
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtmospheric OH reactivity in the western United States determined from comprehensive gas-phase measurements during WE-CAN†
Wade
Permar
 *a,
Lixu
Jin
a,
Qiaoyun
Peng
b,
Katelyn
O'Dell
c,
Emily
Lill
d,
Vanessa
Selimovic
a,
Robert J.
Yokelson
a,
Rebecca S.
Hornbrook
e,
Alan J.
Hills
e,
Eric C.
Apel
e,
I-Ting
Ku
d,
Yong
Zhou
d,
Barkley C.
Sive
f,
Amy P.
Sullivan
d,
Jeffrey L.
Collett
Jr.
d,
Brett B.
Palm
e,
Joel A.
Thornton
b,
Frank
Flocke
e,
Emily V.
Fischer
d and
Lu
Hu
*a
*a,
Lixu
Jin
a,
Qiaoyun
Peng
b,
Katelyn
O'Dell
c,
Emily
Lill
d,
Vanessa
Selimovic
a,
Robert J.
Yokelson
a,
Rebecca S.
Hornbrook
e,
Alan J.
Hills
e,
Eric C.
Apel
e,
I-Ting
Ku
d,
Yong
Zhou
d,
Barkley C.
Sive
f,
Amy P.
Sullivan
d,
Jeffrey L.
Collett
Jr.
d,
Brett B.
Palm
e,
Joel A.
Thornton
b,
Frank
Flocke
e,
Emily V.
Fischer
d and
Lu
Hu
*a
aDepartment of Chemistry and Biochemistry, University of Montana, Missoula, MT, USA. E-mail: wade.permar@umontana.edu; lu.hu@mso.umt.edu
bDepartment of Atmospheric Sciences, University of Washington, Seattle, WA, USA
cDepartment of Environmental and Occupational Health, Milken Institute School of Public Health, George Washington University, Washington, DC, USA
dDepartment of Atmospheric Science, Colorado State University, Fort Collins, CO, USA
eAtmospheric Chemistry Observations & Modeling Laboratory, National Center for Atmospheric Research, Boulder, CO, USA
fAir Resources Division, National Park Service, Denver, CO, USA
First published on 31st October 2022
Abstract
Wildfire smoke contains numerous different reactive organic gases, many of which have only recently been identified and quantified. Consequently, their relative importance as an oxidant sink is poorly constrained, resulting in incomplete representation in both global chemical transport models (CTMs) and explicit chemical mechanisms. Leveraging 160 gas-phase measurements made during the Western Wildfire Experiment for Cloud Chemistry, Aerosol Absorption, and Nitrogen (WE-CAN) aircraft campaign, we calculate OH reactivities (OHRs) for western U.S. wildfire emissions, smoke aged >3 days, smoke-impacted and low/no smoke-impacted urban atmospheres, and the clean free troposphere. VOCs were found to account for ∼80% of the total calculated OHR in wildfire emissions, with at least half of the field VOC OHR not currently implemented for biomass burning (BB) emissions in the commonly used GEOS-Chem CTM. To improve the representation of OHR, we recommend CTMs implement furan-containing species, butadienes, and monoterpenes for BB. The Master Chemical Mechanism (MCM) was found to account for 88% of VOC OHR in wildfire emissions and captures its observed decay in the first few hours of aging, indicating that most known VOC OH sinks are included in the explicit mechanisms. We find BB smoke enhanced the average total OHR by 53% relative to the low/no smoke urban background, mainly due to the increase in VOCs and CO thus promoting urban ozone production. This work highlights the most important VOC species for daytime BB plume oxidation and provides a roadmap for which species should be prioritized in next-generation CTMs to better predict the downwind air quality and health impacts of BB smoke.
Environmental significanceWildfires emit hundreds of different VOCs into the atmosphere, with the most reactive species playing the largest role in ozone and secondary organic aerosol formation. However, the relative importance of many recently identified and quantified VOCs as oxidant sinks is highly uncertain. This work examines the major OH radical sinks in various western U.S. environments during a wildfire season using the metric of OH reactivity. We find that global chemical transport models may be missing at least half of the VOC OH reactivity in wildfire emissions and provide further recommendations for which species to include in next-generation air quality models to better predict the air quality impacts of wildfire smoke. |
1 Introduction
Wildfires are a major source of volatile organic compounds (VOCs) to the atmosphere,1–3 emitting hundreds to thousands of different organic gas-phase species.4–6 Many of the VOCs emitted by wildfires are known hazardous air pollutants, play an integral role in the formation of secondary organic aerosol (SOA) and ozone (O3) as smoke plumes age, and have long-range impacts on tropospheric oxidation potential.7–10 Increasing evidence has suggested that wildfires can bring VOC-rich air into VOC-limited cities resulting in enhanced ozone production in urban atmospheres.10–12 Although many VOC species have recently been identified and quantified in biomass burning (BB) smoke, their roles as a hydroxyl radical (OH) sink are poorly constrained. Consequently, we do not understand how well current global chemical transport models (CTMs) and explicit chemical mechanisms represent BB VOCs, the amount of reactive carbon they may be missing, and which species should be prioritized to incorporate into the next generations of air quality models. In this work, we use comprehensive VOC and other trace gas measurements made during the Western Wildfire Experiment for Cloud Chemistry, Aerosol Absorption, and Nitrogen (WE-CAN) field campaign to examine the major daytime OH radical sinks over the western United States (U.S.) during an active wildfire season.Both plume scale and global chemical transport models have difficulty predicting the evolution and downwind air quality impacts of BB on NOx,13 VOCs,14 O3,15 and SOA in smoke-impacted environments. During the day, OH radicals are the primary oxidant in BB smoke, though O3 can also play an important role in alkene and terpene oxidation, while in optically dense plumes NO3 is a competitive oxidant of phenolics.16 Major BB OH sources include photolysis of HONO,17 O3, and formaldehyde,18,19 while the major sinks include primary and secondary VOCs, NOx, and CO.11,16,20 As the global mean OH concentration is consistently overestimated across CTMs,21,22 it is likely that significant OH sinks are missing in models.23,24 This in turn affects the overall simulated oxidation chemistry and may partly explain why CTMs underestimate O3 enhancement from BB.15
OH reactivity (OHR) is a measure of the total amount of reactive gases in the atmosphere on the scale of reactivity25–27 and can be calculated from field measurements of speciated reactive gases or directly measured. The calculated OHR is defined as,
| OHR = kOHX[X] | (1) |
The major trace gas contributors to the tOHR can vary significantly between environments, resulting in distinct OHR speciation profiles. For example, a majority of the tOHR in biogenic emissions is accounted for by isoprene, monoterpenes, 2-methyl-3-buten-2-ol (MBO), and isoprene oxidation products such as methyl vinyl ketone (MVK) and methacrolein (MACR),28–31 while the tOHR in urban environments is typically dominated by NOx, CO, and petroleum-linked hydrocarbons such as light alkenes and aromatics.28,32–34 Though less reactive than many primarily-emitted VOCs, formaldehyde and acetaldehyde have also been shown to be substantial OH sinks in most environments. To date, the most comprehensive characterizations of OHR in BB emissions come from laboratory experiments and suggest a much more diverse OHR speciation profile than urban or forest atmospheres due to the large number of reactive organic gases that are emitted. Even though BB emits large amounts of CO2, NOx, CO, and CH4, VOCs are the major OH sink, contributing 80% of the initial tOHR in laboratory burning experiments, despite comprising less than 1% of emissions by mass.35 Many highly reactive individual VOCs or groups have been found to significantly contribute to OHR upon emission, including terpenes, alkenes, furan-containing species (collectively referred to here as furans), formaldehyde, and acetaldehyde.4,5,16,20,35 The OHR profile of aged smoke has been less described in the literature and is often mixed with other emission sources, but typically is characterized by oxygenated VOCs (OVOCs), furans, aromatics, and aliphatic hydrocarbons.11,36
Currently, CTMs such as the commonly used GEOS-Chem, are ultimately limited on the number of VOCs they can explicitly describe due to computational constraints. Consequently, CTMs only implement simplified chemistry for a handful (typically <30) of the most studied individual and lumped species, which were often developed and evaluated for relatively well-studied environments such as urban areas, forests, and to a lesser extent the remote/clean troposphere.24,30,37–40 Recent work suggests the GEOS-Chem CTM underpredicts biogenic VOC reactivity flux by 40–60% in forest environments, with major uncertainties related to a few select known/modeled VOCs, rather than unrepresented species.30 How well these modeled species represent the most important individual OH sinks in wildfire smoke has yet to be assessed.
Alternatively, box models such as the Framework for 0-D Atmospheric Modeling (F0AM) can simulate chemistry in BB plumes for tens of thousands of reactions and species using detailed explicit chemical mechanisms such as the Master Chemical Mechanism (MCM).13,18,19 How well these models represent the tOHR and its evolution downwind is bound by the number of reactive species included in the chemical mechanisms, how well we understand their cascade oxidation, and whether these species are included in model initialization. The extent to which the MCM incorporates most of the reactive species emitted from wildfires is not well known due to historically scarce field observations that fully characterize BB smoke. Laboratory burning experiments suggest that the MCM captures 75% of the initial VOC tOHR in BB emissions measured by the mass spectrometry techniques used in that study, while the remaining missing reactivity likely stems from species whose chemistry has not been extensively studied such as oxygenated aromatics and less abundant furans.41 Here, we use a similar suite of instrumentation to build on the laboratory work done by Coggon et al.41 evaluating how the MCM represents OHR in wildfire emissions, while expanding the analysis to examine how the MCM captures OHR as wildfire smoke ages.
To the best of our knowledge, no other studies have employed such a comprehensive suite of gas-phase measurements to examine speciated OHR in various environments affected by wildfire smoke. By leveraging the detailed gas-phase measurements made during WE-CAN we assess major OH sinks using the metric of calculated OHR in fresh wildfire emissions, smoke aged >3 days, smoke-impacted and low/no smoke-impacted urban environments, and the clean free troposphere over the western U.S. during a wildfire season. WE-CAN was designed to measure most known reactive gases and constituted one of the most comprehensive field measurements of trace gases in these environments to date. Using calculated OHR speciation profiles we highlight the most important daytime plume OH initialized oxidation sinks and then examine how much of the field calculated OHR may be missing in current iterations of the GEOS-Chem CTM and MCM. We later use the Taylor Creek Fire as a case study to evaluate the ability of the F0AM model (using MCM chemistry) to simulate changes in plume OHR as smoke ages. These analyses are used to examine the current knowledge of BB VOC representations in models and provide a roadmap for which VOCs should be incorporated into the next generations of chemical transport models for BB applications.
2 Methods
2.1 WE-CAN aircraft campaign and relevant instruments
The Western Wildfire Experiment for Cloud Chemistry, Aerosol Absorption, and Nitrogen (WE-CAN) field campaign deployed a comprehensive payload of instrumentation aboard the NSF/NCAR C-130 research aircraft to investigate near-field smoke plume chemistry and cloud smoke interactions in the western U.S. from 24 July to 13 September 2018 (https://www.eol.ucar.edu/field_projects/we-can). During the campaign, more than 22 hours of in situ BB plume measurements were made across seven states mostly between 14:00 and 19:00 local time when burning conditions were most active. As part of normal operations, the C-130 also sampled various other environments including 1.2 hours of observations of smoke estimated to have aged >3 days, 4.8 hours of clean free troposphere, and 32 vertical profiles of urban/smoke interaction in Boise, ID (metropolitan area population 750![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). In this work, we use OH reactivity (the inverse of the lifetime of OH) calculated for 160 trace gases measured aboard the C-130 to explore the major daytime OH radical sinks in the western U.S. during a wildfire season.
000). In this work, we use OH reactivity (the inverse of the lifetime of OH) calculated for 160 trace gases measured aboard the C-130 to explore the major daytime OH radical sinks in the western U.S. during a wildfire season.
Table 1 summarizes the trace gas measurements used in this work, including methodology, accuracies, sampling intervals, and references. Organic gases were measured by four complementary instruments during WE-CAN: a proton-transfer-reaction time-of-flight mass spectrometer (PTR-ToF-MS), the Advanced Whole Air Sampler (AWAS), the NCAR Trace Organic Gas Analyzer (TOGA), and an iodide adduct chemical-ionization mass spectrometer (I− CIMS). Here, we utilize 154 measured VOCs following the data selection and data reduction criteria in Permar et al.6 with a few exceptions discussed below.
| Species | Instrument | Accuracy | Sample interval | References |
|---|---|---|---|---|
| a Proton-transfer-reaction time-of-flight mass spectrometer. b Trace organic gas analyzer. c Advanced whole air sampler. d Iodide adduct chemical-ionization mass spectrometer. e Quantum cascade laser instrument. f Cavity ring down spectrometer. | ||||
| VOCs and OVOCs | PTR-ToF-MSa | 15% for directly calibrated species, 50% for species calibrated based on molecular properties | 0.2 or 0.5 s | 6 |
| VOCs | NCAR TOGAb | 15–50% | 28–33 s integrated samples every 100–105 s | 42–45 |
| VOCs | AWASc | 10% | 3–7 s canister samples triggered manually | 46–50 |
| Formic acid, phenol, HCN, HONO | I− CIMSd | 30% | 0.5 s | 17, 51 and 52 |
| CO | QCLe | 1 ppb (2σ) | 1 s | 53 |
| CH4 | WS-CRDf | 3 ppb (2σ) | 1.3 s | — |
| NO, NO2, O3 | Chemiluminescence detector | 3%, 4%, and 2% | 54 and 55 | |
| PAN | NCAR-PAN-CIMS | 12% or 25 ppt | 2 s | 56 and 57 |
Permar et al.6 found notable disagreement between PTR-ToF-MS, TOGA, and AWAS measurements of five species relevant to the OHR in BB smoke: isoprene, propene, furan, methyl furans, and monoterpenes. To limit the error from potentially interfering fragments while preserving temporal resolution in this work, PTR-ToF-MS isoprene and propene measurements were calibrated using TOGA and AWAS mixing ratios, resulting in near 1:1 agreement during the campaign. Similarly, TOGA furan and methyl furans are used for OHR calculations due to TOGA's lower detection limits and lack of potential interfering isomers. As the PTR-ToF-MS measured furan and methyl furans approximately 1.5 and 15 times higher than TOGA, the OHR of these two species in this work may represent a lower bound, while use of PTR-ToF-MS data would result in furans being an even larger OH sink. Finally, we note that PTR-ToF-MS measures the sum of monoterpenes at m/z 137.13, which is ∼5 times higher than sum of camphene, α-pinene, β-pinene/myrcene, and tricyclene measured by TOGA, likely representing missing speciated isomers.4,6 Further details of the measurements used in this work, the corresponding instrument, measurement uncertainty, assumed speciation of PTR-ToF-MS measured ions based on Koss et al.,5 and OH rate constants are in Table S1† and discussed in more detail in Permar et al.6
2.2 Data selections and definitions
We focus our analyses on 5 different environments sampled during WE-CAN, including (1) wildfire emissions near the source, (2) aged wildfire smoke with physical age greater than 3 days, (3) smoke-impacted urban atmospheres, (4) low/no smoke urban atmospheres, and (5) free tropospheric clean conditions. Constraints for each of these environments are as follows.WE-CAN wildfire emission transects are defined as the perpendicular plume transects nearest to a given fire as described in Permar et al.6 This results in OHR calculations for 24 unique fires using 31 emission transects and allows for direct comparisons to the emission factors and emission ratios presented therein. Emissions range in physical age from 27–130 minutes downwind from the source due to plane logistical and safety constraints. While those transects represent the freshest smoke sampled during the campaign, there is sufficient time for some photochemical processing to have occurred between emission and sampling.17,58,58–60 Consequently, for very reactive primary emitted species such as monoterpenes, the calculated OHR for WE-CAN emission transects reported here is likely a lower bound. For OVOCs that are both primary emissions and photochemical products, the calculated OHR represents the culmination of production and loss processes between when smoke was emitted and when it was first sampled by the C-130.
For all smoke plumes analyzed in this work, OH reactivity was calculated from the plume center average mixing ratios determined as the top 5% of CO observations for each transect. This metric was chosen to get a better estimate of the freshest smoke by excluding the more aged plume edges while also remaining consistent with previous WE-CAN literature.13,61,62 Discrete TOGA or AWAS measurements were often not available during this period. In this case, we calculated the plume average normalized excess mixing ratio (NEMR) from the available measurements for each transect and then scaled NEMRs to the center of the plume by multiplying by the average top 5% CO abundance. Absolute mixing ratios at the plume center were then determined by adding back in regional background concentrations.
For the discussion in Sections 3.1 and 3.2, plume center mixing ratios are not background corrected and therefore discussed in terms of absolute reactivity, allowing for more direct comparison to other environments sampled during the campaign. The average tOHR during plume background periods is 1.7 ± 2.5 s−1 or 17% of the average total plume OHR. When comparing to emission speciation profiles in CTMs and box model results in Sections 3.4 and 3.5, OHR is calculated from plume excess mixing ratios. Here, transect excess mixing ratios were determined by subtracting the interpolated background measured on both edges of the plume. Physical ages for each transect were calculated based on the fire location and average wind speeds measured in the plume upwind of that transect.
To investigate the effects of photochemical aging on OH reactivity, a >3 days aged smoke profile was calculated according to the plume aging criteria in O'Dell et al.8 Briefly, ∼1.2 hours of smoke plume measurements were identified and classified as being well-aged based on 2-methylfuran < 0.7 ppt, acrolein < 7.4 ppt, and acrylonitrile > 2.9 ppt. An approximate age of >3 days was calculated for these plumes based on the predicted loss of these tracers against OH (assuming regional background of 2 × 106 molecules per cm3), assuming initial concentrations equal to emission transect observations. The altitudes of the aged smoke samples range from 850 to 5300 m above sea level (asl). Though the calculated smoke age generally agrees well with physical age estimates, this method is sensitive to variability in fire emissions, dilution rates, and oxidant concentrations.8 Consequently, the aging time is a best estimate and reflects smoke having undergone significantly more photochemical processing than the fresh emissions.
In addition to sampling in wildfire plumes, the C-130 sampled vertical profiles during ascents and descents at the Boise, ID, airport (KBOI) where the campaign was based. During WE-CAN, Boise was often impacted by mixed aged smoke transported 1–3 days from regional fires, resulting in the enhancement of many pollutants over the anthropogenic emissions.63 Here, we calculate OHR speciation profiles for the average of 16 smoke-impacted and 16 low/no-smoke ascents/descents following the selection criteria described in Lill et al.63 Ascent and descent start and stop times were defined as the period between the C-130 being in contact with the runway and within the boundary layer as identified from temperature gradients. Ascents and descents were defined as smoke-impacted based on mean mixing ratios of hydrogen cyanide (HCN) > 300 ppt and acetonitrile (CH3CN) > 200 ppt. Note that we use the term low/no smoke to describe the ascents/descents that are below this threshold as some smoke influence is expected in these profiles due to a large number of fires in the region during WE-CAN.
Free troposphere measurements were collected while in transit to and from fires. OH reactivities were calculated for non-smoke impacted free troposphere samples, defined as periods with HCN < 250 ppt, CH3CN < 150 ppt, and altitudes > 4000 m asl. The altitude criteria were selected based on the maximum observed boundary layer height as determined by vertical temperature profiles. This results in ∼4.8 hours of clean free troposphere data allowing for the impacts of wildfire smoke on regional atmospheric composition to be assessed by comparison. Sampling locations for the aged smoke and clean free troposphere observations are shown in Fig. S1† and are generally well distributed across the northwestern U.S.
2.3 OH reactivity calculations
Rate constants for the reaction of individual VOCs with the OH radical (kOH) were assigned for PTR-ToF-MS measured masses, with OH rate constants normalized by the fraction of contributing isomers to each ion mass as determined during laboratory burning experiments following Koss et al.5 Rate constants for the other measured species were retrieved from the NIST chemical kinetics database (https://kinetics.nist.gov). Standard temperature and pressure (STP) conditions were assumed for all kOH values, while trace gas mixing ratios were converted to molecules per cm3 using the field-measured temperature and pressure. OH reactivities for the major OH reactive gases measured during WE-CAN were subsequently calculated following eqn (1), with tOHR defined as the sum of all individual species' OH reactivities during a given measurement period. Similarly, the sum of all VOC OH reactivities is defined as the total calculated VOC OHR (tOHRVOC). When applicable, OHRs are normalized to the observed CO mixing ratio to account for dilution effects in BB plumes.Uncertainty in the total calculated OHR for BB has been estimated as 25% when using PTR-ToF-MS, and is mostly dependent on the speciation of ions and the subsequent assignment of rate constants to each mass.11 Specifically, in addition to uncertainties in the measurements and OH rate constants, many of the ion masses measured by PTR-ToF-MS are attributed to one or more potential isomers. Most isomers have similar chemical structures and therefore similar reaction rates with OH. However, if the isomers have very different chemical structures, their kOH may also vary significantly. As a result, uncertainty in the OH reactivity is dependent on the correct weighting of OH rate constants to each ion mass based on the expected fractional contribution of each isomer. As the identification and rate constants for the major contributors to the total OHR are mostly well-defined, the impacts on the total OHR are small. However, the uncertainty in the calculated OHR for individual species, especially those rarely reported, may be 100% or more.
2.4 BB emissions implemented in GEOS-Chem CTM
To assess how well typical global CTMs represent wildfire emissions, we compare the average VOC OHR speciation profile calculated for WE-CAN emissions with that from the Global Fire Assimilation System version 1.2 (GFAS) inventory as implemented in GEOS-Chem version 13.3.2 (GEOS-Chem + GFAS).64,65 GFAS emissions were retrieved for the 2018 fire season (June–September) over the western U.S. domain (36° N–127° W, 49.5° N–105° W) with updated emission ratios for lumped aldehydes, MEK, formic acid, and acetic acid following Permar et al.6 Only those species implemented in GEOS-Chem were included for further comparison, resulting in 18 explicitly implemented VOCs along with three lumped species (21 GEOS-Chem VOC species are listed in Table S2† with their emission estimates and kOH values). OHR speciation profiles were calculated from GFAS emissions estimates in GgC by converting to molecules per cm3 assuming STP conditions, then dividing each species by the total implemented OHR. kOH values were the same as those used in the observations for consistent comparisons. Additionally, kOH was calculated for the three lumped species by weighting the kOH of corresponding WE-CAN measured species by their emission ratios.2.5 The Framework for 0-D Atmospheric Modeling driven by MCM (F0AM + MCM)
To explore how explicit chemical mechanisms represent BB OHR upon emission and as smoke plumes age, we use the F0AM box model66 to simulate the Taylor Creek Fire sampled during WE-CAN. The Taylor Creek Fire was selected as a case study because it has been well described in the WE-CAN literature,9,13,17,62,67,68 was well sampled in a pseudo-Lagrangian fashion, and had well-defined plume edges with no or minimal mixing of regional smoke and other anthropogenic emissions.F0AM was run using the Master Chemical Mechanism (MCM, v3.3.1), augmented with recently developed furans and phenolic chemistry.20,41 Initial conditions, observational constraints, and model setup generally followed those described in detail by Peng et al.13 Briefly, physical parameters such as photolysis frequencies, temperature, and pressure were constrained to measured values at each model step while CO observations were used to derive a plume dilution correction factor. F0AM was initialized using 50 VOCs in addtion to NO, NO2, HONO, O3, and CO. To facilitate consistent model/observation comparison in this work, minor updates were made to approximately half of the initial VOC concentrations in Peng et al.13 to incorporate campaign measured values for species that were unavailable at the time of their study. When using PTR-ToF-MS data to initialize F0AM, individual VOC mixing ratios were estimated using the fractional contribution of known isomers to the total ion signal in BB emissions (Table S1†).5,6 Initialized values are available in Table S3.† Our model updates do not significantly alter the model results presented by Peng et al.13 The simulation of the detailed gas photochemistry then includes 5832 species in 17224 reactions. The top 100 species contributing to the total model OHR are retrieved and compared to the field calculated OHR in Section 3.5. These top 100 species comprise >99% of the total model OHR.
3 Results and discussion
3.1 Atmospheric OH reactivity in various western U.S. environments
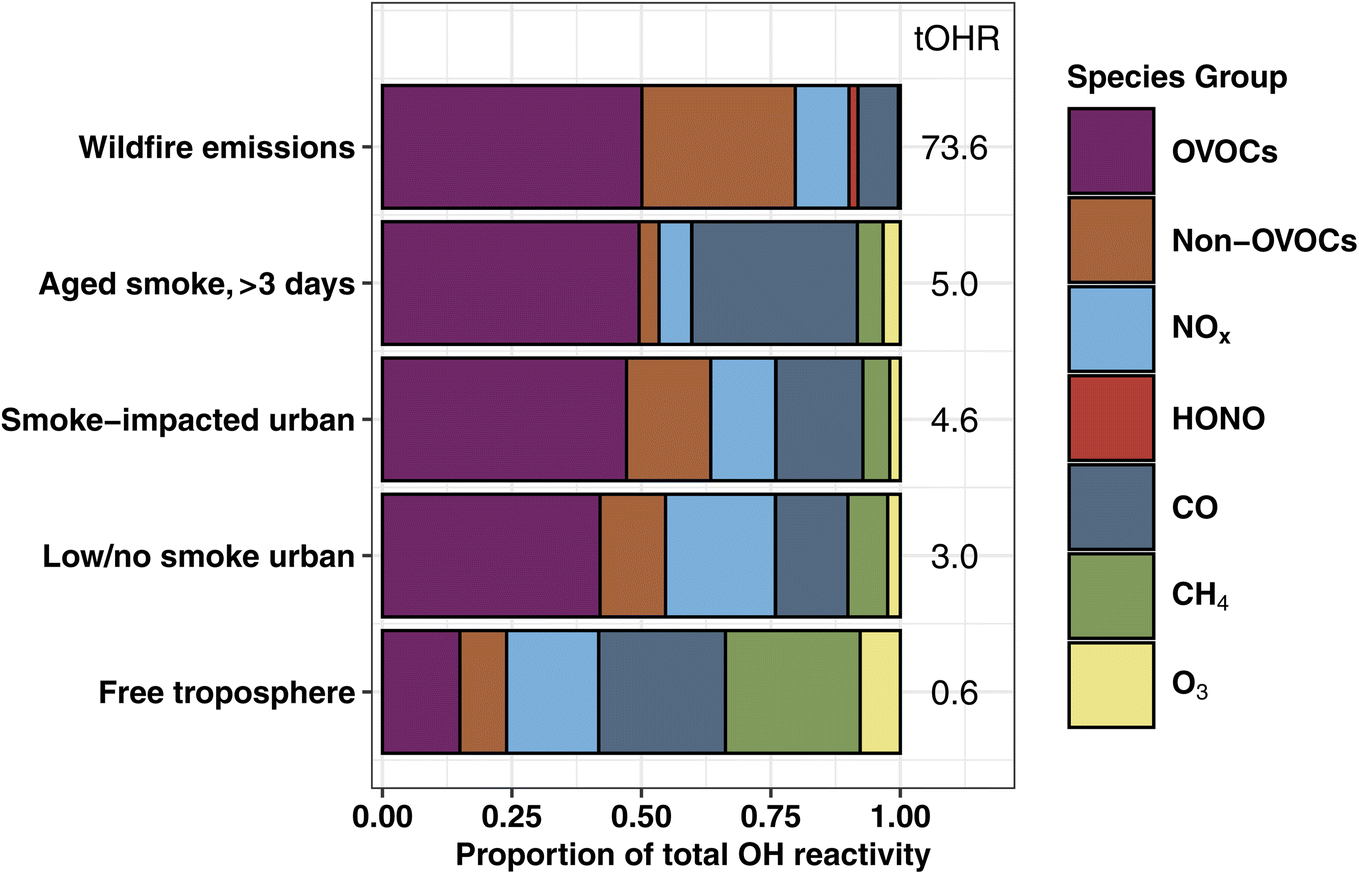 | ||
| Fig. 1 The percent contribution of major reactive gas species/groups to the total calculated OH reactivity for wildfire emissions (31 transects of 24 fires), aged smoke with chemical age >3 days (1.2 hours of samples), atmospheric boundary layer in Boise, ID, separated by smoke-impacted and low/no smoke conditions (32 takeoff/landing profiles), and clean free tropospheric conditions (4.8 hours of data) sampled by the C-130 during WE-CAN. Average calculated OH reactivities (s−1) are shown for each sampling condition to the right of the bars. Note that the low/no smoke profile still likely contains small smoke impacts due to regional fires. | ||
In emission transects the plume center tOHR ranges from 9–199 s−1 (mean 73.6 s−1; Fig. S2†), and plume excess tOHR normalized to CO ranges from 12–41 s−1 ppmCO−1 (mean 27.5 s−1 ppmCO−1). This is at the lower end of laboratory emissions scaled with field observations (98–450 s−1)16 reflecting higher initial CO mixing ratios during the lab study. Previously reported total mOHR values in urban plumes (10–130 s−1) and forested regions (1–70 s−1) vary greatly between environments,69 but are generally similar to or lower than wildfire emissions. This is further illustrated in Fig. 1 where the tOHR of low/no smoke and smoke-impacted urban emissions in Boise, ID, is 16–25 times lower than in wildfire emissions. As discussed in more detail below, wildfire emissions also consist of many different reactive VOCs and consequently have a markedly different OHR speciation profile than urban and forested regions.
The fraction of VOC OHR in aged smoke is ∼26% lower than in the BB emissions (Fig. 1). This loss of the fraction of VOC reactivity is mainly driven by the oxidation of many primarily emitted hydrocarbons (HCs) including propene, ethene, monoterpenes, and C2 substituted furans. The proportion of OVOC OHR remains nearly the same between emissions and aged speciation profiles (∼50%), bolstered in part by the increase in reactivity from formaldehyde and acetic acid. As a result, CO makes up a significantly larger proportion of the plume OHR, from 7.8% of the tOHR in wildfire emissions to 32% in aged smoke, suggesting the increased role of CO in photochemistry in aged smoke.
The amount of missing OHR varies by source and is dependent on the deployed speciated measurements. Using modern analytical instrumentation selected to characterize known gas-phase species, studies have found 25% missing OHR in the global marine boundary layer70 and 0–30% missing OHR in urban environments.28,33,34,71–73 Forested environments have a generally larger missing fraction of 0–75%.25,29,31,74 These studies often have pointed to unmeasured VOCs, particularly OVOCs, as the main contributor to the missing OHR. For BB, Kumar et al.36 suggested 40% missing OHR in the only study to date to report direct OHR field measurements in an agricultural fire-influenced environment, hypothesizing that unmeasured nitrogen-containing VOCs (NVOCs), specifically alkylamines, were likely the main contributor to the missing fraction. During WE-CAN, missing 40% of the OHR in fresh and aged smoke would suggest increases of the average tOHR by 30 s−1 and 2 s−1 respectively. However, 34 NVOCs including 4 alkylamines, account for only 2% of the tOHR and 2.5% of the tOHRVOC (Section 3.2) in WE-CAN emissions, suggesting that in western U.S. wildfires, NVOCs may not be a major contributor to the tOHR. This could point to much greater NVOC emission from agricultural BB because of the higher nitrogen content in crop biomass.36 However, due to the high uncertainty in PTR-ToF-MS NVOC measurements, more detailed speciation and better quantification of NVOCs in BB is needed to fully understand their role as an OH sink.
24 and is elevated over North America and in aged BB smoke.76
BB has little impact on the NOx OHR in Boise, with smoke-impacted profiles having slightly lower NOx OHR than low/no smoke ones (0.58 s−1vs. 0.64 s−1). As a result, the low/no smoke urban atmosphere has a higher fraction of NOx OHR than during smoke-impacted days (21% vs. 13%). This is consistent with our general understanding of aged smoke, where NOx is rapidly depleted due to fast reactions with OH and other primary VOCs (i.e., furans or phenolic compounds) or converted to PAN during transport.10,78
3.2 OH reactivity of VOCs
Here, we use the detailed speciated VOC measurements available during WE-CAN to investigate the contribution of individual VOC species to the OHR speciation profiles discussed above. Our previous work using this dataset found that, in western U.S. wildfires, the 20 most abundantly emitted species account for ∼70% of the total VOC emissions by mass, and ∼81% on a molar basis.6 However, in this study, we find that they account for only 51% of the tOHRVOC.The plume center tOHRVOC for western U.S. wildfire emissions averaged 58 s−1 during WE-CAN, with a range of 7–170 s−1, largely representing different extents of dilution between sampled smoke plumes. Total VOC OHR normalized to CO was found to average 23.5 s−1 ppmCO−1 (range 8.6–31.8 s−1 ppmCO−1), which is lower than observed in previous laboratory studies of biomass burning emissions (61 ± 10 s−1 ppmCO−1).35 Since laboratory studies sample BB smoke only a few seconds after emission while WE-CAN emission transects are 27–130 minutes downwind from each fire, this may represent a loss of the most reactive primary VOCs between emission and sampling by the C-130 as well as differences between laboratory and field burning conditions (typically drier fuels, higher combustion efficiency, and simpler fuel mixtures in the lab).6 The tOHRVOC during WE-CAN then is likely a lower bound due to rapid oxidation of short-lived species such as monoterpenes, isoprene, and many furan containing compounds. Consequently, these species may make up a larger proportion of the tOHR in emissions sampled nearer to the source, while the contribution of photochemically produced OVOCs may be reduced.
Fig. 2 shows the normalized OHR (s−1 ppmCO−1) of the top 10 trace organic gas contributors to the calculated tOHRVOC in western U.S. wildfire emissions (CO abundance for the fire emissions is 2.6 ± 1.7 ppm during WE-CAN). These 10 VOCs account for 62% of the tOHRVOC, while 143 species comprise the remaining 38%. Three VOCs (acetaldehyde, formaldehyde, and propene) each have similar or slightly larger normalized OHR as NOx, collectively accounting for ∼25% of the total calculated trace gas OHR (Section 3.1), and 30% of the tOHRVOC. Importantly, the tOHRVOC of the remaining 143 species/groups not explicitly shown in Fig. 2 is larger than any individual contributor, further emphasizing the complexity of wildfire emissions and the challenge of implementing all observed OHR in atmospheric models.
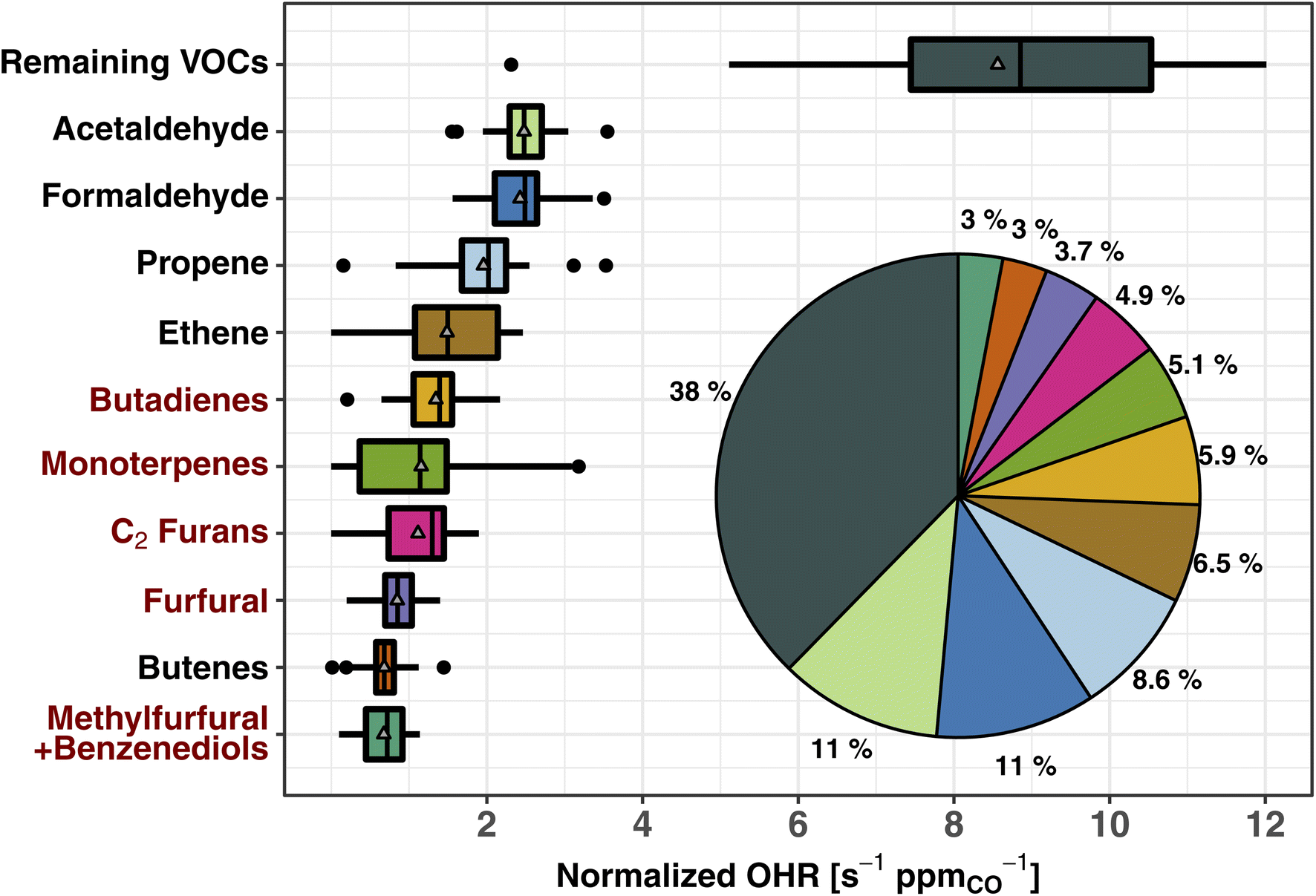 | ||
| Fig. 2 Box and whisker plots of the 10 largest contributors to the total calculated VOC OHR of western U.S. wildfire emissions during WE-CAN. The OHR is normalized to CO to account for BB plume dilution effects. The inset pie chart shows the fraction of plotted species contribution to the total VOC OHR. The remaining VOCs include 143 species not explicitly shown. Pie chart colors correspond to the box plots. Red VOC names correspond to those BB emissions not implemented in the standard GEOS-Chem CTM. Boxes represent the 25th and 75th percentiles, vertical lines as median, whiskers as 1.5 times the interquartile range, points as >1.5 times interquartile range, and triangles as mean. Butadienes include 1,3- and 1,2-butadiene isomers. Monoterpenes are the sum of all ionizable isomers (α-pinene, β-pinene, camphene, etc.). C2 furans include 2,5-dimethylfuran, 2-ethylfuran, and other unidentified C2 substituted furan isomers. Furfurals include 2- and 3-furfural. Methylfurfural and benzenediol isomers are expected to each make up ∼50% of the mass (Table S1†). | ||
Fig. 3 shows the proportion of individual and grouped VOCs to the tOHRVOC. Furan-containing species, alkenes, and aldehydes each make up ∼20% of the plume tOHRVOC upon emission, similar to the VOC OHR profile found by Xu et al.10 during the Fire Influence on Regional to Global Environments and Air Quality (FIREX-AQ) aircraft campaign. Furans, representing 19 species in this work, are important OH sinks in fresh laboratory biomass burning emissions,5,35,79 contributing up to 10% of O3 production in the first four hours since emission.41 Alkene OHR is largely from propene and ethene (15% of the tOHRVOC), while aldehyde OHR is from formaldehyde and acetaldehyde (21%), which are in turn the two largest individual contributors to the plume tOHRVOC. Fig. 3 also shows that reactivity in wildfire emissions is relatively uniformly spread amongst the major contributing groups. This contrasts with more well-studied VOC sources such as biogenic emissions where isoprene, monoterpenes, and isoprene oxidation products such as MVK and MACR are the primary contributors to the tOHR.27,30,31 Similarly, urban tOHR is primarily comprised of ∼50% NOx and CO (Fig. 1), while aromatic and other hydrocarbons make up most of the urban VOC OHR fractions.
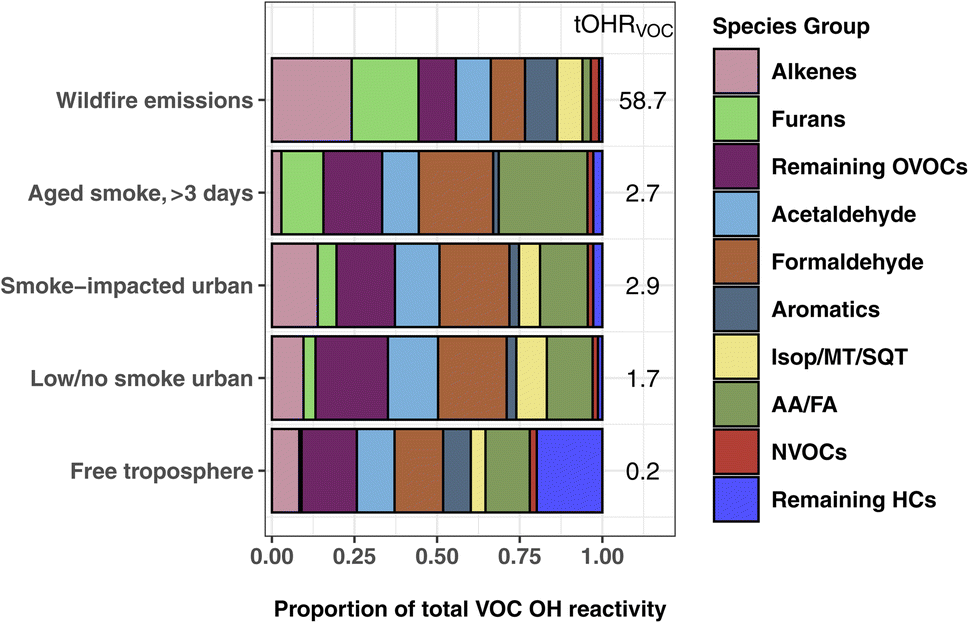 | ||
| Fig. 3 The percent contribution of individual and VOC groups to the total calculated VOC OHR during WE-CAN. WE-CAN wildfire emission average, smoke-impacted urban, low/no smoke urban, and clean free troposphere periods are the same as described in Fig. 1. The Isop/MT/SQT grouping includes isoprene, monoterpenes, and sesquiterpenes. AA/FA are acetic and formic acids. | ||
As smoke plumes age and directly emitted non-methane hydrocarbons are oxidized, OVOCs become an even more dominant OH sink in aged smoke (Fig. 1). In smoke sampled during WE-CAN that had aged >3 days, formaldehyde (23%), acetic acid (measured with glycolaldehyde; 21%), and acetaldehyde (11%) are the largest individual contributors to the tOHRVOC, reflecting their growing importance as an OH sink as smoke is transported downwind and primary VOCs are oxidized. Additionally, formic and acetic acids collectively make up a quarter of the aged smoke tOHRVOC, emphasizing the importance of BB as a source of organic acids and the need to better understand the chemistry leading to their formation and loss. The remaining OVOC fraction in Fig. 3 consists of 36 species contributing 18% of the plume tOHRVOC, with the top 3 VOCs being methylmethacrylate, methanol, and hydroxyacetone (∼13% of the tOHRVOC).
Interestingly, furans still account for 13% of the aged smoke tOHRVOC (Fig. 3), despite their generally short atmospheric lifetime. A closer examination of the aged smoke profile shows that 97% of the furans OHR is accounted for by five species/groups: 5-hydroxy-2-furfural/2-furoic acid (C5H4O3), succinic anhydride (C4H4O3), furanone (C4H4O2), 5-(hydroxymethyl)-2-furfural (C6H6O3), and maleic anhydride (C4H2O3). Maleic and succinic anhydride are long-lived species (>5 days assuming OH concentrations of 1.5 × 106 molecules per cm3) and are known oxidation products of furans and oxygenated aromatics.41,80 Measurement of these anhydrides in the aged plumes sampled during WE-CAN further confirms their relevance as tracers for aged biomass burning plumes as proposed by Coggon et al.41 The remaining three species are generally not well described in the BB literature. We speculate they are likely all photochemically produced from the oxidation of furans though we do not know their exact precursors and chemical pathways. Additionally, speciation of the PTR-ToF-MS measured C5H4O3, C4H4O3, C4H4O2, C6H6O3, and C4H2O3 was based on laboratory BB emissions, meaning that there may be additional isomers and interfering fragments in aged smoke. However, the contribution of these species to the aged plume OH reactivity suggests that furans and their oxidation products could play a sizable role in plume chemistry even far downwind of fires, highlighting the need for further investigations to improve our ability to measure and understand their chemical transformations.
The tOHRVOC in Boise during the aircraft takeoffs/landings was 50% higher (1.6 s−1 to 2.9 s−1) on smoke-impacted days. On these days, the fraction of alkenes and furans were both elevated relative to the urban background, in agreement with our knowledge of BB emissions. Biogenic species including monoterpenes and isoprene make up a larger proportion of the OHR on low/no smoke days due to local and regional biogenic emissions, though they are only a small fraction of the tOHRVOC in Boise (10%). Additionally, on low/no smoke days, formaldehyde and organic acids are significant contributors to the tOHRVOC, suggesting that even during minimal BB impacted periods they play an important role in the local chemistry.
Clean free tropospheric VOC OHR during the study period is largely dominated by OVOCs, including formaldehyde (13%), acetaldehyde (11%), acetic acid/glycolaldehyde (10%), methanol (6%), MVK/MACR/2-butenal (2%), and acetone/propanal (2%), in good agreement with previous studies of the free tropospheric OH budget.24,81 Longer lived alkanes make up most of the remaining hydrocarbons (“remaining HCs” in Fig. 3), and consequently account for 20% of the tOHRVOC in the free troposphere. Methylhexanes, methylheptanes, and propane are the top 3 contributors (50%) of the remaining HCs fraction.
3.3 Implications for urban O3 chemistry
BB has generally been shown to increase O3 production in urban environments, though decreased production may occur under significantly smoky conditions.77,78,82 One way to predict how smoke will impact O3 production is by investigating the O3 production regime of an air mass. Kirchner et al.83 proposed the metric θ = tOHRNOx/tOHRVOC, where tOHRNOx and tOHRVOC are the total NOx and VOC OH reactivity, to define the O3 production regime. When θ is >0.2, O3 production is classified as VOC-limited (or NOx saturated), θ < 0.01 indicates a NOx-limited regime, and θ between 0.01 and 0.2 indicates transitional conditions. Using this metric, Liang et al.11 showed that smoke altered the ozone formation regime in Berkeley, CA and that O3 production was higher during BB-influenced periods as BB VOCs lowered θ below the 0.2 threshold.For the Boise vertical profiles presented here, the influx of BB smoke causes the O3 production regime to shift from being VOC-limited during low/no smoke periods (θ = 0.39) toward a more optimal O3 production regime (θ = 0.20) on smoke-impacted days. This is due to BB smoke adding VOCs and CO to the urban atmosphere, but little additional NOx due to its rapid removal in BB plumes and relatively consistent urban emissions. This impact on O3 production is corroborated by the measured O3 mixing ratio averaging 50 ppb on low/no smoke days and 63 ppb on smoke-impacted days. Recent box modeling work suggests that such urban O3 production on smoky days is largely driven by local photochemistry.12
Wildfire emissions during WE-CAN are typically in the optimal O3 production regime with θ equal to 0.15, while θ in smoke aged >3 days was 0.12. Consequently, fresh and aged wildfire smoke is generally in a more optimal O3 production regime and becomes more NOx-limited with aging and diluting, in agreement with recent studies of BB plume O3 formation.10,19 Incidentally, this analysis suggests that the impact of BB smoke on urban O3 production is in part dependent on the smoke's age. For example, very fresh fire emissions would greatly increase the VOC OHR due to reactive primary species while simultaneously enhancing NOx, while more aged smoke adds less VOC OHR but also little to no NOx. As urban environments are traditionally VOC-limited, the influx of BB smoke will likely enhance ozone production in urban centers which would otherwise typically stay VOC-limited. However, the real-world impacts of BB smoke on urban O3 production could be further complicated due to factors such as smoke-altered photolysis rates,84,85 temperature, and conversion of PAN back to NOx (i.e., PAN mixing ratios increased from 350 ppt on low/no smoke days to 600 ppt on smoke impacted days in Boise during WE-CAN).
3.4 Biomass burning emissions as implemented in the GEOS-Chem CTM
In this section, we probe if any significant initial OH reactivity from BB is missing in current CTM VOC representations to help guide future model development for both modeled and unrepresented VOCs in BB-influenced environments. To this end, we compare the average VOC OHR speciation profiles calculated from WE-CAN wildfire emissions with GFAS emission estimates. We focus on the 21 individual and lumped VOCs included in BB emissions in GEOS-Chem, referred to here as “implemented VOCs”. Using other BB emission inventories including GFED (Global Fire Emissions Database), FINN (FIre INventory from NCAR), and QFED (Quick Fire Emissions Dataset) does not change the conclusions of this work due to emission ratios being mostly similar between the inventories. Note that by using OHR speciation profiles we are not comparing the absolute OHR, which would require further constraints on the BB emission amount. Rather, we assess whether the fractional distribution of OHR agrees between WE-CAN and GEOS-Chem + GFAS emissions and if the 21 implemented species constitute a majority of the field calculated tOHR upon emission.Fig. 4a shows the OHR profile calculated from GEOS-Chem + GFAS emissions. Lumped alkenes are the largest contributor to the calculated OHR (30%), followed by formaldehyde (21%), ethene (17%), and acetaldehyde (13%). Cumulatively, these 4 species make up 81% of the GEOS-Chem + GFAS OHR. Fig. 4b shows the average WE-CAN emission OHR profile filtered to only include implemented/modeled VOCs. We find that field measurements for 17 of the 21 implemented species agree within 3σ of the GEOS-Chem + GFAS OHR proportions, where σ represents the WE-CAN fire-to-fire variability of each VOC's proportion to the tOHRVOC. These 17 species accounts for 93% of the implemented tOHRVOC and indicate that the implemented GFAS species capture the OHR profile observed in wildfire emissions well, in part representing good relative agreement between emission factors (EFs). As mentioned previously, we do not attempt to compare absolute OHR between WE-CAN and GEOS-Chem + GFAS emissions. However, the similar OHR speciation profiles shown here suggest that agreement between the absolute OHR of these 21 implemented species is likely to be most dependent on the amount of dry mass burned rather than EFs. A more detailed analysis of the total BB emission is needed to better quantify how well emission inventories represent absolute wildfire OHR.
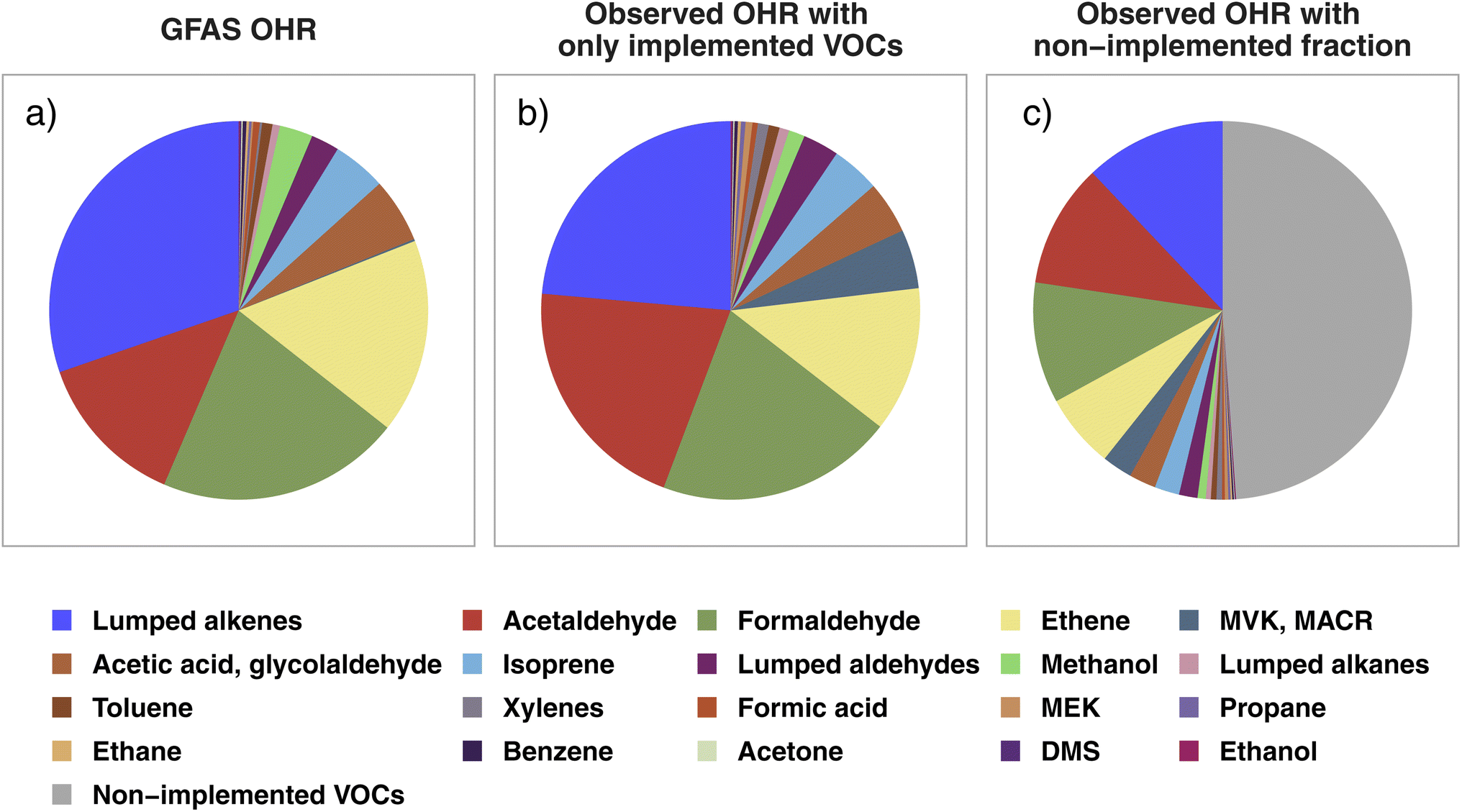 | ||
| Fig. 4 (a) The calculated OHR profile of the 21 GEOS-Chem + GFAS species implemented as biomass burning emissions, estimated as the total regional GFAS emissions during the WE-CAN campaign. (b) The WE-CAN-measured OHR profile for wildfire emissions calculated using only the implemented species in panel a. (c) OHR profile calculated from all VOCs measured during WE-CAN. Colored slices correspond to the implemented VOCs in panels a and b, representing that the 21 implemented species account for 50% of the total VOC OHR in wildfires, while the gray slice is OHR from non-implemented species. The composition of the non-implemented fraction (gray slice) is shown in Fig. S3.† Note that acetic acid and glycolaldehyde emissions in GFAS are summed for better comparison to the field PTR-ToF-MS measurements. | ||
Three of the four species that do not agree in the observed fire-to-fire variability have higher OHR fractions in the observations than in GFAS likely due to the mismatch of measured isomers and model VOC speciation. These include xylenes, MACR, and MEK which are implemented separately in GEOS-Chem (Fig. 4a) but are measured as the sum of C8 aromatics, MACR/MVK/2-butenal, and MEK/butanal/2-methylpropanal during WE-CAN (Fig. 4b). Conversely, the OHR fraction of methanol is lower in the field observations (1.4%) than in GFAS (2.8%).
Although the GEOS-Chem + GFAS OHR profile agrees well with the WE-CAN constraints, Fig. 4c shows that the 21 implemented species account for only ∼50% of the tOHRVOC in western U.S. wildfire emissions. OHR in the non-implemented VOCs is spread across 104 species, with the largest individual contributors being butadienes (12% of the non-implemented fraction), monoterpenes (10%; note that GEOS-Chem does include monoterpene chemistry but only simulates their biogenic emissions), C2 substituted furans (10%), furfural (7%), and methylfurfurals (6%). These species make up 21% of the average tOHRVOC and are the only ones among the top 10 most reactive VOCs in wildfire emissions (Fig. 2) not currently represented in GEOS-Chem.
Grouping the non-implemented fraction discussed above by the same criteria as in Fig. 2 finds that furans comprise 41% of the non-implemented fraction in Fig. 4c, followed by aromatics (18%), other OVOCs (12%), butadienes (12%), and monoterpenes (10%; Fig. S3†). The lack of reactive primary BB VOCs would constitute a missing OH sink in smoke-impacted environments, which is consistent with independent studies suggesting the global model OH is generally too high.21,23 Consequently, implementing butadienes, BB monoterpenes, and speciated or lumped furans is likely to improve model performance, particularly in BB-impacted environments and when the proper model resolution is used.
3.5 Field OH reactivity observations vs. F0AM + MCM, a case study
To assess how well our current knowledge of smoke plume chemistry agrees with field observations, we compare the field-calculated OHR (OHRfield) to the OHR predicted by the F0AM + MCM (OHRmod) for a pseudo-Lagrangian sampled wildfire plume. Unlike CTMs, box models such as the F0AM are less computationally limited and therefore can represent the chemistry of thousands of gases at a time. We use the Taylor Creek Fire (flight track shown in Fig. 5) sampled during WE-CAN as a case study to investigate: (1) to what extent the modeled initial OHR accounts for the observed OHR in wildfire emissions, (2) how well the model captures the observed change in tOHRVOC as plumes age, and (3) if any significant OHR is contributed by secondary species predicted by the model but missing from the field measurements.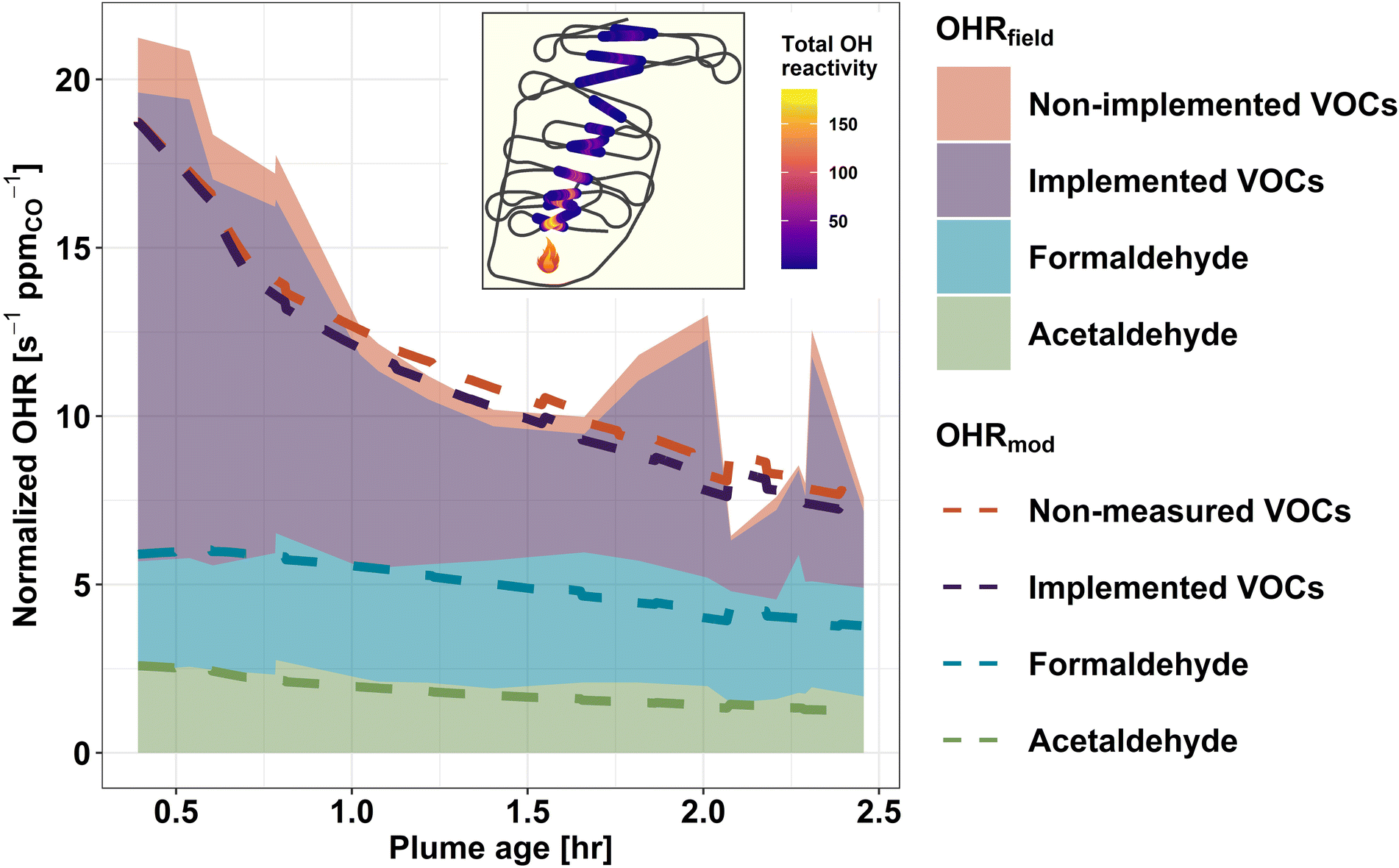 | ||
| Fig. 5 Field and modeled normalized OHR (s−1 ppmCO−1) for the Taylor Creek Fire sampled during WE-CAN with the inset plot showing the corresponding flight track shaded by the total OHR for each plume transect. Shaded regions represent the total calculated OHR from observations, while dashed lines are the model predicted OHR. Formaldehyde and acetaldehyde are shown separately as they are two of the largest contributors to the VOC OHR and important model diagnostics. The purple shading and dashed line represent VOCs measured in the field which are also included in the model. The red shading and dashed line is the combination of measured but not modeled plus modeled but not measured VOCs and show their combined impact is relatively insignificant. | ||
Fig. 5 shows the calculated (shaded regions) and modeled (dashed lines) OH reactivity normalized to CO for the Taylor Creek Fire. The combined green, blue, and purple shading shows the field calculated OHR for acetaldehyde, formaldehyde, and the sum of VOCs that are both measured in the field and present in the top 100 contributors to the OHRmod (implemented VOCs). The red shaded region then represents the combined OHR of species measured in the field but not present in the OHRmod (non-implemented VOCs). Green, blue, and purple dashed lines represent the model OHR for the corresponding VOCs. Similarly, the red dashed line indicates VOCs present in the OHRmod but not measured in the field (non-measured VOC).
F0AM + MCM generally reproduces the OHR of formaldehyde and acetaldehyde well as the plume ages, with their OHRfield and OHRmod agreeing within 2% and 6%, respectively, at initialization and 38% and 30% after 2.5 hours of plume aging (Fig. 5). As the model and field kOH values for these two species are the same, this indicates that the model is simulating their mixing ratios correctly, though maybe under-estimating formaldehyde after 1 hour of aging, in good agreement with previous studies.17,86 Additionally, the field calculated OHR for the two aldehydes can be seen to stay nearly constant with plume age. For formaldehyde, a similar steady state was also observed in some FIREX-AQ sampled plumes, where the plume-to-plume variability was found to be dependent on OH concentrations, with OH-initialized VOC oxidation and photolysis being the main (nearly equal) production and loss pathways.18,86 Conversely, acetaldehyde loss is primarily from reaction with OH radicals.86 Given that acetaldehyde's atmospheric lifetime against OH is ∼1 day, minimal loss is expected within ∼2.5 hours of plume aging,87 thus its near constant OHR in Fig. 5 suggests that it is likely from direct emissions. Due to formaldehyde and acetaldehyde being products of many different VOC oxidation pathways, their accurate representation in the model suggests that much of the overall VOC oxidation chemistry is being simulated correctly.
Upon initialization, the OHRmod is expected to be equal to the OHRfield if all measured species were initialized in the model and kOH values agree between the MCM and weighted rate constants used to calculate the field OHR. Because F0AM is initialized with only 50 of the 154 VOCs used to calculate the OHRfield, excess OHRfield at model initialization represents OH reactivity from species unimplemented in the model and thus constitutes a missing OH sink in the model. Fig. 5 shows that upon initialization the MCM contains 88% of the total field measured VOC OHR, indicating that the MCM is not missing a significant fraction of the initial OHRfield and that most of the known VOC OH sinks are included in the model. This is in good agreement with similar analysis done using FIREX-AQ laboratory measurements that found the MCM captures 75% of the tOHRVOC.41 The slightly higher percentage during WE-CAN is likely explained by the model being initialized by a few additional VOCs during WE-CAN, instrument uncertainty, and potential loss of some reactive species before the C-130 first intercepted the plume. Additionally, when comparing only implemented species, the OHRmod accounts for 95% of OHRfield (purple shading vs. the purple dashed line), which is well within the uncertainty of the field calculated OHR (±25%) and suggests the speciation of PTR-ToF-MS ions and subsequently weighting of kOH values is valid.
Fig. 5 demonstrates that as BB plumes age, oxidation of primary VOCs results in a net decrease in normalized plume OHR, indicative of the oxidation products being generally less reactive towards OH. Good agreement (±20%) between the decay rate of the field calculated and modeled tOHRVOC then suggests that the MCM is accurately representing the bulk of VOC OH oxidation chemistry. Additionally, in the aged plume transects OHR from non-implemented VOCs is seen to decrease slightly (red shading), again pointing to the model capturing most of the OHRfield.
The MCM contains thousands of oxidation products and intermediate species which are formed during the simulation but were not quantified in the field. Deviation of the dashed purple and red lines in Fig. 5 thus represents OHR from secondary VOCs predicted by the MCM but not measured in the field. As the plume ages, this proportion (difference between purple and red dashed lines) increases slightly from 0.5% at t0 to 7.4% at 2.5 hours of aging. This implies that the non-measured modeled OHR is primarily due to oxidation products rather than missing measurements of primary VOC emissions and that the MCM does not generate a significant OH sink of non-measured oxidation products in the plume aging times sampled during WE-CAN. An important implication is that the species used to initialize the model determines the total OH sink for at least the first few hours of plume aging. However, because this may not be sufficient time for the model to generate a sizeable unmeasured pool of OH reactive species, we are unable to extend this analysis to predict whether the WE-CAN payload is missing a reservoir of OH reactive species further downwind. Rather, this analysis shows that the MCM contains chemistry for most of the field-measured OHR, while future studies comparing field-calculated and direct OHR measurements are needed to better understand if there is still significant unmeasured OHR in wildfire smoke.
4 Conclusions
OH reactivity was calculated using 154 VOCs in addtion to NO2, NO, HONO, O3, CH4, and CO measured during the WE-CAN aircraft campaign and assessed in different environments including western U.S. wildfire emissions, smoke aged >3 days, smoke-impacted and low/no smoke urban atmospheres, and the clean free troposphere. Plume center tOHR for wildfire emissions ranges from 9–198 s−1 (12–41 s−1 ppmCO−1), with VOCs accounting for 80 ± 7% of the tOHR. In contrast to more well-studied VOC emission sources, VOC OHR is relatively well spread across many species and groups, with furans, alkenes, and aldehydes each accounting for ∼20% of the tOHRVOC.In smoke aged >3 days, primarily emitted hydrocarbons were found to make up 26% less of the tOHR due to the oxidation of reactive primary emitted species. OVOC and CO become the dominant OH sinks in aged smoke, accounting for 50% and 32% of the tOHR, respectively. Formaldehyde (23% of the tOHRVOC), acetic acid (21%), and acetaldehyde (11%) are the largest individual contributors to the aged smoke tOHRVOC. Notably, furan oxidation products were also found to account for 13% of the aged smoke tOHRVOC, suggesting that furans and their oxidation products play an important role in plume chemistry far downwind of fires. However, the lack of detailed speciation of PTR-ToF-MS data in aged smoke makes it difficult to fully characterize these species, highlighting the need for further investigations of furan oxidation products to improve analytical skills and understanding of their chemistry.
The average calculated clean free troposphere OHR during WE-CAN is 0.7 s−1, with the relatively long-lived species CH4, CO, and O3 accounting for 53% of tOHR. VOCs, mainly formaldehyde, acetaldehyde, acetic acid, and methanol, make up an additional 31% of the tOHR.
Vertical profiles collected during take-off and landing at the Boise, ID, airport allowed for the impacts of smoke on anthropogenic emissions to be explored. During smoke impacted periods, the average tOHR in Boise increases by 53% relative to the typical low/no smoke urban background (from 3.0 s−1 to 4.6 s−1), mainly due to the increase in BB VOCs and CO. NOx OHR remains similar between profiles, consistent with our general understanding of aged BB smoke being NOx depleted and VOC-rich. Consequently, the influx of BB smoke shifts the urban O3 production regime from being VOC-limited during low/no smoke periods to a more optimal O3 production regime on smoke-impacted days.
We use our field results to evaluate how well current iterations of the GEOS-Chem CTM and MCM represent the field-calculated OHR for wildfire emissions. The fractional OHR distribution of 17 of the 21 implemented explicit and lumped VOCs in GEOS-Chem were found to agree well with wildfire emissions measured during WE-CAN, indicating good agreement between the field and inventory emission factors. However, methanol, xylenes, MACR, and MEK distributions did not agree within 3σ likely due to the latter three being implemented separately in GEOS-Chem but measured with their isomers by PTR-ToF-MS. Additionally, the 21 implemented species were found to account for only 50% of the measured tOHRVOC, indicating that even if the model BB emission estimates are correct, they will miss half of the emitted VOC OHR. As the lack of reactive primary BB VOCs would constitute a missing OH sink in smoke-impacted environments, implementation of furan-containing species, butadienes, and monoterpenes (collectively accounting for 62% of the non-implemented VOC OHR) into GEOS-Chem for BB would likely improve model performance.
Using a pseudo-Lagrangian sampled smoke plume as a case study, we find that the F0AM + MCM initiated with 50 field measured VOCs, accounts for ∼90% of the tOHRVOC, suggesting that the MCM is not missing a significant fraction of the tOHRVOC in BB emissions and that most of the known VOC OH sinks are included in the model. Additionally, the F0AM + MCM model was able to simulate the observed decay of the normalized plume tOHRVOC generally within ± 20%, suggesting that the MCM is accurately representing the bulk VOC OH oxidation chemistry.
Our analysis highlights the most important VOC species for daytime BB plume oxidation and assesses how well the current standard GEOS-Chem CTM and MCM capture the observed OHR of wildfire emissions. The results presented here provide a roadmap for which individual VOCs and groups should be prioritized in next-generation air quality models to better predict the downwind air quality and subsequent public health impacts of BB smoke.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The 2018 WE-CAN field campaign was supported by the U.S. National Science Foundation through grants # AGS-1650275 (U of Montana), # AGS-1650786 (Colorado State U), # AGS-1650288 (U of Colorado at Boulder), # AGS-1650493 (U of Wyoming), # AGS-1652688 (U of Washington), # AGS-1748266 (U of Montana), and the National Oceanic and Atmospheric Administration (Award # NA17OAR4310010, Colorado State U). This study was also supported in part by NASA (# 80NSSC20M0166), NSF (EPSCoR Research Infrastructure # 1929210; AGS # 2144896), and Montana NASA EPSCoR Research Initiation Funding. The authors would like to acknowledge high-performance computing resources and support from Cheyenne (https://doi.org/10.5065/D6RX99HX) provided by the NCAR Computational and Information Systems Laboratory, sponsored by the NSF, and the U of Montana's Griz Shared Computing Cluster (GSCC). This material is based upon study supported by the National Center for Atmospheric Research, which is a major facility sponsored by the National Science Foundation under Cooperative Agreement No. 1852977. The data were collected using NSF's Lower Atmosphere Observing Facilities, which are managed and operated by NCAR's Earth Observing Laboratory. We thank Teresa L. Campos, Andrew J. Weinheimer, Denise D. Montzka, and Geoffrey S. Tyndall at the NCAR Atmospheric Chemistry Observations & Modeling Laboratory for provided NO, NO2, O3, CO, and CH4 measurements. All data are available in the WE-CAN data archive (https://data.eol.ucar.edu/master_lists/generated/we-can/).References
- S. K. Akagi, R. J. Yokelson, C. Wiedinmyer, M. J. Alvarado, J. S. Reid, T. Karl, J. D. Crounse and P. O. Wennberg, Emission factors for open and domestic biomass burning for use in atmospheric models, Atmos. Chem. Phys., 2011, 11, 4039–4072 CrossRef CAS.
- M. O. Andreae and P. Merlet, Emission of trace gases and aerosols from biomass burning, Global Biogeochem. Cycles, 2001, 15, 955–966 CrossRef CAS.
- P. J. Crutzen and M. O. Andreae, Biomass Burning in the Tropics: Impact on Atmospheric Chemistry and Biogeochemical Cycles, Science, 1990, 250, 1669–1678 CrossRef CAS PubMed.
- L. E. Hatch, C. N. Jen, N. M. Kreisberg, V. Selimovic, R. J. Yokelson, C. Stamatis, R. A. York, D. Foster, S. L. Stephens, A. H. Goldstein and K. C. Barsanti, Highly Speciated Measurements of Terpenoids Emitted from Laboratory and Mixed-Conifer Forest Prescribed Fires, Environ. Sci. Technol., 2019, 53, 9418–9428 CrossRef CAS PubMed.
- A. R. Koss, K. Sekimoto, J. B. Gilman, V. Selimovic, M. M. Coggon, K. J. Zarzana, B. Yuan, B. M. Lerner, S. S. Brown, J. L. Jimenez, J. Krechmer, J. M. Roberts, C. Warneke, R. J. Yokelson and J. de Gouw, Non-methane organic gas emissions from biomass burning: identification, quantification, and emission factors from PTR-ToF during the FIREX 2016 laboratory experiment, Atmos. Chem. Phys., 2018, 18, 3299–3319 CrossRef CAS.
- W. Permar, Q. Wang, V. Selimovic, C. Wielgasz, R. J. Yokelson, R. S. Hornbrook, A. J. Hills, E. C. Apel, I.-T. Ku, Y. Zhou, B. C. Sive, A. P. Sullivan, J. L. Collett Jr, T. L. Campos, B. B. Palm, Q. Peng, J. A. Thornton, L. A. Garofalo, D. K. Farmer, S. M. Kreidenweis, E. J. T. Levin, P. J. DeMott, F. Flocke, E. V. Fischer and L. Hu, Emissions of Trace Organic Gases from Western U.S. Wildfires Based on WE-CAN Aircraft Measurements, J. Geophys. Res.: Atmos., 2021, 126, e2020JD033838 CAS.
- R. Atkinson and J. Arey, Atmospheric Degradation of Volatile Organic Compounds, Chem. Rev., 2003, 103, 4605–4638 CrossRef CAS PubMed.
- K. O'Dell, R. S. Hornbrook, W. Permar, E. J. T. Levin, L. A. Garofalo, E. C. Apel, N. J. Blake, A. Jarnot, M. A. Pothier, D. K. Farmer, L. Hu, T. Campos, B. Ford, J. R. Pierce and E. V. Fischer, Hazardous Air Pollutants in Fresh and Aged Western US Wildfire Smoke and Implications for Long-Term Exposure, Environ. Sci. Technol., 2020, 54, 11838–11847 CrossRef PubMed.
- B. B. Palm, Q. Peng, C. D. Fredrickson, B. H. Lee, L. A. Garofalo, M. A. Pothier, S. M. Kreidenweis, D. K. Farmer, R. P. Pokhrel, Y. Shen, S. M. Murphy, W. Permar, L. Hu, T. L. Campos, S. R. Hall, K. Ullmann, X. Zhang, F. Flocke, E. V. Fischer and J. A. Thornton, Quantification of organic aerosol and brown carbon evolution in fresh wildfire plumes, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 29469–29477 CrossRef CAS PubMed.
- L. Xu, J. D. Crounse, K. T. Vasquez, H. Allen, P. O. Wennberg, I. Bourgeois, S. S. Brown, P. Campuzano-Jost, M. M. Coggon, J. H. Crawford, J. P. DiGangi, G. S. Diskin, A. Fried, E. M. Gargulinski, J. B. Gilman, G. I. Gkatzelis, H. Guo, J. W. Hair, S. R. Hall, H. A. Halliday, T. F. Hanisco, R. A. Hannun, C. D. Holmes, L. G. Huey, J. L. Jimenez, A. Lamplugh, Y. R. Lee, J. Liao, J. Lindaas, J. A. Neuman, J. B. Nowak, J. Peischl, D. A. Peterson, F. Piel, D. Richter, P. S. Rickly, M. A. Robinson, A. W. Rollins, T. B. Ryerson, K. Sekimoto, V. Selimovic, T. Shingler, A. J. Soja, J. M. S. Clair, D. J. Tanner, K. Ullmann, P. R. Veres, J. Walega, C. Warneke, R. A. Washenfelder, P. Weibring, A. Wisthaler, G. M. Wolfe, C. C. Womack and R. J. Yokelson, Ozone chemistry in western U.S. wildfire plumes, Sci. Adv., 2021, 7, eabl3648 CrossRef CAS PubMed.
- Y. Liang, R. J. Weber, P. K. Misztal, C. N. Jen and A. H. Goldstein, Aging of Volatile Organic Compounds in October 2017 Northern California Wildfire Plumes, Environ. Sci. Technol., 2022, 56, 1557–1567 CrossRef PubMed.
- M. Ninneman and D. A. Jaffe, The impact of wildfire smoke on ozone production in an urban area: insights from field observations and photochemical box modeling, Atmos. Environ., 2021, 267, 118764 CrossRef CAS.
- Q. Peng, B. B. Palm, C. D. Fredrickson, B. H. Lee, S. R. Hall, K. Ullmann, T. Campos, A. J. Weinheimer, E. C. Apel, F. Flocke, W. Permar, L. Hu, L. A. Garofalo, M. A. Pothier, D. K. Farmer, I.-T. Ku, A. P. Sullivan, J. L. Collett, E. Fischer and J. A. Thornton, Observations and Modeling of NOx Photochemistry and Fate in Fresh Wildfire Plumes, ACS Earth Space Chem., 2021, 5, 2652–2667 CrossRef CAS.
- M. Müller, B. E. Anderson, A. J. Beyersdorf, J. H. Crawford, G. S. Diskin, P. Eichler, A. Fried, F. N. Keutsch, T. Mikoviny, K. L. Thornhill, J. G. Walega, A. J. Weinheimer, M. Yang, R. J. Yokelson and A. Wisthaler, In situ measurements and modeling of reactive trace gases in a small biomass burning plume, Atmos. Chem. Phys., 2016, 16, 3813–3824 CrossRef.
- I. Bourgeois, J. Peischl, J. A. Neuman, S. S. Brown, C. R. Thompson, K. C. Aikin, H. M. Allen, H. Angot, E. C. Apel, C. B. Baublitz, J. F. Brewer, P. Campuzano-Jost, R. Commane, J. D. Crounse, B. C. Daube, J. P. DiGangi, G. S. Diskin, L. K. Emmons, A. M. Fiore, G. I. Gkatzelis, A. Hills, R. S. Hornbrook, L. G. Huey, J. L. Jimenez, M. Kim, F. Lacey, K. McKain, L. T. Murray, B. A. Nault, D. D. Parrish, E. Ray, C. Sweeney, D. Tanner, S. C. Wofsy and T. B. Ryerson, Large contribution of biomass burning emissions to ozone throughout the global remote troposphere, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2109628118 CrossRef CAS PubMed.
- Z. C. J. Decker, M. A. Robinson, K. C. Barsanti, I. Bourgeois, M. M. Coggon, J. P. DiGangi, G. S. Diskin, F. M. Flocke, A. Franchin, C. D. Fredrickson, G. I. Gkatzelis, S. R. Hall, H. Halliday, C. D. Holmes, L. G. Huey, Y. R. Lee, J. Lindaas, A. M. Middlebrook, D. D. Montzka, R. Moore, J. A. Neuman, J. B. Nowak, B. B. Palm, J. Peischl, F. Piel, P. S. Rickly, A. W. Rollins, T. B. Ryerson, R. H. Schwantes, K. Sekimoto, L. Thornhill, J. A. Thornton, G. S. Tyndall, K. Ullmann, P. Van Rooy, P. R. Veres, C. Warneke, R. A. Washenfelder, A. J. Weinheimer, E. Wiggins, E. Winstead, A. Wisthaler, C. Womack and S. S. Brown, Nighttime and daytime dark oxidation chemistry in wildfire plumes: an observation and model analysis of FIREX-AQ aircraft data, Atmos. Chem. Phys., 2021, 21, 16293–16317 CrossRef CAS.
- Q. Peng, B. B. Palm, K. E. Melander, B. H. Lee, S. R. Hall, K. Ullmann, T. Campos, A. J. Weinheimer, E. C. Apel, R. S. Hornbrook, A. J. Hills, D. D. Montzka, F. Flocke, L. Hu, W. Permar, C. Wielgasz, J. Lindaas, I. B. Pollack, E. V. Fischer, T. H. Bertram and J. A. Thornton, HONO Emissions from Western U.S. Wildfires Provide Dominant Radical Source in Fresh Wildfire Smoke, Environ. Sci. Technol., 2020, 54, 5954–5963 CrossRef CAS PubMed.
- J. Liao, G. M. Wolfe, R. A. Hannun, J. M. St. Clair, T. F. Hanisco, J. B. Gilman, A. Lamplugh, V. Selimovic, G. S. Diskin, J. B. Nowak, H. S. Halliday, J. P. DiGangi, S. R. Hall, K. Ullmann, C. D. Holmes, C. H. Fite, A. Agastra, T. B. Ryerson, J. Peischl, I. Bourgeois, C. Warneke, M. M. Coggon, G. I. Gkatzelis, K. Sekimoto, A. Fried, D. Richter, P. Weibring, E. C. Apel, R. S. Hornbrook, S. S. Brown, C. C. Womack, M. A. Robinson, R. A. Washenfelder, P. R. Veres and J. A. Neuman, Formaldehyde evolution in US wildfire plumes during the Fire Influence on Regional to Global Environments and Air Quality experiment (FIREX-AQ), Atmos. Chem. Phys., 2021, 21, 18319–18331 CrossRef CAS.
- M. A. Robinson, Z. C. J. Decker, K. C. Barsanti, M. M. Coggon, F. M. Flocke, A. Franchin, C. D. Fredrickson, J. B. Gilman, G. I. Gkatzelis, C. D. Holmes, A. Lamplugh, A. Lavi, A. M. Middlebrook, D. M. Montzka, B. B. Palm, J. Peischl, B. Pierce, R. H. Schwantes, K. Sekimoto, V. Selimovic, G. S. Tyndall, J. A. Thornton, P. Van Rooy, C. Warneke, A. J. Weinheimer and S. S. Brown, Variability and Time of Day Dependence of Ozone Photochemistry in Western Wildfire Plumes, Environ. Sci. Technol., 2021, 55, 10280–10290 CrossRef CAS PubMed.
- Z. C. J. Decker, K. J. Zarzana, M. Coggon, K.-E. Min, I. Pollack, T. B. Ryerson, J. Peischl, P. Edwards, W. P. Dubé, M. Z. Markovic, J. M. Roberts, P. R. Veres, M. Graus, C. Warneke, J. de Gouw, L. E. Hatch, K. C. Barsanti and S. S. Brown, Nighttime Chemical Transformation in Biomass Burning Plumes: A Box Model Analysis Initialized with Aircraft Observations, Environ. Sci. Technol., 2019, 53, 2529–2538 CrossRef CAS PubMed.
- L. T. Murray, A. M. Fiore, D. T. Shindell, V. Naik and L. W. Horowitz, Large uncertainties in global hydroxyl projections tied to fate of reactive nitrogen and carbon, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2115204118 CrossRef CAS PubMed.
- V. Naik, A. Voulgarakis, A. M. Fiore, L. W. Horowitz, J.-F. Lamarque, M. Lin, M. J. Prather, P. J. Young, D. Bergmann, P. J. Cameron-Smith, I. Cionni, W. J. Collins, S. B. Dalsøren, R. Doherty, V. Eyring, G. Faluvegi, G. A. Folberth, B. Josse, Y. H. Lee, I. A. MacKenzie, T. Nagashima, T. P. C. van Noije, D. A. Plummer, M. Righi, S. T. Rumbold, R. Skeie, D. T. Shindell, D. S. Stevenson, S. Strode, K. Sudo, S. Szopa and G. Zeng, Preindustrial to present-day changes in tropospheric hydroxyl radical and methane lifetime from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Atmos. Chem. Phys., 2013, 13, 5277–5298 CrossRef.
- J. Mao, X. Ren, W. H. Brune, J. R. Olson, J. H. Crawford, A. Fried, L. G. Huey, R. C. Cohen, B. Heikes, H. B. Singh, D. R. Blake, G. W. Sachse, G. S. Diskin, S. R. Hall and R. E. Shetter, Airborne measurement of OH reactivity during INTEX-B, Atmos. Chem. Phys., 2009, 9, 163–173 CrossRef CAS.
- K. R. Travis, C. L. Heald, H. M. Allen, E. C. Apel, S. R. Arnold, D. R. Blake, W. H. Brune, X. Chen, R. Commane, J. D. Crounse, B. C. Daube, G. S. Diskin, J. W. Elkins, M. J. Evans, S. R. Hall, E. J. Hintsa, R. S. Hornbrook, P. S. Kasibhatla, M. J. Kim, G. Luo, K. McKain, D. B. Millet, F. L. Moore, J. Peischl, T. B. Ryerson, T. Sherwen, A. B. Thames, K. Ullmann, X. Wang, P. O. Wennberg, G. M. Wolfe and F. Yu, Constraining remote oxidation capacity with ATom observations, Atmos. Chem. Phys., 2020, 20, 7753–7781 CrossRef CAS PubMed.
- P. Di Carlo, W. H. Brune, M. Martinez, H. Harder, R. Lesher, X. Ren, T. Thornberry, M. A. Carroll, V. Young, P. B. Shepson, D. Riemer, E. Apel and C. Campbell, Missing OH Reactivity in a Forest: Evidence for Unknown Reactive Biogenic VOCs, Science, 2004, 304, 722–725 CrossRef CAS PubMed.
- T. A. Kovacs, W. H. Brune, H. Harder, M. Martinez, J. B. Simpas, G. J. Frost, E. Williams, T. Jobson, C. Stroud, V. Young, A. Fried and B. Wert, Direct measurements of urban OH reactivity during Nashville SOS in summer 1999, J. Environ. Monit., 2003, 5, 68–74 RSC.
- D. Sanchez, R. Seco, D. Gu, A. Guenther, J. Mak, Y. Lee, D. Kim, J. Ahn, D. Blake, S. Herndon, D. Jeong, J. T. Sullivan, T. Mcgee, R. Park and S. Kim, Contributions to OH reactivity from unexplored volatile organic compounds measured by PTR-ToF-MS – a case study in a suburban forest of the Seoul metropolitan area during the Korea–United States Air Quality Study (KORUS-AQ) 2016, Atmos. Chem. Phys., 2021, 21, 6331–6345 CrossRef CAS.
- C. L. Heald, J. de Gouw, A. H. Goldstein, A. B. Guenther, P. L. Hayes, W. Hu, G. Isaacman-VanWertz, J. L. Jimenez, F. N. Keutsch, A. R. Koss, P. K. Misztal, B. Rappenglück, J. M. Roberts, P. S. Stevens, R. A. Washenfelder, C. Warneke and C. J. Young, Contrasting Reactive Organic Carbon Observations in the Southeast United States (SOAS) and Southern California (CalNex), Environ. Sci. Technol., 2020, 54, 14923–14935 CrossRef CAS PubMed.
- J. F. Hunter, D. A. Day, B. B. Palm, R. L. N. Yatavelli, A. W. H. Chan, L. Kaser, L. Cappellin, P. L. Hayes, E. S. Cross, A. J. Carrasquillo, P. Campuzano-Jost, H. Stark, Y. Zhao, T. Hohaus, J. N. Smith, A. Hansel, T. Karl, A. H. Goldstein, A. Guenther, D. R. Worsnop, J. A. Thornton, C. L. Heald, J. L. Jimenez and J. H. Kroll, Comprehensive characterization of atmospheric organic carbon at a forested site, Nat. Geosci., 2017, 10, 748–753 CrossRef CAS.
- D. B. Millet, H. D. Alwe, X. Chen, M. J. Deventer, T. J. Griffis, R. Holzinger, S. B. Bertman, P. S. Rickly, P. S. Stevens, T. Léonardis, N. Locoge, S. Dusanter, G. S. Tyndall, S. L. Alvarez, M. H. Erickson and J. H. Flynn, Bidirectional Ecosystem–Atmosphere Fluxes of Volatile Organic Compounds across the Mass Spectrum: How Many Matter?, ACS Earth Space Chem., 2018, 2, 764–777 CrossRef CAS PubMed.
- V. Sinha, J. Williams, J. Lelieveld, T. M. Ruuskanen, M. K. Kajos, J. Patokoski, H. Hellen, H. Hakola, D. Mogensen, M. Boy, J. Rinne and M. Kulmala, OH Reactivity Measurements within a Boreal Forest: Evidence for Unknown Reactive Emissions, Environ. Sci. Technol., 2010, 44, 6614–6620 CrossRef CAS PubMed.
- S. Lou, F. Holland, F. Rohrer, K. Lu, B. Bohn, T. Brauers, C. C. Chang, H. Fuchs, R. Häseler, K. Kita, Y. Kondo, X. Li, M. Shao, L. Zeng, A. Wahner, Y. Zhang, W. Wang and A. Hofzumahaus, Atmospheric OH reactivities in the Pearl River Delta – China in summer 2006: measurement and model results, Atmos. Chem. Phys., 2010, 10, 11243–11260 CrossRef CAS.
- J. Mao, X. Ren, S. Chen, W. H. Brune, Z. Chen, M. Martinez, H. Harder, B. Lefer, B. Rappenglück, J. Flynn and M. Leuchner, Atmospheric oxidation capacity in the summer of Houston 2006: comparison with summer measurements in other metropolitan studies, Atmos. Environ., 2010, 44, 4107–4115 CrossRef CAS.
- X. Ren, H. Harder, M. Martinez, R. L. Lesher, A. Oliger, J. B. Simpas, W. H. Brune, J. J. Schwab, K. L. Demerjian, Y. He, X. Zhou and H. Gao, OH and HO2 chemistry in the urban atmosphere of New York City, Atmos. Environ., 2003, 37, 3639–3651 CrossRef CAS.
- J. B. Gilman, B. M. Lerner, W. C. Kuster, P. D. Goldan, C. Warneke, P. R. Veres, J. M. Roberts, J. A. de Gouw, I. R. Burling and R. J. Yokelson, Biomass burning emissions and potential air quality impacts of volatile organic compounds and other trace gases from fuels common in the US, Atmos. Chem. Phys., 2015, 15, 13915–13938 CrossRef CAS.
- V. Kumar, B. P. Chandra and V. Sinha, Large unexplained suite of chemically reactive compounds present in ambient air due to biomass fires, Sci. Rep., 2018, 8, 626 CrossRef CAS PubMed.
- V. Ferracci, I. Heimann, N. L. Abraham, J. A. Pyle and A. T. Archibald, Global modelling of the total OH reactivity: investigations on the “missing” OH sink and its atmospheric implications, Atmos. Chem. Phys., 2018, 18, 7109–7129 CrossRef CAS.
- J. Lelieveld, S. Gromov, A. Pozzer and D. Taraborrelli, Global tropospheric hydroxyl distribution, budget and reactivity, Atmos. Chem. Phys., 2016, 16, 12477–12493 CrossRef CAS.
- S. A. Safieddine, C. L. Heald and B. H. Henderson, The global nonmethane reactive organic carbon budget: a modeling perspective, Geophys. Res. Lett., 2017, 44, 3897–3906 CrossRef CAS.
- T. S. Carter, C. L. Heald, J. H. Kroll, E. C. Apel, D. Blake, M. Coggon, A. Edtbauer, G. Gkatzelis, R. S. Hornbrook, J. Peischl, E. Y. Pfannerstill, F. Piel, N. G. Reijrink, A. Ringsdorf, C. Warneke, J. Williams, A. Wisthaler and L. Xu, An Improved Representation of Fire Non-Methane Organic Gases (NMOGs) in Models: Emissions to Reactivity, Atmos. Chem. Phys., 2022, 2022, 1–34 Search PubMed.
- M. M. Coggon, C. Y. Lim, A. R. Koss, K. Sekimoto, B. Yuan, J. B. Gilman, D. H. Hagan, V. Selimovic, K. J. Zarzana, S. S. Brown, J. M. Roberts, M. Müller, R. Yokelson, A. Wisthaler, J. E. Krechmer, J. L. Jimenez, C. Cappa, J. H. Kroll, J. de Gouw and C. Warneke, OH chemistry of non-methane organic gases (NMOGs) emitted from laboratory and ambient biomass burning smoke: evaluating the influence of furans and oxygenated aromatics on ozone and secondary NMOG formation, Atmos. Chem. Phys., 2019, 19, 14875–14899 CrossRef CAS.
- E. C. Apel, A. J. Hills, R. Lueb, S. Zindel, S. Eisele and D. D. Riemer, A fast-GC/MS system to measure C2 to C4 carbonyls and methanol aboard aircraft, J. Geophys. Res.: Atmos., 2003, 108, 8794 CrossRef.
- E. C. Apel, L. K. Emmons, T. Karl, F. Flocke, A. J. Hills, S. Madronich, J. Lee-Taylor, A. Fried, P. Weibring, J. Walega, D. Richter, X. Tie, L. Mauldin, T. Campos, A. Weinheimer, D. Knapp, B. Sive, L. Kleinman, S. Springston, R. Zaveri, J. Ortega, P. Voss, D. Blake, A. Baker, C. Warneke, D. Welsh-Bon, J. de Gouw, J. Zheng, R. Zhang, J. Rudolph, W. Junkermann and D. D. Riemer, Chemical evolution of volatile organic compounds in the outflow of the Mexico City Metropolitan area, Atmos. Chem. Phys., 2010, 10, 2353–2375 CrossRef CAS.
- E. C. Apel, R. S. Hornbrook, A. J. Hills, N. J. Blake, M. C. Barth, A. Weinheimer, C. Cantrell, S. A. Rutledge, B. Basarab, J. Crawford, G. Diskin, C. R. Homeyer, T. Campos, F. Flocke, A. Fried, D. R. Blake, W. Brune, I. Pollack, J. Peischl, T. Ryerson, P. O. Wennberg, J. D. Crounse, A. Wisthaler, T. Mikoviny, G. Huey, B. Heikes, D. O'Sullivan and D. D. Riemer, Upper tropospheric ozone production from lightning NOx-impacted convection: smoke ingestion case study from the DC3 campaign, J. Geophys. Res.: Atmos., 2015, 120, 2505–2523 CrossRef CAS.
- R. S. Hornbrook, D. R. Blake, G. S. Diskin, A. Fried, H. E. Fuelberg, S. Meinardi, T. Mikoviny, D. Richter, G. W. Sachse, S. A. Vay, J. Walega, P. Weibring, A. J. Weinheimer, C. Wiedinmyer, A. Wisthaler, A. Hills, D. D. Riemer and E. C. Apel, Observations of nonmethane organic compounds during ARCTAS; part 1: biomass burning emissions and plume enhancements, Atmos. Chem. Phys., 2011, 11, 11103–11130 CrossRef CAS.
- S. J. Andrews, L. J. Carpenter, E. C. Apel, E. Atlas, V. Donets, J. R. Hopkins, R. S. Hornbrook, A. C. Lewis, R. T. Lidster, R. Lueb, J. Minaeian, M. Navarro, S. Punjabi, D. Riemer and S. Schauffler, A comparison of very short lived halocarbon (VSLS) and DMS aircraft measurements in the tropical west Pacific from CAST, ATTREX and CONTRAST, Atmos. Meas. Tech., 2016, 9, 5213–5225 CrossRef.
- K. B. Benedict, Y. Zhou, B. C. Sive, A. J. Prenni, K. A. Gebhart, E. V. Fischer, A. Evanoski-Cole, A. P. Sullivan, S. Callahan, B. A. Schichtel, H. Mao, Y. Zhou and J. L. Collett Jr, Volatile organic compounds and ozone in Rocky Mountain National Park during FRAPPÉ, Atmos. Chem. Phys., 2019, 19, 499–521 CrossRef CAS.
- K. B. Benedict, A. J. Prenni, M. M. H. El-Sayed, A. Hecobian, Y. Zhou, K. A. Gebhart, B. C. Sive, B. A. Schichtel and J. L. Collett, Volatile organic compounds and ozone at four national parks in the southwestern United States, Atmos. Environ., 2020, 239, 117783 CrossRef CAS.
- R. S. Russo, Y. Zhou, M. L. White, H. Mao, R. Talbot and B. C. Sive, Multi-year (2004–2008) record of nonmethane hydrocarbons and halocarbons in New England: seasonal variations and regional sources, Atmos. Chem. Phys., 2010, 10, 4909–4929 CrossRef CAS.
- Y. Zhou, D. Shively, H. Mao, R. S. Russo, B. Pape, R. N. Mower, R. Talbot and B. C. Sive, Air Toxic Emissions from Snowmobiles in Yellowstone National Park, Environ. Sci. Technol., 2010, 44, 222–228 CrossRef CAS PubMed.
- B. H. Lee, F. D. Lopez-Hilfiker, P. R. Veres, E. E. McDuffie, D. L. Fibiger, T. L. Sparks, C. J. Ebben, J. R. Green, J. C. Schroder, P. Campuzano-Jost, S. Iyer, E. L. D'Ambro, S. Schobesberger, S. S. Brown, P. J. Wooldridge, R. C. Cohen, M. N. Fiddler, S. Bililign, J. L. Jimenez, T. Kurtén, A. J. Weinheimer, L. Jaegle and J. A. Thornton, Flight Deployment of a High-Resolution Time-of-Flight Chemical Ionization Mass Spectrometer: Observations of Reactive Halogen and Nitrogen Oxide Species, J. Geophys. Res.: Atmos., 2018, 123, 7670–7686 CAS.
- B. B. Palm, X. Liu, J. L. Jimenez and J. A. Thornton, Performance of a new coaxial ion–molecule reaction region for low-pressure chemical ionization mass spectrometry with reduced instrument wall interactions, Atmos. Meas. Tech., 2019, 12, 5829–5844 CrossRef CAS.
- B. Lebegue, M. Schmidt, M. Ramonet, B. Wastine, C. Yver Kwok, O. Laurent, S. Belviso, A. Guemri, C. Philippon, J. Smith and S. Conil, Comparison of nitrous oxide (N2O) analyzers for high-precision measurements of atmospheric mole fractions, Atmos. Meas. Tech., 2016, 9, 1221–1238 CrossRef CAS.
- B. A. Ridley, F. E. Grahek and J. G. Walega, A Small High-Sensitivity, Medium-Response Ozone Detector Suitable for Measurements from Light Aircraft, J. Atmos. Ocean. Tech., 1992, 9, 142–148 CrossRef.
- B. A. Ridley and F. E. Grahek, A Small, Low Flow, High Sensitivity Reaction Vessel for NO Chemiluminescence Detectors, J. Atmos. Ocean. Tech., 1990, 7, 307–311 CrossRef.
- D. L. Slusher, L. G. Huey, D. J. Tanner, F. M. Flocke and J. M. Roberts, A thermal dissociation–chemical ionization mass spectrometry (TD-CIMS) technique for the simultaneous measurement of peroxyacyl nitrates and dinitrogen pentoxide, J. Geophys. Res.: Atmos., 2004, 109, D19315 CrossRef.
- W. Zheng, F. M. Flocke, G. S. Tyndall, A. Swanson, J. J. Orlando, J. M. Roberts, L. G. Huey and D. J. Tanner, Characterization of a thermal decomposition chemical ionization mass spectrometer for the measurement of peroxy acyl nitrates (PANs) in the atmosphere, Atmos. Chem. Phys., 2011, 11, 6529–6547 CrossRef CAS.
- S. K. Akagi, J. S. Craven, J. W. Taylor, G. R. McMeeking, R. J. Yokelson, I. R. Burling, S. P. Urbanski, C. E. Wold, J. H. Seinfeld, H. Coe, M. J. Alvarado and D. R. Weise, Evolution of trace gases and particles emitted by a chaparral fire in California, Atmos. Chem. Phys., 2012, 12, 1397–1421 CrossRef CAS.
- P. V. Hobbs, P. Sinha, R. J. Yokelson, T. J. Christian, D. R. Blake, S. Gao, T. W. Kirchstetter, T. Novakov and P. Pilewskie, Evolution of gases and particles from a savanna fire in South Africa, J. Geophys. Res.: Atmos., 2008, 108, 8485 Search PubMed.
- J. Lindaas, I. B. Pollack, L. A. Garofalo, M. A. Pothier, D. K. Farmer, S. M. Kreidenweis, T. L. Campos, F. Flocke, A. J. Weinheimer, D. D. Montzka, G. S. Tyndall, B. B. Palm, Q. Peng, J. A. Thornton, W. Permar, C. Wielgasz, L. Hu, R. D. Ottmar, J. C. Restaino, A. T. Hudak, I.-T. Ku, Y. Zhou, B. C. Sive, A. Sullivan, J. L. Collett Jr and E. V. Fischer, Emissions of Reactive Nitrogen from Western U.S. Wildfires during Summer 2018, J. Geophys. Res.: Atmos., 2021, 126, e2020JD032657 CAS.
- Z. C. J. Decker, S. Wang, I. Bourgeois, P. Campuzano Jost, M. M. Coggon, J. P. DiGangi, G. S. Diskin, F. M. Flocke, A. Franchin, C. D. Fredrickson, G. I. Gkatzelis, S. R. Hall, H. Halliday, K. Hayden, C. D. Holmes, L. G. Huey, J. L. Jimenez, Y. R. Lee, J. Lindaas, A. M. Middlebrook, D. D. Montzka, J. A. Neuman, J. B. Nowak, D. Pagonis, B. B. Palm, J. Peischl, F. Piel, P. S. Rickly, M. A. Robinson, A. W. Rollins, T. B. Ryerson, K. Sekimoto, J. A. Thornton, G. S. Tyndall, K. Ullmann, P. R. Veres, C. Warneke, R. A. Washenfelder, A. J. Weinheimer, A. Wisthaler, C. Womack and S. S. Brown, Novel Analysis to Quantify Plume Crosswind Heterogeneity Applied to Biomass Burning Smoke, Environ. Sci. Technol., 2021, 55, 15646–15657 CrossRef CAS PubMed.
- B. B. Palm, Q. Peng, S. R. Hall, K. Ullmann, T. L. Campos, A. Weinheimer, D. Montzka, G. Tyndall, W. Permar, L. Hu, F. Flocke, E. V. Fischer and J. A. Thornton, Spatially Resolved Photochemistry Impacts Emissions Estimates in Fresh Wildfire Plumes, Geophys. Res. Lett., 2021, 48, e2021GL095443 CrossRef CAS.
- E. Lill, J. Lindaas, J. F. Juncosa Calahorrano, T. Campos, F. Flocke, E. C. Apel, R. S. Hornbrook, A. Hills, A. Jarnot, N. Blake, W. Permar, L. Hu, A. Weinheimer, G. Tyndall, D. D. e Montzka, S. R. Hall, K. Ullmann, J. Thornton, B. B. Palm, Q. Peng, I. Pollack and E. V. Fischer, Wildfire-driven changes in the abundance of gas-phase pollutants in the city of Boise, ID during summer 2018, Atmos. Pollut. Res., 2022, 13, 101269 CrossRef CAS.
- I. Bey, D. J. Jacob, R. M. Yantosca, J. A. Logan, B. D. Field, A. M. Fiore, Q. Li, H. Y. Liu, L. J. Mickley and M. G. Schultz, Global modeling of tropospheric chemistry with assimilated meteorology: model description and evaluation, J. Geophys. Res.: Atmos., 2001, 106, 23073–23095 CrossRef CAS.
- J. W. Kaiser, A. Heil, M. O. Andreae, A. Benedetti, N. Chubarova, L. Jones, J.-J. Morcrette, M. Razinger, M. G. Schultz, M. Suttie and G. R. van der Werf, Biomass burning emissions estimated with a global fire assimilation system based on observed fire radiative power, Biogeosciences, 2012, 9, 527–554 CrossRef CAS.
- G. M. Wolfe, M. R. Marvin, S. J. Roberts, K. R. Travis and J. Liao, The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 2016, 9, 3309–3319 CrossRef.
- L. A. Garofalo, M. A. Pothier, E. J. T. Levin, T. Campos, S. M. Kreidenweis and D. K. Farmer, Emission and Evolution of Submicron Organic Aerosol in Smoke from Wildfires in the Western United States, ACS Earth Space Chem., 2019, 3, 1237–1247 CrossRef CAS.
- J. Lindaas, I. B. Pollack, J. J. Calahorrano, K. O'Dell, L. A. Garofalo, M. A. Pothier, D. K. Farmer, S. M. Kreidenweis, T. Campos, F. Flocke, A. J. Weinheimer, D. D. Montzka, G. S. Tyndall, E. C. Apel, A. J. Hills, R. S. Hornbrook, B. B. Palm, Q. Peng, J. A. Thornton, W. Permar, C. Wielgasz, L. Hu, J. R. Pierce, J. L. Collett Jr, A. P. Sullivan and E. V. Fischer, Empirical Insights into the Fate of Ammonia in Western U.S. Wildfire Smoke Plumes, J. Geophys. Res.: Atmos., 2021, 126, e2020JD033730 CAS.
- Y. Yang, M. Shao, X. Wang, A. C. Nölscher, S. Kessel, A. Guenther and J. Williams, Towards a quantitative understanding of total OH reactivity: a review, Atmos. Environ., 2016, 134, 147–161 CrossRef CAS.
- A. B. Thames, W. H. Brune, D. O. Miller, H. M. Allen, E. C. Apel, D. R. Blake, T. P. Bui, R. Commane, J. D. Crounse, B. C. Daube, G. S. Diskin, J. P. DiGangi, J. W. Elkins, S. R. Hall, T. F. Hanisco, R. A. Hannun, E. Hintsa, R. S. Hornbrook, M. J. Kim, K. McKain, F. L. Moore, J. M. Nicely, J. Peischl, T. B. Ryerson, J. M. S. Clair, C. Sweeney, A. Teng, C. R. Thompson, K. Ullmann, P. O. Wennberg and G. M. Wolfe, Missing OH reactivity in the global marine boundary layer, Atmos. Chem. Phys., 2020, 20, 4013–4029 CrossRef CAS.
- R. F. Hansen, M. Blocquet, C. Schoemaecker, T. Léonardis, N. Locoge, C. Fittschen, B. Hanoune, P. S. Stevens, V. Sinha and S. Dusanter, Intercomparison of the comparative reactivity method (CRM) and pump–probe technique for measuring total OH reactivity in an urban environment, Atmos. Meas. Tech., 2015, 8, 4243–4264 CrossRef.
- T. R. Shirley, W. H. Brune, X. Ren, J. Mao, R. Lesher, B. Cardenas, R. Volkamer, L. T. Molina, M. J. Molina, B. Lamb, E. Velasco, T. Jobson and M. Alexander, Atmospheric oxidation in the Mexico City Metropolitan Area (MCMA) during April 2003, Atmos. Chem. Phys., 2006, 6, 2753–2765 CrossRef CAS.
- A. Yoshino, Y. Sadanaga, K. Watanabe, S. Kato, Y. Miyakawa, J. Matsumoto and Y. Kajii, Measurement of total OH reactivity by laser-induced pump and probe technique—comprehensive observations in the urban atmosphere of Tokyo, Atmos. Environ., 2006, 40, 7869–7881 CrossRef CAS.
- A. C. Nölscher, A. M. Yañez-Serrano, S. Wolff, A. C. de Araujo, J. V. Lavrič, J. Kesselmeier and J. Williams, Unexpected seasonality in quantity and composition of Amazon rainforest air reactivity, Nat. Commun., 2016, 7, 10383 CrossRef PubMed.
- W. H. Brune, X. Ren, L. Zhang, J. Mao, D. O. Miller, B. E. Anderson, D. R. Blake, R. C. Cohen, G. S. Diskin, S. R. Hall, T. F. Hanisco, L. G. Huey, B. A. Nault, J. Peischl, I. Pollack, T. B. Ryerson, T. Shingler, A. Sorooshian, K. Ullmann, A. Wisthaler and P. J. Wooldridge, Atmospheric oxidation in the presence of clouds during the Deep Convective Clouds and Chemistry (DC3) study, Atmos. Chem. Phys., 2018, 18, 14493–14510 CrossRef CAS.
- J. A. Snow, B. G. Heikes, H. Shen, D. W. O’Sullivan, A. Fried and J. Walega, Hydrogen peroxide, methyl hydroperoxide, and formaldehyde over North America and the North Atlantic, J. Geophys. Res.: Atmos., 2007, 122, D12S07 Search PubMed.
- C. D. McClure and D. A. Jaffe, US particulate matter air quality improves except in wildfire-prone areas, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7901–7906 CrossRef CAS PubMed.
- D. A. Jaffe and N. L. Wigder, Ozone production from wildfires: a critical review, Atmos. Environ., 2012, 51, 1–10 CrossRef CAS.
- L. E. Hatch, R. J. Yokelson, C. E. Stockwell, P. R. Veres, I. J. Simpson, D. R. Blake, J. J. Orlando and K. C. Barsanti, Multi-instrument comparison and compilation of non-methane organic gas emissions from biomass burning and implications for smoke-derived secondary organic aerosol precursors, Atmos. Chem. Phys., 2017, 17, 1471–1489 CrossRef CAS.
- J. Jiang, W. P. L. Carter, D. R. Cocker and K. C. Barsanti, Development and Evaluation of a Detailed Mechanism for Gas-Phase Atmospheric Reactions of Furans, ACS Earth Space Chem., 2020, 4, 1254–1268 CrossRef CAS.
- X. Chen, D. B. Millet, H. B. Singh, A. Wisthaler, E. C. Apel, E. L. Atlas, D. R. Blake, I. Bourgeois, S. S. Brown, J. D. Crounse, J. A. de Gouw, F. M. Flocke, A. Fried, B. G. Heikes, R. S. Hornbrook, T. Mikoviny, K.-E. Min, M. Müller, J. A. Neuman, D. W. O'Sullivan, J. Peischl, G. G. Pfister, D. Richter, J. M. Roberts, T. B. Ryerson, S. R. Shertz, C. R. Thompson, V. Treadaway, P. R. Veres, J. Walega, C. Warneke, R. A. Washenfelder, P. Weibring and B. Yuan, On the sources and sinks of atmospheric VOCs: an integrated analysis of recent aircraft campaigns over North America, Atmos. Chem. Phys., 2019, 19, 9097–9123 CrossRef CAS PubMed.
- S. A. McKeen, G. Wotawa, D. D. Parrish, J. S. Holloway, M. P. Buhr, G. Hübler, F. C. Fehsenfeld and J. F. Meagher, Ozone production from Canadian wildfires during June and July of 1995, J. Geophys. Res.: Atmos., 2002, 107, ACH 7-1–ACH 7-25 CrossRef.
- F. Kirchner, F. Jeanneret, A. Clappier, B. Krüger, H. van den Bergh and B. Calpini, Total VOC reactivity in the planetary boundary layer: 2. A new indicator for determining the sensitivity of the ozone production to VOC and NOx, J. Geophys. Res.: Atmos., 2001, 106, 3095–3110 CrossRef CAS.
- C. E. Buysse, A. Kaulfus, U. Nair and D. A. Jaffe, Relationships between Particulate Matter, Ozone, and Nitrogen Oxides during Urban Smoke Events in the Western US, Environ. Sci. Technol., 2019, 53, 12519–12528 CrossRef CAS PubMed.
- V. Selimovic, R. J. Yokelson, G. R. McMeeking and S. Coefield, Aerosol Mass and Optical Properties, Smoke Influence on O3, and High NO3 Production Rates in a Western U.S. City Impacted by Wildfires, J. Geophys. Res.: Atmos., 2020, 125, e2020JD032791 CAS.
- G. M. Wolfe, T. F. Hanisco, H. L. Arkinson, D. R. Blake, A. Wisthaler, T. Mikoviny, T. B. Ryerson, I. Pollack, J. Peischl, P. O. Wennberg, J. D. Crounse, J. M. St. Clair, A. Teng, L. G. Huey, X. Liu, A. Fried, P. Weibring, D. Richter, J. Walega, S. R. Hall, K. Ullmann, J. L. Jimenez, P. Campuzano-Jost, T. P. Bui, G. Diskin, J. R. Podolske, G. Sachse and R. C. Cohen, Photochemical evolution of the 2013 California Rim Fire: synergistic impacts of reactive hydrocarbons and enhanced oxidants, Atmos. Chem. Phys., 2022, 22, 4253–4275 CrossRef CAS.
- D. B. Millet, A. Guenther, D. A. Siegel, N. B. Nelson, H. B. Singh, J. A. de Gouw, C. Warneke, J. Williams, G. Eerdekens, V. Sinha, T. Karl, F. Flocke, E. Apel, D. D. Riemer, P. I. Palmer and M. Barkley, Global atmospheric budget of acetaldehyde: 3-D model analysis and constraints from in-situ and satellite observations, Atmos. Chem. Phys., 2010, 10, 3405–3425 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ea00063f |
| This journal is © The Royal Society of Chemistry 2023 |