
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient asymmetrical silicon–metal dimer electrocatalysts for the nitrogen reduction reaction†
Chuangwei
Liu
 a,
Haoren
Zheng
a,
Tianyi
Wang
ab,
Xiaoli
Zhang
c,
Zhongyuan
Guo
*b and
Hao
Li
*b
a,
Haoren
Zheng
a,
Tianyi
Wang
ab,
Xiaoli
Zhang
c,
Zhongyuan
Guo
*b and
Hao
Li
*b
aKey Lab for Anisotropy and Texture of Materials, School of Materials Science and Engineering, Northeastern University, Shenyang 110819, China
bAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, Sendai 980-8577, Japan. E-mail: zhongyuanguo2022@163.com; li.hao.b8@tohoku.ac.jp
cSchool of Material Science and Engineering, Zhengzhou University, Zhengzhou 450001, China
First published on 14th April 2023
Abstract
The electrocatalytic nitrogen reduction reaction (ENRR) has been regarded as an eco-friendly and feasible substitute for the Haber–Bosch method. Identifying the effective catalysts for the ENRR is an extremely important prerequisite but challenging. Herein, asymmetrical silicon–metal dimer catalysts doped into g-C3N4 nanosheets with nitrogen vacancies (SiM@C3N4) were designed to address nitrogen activation and reduction. The concept catalysts of SiM@C3N4 can combine the advantages of silicon-based and metal-based catalysts during the ENRR. Among the catalysts investigated, SiMo@C3N4 and SiRu@C3N4 exhibited the highest activities towards the ENRR with ultra-low onset potentials of −0.20 and −0.39 V; meanwhile, they suppressed the competing hydrogen evolution reaction (HER) due to the relative difficulty in releasing hydrogen. Additionally, SiRu@C3N4 is demonstrated to possess strong hydrophobicity, which is greatly beneficial to the production of ammonia. This research provides insights into asymmetrical silicon–metal dimer catalysts and reveals a new method for developing dual-atom electrocatalysts. This asymmetrical dimer strategy can be applied in other electrocatalytic reactions for energy conversion.
1. Introduction
The electrocatalytic nitrogen reduction reaction (ENRR) has been considered to be the most promising route for artificial N2 fixation under ambient conditions, as it can convert N2 into NH3 using renewable energy sources (wind and solar energies) on the surface of electrocatalysts.1–5 Although tremendous efforts have been made, the performance of electrocatalysts as the core part of electrocatalytic systems is still far from that required for realizing a pragmatic application.6 Now, our societies rely heavily on the century-old Haber–Bosch (HB) method for industrial NH3 production, which consumes considerable energy and releases a tremendous amount of CO2 annually.7 To cope with the global energy crisis and climate change, high-performance ENRR catalysts are urgently needed to substitute for the HB route.In recent years, metal-free ENRR catalysts have been attracting attention due to their application potential.3,5,8–14 Unlike metal-based electrocatalysts, metal-free electrocatalysts can circumvent the intrinsic shortcoming of the strong *H affinity on metal-based ones, which is conducive to increasing Faradaic efficiency (FE). Moreover, metal-free electrocatalysts possess the advantages of low cost and high modification versatility, which make them suitable for pragmatic applications. For example, Sun et al.15–18 experimentally modified carbon-based materials via O, F, and S heteroatoms for N2 electroreduction to NH3 and achieved comparable performance with that of metal-based electrocatalysts. In addition, metal-free Si-based catalysts have begun to arouse research interests in relation to the ENRR. For instance, Li et al.19 experimentally investigated atomically dispersed Si atoms on carbon nanolayers for N2 fixation and found that isoelectric Si heteroatoms can function as metal-free single-atom-based catalysts to greatly improve the FE of the pristine carbon support, with comparable results to atomically dispersed metal-based catalysts. Our previous studies also investigated the potential of Si-based electrocatalysts for the ENRR from a theoretical viewpoint, of note,13,20,21 finding the relatively lower affinity of N2 on Si sites compared with that on transition metal sites, which could adversely affect the subsequent activation and hydrogenation of *N2. However, low-coordination Si atoms can act as a Lewis acid to interact with N2 as a Lewis base with under-occupied p-orbitals via an electron acceptance-donation process.22 Compared with the carbon atom sites on the carbon-based catalysts mentioned above, the lower electronegativity of Si can contribute to more electron back-donation into the 1π* anti-bonding orbitals of *N2.
The atomically dispersed dual-atom catalysts (DACs) have garnered extensive research interest in heterogeneous catalysis due to their unique electronic structures, maximum atom-utilization efficiencies, and flexible reaction sites.23–25 Especially, the heteronuclear DACs with asymmetric active sites, as a new rising star, have opened a novel pathway in atomic catalysis, which will advance the material design to address critical challenges in energy conversion. For example, Wang et al. designed heteronuclear DACs to break through the restriction of scaling relations on CO2 catalytic activity by combining two kinds of metal atoms embedded in the 2D C2N support.26 Moreover, Du et al. proposed metal Cu and non-metal B atoms supported on g-C3N4 (Cu-B@g-C3N4) for CO dimerization, which effectively reduced the CO dimerization free energy barrier due to the asymmetric synergy of the Cu–B center and suppresses the parasitic hydrogen evolution reaction (HER) caused by the synergistic dCu–pB coupling.27 Therefore, based on the aforementioned discussions, in this work, we hypothesized that the dual-atom silicon (Si)–metal (M) dimers on supports could be a viable way to increase the affinity of N2 on catalysts and achieve an excellent catalytic performance in the ENRR on an asymmetric active center. The chosen supporter was g-C3N4 due to its facile synthesis methods, large surface, and porous structure.28 And g-C3N4 has been widely employed in photocatalysis and electrocatalysis.29–35 More importantly, as for Si–M dimers, the comparably easily formed N vacancy sites (NV) during the synthesis of g-C3N4 are suitable anchoring sites, and have been reported to facilitate electrocatalysis on doped g-C3N4.36–38 Based on previous reports on efficient metal-based catalysts for the ENRR,39–46 here we selected Fe, Co, Ni, Nb, Mo, Ru, Rh, W, and Re metal atoms for the design of the Si–M dimer on g-C3N4. The finally designed dimer catalysts were denoted as SiM@C3N4, and the corresponding single-atom Si/M-doped catalysts were denoted as Si@C3N4 or M@C3N4 (more information regarding the catalyst models is provided in the ESI†). After comprehensive calculations, it was found that the asymmetric dimer catalysts (SiM@C3N4) can not only improve the capture of N2 in the horizontal mode but also boost the subsequent hydrogenation process. Among the concept catalyst models, SiMo@C3N4 and SiRu@C3N4 are demonstrated to have ultra-low onset potentials of −0.20 and −0.39 V, respectively, towards the ENRR. Meanwhile, the HER would be suppressed on these two dimer catalysts due to larger free-energy barriers for H2 release from active sites (*H + H+ + e− → H2). This work for the first time explores asymmetrical dimer catalysts composed of metals and non-metals for the ENRR, which not only combine the advantages of two types of external atoms but also offer a novel avenue for designing high-performance artificial N2 fixation catalysts.
2. Calculation details
Spin-polarized density functional theory (DFT) calculations were performed using the DMol3 code to study the structure properties of Si–M dimers on g-C3N4 and their corresponding ENRR activity. The revised Perdew–Burk–Ernzerhof (RPBE) generalized gradient approximation (GGA) was used to describe the electron exchange–correlation. The DFT semi-core pseudopots (dspp) pseudopotentials and double numerical basis sets with polarization functions (DNP) were adopted in all calculations. The Tkatchenko–Scheffler (TS) scheme was employed to illustrate the van der Waals (VDW) interactions between catalysts and adsorbates.47 The real-space global cutoff radius was set to be 5.2 Å. A (3 × 3 × 1) k-point set within the Monkhorst–Pack scheme was used to sample the Brillouin zone. Moreover, the solvation effect was also considered using the COSMO scheme with an H2O dielectric constant of 78.54 for the ENRR. All structural optimizations were performed until the energy tolerance and residual force were smaller than 10−5 Ha and 0.002 Ha Å−1, respectively, and SCF tolerance was set as 10−6 Ha. On the basis of the computational hydrogen electrode (CHE) model developed by Nørskov et al.,48,49 the free energy of each elementary reaction at 298.15 K and pH = 0 without the extra potential was calculated as follows:| ΔG = ΔEDFT + ΔEZPE − TΔS |
3. Results and discussion
3.1 N2 adsorption and reduction on Si@C3N4
Before examining the ENRR performance of SiM@C3N4, we started with investigating the N2 adsorption and reduction on single-atom Si-doped g-C3N4 (Si@C3N4) for comparison purposes with the subsequent studies on SiM@C3N4. In g-C3N4, there are three types of N sites, as shown in Fig. 1a. According to the relevant report on defective g-C3N4,31,52 Nv1 is the common and easily formed defective site on the plane of g-C3N4, which is an ideal anchoring site for heteroatom doping. Therefore, a single-atom Si heteroatom is embedded into this defect and then the designed Si@C3N4 is optimized, as shown in Fig. 1a for the subsequent calculations and comparative analysis. According to the binding energy (Eb) equation: Eb = Ecat. − ESi − Er, where Ecat., ESi, and Er represent the total electronic energies of the catalyst designed, atomic Si in the gas phrase, and the remaining part of the catalyst with atoms constrained in the lattice position, we obtained Eb,Si = −6.51 eV on g-C3N4, suggesting the strong binding strength of Si on the defective g-C3N4 catalyst. Then compared with the cohesive energy of Si (Eco,Si = −4.55 eV), one can see the excellent stability of single-atom Si@C3N4 catalysts under ambient conditions. Moreover, the formation energy (Ef) of Si@C3N4 was calculated as Ef = ½EN2 + ESi@C3N4 − μSi − Eg-C3N4 = −0.08 eV, where EN2, ESi@C3N4, and Eg-C3N4 represent the total electronic energies of the N2 molecule, the Si@C3N4 catalyst, and the pristine g-C3N4 nanosheet, respectively. μSi refers to the chemical potential of Si, obtained from 1/n*ESi-cell, suggesting the easy in-laboratory synthesis. Also, the interstitial Si-doping configuration, as shown in Fig. S2 (ESI†), was considered. A comparison between the two formation energies indicated that Si@C3N4 is more energetically favorable. Therefore, the subsequent concept catalysts of Si–M dimers were built on two Nv1 sites, named SiM@C3N4.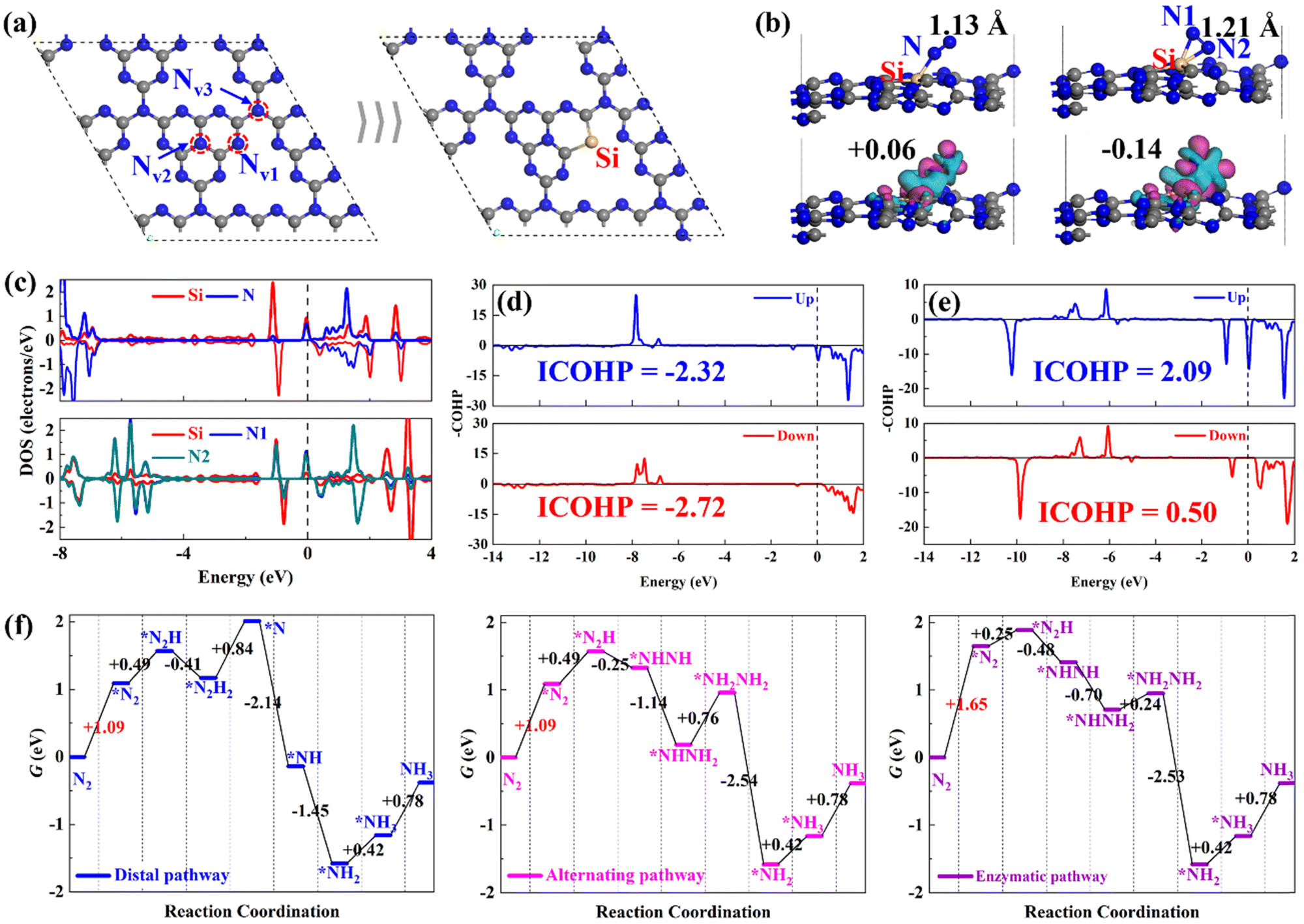 | ||
| Fig. 1 Single Si dopped in g-C3N4 and the corresponding electronic structure and energy analyses: (a) schematic diagram of Si@C3N4 formation through Nv1; (b) two adsorption configurations and their corresponding charge density difference induced by N2 adsorption with the red and cyan color representing the electron accumulation and loss, respectively (isosurface = 0.01 a.u.). Black values show the Hirshfeld charges of adsorbed N2; (c) the Si–N electron interaction by DOS analysis; the –COHP and integrated –COHP analyses of the N–N bond from the end-on (d) and (e) side-on N2 adsorption configurations; and (f) free energy evolutions through different reaction pathways. Color scheme: Si (yellow), N (blue), and C (grey). | ||
Then, we examined the adsorption of N2 on Si@C3N4. Fig. 1b exhibits two adsorption configurations of N2, i.e., end-on and side-on modes, and their corresponding N2-induced charge density difference (CDD). The CDD shows that there exists noticeable electron transfer, i.e., electron acceptance–backdonation, between the Si site and N2, and the Hirshfeld charge analysis shows that N2 obtains more electrons from the catalyst in the side-on mode. The DOS results, shown in Fig. 1c, explain the electron interaction between Si and N: (1) N2 is polarized after adsorption and (2) the electron interaction is stronger in the side-on mode. To obtain an intuitive comparison on the degree of activation of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, the –COHP calculations were conducted for the end-on and side-on models, as shown in Fig. 1d and 1e. The –COHP analysis shows that there are more electrons occupied in the 1π* anti-bonding orbitals below the Fermi level in the side-on configuration, leading to the activation of N2. The analysis of the N–N bond length, as displayed in Fig. 1b, describes the NN triple bond elongation to 1.13 and 1.21 Å, also in agreement with the –COHP results. However, the polarized *N2 with more electrons occupying the 1π* orbital will increase the electronic energy. Fig. 1f shows the free energy evolutions of the ENRR starting from free N2 to the NH3 product (reaction mechanisms53,54 refer to Fig. S1, ESI†), from which we can see that the adsorption of N2 presents a significant barrier to the reaction, with relatively large free energy changes of +1.09 and +1.65 eV. Such significant free energy changes are difficult to overcome under ambient conditions, indicating that the ENRR will not occur on Si@C3N4. Enhancing the N2 adsorption will boost the ENRR performance.
N, the –COHP calculations were conducted for the end-on and side-on models, as shown in Fig. 1d and 1e. The –COHP analysis shows that there are more electrons occupied in the 1π* anti-bonding orbitals below the Fermi level in the side-on configuration, leading to the activation of N2. The analysis of the N–N bond length, as displayed in Fig. 1b, describes the NN triple bond elongation to 1.13 and 1.21 Å, also in agreement with the –COHP results. However, the polarized *N2 with more electrons occupying the 1π* orbital will increase the electronic energy. Fig. 1f shows the free energy evolutions of the ENRR starting from free N2 to the NH3 product (reaction mechanisms53,54 refer to Fig. S1, ESI†), from which we can see that the adsorption of N2 presents a significant barrier to the reaction, with relatively large free energy changes of +1.09 and +1.65 eV. Such significant free energy changes are difficult to overcome under ambient conditions, indicating that the ENRR will not occur on Si@C3N4. Enhancing the N2 adsorption will boost the ENRR performance.
3.2 N2 adsorption on SiM@C3N4
Here, asymmetric dimer electrocatalysts (SiM@C3N4) were designed to address the low affinity of N2 on active sites. We chose some high-performance ENRR transition metals (TMs) to cooperate with Si to capture N2, and the metal elements selected are shown in Fig. 2a. Fig. 2b displays the design scheme of concept-SiM@C3N4 which involves using two NV1 vacancies on g-C3N4. The optimized SiM@C3N4 catalysts are shown in Fig. S3 (ESI†). According to Eb and (Eco.Si + Eco.M.) analyses of dimers (see Table S1, ESI†), it is anticipated that these SiM@C3N4 concept models all exhibit strong stability under ambient conditions.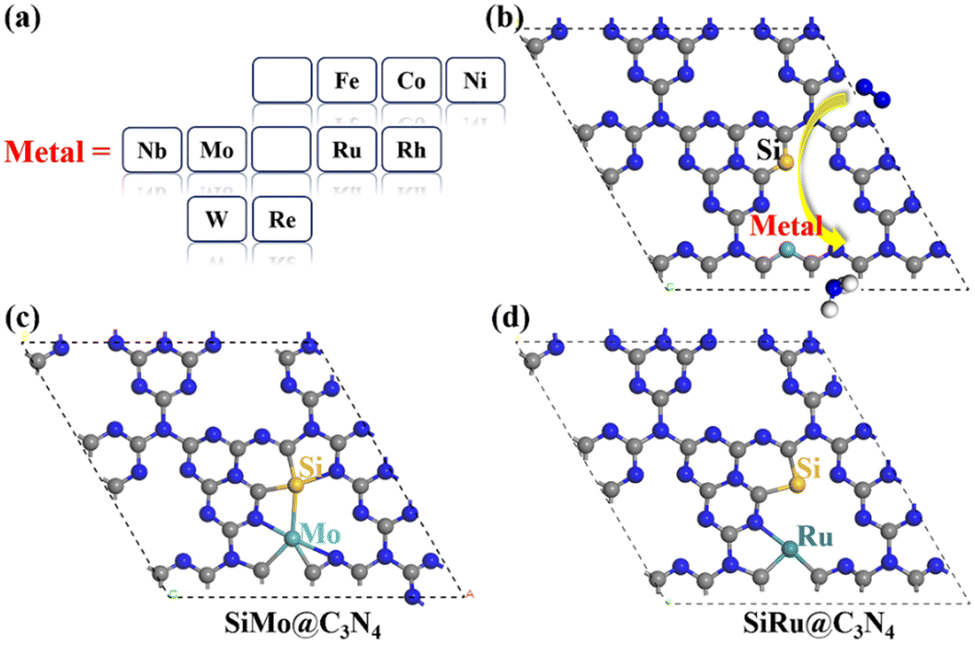 | ||
| Fig. 2 (a) Chosen metal elements for the concept models of SiM@C3N4; (b) the design strategy of SiM@C3N4 for ENRR; (c and d) potential high-performance catalysts: SiMo@C3N4 and SiRu@C3N4. Color scheme: Si (yellow), H (white), N (blue), and C (grey). | ||
Then N2 adsorption on SiM@C3N4 was examined. Fig. 2c and 2d display two dimer models, i.e., SiMo@C3N4 and SiRu@C3N4. They all have strong N2 adsorption ability with the adsorption free energy (ΔG*N2) less than 0.50 eV (see Fig. 3), indicating the occurrence possibility of N2 adsorption under ambient conditions.55,56Fig. 3 displays the side-on adsorption configurations of N2 on SiM@C3N4 except for SiRe@C3N4, because N2 can only be captured in the end-on model at the Re site after geometry optimization as shown in Fig. S4 (ESI†). Among nine dimer concept-electrocatalysts, based on Eads and adsorption-free energies (ΔG*N2), only two dimer catalysts were shown to have good N2 adsorption ability, i.e., SiMo@C3N4 and SiRu@C3N4, with ΔG*N2 of 0.17 and 0.28 eV, respectively. Moreover, the side-on adsorption modes on SiMo@C3N4 and SiRu@C3N4 well-activated the adsorbed *N2 with the extended NN lengths of 1.21 and 1.20 Å, respectively. However, other dimers showed ΔG*N2 > 0.50 eV, indicating that N2 adsorption under ambient conditions is challenging to overcome, although they all exhibited the activated characteristics due to the elongated N–N bond. To further analyze the adsorption process of N2 on SiM@C3N4, the kinetic barriers were analyzed as shown in Fig. S5 (ESI†), from which it was noticed that N2 adsorption barriers were all less than 0.50 eV except for SiNi@C3N4. SiMo@C3N4 and SiRu@C3N4 with the most energetically favorable *N2 configurations demonstrated quite small reaction barriers of 0.23 and 0.24 eV, respectively. Therefore, from the perspective of adsorption thermodynamics and kinetics, N2 can be readily activated on SiMo@C3N4 and SiRu@C3N4 under ambient conditions.
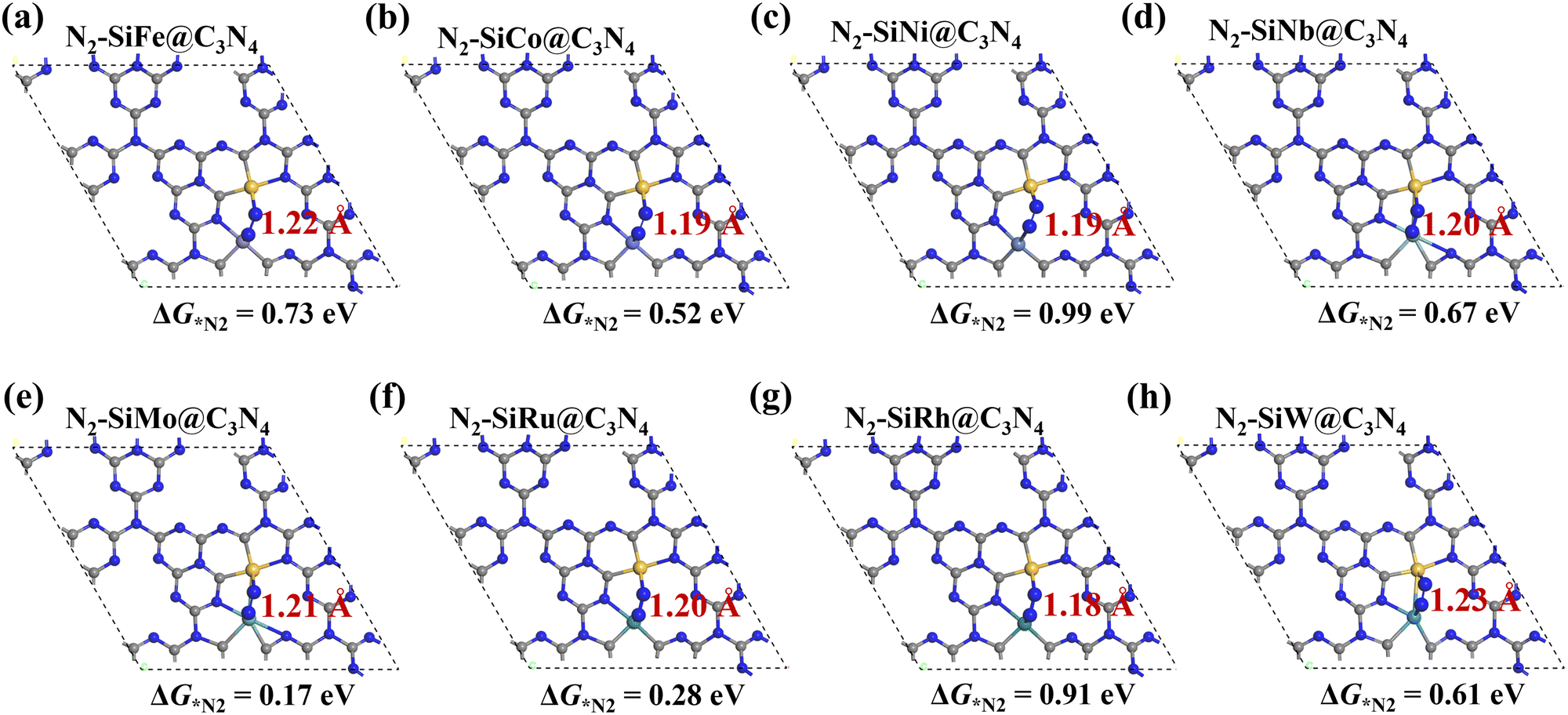 | ||
| Fig. 3 (a–h) Side-on adsorption configurations of *N2 on SiM@C3N4 with adsorption-free energies (ΔG*N2) and N–N bond lengths. N2 adsorption configuration on SiRe@C3N4 is shown in Fig. S4 (ESI†). | ||
3.3 ENRR on SiMo@C3N4 and SiRu@C3N4
Following the discussion on N2 adsorption above, we examined the ENRR performance on SiMo@C3N4 and SiRu@C3N4. According to relevant reports,57,58 when N2 is captured on dimer catalyts in a side-on mode, the ENRR generally can occur through two mechanisms, i.e., enzymatic and consecutive ones, which is schematically depicted in Fig. S6 (ESI†). Therefore, the ENRR on SiMo@C3N4 and SiRu@C3N4 was investigated according to the proposed mechanisms computationally.Fig. 4 and Fig. S7 (ESI†) show the enzymatic and consecutive pathways on SiMo@C3N4 and SiRu@C3N4, respectively. For the first hydrogenation reduction step of *N2 + H+ + e− → *N2H, there are two possible hydrogenation ways through the attack of the H+/e− pair (shown in Fig. S8, ESI†): one is to attack the N adatom bonded to Si and another is to attack the N adatom connected to the metal site. Based on free-energy analyses, the H+/e− pair energetically first tends to attack the N adatom bonded to the metal site. Likewise, on SiMo@C3N4, *NH3 species also tend to form on the metal site; however, for SiRu@C3N4, *NH2 and *NH3 species can only form on the Si site. Fig. 5 demonstrates their corresponding free energy evolutions along enzymatic and consecutive pathways, from which we can notice that the PDS is the first hydrogenation step. Due to the well-activated N–N bond of *N2, free-energy changes during the first hydrogenation steps on these two asymmetrical dimer centers, i.e., SiMo@C3N4 and SiRu@C3N4, are pretty low. The limiting potentials (UL) on SiMo@C3N4 and SiRu@C3N4 are merely −0.20 and −0.39 V, respectively, for the ENRR, with the responding overpotentials (η) of 0.04 and 0.23 V, respectively. Such low overpotentials indicate that the ENRR can be boosted under an ultra-low external potential. Moreover, these two dimers show comparable and even better performance than the reported metal-based electrocatalysts (FeV@C2N:59UL = −0.17 V; NbB:60UL = −0.40 V; MnB:60UL = −0.33 V; Mo and W-doped Au-based single-atom alloyes:61UL = −0.30 V; Mo@BM-β12:62UL = −0.26 V; and Mn@BM-β12:62UL = −0.32 V;), even outperform some 2D Mxenes63 (Mo2C(OH)2: UL = −0.62 V; V2C(OH)2: UL = −0.71 V; and Cr2C(OH)2: UL = −0.78 V) and TM single-atom catalysts45,64 (Mo1/pyrrolic-N3-G: UL = −0.49 V, Re1/pyrrolic-N3-G: UL = −0.51 V; and Nb@P3-Ars: UL = −0.52 V), exhibiting a great promising prospect towards electrocatalytic nitrogen fixation.
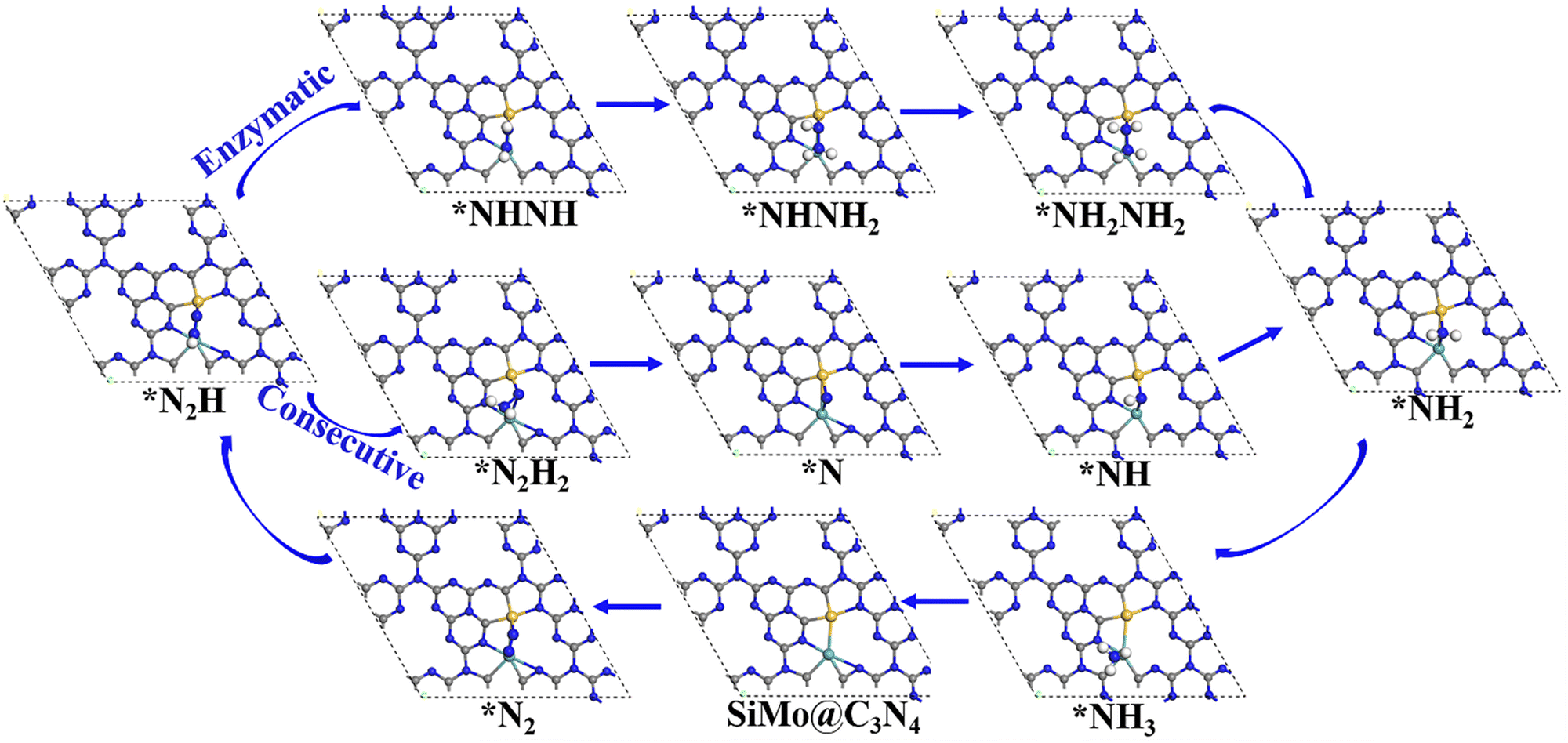 | ||
| Fig. 4 The enzymatic and consecutive pathways of the ENRR on SiMo@C3N4, and the ENRR on SiRu@C3N4 is shown in Fig. S7 (ESI†). | ||
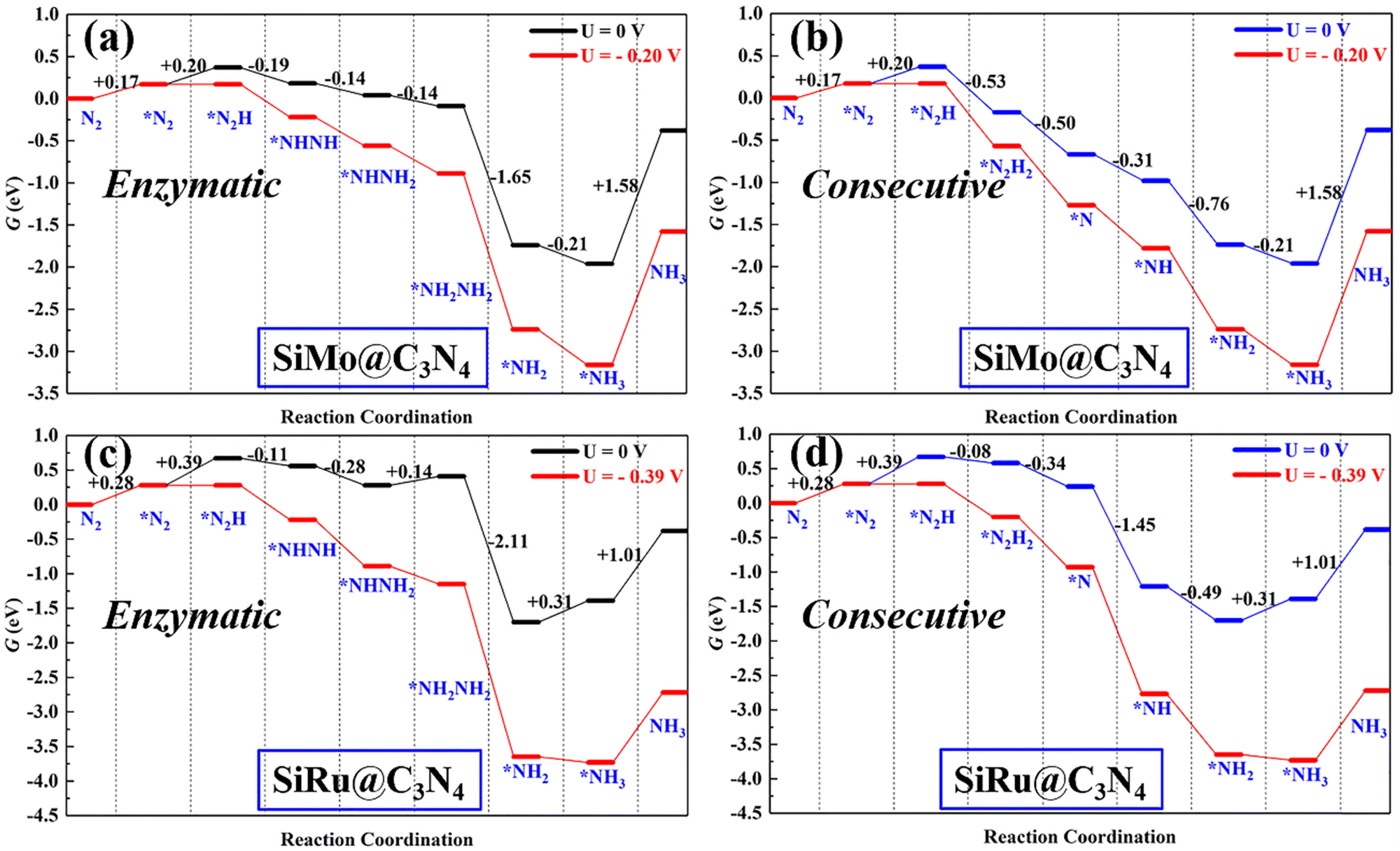 | ||
| Fig. 5 The free energy evolutions of enzymatic and consecutive pathways on (a and b) SiMo@C3N4 and (c and d) SiRu@C3N4 with and without the applied potential (UL). | ||
ENRR on Mo@C3N4 and Ru@C3N4
Single-atom catalysts (SACs) have been the front line for upgrading ENRR electrocatalysts due to their peculiar properties, such as high-atom utilization, low-coordination sites, and tunable electronic properties. However, sometimes SACs suffer from the limitation of scaling relationship during the ENRR, i.e., less flexibility to meet all energetic requirements of multiple proton-coupled electron-transfer steps by tuning the electronic structures. Therefore, dual active sites may provide a solution to this issue.For comparison with dimer catalysts, here ENRR performance on TM single-atom Mo@C3N4 and Ru@C3N4 (see Fig. S9–S11, ESI†) catalysts was also investigated. First, N2 adsorption on Mo@C3N4 and Ru@C3N4 was also studied and is shown in Fig. S10 (ESI†). One can find that, on Mo@C3N4, the end-on adsorption configuration with ΔG*N2 = −0.20 eV is more favorable energetically than the side-on mode with ΔG*N2 = 0.56 eV, while as for Ru@C3N4, N2 can be chemically adsorbed on the Ru site only in the end-on mode with ΔG*N2 = 0.33 eV. Therefore, tasking the N2 end-on adsorption modes on both SACs as starting points, the ENRR along the distal and alternating pathway was investigated as shown in Fig. S11 (ESI†). Their corresponding free-energy evolutions are presented in Fig. 6, from which again we noticed that the first hydrogenation step (*N2 + H+ + e− → *N2H) is the PDS, with ΔG of 0.78 and 0.89 eV, for Mo@C3N4 and Ru@C3N4, respectively. As a result, the limiting potentials on Mo@C3N4 and Ru@C3N4 are −0.78 and −0.89 V, respectively, which are distinctly greater than those on SiMo@C3N4 and SiRu@C3N4 dimer catalysts. Generally, the first hydrogenation step is determined from the activation degree of *N2 on electrocatalysts. This suggests that dimer catalysts possess a superb activity in the ENRR than SACs, demonstrating the synergistic advantage of the combination of Si and metal sites (Mo/Ru) in N2 adsorption and activation.
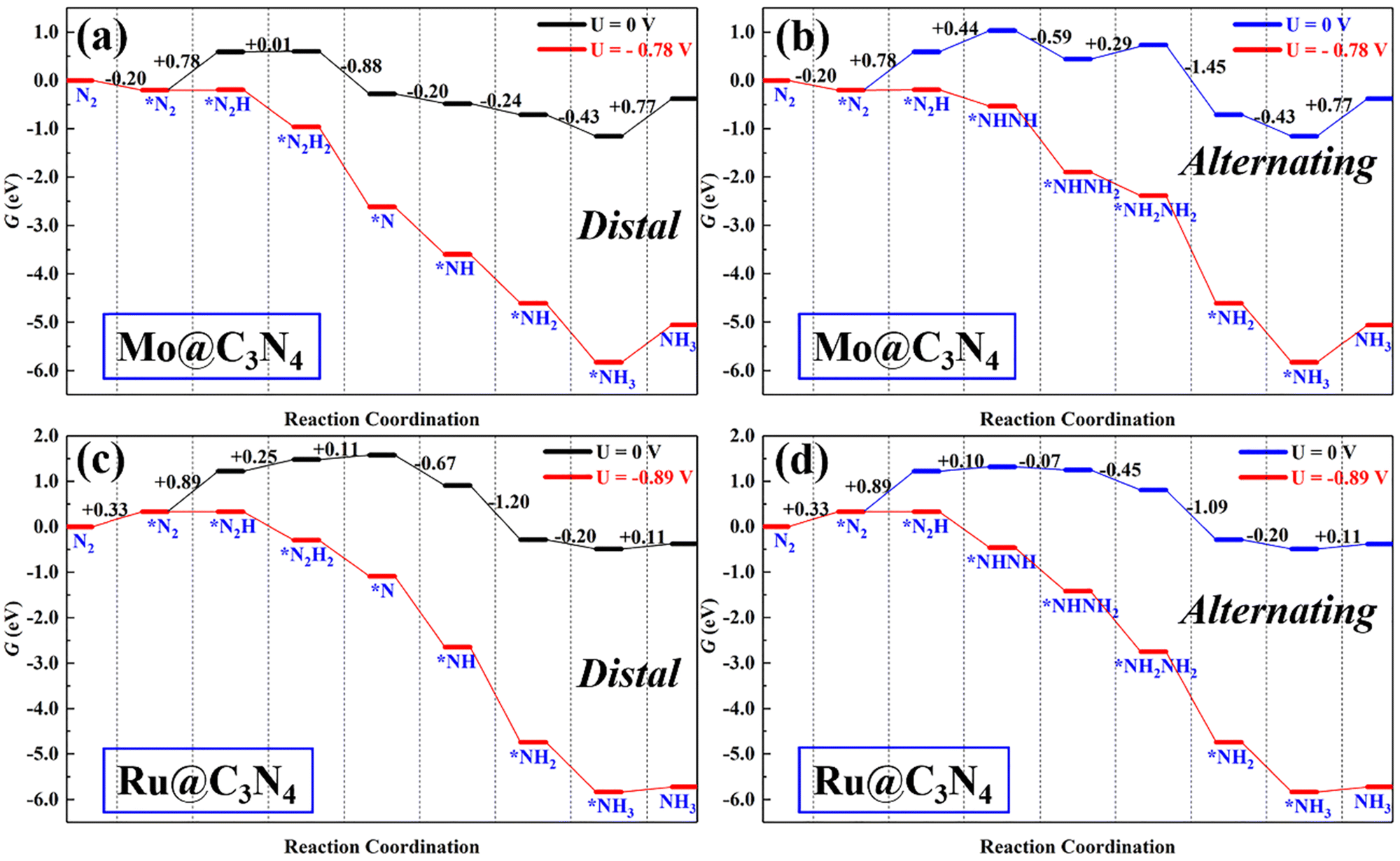 | ||
| Fig. 6 The free energy evolution of the ENRR on (a and b) Mo@C3N4 and (c and d) Ru@C3N4. | ||
3.5 Origin of high-performance dimers
For studying the high-performance origin of SiMo@C3N4 and SiRu@C3N4, we investigated their electronic structures, as shown in Fig. 7. First, the calculated charge density difference (Fig. 7a) suggests that N2 has a stronger electron exchange on SiM@C3N4 than on M@C3N4, and the Hirshfeld charge analysis (Table S2, ESI†) proved it quantitatively. The DOS data demonstrate the electron interaction around the Fermi level, and the strong electron exchange between N2 and SiM@C3N4 resulted in splitting of the 1π molecular-orbital (MO). As for the N2 adsorption on Mo@C3N4 and Ru@C3N4, the DOS overlap between the metal site and N2 below the Fermi level was very small, suggesting a weaker electron interaction. This is also in accordance with the N–N bond length and electron density analyses (Fig. 7a). The COHP, offering an atom pair's chemical bonding and anti-bonding information, is a visual bonding indicator. Thus, the COHP analysis was performed for *N2, as shown in Fig. 7c. Indeed, a larger electron occupation in the 1π* orbital for side-on adsorption configurations on SiM@C3N4 was observed, and ICOHP results are on the order of −11.72 (*N2 on SiRu@C3N4) < −10.32 (*N2 on SiMo@C3N4), −4.37 (*N2 on Ru@C3N4) < −4.08 (*N2 on Mo@C3N4), quantitatively explaining the activation degree. The larger activation degree of N2 is conducive to decreasing the free energy of the first hydrogenation step.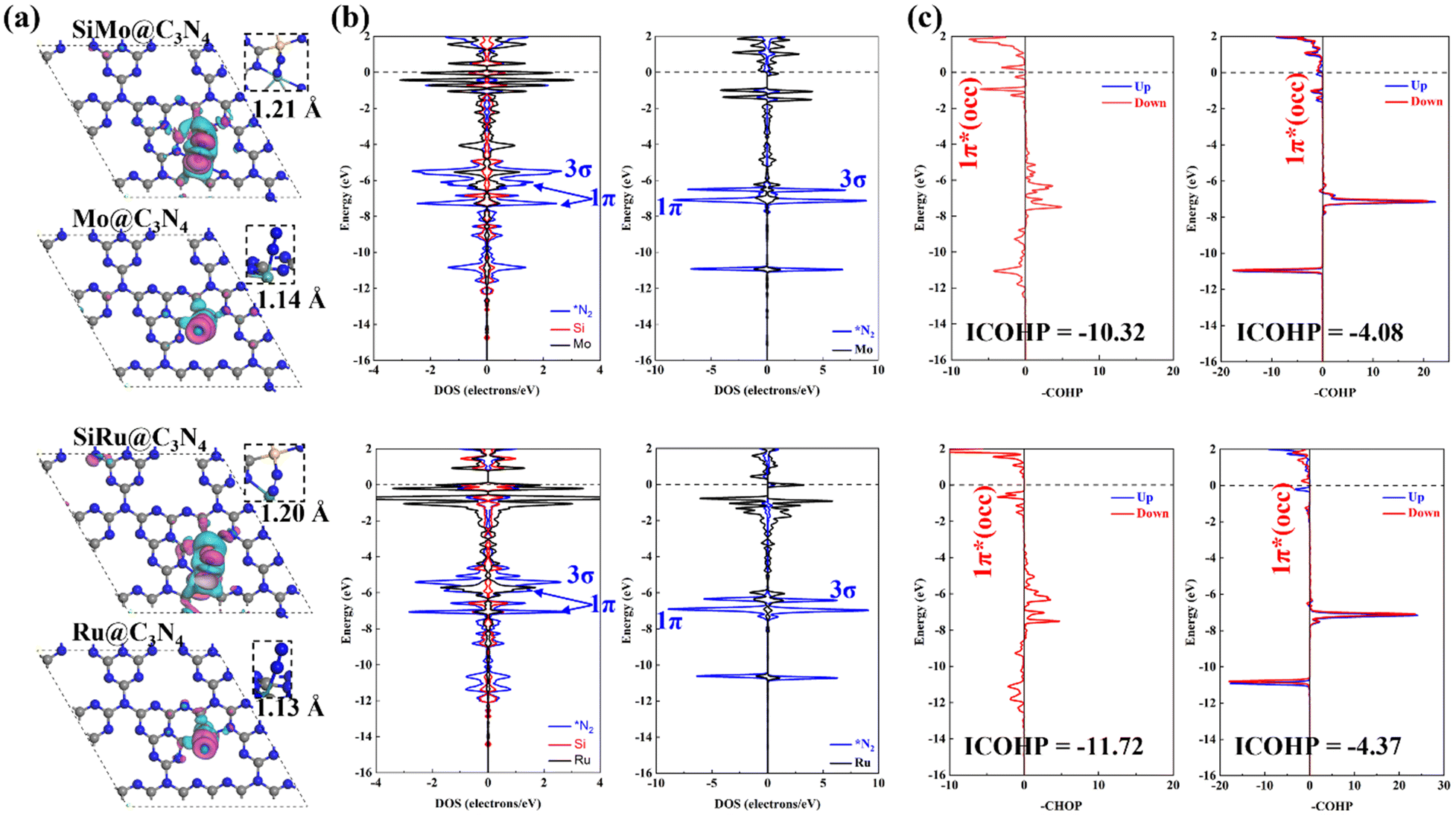 | ||
| Fig. 7 Analyses of the electronic structure: (a) the DFT-calculated charge density difference induced by N2 adsorption with the N–N bond length, and the red and cyan represent the electron accumulation and loss, respectively (isosurface = 0.01 a.u.); (b) DOS analyses for *N2, Si, Mo, and Ru of four concept-models; and (c) the –COHP analyses for *N2, where “1π* (occ)” refers to the occupation of 1π* anti-bonding orbitals. | ||
Besides, the electron transfer between reaction intermediates and active sites also plays a crucial role during the hydrogenation process. Thus, we analyzed the Hirshfeld charge evolution of the ENRR on SiM@C3N4 and M@C3N4 (M = Mo and Ru) as shown in Fig. S13 (ESI†). Here, we analyzed Hirshfeld charges of four moieties on SiM@C3N4 (M = Mo and Ru) and three moieties on M@C3N4 (M = Mo and Ru): NxHy, (the reaction intermediates), Si (the non-metal site), Mo/Ru (the metal site) and defective g-C3N4 (the supporter structure). First, on SiMo@C3N4 (see Fig. S13a, ESI†), the charge evolution of NxHy was different from that of g-C3N4, and obvious charge oscillation for both NxHy and g-C3N4 could be observed. However, the charges on active sites (Si and Mo) oscillate slightly, and even the charge on Mo remains almost unchanged along the reaction coordinate. These findings suggest that g-C3N4 serves as the electron reservoir, and the active sites (Si and Mo) act as the electron adaptors. Similar results can also be obtained on SiRu@C3N4 (see Fig. S13c, ESI†). Additionally, these two active sites of SiM@C3N4 (M = Mo and Ru) could slightly regulate the charges on NxHy as a mediator during the synthesis of second ammonia. But, as for single-atom Mo@C3N4 and Ru@C3N4 (see Fig. S13b and d, ESI†), only the metal site can transfer electrons back and forth between g-C3N4 and NxHy intermediates. And the active site (Mo/Ru) mainly acts as an electron adaptor during the ENRR process because of almost unchanged Hirshfeld charges on them.
3.6 Hydrogen evolution reaction
The HER is parasitic in electrocatalysis, which can primarily consume electrons and reduce the selectivity of ammonia during the ENRR. The HER is a common issue in electrocatalysis, and many electro-chemical reduction reactions, including but not limited to carbon dioxide reduction,65,66 nitrate reduction,67 and nitrogen reduction. A suitable catalyst should not only have high activity but also possess decent selectivity. Here, adsorption of *H on two potential active sites of dimer catalysts, i.e., Si and M (M = Mo/Ru), was examined as shown in Fig. 8a and 8b. We can find that ΔGmaxHER on SiMo@C3N4 is larger than ΔGmaxNRR (0.20 eV), and ΔGmaxHER on SiRu@C3N4 is also larger than ΔGmaxNRR (0.39 eV), which suggests that the HER will be suppressed due to the hindered Heyrovskey step (*H + H+ + e− → H2). Therefore, the ENRR is energetically more favorable compared to the HER. Therefore, SiMo@C3N4 and SiRu@C3N4 would have high ammonia selectivity.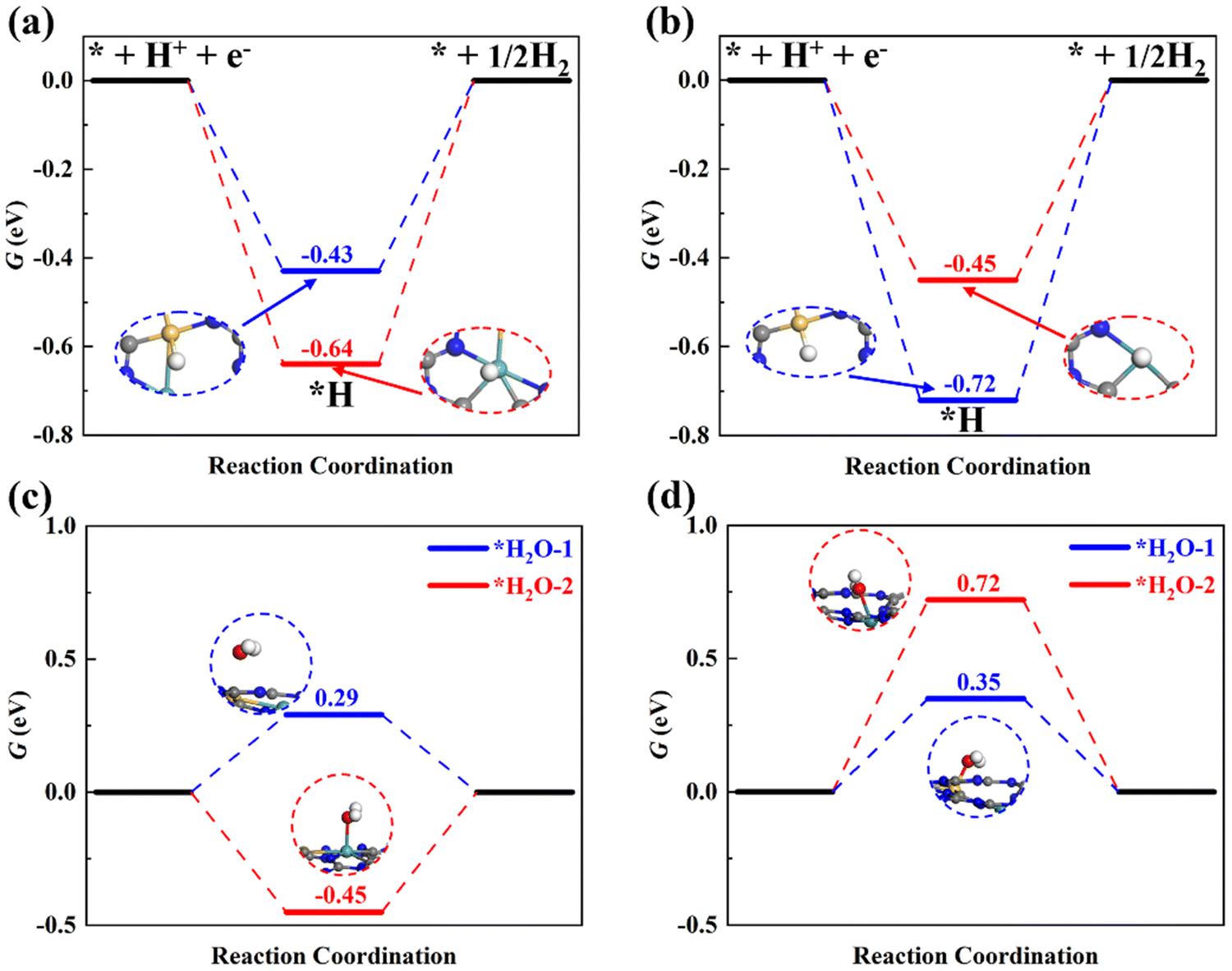 | ||
| Fig. 8 Hydrogen evolution reaction on (a) SiMo@C3N4 and (b) SiRu@C3N4; the adsorption free energies of H2O on (c) SiMo@C3N4 and (d) SiRu@C3N4. Note that there are two potential active sites on dimer catalysts for hydrogen and water adsorption. | ||
In addition, the water molecules in electrolytes may affect the N2 adsorption. Therefore, water adsorption was also studied, as shown in Fig. 8c and d. In comparison with the N2 adsorption, we can notice that the Mo site on SiMo@C3N4 will be occupied by H2O due to a lower adsorption free-energy of −0.45 eV, which will adversely influence the nitrogen activation and reduction; while SiRu@C3N4 tends to adsorb N2 rather than H2O due to the lower adsorption free energy (0.33 eV) than that (0.35 and 0.72 eV) of H2O. This indicates that SiRu@C3N4 will exhibit high activity and selectivity towards the ENRR, while SiMo@C3N4 will favor the ENRR in electrolytes with a low concentration of H2O, for example, ionic liquids.68 To evaluate the synthetic availability of SiMo@C3N4 and SiRu@C3N4 with high activity in the laboratory, their formation energy was analyzed, which was found to be 2.19 and 1.36 eV, respectively, which means that both of them can be synthesized using some precursors and SiRu@C3N4 is easier to synthesize. Meanwhile, we designed another two models, SiMo-C3N4 and SiRu-C3N4 (see Fig. S14, ESI†), which are derived by directly doping two atoms into the holes of g-C3N4 nanosheets. By comparing their formation energies (Table S3, ESI†), we can see that the concept models of SiMo@C3N4 and SiRu@C3N4 are easier to synthesize. Therefore, again it is shown that the Nv site is a perfect anchoring site for atomic catalysts.
Conclusion
To sum up, asymmetrical dimer electrocatalysts were designed to realize the ENRR efficiently. Among the investigated SiM@C3N4 catalyst models, SiMo@C3N4 and SiRu@C3N4 exhibited the highest activity towards the ENRR with strong N2 adsorption abilities and ultra-low onset potentials of −0.20 and −0.39 V, respectively. Moreover, these catalysts are capable of suppressing the competitive HER. Especially, SiRu@C3N4 exhibiting strong hydrophobicity possesses high NH3 selectivity. This design strategy not only addresses the issue of low adsorption and activation ability towards N2 on Si but also opens up a new avenue for designing asymmetrical dimer electrocatalysts for the ENRR, with the advantage of metal-based and non-metal-based catalysts.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge the Center for Computational Materials Science, Institute for Materials Research, Tohoku University, for the use of MASAMUNE-IMR (project no. 202212-SCKXX-0204) and the Institute for Solid State Physics (ISSP) at the University of Tokyo for the use of their supercomputers. This work was supported by the China BaoWu Low Carbon Metallurgical Innovation Foundation (no. BWLCF202113), the Fundamental Research Funds for the Central Universities (no. N2202012), JSPS KAKENHI (No. JP23K13703), and the Iwatani Naoji Foundation. The authors also thank Beijing PARATERA Tech Co., Ltd., for providing HPC resources.References
- X. Zhu, S. Mou, Q. Peng, Q. Liu, Y. Luo, G. Chen, S. Gao and X. Sun, J. Mater. Chem. A, 2020, 8, 1545–1556 RSC.
- D. Hao, Y. Liu, S. Gao, H. Arandiyan, X. Bai, Q. Kong, W. Wei, P. K. Shen and B.-J. Ni, Mater. Today, 2021, 46, 212–233 CrossRef CAS.
- Y. Wen, H. Zhu, J. Hao, S. Lu, W. Zong, F. Lai, P. Ma, W. Dong, T. Liu and M. Du, Appl. Catal., B, 2021, 292, 120144 CrossRef CAS.
- W. Liao, L. Qi, Y. Wang, J. Qin, G. Liu, S. Liang, H. He and L. Jiang, Adv. Funct. Mater., 2021, 2009151 CrossRef CAS.
- W. Zhang, J. Low, R. Long and Y. Xiong, Energy Chem., 2020, 2, 100040 CrossRef.
- L. Niu, L. An, X. Wang and Z. Sun, J. Energy Chem., 2021, 61, 304–318 CrossRef CAS.
- H. Liu, Chin. J. Catal., 2014, 35, 1619–1640 CrossRef CAS.
- L. Shi, Q. Li, C. Ling, Y. Zhang, Y. Ouyang, X. Bai and J. Wang, J. Mater. Chem. A, 2019, 7, 4865–4871 RSC.
- J. Zhao, X. Ren, X. Li, D. Fan, X. Sun, H. Ma, Q. Wei and D. Wu, Nanoscale, 2019, 11, 4231–4235 RSC.
- P. Song, L. Kang, H. Wang, R. Guo and R. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 12408–12414 CrossRef CAS PubMed.
- Y. Li, D. Gao, S. Zhao, Y. Xiao, Z. Guo, Y. Fang, J. Lin, Z. Liu, Y. Huang, K. Guo and C. Tang, Chem. Eng. J., 2021, 410, 128419 CrossRef CAS.
- Z. Guo, S. Qiu, H. Li, Y. Xu, S. J. Langford and C. Sun, Diamond Relat. Mater., 2021, 111, 108210 CrossRef CAS.
- Z. Guo, S. Qiu, H. Li, Y. Xu, S. J. Langford and C. Sun, Phys. Chem. Chem. Phys., 2020, 22, 21761–21767 RSC.
- C. Liu, Q. Li, C. Wu, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Am. Chem. Soc., 2019, 141, 2884–2888 CrossRef CAS PubMed.
- J. Zhao, B. Wang, Q. Zhou, H. Wang, X. Li, H. Chen, Q. Wei, D. Wu, Y. Luo, J. You, F. Gong and X. Sun, Chem. Commun., 2019, 55, 4997–5000 RSC.
- J. Zhao, J. Yang, L. Ji, H. Wang, H. Chen, Z. Niu, Q. Liu, T. Li, G. Cui and X. Sun, Chem. Commun., 2019, 55, 4266–4269 RSC.
- L. Xia, X. Wu, Y. Wang, Z. Niu, Q. Liu, T. Li, X. Shi, A. M. Asiri and X. Sun, Small Methods, 2018, 3, 1800251 CrossRef.
- L. Xia, J. Yang, H. Wang, R. Zhao, H. Chen, W. Fang, A. M. Asiri, F. Xie, G. Cui and X. Sun, Chem. Commun., 2019, 55, 3371–3374 RSC.
- G.-Y. Zhai, D. Xu, S.-N. Zhang, Z.-H. Xue, H. Su, Q.-Y. Yu, H.-H. Wang, X. Lin, Y.-X. Lin, L.-H. Sun, X.-H. Li and J.-S. Chen, Adv. Funct. Mater., 2020, 30, 2005779 CrossRef CAS.
- Z. Guo, S. Qiu, H. Li, Y. Xu, S. J. Langford and C. Sun, ChemCatChem, 2021, 13, 1239–1245 CrossRef CAS.
- Z. Guo, T. Wang, H. Liu, S. Qiu, X. Zhang, Y. Xu, S. J. Langford and C. Sun, Nanoscale, 2022, 14, 5782–5793 RSC.
- Y. Luo, M. Li, Y. Dai, X. Zhang, R. Zhao, F. Jiang, C. Ling and Y. Huang, J. Mater. Chem. A, 2021, 9, 15217–15225 RSC.
- T. He, A. R. P. Santiago, Y. Kong, M. A. Ahsan, R. Luque, A. Du and H. Pan, Small, 2022, 18, 2106091 CrossRef CAS PubMed.
- X. Sun, Y. Qiu, B. Jiang, Z. Chen, C. Zhao, H. Zhou, L. Yang, L. Fan, Y. Zhang and N. Zhang, Nat. Commun., 2023, 14, 291 CrossRef CAS PubMed.
- D. Wu, B. He, Y. Wang, P. Lv, D. Ma and Y. Jia, J. Phys. D: Appl. Phys., 2022, 55, 203001 CrossRef.
- Y. Ouyang, L. Shi, X. Bai, Q. Li and J. Wang, Chem. Sci., 2020, 11, 1807–1813 RSC.
- T. He, K. Reuter and A. Du, J. Mater. Chem. A, 2020, 8, 599–606 RSC.
- M. Groenewolt and M. Antonietti, Adv. Mater., 2005, 17, 1789–1792 CrossRef CAS.
- W. Wang, H. Zhang, S. Zhang, Y. Liu, G. Wang, C. Sun and H. Zhao, Angew. Chem., Int. Ed., 2019, 58, 16644–16650 CrossRef CAS PubMed.
- H. Ma, Z. Shi, S. Li and N. Liu, Appl. Surf. Sci., 2016, 379, 309–315 CrossRef CAS.
- C. Ren, Y. Zhang, Y. Li, Y. Zhang, S. Huang, W. Lin and K. Ding, J. Phys. Chem. C, 2019, 123, 17296–17305 CrossRef CAS.
- C. Lv, Y. Qian, C. Yan, Y. Ding, Y. Liu, G. Chen and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 10246–10250 CrossRef CAS PubMed.
- Z. Chen, J. Zhao, C. R. Cabrera and Z. Chen, Small Methods, 2018, 3, 1800368 CrossRef.
- X. Li, X. Sun, L. Zhang, S. Sun and W. Wang, J. Mater. Chem. A, 2018, 6, 3005–3011 RSC.
- G. Dong, W. Ho and C. Wang, J. Mater. Chem. A, 2015, 3, 23435–23441 RSC.
- K. Chu, Q.-q Li, Y.-p Liu, J. Wang and Y.-h Cheng, Appl. Catal., B, 2020, 267, 118693 CrossRef CAS.
- K. Wang, Q. Li, B. Liu, B. Cheng, W. Ho and J. Yu, Appl. Catal., B, 2015, 176–177, 44–52 CAS.
- H. Xie, Y. Zheng, X. Guo, Y. Liu, Z. Zhang, J. Zhao, W. Zhang, Y. Wang and Y. Huang, ACS Sustainable Chem. Eng., 2021, 9, 6788–6798 CrossRef CAS.
- D. Jiao, Y. Liu, Q. Cai and J. Zhao, J. Mater. Chem. A, 2021, 9, 1240–1251 RSC.
- J. Zhang, X. Tian, M. Liu, H. Guo, J. Zhou, Q. Fang, Z. Liu, Q. Wu and J. Lou, J. Am. Chem. Soc., 2019, 141, 19269–19275 CrossRef CAS PubMed.
- S. Mukherjee, X. Yang, W. Shan, W. Samarakoon, S. Karakalos, D. A. Cullen, K. More, M. Wang, Z. Feng, G. Wang and G. Wu, Small Methods, 2020, 4, 1900821 CrossRef CAS.
- Z. Zhang and X. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 56987–56994 CrossRef CAS PubMed.
- Q. Li, L. He, C. Sun and X. Zhang, J. Phys. Chem. C, 2017, 121, 27563–27568 CrossRef CAS.
- C. Liu, Q. Li, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Mater. Chem. A, 2019, 7, 4771–4776 RSC.
- W. Zhao, L. Chen, W. Zhang and J. Yang, J. Mater. Chem. A, 2021, 9, 6547–6554 RSC.
- D. Ma, Z. Zeng, L. Liu and Y. Jia, J. Energy Chem., 2021, 54, 501–509 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- H.-J. Chun, V. Apaja, A. Clayborne, K. Honkala and J. Greeley, ACS Catal., 2017, 7, 3869–3882 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- Q. Tay, P. Kanhere, C. F. Ng, S. Chen, S. Chakraborty, A. C. H. Huan, T. C. Sum, R. Ahuja and Z. Chen, Chem. Mater., 2015, 27, 4930–4933 CrossRef CAS.
- X. Cui, C. Tang and Q. Zhang, Adv. Energy Mater., 2018, 8, 1800369 CrossRef.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- Q. Liu, S. Wang, G. Chen, Q. Liu and X. Kong, Inorg. Chem., 2019, 58, 11843–11849 CrossRef CAS PubMed.
- Y. Abghoui and E. Skúlason, Catal. Today, 2017, 286, 69–77 CrossRef CAS.
- L. J. Arachchige, Y. Xu, Z. Dai, X. L. Zhang, F. Wang and C. Sun, J. Mater. Sci. Technol., 2021, 77, 244–251 CrossRef CAS.
- D. Ma, Z. Zeng, L. Liu, X. Huang and Y. Jia, J. Phys. Chem. C, 2019, 123, 19066–19076 CrossRef CAS.
- W. Zhang and B.-W. Zhang, Nano-Micro Lett., 2021, 13, 106 CrossRef CAS PubMed.
- M. Zafari, A. S. Nissimagoudar, M. Umer, G. Lee and K. S. Kim, J. Mater. Chem. A, 2021, 9, 9203–9213 RSC.
- G. Zheng, Y. Li, X. Qian, G. Yao, Z. Tian, X. Zhang and L. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 16336–16344 CrossRef CAS PubMed.
- L. Xu, L.-M. Yang and E. Ganz, ACS Appl. Mater. Interfaces, 2021, 13, 14091–14101 CrossRef CAS PubMed.
- X. Lv, L. Kou and T. Frauenheim, ACS Appl. Mater. Interfaces, 2021, 13, 14283–14290 CrossRef CAS PubMed.
- R. Song, J. Yang, M. Wang, Z. Shi, X. Zhu, X. Zhang, M. He, G. Liu, G. Qiao and Z. Xu, ACS Omega, 2021, 6, 8662–8671 CrossRef CAS PubMed.
- C. Liu, T. Wang, D. Hao, Q. Li, S. Li and C. Sun, J. Mater. Sci. Technol., 2022, 110, 96–102 CrossRef.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed.
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC.
- F. Zhou, L. M. Azofra, M. Ali, M. Kar, A. N. Simonov, C. McDonnell-Worth, C. Sun, X. Zhang and D. R. MacFarlane, Energy Environ. Sci., 2017, 10, 2516–2520 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp05959b |
| This journal is © the Owner Societies 2023 |