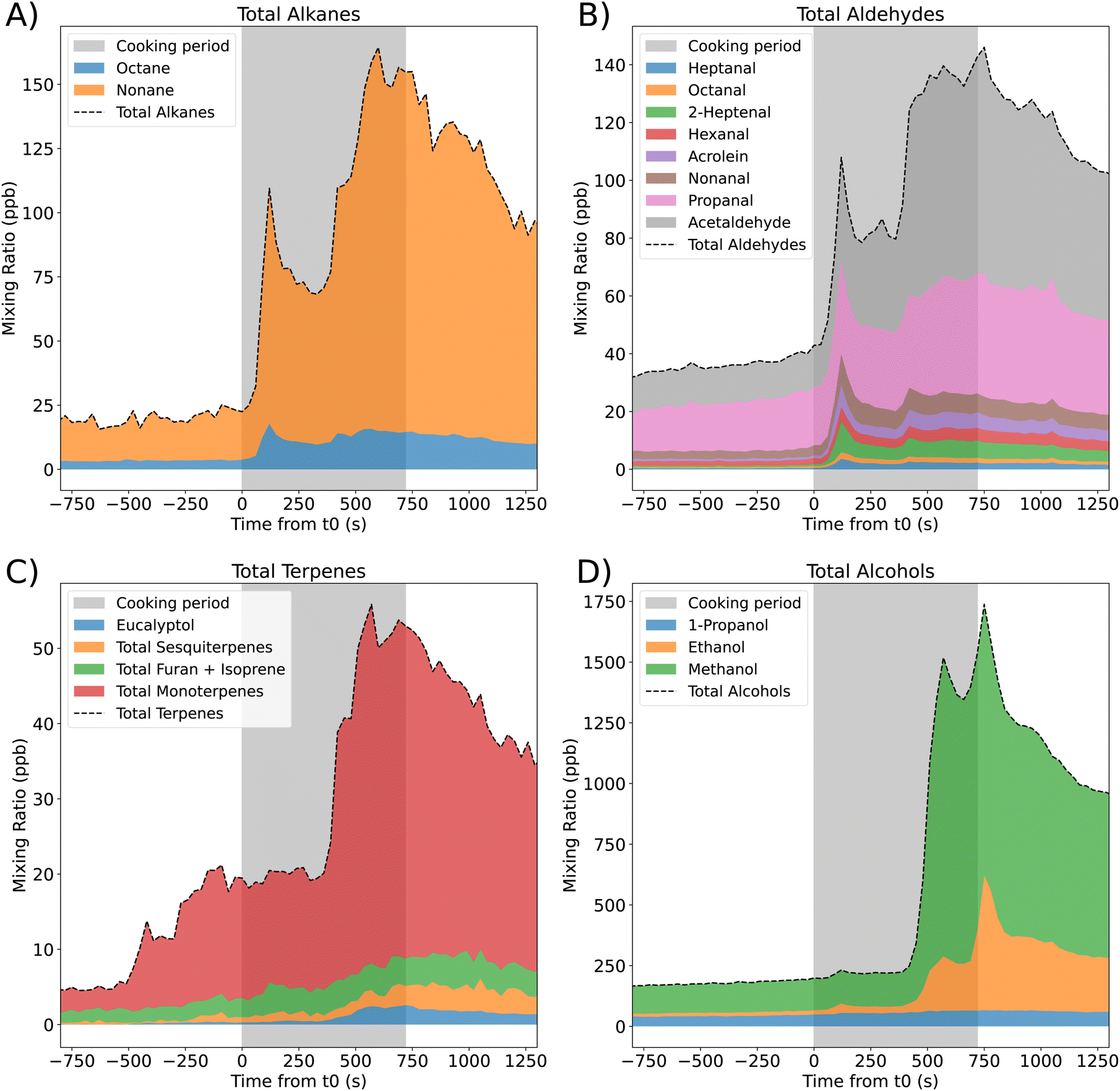 | ||
| Fig. 1 Mixing ratios of alkanes (A), aldehydes (B), terpenes (C) and alcohols (D) measured by SIFT-MS during the cooking experiments. The grey shaded area signifies the time period when cooking took place, with 0 s (t0) being the time point when the oil was first added to a hot pan. Total mixing ratios for each group of chemical species are shown, with the different colours indicating the contribution from each individual species. Species whose mixing ratio increased by less that 1 ppb during cooking are excluded from the figure for clarity. Data shows the average mixing ratios from the two stir-fry repeats. | ||
Different species peak at different times during cooking. For terpenes, there are two timepoints where the concentration increases rapidly, and the increase is largely due to increases in total monoterpenes. The first increase in monoterpene concentration happens at ≈−500 s, which corresponds to when spice preparation begins. This suggests that one or more of the spices (garlic, ginger, and chilli) used in this recipe emit monoterpenes, which is in agreement with previous work.22 The second peak occurs later in the cooking process, when there is also a small increase in sesquiterpene and eucalyptol mixing ratios, coinciding with when spices are added to the hot pan.
Isoprene and furan cannot be distinguished by the SIFT-MS, therefore, are measured together. The concentration of this combined species increases gradually over the full cooking period. Emission of isoprene from the breath of one adult is approximately 5.4 × 106 molecules per cm3 per s,44 while the emission rate observed during the cooking period is approximately 4 × 107 molecules per cm3 per s. As furan is likely to be released during cooking,46 the higher emission rate seen here is reasonable. However, it is not possible to attribute individual emission rates to the two species in this case.
Alcohols (Fig. 1D) are by far the most abundantly emitted species, with a maximum total mixing ratio of over 1500 ppb, of which the majority is methanol. The next most important chemical group is alkanes, dominated by nonane, which reaches a maximum mixing ratio of ≈170 ppb towards the end of the cooking period (Fig. 1A). A wide range of aldehydes (Fig. 1B) are produced throughout the cooking period, with acetaldehyde and propanal being the dominant species. However, there is also appreciable contribution from acrolein, hexanal, 2-heptenal and nonanal.
In the larger-scale HOMEChem experiments,23 VOC emissions were measured when a vegetable stir-fry was cooked.5 The most common VOCs observed during HOMEChem were methanol and ethanol as we observed here, however, in DOMESTIC, we observed higher methanol than ethanol, which is the opposite finding to the HOMEChem campaign. The PTR-ToF-MS (proton transfer reaction time-of-flight mass spectrometry) used in the HOMEChem experiments did not measure alkanes (due to their low proton affinity), so they are not included in the emission ranking. However, 4 of the top 8 emitted species are aldehydes (acetaldehyde, propanal, acrolein and butanal), similar to those seen in the DOMESTIC campaign. This suggests that these species are largely attributable to the stir-frying process and non-meat ingredients, as they are emitted, whether or not meat is included.
Monoterpenes account for 91% of the total terpene mixing ratio at the peak (Fig. 1C). However, because SIFT-MS is unable to distinguish between compounds of the same mass, it is not possible to tell which specific monoterpenes contribute to this peak. To qualitatively determine the contributing monoterpenes to this peak (with a molecular weight of 136 g mol−1), canister samples were collected during the campaign, 720–780 s after t0, corresponding to peak terpene mixing ratios. Post-campaign analysis of the canisters by GC-MS revealed that the average monoterpene proportions on the two cooking-only days were 63% camphene, 19% α-pinene and 18% limonene.
In addition to the chemical groups shown in Fig. 1, smaller concentrations of sulfides, aromatics and ketones were also identified, and their concentrations were dominated by dimethyl trisulfide, trimethylbenzenes, and acetone, respectively.
3.2 Timing of emission
The real-time nature of SIFT-MS measurements allows the identification of specific processes in the stir-fry cooking activity. In Fig. 2, the mixing ratios of total monoterpenes, and assorted alkanes, aldehydes and alcohols are shown, overlying shaded areas which indicate when different steps of the stir-fry cooking took place. The SIFT-MS data may lag behind the cooking step timestamps slightly due to the residence times of gases in the sample lines, and due to differences in polarity and ‘stickiness’ of species to the lines. However, the lag is likely to be no more than a few seconds. The data in Fig. 2 are for the average stir-fry.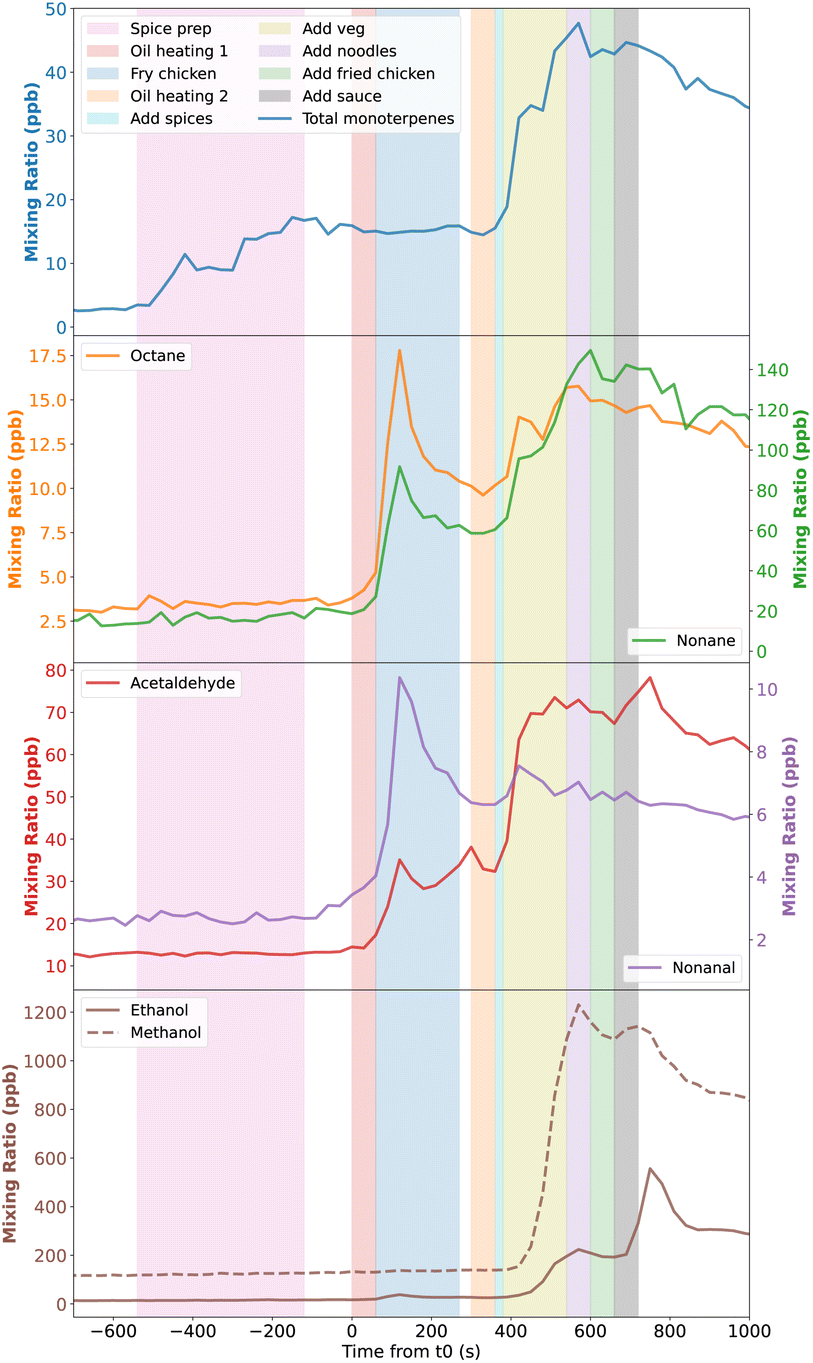 | ||
| Fig. 2 Mixing ratios of multiple species seen during the average stir-fry experiment, averaged across the two stir-fry repeats. t0 is the time when oil is added to a hot pan. Shaded areas indicate the different steps of the stir-fry cooking process. | ||
Monoterpenes increased in concentration during the spice preparation, when 10 g each of fresh ginger, garlic, and chilli were peeled and chopped in a mini-chopper. There was an additional spike in concentration when the spices were added to the hot pan at 360 s. Interestingly, despite the stir-fry sauce containing garlic, ginger and chilli, no monoterpene emissions were seen when the sauce is added to the pan. This suggests that the freshness or level of processing of the spices may affect their emission profiles. Alternatively, it may be that the spices do not reach as high a temperature when added to the pan in the sauce, compared to when they are just added to hot oil, and monoterpene emission may be temperature dependent. Emissions of monoterpenes from vegetation shows a temperature dependence, with increasing temperature resulting in greater emissions.47,48 Whether this directly translates to cooking of spices is not fully known, as the plant-based emissions of monoterpenes are due to enzymatic activity, which reduces above a certain temperature threshold,47,49 and via release from storage pools as a result of mechanical damage.50 During cooking, the release of monoterpenes is not a physiological enzymatic process so is not limited by denaturation, and is likely to just be from storage pools, which may be depleted during the sauce manufacturing. Therefore, the emission may be less from the sauce than from the relatively unprocessed spices.
Alkanes and aldehydes show similar profiles to each other, and emissions appear to be linked to when oil is heated, particularly when the first batch of oil is heated at t = 0 s. However, when heating the second batch of oil, the increase in octane, nonane and nonanal is less pronounced, and continues for longer, than for the first batch of oil. This could be because the oil remains in the pan for the rest of the cooking process, likely still emitting these species until the end of the experiment. Conversely, acetaldehyde shows a relatively small first concentration peak, when compared to the second peak. This suggests that the larger alkane/aldehyde species shown in Fig. 2 may be more associated with heating of oil, but that smaller acetaldehyde may also be emitted from a wider range of ingredients, thus giving the larger second peak when more ingredients are in the hot pan. Emissions of alkanes and aldehydes during the heating of various cooking oils have been observed in past studies,21,51 though not with the time-resolution provided by the SIFT-MS used during the DOMESTIC campaign.
Finally, methanol and ethanol are emitted much later in the cooking process, specifically, when the mixed vegetables (see Table 1) are added to the pan at 380 s. This is in agreement with Klein et al.,21 where the emissions from shallow frying vegetables were dominated by alcohols, compared to cooking meats or oils alone, when the emissions were dominated by aldehydes. For ethanol (and methanol to a lesser extent), Fig. 2 shows that there is also a second peak in concentration when the sauce is added into the pan, suggesting that components of the sauce also release alcohols.
The measured concentrations for methanol were not considered further in this study as there were extremely high background emissions occurring at all times (giving background mixing ratios of up to 400 ppb, compared to ≈40 ppb and ≈29 ppb values reported in previous studies of unoccupied residences5,52), including during periods of inactivity. This cannot be seen in Fig. 1 and 2 as the activity period shown followed a period of venting of the space, which lowered the methanol mixing ratios to the outdoor levels (<10 ppb) due to the increased ventilation. However, as the background emission rates were so high (10–16 mg h−1), it is not possible to reliably differentiate between the background and activity emissions.
It is likely that the relatively new building materials (≈1 year old) used to fit out the container were still off-gassing at significant rates. Methanol has been shown to be emitted from newly-made processed wooden boards, with very high emission rates, which decrease over time. For example, in Brown, 1999,53 it was shown that new office furniture made of processed wood emits methanol at a rate of 5000 μg h−1 in the first 4 h, dropping to 3400 μg h−1 over the first day. Applying these emission rates to the area of the DOMESTIC container walls (≈23 m2), which are made of processed wood, gives total emission of 115 mg h−1 and 78 mg h−1 for the two time points. These values are even higher than those estimated during the DOMESTIC campaign, which is expected due to the ageing of the materials. Therefore, it is feasible that a major component of the methanol emissions were attributable to off-gassing of the wall materials.
3.3 The base case simulation scenario
Using the data collected during experiments in the DOMESTIC container, emission rates were calculated and input into INCHEM-Py to simulate an average stir-fry meal. In total, 15 of the emitted species measured by the SIFT-MS were present in the MCM and used in INCHEM-Py. The 15 species included alkanes, aldehydes, monoterpenes and ethanol, and the simulated emissions/concentrations of each are shown in Fig. S5.† Due to the uncertainties around methanol concentrations (as described above), and the fact that methanol made negligible differences to the simulated chemistry (Fig. S6†), it was excluded from the simulations. To simulate the monoterpenes, the ratios determined by canister samples and GC-MS were used to split the total monoterpene emission rate between camphene (63%), α-pinene (19%) and limonene (18%). Emissions were simulated in an ‘average kitchen’ as outlined in Section 2.5.1 above. The combination of these experimentally-derived emissions and the average kitchen properties constitutes the base case cooking scenario.The effect of the base case cooking scenario on oxidants and specific secondary products are shown in Fig. 3 by the pink and purple circles in the grey shaded areas. For the background (BG, no cooking emissions) and stir-fry cooking (SF, cooking emissions included) simulations, the average concentration or mixing ratio of each species are calculated over a 3.25 h period, beginning 15 min before t0 (to include the period of spice preparation), and extending to 3 hours after t0. When comparing the average values in the SF simulation to the BG simulation, there is a decrease in the average values of O3 (≈50 ppt), OH (≈5 × 105 molecules per cm3) and NO3 (≈0.2 ppt, not shown), and an increase in the average concentrations of RO2 and HO2. This results from the oxidation of the cooking-related VOCs by OH, O3 and NO3, which serves to deplete the oxidants, while producing RO2/HO2 species.
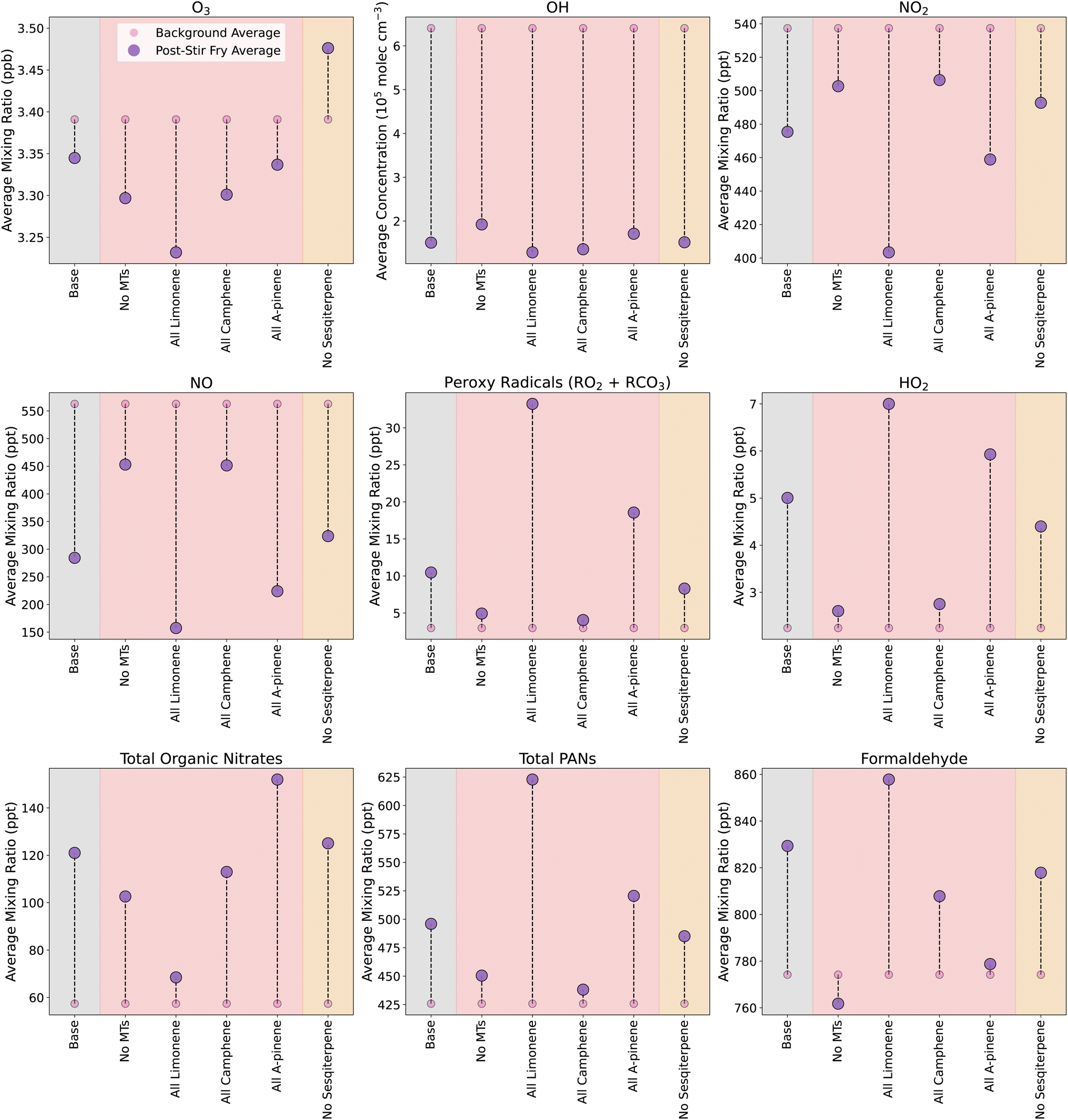 | ||
| Fig. 3 Change in average mixing ratios or concentrations of secondary species following cooking and for different conditions. Small pink circles give the average background level without any cooking, and the large purple circles show the average concentration/mixing ratio following the specific simulated cooking scenario. Different coloured shaded areas correspond to simulations when different emission groups are varied: grey = standard, base case stir-fry, red = scaled monoterpene emissions, orange = sesquiterpene emissions omitted. Averages are calculated from 15 min before t0, to 3 hours after. | ||
In addition, the SF simulation also shows increases (compared to BG) in the average mixing ratios of total PAN species (≈50 ppt) via reaction (4), organic nitrates (≈60 ppt) via reaction (2), and formaldehyde (≈60 ppt) via reaction (3).
Fig. 3 shows how the formation or depletion of oxidants, peroxy radicals and secondary products are of a much smaller magnitude (10 s–100 s of ppt) compared to the species that are directly emitted by the cooking process (10–100 s of ppb). However, when timescales for total directly emitted species and total secondary products of interest are compared, the secondaries are elevated compared to background levels for much longer (Fig. S7†). Following the initial peak, the primary emissions reach 50% and 10% of peak by ≈35 min and 2 h 20 min, respectively. In contrast, the secondaries reach 50% and 10% of post-cooking peak concentrations by ≈1 h 50 min and 4 h 40 min, respectively. Therefore, while the absolute concentration of secondaries is much smaller than the primary emissions, the relative rise in secondary concentration compared to backgrounds is greater for longer.
The changes in formaldehyde concentrations as a result of secondary chemistry are below the safe exposure limits in this case. However, there is very little known about safe exposure limits for PANs and organic nitrates. While both are linked to detrimental health effects, further toxicology is required to determine safe exposure limits. In a paper by Vyskocil et al., 1998,54 it was suggested that an exposure to 0.64 mg m−3 (≈120 ppb) of PAN was sufficient to result in eye irritation in humans, well above the concentrations we observed in our modelled scenarios. That said, long-term, chronic exposure to low-levels of PANs has not been investigated. For organic nitrates, there are no data on exposure limits, only a link between exposure and detrimental effects. In addition to a lack of exposure limit data for either species, there is also no current understanding of the additive effects of low concentrations of multiple species that are inhaled. Therefore, this work provides insight into how the different secondary species are produced and, as further health-related work is carried out, could be used to inform mechanisms for reducing exposure. For example, cooking with less spices that would reduce monoterpene emissions and secondary product formation.
3.4 Sensitivity to terpene composition
In this section, we investigate the impact of removing different terpene emissions during the simulated cooking activities, so we can assess the impact of each species on the secondary chemistry. Removing the monoterpenes had a large impact on secondary species concentrations, as shown in Fig. 3. Compared to the base case SF, removing monoterpene emissions resulted in decreased formation of RO2 (74% less), HO2 (87% less), organic nitrates (29% less) and total PAN species (65% less) when cooking emissions were included. In addition, the average formaldehyde mixing ratio decreased slightly by ≈12 ppt.To investigate the impact of individual monoterpenes, simulations were run where the total monoterpene emission from the base case was maintained, but assumed to either be all limonene, camphene or α-pinene. When all the monoterpenes were assumed to be limonene, the formation of HO2 and RO2 was increased by 72% and over 300%, respectively, compared to in the base case. This is because limonene reacts more readily with OH and O3 than camphene and α-pinene. Similarly, compared to the base case SF, the average concentration was elevated by 30 ppt for formaldehyde (52% increase) and ≈100 ppt of total PAN species (180% increase), while the average concentration of organic nitrates was greatly reduced (≈50 ppt/83% less than base case).
The efficient production of RO2 species by limonene, and the subsequent oxidation pathways, also results in the extra depletion of NO seen in Fig. 3, via reactions (1) & (2).
When the monoterpenes were assumed to be camphene, the deviations from the baseline cooking scenario were less pronounced than for the more reactive limonene case. OH depletion by camphene was similar to the limonene-only scenario. The formation of HO2 and RO2 in the camphene-only case was similar to the case with no monoterpenes, although formaldehyde and organic nitrate formation was enhanced when only camphene was included.
When all monoterpenes were assumed to be α-pinene, there was increased production of RO2, HO2, organic nitrates and PANs, but a reduction in formaldehyde formation, when compared to the base case. Therefore, the composition of monoterpenes in cooking emissions appears to affect the secondary chemistry, with limonene driving high PAN and formaldehyde production, while α-pinene produces relatively less PANs and formaldehyde and, instead, favours the formation of organic nitrates. Both of these species can drive substantial secondary chemistry, whereas, camphene on its own results in fewer secondary chemistry effects in all the investigated oxidants and secondary species.
The relative effects of different monoterpenes on secondary chemistry has also been discussed in the context of cleaning product formulations in ref. 55. When comparing the formaldehyde production potential of limonene and α-pinene, limonene produced almost twice as much formaldehyde as α-pinene. However, when the emission was a 50:50 mix of limonene and α-pinene, the formaldehyde production was even greater than either single monoterpene, despite the total monoterpene emission being kept constant. The study concluded that this is due to the combination of oxidation reaction rate coefficients, and showed how different combinations of species may have unexpected results on secondary chemistry, due to the complex interplay between the different degradation pathways.
In the base case cooking scenario, O3 concentrations were depleted through reactions with the combination of sesquiterpenes (β-caryophyllene) and monoterpenes. The reaction rates for O3 (kO3) with β-caryophyllene and the monoterpenes are given in Table 2, and show that β-caryophyllene has, by far, the highest kO3. This explains why, when monoterpenes were omitted, the resulting ozone concentration was 2× lower than the base case, as the β-caryophyllene could react freely with ozone, at a high rate, with less competition, whilst also producing more OH through ozonolysis than in the base case. However, when all the monoterpenes were considered to be limonene, the ozone depletion was greater still. Although the addition of limonene competed with β-caryophyllene for O3, the relatively high kO3 for limonene, and the high concentration of limonene, still resulted in high levels of O3 being used up. This effect was not seen when all monoterpenes were assumed to be camphene. In this case, although the camphene concentration was high, the kO3 was much lower, so that the ozone depletion was almost entirely due to β-caryophyllene, resulting in a depletion similar to when monoterpenes are omitted completely.
| Species | k O3 | OH yield | k OH | OH prod:lossb |
|---|---|---|---|---|
|
a Rate coefficients have units of cm3 per molecule per s.
b OH prod:loss ratio calculated as kO3 × OH yield/kOH.
|
||||
| β-Caryophyllene | 1.20 × 10−14 | 0.08 | 1.97 × 10−10 | 4.87 × 10−6 |
| Limonene | 2.02 × 10−16 | 0.87 | 1.68 × 10−10 | 1.14 × 10−6 |
| α-Pinene | 9.06 × 10−17 | 0.80 | 5.39 × 10−11 | 1.42 × 10−6 |
| Camphene | 9.06 × 10−19 | 0.18 | 5.33 × 10−11 | 3.06 × 10−9 |
In the case where α-pinene was the monoterpene included, O3 and OH were depleted less than in the no monoterpenes and camphene-only scenarios. However, RO2 formation was considerably higher, meaning that there were still high levels of α-pinene oxidation occurring. This observation was likely due to a combination of increased OH formation following ozonolysis, and a kO3 that is high enough to compete with sesquiterpenes, but not high enough to deplete the O3 as much as in the limonene case. In ref. 55, the balance between kO3 values, OH yields from VOC oxidation by O3, and OH reaction rate coefficients (kOH) for different monoterpenes was discussed. It was shown that α-pinene was the most efficient producer of OH, when compared to β-pinene and limonene. This is shown in Table 2, along with data for camphene. The OH formed will be able to oxidise the VOCs to RO2 more efficiently, hence, the enhanced formation of RO2 when the monoterpenes are assumed to be α-pinene.
3.5 Changing the levels of oxidants
The secondary chemistry resulting from cooking a chicken and vegetable stir-fry is affected by ingredient composition, as discussed above, and it is also affected by the ambient conditions of the room. These conditions are highly variable, depending on factors such as light levels in the room, the ACR, the surfaces in the room and the number of people present. Therefore, a sensitivity study has been performed to investigate how the secondary chemistry resulting from the average stir-fry presented above is affected by these factors. Table 3 outlines the model parameters used in the sensitivity study, with the base case (Base) parameters shown in the first row. The sensitivity study investigates the effects of increasing (hiSAV) and decreasing (loSAV) the SAV of the kitchen, increasing (GlassC) and decreasing (LEWF) the emissivity of the glass, cooking in winter (Winter) or in the evening (Evening), increasing the number of people in the kitchen to two adults and two children (Family), increasing the ACR (hiACR), and changing the outdoor oxidant concentrations, by considering data from a polluted area during a heatwave in Milan, when outdoor O3 and NOx concentrations were elevated (Polluted). Each scenario is named according to the parameter changed and consists of a background (BG) and stir-fry (SF) simulation. The name will be used to refer to each scenario in the following sections, along with the -BG or -SF suffix, to indicate whether cooking emissions are included or not.| Scenario | SA/V (cm−1) | Glass | Date (DD/MM) | Time (hh:mm) | Peoplea | AER (h−1) | Locationb |
|---|---|---|---|---|---|---|---|
| a Number of people in the kitchen, where a = adult and c = child. b Location of where the diurnally varying outdoor concentrations of ozone, NO and NO2 were measured. c Data downloaded from the European Air Quality Database41 for the London suburban background monitoring station (GB0586A, 0.070766, 51.45258). | |||||||
| Base | 0.0253 | LE | 21/06 | 13:00 | 1a | 0.5 | London Suburbanc |
| hiSAV | 0.0292 | LE | 21/06 | 13:00 | 1a | 0.5 | London Suburbanc |
| loSAV | 0.0212 | LE | 21/06 | 13:00 | 1a | 0.5 | London Suburbanc |
| LEWF | 0.0253 | LEWF | 21/06 | 13:00 | 1a | 0.5 | London Suburbanc |
| GlassC | 0.0253 | GlassC | 21/06 | 13:00 | 1a | 0.5 | London Suburbanc |
| Winter | 0.0253 | LE | 21/12 | 13:00 | 1a | 0.5 | London Suburbanc |
| Evening | 0.0253 | LE | 21/06 | 18:30 | 1a | 0.5 | London Suburbanc |
| Family | 0.0253 | LE | 21/06 | 13:00 | 2a/2c | 0.5 | London Suburbanc |
| hiACR | 0.0253 | LE | 21/06 | 13:00 | 1a | 2.0 | London Suburbanc |
| Polluted | 0.0253 | LE | 21/06 | 13:00 | 1a | 0.5 | Milan56 |
Fig. 4 shows the concentrations of oxidants and secondary species under different ambient conditions, in both background (BG, small pink circles) and stir-fry (SF, larger purple circles) model runs. The BG and SF concentrations are averages over a 3.25 h period, starting 15 min before t0. The base case is shown in the grey shaded area, and is the same as in Fig. 3. The base case conditions are outlined in Section 2.5.1 and Table 3.
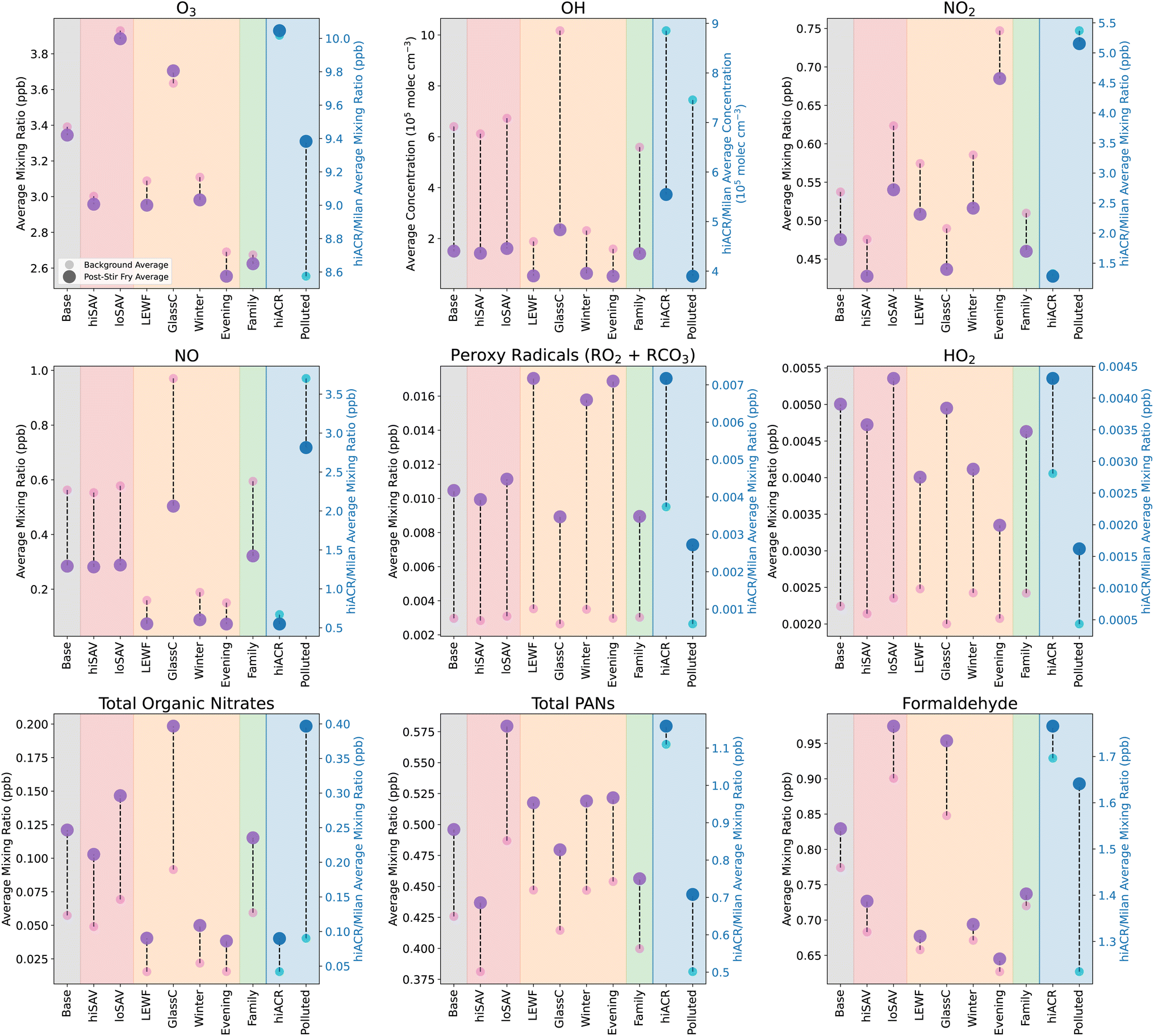 | ||
| Fig. 4 Change in average mixing ratios or concentrations of secondary species following cooking, when the ambient conditions are varied. Small pink/blue circles give the average background levels without any cooking, and the large purple/dark blue circles show the average concentration/mixing ratio of species following the simulated cooking activity. Different coloured shaded areas correspond to simulations when ambient conditions are varied: grey = standard base case stir-fry, red = varying surface areas, orange = varying light conditions, green = varying occupancy and blue = varying air change rate. The concentrations in the blue shaded area correspond to the right hand y axis, while all the other concentrations correspond to the left hand y axis. Averages are calculated from 15 min before t0, to 3 hours after. Scenario names correspond to those in Table 3. | ||
Fig. 4 shows that loSAV-BG had higher concentrations of OH, NO2, O3, and formaldehyde, than hiSAV-BG or Base-BG, partly due to having a smaller area for surface deposition. Lower depositon rates mean there is more ozone available for reaction, but also, a smaller chance of the organic nitrates, PANs and formaldehyde that are formed from VOC oxidation depositing onto surfaces. The opposite was seen in the hiSAV scenario. As shown in Fig. 4, compared to Base-BG, the hiSAV concentrations of O3 and NO2 were decreased, as were the resulting secondary formaldehyde and PAN background concentrations.
To compare the effects of different SAVs on the secondary chemistry following the average stir-fry cooking, the formation of secondaries in the different SAV scenarios was compared to formation in the base case scenario, to give a relative formation for each secondary species. The relative formation was calculated as follows:
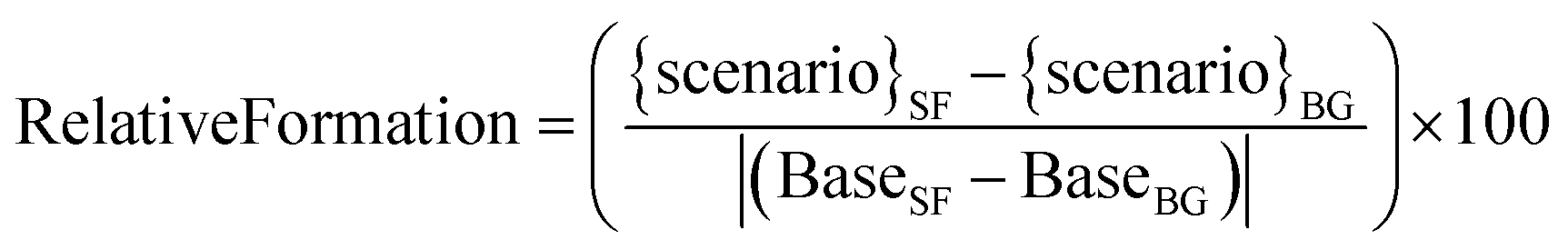 | (7) |
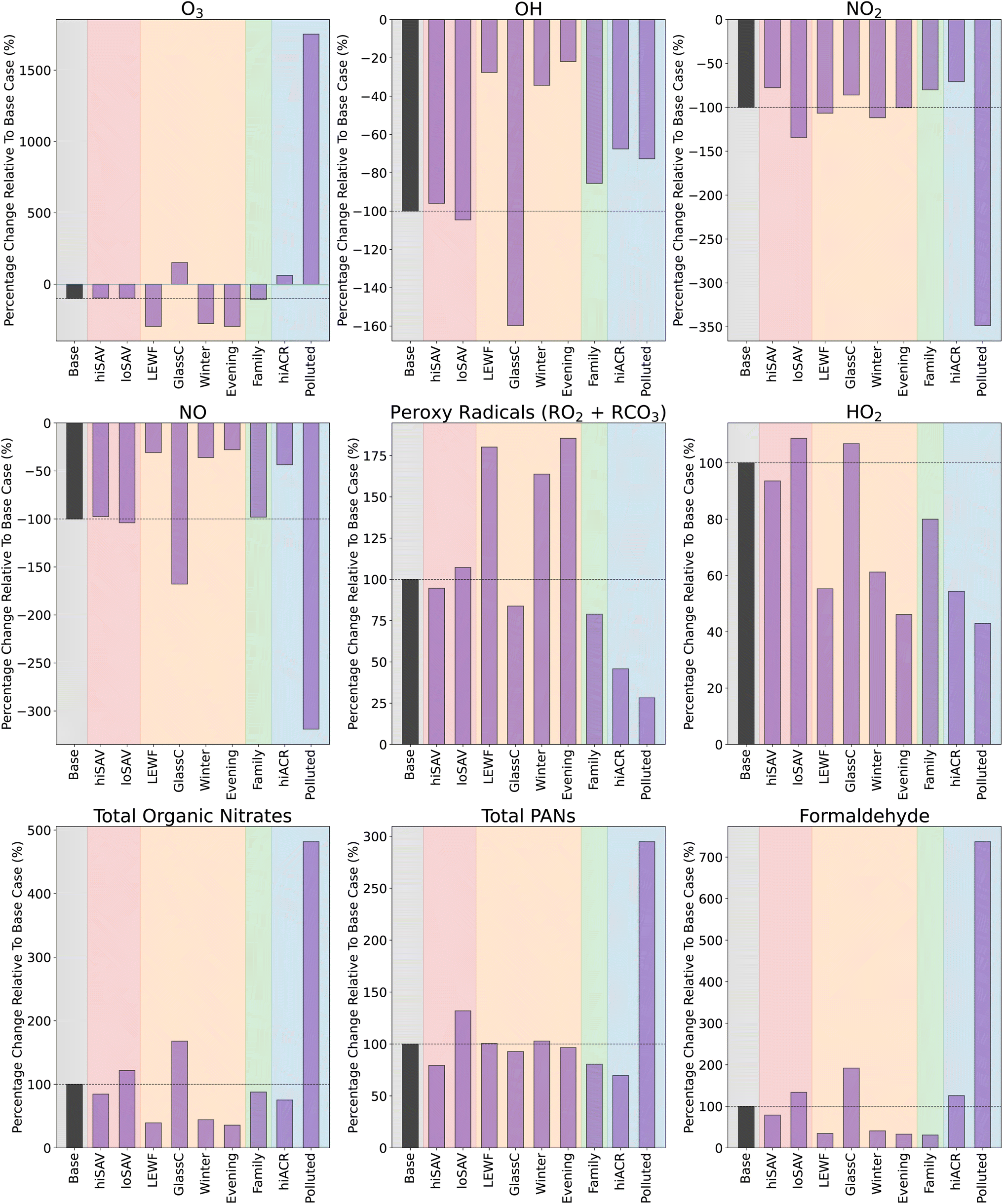 | ||
| Fig. 5 Percentage difference in secondary production formation compared to base case. | ||
As shown in Fig. 5, the cooking-related change in concentrations of organic nitrates, PANs and formaldehyde in the hiSAV scenario were 85%, 79% and 79% of the change seen in the Base case, respectively. Therefore, this indicates a reduction in formation, compared to the base case. Conversely, in the loSAV scenario, there was increased formation of organic nitrates, sum of PANs and formaldehyde compared to base case, shown by relative percentages of 122%, 132% and 134%, respectively.
For Evening-BG, the concentration of NO2 is elevated compared to Base-BG, as the time coincides with evening rush hour and, consequently, higher NO2 concentrations outdoors. However, the lower light levels compared to midday mean that the NO2 is not photolysed, so NO, O3 and RO2 concentrations remain low, leading to little formation of organic nitrates and formaldehyde. Conversely, the elevated NO2 results in slightly more PAN formation, and higher nitrous acid (HNO2) concentrations than Base-BG, as the dominant production mechanism for HNO2 is as follows:
| NO2 + surface → HNO2. | (8) |
Coupled, with reduced photolysis, this results in elevated background HNO2 and lower OH concentrations in the evening, compared to at midday (base case). Increasing the light levels (GlassC-BG) had the opposite effect and resulted in higher background concentrations of O3, OH, NO, organic nitrates and formaldehyde, and decreased background concentrations of NO2, RO2, PANs and HNO2, when compared to the base case.
It is important to note that in this study, the indoor NO2 concentrations are based only on outdoor values and ventilation rates, without any influence from any indoor appliances. This is reasonable for simulating kitchens that do not have gas appliances, such as gas stoves. However, gas stoves are known to increase both background and cooking concentrations of indoor NO2. These increases can be significant, up to 10 s of ppb during cooking,59 which would likely affect the secondary chemistry by altering the rates of NO2-dependent reactions. Therefore, this would need to be a consideration for simulating scenarios containing gas appliances.
The ambient light levels in a kitchen have a large impact on the background oxidant and secondary species concentrations, and further effects were also seen when cooking emissions were included. When VOCs were added into the simulation in high light conditions (GlassC-SF), the high OH background concentration facilitates efficient VOC oxidation, which produces enhanced concentrations of RO2 and HO2. However, because the background concentration of NO is so much higher in GlassC-BG compared to Base-BG (4), the RO2 is quickly depleted by the NO, to produce large amounts of organic nitrates and formaldehyde. Compared to the base case, the relative formation of organic nitrates and formaldehyde is 168% and 192%, as shown in Fig. 5. However, Fig. 5 also shows that increased light levels had only a very small effect on PAN formation.
In terms of the secondary products formed when the cooking emissions are included, Fig. 5 shows that increasing the number of people has little effect on organic nitrate and PAN formation, but reduces the formation of formaldehyde by 69%, compared to the base case. This is due to the lower availability of O3 to oxidise the monoterpenes to formaldehyde.
| NO2 + O3 → NO3 + O2. | (9) |
For secondary species such as NO, RO2, HO2 and organic nitrates, the background concentrations between the hiACR and base cases are very similar. This means that, despite the increased oxidant concentrations, the oxidised species are unable to accumulate due to the increased loss to outdoors through ventilation. The exceptions to this are PAN species and formaldehyde, which have much higher background concentrations in the hiACR case compared to the base case. This is because these species are not only created as secondary species from cooking, they also have significant ambient outdoor concentrations. The outdoor mixing ratio of formaldehyde in our simulation is 2.4 ppb, while the peak indoor value following cooking is ≈1.9 ppb. Therefore, with a higher ACR, the net movement of species is still towards indoors. Similarly, the outdoor mixing ratio of PAN is 2.2 ppb, while the maximum indoor value during cooking is only 1.2 ppb.
In the base case, the kitchen is assumed to be in a suburban London location. To investigate how changing the outdoor oxidant concentrations will affect the secondary chemistry of the present cooking simulation, the outdoor diurnal profiles of O3, NO and NO2 were changed, by using data from Milan, during a particularly polluted time period in August 2003 (Fig. S8†).56 The Polluted scenario in Fig. 4 shows the mixing ratios and concentrations under these conditions, with and without the stir-fry emissions. As expected, compared to the base case, the indoor concentrations of O3, NO and NO2 in the Polluted scenario are considerably higher. Consequently, NO3 is also higher (not shown), as the increased mixing ratios of NO2 and O3 result in a higher reaction rate of reaction (9). In addition, reaction (8) increases, resulting in higher HNO2 concentrations (not shown) and, subsequently, higher OH concentrations, compared to the base case. Despite the increase in oxidant concentrations, background RO2 concentrations are decreased, due to the increased NO, compared to the base case. This gives greater background concentrations of formaldehyde and organic nitrates.
When cooking emissions are included, there is only a small increase in the RO2 average mixing ratios of ≈2 ppt (compared to ≈7 ppt in the base case). This is because, as shown in Fig. 5, there is much greater production of organic nitrates (400% increase), formaldehyde (over 600% increase) and total PANs (200% increase) in the Polluted case, compared to the base case, due to the higher NO concentrations. Fig. 5 shows that the Polluted scenario shows the greatest production of harmful secondary products, compared to all the other scenarios.
4 Conclusions
In this study, we show how cooking a chicken and vegetable stir-fry results in the direct emission of a wide range of VOCs, of which the majority of the total emission is alcohols. However, the modelling study has shown that the monoterpene emissions account for the majority of the formation of potentially harmful secondary products, including organic nitrates, total PANs and formaldehyde. Additionally, a sensitivity study identified the outdoor oxidant concentration as the most important factor in determining the extent of the formation of harmful secondaries.Due to the dependence of indoor secondary chemistry on outdoor oxidant species concentrations, we would also expect that climate change may have a detrimental effect on secondary product formation indoors, as outdoor ozone concentrations are expected to increase.60 Coupled with this, rising temperatures may result in increased window-opening allowing more outdoor ozone indoors, which may then have a detrimental effect on the indoor air quality following activities such as cooking and cleaning, by enhancing formation of secondary species. This finding is relevant both in a domestic setting, but should also be a consideration for larger-scale cooking locations such as restaurants, in order to ensure appropriate mitigation measures are in place to protect the health of workers. Finally, there is also the potential for increased impacts on outdoor air quality, as some of the emissions generated indoors make their way into the ambient environment.
In the future, this work would benefit from further experimental studies in different room environments, to validate the sensitivity analyses. In addition, further in-depth characterisation of the indoor air pollutant concentrations (e.g. background VOC concentrations, real-time indoor and outdoor oxidant concentrations, more detailed monoterpene characterisation over time etc.) would allow improved translation of experimental parameters into INCHEM-Py, thus allowing temporal comparison between experiments and simulations. Not only would this give better insight into the evolution of secondary chemistry, rather than the average effects shown in this work, it would also provide model benchmarking, and highlight any potential processes that are currently not included in the model (e.g. re-emission of VOCs from surfaces, following deposition).
Author contributions
Helen Davies: methodology, investigation, formal analysis, data curation, writing – original draft, writing – review and editing, visualisation. Catherine O'Leary: methodology, investigation, formal analysis, data curation, data curation, writing – original draft, writing – review and editing. Terry Dillon: methodology, investigation, writing – review and editing. David Shaw: methodology, software. Marvin Shaw: methodology, investigation, writing – review and editing. Archit Mehra: methodology, investigation. Gavin Philips: methodology, supervision. Nicola Carslaw: conceptualisation, methodology, writing – original draft, writing – review and editing, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank James R. Hopkins for his assistance with the offline experiments, and Steven Andrews for his support with SIFT-MS calibrations. The authors would like to acknowledge the source of funding as EPSRC, grant reference EP/T014474/1.Notes and references
- N. E. Klepeis, W. C. Nelson, W. R. Ott, J. P. Robinson, A. M. Tsang and P. Switzer, et al., The National Human Activity Pattern Survey (NHAPS): a resource for assessing exposure to environmental pollutants, J. Exposure Sci. Environ. Epidemiol., 2001, 11(3), 231–252 CrossRef CAS PubMed.
- C. J. Weschler, Changes in indoor pollutants since the 1950s, Atmos. Environ., 2009, 43(1), 153–169 CrossRef CAS.
- A. M. Yeoman, M. Shaw, N. Carslaw, T. Murrells, N. Passant and A. C. Lewis, Simplified speciation and atmospheric volatile organic compound emission rates from non-aerosol personal care products, Indoor Air, 2020, 30(3), 459–472 CrossRef CAS PubMed.
- H. Destaillats, R. L. Maddalena, B. C. Singer, A. T. Hodgson and T. E. McKone, Indoor pollutants emitted by office equipment: A review of reported data and information needs, Atmos. Environ., 2008, 42(7), 1371–1388 CrossRef CAS.
- C. Arata, P. K. Misztal, Y. Tian, D. M. Lunderberg, K. Kristensen and A. Novoselac, et al., Volatile organic compound emissions during HOMEChem, Indoor Air, 2021, 31(6), 2099–2117 CrossRef CAS PubMed.
- W. W. Nazaroff and C. J. Weschler, Cleaning products and air fresheners: Exposure to primary and secondary air pollutants, Atmos. Environ., 2004, 38(18), 2841–2865 CrossRef CAS.
- X. Tang, P. K. Misztal, W. W. Nazaroff and A. H. Goldstein, Volatile organic compound emissions from humans indoors, Environ. Sci. Technol., 2016, 50(23), 12686–12694 CrossRef CAS PubMed.
- T. Salthammer, S. Mentese and R. Marutzky, Formaldehyde in the indoor environment, Chem. Rev., 2010, 110(4), 2536–2572 CrossRef CAS PubMed.
- M. Takhar, Y. Li, J. C. Ditto and A. W. H. Chan, Formation pathways of aldehydes from heated cooking oils, Environ. Sci.: Processes Impacts, 2022, 25(2), 165–175 RSC.
- G. D. Leikauf, Hazardous air pollutants and asthma, Environ. Health Perspect., 2002, 110(4), 505 CrossRef CAS PubMed.
- World Health Organization, WHO Guidelines for Indoor Air Quality: Selected Pollutants, World Health Organization. Regional Office for Europe, 2010 Search PubMed.
- G. D. Nielsen, S. T. Larsen and P. Wolkoff, Recent trend in risk assessment of formaldehyde exposures from indoor air, Arch. Toxicol., 2012, 87(1), 73–98 CrossRef PubMed.
- N. Carslaw and D. Shaw, Secondary product creation potential (SPCP): a metric for assessing the potential impact of indoor air pollution on human health, Environ. Sci.: Processes Impacts, 2019, 21(8), 1313–1322 RSC.
- S. Wieck, O. Olsson, K. Kummerer and U. Klaschka, Fragrance allergens in household detergents, Regul. Toxicol. Pharmacol., 2018, 97, 163–169 CrossRef CAS PubMed.
- S. M. Saunders, M. E. Jenkin, R. G. Derwent and M. J. Pilling, Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of nonaromatic volatile organic compounds, Atmos. Chem. Phys., 2003, 3(1), 161–180 CrossRef CAS.
- N. Carslaw, A new detailed chemical model for indoor air pollution, Atmos. Environ., 2007, 41(6), 1164–1179 CrossRef CAS.
- N. Carslaw, A mechanistic study of limonene oxidation products and pathways following cleaning activities, Atmos. Environ., 2013, 80, 507–513 CrossRef CAS.
- R. Olsen, P. Molander, S. Ovrebo, D. G. Ellingsen, S. Thorud and Y. Thomassen, et al., Reaction of glyoxal with 2-deoxyguanosine, 2-deoxyadenosine, 2-deoxycytidine, cytidine, thymidine, and calf thymus DNA: Identification of DNA adducts, Chem. Res. Toxicol., 2005, 18(4), 730–739 Search PubMed.
- G. Zhang, Y. Mu, L. Zhou, C. Zhang, Y. Zhang and J. Liu, et al., Summertime distributions of peroxyacetyl nitrate (PAN) and peroxypropionyl nitrate (PPN) in Beijing: Understanding the sources and major sink of PAN, Atmos. Environ., 2015, 103, 289–296 CrossRef CAS.
- K. L. Abdullahi, J. M. Delgado-Saborit and R. M. Harrison, Emissions and indoor concentrations of particulate matter and its specific chemical components from cooking: A review, Atmos. Environ., 2013, 71, 260–294 CrossRef CAS.
- F. Klein, S. M. Platt, N. J. Farren, A. Detournay, E. A. Bruns and C. Bozzetti, et al., Characterization of Gas-Phase Organics Using Proton Transfer Reaction Time-of-Flight Mass Spectrometry: Cooking Emissions, Environ. Sci. Technol., 2016, 50(3), 1243–1250 CrossRef CAS PubMed.
- F. Klein, N. J. Farren, C. Bozzetti, K. R. Daellenbach, D. Kilic and N. K. Kumar, et al., Indoor terpene emissions from cooking with herbs and pepper and their secondary organic aerosol production potential OPEN, Sci. Rep., 2016, 6(1), 36623 CrossRef CAS PubMed.
- D. K. Farmer, M. E. Vance, J. P. D. Abbatt, A. Abeleira, M. R. Alves and C. Arata, et al., Overview of HOMEChem: House Observations of Microbial and Environmental Chemistry, Environ. Sci.: Processes Impacts, 2019, 21, 1280–1300 RSC.
- S. Patel, S. Sankhyan, E. K. Boedicker, P. F. Decarlo, D. K. Farmer and A. H. Goldstein, et al., Indoor Particulate Matter during HOMEChem: Concentrations, Size Distributions, and Exposures, Environ. Sci. Technol., 2020, 54, 7107–7116 CrossRef CAS PubMed.
- J. Q. Koenig, D. S. Covert and W. E. Pierson, Effects of inhalation of acidic compounds on pulmonary function in allergic adolescent subjects, Environ. Health Perspect., 1989, 79, 173–178 CrossRef CAS PubMed.
- T. Berkemeier, M. Ammann, T. F. Mentel, U. Poschl and M. Shiraiwa, Organic Nitrate Contribution to New Particle Formation and Growth in Secondary Organic Aerosols from α -Pinene Ozonolysis, Environ. Sci. Technol., 2016, 50(12), 6334–6342 CrossRef CAS PubMed.
- G. D. Nielsen and P. Wolkoff, Cancer effects of formaldehyde: a proposal for an indoor air guideline value, Arch. Toxicol., 2010, 84(6), 423–446 CrossRef CAS PubMed.
- P. Wolkoff and G. D. Nielsen, Non-cancer effects of formaldehyde and relevance for setting an indoor air guideline, Environ. Int., 2010, 36(7), 788–799 CrossRef CAS PubMed.
- D. Shaw and N. Carslaw, INCHEM-Py: An open source Python box model for indoor air chemistry, J. Open Source Softw., 2021, 6(63), 3224 CrossRef.
- R. L. Wagner, N. J. Farren, J. Davison, S. Young, J. R. Hopkins and A. C. Lewis, et al., Application of a mobile laboratory using a selected-ion flow-tube mass spectrometer (SIFT-MS) for characterisation of volatile organic compounds and atmospheric trace gases, Atmos. Meas. Tech., 2021, 14(9), 6083–6100 CrossRef CAS.
- D. Smith, P. Španěl, M. Hamdan and G. Wellcome, The Novel Selected-ion Flow Tube Approach to Trace Gas Analysis of Air and Breath, Rapid Commun. Mass Spectrom., 1996, 10, 1183–1184 CrossRef CAS PubMed.
- D. Smith and P. Španěl, Selected ion flow tube mass spectrometry (SIFT-MS) for on-line trace gas analysis, Mass Spectrom. Rev., 2005, 24(5), 661–700 CrossRef CAS PubMed.
- C. O'Leary, Y. de Kluizenaar, P. Jacobs, W. Borsboom, I. Hall and B. Jones, Investigating measurements of fine particle (PM2.5) emissions from the cooking of meals and mitigating exposure using a cooker hood, Indoor Air, 2019, 29(3), 423–438 CrossRef PubMed.
- D. R. Shaw, T. J. Carter, H. L. Davies, E. Harding-Smith, E. C. Crocker, G. Beel, et al., INCHEM-Py v1.2: A Community Box Model for Indoor Air Chemistry, DOI:10.5194/egusphere-2023-1328.
- M. E. Jenkin, S. M. Saunders and M. J. Pilling, The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 1997, 31(1), 81–104 CrossRef CAS.
- M. E. Jenkin, S. M. Saunders, V. Wagner and M. J. Pilling, Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 2003, 3(1), 181–193 CrossRef CAS.
- C. Bloss, V. Wagner, M. E. Jenkin, R. Volkamer, W. J. Bloss and J. D. Lee, et al., Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys., 2005, 5(3), 641–664 CrossRef CAS.
- Master Chemical Mechanism. MCM, Date accessed: 20-04-2023. available from: http://mcm.york.ac.uk/roots.htt.
- T. J. Carter, D. G. Poppendieck, D. Shaw and N. Carslaw, A Modelling Study of Indoor Air Chemistry: The Surface Interactions of Ozone and Hydrogen Peroxide, Atmos. Environ., 2023, 297, 119598 CrossRef CAS.
- A. Manuja, J. Ritchie, K. Buch, Y. Wu, C. M. A. Eichler and J. C. Little, et al., Total surface area in indoor environments, Environ. Sci.: Processes Impacts, 2019, 21(8), 1384–1392 RSC.
- European Environmental Agency, European Air Quality Portal, Date accessed: 03-03-2023. available from: https://eeadmz1-cws-wp-air02.azurewebsites.net/.
- W. W. Nazaroff, Residential air-change rates: A critical review, Indoor Air, 2021, 31(2), 282–313 CrossRef PubMed.
- M. Blocquet, F. Guo, M. Mendez, M. Ward, S. Coudert and S. Batut, et al., Impact of the spectral and spatial properties of natural light on indoor gas-phase chemistry: Experimental and modeling study, Indoor Air, 2018, 28(3), 426–440 CrossRef CAS PubMed.
- M. Kruza and N. Carslaw, How do breath and skin emissions impact indoor air chemistry?, Indoor Air, 2019, 29(3), 369–379 CrossRef CAS PubMed.
- C. J. Weschler, A. Wisthaler, S. Cowlin, G. Tamas, P. Strom-Tejsen and A. T. Hodgson, et al., Ozone-Initiated Chemistry in an Occupied Simulated Aircraft Cabin, Environ. Sci. Technol., 2007, 41(17), 6177–6184 CrossRef CAS PubMed.
- D. C. Zhang, J. J. Liu, L. Z. Jia, P. Wang and X. Han, Speciation of VOCs in the cooking fumes from five edible oils and their corresponding health risk assessments, Atmos. Environ., 2019, 211, 6–17 CrossRef CAS.
- A. B. Guenther, P. R. Zimmerman, P. C. Harley, R. K. Monson and R. Fall, Isoprene and monoterpene emission rate variability: Model evaluations and sensitivity analyses, J. Geophys. Res.: Atmos., 1993, 98(D7), 12609–12617 CrossRef.
- A. Guenther, Seasonal and spatial variations in natural volatile organic compound emissions, Ecol. Appl., 1997, 7(1), 34–45 CrossRef.
- F. Loreto, A. Forster, M. Durr, O. Csiky and G. Seufert, On the monoterpene emission under heat stress and on the increased thermotolerance of leaves of Quercus ilex L. fumigated with selected monoterpenes, Plant, Cell Environ., 1998, 21(1), 101–107 CrossRef CAS.
- M. Lupke, M. Leuchner, R. Steinbrecher and A. Menzel, Quantification of monoterpene emission sources of a conifer species in response to experimental drought, AoB Plants, 2017, 9(5), plx045 CrossRef PubMed.
- H. R. Katragadda, A. Fullana, S. Sidhu and Á. A. Carbonell-Barrachina, Emissions of volatile aldehydes from heated cooking oils, Food Chem., 2010, 120(1), 59–65 CrossRef CAS.
- Y. Liu, P. K. Misztal, J. Xiong, Y. Tian, C. Arata and R. J. Weber, et al., Characterizing sources and emissions of volatile organic compounds in a northern California residence using space- and time-resolved measurements, Indoor Air, 2019, 29, 12562 CrossRef PubMed.
- S. K. Brown, Chamber Assessment of Formaldehyde and VOC Emissions from Wood-Based Panels, Indoor Air, 1999, 9(3), 209–215 CrossRef CAS PubMed.
- A. Vyskocil, C. Viau and S. Lamy, Peroxyacetyl nitrate: review of toxicity, Hum. Exp. Toxicol., 1998, 17(4), 212–220 CrossRef CAS PubMed.
- N. Carslaw and D. Shaw, Modification of cleaning product formulations could improve indoor air quality, Indoor Air, 2022, 32(3), e13021 CrossRef CAS PubMed.
- A. C. Terry, N. Carslaw, M. Ashmore, S. Dimitroulopoulou and D. C. Carslaw, Occupant exposure to indoor air pollutants in modern European offices: An integrated modelling approach, Atmos. Environ., 2014, 82, 9–16 CrossRef CAS.
- Z. Wang, D. Shaw, T. Kahan, C. Schoemaecker and N. Carslaw, A modeling study of the impact of photolysis on indoor air quality, Indoor Air, 2022, 32(6), e13054 CAS.
- H. Sacht, L. Braganca, M. Almeida, J. H. Nascimento and R. Caram, Spectrophotometric Characterization of Simple Glazings for a Modular Facade, Energy Procedia, 2016, 96, 965–972 CrossRef.
- S. Zhou, C. J. Young, T. C. VandenBoer, S. F. Kowal and T. F. Kahan, Timeresolved measurements of nitric oxide, nitrogen dioxide, and nitrous acid in an occupied New York home, Environ. Sci. Technol., 2018, 52(15), 8355–8364 CrossRef CAS PubMed.
- J. J. Zhang, Y. Wei and Z. Fang, Ozone Pollution: A Major Health Hazard Worldwide, Front. Immunol., 2019, 10(10), 2518 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3em00167a |
| ‡ Current address: Faculty of Science and Engineering, Maastricht University, Maastricht, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2023 |