
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly chemoselective homologative assembly of the α-substituted methylsulfinamide motif from N-sulfinylamines†
Monika
Malik
a,
Raffaele
Senatore
a,
Davide
Castiglione
b,
Alexander
Roller-Prado
c and
Vittorio
Pace
 *ab
*ab
aDepartment of Pharmaceutical Sciences, Division of Pharmaceutical Chemistry, University of Vienna, Josef-Holaubek-Platz 2, 1090, Vienna, Austria. E-mail: vittorio.pace@univie.ac.at
bDepartment of Chemistry, Via Giuria 7, University of Turin, Turin 10125, Italy
cDepartment of Inorganic Chemistry – Functional Materials, University of Vienna, Waehringerstrasse 42, 1090, Vienna, Austria
First published on 21st August 2023
Abstract
α-Substituted methylsulfinamide are prepared through the homologation of electrophilic N-sulfinylamines with Li-CHXY reagents. The transformation takes place under full chemocontrol and exhibits good flexibility for preparing both N-aryl and N-alkyl analogues. Various sensitive functionalities can be accommodated on the starting materials, thus documenting a wide reaction scope.
The sulfinamide moiety constitutes an important organic framework of considerable interest across the chemical sciences,1 also exhibited in natural products.2 It is indeed a key representative example of sulfur(IV) entities featuring a unique reactivity profile imparted by the N–S delocalization. The constitutive chirality of the sulfur atom3 guided the development of enantiopure analogues – illustrated by the venerable Ellmann's t-butylsulfinamide4 (Scheme 1, box) – nowadays belonging to the synthetic chemist's toolbox.5 Moreover, they can be engaged in oxidative transformations en route to sulfonamides and aza-analogues, widely expressed in biologically active substances.6 The retrosynthetic analysis of the cluster enables individuating three main disconnections levered on: (i) N–S or, (ii) C–S bond forging events and, (iii) oxidation of low-valent sulfur species (sulfenamides).1,7 The notoriously not ideal manipulation of these latter reagents or, in general, sulfur(ii) species (e.g. RSH),8 makes preferable adopting logics paved on the construction of C–S linkages.9 Inspired by his seminal work on heterocumulenes (carbon dioxide, isocyanates and isothiocyanates),10 Gilman in 1926 successfully prepared sulfinamides by treating readily accessible sulfinylamines (i.e. monoaza analogues of SO2)11 and Grignard reagents.12 While considered intuitive, the strategy may suffer from low chemocontrol due to the use of such hard nucleophiles and intrinsic high reactivity of the sp2-hybridized sulfur atom towards moisture.13 Notwithstanding, in recent years Willis uncovered the innate potential of both the tactic and the versatility of sulfinylamines, thus designing a plethora of elegant transformations conducting to versatile sulfur(VI) materials14 which complemented the well-studied application of sulfinylamines in cycloadditions and ene reactions.15 Among the fundamental achievements of Willis’ work, it has to be emphasized the efficient transformations conducted on reluctant alkyl-type sulfinylamines through the judicious installation of sterically hindered substituents on nitrogen (i.e. N-trityl) thus, resulting in a remarkable extension of the scope of the methodology.14a,16 Analogous systems could also undergo Ni(II)-catalyzed (hetero)arylation – giving a series of sulfinamides analogues – when employing boroxines.17 Moreover, Bolm showed the metal-free addition of aryldiazoniums to N-tritylsulfinylamines for preparing sulfonimidamides.18 As a consequence of the challenging installation of C-sp3 hybridized elements under nucleophilic regime, Li and Zhao introduced in 2022 a photoredox protocol enabling the successful transfer of various functionalized alkyl elements released from 4-substituted-1,4-dihydropyridines.19 Intrigued by the lack of precedents in literature regarding substituted α-methylsulfinamides and cognizant they would be suited for subsequent functionalization owing to the constitutive electrophilic vicinal sulfur and carbon sites,20 we argued they could be obtained – in one synthetic operation – via the addition of a nucleophilic homologating synthon – i.e. [M]CH2X21 – to sulfinylamines. To be productive, the strategy had to overcome the low chemocontrol potentially associated with the addition of hard nucleophiles. In this context, previously reported carbanions additions to sulfinylamines have been performed on relatively not functionalized systems, thus making the proposed tactic risky in terms of chemocontrol.22
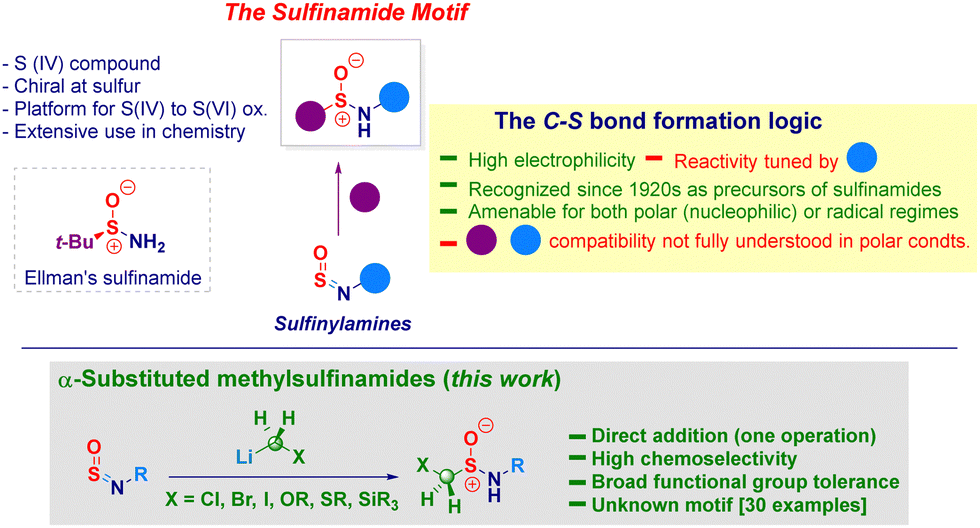 | ||
| Scheme 1 General context of the presented work. | ||
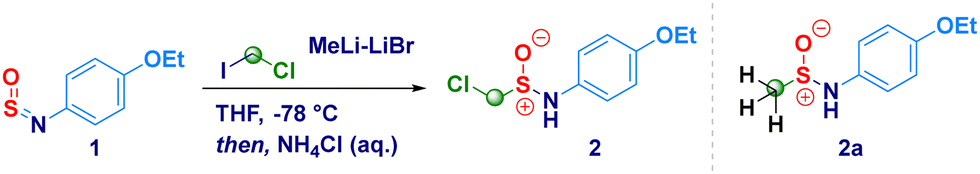
4-Ethoxy-N-sulfinylbenzenamine (1) was selected as the model substrate for accomplishing the addition of LiCH2Cl generated in situ from chloroiodomethane and MeLi–LiBr (Table 1).23 By running the reaction at −78 °C in diethyl ether, we were pleased to observe the formation of the homologated product (2) in 78% yield (Table 1, entry 1). The employment of less coordinating solvents (known to destabilize carbenoids)24 – frequently used as alternative to classic ethereal solvents – such as t-butyl methyl ether (TBME), cyclopentyl methyl ether (CPME)25 and 2-methyltetrahydrofuran (2-MeTHF),26 resulted in lower yields (entries 2–4). By running the reaction in THF – a stronger coordinating solvent able to tame the degradative Kirmse's α-elimination of carbenoids24 – 2 was obtained in a very good 85% yield after 1 h (Table 1, entry 5); in agreement with the limited existing time of LiCH2Cl,27 the increase of the reaction time up to 2 hours resulted in no further improvement (Table 1, entry 6). Conversely, the reaction did not reach completion when kept for 30 min (53% yield, entry 7). The noticeable instability of compound 2 during the chromatographic purification [SiO2 eventually deactivated with triethylamine (2% v/v) or TMSCl (1% v/v)] or neutral alumina (Brockmann degree III),28 suggested us to maximize the rate of conversion, as judged by 1H-NMR analysis. To this end, it was essential to use of 2.8 equiv. of carbenoids; in fact, lowering the loading to 1.8 equiv. [generated from ICH2Cl (2.0 equiv.) and MeLi–LiBr (1.8 equiv.)], not only resulted in a less efficient process – 46% conversion – but, also the attack of the methyl carbanion to the sulfur atom of the sulfinylamine was observed thus, furnishing 2a as a side product (entry 8). The same trend was also noticed when using 2.3 equiv. of carbenoid, though at a less extent (entry 9). Some additional aspects merit mention: (a) the less nucleophilic magnesium carbenoid - generated with i-PrMgCl-LiCl – did not promote the reaction regardless the adoption of Barbier or non-Barbier conditions (entry 10); (b) the use of the lithium carbenoid requires Barbier-type conditions, presumably because of the aforementioned intrinsic instability (entry 11); (c) at a higher temperature (−50 °C), reaction efficiency dropped significantly, as a consequence of the thermal sensitivity of the carbenoid (entry 12).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Homologating agenta (equiv.) | Solvent | Reaction time (h) | Temperature [°C] | Yield of 2b (%) |
| a Unless otherwise stated, reactions were run with LiCH2X. b Isolated yield. c The 1H-NMR analysis of the reaction crude indicated 46% conversion – calculated as mol(2)/[mol(1) + mol(2)] × 100; 1,3,5-trimethylbenzene internal standard; by running chromatography on neutral Al2O3, 2 was recovered in 28% yield; Compound 2a was formed as side product in 20% yield. d 62% conversion; 46% yield (purification on silica gel); 39% yield (purification on neutral Al2O3) for compound 2, respectively; Compound 2a was formed as side product in 17% yield. e Reactions run starting from ICH2Cl and i-PrMgCl-LiCl under both Barbier and non-Barbier conditions. f Non-Barbier conditions. | |||||
| 1 | LiCH2Cl (2.8) | Et2O | 1 | −78 | 78 |
| 2 | LiCH2Cl (2.8) | TBME | 1 | −78 | 62 |
| 3 | LiCH2Cl (2.8) | CPME | 1 | −78 | 55 |
| 4 | LiCH2Cl (2.8) | MeTHF | 1 | −78 | 43 |
| 5 | LiCH2Cl (2.8) | THF | 1 | −78 | 85 |
| 6 | LiCH2Cl (2.8) | THF | 2 | −78 | 83 |
| 7 | LiCH2Cl (2.8) | THF | 0.5 | −78 | 53 |
| 8c | LiCH2Cl (1.8) | THF | 1 | −78 | 34 |
| 9d | LiCH2Cl (2.3) | THF | 1 | −78 | 46 |
| 10e | i-PrMgCH2Cl LiCl (2.8) | THF | 1 | −78 | — |
| 11f | LiCH2Cl (2.8) | THF | 1 | −78 | — |
| 12 | LiCH2Cl (2.8) | THF | 1 | −50 | 42 |
With the optimal conditions in hand, we then examined the scope of the method (Scheme 2). The protocol proved to be flexible, thus enabling the efficient transformation of a series of N-sulfinylanilines into the corresponding chloromethylated products. The high electrophilicity of the sulfur atom uniformly allowed the nucleophilic attack of the carbenoid, thus making negligible the electronic and/or steric behavior of substituents on nitrogen. Electron-rich rings (2–3) were competent substrates regardless the position of the ethereal fragment, including the isosteric pharmaceutically relevant trifluoromethoxy- analogue (4).29 Simple alkyl groups installed on the aromatic ring (5–6) were not detrimental for the process; remarkably, the protocol was further validated by embodying two ethyl moieties at the sterically demanding ortho positions (2,6-diethyl-), thus, obtaining 6 in an excellent 85% yield. Unsaturated – sp2 and sp – carbon-centered functionalities (vinyl and ethynyl), susceptible of Simmons–Smith type cyclopropanation with carbenoids,30 fully preserved the chemical integrity, furnishing exclusively the desired adducts 7 and 8. It should be highlighted that the (basic) nucleophilic carbenoid did not deprotonate the acidic terminal alkyne. Besides the unsubstituted analogue 9, the introduction of halogens was not detrimental for the genuine chemocontrol of the protocol. Thus, chloro-derivatives 10 and 11 could be easily prepared, as well as, the synthetically useful (see below) bromo-(12), iodo-(13) and fluoro-(14) analogues. No concomitant aromatic lithiation – potentially occurring on bromo- and iodo-derivatives31 – was noticed under the reaction conditions during the carbenoid generation event from the dihalomethane. The chemoselectivity of the protocol was further deduced by engaging N-sulfinylamines containing sensitive functionalities potentially reactive under the adopted conditions. Not only the challenging nitro group remained untouched (15) but, also when electrophilic cyano-(16) and ester (17, regioisomer of the common local anaesthetic drug benzocaine) were accommodated on the aromatic ring, the exclusive occurring process was the sulfur halomethylation. This outcome is particularly relevant in view of the established use of esters as placeholders for halomethylenic fragments under nucleophilic regime.32 Presumably, N-sulfinylanilines are stronger electrophiles than esters, thus enabling the selective attack at the sulfur atom instead of the carboxylic carbon. The presence of nitrogen-centered functionalities [i.e. pyrrolidine (18) or phenyl-diazo (19)] further expanded the reaction scope: notably, the homologative process en route to 19 was scalable to 20 mmol in comparable efficiency, thus furnishing pure crystals suitable for X-ray analysis (see ESI†). Moreover, the methodology could be validated also in the case of the sulfonyl-substituted analogue (20). Not only chloromethylating agents could be employed for the homologation but, also the analogous bromo-derivative conveniently prepared from bromoiodomethane:33 accordingly, structures 21–25 were obtained under comparable chemocontrol, thus further highlighting the tolerance for strong electrophiles such as cyano (24) and ester (25) functionalities. The α-iodomethylation (with LiCH2I generated from CH2I2) furnished analogue 26 featuring two exceptional high electrophilic vicinal sites (sulfur and methylene). The robust stabilizing effect imparted by the sterically hindered trityl group on the nitrogen,14a enabled the extension of the protocol to aliphatic N-sulfinylamines suitable for homologation with both mono-halo (27) and di-halo carbenoids (28–29), being the latter generated through deprotonation of the corresponding dihalomethane.34 With the aim to gain full insight into the potential of homologative transformations of N-sulfinylanilines, we next turned our attention towards non-halogenated C1-lithiated agents. In this sense, LiCH2OEt35 (30) and LiCH2SMe36 (31) – both generated via Yus’ reductive lithiation methodology in the presence of Li metal and DTBB37 – could be efficiently added without altering the chemoselectivity profile. Though sceptical at the beginning as a consequence of the facile Cl-Li exchange under reductive conditions, we were delighted to observe exclusively the formation of the substituted α-methyllithium species (from ClCH2OEt or ClCH2SMe) in the presence of potentially exchangeable chlorine atom on the aromatic nuclei. Finally, by running the reaction with the commercially available LiCH2SiMe3, the stable α-silylmethyl analogue 32 was prepared in high yield (Scheme 3).
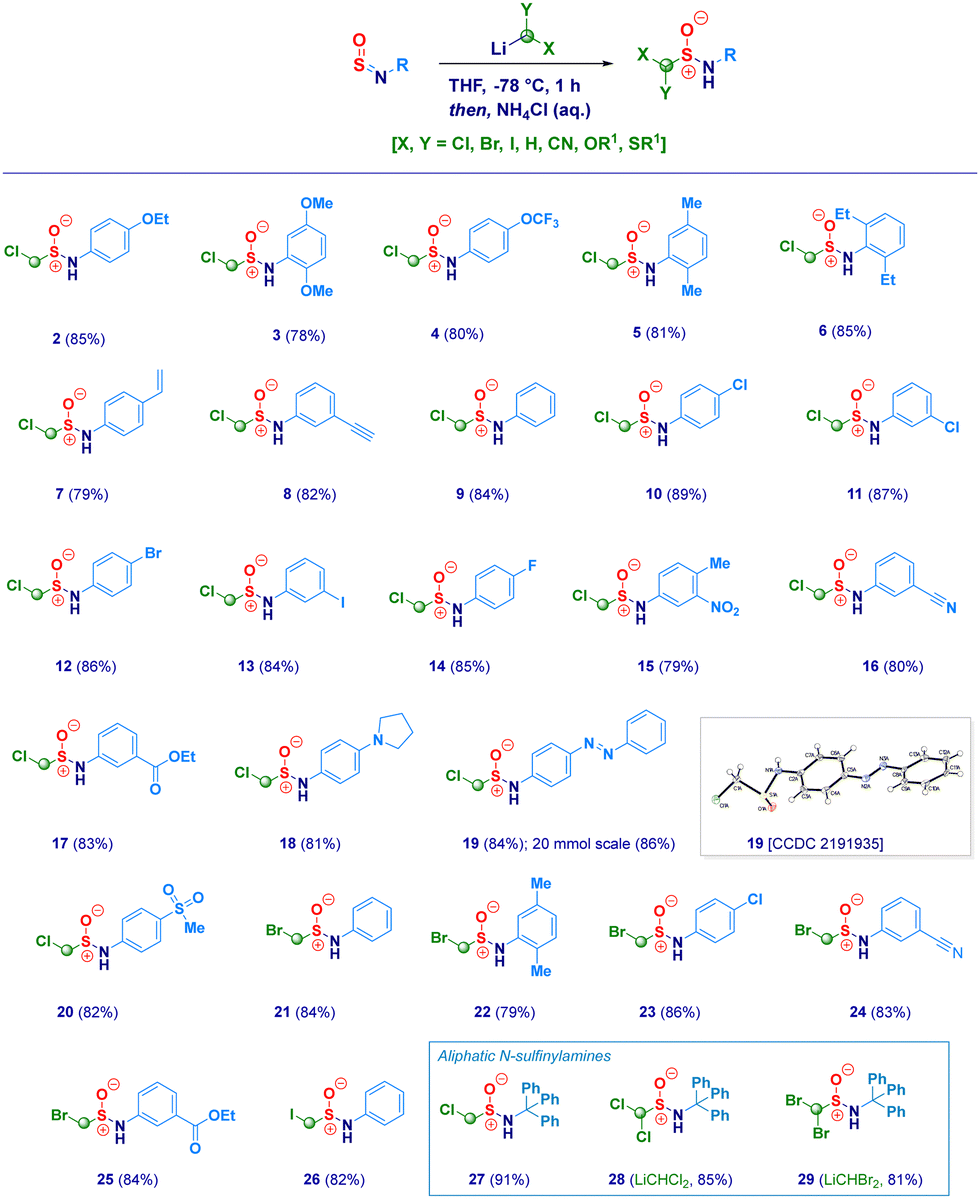 | ||
| Scheme 2 Homologation of N-sulfinylamines with halogenated α-methyllithiums. | ||
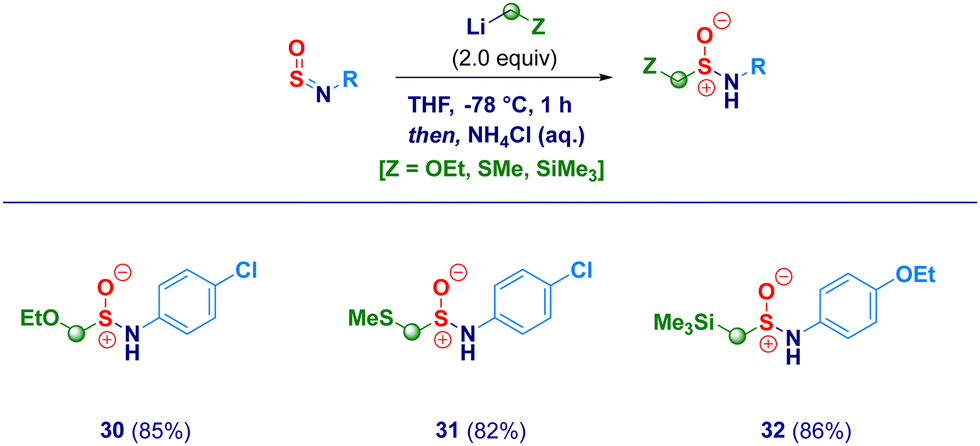 | ||
| Scheme 3 Homologation of N-sulfinylamines with non-halogenated agents. | ||
Finally, we briefly screened the synthetic behavior of the newly introduced α-halomethyl sulfinamide motif through transformations realized on both the methylene unit and on the aromatic ring (Scheme 4). The treatment of derivative 6 with a thiol (PhSH) under basic conditions afforded the expected nucleophilic substitution product 33 (path a), thus remarking the electrophilic nature of the methylene introduced via homologation. Under Pd-catalytic conditions – halogenated arenes [iodo (13) and bromo (12)] – smoothly coupled with an organolithium (Feringa – Fañanás-Mastral protocol,3834) or with an amine (morpholine, 35) in Buchwald-Hartwig mode (paths b and c).39 In summary, we have developed a straightforward synthesis of previously undisclosed α-substituted methylsulfinamides via a homologative tactic levered on the nucleophilic transfer of a formal –CH2X (or CHXY) reagent to the electrophilic sulfur atom of N-sulfinylamines. Upon tuning the nucleophiles genesis, a series of functionalized methyl fragments can be efficiently added, thus converting the starting materials into the homologues. Full chemocontrol is uniformly exhibited by the process, as showcased by challenging N-sulfinylamines embodying chemical moieties susceptible of nucleophilic attack such as ester, halogen, nitro, nitrile, olefin or alkynyl functionalities. This one-step, high yielding methodology is flexible for being applied to the synthesis of more elusive N-alkyl type analogues.
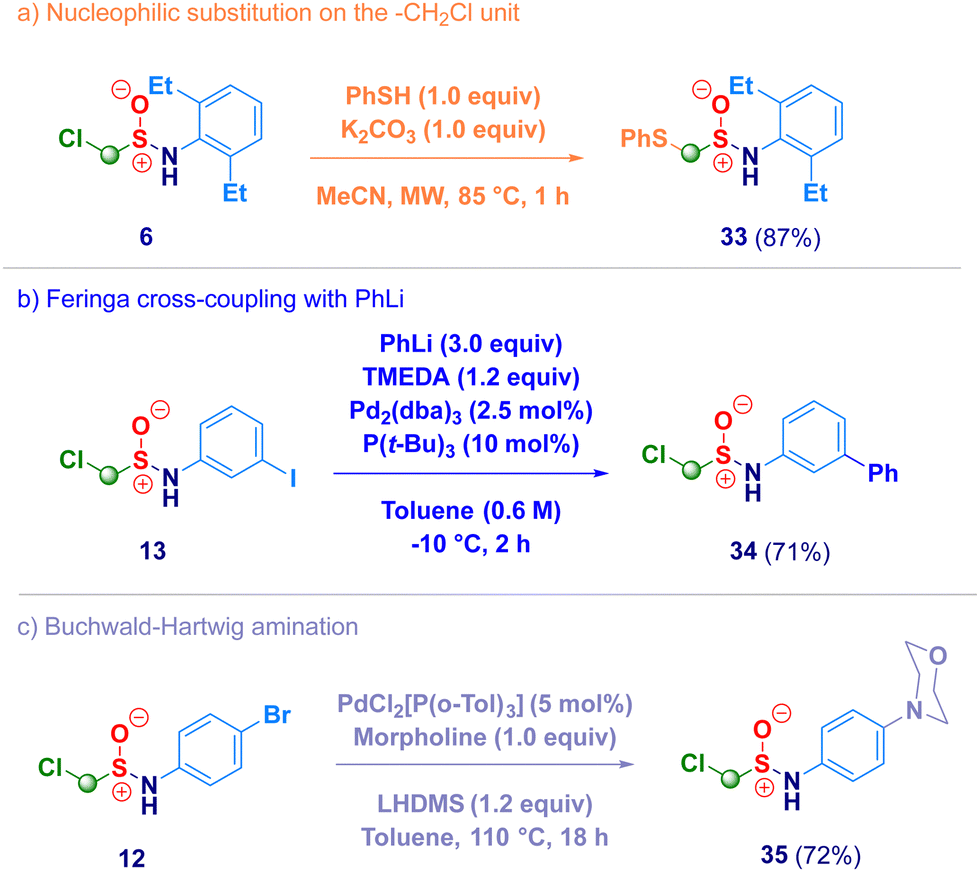 | ||
| Scheme 4 Derivatization of α-halomethylsulfinamides. | ||
M. M. and R. S. performed the experiments and contributed equally. D. C. assisted and completed the experimental work. A. R.-P. realized X-ray analysis. V. P. directed the project and wrote the manuscript.
We thank the University of Vienna, the University of Turin and All4Labels Group (Hamburg, Germany) for generous funding. The support from Project CH4.0 under MUR (Italian Ministry for the University) program “Dipartimenti di Eccellenza 2023–2027” (CUP: D13C22003520001) is acknowledged.
Conflicts of interest
No conflict to declare.Notes and references
- For a review, see: Q. Zhang, J. Xi, H. Ze and Z. Qingle, Synthesis, 2021, 2570 CAS.
- J. J. Petkowski, W. Bains and S. Seager, J. Nat. Prod., 2018, 81, 423 CrossRef CAS PubMed.
- (a) X. Zhang, F. Wang and C.-H. Tan, JACS Au, 2023, 3, 700 CrossRef CAS PubMed; (b) E. Wojaczyńska and J. Wojaczyński, Chem. Rev., 2020, 120, 4578 CrossRef PubMed.
- a) M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600 CrossRef CAS PubMed.
- Y. Aota, T. Kano and K. Maruoka, J. Am. Chem. Soc., 2019, 141, 19263 CrossRef CAS PubMed.
- (a) U. Lücking, Org. Chem. Front., 2019, 6, 1319 RSC; (b) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed; (c) P. K. Chinthakindi, T. Naicker, N. Thota, T. Govender, H. G. Kruger and P. I. Arvidsson, Angew. Chem., Int. Ed., 2017, 56, 4100 CrossRef CAS PubMed; (d) Z. P. Shultz, T. Scattolin, L. Wojtas and J. M. Lopchuk, Nat. Synth., 2022, 1, 170 CrossRef PubMed; (e) P. Mäder and L. Kattner, J. Med. Chem., 2020, 63, 14243 CrossRef PubMed; (f) M. Feng, B. Tang, H. S. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef CAS PubMed.
- S. Chatterjee, S. Makai and B. Morandi, Angew. Chem., Int. Ed., 2021, 60, 758 CrossRef CAS PubMed.
- C.-W. Feng, D.-Y. Wang, H.-L. Lu, Z.-W. Xi, Y.-M. Shen and J. Cao, Org. Lett., 2022, 24, 4485 CrossRef CAS PubMed.
- P. K. T. Lo, G. A. Oliver and M. C. Willis, J. Org. Chem., 2020, 85, 5753 CrossRef CAS PubMed.
- (a) H. Gilman and C. R. Kinney, J. Am. Chem. Soc., 1924, 46, 493 CrossRef CAS See also: ; (b) V. Pace, S. Monticelli, K. de la Vega-Hernandez and L. Castoldi, Org. Biomol. Chem., 2016, 14, 7848 RSC; (c) K. de la Vega-Hernández, R. Senatore, M. Miele, E. Urban, W. Holzer and V. Pace, Org. Biomol. Chem., 2019, 17, 1970 RSC.
- T. Q. Davies and M. C. Willis, Chem. – Eur. J., 2021, 27, 8918 CrossRef CAS PubMed.
- (a) H. Gilman and H. L. Morris, J. Am. Chem. Soc., 1926, 48, 2399 CrossRef CAS; (b) A. Sonn and E. Schmidt, Ber. Dtsch. Chem. Ges., 1924, 57, 1355 CrossRef; (c) D. Klamann, C. Sass and M. Zelenka, Chem. Ber., 1959, 92, 1910 CrossRef CAS.
- G. Kresze, A. Maschke, R. Albrecht, K. Bederke, H. P. Patzschke, H. Smalla and A. Trede, Angew. Chem., Int. Ed. Engl., 1962, 1, 89 CrossRef.
- (a) T. Q. Davies, A. Hall and M. C. Willis, Angew. Chem., Int. Ed., 2017, 56, 14937 CrossRef CAS PubMed; (b) Z.-X. Zhang, T. Q. Davies and M. C. Willis, J. Am. Chem. Soc., 2019, 141, 13022 CrossRef CAS PubMed; (c) L. Bayeh, P. Q. Le and U. K. Tambar, Nature, 2017, 547, 196 CrossRef CAS PubMed.
- (a) G. A. Oliver, M. N. Loch, A. U. Augustin, P. Steinbach, M. Sharique, U. K. Tambar, P. G. Jones, C. Bannwarth and D. B. Werz, Angew. Chem., Int. Ed., 2021, 60, 25825 CrossRef CAS PubMed; (b) S. M. Weinreb, Acc. Chem. Res., 1988, 21, 313 CrossRef CAS.
- T. Q. Davies, M. J. Tilby, J. Ren, N. A. Parker, D. Skolc, A. Hall, F. Duarte and M. C. Willis, J. Am. Chem. Soc., 2020, 142, 15445 CrossRef CAS PubMed.
- P. K. T. Lo and M. C. Willis, J. Am. Chem. Soc., 2021, 143, 15576 CrossRef CAS PubMed.
- M. Bremerich, C. M. Conrads, T. Langletz and C. Bolm, Angew. Chem., Int. Ed., 2019, 58, 19014 CrossRef CAS PubMed.
- L. Li, S.-q Zhang, Y. Chen, X. Cui, G. Zhao, Z. Tang and G.-x Li, ACS Catal., 2022, 12, 15334 CrossRef CAS.
- J. Börgel and T. Ritter, Chemistry, 2020, 6, 1877 CrossRef.
- L. Castoldi, S. Monticelli, R. Senatore, L. Ielo and V. Pace, Chem. Commun., 2018, 54, 6692 RSC.
- (a) J. G. Tillett, in Sulphinic Acids, Esters and Derivatives, ed. S. Patai, 1990, p. 603 Search PubMed; (b) K. Ruitenberg and P. Vermeer, J. Organomet. Chem., 1983, 256, 175 CrossRef CAS; (c) I. Sen, D. P. Kloer, R. G. Hall and S. Pal, Synthesis, 2013, 3018 CAS; (d) K. K. Andersen and J. B. Biasotti, J. Am. Chem. Soc., 1971, 93, 1178 CrossRef CAS; (e) W. T. Smith Jr., P. A. Thio and M. Grasley, J. Org. Chem., 1962, 27, 692 CrossRef; (f) J.-B. Baudin and S. A. Julia, Tetrahedron Lett., 1986, 27, 837 CrossRef CAS; (g) H. A. Selling and H. J. Mak, Synth. Commun., 1976, 6, 129 CrossRef CAS; (h) R. L. Pennington, X. Sha and S. B. King, Bioorg. Med. Chem. Lett., 2005, 15, 2331 CrossRef CAS PubMed.
- (a) V. Pace, W. Holzer and N. De Kimpe, Chem. Rec., 2016, 16, 2061 CrossRef CAS PubMed; (b) Q. Xie and G. Dong, J. Am. Chem. Soc., 2022, 144, 8498 CrossRef CAS PubMed.
- L. Ielo, M. Miele, V. Pillari, R. Senatore, S. Mirabile, R. Gitto, W. Holzer, A. R. Alcántara and V. Pace, Org. Biomol. Chem., 2021, 19, 2038 RSC.
- U. Azzena, M. Carraro, L. Pisano, S. Monticelli, R. Bartolotta and V. Pace, ChemSusChem, 2019, 12, 40 CrossRef CAS PubMed.
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369 CrossRef CAS PubMed.
- (a) V. Pace, L. Castoldi, S. Monticelli, M. Rui and S. Collina, Synlett, 2017, 879 CrossRef CAS; (b) P. Musci, M. Colella, A. Sivo, G. Romanazzi, R. Luisi and L. Degennaro, Org. Lett., 2020, 22, 3623 CrossRef CAS PubMed.
- L. Castoldi, W. Holzer, T. Langer and V. Pace, Chem. Commun., 2017, 53, 9498 RSC.
- R. Britton, V. Gouverneur, J.-H. Lin, M. Meanwell, C. Ni, G. Pupo, J.-C. Xiao and J. Hu, Nat. Rev. Methods Primers, 2021, 1, 47 CrossRef CAS.
- (a) Z. Ke, Y. Zhou, H. Gao, C. Zhao and D. L. Phillips, Chem. – Eur. J., 2007, 13, 6724 CrossRef CAS PubMed; (b) M. Colella, A. Tota, A. Großjohann, C. Carlucci, A. Aramini, N. S. Sheikh, L. Degennaro and R. Luisi, Chem. Commun., 2019, 55, 8430 RSC.
- Lithium Compounds in Organic Synthesis: From Fundamentals to Applications, ed., R. Luisi and V. Capriati, Wiley-VCH, Weinheim, 2014 Search PubMed.
- (a) V. Pace, W. Holzer, G. Verniest, A. R. Alcántara and N. De Kimpe, Adv. Synth. Catal., 2013, 355, 919 CrossRef CAS; (b) V. Pace, L. Castoldi and W. Holzer, J. Org. Chem., 2013, 78, 7764 CrossRef CAS PubMed; (c) M. Miele, A. Citarella, T. Langer, E. Urban, M. Zehl, W. Holzer, L. Ielo and V. Pace, Org. Lett., 2020, 22, 7629 CrossRef CAS PubMed; (d) M. Miele, R. D’Orsi, V. Sridharan, W. Holzer and V. Pace, Chem. Commun., 2019, 55, 12960 RSC.
- (a) R. Senatore, M. Malik, T. Langer, W. Holzer and V. Pace, Angew. Chem., Int. Ed., 2021, 60, 24854 CrossRef CAS PubMed; (b) V. Pace, A. Pelosi, D. Antermite, O. Rosati, M. Curini and W. Holzer, Chem. Commun., 2016, 52, 2639 RSC.
- (a) S. Touqeer, R. Senatore, M. Malik, E. Urban and V. Pace, Adv. Synth. Catal., 2020, 362, 5056 CrossRef CAS; (b) V. Pace, L. Castoldi, A. D. Mamuye and W. Holzer, Synthesis, 2014, 2897 CrossRef.
- V. Pace, I. Murgia, S. Westermayer, T. Langer and W. Holzer, Chem. Commun., 2016, 52, 7584 RSC.
- R. Senatore, L. Ielo, E. Urban, W. Holzer and V. Pace, Eur. J. Org. Chem., 2018, 2466 CrossRef CAS.
- F. F. Huerta, C. Gómez, A. Guijarro and M. Yus, Tetrahedron, 1995, 51, 3375 CrossRef CAS.
- M. Giannerini, M. Fañanás-Mastral and B. L. Feringa, Nat. Chem., 2013, 5, 667 CrossRef CAS PubMed.
- P. A. Forero-Cortés and A. M. Haydl, Org. Proc. Res. Dev., 2019, 23, 1478 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2191935. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc03326k |
| This journal is © The Royal Society of Chemistry 2023 |