
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deeper mechanistic insights into epitaxial nickelate electrocatalysts for the oxygen evolution reaction
Ellen M.
Kiens†
 a,
Min-Ju
Choi†
b,
Luhan
Wei
c,
Qiyang
Lu
*cd,
Le
Wang
*b and
Christoph
Baeumer
*ae
a,
Min-Ju
Choi†
b,
Luhan
Wei
c,
Qiyang
Lu
*cd,
Le
Wang
*b and
Christoph
Baeumer
*ae
aMESA+ Institute for Nanotechnology, University of Twente, Faculty of Science and Technology, P.O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: c.baeumer@utwente.nl
bPhysical and Computational Sciences Directorate, Pacific Northwest National Laboratory, Richland, WA 99354, USA. E-mail: le.wang@pnnl.gov
cSchool of Engineering, Westlake University, Hangzhou 310030, P. R. China. E-mail: luqiyang@westlake.edu.cn
dResearch Center for Industries of the Future, Westlake University, Hangzhou 310030, Zhejiang, P. R. China
ePeter Gruenberg Institute and JARA-FIT, Forschungszentrum Juelich GmbH, 52425 Juelich, Germany
First published on 2nd March 2023
Abstract
Mass production of green hydrogen via water electrolysis requires advancements in the performance of electrocatalysts, especially for the oxygen evolution reaction. In this feature article, we highlight how epitaxial nickelates act as model systems to identify atomic-level composition–structure–property–activity relationships, capture dynamic changes under operating conditions, and reveal reaction and failure mechanisms. These insights guide advanced electrocatalyst design with tailored functionality and superior performance. We conclude with an outlook for future developments via operando characterization and multilayer electrocatalyst design.
 Ellen M. Kiens | Ellen Kiens obtained a MSc degree in Applied Physics and Chemical Engineering at the University of Twente. She is currently pursuing a PhD at the MESA+ Institute of Nanotechnology. Her research is focused on the manipulation of electronic and structural properties of ultrathin layers, using an epitaxial multilayered approach, and thereby finding a relationship between these properties and electrochemical activity and stability. Her interests lie within the characterization of the (electronic) structure of the pre-catalyst as well as in operando conditions to develop further understanding of the formation and stability of active sites in perovskite catalysts. |
 Min-Ju Choi | Dr Min-Ju Choi is a postdoctoral research associate at Pacific Northwest National Laboratory (PNNL). She received her PhD degree from the Department of Materials Science and Engineering at Seoul National University in 2020. Her research focuses on the design and synthesis of epitaxial thin films to deepen the understanding of energy conversion and storage devices. |
 Luhan Wei | Luhan Wei received his BS degree in Functional Materials from Northeastern University in China in 2021. Currently, he is pursuing PhD degree under the supervision of Prof. Qiyang Lu at the School of Engineering of Westlake University. His research interests primarily lie in using spectro-electrochemistry, physical chemistry modeling and in situ/operando characterizations to examine the kinetics of cation (de)-intercalation behavior in electrode materials. He is also interested in the connection between material properties, such as defects and surface reconstructions, and the electrocatalytic performance of oxygen evolution reaction (OER) in thin film nickelate systems. |
 Qiyang Lu | Dr Qiyang Lu is currently an assistant professor in School of Engineering at Westlake University. He received his bachelor's degree from Tsinghua University and his PhD degree from Massachusetts Institute of Technology, both in Materials Science and Engineering. He was the recipient of several awards, including the Best PhD Thesis Award from MIT and Ross Coffin Purdy award from American Ceramic Society. His current research interest is in the field of solid state ionics, mainly focused on designing mixed electronic and ionic conducting oxide thin films for reversible solid state electrochemical cells, electrocatalysts and ionotronic devices. |
 Le Wang | Dr Le Wang is a materials scientist in the team of Atomically Precise Materials at PNNL. He received his PhD degree from the Institute of Physics Chinese Academy of Sciences in 2014. After that, he worked at Nanyang Technological University, Singapore as a research fellow. He joined PNNL as a Postdoc in 2017 and became a staff scientist in March 2020. His research interest focuses on the synthesis of epitaxial oxide thin films and heterostructures and exploring their potential in electronics, ion diffusion/transport, and electrocatalysis. |
 Christoph Baeumer | Dr Christoph Baeumer is an assistant professor at the University of Twente and guest scientist at Forschungszentrum Jülich. He obtained his MSc in Materials Science from the University of Illinois, his PhD in Physics from RWTH Aachen University and Forschungszentrum Jülich and he was a Marie Skłodowska Curie Fellow at Stanford University (2018–2020). His Electrochemical Thin Films and Interfaces group focuses on operando characterization and fundamental structure-function relations in model electrocatalysts. He received several awards and personal grants, including the Helmholtz Doctoral Prize (2017) and the ERC Starting Grant (2022). |
1. Introduction
Mass production of green hydrogen through water electrolysis and electrochemical CO2 reduction are widely anticipated pathways to reach carbon neutrality. Yet both hydrogen generation and CO2 reduction require an oxidation reaction at the anode, which is typically the oxygen evolution reaction (OER). The OER is kinetically complex and often requires a high overpotential, necessitating highly-active electrocatalysts, which must be low cost and also stable under reaction conditions.1–5 To date, the understanding of the complicated reaction and failure mechanisms remains incomplete, which impedes the development of electrocatalysts with tailored functionality and superior performance.An attractive material class for OER electrocatalysts contains the perovskite oxides of the general formula ABO3, with rare earth or alkaline earth elements as A-site cations and transition metals (TMs) as B-site cations.6 Among perovskite oxides, nickelates (RNiO3, where R represents a rare-earth lanthanide element) have garnered special attention, because of comparably high activity7 and suitable earth-abundance. Their excellent performance can be attributed to the following unique characteristics: (1) relatively weaker bond strength between Ni and OH* compared to other first-row TMs,8 (2) eg electron filling close to unity,9 (3) small or negative charge transfer energy,10 and (4) strong hybridization between Ni 3d and O 2p orbitals.11 These electronic structure descriptors, to which we will return below, identify perovskite nickelates as one of the most promising class of OER electrocatalysts.
Recent experimental observations have revealed that perovskite oxide electrocatalysts – including nickelates – show a range of dynamic changes rather than staying static during OER.12–15 The complicated modifications observed for both surfaces and bulk of nickelate electrocatalysts include the exchange of lattice oxygen and oxygen species between the electrocatalyst and the electrolyte (so-called lattice oxygen mechanisms), the formation and annihilation of ionic defects, which can also be regarded as ion (de-)intercalation processes similar to the electrode materials for aqueous batteries, as well as dynamic surface reconstructions. These dynamic changes during the OER can greatly impact the electrocatalytic activity and stability of nickelate electrocatalysts.
This situation is further complicated by the observation that enhanced catalyst activity seemingly comes at the cost of a decreased stability and lifetime of the catalyst,15–20 which implies that good catalyst materials typically degrade very quickly, resulting in a rapid loss of the desired catalytic activity. This inverse correlation between stability and activity of catalyst materials involved in electrochemical processes presents a major obstacle for applications.
Researchers therefore face the challenge of identifying specific factors in the composition, crystal structure, and electronic structure of (nickelate) electrocatalysts that lead to a refined explanation of these dynamic processes, which would enable the design of catalysts with optimized activity and stability. Conventionally, electrocatalysts are fabricated as (nano) particles or electrodeposited thin films. These typically have heterogeneous surface composition, ill-defined crystallographic surface phase and exposed facets, which makes extracting intrinsic activity and stability trends difficult. In contrast, epitaxial thin films with well-defined microstructures and surfaces/interfaces offer a valuable platform for elucidating the origins of these dynamic processes.6,21,22
In this feature article, we highlight the recent progress in the field on the mechanistic understanding obtained by using epitaxial nickelate OER electrocatalysts. We start by summarizing the current state-of-the-art regarding electrocatalyst activity trends, the role of lattice oxygen participation, and the formation of defects and new “active” phases. We introduce epitaxial thin films as a valuable and informative model platform to identify descriptive structure–property–functionality relationships. We further discuss the remaining unresolved questions and challenges confronting the utilization of the epitaxial nickelate system. We end this review by projecting future directions that enable deeper insights into the dynamic changes of OER electrocatalysts through operando characterization, and ultimate tuning of material properties and functionality using multilayer approaches.
2. The quest for mechanistic understanding of dynamic changes during OER
The classical empirical description of electrocatalytic reactions including the OER assumes a reaction at a solid electrode/liquid electrolyte interface with static composition and structure of the solid. Only little attention has been paid to the “internal life” of the electrocatalysts, i.e., the composition and electronic structure of the surface and subsurface, which may change as a function of potential and time, especially for TM oxide (TMO) based electrocatalysts. There has been mounting evidence that shows the surface and the “bulk” (at least several nanometers from the interface into the electrocatalysts) can participate in the electrochemical reaction, and often determines the kinetics and performance of these electrocatalysts.15,23–27 Therefore, a holistic picture considering the solid/liquid interfaces, as well as the constituent ions and ionic defects of TMOs, becomes important for understanding the reaction mechanism and guiding the predictive design of nickelate OER electrocatalysts. Due to the participation of electrocatalyst “bulk” during the electrochemical reactions, a wide range of dynamic mechanisms and phenomena have been observed. These dynamic behaviors of nickelate electrocatalysts indicate that at the OER condition (e.g., E ≥ 1.55 V vs. RHE (Reversible Hydrogen Electrode)), both the surface and the near-surface region of these electrocatalysts can be very different from the “pre-catalyst” (the parent perovskite oxide before any electrochemical treatment) in both chemical composition and crystal structure. During extended operation, the compositional or structural dynamic changes may further evolve. These dynamic changes, potentially driven by the reorganization of cationic and anionic defects, will drastically alter the electronic structure and active sites of the electrocatalysts. In this section, we will first briefly summarize bulk electronic descriptors of the OER activity that have been shown to be relevant for perovskite oxides. Next, we will review three important aspects regarding the dynamic changes of nickelate electrocatalysts, which are (1) the role of lattice oxygen in OER mechanisms (so-called lattice oxygen mechanism); (2) formation of ionic point defects (oxygen vacancies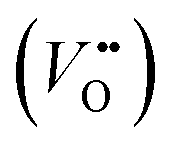 /protons (H+)); (3) formation of secondary phases at the near-surface region. Schematics of these three dynamic behaviors are summarized in Fig. 1. To conclude the section, we describe why the use of epitaxial thin films as model systems can help answer unresolved questions in the field.
/protons (H+)); (3) formation of secondary phases at the near-surface region. Schematics of these three dynamic behaviors are summarized in Fig. 1. To conclude the section, we describe why the use of epitaxial thin films as model systems can help answer unresolved questions in the field.
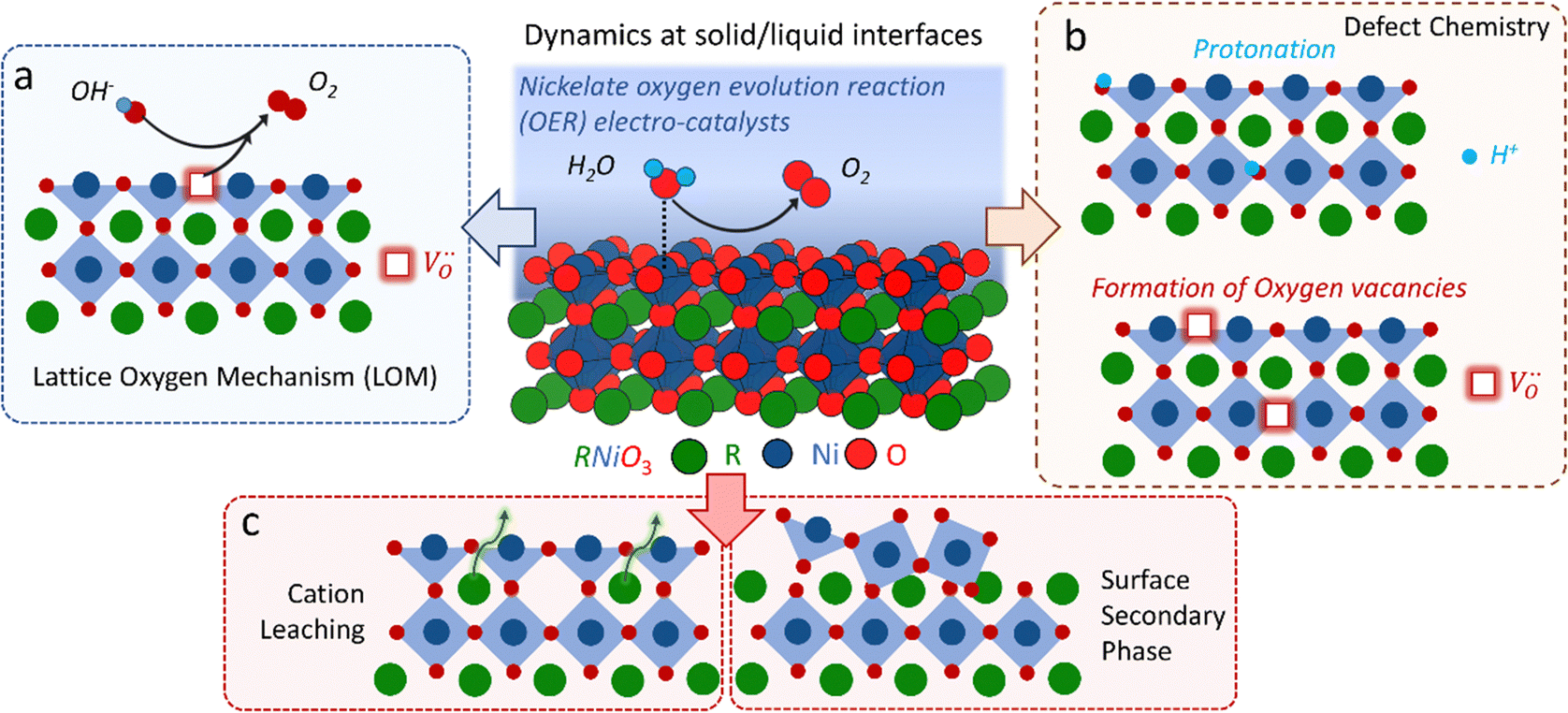 | ||
| Fig. 1 Dynamic changes of nickelate OER electrocatalysts with the perovskite crystal structure. (a) Lattice oxygen mechanism; (b) formation of ionic point defects; (c) surface compositional change and formation of secondary phases. | ||
2.1 Bulk descriptors of the OER
As the OER activity and reaction kinetics depend on the electronic structure of the perovskite oxide electrocatalysts, several approximate descriptors relating bulk electronic structure and catalytic activity have been identified, such as the orbital filling of the frontier orbitals and relative orbital positions.9,10,28,29 For example, the bonding strength of intermediates is determined by the filling of the antibonding orbital formed between the oxygen species of the adsorbates and TM.29 The formation and filling of this antibonding orbital are highly dependent on the eg orbital filling of the TM cations. Hence, the activity of perovskite oxides towards the OER was proposed to follow a volcano-like dependence on the eg orbital filling, with the optimum close to unity, following the Sabatier principle.9,29 Not only the occupancy of the eg orbitals influences the binding of intermediates, their chemisorption is also determined by the geometry of the occupied orbitals. The d3z2−r2 orbital points towards the adsorbates leading to more spatial overlap with the adsorbate O 2p orbitals compared to the dx2−y2 orbitals.30 Hence, a preferred occupancy of these orbitals, e.g. through orbital polarization lifting the degeneracy of the eg energy states, will strongly influence the chemisorption and the OER activity, as will be discussed later. In addition to the occupancy and degeneracy of the eg bands, their relative positions also influence the chemisorption and OER activity. For example, suppressing Jahn–Teller distortions in Mn-containing catalysts with spinel structure lifts up the occupied degenerated eg orbitals to higher energy, thereby leading to stronger overlap with the O 2p adsorbate orbitals when compared to Jahn–Teller distorted catalysts, which enhances OER activity.31,32 The relative positions of the TM 3d and O 2p have been proposed as a descriptor for OER activities as well. TMOs with perovskite structure typically are materials with mixed ionic and covalent character with a large degree of TM–oxygen orbital mixing (often referred to as TM–oxygen hybridization). The charge transfer energy, defined as the energy difference between the O 2p non-bonding and the TM 3d orbitals, is a measure of the covalency and believed to correlate with the OER activity in a linear manner. Low charge transfer energy, corresponding to high covalency, favors the OER activity.10,28 Other descriptors for the OER activity are closely related to defects in the catalyst bulk, such as distortion and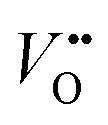 .12,33
.12,33
2.2 The role of lattice oxygen in OER mechanisms
Conventionally, the OER in alkaline electrolyte is believed to occur by the so-called adsorbate evolution mechanism (AEM), which means that the reaction follows a consecutive reaction pathway via OH* → O* → OOH* → O2(g) (* denotes adsorbed species) with TM cations as the main adsorption and reaction sites. In the AEM picture, the lattice oxygen anions of the perovskite oxides are regarded as inert building blocks that do not directly participate in the reaction. In 2016, several groups introduced the lattice oxygen mechanism (LOM) based on theoretical and experimental work.23,34,35 It differs from the AEM by the consideration that the O2 molecule reaction product can form by taking one oxygen from the lattice and leaving an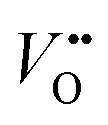 as shown in Fig. 1a. Interestingly, the authors used LaNiO3 nickelate as a model system for their first computational study.35 They showed that for LaNiO3, the reaction barrier (theoretical overpotential) was much lower for the LOM compared to the AEM. In a later contribution, the same group showed that taking the LOM into account will drastically alter the volcano plot of activity vs. B-site 3d TM in LaBO3 (B = V, Cr, Mn, Co, Ni and Cu).36 Considering the LOM puts LaNiO3 at the top of the volcano plot of ternary perovskite oxides. Although the LOM was explained by using LaNiO3 as a computational model system, the first experimental demonstration of this new mechanism was on perovskite cobaltates (e.g., La1−xSrxCoO3−δ).23,34 Grimaud et al.23 directly showed the evolution of O2 species involving lattice oxygen by using in situ18O isotope labelling mass spectrometry, and Mefford et al.34 showed that the activity scales with the density of
as shown in Fig. 1a. Interestingly, the authors used LaNiO3 nickelate as a model system for their first computational study.35 They showed that for LaNiO3, the reaction barrier (theoretical overpotential) was much lower for the LOM compared to the AEM. In a later contribution, the same group showed that taking the LOM into account will drastically alter the volcano plot of activity vs. B-site 3d TM in LaBO3 (B = V, Cr, Mn, Co, Ni and Cu).36 Considering the LOM puts LaNiO3 at the top of the volcano plot of ternary perovskite oxides. Although the LOM was explained by using LaNiO3 as a computational model system, the first experimental demonstration of this new mechanism was on perovskite cobaltates (e.g., La1−xSrxCoO3−δ).23,34 Grimaud et al.23 directly showed the evolution of O2 species involving lattice oxygen by using in situ18O isotope labelling mass spectrometry, and Mefford et al.34 showed that the activity scales with the density of 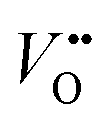 . Presumably, following the computational volcano plot we should see a more important role of lattice oxygen for nickelates compared with cobaltates. However, direct probing on the fraction of AEM/LOM in the total reaction and a comparison between perovskites with different B-site cations is still lacking. Furthermore, for the LOM, it remains yet to be clarified whether only the surface lattice oxygen anions can evolve to oxygen gas molecules, or whether there is a “critical thickness” where all oxygen anions are active within a certain layer. Another important aspect of the LOM is the formation of
. Presumably, following the computational volcano plot we should see a more important role of lattice oxygen for nickelates compared with cobaltates. However, direct probing on the fraction of AEM/LOM in the total reaction and a comparison between perovskites with different B-site cations is still lacking. Furthermore, for the LOM, it remains yet to be clarified whether only the surface lattice oxygen anions can evolve to oxygen gas molecules, or whether there is a “critical thickness” where all oxygen anions are active within a certain layer. Another important aspect of the LOM is the formation of 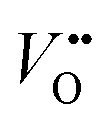 , which is one of the intermediate steps of the OER. Therefore, the ease of
, which is one of the intermediate steps of the OER. Therefore, the ease of 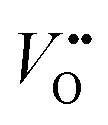 formation is of great importance for the activation of the LOM. This highlights the role of ionic point defects in the reaction pathways and activity of nickelates. However, the understanding of the defect chemistry of nickelates under the OER condition, e.g., a quantitative correlation between the
formation is of great importance for the activation of the LOM. This highlights the role of ionic point defects in the reaction pathways and activity of nickelates. However, the understanding of the defect chemistry of nickelates under the OER condition, e.g., a quantitative correlation between the 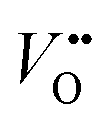 concentration (ideally with depth resolution) and the applied potential, is still incomplete.
concentration (ideally with depth resolution) and the applied potential, is still incomplete.
2.3 Formation of ionic point defects
Experimental evidence shows that nickelates always contain non-negligible amounts of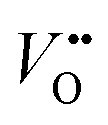 , and that oxygen exchange can occur under sufficient electrochemical driving forces.37 Compared to
, and that oxygen exchange can occur under sufficient electrochemical driving forces.37 Compared to 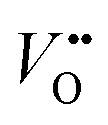 , H+ incorporation (protonation) can occur more readily in nickelates in aqueous solution since the hydration process is spontaneous and can compensate the total energy cost for protonation reaction. Recent work by Tang et al.38 shows experimental evidence that
, H+ incorporation (protonation) can occur more readily in nickelates in aqueous solution since the hydration process is spontaneous and can compensate the total energy cost for protonation reaction. Recent work by Tang et al.38 shows experimental evidence that  and H+ can co-exist in a perovskite thin film electrocatalyst in alkaline electrolyte. Furthermore, extensive studies show that protonation can be triggered either chemically or electrochemically in nickelate thin films, and that the incorporation of H+ can completely change the electrical conductivity (from electronic/metallic to ionic/insulating) and lattice constant of nickelates.38 Therefore, the role of point defects as schematically shown in Fig. 1b should be considered when examining the electrocatalytic activity. However, there are several open questions on the room-temperature defect chemical equilibrium and dynamics. For instance, the equivalent of a “Brouwer diagram”, i.e., a quantitative relationship between defect concentration and applied electrochemical driving force (or the activity of oxygen) is still lacking for RNiO3 in contact with a liquid electrolyte system. This gap of understanding greatly impedes the inclusion of a “defect concentration” term in the modeling of electrochemical reaction kinetics, which has already been considered and widely adopted for treating high-temperature electrochemical reaction kinetics. Furthermore, a detailed study of the defect concentration profile from the solid/liquid interface to the bulk is still lacking. A quantitative understanding of the depth of the near-surface region that can form either
and H+ can co-exist in a perovskite thin film electrocatalyst in alkaline electrolyte. Furthermore, extensive studies show that protonation can be triggered either chemically or electrochemically in nickelate thin films, and that the incorporation of H+ can completely change the electrical conductivity (from electronic/metallic to ionic/insulating) and lattice constant of nickelates.38 Therefore, the role of point defects as schematically shown in Fig. 1b should be considered when examining the electrocatalytic activity. However, there are several open questions on the room-temperature defect chemical equilibrium and dynamics. For instance, the equivalent of a “Brouwer diagram”, i.e., a quantitative relationship between defect concentration and applied electrochemical driving force (or the activity of oxygen) is still lacking for RNiO3 in contact with a liquid electrolyte system. This gap of understanding greatly impedes the inclusion of a “defect concentration” term in the modeling of electrochemical reaction kinetics, which has already been considered and widely adopted for treating high-temperature electrochemical reaction kinetics. Furthermore, a detailed study of the defect concentration profile from the solid/liquid interface to the bulk is still lacking. A quantitative understanding of the depth of the near-surface region that can form either 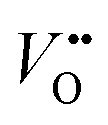 or H+ (or both), and its dependence on the electrochemical driving force and chemical environment, will be important for implementing “defect engineering” in nickelate electrocatalysts.
or H+ (or both), and its dependence on the electrochemical driving force and chemical environment, will be important for implementing “defect engineering” in nickelate electrocatalysts.
2.4 Formation of secondary phases at the near-surface region
It is well accepted that the near-surface region of TMO-based electrocatalysts can undergo substantial changes in chemistry and phase under sufficiently large electrochemical driving forces. The detailed mechanism or the degree of this dynamic change has been shown to be dependent on the composition and electronic structure of the “pre-catalyst”, as indicated in Fig. 1c. For instance, it has been reported that the cation leaching process, to which we will return later, can be directly correlated with the electronic structure descriptors.39 Effectively, for most TMO-based electrocatalysts, the surface becomes a secondary phase under OER conditions, which must be treated differently from the bulk. For nickelates, the surface secondary phase formation might be very subtle. The identification of the dynamic changes thus requires characterizations with high spatial and chemical resolution, which has been demonstrated by our own work (Baeumer et al.14) and will be discussed in detail later in this review. Although the surface morphology change might be subtle, the altered properties, especially the electronic structure, of the surface secondary phase will directly determine the OER overpotential. Nevertheless, the open question here is how to predict the formation of secondary phases based on the key properties (or descriptors) of the “pre-catalyst”, which might include both the bulk properties (electronic structure, chemical composition, etc.) and the surface properties (surface termination, exposed crystal facet, etc.).2.5 The need for epitaxial thin films as model system for studying dynamic changes
In order to resolve the dynamic changes of nickelate electrocatalysts during the OER, we propose that epitaxial thin films with well-controlled geometry, composition, surface termination and strain state are a valuable platform. Such films can be constructed with up to atomic precision using deposition techniques such as pulsed laser deposition (PLD) and oxide molecular beam epitaxy (MBE) with in situ growth monitoring as described in more detail elsewhere.22,40–43 The versatility and control provided by these techniques allows for fabrication of epitaxial thin films as a clean model system without the use of carbon additives or binders, and enables fabrication of materials distinguished by the absence of grain boundaries and complex morphologies. Moreover, control of surface termination and exposed crystallographic facet allows for direct comparison to density functional theory (DFT) calculations. The full control of nickelate samples from the bulk to the surface is of vital importance to develop understanding of intricate connections between multiple phenomena. Furthermore, extracting relevant information from advanced characterization tools can be easier on model epitaxial thin films compared to powder samples, as discussed in Section 4.1. The advantages of using nickelate epitaxial thin films will be showcased by focusing on our own work as examples in this review.3. Epitaxial thin films as a well-controlled and informative platform for elucidating structure–property–function relationships and factors governing the dynamic changes during OER
Below, we discuss recent progress on structure–property–function relationships in epitaxial perovskite nickelate electrocatalysts for the OER. First, we discuss how epitaxial strain can be used to fine-tune crystal field splitting and electronic–structure OER descriptors like the covalency. Then, we turn to modifying the surface composition selectively with atomic layer control to assess the influence of the surface termination for the transformation toward an OER-active surface phase. We finish the chapter with a discussion of compositional variation on the perovskite A- and B-site, which help optimize the electrocatalytic performance and reveal fundamental composition-activity trends.3.1 Epitaxial strain to fine-tune electronic structure and orbital filling
Strain can be introduced by depositing pseudomorphic epitaxial films on single-crystal substrates with different lattice constants. The effect of strain on the OER activity of NdNiO3 was studied in our previous work (Wang et al.44). We used PLD to deposit 5 nm thick NdNiO3 thin films on four single-crystal substrates: SrLaAlO4, LaAlO3, NdGaO3, and SrTiO3. For these substrates, the in-plane misfit strain of NdNiO3 films varies from −1.39% (compressive) to +2.49% (tensile) depending on the substrate, as deduced from the pseudocubic lattice constants of bulk NdNiO3 and the four substrates (Fig. 2a). A typical reciprocal space map shown in the inset of Fig. 2c reveals a coherent growth of NdNiO3 films on these substrates. Fig. 2b shows the X-ray diffraction (XRD) θ−2θ scans around (002) peaks of NdNiO3 films and the respective substrates. The peak shift as a function of substrate lattice parameter indicates that the substrate-induced in-plane misfit strain is accommodated by the elastic deformation (out-of-plane expansion or contraction) of the NdNiO3 films. Fig. 2c summarizes the experimentally obtained and calculated out-of-plane lattice constants of NdNiO3 films as a function of strain. It can be seen that the calculated out-of-plane lattice constant of NdNiO3 matches well with the experimental value for the film grown on LaAlO3, i.e. the substrate inducing the minimum in-plane strain. This indicates a very low concentration of point defects such as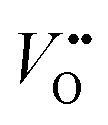 exist in NdNiO3 on LaAlO3. On the other hand, the experimental out-of-plane lattice constants of NdNiO3 films grown on the remaining substrates are larger than the calculated values which suggests the formation of point defects due to the larger lattice mismatch. Besides showing the largest difference between the experimental and calculated out-of-plane lattice constants, NdNiO3 on SrTiO3 exhibits the highest resistivity at room temperature and the lowest intensity of the O K pre-edge observed in X-ray absorption spectroscopy, as shown in Fig. 2d. These results suggest the highest
exist in NdNiO3 on LaAlO3. On the other hand, the experimental out-of-plane lattice constants of NdNiO3 films grown on the remaining substrates are larger than the calculated values which suggests the formation of point defects due to the larger lattice mismatch. Besides showing the largest difference between the experimental and calculated out-of-plane lattice constants, NdNiO3 on SrTiO3 exhibits the highest resistivity at room temperature and the lowest intensity of the O K pre-edge observed in X-ray absorption spectroscopy, as shown in Fig. 2d. These results suggest the highest 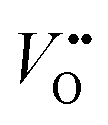 concentration is present in NdNiO3 on SrTiO3 (Fig. 2d). The orbital occupancy of strained NdNiO3 films was probed by X-ray linear dichroism (XLD = Eab − Ec), a technique that uses differential absorption of horizontally and vertically linear polarized light to examine electronic properties. Fig. 2e shows the XLD results of NdNiO3 films as a function of strain. For the NdNiO3 films under compressive strain, a positive XLD value was obtained, indicating preferential d3z2−r2 orbital occupancy. The negative integrated XLD intensity for NdNiO3 films under tensile strain indicates the preferred occupancy of the dx2−y2 orbital, resulting in stronger Ni–O chemisorption and lower OER activity (Fig. 2f). This is the reason why the NdNiO3 films under compressive strain show higher OER activity than the NdNiO3 films under tensile strain. Bak et al.33 probed the effect of octahedral distortions on the OER activity of perovskite nickelates, which also depend on the strain. They found that strong distortion of oxygen octahedra results in a considerable increase in electronic states near the Fermi level (EF), making the charge transfer between TM and oxygen much easier, thereby enhancing OER activity.33 This is different for Mn-containing catalysts with spinel structure, in which the OER activity increases with the suppression of the Jahn–Teller distortion.31,32 On the other hand, when a tensile strain large enough to generate
concentration is present in NdNiO3 on SrTiO3 (Fig. 2d). The orbital occupancy of strained NdNiO3 films was probed by X-ray linear dichroism (XLD = Eab − Ec), a technique that uses differential absorption of horizontally and vertically linear polarized light to examine electronic properties. Fig. 2e shows the XLD results of NdNiO3 films as a function of strain. For the NdNiO3 films under compressive strain, a positive XLD value was obtained, indicating preferential d3z2−r2 orbital occupancy. The negative integrated XLD intensity for NdNiO3 films under tensile strain indicates the preferred occupancy of the dx2−y2 orbital, resulting in stronger Ni–O chemisorption and lower OER activity (Fig. 2f). This is the reason why the NdNiO3 films under compressive strain show higher OER activity than the NdNiO3 films under tensile strain. Bak et al.33 probed the effect of octahedral distortions on the OER activity of perovskite nickelates, which also depend on the strain. They found that strong distortion of oxygen octahedra results in a considerable increase in electronic states near the Fermi level (EF), making the charge transfer between TM and oxygen much easier, thereby enhancing OER activity.33 This is different for Mn-containing catalysts with spinel structure, in which the OER activity increases with the suppression of the Jahn–Teller distortion.31,32 On the other hand, when a tensile strain large enough to generate 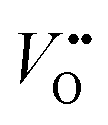 is applied, the Ni valence can be reduced from Ni3+ to Ni2+. This leads to eg filling slightly larger than one, which can contribute to the enhanced OER activity observed in NdNiO3 on SrTiO3. Similar experiments were also performed on other TMOs, demonstrating that strain-induced structural distortion and oxygen deficiencies modify the OER activity of epitaxial TMO films.42,45
is applied, the Ni valence can be reduced from Ni3+ to Ni2+. This leads to eg filling slightly larger than one, which can contribute to the enhanced OER activity observed in NdNiO3 on SrTiO3. Similar experiments were also performed on other TMOs, demonstrating that strain-induced structural distortion and oxygen deficiencies modify the OER activity of epitaxial TMO films.42,45
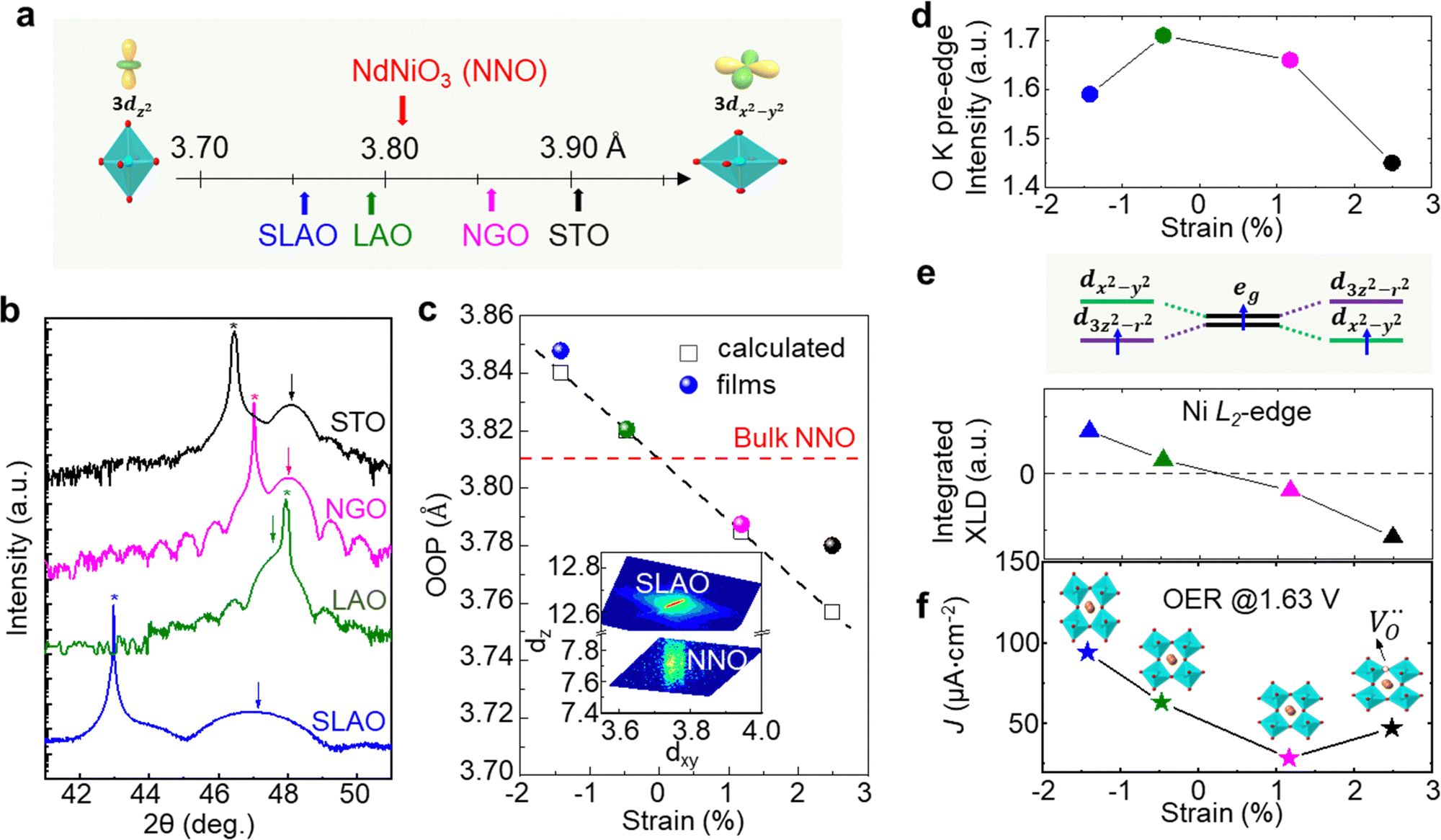 | ||
| Fig. 2 (a) Pseudo-cubic lattice parameters for bulk NdNiO3 and single crystalline substrates, including SrLaAlO4 (SLAO), LaAlO3 (LAO), NdGaO3 (NGO), and SrTiO3 (STO). The inset shows a schematic diagram of octahedral distortion induced by bi-axial compressive and tensile strain. (b) XRD θ−2θ scans around the (002) diffraction peak of 5 nm NNO films deposited on different substrates. Film and substrate peaks are noted by arrows and stars, respectively. (c) Experimentally obtained out-of-plane lattice parameter values (colored circles) of NdNiO3 films as a function of strain. Strained out-of-plane lattice parameters of NdNiO3 films estimated from the Young's modulus are presented by black open squares. The inset displays a reciprocal space map of NdNiO3 on SLAO. (d) O K pre-edge intensity of NdNiO3 films as a function of strain. (e) Schematic electronic configuration of eg orbital and integrated XLD as a function of strain. (f) OER current density at 1.63 V vs. RHE of NdNiO3 films under different biaxial strains. The inset in (f) shows schematic illustrations of octahedral distortions induced by strain. Reprinted (adapted) with permission from ref. 41. Copyright 2019, American Chemical Society. | ||
3.2 Tuning surface termination towards active surface phase formation
As discussed in Part 2, nickelate OER electrocatalysts often go through dynamic changes of the structure and chemical composition, especially at the electrochemical (solid/liquid) interface and the near-surface region of the oxide. While conventional particulate electrocatalysts (even nanoparticles) can be difficult to control precisely, our recent work (Baeumer et al.14) shows that epitaxial nickelate thin films, with the capability of controlling the surface termination and composition with atomic precision, can be a powerful platform to obtain detailed understanding on the dynamic changes during the OER. We employed standing-wave X-ray photoemission spectroscopy (SW-XPS), which is an emerging technique capable of depth-profiling the chemical environment in the near-surface region with 1–2 Å depth resolution, to reveal details of the surface chemical composition of LaNiO3. Epitaxial layers are an ideal system for this spectroscopic technique because of the opportunity to create smooth films supported superlattices functioning as X-ray mirrors (Fig. 3a). SW-XPS analysis of LaNiO3 prepared in different growth conditions revealed predominant NiO2 and LaO termination in Fig. 3b and c, respectively. Electrochemical characterization of the same samples revealed that NiO2-terminated LaNiO3 thin films show much higher OER activity compared to LaNiO3 samples with LaO-termination (Fig. 3e). Importantly, the surface morphology of samples before and after determining the OER activity was very similar, both showing atomically smooth terraces (Fig. 3d). Furthermore, redox peaks in cyclic voltammetry tests were only observed in the NiO2-terminated LaNiO3. Their peak area (which corresponds to the charge transferred across the solid/liquid interface) increases with Ni coverage of the first surface layer of LaNiO3. Combining experimental evidence from SW-XPS and electrochemical measurement showed that the different activity can be directly related to the different composition of the surface termination layer. With the insights from both experimental evidence and computational support, the dynamic changes of LaNiO3 surfaces were clarified. The difference in surface composition evolves to different surface secondary phases, i.e., NiOxHy (oxy-hydroxide like) and La(OH)3 at sufficiently high potential, with the former much more active than the latter. Therefore, this work proves that a very subtle difference in chemical composition (only the first atomic surface layer) could lead to a very large difference in activity, which can be more readily captured by using well-controlled epitaxial thin films as model systems.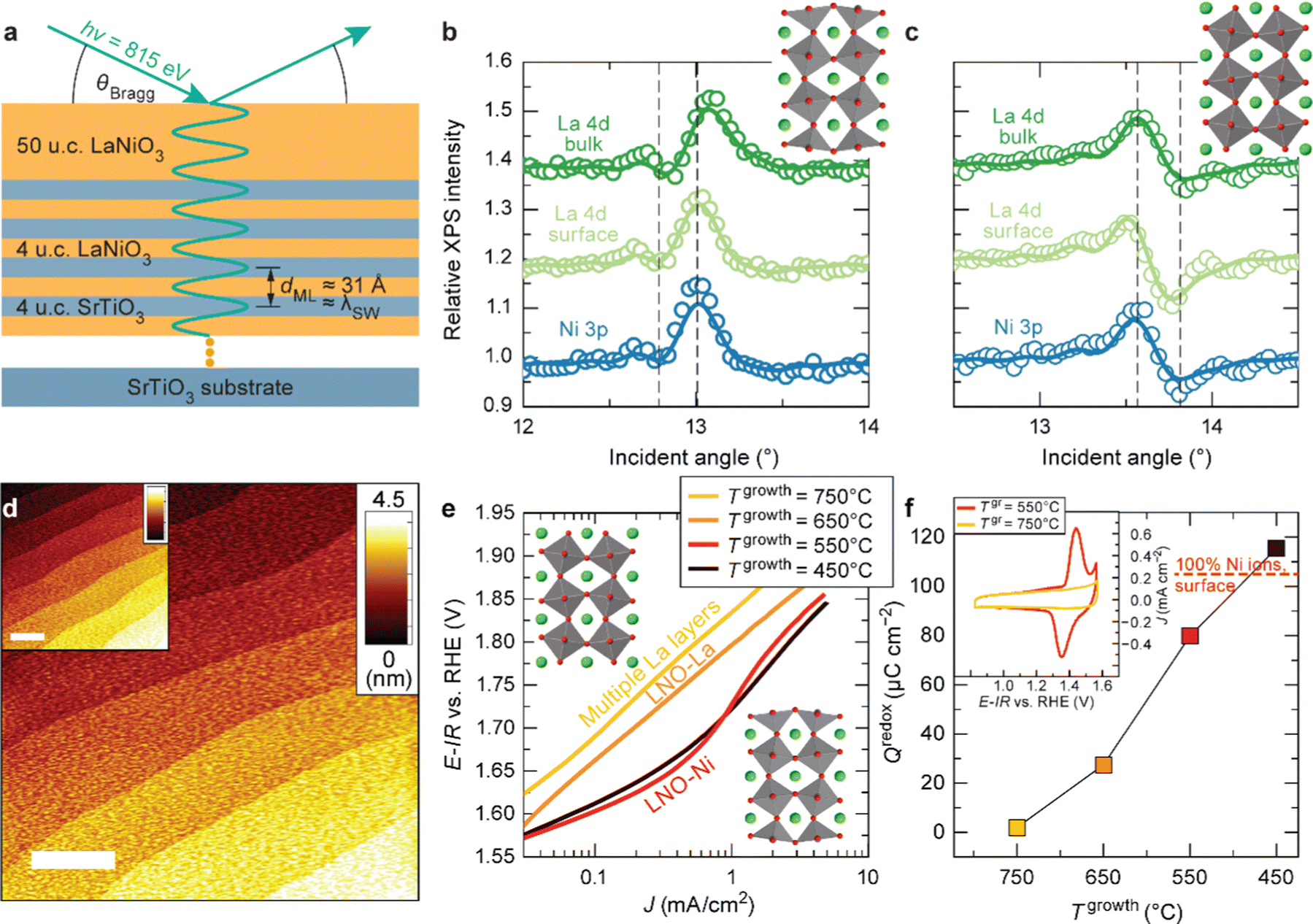 | ||
| Fig. 3 (a) Schematic diagram of LaNiO3 thin films on a superlattice of LaNiO3 (4 unit cells (u.c.)) and SrTiO3 (4 u.c.). SW-XPS rocking curves of La 4d and Ni 3p for LaNiO3 top layers grown at (b) 450 and (c) 650 °C. Atomic force microscopy images for (d) as-deposited LaNiO3 film (20 nm) grown at 550 °C and (inset) after cyclic voltammetry (CV). (e) Tafel-like plot for LaNiO3 films with different surface termination. (f) Redox charge for conversion from Ni2+ to Ni3+ during CV. (inset) CV curves in the Ni2+–Ni3+ redox area for LaNiO3 films deposited at 550 and 750 °C. Reproduced with permission from ref. 10. | ||
3.3 Optimizing electrocatalyst functionality via cation substitution
Cation substitution is a common strategy to tune the electronic structure of nickelates for optimizing OER activity. Taking LaNiO3 (Fig. 4a) as an example, the cation substitution can be classified based on the doping sites, namely A-site, B-site, or dual sites. Although A-site cations in nickelates are not considered as active sites for OER, they can indirectly contribute to the OER by altering lattice distortion,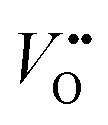 concentration, Ni valence state, and the Ni 3d and O 2p hybridization. According to the doping elements, the A-site substitution can be divided to isovalent or aliovalent substitution. In 2018, we (Wang et al.46) used PLD to synthesize a series of RNiO3 (where R = La, La0.5Nd0.5, La0.2Nd0.8, Nd, Nd0.5Sm0.5, Sm, and Gd) thin films on SrTiO3 substrates and investigated the effect of A-site isovalent substitution on OER activity. We found that decreasing the ionic radius of A-site elements in RNiO3 leads to a decrease in the electronic conductivity, which is consistent with a decrease in Ni–O bond covalency due to bending of the Ni–O–Ni angle. Moreover, reducing the ionic radius of A-site elements induces the formation of
concentration, Ni valence state, and the Ni 3d and O 2p hybridization. According to the doping elements, the A-site substitution can be divided to isovalent or aliovalent substitution. In 2018, we (Wang et al.46) used PLD to synthesize a series of RNiO3 (where R = La, La0.5Nd0.5, La0.2Nd0.8, Nd, Nd0.5Sm0.5, Sm, and Gd) thin films on SrTiO3 substrates and investigated the effect of A-site isovalent substitution on OER activity. We found that decreasing the ionic radius of A-site elements in RNiO3 leads to a decrease in the electronic conductivity, which is consistent with a decrease in Ni–O bond covalency due to bending of the Ni–O–Ni angle. Moreover, reducing the ionic radius of A-site elements induces the formation of 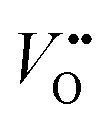 (the decrease of the O K pre-edge intensity), which increases the average occupancy of the antibonding eg orbital and lowers EF. This is favorable for the OER activity (Fig. 4c). One year later, we (Liu, Wang et al.47) used MBE to deposit a set of La1−xSrxNiO3 (x = 0, 0.12, 0.25, 0.5) thin films on LaAlO3 substrates and investigated the effect of A-site aliovalent substitution on the OER activity. Cyclic voltammetry curves of La1−xSrxNiO3 thin films in Fig. 4d show a clear trend towards improved OER activity as x increases, with x = 0.5 showing the most active OER activity. DFT calculations (Fig. 4e) clearly demonstrate that Sr doping in LaNiO3 induces an upward energy shift of O 2p band relative to EF, strengthening Ni 3d–O 2p hybridization and decreasing the charge transfer energy. Both contribute to enhancing the OER activity.
(the decrease of the O K pre-edge intensity), which increases the average occupancy of the antibonding eg orbital and lowers EF. This is favorable for the OER activity (Fig. 4c). One year later, we (Liu, Wang et al.47) used MBE to deposit a set of La1−xSrxNiO3 (x = 0, 0.12, 0.25, 0.5) thin films on LaAlO3 substrates and investigated the effect of A-site aliovalent substitution on the OER activity. Cyclic voltammetry curves of La1−xSrxNiO3 thin films in Fig. 4d show a clear trend towards improved OER activity as x increases, with x = 0.5 showing the most active OER activity. DFT calculations (Fig. 4e) clearly demonstrate that Sr doping in LaNiO3 induces an upward energy shift of O 2p band relative to EF, strengthening Ni 3d–O 2p hybridization and decreasing the charge transfer energy. Both contribute to enhancing the OER activity.
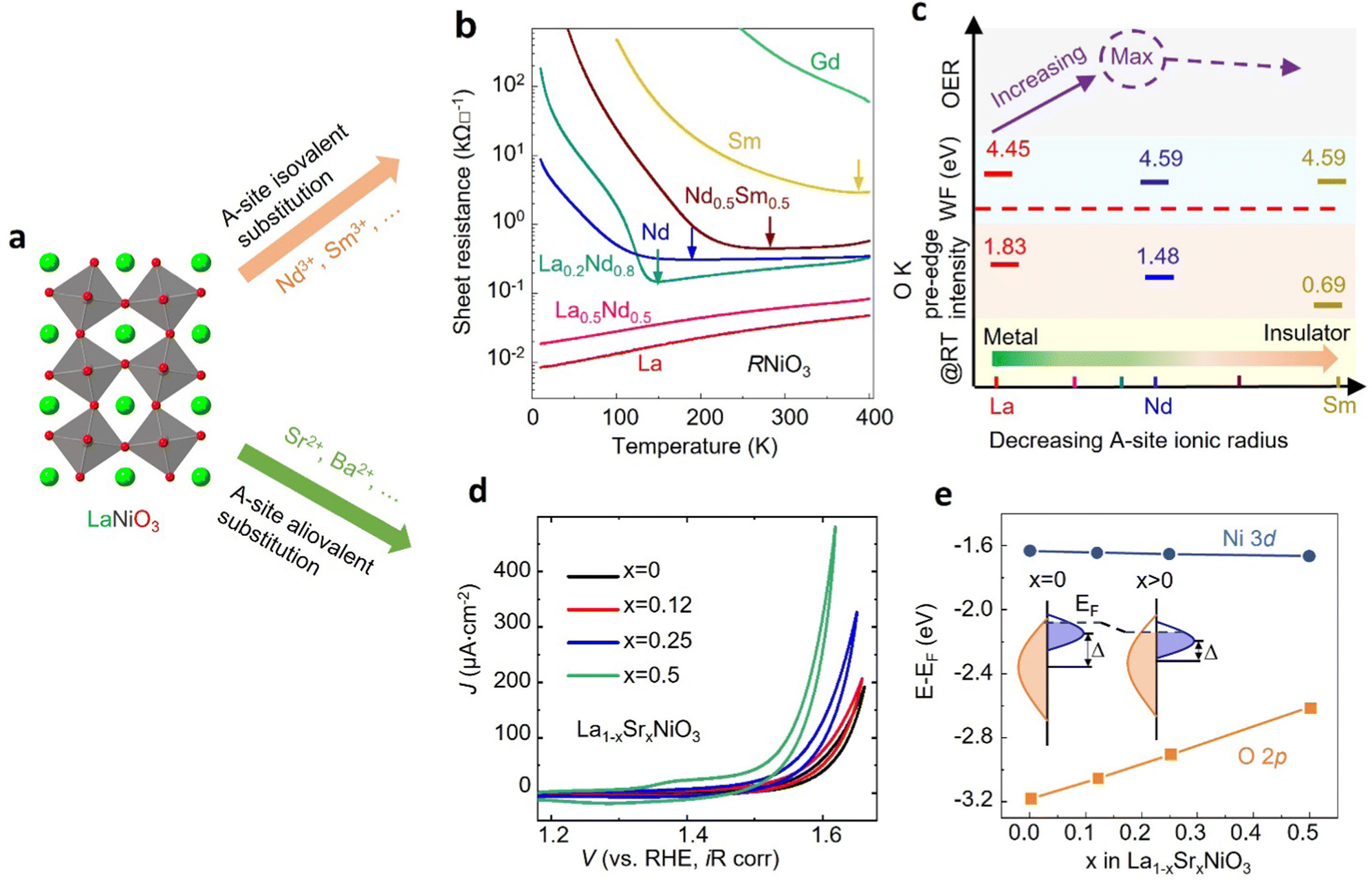 | ||
| Fig. 4 (a) Schematic illustration of LaNiO3 crystal structure. (b) Sheet resistance as a function of temperature for perovskite nickelate thin films with different A-site cations grown on SrTiO3 substrate. (c) The trends in the OER activity, work function, O K pre-edge intensity, and room temperature conductivity with decreasing the ionic radius of A-site cations in nickelates. Reproduced with permission from ref. 43. Copyright 2018, Wiley-VCH. (d) CV curves of La1−xSrxNiO3 thin films with x = 0, 0.12, 0.25, and 0.5. (e) Calculated average onsite energies of Ni 3d and O 2p orbitals with EF as a reference. The inset shows a schematic energy band diagram of LaNiO3 (x = 0) and La1−xSrxNiO3 (x > 0). Δ denotes the charge transfer energy. Reproduced with permission from ref. 44. Copyright 2019, Wiley-VCH. | ||
Unlike A-site cations, the B-site cation, i.e., the Ni site of RNiO3 nickelates, is believed to directly participate in the electrochemical reaction, either as active site for OER or through the covalent interaction with the active oxygen site. Recently, we (Wang et al.48) further explored the effect of B-site substitution, namely partially replacing Ni with other transition metals, on the OER activity. The electronegativity of the first-row transition metals increases when moving from left to right across the periodic table. Thus, Ni is the most electronegative element compared to other four cations Cr, Mn, Fe, and Co (Fig. 5a). Partially replacing Ni with Fe (Fig. 5b), or Cr, Mn, Co is expected to reduce the Ni valence state, which was verified in our work (Wang et al.48). A whole set of epitaxial LaNi1−xFexO3 (x = 0, 0.12, 0.25, 0.33, 0.375, 0.5, 1) solid solutions were synthesized by MBE. Chemical compositions and electronic structures were directly characterized by in situ XPS without air exposure. Fig. 5c clearly shows that, with doping Fe in LaNiO3, charge transfer from Fe to Ni occurs in these solid solutions, reducing Ni from Ni3+ to Ni2+ and oxidizing Fe from Fe3+ to Fe4+. DFT calculations indicate the high-valent Fe4+ states can significantly contribute to the density of states around EF (Fig. 5e), increasing the total TM 3d bandwidth via d-orbital coupling in Ni–O–Fe bridges. This change in electronic structure decreases the energetic cost to accept/donate electrons at the adsorbate–electrocatalyst interface, boosting the OER activity (Fig. 5d). Moreover, the Ni2+/Ni3+ gradually increases with x in LaNi1−xFexO3, which reduces the Ni 3d bandwidth and finally results in the bandgap opening. The decrease of total TM 3d–O 2p orbital hybridization induces a quick drop in OER activity at x = 0.5. Thereby, a volcano-like OER catalytic trend was observed with 37.5% Fe substitution in LaNi1−xFexO3 as the most active composition.
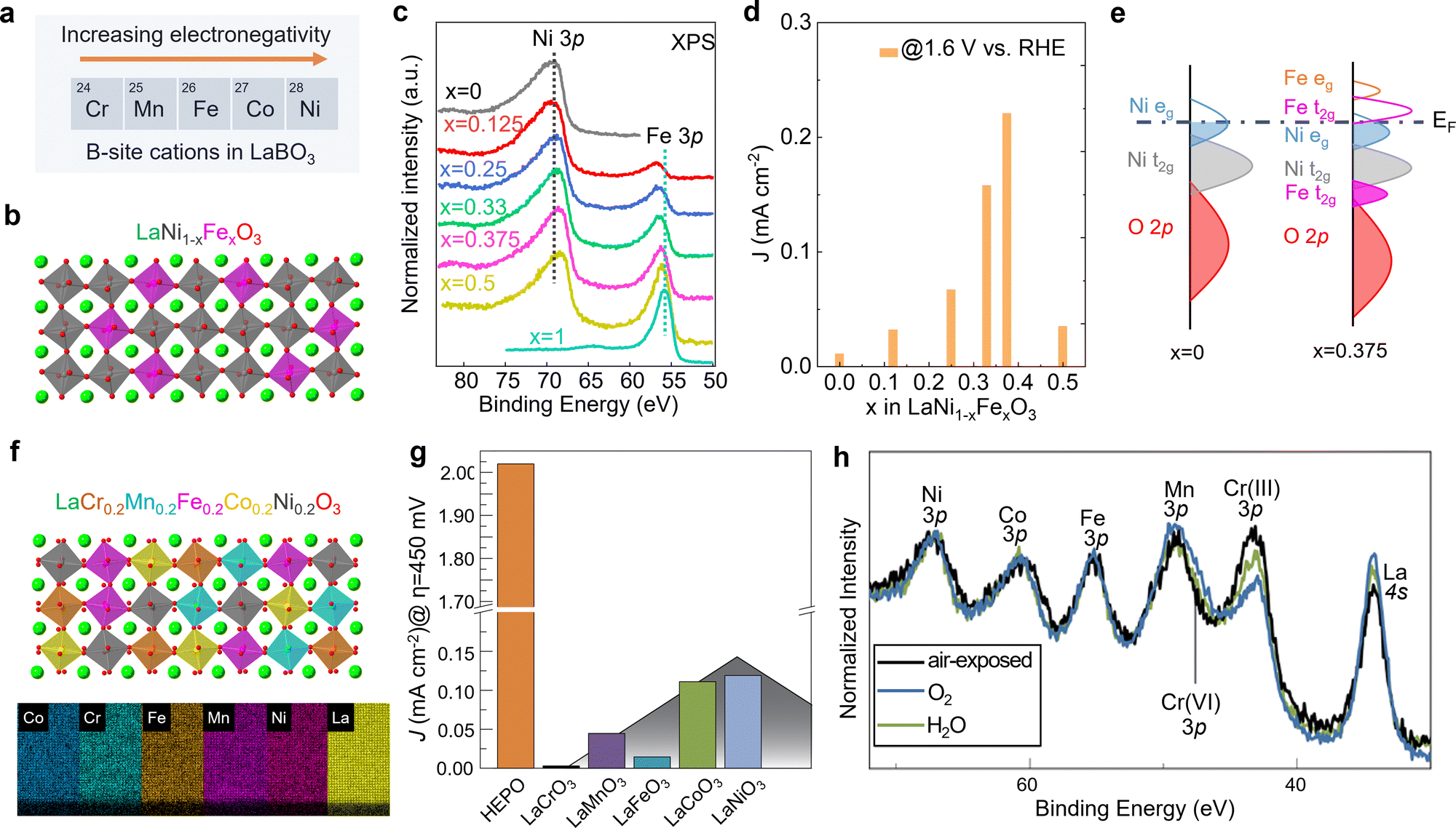 | ||
| Fig. 5 (a) Five common first-row transition metals used as B-site cations in LaBO3 and the trend of electronegativity. (b) Schematic illustration of LaNi1−xFexO3 crystal structure. (c) In situ Ni 3p and Fe 3p core level XPS spectra of LaNi1−xFexO3 films with x = 0, 0.125, 0.25, 0.33, 0.375, and 0.5. (d) OER current density of LaNi1−xFexO3 films measured at 1.6 V vs. RHE. (e) Schematic energy band diagram with corresponding electronic structure near EF. Reprinted (adapted) with permission from ref. 32. Copyright 2021 American Chemical Society. (f) Schematic illustration of LaCr0.2Mn0.2Fe0.2Co0.2Ni0.2O3 and energy-dispersive X-ray spectroscopy maps of a LaCr0.2Mn0.2Fe0.2Co0.2Ni0.2O3 high entropy perovskite oxide (HEPO) thin film. Scale bar: 5 nm. (g) OER current density of HEPO and five parent perovskites thin films measured at 1.68 V vs. RHE. (h) TM 3p core level ambient pressure XPS spectra of the HEPO thin film in two different atmospheres. (f–h) Reprinted (adapted) with permission from ref. 49. | ||
The potential beneficial effect of substitutional doping can be maximized using multiple elements on the same lattice site by using a solid–solution approach. Such multi-compositional perovskite oxides are frequently referred to as high-entropy perovskite oxides due to the increased configurational entropy induced by the mixing of several elements on the same crystallographic lattice site. These oxides may benefit from compositionally-fine-tuned electronic and atomic structure and the synergistic effects of multiple cations simultaneously/consecutively participating in electrocatalytic reactions. Furthermore, the high entropy may lead to phase stabilization according to the free energy of formation, resulting in increased stability against dissolution.50 We recently found that (001)-oriented LaCr0.2Mn0.2Fe0.2Co0.2Ni0.2O3−δ epitaxial thin films outperformed the OER activity of all parent perovskites with single B-site cation by a factor of 16 to 670 (Fig. 5g).49 We speculated that this enhancement of electrocatalytic activity may be the result of the synergistic behavior of multiple cations, since the adsorption and binding of water molecules to the surface has been shown to induce an appreciable change in oxidation state of Cr, Co, and Ni simultaneously (Fig. 5h). The electronic structure descriptors discussed previously cannot explain the observed activity trend. Therefore, more experimental and theoretical work are needed to capture the complex composition–property–function relationships in this multi-dimensional space.50
4. Outlook
4.1 Operando characterization for deeper mechanistic insights
Solid–solid, solid–gas and especially solid–liquid interfaces continuously evolve as a function of applied potentials, ionic concentrations, and time. For electrocatalysts, this implies that true active phases and reaction sites do not “pre-exist” in as-prepared “pre-catalyst” surfaces, but evolve under reaction conditions.51 This will be discussed below in more detail for the case of NiO2-terminated LaNiO3 from the example in Section 3.2. To achieve further understanding of the structure–property–function relationships governing electrocatalyst materials discovery, characterization of chemical composition, atomic arrangement and electronic structure of the interface under operating conditions is therefore necessary.The term “operando” refers to measuring material properties under working conditions, i.e., while the reaction of interest occurs, with simultaneous measurement of reaction rate.52,53Operando characterization goes well beyond in situ or ex situ characterization of the as-prepared or even the operated surfaces because the catalyst changes reversibly or irreversibly as a function of potential. Therefore, only operando measurements can shed light on the surface properties actually giving rise to electrocatalytic activity. In this section, we will provide an overview of operando characterization techniques recently employed for epitaxial thin film electrocatalysts and techniques that exhibit high potential for impactful future use. We include a discussion why epitaxial thin-film samples are particularly useful to extract the relevant information.
Typical surface transformation giving rise to the electrocatalytic activity are confined to the surface,14 yet many operando characterization techniques are bulk-sensitive, i.e., averaging the information from a thin surface layer with the underlying volume of the electrocatalyst. This hampers sensitivity to small structural or chemical changes in the surface layer. Fig. 6a and b shows a guiding principle to extract surface-sensitive information from a nominally bulk-sensitive technique, described in more detail in ref. 14 and 54. The relative intensity of a thin surface layer depends on the total thickness of the electrode. Using epitaxial thin films with unit-cell thickness control, one can extract the surface signal from a series of operando experiments with various sample thicknesses. We employed this technique successfully using bulk-sensitive UV-Vis spectroscopy in transmission geometry (Fig. 6c). The optical density of the electrocatalyst at a characteristic photon wavelength changes reversibly at the previously discussed Ni redox potential. Comparison to reference samples and systematic thickness-dependent measurements revealed a phase transition of a unit-cell-thin layer to a Ni oxyhydroxide-like layer.14 This transformation towards an edge-sharing octahedral arrangement of the Ni atoms in a sub-nanometer surface layer is key for the high electrocatalytic activity observed for NiO2-termination because it does not occur for LaO-termination.
 | ||
| Fig. 6 (a) Schematic for extracting surface-sensitive information from bulk-sensitive techniques. A representative perovskite oxide electrocatalyst (red corner-sharing octahedra) exposed to an electrolyte develops a structurally and electronically different surface layer when a potential is applied to drive the oxygen evolution reaction (OER), in this example involving transformation of the top unit cell (violet edge-sharing octahedra), inspired by (ref. 10 and 49). (b) The relative intensity of a 0.8 nm surface layer measured by a bulk-sensitive technique like XAS or UV-Vis spectroscopy increases with decreasing total thickness of the electrode. Reprinted from C. Baeumer, J. Appl. Phys., 2021, 129, 170901, with the permission of AIP Publishing (c) cyclic voltammetry in the pre-OER region for Ni-terminated LaNiO3 (LNO–Ni), along optical density change at λ = 500 nm during potential holds. Reproduced with permission from ref. 10. (d) Surface roughening during cycling up to increasingly higher potentials in and 0.5 M H2SO4 (scale bars are 400 nm). Used with permission of Royal Society of Chemistry, from “Probing the Stability of SrIrO3 During Active Water Electrolysis via Operando Atomic Force Microscopy”, A. R. Akbashev et al., Energy Environ. Sci., 2023, 14. (e) Electrocatalyst changes with applied potential tracked by Mn L-edge XAS of electrodeposited MnOx films in 0.1 M KOH cycled from 1.65 V to 0.50 V and back to 1.65 V vs. RHE. Reproduced (adapted) with permission from ref. 36. Copyright 2017, American Chemical Society. (f) Sample geometry used for SW-XPS analysis. (g) Comparison of experimental rocking curves for various XPS core-level intensities with calculations based on the optimized sample configuration, which allows extraction of sub-nanometer-resolution depth profiles of atomic concentrations. (f and g) Reproduced with permission from ref. 55. Copyright 2014, Springer Nature. (h) Optical conductivity spectra of a La0.5Sr0.5FeO3−δ thin film, recorded at various potentials. The inset shows a schematic representation of the chamber used for the measurements. Reprinted (adapted) with permission from Y. Q. Tang et al., ACS Appl. Mater. Interfaces, 2022, 14, 18486–18497. Copyright 2022 American Chemical Society. (i) ETEM investigation of the La0.6Sr0.4CoO3−δ/H2O interface, showing a representative images of a time sequence in 0.5 Pa of H2O. Highly mobile adatoms are highlighted by white arrows. Reproduced with permission from ref. 9. (j) (01L) and (00L) crystal truncation rods measured at 0.5 V (blue) and 1.5 V vs. RHE (red). Open points and solid lines denote the experimentally measured intensities and the best-fit results from the fitting process, respectively. Used with permission of Royal Society of Chemistry, from “Towards identifying the active sites on RuO2 (110) in catalyzing oxygen evolution”, R. R. Rao et al., Energy Environ. Sci., 2017, 10, 2626–2637. | ||
A similar approach to tune bulk-sensitive methods towards surface-sensitivity may be applied to X-ray absorption spectroscopy (XAS). XAS allows extraction of oxidation states, coordination geometry and number, and elemental composition from peak shapes, positions, intensities, and spin–orbit splitting.56 In the past, operando XAS was already employed, for example, to track valence changes at OER-relevant voltages of a layered manganese oxide film prepared by electrodeposition (Fig. 6e).55 X-ray absorption spectra revealed reversible yet hysteretic Mn redox. Oxidation to a mixed Mn3+/4+ valence preceded the oxygen evolution, and additional analysis of the O spectra showed changes in Mn–O hybridization with different applied potentials, where an eg occupancy near one likely corresponds to the highest hybridization. In the future, operando XAS may therefore aid in identifying mechanistic OER activity trends based on the electronic structure of epitaxial nickelates under operating conditions. Yet, for epitaxial thin films, designing appropriate experimental cells is more challenging compared to conventional electrocatalyst materials because fabrication on X-ray-transparent membranes or a cell design with very thin liquid compartments are necessary to minimize attenuation of incoming and outgoing X-ray, as discussed in more detail elsewhere.51
In addition to studying activity trends, characterization of degradation pathways becomes increasingly important for selecting ideal electrocatalyst materials since an inverse activity–stability relationship seems to exist as introduced previously. Akbashev et al.57 recently investigated the degradation of SrIrO3, a highly active electrocatalyst, during the oxygen evolution reaction (OER) using operando electrochemical atomic force microscopy (EC-AFM). This material serves as a model system for degradation studies of perovskite ABO3 oxides, exhibiting both A-site cation leaching and transition metal (B-site) dissolution. EC-AFM can track surface morphology changes and dissolution, and epitaxial thin films are particularly suited for mechanistic studies because they provide a smooth, step-terraced surface morphology with unit-cell precise thickness control of the as-prepared state. For the epitaxial SrIrO3, the thickness remained identical during operation in H2SO4 up to 2 V vs. RHE, implying that no dissolution occurred in these conditions and in contrast to severe dissolution in alkaline media.58 In H2SO4, particulates appear at ∼1.17 V vs. RHE and disappear at 1.5–1.6 V vs. RHE, as shown in Fig. 6d. These particulates affect the surface roughness but the step-terraced morphology remains. The authors proposed that these particulates are SrSO4, and discuss the formation and removal of the particulates in the context of Sr leaching observed by XPS and mass spectroscopy. Importantly, they also conclude that the stability of perovskite oxide electrocatalysts under operating conditions might be improved by suppressing A-site leaching a common degradation mechanism to which we will return below. For iridium based electrocatalysts, dissolution of the A-site element compared to Ir is highly dependent on their structure.59 Electrocatalysts composed of strong edge- and face-sharing octahedra are more robust against A-site dissolution.60 Furthermore, balancing A- and B-site dissolution by B-site substitution can help to maintain crystal structure and prevent catalyst degradation.61Operando characterization may aid to identify these degradation mechanisms and related material properties to ultimately design more efficient and robust catalysts.
As previously discussed, the electrocatalytic performance is not only controlled by the surface but also by the defect chemistry of the underlying layer, which also changes during operation due to parasitic (de-)intercalation reactions. Therefore, operando probes for the type and concentration of point defects such as  and protons are needed. Tang et al.38 recently developed an operando spectroscopic ellipsometry approach, allowing the tracking of the concentration of holes as a function of the intercalation potential (Fig. 6h). They characterized epitaxial La0.5Sr0.5FeO3−δ thin films and indirectly quantified the concentration of electron holes for different electrochemical potentials using the Fe oxidation state. Combining the experimental results with a defect chemistry model, they extracted estimates for the concentration of H+ and
and protons are needed. Tang et al.38 recently developed an operando spectroscopic ellipsometry approach, allowing the tracking of the concentration of holes as a function of the intercalation potential (Fig. 6h). They characterized epitaxial La0.5Sr0.5FeO3−δ thin films and indirectly quantified the concentration of electron holes for different electrochemical potentials using the Fe oxidation state. Combining the experimental results with a defect chemistry model, they extracted estimates for the concentration of H+ and 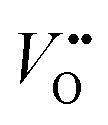 in the epitaxial layer as a function of potential. This approach may help to tune electrocatalyst properties through property-control by ionic intercalation, and can reveal the changes in electronic structure during electrochemical preconditioning.
in the epitaxial layer as a function of potential. This approach may help to tune electrocatalyst properties through property-control by ionic intercalation, and can reveal the changes in electronic structure during electrochemical preconditioning.
We now turn towards future pathways for operando characterization that may lead to even deeper analysis or higher sensitivity and interface-specificity. As discussed in the previous section, SW-XPS is capable of depth-profiling the chemical environment in the near-surface region with 1–2 Å depth resolution. Nemšák et al.62 reported a strategy to extend SW-XPS to solid–liquid interfaces using an ∼1 nm thick hydrated layer on α-Fe2O3, combining ambient-pressure XPS and SW-XPS. They found that SW-XPS at the solid–liquid interface allows sub-nm resolution of the spatial arrangement of chemical species, in this case the position of Cs and Na cations in the hydrated layer, as well as local potential energy variations along the direction perpendicular to the interface (Fig. 6f and g). Later, the same research group followed up with a pioneering operando study, revealing that SW-XPS is even possible in the presence of an electrolyte meniscus with ∼20 nm thickness while applying electrochemical potentials.63 In this example, they demonstrated the oxidation and hydroxylation of an 8 nm thick Ni film while maintaining sub-nanometer depth resolution using the increasingly popular dip-and-pull geometry, which was introduced in the seminal paper by Axnanda, Crumlin et al.64 The same depth resolution can be now achieved using near total reflection geometry, which is especially attractive because it provides all the strengths of a standard SW-XPS without the need of an artificial multilayer mirror, making the technique more broadly applicable.65
For atomic-level spatial resolution, environmental transmission electron microscopy (ETEM) is a key development as it enables the direct imaging of surface processes on the atomic scale under exposure to ambient gases. When operated in H2O conditions, a thin water layer condensates on the perovskite surface, as pioneered in the group of Jooss.66 The primary electron beam of the TEM causes anodic polarization of the perovskite catalyst, resulting in close to relevant OER potentials. Weber et al.13 and Lole et al.66 recently used this technique to reveal the dynamics at the La0.6Sr0.4CoO3−δ/H2O and the La0.6Sr0.4MnO3−δ/H2O interfaces, where the TM ions are found to be highly mobile under OER potentials; a snapshot of the time sequence for this interface is shown in Fig. 6i.13 Such dynamics present an important future contribution for our understanding of electrocatalytic activity at continuously transforming electrocatalyst surfaces.
Lastly, we suspect that surface XRD and the investigation of crystal truncation rods (CTR) will lead to new fundamental insights when employed for epitaxial thin film electrocatalysts. CTR analysis was already successfully employed to study the surface structure and binding of reaction intermediates at single crystal surfaces, as showcased for the RuO2 surface in Fig. 6j, where the diffraction data was fitted with a surface model including the presence of an *OO intermediate on specific Ru sites.67,68 Such analyses, especially when combined with ab initio modelling and microkinetic modelling, may therefore lead to a more complete picture of the reaction intermediates preceding the rate determining step and potential-dependent surface structures and coverages, all of which are urgently needed for enhanced mechanistic understanding of OER electrocatalysts.
4.2 Layer-by-layer control of functionality and performance
To further enhance the performance of nickelates and perovskite electrocatalysts in general, both the activity and stability must be considered because of the typically-observed inverse activity–stability relationship. As stated before, epitaxial thin films are a valuable platform to develop structure–property–function relationships that might provide design rules to optimize both activity and stability simultaneously. It was recently shown that this inverse activity–stability relationship is not universal and can be overcome by careful inspection of facet-dependent activity and stability. For example, for LaNiO3, the (111) facet is both more active and more stable than other facets, a trend that could be revealed studying epitaxial thin films synthesized on differently oriented substrates.69 Epitaxial thin films also enable the fabrication of material combinations with hybrid properties by interfacing different perovskite oxides with unit cell thickness control.22 Such heterostructures offer even more degrees of freedom to engineer stable and active electrocatalysts that overcome the inverse stability–activity relationship.As previously discussed, the electrochemical properties of epitaxial thin films are influenced by the surface layer as well as the subsurface layer. Engineering of the subsurface layer allows for optimization of the activity and stability. For example, electronic transport in thin film or heterostructure electrodes can be manipulated by engineering the interfacial depletion layers and corresponding potential barriers. Baniecki et al.70 showed that buried epitaxial layers can be exploited to engineer the depletion layer and enhance the overall performance of epitaxial catalysts. Heterostructures of La-doped BaSnO3 and La-doped SrTiO3 on Nb-doped SrTiO3 substrates were used as support of few-unit-cell thin La0.7Sr0.3CoO3−δ catalysts layers. The introduction of La-doped BaSnO3 and La-doped SrTiO3 underneath the electrochemically active La0.7Sr0.3CoO3−δ results in a narrower depletion layer and facilitates p-type character in the catalyst that greatly enhances its activity. This results in fast transport kinetics, even accompanied by enhanced stability as shown in Fig. 7c and d. Furthermore, engineering of the band edge has also been found to greatly enhance the performance of photoanodes in photoelectrochemical water-splitting. For example, the introduction of subsurface dipoles by placement of 1 unit cell LaAlO3 in SrTiO3 leads to improved efficiency.71
 | ||
| Fig. 7 Epitaxial multilayers offer opportunities to engineer both active and stable OER catalysts using various methods. (a) Hybrid structures can prevent catalyst dissolution. (b) Subsurface layers of SrRuO3 (SRO) can activate SrTiO3 (STO). Subscripts indicate the number of unit cells of STO (SRO) in the surface (subsurface). The current density of SrTiO3 with an inactive subsurface layer of LaTiO3 (LTO) and of a thin film of SrRuO3 over time are shown for comparison. Used with permission of Royal Society of Chemistry, from “Activation of ultrathin SrTiO3 with subsurface SrRuO3 for the oxygen evolution reaction”, A. R. Akbashev et al., Energy Environ. Sci., 2018, 11, 1762–1769 (ref. 54. (c and d) Band engineering using layers of La doped BaSnO3 (BLSO) and La-doped SrTiO3 (LSTO) facilitates charge transport and enhances the lifetime of epitaxial catalysts grown on Nb-doped SrTiO3 (NSTO). Reproduced with permission from ref. 63. Copyright 2019, Wiley-VCH. (e) Interfacial engineering allows for modification of surface properties without changing the chemical composition of the catalyst. (f) Interfacial engineering tunes the Co–O covalency of hybrid bilayers of 2–8 unit cells (u.c.) LaCoO3 on 20 nm La0.6Sr0.4CoO3 as shown using X-ray photoelectron of the valence band. Reproduced with permission from ref. 65. (g) Interfacial engineering results in a change in Co oxidation state near an epitaxial interface as observed with high angle annular dark field (HAADF) scanning transmission electron microscopy (STEM) with electron energy loss spectroscopy (EELS). Reprinted figure with permission from Araizi-Kanoutas et al., Phys. Rev. Mater., 2020, 4, 026001. Copyright 2020 by the American Physical Society (ref. 68). | ||
In addition to the manipulation of band bending at the solid/liquid interface, electronic properties of the active surface can be tuned in epitaxial multilayers as schematically shown in Fig. 7e. In our work (Heymann et al.72), we showed that the enhanced Co–O covalency at the surface of hybrid epitaxial bilayers of La0.6Sr0.4CoO3 and LaCoO3 influences its activity. We evaluated valence band spectra of hybrid bilayers of 2–8 unit cells LaCoO3 on top of 20 nm La0.6Sr0.4CoO3 and their parent compounds, as shown in Fig. 7f. The valence band of La0.6Sr0.4CoO3 shows more overlap between the O 2p (blue) and Co 3d (red) contribution compared to LaCoO3, which indicates higher Co–O covalency for La0.6Sr0.4CoO3. The spectra obtained for the hybrid bilayers also show large overlap between O 2p and Co 3d bands (more than expected based on a linear combination of the parent spectra) indicating interfacial hybridization in the bilayer catalysts. As Mott–Schottky analysis indicates hole accumulation for all investigated bilayers and both parent compounds in the OER regime, the observed activity of the fabricated bilayers is attributed to the increased Co–O covalency obtained by interfacial hybridization, rather than band bending at the solid/liquid interface.
Similar interfacial hybridization effects are exploited by Akbashev et al.58 to activate OER-inactive SrTiO3 by burying highly active but unstable ultra-thin SrRuO3 layers in the subsurface. The placement of such an active subsurface layer in a stable host perovskite is shown schematically in Fig. 7a. The introduction of a SrRuO3 layer in SrTiO3 as a host material is expected to result in Ru 4d states that hybridize with SrTiO3 electronic states and lie within the band gap of SrTiO3, as predicted by DFT calculations. The authors show that as little as one unit cell of SrRuO3 in the subsurface greatly enhances the activity of the catalyst compared to SrTiO3, as indicated in Fig. 7b. Furthermore, they show that the lifetime of the multilayered catalyst is enhanced compared to bare SrRuO3 because the dissolution of SrRuO3 is suppressed by the SrTiO3 surface layer. The stability of the multilayer greatly increases as the SrTiO3 capping layer thickness is increased from 1 to 2 unit cells, while its activity decreases with increasing capping layer thickness. Hence, a clear trade-off between activity and stability seems to exist in these hybrid multilayered catalysts. Other promising material combinations to achieve an enhancement of activity by subsurface doping have been identified computationally by Zhang et al.73
A similar method of enhancing activity by the introduction of a subsurface layer is applicable to epitaxial catalysts for the oxygen reduction reaction. Eom et al.74 showed that single unit cell thick layers of SrMnO3 in LaMnO3 can greatly enhance its activity. Changing the A-site cation from La to Sr affects the electronic configuration of Mn and therefore the activity of the catalyst. The authors found that a layer of SrMnO3 in the subsurface shows a greater enhancement in activity compared to placement of this layer at the surface. The lower activity of the catalyst when Sr is present in the surface layer is attributed to SrO formation upon exposure of the catalyst to oxygen, as observed by ambient pressure X-ray photoelectron spectroscopy. This work serves as a demonstration of how the introduction of different sub-surface layers may activate a catalyst without changing the chemical composition at the surface and therefore compromising its stability.
In addition to the stabilization of active catalysts by the prevention of dissolution with capping layers, or activation of inactive catalysts by placement of more active subsurface layers, as shown in Fig. 7a, interfacial engineering offers even more opportunities to tune the properties of stable catalysts to enhance activity, as shown in Fig. 7e. For example, the oxidation state of B-site atoms in epitaxial perovskites changes near the interface of perovskites with a different B-site atom, as shown in Fig. 7g.75 This offers opportunities to manipulate the oxidation state, structural distortions, orbital filling and covalency of ultrathin catalytic layers without changing their chemical composition. As the chemical composition remains unchanged, this is a promising strategy to overcome the observed inverse activity–stability relationship by activating chemically stable surfaces through remote doping, which may be extended from these Co- and Ru-based examples to epitaxial nickelate layers. Due to the control of the growth of epitaxial multilayers, these films serve as promising model systems to provide guidelines for the design of catalytic core–shell nanoparticles that show superior stability and activity.
In conclusion, epitaxial nickelate layers have emerged as an ideal model to study intrinsic structure–property–functionality relationships, which will be exploited by translating materials design criteria towards high-surface-area synthesis pathways. It was found that engineering the strain state, the crystallographic facet and the surface composition can greatly affect the catalytic activity. Further optimization can be achieved through substitutional control of the composition and resulting electronic structure. The next steps forward will likely be achieved by designing future generation catalysts with the insights from operando characterization of the active state of the surface and subsurface (defect) structure and composition, and by interfacing multiple electrocatalysts in a core–shell structure to enhance functionality without changing the surface chemical composition.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. K. Acknowledges funding by the Netherlands Ministry of Economic Affairs' Top Consortia for Knowledge and Innovation (TKIs) Allowance (CHEMIE.PGT.2021.007). C. B. has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program under grant agreement No. 101040669 – Interfaces at Work. M. C. and L. W. acknowledge funding support by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering under Award #10122. Q. L. acknowledges the startup funding provided by the School of Engineering, Westlake University, funding support from the Research Center for Industries of the Future, Westlake University and the National Natural Science Foundation of China (NSFC, Grant No. 52202148).References
- S. Yuan, J. Y. Peng, B. Cai, Z. H. Huang, A. T. Garcia-Esparza, D. Sokaras, Y. R. Zhang, L. Giordano, K. Akkiraju, Y. G. Zhu, R. Hubner, X. D. Zou, Y. Roman-Leshkov and Y. Shao-Horn, Nat. Mater., 2022, 21, 673–680 CrossRef CAS PubMed.
- J. T. Mefford, A. R. Akbashev, M. K. Kang, C. L. Bentley, W. E. Gent, H. T. D. Deng, D. H. Alsem, Y. S. Yu, N. J. Salmon, D. A. Shapiro, P. R. Unwin and W. C. Chueh, Nature, 2021, 593, 67–73 CrossRef CAS PubMed.
- Y. L. Pan, X. M. Xu, Y. J. Zhong, L. Ge, Y. B. Chen, J. P. M. Veder, D. Q. Guan, R. O'Hayre, M. R. Li, G. X. Wang, H. Wang, W. Zhou and Z. P. Shao, Nat. Commun., 2020, 11, 2002 CrossRef CAS PubMed.
- H. Park, J. W. Bae, T. H. Lee, I. J. Park, C. Kim, M. G. Lee, S. A. Lee, J. W. Yang, M. J. Choi, S. H. Hong, S. Y. Kim, S. H. Ahn, J. Y. Kim, H. S. Kim and H. W. Jang, Small, 2022, 18, 2105611 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- D. Antipin and M. Risch, J. Phys. Energy, 2020, 2, 032003 CrossRef CAS.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martinez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Norskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- J. O. Bockris and T. Otagawa, J. Electrochem. Soc., 1984, 131, 290–302 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- W. T. Hong, K. A. Stoerzinger, Y. L. Lee, L. Giordano, A. Grimaud, A. M. Johnson, J. Hwang, E. J. Crumlin, W. L. Yang and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 2190–2200 RSC.
- J. Suntivich, W. T. Hong, Y. L. Lee, J. M. Rondinelli, W. L. Yang, J. B. Goodenough, B. Dabrowski, J. W. Freeland and Y. Shao-Horn, J. Phys. Chem. C, 2014, 118, 1856–1863 CrossRef CAS.
- T. G. Yun, Y. Sim, Y. Lim, D. Kim, J. S. An, H. Y. D. Lee, Y. G. Du and S. Y. Chung, Mater. Today, 2022, 58, 221–237 CrossRef CAS.
- M. L. Weber, G. Lole, A. Kormanyos, A. Schwiers, L. Heymann, F. D. Speck, T. Meyer, R. Dittmann, S. Cherevko, C. Jooss, C. Baeumer and F. Gunkel, J. Am. Chem. Soc., 2022, 144, 17966–17979 CrossRef CAS PubMed.
- C. Baeumer, J. Li, Q. Y. Lu, A. Y. L. Liang, L. Jin, H. P. Martins, T. Duchon, M. Gloss, S. M. Gericke, M. A. Wohlgemuth, M. Giesen, E. E. Penn, R. Dittmann, F. Gunkel, R. Waser, M. Bajdich, S. Nemsak, J. T. Mefford and W. L. C. Chueh, Nat. Mater., 2021, 20, 674–682 CrossRef CAS PubMed.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B. J. Kim, J. Durst, F. Bozza, T. Graule, R. Schaublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925–931 CrossRef CAS PubMed.
- N. Danilovic, R. Subbaraman, K. C. Chang, S. H. Chang, Y. J. J. Kang, J. Snyder, A. P. Paulikas, D. Strmcnik, Y. T. Kim, D. Myers, V. R. Stamenkovic and N. M. Markovic, J. Phys. Chem. Lett., 2014, 5, 2474–2478 CrossRef CAS PubMed.
- T. Binninger, R. Mohamed, K. Waltar, E. Fabbri, P. Levecque, R. Kotz and T. J. Schmidt, Sci. Rep., 2015, 5, 12167 CrossRef CAS PubMed.
- E. Fabbri, A. Habereder, K. Waltar, R. Kotz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- C. Z. Yang, G. Rousse, K. L. Svane, P. E. Pearce, A. M. Abakumov, M. Deschamps, G. Cibin, A. V. Chadwick, D. A. Dalla Corte, H. A. Hansen, T. Vegge, J. M. Tarascon and A. Grimaud, Nat. Commun., 2020, 11, 1378 CrossRef CAS PubMed.
- S. H. Chang, N. Danilovic, K. C. Chang, R. Subbaraman, A. P. Paulikas, D. D. Fong, M. J. Highland, P. M. Baldo, V. R. Stamenkovic, J. W. Freeland, J. A. Eastman and N. M. Markovic, Nat. Commun., 2014, 5, 4191 CrossRef CAS PubMed.
- P. Adiga and K. A. Stoerzinger, J. Vac. Sci. Technol., A, 2022, 40, 010801 CrossRef CAS.
- M. L. Weber and F. Gunkel, J. Phys. Energy, 2019, 1, 031001 CrossRef CAS.
- A. Grimaud, O. Diaz-Morales, B. H. Han, W. T. Hong, Y. L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- H. W. Chen, M. D. Dong, Y. Hu, T. Lin, Q. H. Zhang, E. J. Guo, L. Gu, J. Wu and Q. Y. Lu, Nano Lett., 2022, 22, 8983–8990 CrossRef CAS PubMed.
- D. Y. Chung, P. P. Lopes, P. F. B. D. Martins, H. Y. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik, Y. S. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 550 CrossRef.
- A. Boucly, L. Artiglia, E. Fabbri, D. Palagin, D. Aegerter, D. Pergolesi, Z. Novotny, N. Comini, J. T. Diulus, T. Huthwelker, M. Ammann and T. J. Schmidt, J. Mater. Chem. A, 2022, 10, 2434–2444 RSC.
- C. E. Beall, E. Fabbri and T. J. Schmidt, ACS Catal., 2021, 11, 3094–3114 CrossRef CAS.
- I. Yamada, A. Takamatsu, K. Asai, T. Shirakawa, H. Ohzuku, A. Seno, T. Uchimura, H. Fujii, S. Kawaguchi, K. Wada, H. Ikeno and S. Yagi, J. Phys. Chem. C, 2018, 122, 27885–27892 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 647 CrossRef CAS.
- J. R. Petrie, V. R. Cooper, J. W. Freeland, T. L. Meyer, Z. Y. Zhang, D. A. Lutterman and H. N. Lee, J. Am. Chem. Soc., 2016, 138, 2488–2491 CrossRef CAS PubMed.
- J. Ge, W. Zhang, J. Tu, T. Xia, S. P. Chen and G. Xie, Small, 2020, 16, 2001856 CrossRef CAS PubMed.
- S. Hirai, S. Yagi, A. Seno, M. Fujioka, T. Ohno and T. Matsuda, RSC Adv., 2016, 6, 2019–2023 RSC.
- J. Bak, H. B. Bae and S. Y. Chung, Nat. Commun., 2019, 10, 2713 CrossRef PubMed.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- X. Rong, J. Parolin and A. M. Kolpak, ACS Catal., 2016, 6, 1153–1158 CrossRef CAS.
- J. S. Yoo, X. Rong, Y. S. Liu and A. M. Kolpak, ACS Catal., 2018, 8, 4628–4636 CrossRef CAS.
- R. Merkle, M. F. Hoedl, G. Raimondi, R. Zohourian and J. Maier, Annu. Rev. Mater. Res., 2021, 51, 461–493 CrossRef CAS.
- Y. Q. Tang, F. Chiabrera, A. Morata, A. Cavallaro, M. O. Liedke, H. Avireddy, M. Maller, M. Butterling, A. Wagner, M. Stchakovsky, F. Baiutti, A. Aguadero and A. Tarancon, ACS Appl. Mater. Interfaces, 2022, 14, 18486–18497 CrossRef CAS PubMed.
- B. H. Han, K. A. Stoerzinger, V. Tileli, A. D. Gamalski, E. A. Stach and Y. Shao-Horn, Nat. Mater., 2017, 16, 121–126 CrossRef CAS PubMed.
- J. S. Liu, E. D. Jia, K. A. Stoerzinger, L. Wang, Y. N. Wang, Z. Z. Yang, D. W. Shen, M. H. Engelhard, M. E. Bowden, Z. H. Zhu, S. A. Chambers and Y. G. Du, J. Phys. Chem. C, 2020, 124, 15386–15390 CrossRef CAS.
- J. H. Song, X. Jin, S. Zhang, X. Y. Jiang, J. F. Yang, Z. Li, J. Zhou, H. G. Zhang and Y. F. Nie, ACS Appl. Energy Mater., 2021, 4, 12535–12542 CrossRef CAS.
- J. R. Petrie, H. Jeen, S. C. Barron, T. L. Meyer and H. N. Lee, J. Am. Chem. Soc., 2016, 138, 7252–7255 CrossRef CAS PubMed.
- M. J. Choi, T. L. Kim, J. K. Kim, T. H. Lee, S. A. Lee, C. Kim, K. Hong, C. W. Bark, K. T. Ko and H. W. Jang, Nano Lett., 2020, 20, 8040–8045 CrossRef CAS PubMed.
- L. Wang, K. A. Stoerzinger, L. Chang, X. M. Yin, Y. Y. Li, C. S. Tang, E. D. Jia, M. E. Bowden, Z. Z. Yang, A. Abdelsamie, L. You, R. Guo, J. S. Chen, A. Rusydi, J. L. Wang, S. A. Chambers and Y. G. Du, ACS Appl. Mater. Interfaces, 2019, 11, 12941–12947 CrossRef CAS PubMed.
- K. A. Stoerzinger, W. S. Choi, H. Jeen, H. N. Lee and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 487–492 CrossRef CAS PubMed.
- L. Wang, K. A. Stoerzinger, L. Chang, J. L. Zhao, Y. Y. Li, C. S. Tang, X. M. Yin, M. E. Bowden, Z. Z. Yang, H. Z. Guo, L. You, R. Guo, J. Wang, K. Ibrahim, J. S. Chen, A. Rusydi, J. L. Wang, S. A. Chambers and Y. G. Du, Adv. Funct. Mater., 2018, 28, 1803712 CrossRef.
- J. S. Liu, E. D. Jia, L. Wang, K. A. Stoerzinger, H. Zhou, C. S. Tang, X. M. Yin, X. He, E. Bousquet, M. E. Bowden, A. T. S. Wee, S. A. Chambers and Y. G. Du, Adv. Sci., 2019, 6, 1901073 CrossRef CAS PubMed.
- L. Wang, P. Adiga, J. L. Zhao, W. S. Samarakoon, K. A. Stoerzinger, S. R. Spurgeon, B. E. Matthews, M. E. Bowden, P. V. Sushko, T. C. Kaspar, G. E. Sterbinsky, S. M. Heald, H. Wang, L. W. Wangoh, J. P. Wu, E. J. Guo, H. J. Qian, J. O. Wang, T. Varga, S. Thevuthasan, Z. X. Feng, W. L. Yang, Y. G. Du and S. A. Chambers, Nano Lett., 2021, 21, 8324–8331 CrossRef CAS PubMed.
- M. V. Kante, M. L. Weber, S. Ni, I. C. G. van den Bosch, E. van der Minne, L. Heymann, L. J. Falling, N. Gauquelin, M. Tsvetanova, D. M. Cunha, G. Koster, F. Gunkel, S. Nemšák, H. Hahn, L. Velasco and C. Baeumer, ACS Nano, 2023 DOI:10.1021/acsnano.2c08096.
- L. Wang, M. D. Hossain, Y. Du and S. A. Chamber, Nano Today, 2022, 47, 101697 CrossRef CAS.
- R. R. Rao, I. C. G. Van den Bosch and C. Baeumer, Operando X-ray characterization of interfacial charge transfer and structural rearrangements, in Encyclopedia of Solid-Liquid Interfaces, ed. K. Wandelt and G. Bussetti, Elsevier Inc., 1st edn, 2023, DOI:10.1016/B978-0-323-85669-0.00068-4.
- B. M. Weckhuysen, Chem. Commun., 2002, 97–110 RSC.
- M. A. Banares and I. E. Wachs, J. Raman Spectrosc., 2002, 33, 359–380 CrossRef CAS.
- C. Baeumer, J. Appl. Phys., 2021, 129, 170901 CrossRef CAS.
- M. Risch, K. A. Stoerzinger, B. H. Han, T. Z. Regier, D. Peak, S. Y. Sayed, C. Wei, Z. C. Xu and Y. Shao-Horn, J. Phys. Chem. C, 2017, 121, 17682–17692 CrossRef CAS.
- M. Risch, D. M. Morales, J. Villalobos and D. Antipin, Angew. Chem., Int. Ed., 2022, 61, e202211949 CrossRef CAS PubMed.
- A. R. Akbashev, V. Roddatis, C. Baeumer, T. Liu, J. T. Mefford and W. C. Chueh, Energy Environ. Sci., 2023, 16, 513–522 RSC.
- A. R. Akbashev, L. Zhang, J. T. Mefford, J. Park, B. Butz, H. Luftman, W. C. Chueh and A. Vojvodic, Energy Environ. Sci., 2018, 11, 1762–1769 RSC.
- N. Li, L. Cai, C. Wang, Y. Lin, J. Z. Huang, H. Y. Sheng, H. B. Pan, W. Zhang, Q. Q. Ji, H. L. Duan, W. Hu, W. H. Zhang, F. C. Hu, H. Tan, Z. H. Sun, B. Song, S. Jin and W. S. Yan, J. Am. Chem. Soc., 2021, 143, 18001–18009 CrossRef CAS PubMed.
- C. W. Song, J. Lim, H. B. Bae and S. Y. Chung, Energy Environ. Sci., 2020, 13, 4178–4188 RSC.
- S. Hirai, S. Yagi, H. C. Oh, Y. Sato, W. Liu, E. P. Liu, W. T. Chen, A. Miura, M. Nagao, T. Ohno and T. Matsuda, RSC Adv., 2022, 12, 24427–24438 RSC.
- S. Nemsak, A. Shavorskiy, O. Karslioglu, I. Zegkinoglou, A. Rattanachata, C. S. Conlon, A. Keqi, P. K. Greene, E. C. Burks, F. Salmassi, E. M. Gullikson, S. H. Yang, K. Liu, H. Bluhm and C. S. Fadley, Nat. Commun., 2014, 5, 5441 CrossRef PubMed.
- O. Karslioglu, S. Nemsak, I. Zegkinoglou, A. Shavorskiy, M. Hartl, F. Salmassi, E. M. Gullikson, M. L. Ng, C. Rameshan, B. Rude, D. Bianculli, A. A. Cordones, S. Axnanda, E. J. Crumlin, P. N. Ross, C. M. Schneider, Z. Hussain, Z. Liu, C. S. Fadley and H. Bluhm, Faraday Discuss., 2015, 180, 35–53 RSC.
- S. Axnanda, E. J. Crumlin, B. H. Mao, S. Rani, R. Chang, P. G. Karlsson, M. O. M. Edwards, M. Lundqvist, R. Moberg, P. Ross, Z. Hussain and Z. Liu, Sci. Rep., 2015, 5, 9788 CrossRef CAS PubMed.
- H. P. Martins, G. Conti, I. Cordova, L. Falling, H. Kersell, F. Salmassi, E. Gullikson, I. Vishik, C. Baeumer, P. Naulleau, C. M. Schneider and S. Nemsak, J. Phys. D: Appl. Phys., 2021, 54, 464002 CrossRef CAS.
- G. Lole, V. Roddatis, U. Ross, M. Risch, T. Meyer, L. Rump, J. Geppert, G. Wartner, P. Blöchl and C. Jooss, Commun. Mater., 2020, 1, 68 CrossRef.
- R. R. Rao, M. J. Kolb, N. B. Halck, A. F. Pedersen, A. Mehta, H. You, K. A. Stoerzinger, Z. X. Feng, H. A. Hansen, H. Zhou, L. Giordano, J. Rossmeisl, T. Vegge, I. Chorkendorff, I. E. L. Stephens and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 2626–2637 RSC.
- R. R. Rao, M. J. Kolb, L. Giordano, A. F. Pedersen, Y. Katayama, J. Hwang, A. Mehta, H. You, J. R. Lunger, H. Zhou, N. B. Halck, T. Vegge, I. Chorkendorff, I. E. L. Stephens and Y. Shao-Horn, Nat. Catal., 2020, 3, 516–525 CrossRef CAS.
- M. Wohlgemuth, M. L. Weber, L. Heymann, C. Baeumer and F. Gunkel, Front. Chem., 2022, 10, 913419 CrossRef CAS PubMed.
- J. D. Baniecki, H. Yamaguchi, C. Harnagea, D. Ricinschi, Z. Q. Gu, J. E. Spanier, T. Yamazaki and H. Aso, Adv. Energy Mater., 2019, 9, 1803846 CrossRef.
- Y. Hikita, K. Nishio, L. C. Seitz, P. Chakthranont, T. Tachikawa, T. F. Jaramillo and H. Y. Hwang, Adv. Energy Mater., 2016, 6, 1502154 CrossRef.
- L. Heymann, M. L. Weber, M. Wohlgemuth, M. Risch, R. Dittmann, C. Baeumer and F. Gunkel, ACS Appl. Mater. Interfaces, 2022, 14, 14129–14136 CrossRef CAS PubMed.
- L. Zhang, A. S. Raman and A. Vojvodic, Chem. Mater., 2020, 32, 5569–5578 CrossRef CAS.
- C. J. Eom, D. Y. Kuo, C. Adamo, E. J. Moon, S. J. May, E. J. Crumlin, D. G. Schlom and J. Suntivich, Nat. Commun., 2018, 9, 4034 CrossRef PubMed.
- G. Araizi-Kanoutas, J. Geessinck, N. Gauquelin, S. Smit, X. H. Verbeek, S. K. Mishra, P. Bencok, C. Schlueter, T. L. Lee, D. Krishnan, J. Fatermans, J. Verbeeck, G. Rijnders, G. Koster and M. S. Golden, Phys. Rev. Mater., 2020, 4, 026001 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |