
Synthesis of graphdiyne on a copper substrate via a self-coupling reaction†
Jiasheng
Wu‡
,
Jie
Liu‡
,
Jizhe
Liang
,
Yijie
Zhang
,
Xiaoli
Zhao
and
Chunxue
Yuan
 *
*
College of Materials Science and Engineering, Tongji University, Shanghai, China
First published on 1st December 2022
Abstract
A new strategy is proposed to prepare graphdiyne via the self-coupling reaction of hexakis(bromoethynyl)benzene (hBEP) with alkynyl bromide groups. Prominently, the reaction can proceed moderately in 12 h at room temperature. The as-synthesized film displays a porous structure, excellent chemical characteristics, and an admirable catalytic performance for the hydrogen/oxygen evolution reaction (HER/OER).
Carbon-based allotropes have attracted great attention all over the world, and carbon materials like diamond, graphite, fullerene, carbon nanotubes and graphene have already exhibited excellent and diverse properties.1–3 It is worth noting that these materials generally involve sp3 and sp2 carbon atoms. Compared with other carbon materials, graphdiyne (GDY) is constructed from sp and sp2 hybridized carbon atoms.4,5 The introduction of the sp hybrid status can form highly conjugated –C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– bonds with the advantages of a linear structure and no cis–trans isomerism. The extended π-conjugation and the periodic arrangement of benzene rings and diacetylenic linkages (–CC–CC–) endow graphdiyne with an excellent porous structure, a natural band gap and a high carrier mobility (up to 104–105 cm2 V−1 s−1).6–8 To satisfy the versatile demand for this material, its pore size, band gap, and even the connection patterns of hybridized carbon atoms can be tailored or adjusted, such as in β-GDY, HsGDY and Cl-GDY.9–12 Moreover, numerous delicate forms of GDY have been fabricated, such as nanowires, nanowalls, nanosheets, nanotube arrays, etc.12–15 The multiformity and versatility of graphdiyne have revealed its promising potential in catalysis, energy storage, photodetectors and water purification.6,10,14,16–22
C– bonds with the advantages of a linear structure and no cis–trans isomerism. The extended π-conjugation and the periodic arrangement of benzene rings and diacetylenic linkages (–CC–CC–) endow graphdiyne with an excellent porous structure, a natural band gap and a high carrier mobility (up to 104–105 cm2 V−1 s−1).6–8 To satisfy the versatile demand for this material, its pore size, band gap, and even the connection patterns of hybridized carbon atoms can be tailored or adjusted, such as in β-GDY, HsGDY and Cl-GDY.9–12 Moreover, numerous delicate forms of GDY have been fabricated, such as nanowires, nanowalls, nanosheets, nanotube arrays, etc.12–15 The multiformity and versatility of graphdiyne have revealed its promising potential in catalysis, energy storage, photodetectors and water purification.6,10,14,16–22
In 1987, Baughman et al. first proposed the stable existence of graphyne.23 However, its feasible preparation remained a struggle until the ground-breaking study of Li and co-workers in 2010.24 They synthesized a large-scale GDY film using HEB via a cross-coupling reaction in 72 hours. Since then, research into graphdiyne has made huge progress from theoretical predictions to practical applications, and the reaction time has been shortened to about 20 hours.17 So far, wet chemistry has become the main strategy for synthesizing graphdiyne.8
Scholars usually make use of the molecules containing terminal alkynyl groups according to the Glaser, Glaser–Hay and Eglinton coupling reactions.11,13,14,20,24 The homocoupling reactions of such monomers (like TEE, TEB and HEB) can proceed efficiently, and the yield of the final products is commonly high. However, the terminal alkynyl groups are so sensitive to oxygen that side reactions and defects occur, and extra heating is frequently required. In addition, depending on the active functional groups, another choice of wet chemistry is to utilize the homocoupling reaction of terminal alkynylsilane groups as in Hiyama coupling.25 The introduction of trimethylsilyl primarily improves the stability of the molecules (like HEB-TMS), and they can directly form conjugated diacetylene bonds without deprotection treatment.26,27 However, the reactivity of the monomers is decreased accordingly, which leads to the generation of various byproducts and the yield of the final products is ultimately reduced. Therefore, it is urgent to find new types of functional group that have both high reactivity and good stability.
In previous work, we designed a novel monomer tBEP and prepared the graphdiyne-like carbon skeleton on Au (111) under ultrahigh vacuum.28 Guided by this idea, we further synthesized the HsGDY film with uniformly distributed pores using tBEP as the precursor, achieving the successful transformation from on-surface synthesis to solution synthesis.16 It is worth noting that the terminal alkynyl bromide groups of tBEP display a greater stability and higher selectivity than the terminal alkynyl, which results in the more moderate reaction conditions and a reduced reaction time. Combining these studies, terminal alkynyl bromide groups are promising functional groups to accomplish the controllable mass preparation of GDY in solution.
In this work, we designed a new precursor hBEP and synthesized a large-area GDY film on a copper substrate under the conditions of pyridine and an NaOH aqueous solution. The obtained GDY is a free-standing transparent dark film with a three-dimensional porous morphology. As confirmed using EDS, XPS, Raman and FT-IR spectroscopy, the chemical structure of film is proved. Furthermore, the electrochemical catalytic activity of our as-prepared GDY is tested through the HER/OER.
The synthetic routes for the monomer hBEP are presented in Scheme S1 (ESI†). The copper foil used (3.5 × 2 cm2) was sonicated with 1 M HCl aqueous solution, deionized water, acetone and ethanol, respectively, for 15 min, and dried under nitrogen. The pretreated copper foil and 20 mg hBEP were added to a three-necked flask. Subsequently, 20 mL pyridine and 2 mL 0.1 M NaOH aqueous solution were injected into the flask separately and the whole flask was stirred under a nitrogen atmosphere at r.t. After 12 h, acetone and ethanol were used in turn to wash the surface of the retrieved copper foil. Whereafter, the prepared sample was immersed in 1 M HCl aqueous solution until the copper foil was completely dissolved. Eventually, a nearly transparent black film was obtained.
In the composite alkaline environment of pyridine and NaOH aqueous solution, the dehalogenative homocoupling reaction proceeded between the hBEP monomers (Fig. 1, top), and eventually the GDY film was obtained on the surface of the copper foil (Fig. 1, bottom left).16,24 After the reaction, the copper foil turned red-brown, and showed a clear luster. As shown in Fig. 1 (bottom right) the obtained GDY film is a nearly transparent dark film with an area of 7 cm2, which can be held stably without the support of the copper foil. To further examine the microscopic morphology of the as-prepared film, SEM was employed. An obvious and continuous two-dimensional structure can be observed, and enormous pores are spread randomly all over the material (Fig. 2a). The diameter of these pores ranges from tens of nanometers to several hundred nanometers, as shown in Fig. 2b and c. These characteristics result in a unique macro/mesopore second-order structure of the as-grown film to provide plentiful vacancies and channels for iron storage and separation. During this reaction, the copper foil acted not only as an efficient metal catalyst but also as a template for the formation to achieve a flat and smooth film.13 Once the monomers touch the copper foil, the C–Br bonds are directly fractured to constitute the conjugated diacetylene structure.28 As the reaction proceeded, the monomers connected with each other and gradually spread over the template. In fact, the ideal GDY material should be a two-dimensional planar material containing uniformly distributed pores. However, the film we obtained is more like a three-dimensional material comprised of graphdiyne multilayers. This is because, at a certain moment, the formed product would hinder the continuous growth of its plane. This obstacle forced the subsequent fragment to grow out of the plane, which eventually caused the film to be a three-dimensional porous material rather than a two-dimensional planar material.
 | ||
| Fig. 1 (Top) Schematic diagram of the dehalogenative homocoupling synthetic route and the chemical structure of the GDY film. (Bottom) Optical images of the film on (left) and removed from (right) the copper foil. | ||
 | ||
| Fig. 2 SEM and EDS mapping images of GDY: (a–c) SEM images of the GDY film; and (d–g) EDS mapping images of C + O (e), C (f), and O (g) of the GDY film. | ||
As a reliable technique, energy dispersive spectrometry (EDS) was applied to analyze the elemental composition of the sample. As displayed in Fig. 2e–f, carbon is evenly and densely located throughout the film, while oxygen is scattered sporadically. Given the chemical structure of the precursor, the resulting film should be made entirely of carbon. However, the presence of holes and defects leads to unavoidable oxygen atoms. In addition, the content of elemental carbon reaches 74.44% (Fig. S2, ESI†), verifying that the GDY film is a typical carbon-rich material.
Raman spectroscopy is an effective tool to research systemically the chemical bonding structure of carbon materials and characterize the quality and uniformity of the products. As illustrated in Fig. 3a, there are four major peaks for the GDY we synthesized. The peak around 1390 cm−1 belongs to the D-band, which is a characteristic signal for carbon nanomaterials. It generally derives from the breathing vibration of the aromatic domain and the broken symmetry due to the defects, disordered carbon and irregular edges of the sample.24 The G-band at 1592 cm−1 is another typical Raman peak of graphene carbon, which is attributed to the skeletal vibration of abundant benzene rings. The peaks located at 1953 cm−1 and 2225 cm−1 correspond to the different vibration modes of the conjugated diacetylene and carbon–carbon triple bonds between the sp2 atoms, respectively.29,30 Besides, FT-IR spectra are a powerful complement to detect the asymmetrical vibrations of functional groups in the materials. As described in Fig. 3b, two major signal peaks are presented in the FT-IR spectra of GDY. The peak at 1569 cm−1 can be assigned to the skeletal vibration of the aromatic nucleus. This result corresponds with the G-band of the Raman spectrum. Meanwhile, the band at 2175 cm−1 is a typical signal peak related to stretching vibration of the alkynyl bond, and accords with the results of the Raman spectra. These results indicate the presence of benzene rings and conjugated butadiyne bonds. There is no peak generated by the vibration of C–Br from the monomers, which proves that the dehalogenative reaction is complete. Besides, the broad intense peak at 3332 cm−1 is assigned to the stretching vibration of O–H groups.31 These functional groups mainly come from the deionized water and ethanol absorbed by the various pores of the film. While owing to the perfect symmetrical structure of GDY, the intensities of the FT-IR peaks are weaker than those of the Raman peaks.31 Combined with the results mentioned above, hBEP is consumed in the reaction and the functional groups of GDY are detected accurately, as we envisaged.
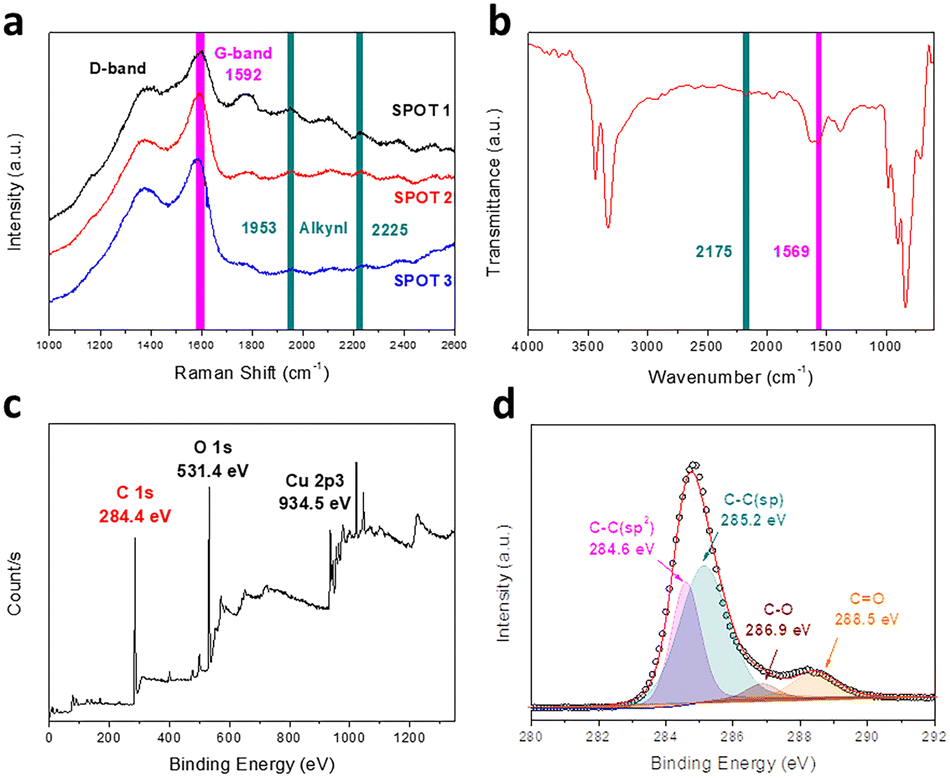 | ||
| Fig. 3 Chemical structure characterization of the as-grown GDY film. (a) Raman spectra obtained at three random points on the film. (b) FT-IR spectrum of the GDY film on the substrate. (c and d) XPS total spectrum (c) and high resolution C 1s spectrum (d). | ||
Through Raman and FT-IR spectroscopy, we can acquire abundant information about the functional groups contained in GDY. To fully confirm the types of carbon hybridization and measure their relative contents, however, X-ray photoelectron spectroscopy (XPS) is a powerful and reliable technique.32Fig. 3c shows the full XPS spectrum of the GDY film, which contains three major peaks at 284.4 eV (C 1s), 531.4 eV (O 1s) and 934.5 eV (Cu 2p3). It is worth noting that the XPS spectra verify the existence of elemental Cu, in agreement with the results of EDS analysis, which mainly come from the copper foil, whose content is quite low. After the background was subtracted and deconvolution was performed, the chemical environment of C 1s was discerned, as shown in the high-resolution scan of Fig. 3d. In detail, the peak of C 1s is composed of four sub-peaks: 284.6 eV, 285.2 eV, 286.9 eV and 288.5 eV. The peaks at 284.6 eV and 285.2 eV are, respectively, assigned to C–C (sp2) and C–C (sp), which indicate the presence of benzene rings and acetylene bonds. Furthermore, the area ratio of the sp and sp2 sub-peaks is close to 2, demonstrating that the aromatic rings are connected with diacetylene bonds, which conforms to the theoretical structure of GDY. The peaks observed at 286.9 eV and 288.5 eV manifest the essentially equivalent binding energies of C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. Subsequently, to further study the chemical status of oxygen, the O 1s peak was also deconvoluted (Fig. S3, ESI†) and the spectra display the existence of carbonyl (CO), ester (OC–O) and hydroxyl (O–H).29 There are two major sources of oxygen atoms in the sample. One is the absorption of the air due to the porous network, and the other is the slight doping of oxygen at the edges and defects of the film.20 These doped oxygen atoms may have a complicated influence on GDY. On one hand, these atoms may cause elongation of the surrounding bonds, lead to the irregularity of the structure and even break the planar conjugated π system. On the other hand, the electronegativity and hydrophilicity of oxygen-containing groups may also make it easy for GDY to attach to other compounds, like metal ions, and stably disperse in an aqueous environment.
O, respectively. Subsequently, to further study the chemical status of oxygen, the O 1s peak was also deconvoluted (Fig. S3, ESI†) and the spectra display the existence of carbonyl (CO), ester (OC–O) and hydroxyl (O–H).29 There are two major sources of oxygen atoms in the sample. One is the absorption of the air due to the porous network, and the other is the slight doping of oxygen at the edges and defects of the film.20 These doped oxygen atoms may have a complicated influence on GDY. On one hand, these atoms may cause elongation of the surrounding bonds, lead to the irregularity of the structure and even break the planar conjugated π system. On the other hand, the electronegativity and hydrophilicity of oxygen-containing groups may also make it easy for GDY to attach to other compounds, like metal ions, and stably disperse in an aqueous environment.
The planar conjugated structure constructed by aromatic rings and diacetylene endows GDY with an extremely high carrier mobility.33,34 Meanwhile, ion storage and molecular reactions are provided by the enormous tunnels and interspaces of the porous networks and macro/mesopore second-order structure.19,35 In this work, the catalytic performance for the HER and the OER of the as-prepared GDY was also investigated using a three-electrode system in 1 M KOH aqueous solution, and the testing data were adjusted to the reversible hydrogen electrode (RHE) in light of the Nernst equation. It is noteworthy that the GDY grown on the copper foil was used directly as the working electrode without any extra treatment. As depicted in Fig. 4a and b, when catalyzing the HER, the overpotential required for GDY is 458 mV (j = 10 mA cm−2) and the corresponding Tafel slope is 77 mV dec−1, respectively. For the OER, GDY exhibits an overpotential of 386 mV (j = 10 mA cm−2) and a Tafel slope of 330 mV dec−1 (Fig. 4c and d), respectively. The HER/OER activity is similar to that in previous work (173 mV/445 mV) and slightly better than the HsGDY (531 mV/646 mV) we synthesized previously.16,31 Compared with HsGDY, the proportion and density of acetylene bonds in GDY are higher, where are the active sites for HER and OER. Thus, GDY exhibits a better HER/OER catalytic performance than HsGDY and promisingly achieves a better performance when combined with other metal atoms.36
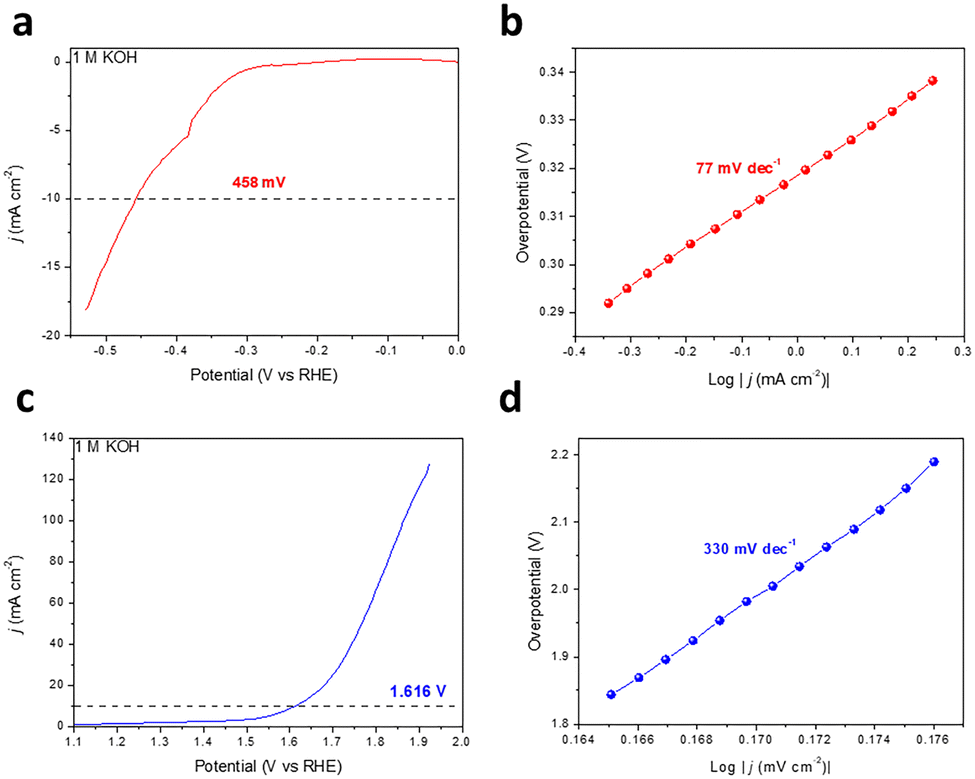 | ||
| Fig. 4 HER/OER catalytic properties of as-prepared GDY. (a) LSV curve for the HER, and (b) the corresponding Tafel plot of the HER. (c) LSV curve for the OER, and (d) the corresponding Tafel plot of the OER. | ||
Combined with the traditional synthesis strategies for GDY and our previous studies regarding terminal alkynyl bromide groups, we designed a novel monomer hBEP to prepare a GDY film on copper foil via a dehalogenative homocoupling reaction. The obtained GDY film is a free-standing porous carbon material with a meso/macropore second-order structure. Qualitative measurements indicate the presence of abundant benzene rings and diacetylene bonds, and quantitative analysis shows that the aromatic rings are truly connected by diacetylene bonds. XPS data prove that GDY contains sp and sp2 hybridized carbon atoms and its proportion is nearly 2, which is consistent with the theoretical model of GDY. Based on the faint stability and high reactivity of terminal alkynyl bromide groups, our approach can proceed at r.t. without the need for extra heating, and the reaction time is significantly shortened to 12 h. In addition, the catalytic performance of GDY for the HER and the OER was also measured. GDY displays a similar electrochemical activity to that in former investigations.33–38 Integrated with our previous studies, we propose not only a novel synthesis route for large-scale film formation but a potential method for the massive industrial preparation of GDY and its analogues.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Opening Test Fund of Large Instrument and Equipment of Tongji University (No. 2022GX070).Notes and references
- F. Diederich and C. Thilgen, Science, 1996, 271, 317–324 CrossRef CAS.
- A. D. Franklin, Nature, 2013, 498, 443–444 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- M. M. Haley, S. C. Brand and J. J. Pak, Angew. Chem., Ed. Engl., 1997, 36, 836–838 Search PubMed.
- Y. Fang, Y. Liu, L. Qi, Y. Xue and Y. Li, Chem. Soc. Rev., 2022, 51, 2681–2709 RSC.
- Y. Li, Z. Zuo and Y. Li, Handb. Carbon-Based Nanomater., 2021, 461–516 CAS.
- M. Inagaki and F. Kang, J. Mater. Chem. A, 2014, 2, 13193–13206 RSC.
- X. Gao, H. Liu, D. Wang and J. Zhang, Chem. Soc. Rev., 2019, 48, 908–936 RSC.
- N. Wang, J. He, Z. Tu, Z. Yang, F. Zhao, X. Li, C. Huang, K. Wang, T. Jiu, Y. Yi and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 10740–10745 CrossRef CAS PubMed.
- J. Li, Z. Xie, Y. Xiong, Z. Li, Q. Huang, S. Zhang, J. Zhou, R. Liu, X. Gao, C. Chen, L. Tong, J. Zhang and Z. Liu, Adv. Mater., 2017, 29, 1700421 CrossRef PubMed.
- J. Li, Y. Xiong, Z. Xie, X. Gao, J. Zhou, C. Yin, L. Tong, C. Chen, Z. Liu and J. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 2734–2739 CrossRef CAS PubMed.
- S. Zhuo, Y. Shi, L. Liu, R. Li, L. Shi, D. H. Anjum, Y. Han and P. Wang, Nat. Commun., 2018, 9, 3132 CrossRef PubMed.
- J. Zhou, X. Gao, R. Liu, Z. Xie, J. Yang, S. Zhang, G. Zhang, H. Liu, Y. Li, J. Zhang and Z. Liu, J. Am. Chem. Soc., 2015, 137, 7596–7599 CrossRef CAS PubMed.
- X. Gao, J. Zhou, R. Du, Z. Xie, S. Deng, R. Liu, Z. Liu and J. Zhang, Adv. Mater., 2016, 28, 168–173 CrossRef CAS PubMed.
- T. Wang, J. Huang, H. Lv, Q. Fan, L. Feng, Z. Tao, H. Ju, X. Wu, S. L. Tait and J. Zhu, J. Am. Chem. Soc., 2018, 140, 13421–13428 CrossRef CAS PubMed.
- J. Wu, J. Liang, Y. Zhang, X. Zhao and C. Yuan, Chem. Commun., 2021, 57, 5036–5039 RSC.
- H. Yu, Y. Xue and Y. Li, Adv. Mater., 2019, 31, 1803101 CrossRef CAS PubMed.
- W. Ding, M. Sun, Z. Zhang, X. Lin and B. Gao, Ultrason. Sonochem., 2020, 61, 104850 CrossRef CAS PubMed.
- C. Yang, Y. Li, Y. Chen, Q. Li, L. Wu and X. Cui, Small, 2019, 15, 1804710 CrossRef PubMed.
- J. He, N. Wang, Z. Cui, H. Du, L. Fu, C. Huang, Z. Yang, X. Shen, Y. Yi, Z. Tu and Y. Li, Nat. Commun., 2017, 8, 1172 CrossRef PubMed.
- Z. Zuo, H. Shang, Y. Chen, J. Li, H. Liu, Y. Li and Y. Li, Chem. Commun., 2017, 53, 8074–8077 RSC.
- F. He and Y. Li, CCS Chem., 2022, 1–23 Search PubMed.
- R. H. Baughman, H. Eckhardt and M. Kertesz, J. Chem. Phys., 1987, 87, 6687–6699 CrossRef CAS.
- G. Li, Y. Li, H. Liu, Y. Guo, Y. Li and D. Zhu, Chem. Commun., 2010, 46, 3256–3258 RSC.
- K. Ikegashira, Y. Nishihara, K. Hirabayashi, A. Mori and T. Hiyama, Chem. Commun., 1997, 1039–1040 RSC.
- J. Zhou, Z. Xie, R. Liu, X. Gao, J. Li, Y. Xiong, L. Tong, J. Zhang and Z. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 2632–2637 CrossRef CAS PubMed.
- C. Huang, Y. Li, N. Wang, Y. Xue, Z. Zuo, H. Liu and Y. Li, Chem. Rev., 2018, 118, 7744–7803 CrossRef CAS PubMed.
- Q. Sun, L. Cai, H. Ma, C. Yuan and W. Xu, ACS Nano, 2016, 10, 7023–7030 CrossRef CAS PubMed.
- Q. Yang, Y. Guo, J. Gu, N. Li, C. Wang, Z. Liu, X. Li, Z. Huang, S. Wei, S. Xu, L. Song, J. Fan, Z. Chen, J. Qiu and C. Zhi, Nano Energy, 2020, 78, 105283 CrossRef CAS.
- S. Zhang, J. Wang, Z. Li, R. Zhao, L. Tong, Z. Liu, J. Zhang and Z. Liu, J. Phys. Chem. C, 2016, 120, 10605–10613 CrossRef CAS.
- H. Yu, Y. Xue, L. Hui, F. He, C. Zhang, Y. Liu, Y. Fang, C. Xing, Y. Li, H. Liu and Y. Li, Nano Energy, 2019, 64, 103928 CrossRef CAS.
- H. Bao, L. Wang, C. Li and J. Luo, ACS Appl. Mater. Interfaces, 2019, 11, 2717–2729 CrossRef CAS PubMed.
- Y. Liu, Y. Gao, F. He, Y. Xue and Y. Li, CCS Chem., 2022, 1–11 Search PubMed.
- X. Zheng, Y. Xue, C. Zhang and Y. Li, CCS Chem., 2022, 1–10 Search PubMed.
- Y. Gao, Y. Xue, L. Qi, C. Xing, X. Zheng, F. He and Y. Li, Nat. Commun., 2022, 13, 5227 CrossRef CAS PubMed.
- Y. Gao, Y. Xue, F. He and Y. Li, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, 2206946119 CrossRef PubMed.
- L. Hui, Y. Xue, H. Yu, Y. Liu, Y. Fang, C. Xing, B. Huang and Y. Li, J. Am. Chem. Soc., 2019, 141, 10677–10683 CrossRef CAS PubMed.
- Y. Xue, B. Huang, Y. Yi, Y. Guo, Z. Zuo, Y. Li, Z. Jia, H. Liu and Y. Li, Nat. Commun., 2018, 9, 1460 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04384j |
| ‡ These authors contributed equally. |
| This journal is © the Owner Societies 2023 |