
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Well-defined electrochemical switching of amphiphilic glycolated poly(3,4-ethylenedioxythiophene)†‡
Renata
Rybakiewicz-Sekita
 *ab,
Maciej
Gryszel
ac,
Gaurav
Pathak
c,
Roman
Gańczarczyk
a,
Mary Jocelyn
Donahue
c and
Eric Daniel
Głowacki
*ad
*ab,
Maciej
Gryszel
ac,
Gaurav
Pathak
c,
Roman
Gańczarczyk
a,
Mary Jocelyn
Donahue
c and
Eric Daniel
Głowacki
*ad
aWarsaw University of Technology, Faculty of Chemistry, Noakowskiego 3, 00-664, Warsaw, Poland. E-mail: eric.glowacki@pw.edu.pl
bFaculty of Mathematics and Natural Sciences, School of Sciences, Institute of Chemical Sciences, Cardinal Stefan Wyszynski University in Warsaw, Wóycickiego 1/3, 01-815, Warsaw, Poland. E-mail: r.rybakiewicz@uksw.edu.pl
cLaboratory of Organic Electronics, ITN Campus Norrköping, Linköping University, SE-60174, Norrköping, Sweden
dBioelectronics Materials and Devices Lab, Central European Institute of Technology, Brno University of Technology, Purkyňova 123, 61200, Brno, Czech Republic. E-mail: eric.daniel.glowacki@ceitec.vutbr.cz
First published on 9th November 2022
Abstract
The approach of using polyether, aka glycol, side chains to afford amphiphilicity to conducting polymers has recently emerged as a powerful technique for next-generation materials for bioelectronics and electrochemical devices. Herein we apply this synthetic logic to the archetypical conducting polymer poly(3,4-ethylenedioxythiophene), PEDOT, to generate a glycolated PEDOT analogue, G-PEDOT. We report on the electropolymerization of this material, and its electrochemical properties: including spectroelectrochemistry, electrochemical capacitance, and operation of microelectrodes and electrochemical transistors. While in many respects performing like PEDOT, G-PEDOT has electrochemical switching within lower potentials with complete de-doping at lower potentials, affording transistors with higher on/off ratios than PEDOT, and electrochromic switching within a smaller electrochemical window. Overall, G-PEDOT emerges as a useful, functional alternative to other PEDOT derivatives, and could be a building block in copolymers.
1. Introduction
Conducting polymers are a class of multifunctional electronic materials possessing hybrid electronic and ionic conductivity, chemical synthetic tunability, and unique electrochemical, catalytic, and optoelectronic properties. Conducting polymers can be divided into two families: hydrophobic and hydrophilic. The former is synthesized and processed from organic solvents such as toluene, chloroform, etc. Thin films of such polymers can be used in many solid-state devices like light-emitting diodes and photovoltaic cells. The other class of conducting polymers are hydrophilic and are processed and often used in aqueous conditions. This class is practically relevant in bioelectronics, biosensors,1,2 and electrolytic capacitors.3 Recently, amphiphilic conducting polymers have emerged, featuring polyether-based side chains commonly referred to as glycol side chains.4–6 Glycol side chains afford hydrophilicity, but also affinity for organic solvents such as chloroform. The balance between hydrophilicity and hydrophobicity can be therefore tuned using glycol side chains, or combinations of glycol and alkyl side chains.6,7 This principle of tailoring the processability (from organic or aqueous solvent systems) and wettability using glycol side chains has been applied to yield next-generation conducting polymers for electrochemical transistors, ECTs,8 as well as electrochromic devices.9 Both applications rely on reversible electrochemical redox reactions to modulate the doping of the polymer, and require good ion mobility and efficient exchange of ions between the aqueous surroundings and the polymer film. Side chains play an important role in running wettability and ionic transport. ECT devices are used for biosensing applications, and as transducers for amplifying biopotential signals.10,11 High-performance implantable neural recording arrays based on ECT pixels are a major area of research.12 ECTs require fast and reversible electrochemical redox reactions to modulate the conductivity of the polymer, simultaneously, the polymers must maintain good electrical conductivity and robustness with respect to dissolution or delamination. Poly(3,4-ethylenedioxythiophene), PEDOT, has become one of the most widespread and successful conducting polymer materials, and dominates in ECT research and applications. Most commonly it is deployed with the polyelectrolyte poly(styrene sulfonate) acting as the counter ion, to yield the composite PEDOT:PSS. While many glycol-containing homo- and heteropolymers have begun to compete with PEDOT in ECT and other bioelectronics applications,7,13–15 surprisingly a glycolated derivative of PEDOT itself has not been explored in these contexts. The synthesis of a glycolated PEDOT was reported in 2000, with a preliminary evaluation of electrochemical properties.16 This encouraged us to revisit this concept in detail, and perform measurements relevant to modern applications. The motivation of this work was to synthesize a simple glycol side-chain bearing PEDOT and test its electrochemical, spectroelectrochemical, and electrical properties in microelectrode and transistor geometry, in order to inform the growing body of knowledge about amphiphilic conducting polymers. We successfully demonstrate electropolymerization to produce useful G-PEDOT layers and devices. We also test the possibility of solution processing from organic solvents, finding negative results. What emerges is that G-PEDOT behaves much like the well-known PEDOT, but with advantages of more defined electrochemical doping/dedoping within a smaller electrochemical window.2. Experimental section
2.1. Materials
2,3-Dihydrothieno[3,4-b][1,4]dioxin-2-yl methanol (hydroxymethyl EDOT), copper(I) iodide, 1-(2-bromoethoxy)-2-(2-methoxyethoxy)ethane, potassium tert-butoxide, dry pyridine, poly(3,4-ethylenedioxythiophene)–poly(styrenesulfonate) (3–4 wt% dispersion in H2O), iron(III) chloride anhydrous, acetonitrile, ammonia, hydrazine, dimethylformamide were purchased from Aldrich. Preparative column chromatography was performed on a glass column with Merck silica gel, 230–400 mesh.2.2. Synthesis
The investigated EDOT derivative containing oligo(oxyethylene) substituent, abbreviated as G-EDOT was prepared by modified Ullmann coupling17 between 2,3-dihydrothieno[3,4-b]-l,4-dioxin-2-yl methanol (HO-EDOT) and 1-(2-bromoethoxy)-2-(2-methoxyethoxy)ethane, using a modification of the procedure previously reported.18 (see Fig. 1a). HO-EDOT can be prepared starting from inexpensive thiodiglycolic acid19 but recently it has become commercially available. The detailed synthetic protocol for G-EDOT, containing spectroscopic characterization data, can be found below.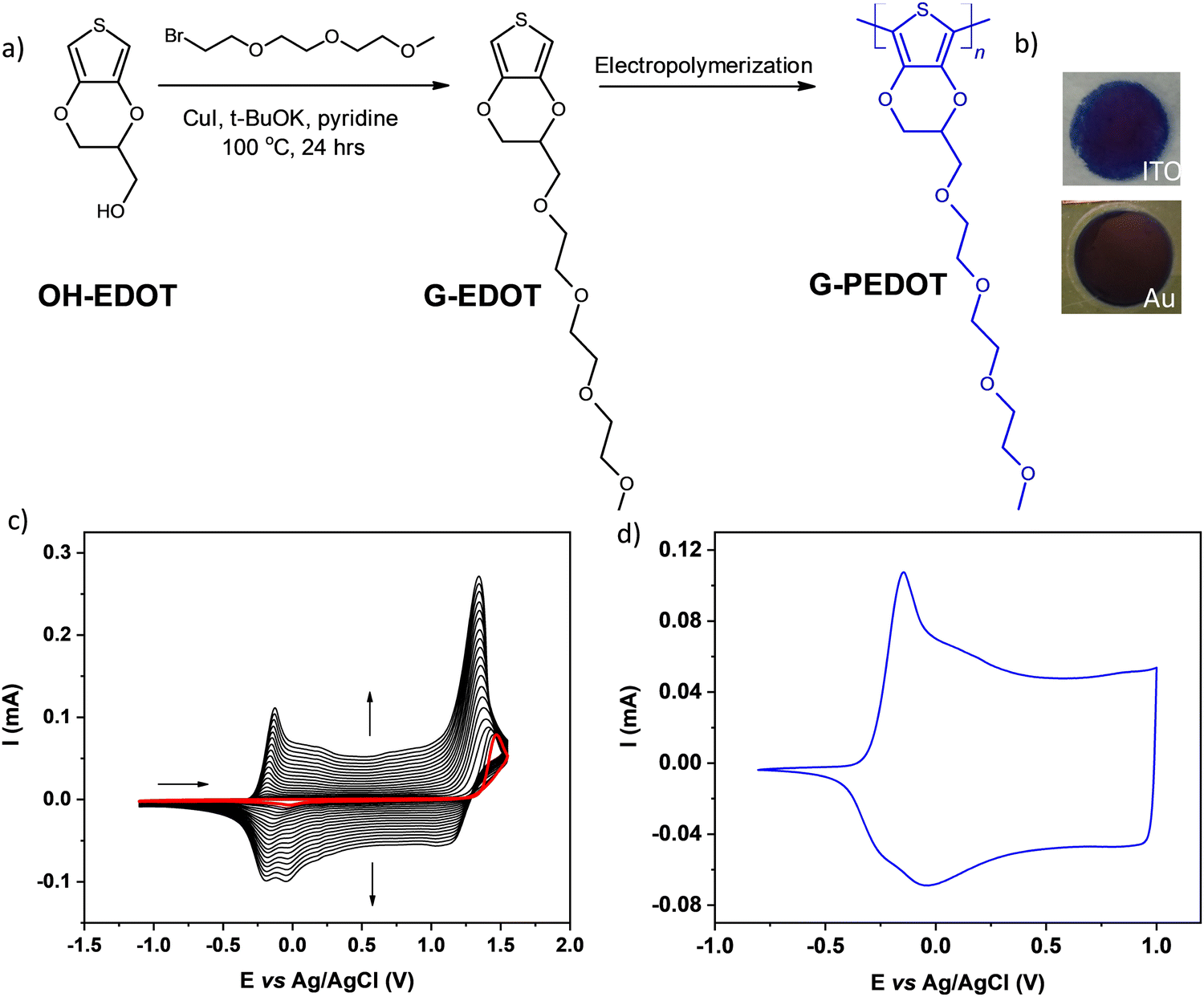 | ||
| Fig. 1 (a) Synthetic route for the preparation of G-EDOT and G-PEDOT. (b) Photographs of 1 cm2 areas of G-PEDOT polymerized on ITO and gold-coated PET substrates. (c) Potentiodynamic electropolymerization of G-EDOT on a platinum electrode immersed in 0.1 M TBAPF6/CH3CN electrolyte containing 5 × 10−3 M of the monomer, first scan is red. Scan rate 100 mV s−1. (d) CV scan of a thin film of G-PEDOT electropolymerized on a Pt electrode. Electrolyte 0.1 M TBAPF6/CH3CN, scan rate 100 mV s−1. | ||
1H NMR (400 MHz, CDCl3) δ 6.33–6.31 (m, 2H), 4.35–4.29 (m, 1H), 4.25 (dd, J = 11.7, 2.3 Hz, 1H), 4.06 (dd, J = 11.7, 7.5 Hz, 1H), 3.77 (dd, J = 10.6, 5.1 Hz, 1H), 3.71–3.63 (m, 11H), 3.56–3.52 (m, 2H), 3.37 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 141.72, 141.64, 99.80, 99.70, 72.77, 72.07, 71.35, 70.79, 70.77, 70.70, 70.69, 69.78, 66.28, 59.19. UV-Vis (CH2Cl2): λmax = 258 nm.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 molar ratio during the overnight room temperature polymerization in acetonitrile. Finally, the G-PEDOT film was rinsed with methanol several times to remove the oxidative agent and dried. Subsequently, the G-PEDOT was soaked in three different solvent mixtures for 48 hours at room temperature: DMF, methanol–ammonia (5:1), and methanol–hydrazine (5:1). Nevertheless, the attempts of chemical reduction followed by solution uptake did not allow for solution processing effectively enough for the next step characterization. Due to the very low solubility of this chemically polymerized and reduced polymer, the results provided in this work have been limited to the electropolymerized G-PEDOT films.
:4 molar ratio during the overnight room temperature polymerization in acetonitrile. Finally, the G-PEDOT film was rinsed with methanol several times to remove the oxidative agent and dried. Subsequently, the G-PEDOT was soaked in three different solvent mixtures for 48 hours at room temperature: DMF, methanol–ammonia (5:1), and methanol–hydrazine (5:1). Nevertheless, the attempts of chemical reduction followed by solution uptake did not allow for solution processing effectively enough for the next step characterization. Due to the very low solubility of this chemically polymerized and reduced polymer, the results provided in this work have been limited to the electropolymerized G-PEDOT films.
2.3. Characterization techniques
1HNMR and 13CNMR spectra were recorded in deuterated chloroform (CDCl3) on a Bruker 400 MHz Instrument spectrometer. UV-Vis absorption spectra of the obtained compounds dissolved in dichloromethane, as well as of the polymer thin films deposited on an ITO electrode, were recorded on a Varian Cary 5000. Electrochemical measurements were conducted using an Ivium Vertex One potentiostat. For electrochemical polymerization, the monomer concentration was 5 × 10−3 M in an electrolyte consisting of 0.1 M TBAPF6 dissolved in deoxygenated CH3CN. The electropolymerization was performed in a single-compartment, three-electrode electrochemical cell with a platinum counter electrode and an Ag/AgCl pseudoreference electrode. Thin films of G-PEDOT and PEDOT (electropolymerization products of G-EDOT and EDOT, respectively) were deposited either on a platinum disk (for CV) or on an ITO (indium–tin oxide) electrode (for UV-vis-NIR spectroelectrochemical investigations), or gold-coated PET or glass. Whereas CV scans for PEDOT:PSS were performed for layers deposited on glass/ITO by drop casting of its water solution (2 μL of ∼0.5 wt% dispersion in water on glass/ITO area of 0.05 cm2). The spectroelectrochemical experiments were carried out in an anhydrous, degassed solution of 0.1 M TBAPF6 in CH3CN and in a deoxygenated 0.1 M KCl aqueous solution as the supporting electrolyte with platinum and Ag/AgCl as counter and pseudoreference electrodes, respectively.2.4. Deposition of G-PEDOT for capacitance determination
To calculate electrochemical capacitance, all G-PEDOT depositions and electrochemical characterizations were done with Ivium technologies Vertex One potentiostat and a PEC 15 mL double-sided electrochemical cell (Redox.me). To facilitate deposition of layers of linearly increasing thickness, galvanostatic electropolymerization (2 electrode arrangement, with Pt coil as a counter electrode) was used, with a constant current of 10 μA (0.2 mA cm−2) and varying deposition time (75, 150, 225 or 300 s). The electrolyte was composed of 3.14 mM (1 mg mL−1) of the monomer and 100 mM HClO4, dissolved in deionized water. ITO coated glass (Kintec, 15 Ohm sq−1, 2 × 2 cm pieces) was used as a substrate, after cleaning by sequential sonication in acetone, isopropanol, and DI water and limiting the active area to 0.05 cm2 by application of a self-adhesive PVC foil with a 2.5 mm ∅ opening. Electrical contact to the working electrode terminal was provided by copper tape. Thickness of layers was measured with a Dektak XT Bruker profilometer. The capacitance calculation was based on the integrals of cyclic voltammograms, recorded in deoxygenated 0.1 M KCl water solution with the scan rate of 100 mV s−1 and a scanning range of −0.1 V to +0.3 V vs. Ag/AgCl reference electrode. A Pt coil was used as a counter electrode.2.5. Fabrication and measurement of PEDOT and G-PEDOT microelectrode arrays (6 pieces, 50 × 50 μm sized electrodes) and preparation of vertical electrochemical transistors (vECT)
Devices were based on 3 × 1′′ glass slides, cleaned by sequential sonication in acetone, isopropanol, 2% Hellmanex, and deionized water. The surface was treated with oxygen plasma (200 W, 600 s, Diener electronic GmbH) before deposition of a base layer of 2 μm parylene-c by CVD (Diener electronic GmbH). Then, 3 nm Pd sticking layer and 100 nm Au layer were deposited by sputtering (Vaksis 3M sputtering system) using a DC magnetron for Au and a RF magnetron for Pd (13.56 MHz), with 100 W power and 3.8–4.0 mTorr pressure of pure argon. The conductive leads between the contact pads and the microelectrodes were patterned using Microposit S1818 G2 photoresist, MF-319 developer and a KI/I2 wet etchant that removed both the Au and the Pd layers. Later, samples were encapsulated with a 2 μm thick layer of parylene-c with 3-(trimethoxysilyl)propyl methacrylate present in the CVD chamber as an adhesion promoter. The openings for microelectrodes and contact pads were patterned by AZ 10XT photoresist, AZ developer and RIE (150 W, O2/CF4 500/100 sccm). The residual resist was removed by acetone, followed by quick rinsing with isopropanol and deionized water yielding the final substrates shown in Fig. S3 (ESI‡).For fair comparison of properties of PEDOT and G-PEDOT, deposition of layers was performed by galvanostatic electropolymerization (2-electrode arrangement) with the same t and I for both monomers. Two sets of samples were prepared, with electrodeposition charge of 2 μC (100 s, 20 nA = 0.8 mA cm−2) and 4 μC (80 s, 50 nA = 2 mA cm−2). The electrolyte (5 mM of a monomer and 100 mM LiClO4 in acetonitrile) was loaded to a modified 1 mL plastic syringe (with a small Pt plate confined inside to serve as a counter electrode) by adjusting the pressure with another syringe connected by a flexible tubing. The sample was precisely positioned under the syringe tip to achieve the electrolytic contact without any mechanical connection. The electrical contact to the working electrode terminal was carefully established with a needle probe placed on the contact pad.
Electrochemical impedance of microelectrodes was measured in a similar way, with a syringe (designated for water-based solutions) confining a platinum plate as a counter electrode and a Ag/AgCl pseudoreference electrode, filled with the electrolyte (Phosphate buffered saline (10 mM phosphate; 138 mM NaCl, 2.7 mM KCl) of pH 7.4) by adjusting the pressure with another syringe connected by a flexible tubing. EIS spectra were recorded within 100 kHz to 1 Hz range (with 20 points per decade) using a sine probing AC voltage of 30 mV amplitude at a DC bias of 0 V vs. the reference electrode.
Fabrication of vECTs was done using the same procedures for parylene deposition and metal patterning as for the metal electrodes, with the additional step of depositing a parylene interlayer. This interlayer has a thickness of 1 μm and defines the vECT channel length, L. The final W/L = 250 μm/1 μm. For vECTs, the active polymer layer was deposited by electropolymerization by short circuiting the source and drain electrodes together to function as the working electrode. A platinum mesh and an Ag/AgCl pellet were used as the counter and reference electrodes, respectively. The electrolyte of 10 mM monomer in 100 mM tetrabutyl ammonium perchlorate was confined in a glass well over the source/drain electrode area. Electropolymerization was done using CV, with a scan rate of 100 mV s−1 and a voltage range of −300 mV to +1000 mV. A total of three cycles were used to complete the polymer channel.
3. Results and discussion
3.1 Electropolymerization and spectroelectrochemistry
For preliminary CV characterization G-PEDOT layers were deposited on a platinum disk. In the investigated potential range during the first scan, red colored in Fig. 1c, the irreversible peak is observed at 1.57 V and one reduction wave at 0.02 V in the reverse scan. This voltammogram was very similar to that reported for EDOT,21 however, its anodic peak maximum is shifted towards higher potentials reflecting slightly more difficult oxidation of G-EDOT. A representative cyclic voltammogram of G-PEDOT on the platinum electrode (Fig. 1d), prepared by potential scanning, consists of the principle oxidative peak in the anodic part at −0.14 V. In the cathodic part of the voltammogram the broad reduction peak is observed at −0.04 V. The introduction of the oligo(oxyethylene) chain on the poly(3,4-ethylenedioxythiophene) backbone leads to CV shape changes compared to the ones reported for PEDOT.22,23 In particular for G-PEDOT, at cathodic potentials < 0 V currents become very low, indicating it is electrochemically depleted, while PEDOT retains significant currents in this potential region.16 This comparison will become more apparent in aqueous electrochemistry differences seen during ECT characterization. Consistent with this electrochemical switching, G-PEDOT shows marked electrochromic color changes of the deposited polymer film of G-PEDOT cycled between reduced and oxidized forms, from deep-blue (reduced state) to transparent (oxidized state) (Fig. 2).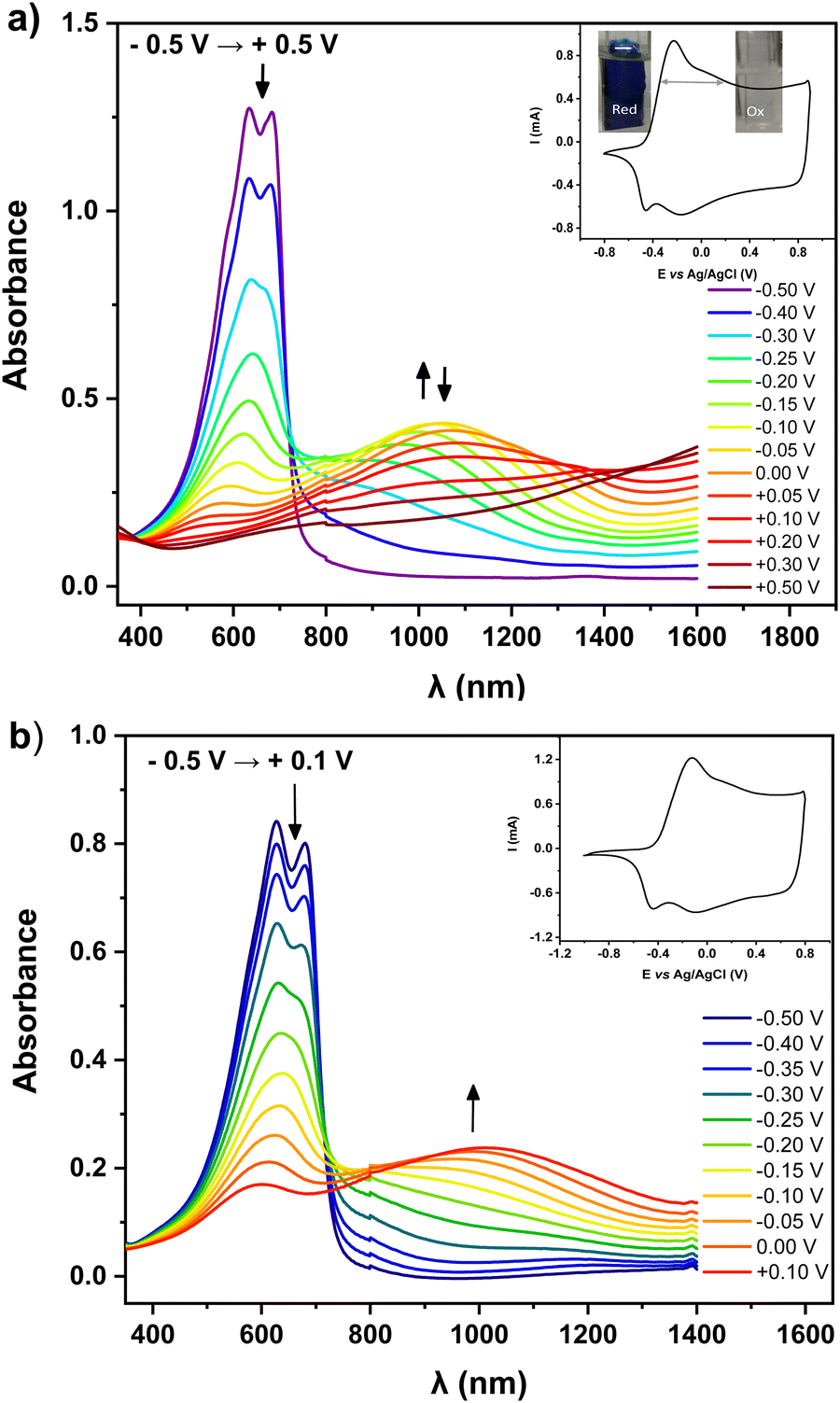 | ||
| Fig. 2 CV scans and UV-vis-NIR spectra of thin films of G-PEDOT deposited on an ITO substrate, recorded for increasing electrode potential vs. Ag/AgCl pseudo-reference wire, under an inert atmosphere of argon. (a) Electrolyte: 0.1 M TBAPF6/CH3CN; (b) electrolyte: 0.1 M KCl/H2O. | ||
To quantify optical changes in detail, in situ spectroelectrochemistry was applied. Spectroelectrochemical measurements of the polymer film deposited on indium tin oxide (ITO) are shown in Fig. 2a and b, for organic and aqueous electrolyte conditions, respectively. UV-Vis-NIR spectral changes of G-PEDOT in acetonitrile using 0.1 M TBAPF6 as an electrolyte and in 0.1 M KCl aqueous solution, induced by the oxidation process, are compared (see Table 1). In the first case, the polymer film was biased at different potentials from E = −0.5 V to E = +0.5 V vs. Ag/AgCl. As seen in Fig. 2a, G-PEDOT film exhibit a dual band with a maximum of 634 nm and 683 nm in the reduced form (E = −0.5 V). Upon electrochemical oxidation, the intensity of these peaks is gradually lost, while a new broad band at about 1028 nm at E = −0.1 V indicating the formation of charge carriers starts to appear and intensify. At the potential of E = +0.5 V, the featureless absorption tail can be observed, extending towards the NIR part of the spectrum.
| Polymer/electrolyte | Reduced form λmax [nm] | Oxidized form λmax [nm] | ΔT% at λ = 600 nm |
|---|---|---|---|
| G-PEDOT in ACN | 634, 683 (−0.5 V) | 610, 1028 (−0.1 V) | 64.8 |
| G-PEDOT in H2O | 627, 680 (−0.5 V) | 601, 1012 (+0.1 V) | 49.1 |
| PEDOT in ACN | 621 (−0.6 V) | 607, 927 (+0.1 V) | 47.3 |
| PEDOT in H2O | 619 (−0.9 V) | 599, 925 (+0.1 V) | 37.5 |
Similar to the organic electrolyte, the oxidation process of the G-PEDOT film in an aqueous solution (Fig. 2b) gives rise to spectral changes characteristic of conducting polymer oxidative doping,24 consistent with its cyclic voltammogram. However, the position of the registered peaks is bathochromically shifted with respect to the corresponding band in Fig. 2a. Its oxidative doping in aqueous solution starts at E = −0.5 V. The increase of the potential results in an apparent decrease in intensity of the dual band located at 627 nm and 680 nm with the simultaneous growth of one broad band with a maximum at 1012 nm at E = +0.1 V. The spectral features during oxidation/reduction of the G-PEDOT are very similar to what has been published previously for poly(3,4-ethylenedioxythiophene) derivatives and can be ascribed to radical cationic and dicationic forms of the polymer chain i.e. in terms of the solid-state physics to positive polarons and bipolarons.25–28 However, in contrast to PEDOT, the G-PEDOT is dedoped in a smaller voltage window. To confirm this, we did control experiments with PEDOT films electropolymerized and measured in the same way (Fig. S1, ESI‡). G-PEDOT rapidly dedopes at negative polarizations in aqueous electrolyte, and by −0.5 V is completely dedoped, evidenced by the saturation of the peaks 627–680 nm (absorption of the neutral form). This absorption in PEDOT, in contrast, does not saturate until −0.9 V.
The doped and dedoped forms of both polymers are strongly coupled to their coloring and bleaching states. Therefore, in this study, the optical contrast (ΔT%)29 of the produced layers, defined as the change in the percentage of transmittance between the oxidized and reduced states of G-PEDOT and PEDOT, was determined. A comparison of the optical contrast of the layers at 600 nm (Fig. S2, ESI‡) showed that the presence of the glycol side chain in the G-PEDOT structure had a larger effect on the optical contrast of the film (Table 1). The G-PEDOT had around 18% and 12% more optical contrast than the PEDOT layers in organic and aqueous solvents, respectively. Such an improvement was attributed to the loose packing of the G-PEDOT chains because of the glycol chain presence, leading to more accessible doping sites and the facile ion movement during the redox switching. Simultaneously, this implies that PEDOT layers are opaquer, and this makes it difficult to effectively change the color between the oxidized and reduced states. Finally, the apparent smaller difference in the potential window needed to reduce and de-dope G-PEDOT versus PEDOT will be shown to be consequential in the on/off voltages for ECTs, discussed in Section 3.3.
3.2 Electrochemical capacitance
Many applications of conducting polymers rely on high electrochemical capacitance. For these, the figures of merit are areal and volumetric capacitance. To test capacitance, we electropolymerized different thicknesses of G-PEDOT on ITO, an electrochemically inert substrate30 with low electrochemical capacitance. The film thickness was verified by profilometry and then capacitance was calculated from cyclic voltammetry curves in a nonfaradaic potential window of −0.1 to 0.3 V versus Ag/AgCl in 0.1 M KCl electrolyte (Fig. 3). The areal capacitance increases linearly with thickness, which is a characteristic property of a volumetric capacitor (Fig. 3a, thickness as a function of polymerization time is plotted in Fig. S3, ESI‡). The values of areal capacitance μF cm−2 and volumetric capacitance μF cm−3 are summarized in Table 2. Volumetric capacitance is similar to reported values for PEDOT:PSS formulations.31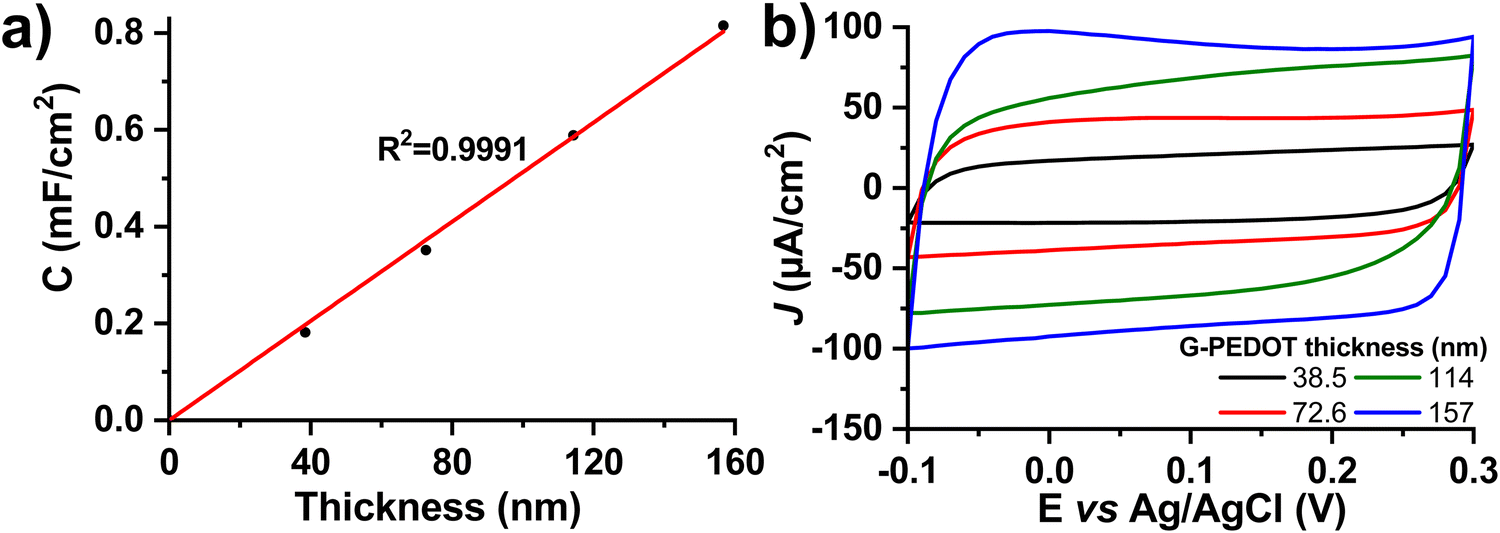 | ||
| Fig. 3 Electrochemical capacitance of G-PEDOT. (a) Linear dependence of areal capacitance on the layer thickness. (b) Cyclic voltammograms (0.1 M KCl, 100 mV s−1) used for calculation of the capacitance. | ||
| Deposition time (s) | Thickness (nm) | Areal capacitance (μF cm−2) | Volumetric capacitance (F cm−3) |
|---|---|---|---|
| 0 (= bare ITO) | 0.0 | 10 | N/A |
| 75 | 38.5 | 181 | 47.2 |
| 150 | 72.6 | 351 | 48.3 |
| 225 | 114 | 588 | 51.5 |
| 300 | 157 | 815 | 52.0 |
3.3 Microelectrodes and electrochemical transistors – comparison with PEDOT
Applications in bioelectronics devices for sensing and neural recording are one of the major focuses of research in PEDOT and new conducting polymers. Therefore, we compared electrochemically polymerized PEDOT with G-PEDOT in microelectrodes, as well as ECTs. Gold microelectrodes 50 × 50 μm in size were used as the substrate for galvanostatic electropolymerization (photomicrograph in Fig. S4, ESI‡), to have comparable films synthesized with the same passed charge (samples were prepared with 2 or 4 μC total passed charge). Cyclic voltammetry of the resultant microelectrodes shows essentially the same behavior and electrochemical capacity for both PEDOT and G-PEDOT (Fig. 4a and b). The microelectrodes demonstrate stability over 4000 cycles. Electrochemical impedance of the microelectrodes (Fig. 4c and d) reveals that modification with electropolymers significantly reduces impedance with respect to the metal microelectrode, and that the impedance traces for both PEDOT and G-PEDOT are essentially the same. An impedance around 4 kOhm @ 1 kHz is a relatively competitive value for a microelectrode of this size,32 and compares favorably with other examples of PEDOT-coated microelectrodes.33 The addition of the glycol chain therefore does not significantly affect the performance in this application with respect to the unfunctionalized PEDOT. All samples were subjected to an accelerated aging test at 60 °C over 41 days, and PEDOT and G-PEDOT remained comparably stable. ESI‡ Fig. S5 and S6 show the measurements from more days during the same aging test shown in Fig. 4, and results from thinner films prepared with 2 μC total passed charge.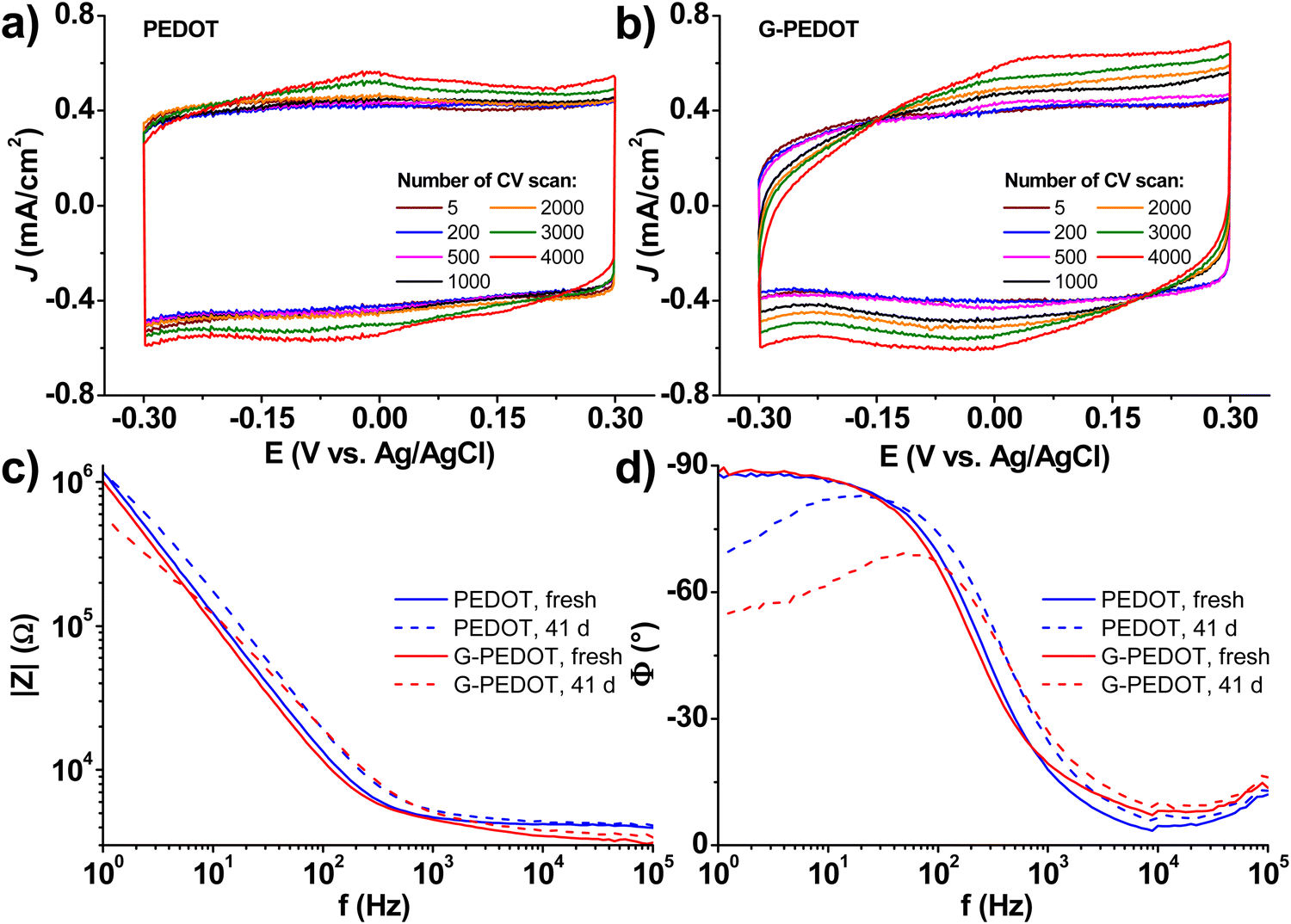 | ||
| Fig. 4 Electrochemistry of PEDOT and G-PEDOT microelectrodes (50 × 50 μm) prepared by galvanostatic electropolymerization (2 μC: 20 nA, 100 s) (a and b): selected cyclic voltammograms (scan rate = 100 mV s−1) over 4000 cycles (electrochemical stability test); (c and d): electrochemical impedance spectra (bode plots, averaged over 2–3 different specimens) as prepared and after 41 days of accelerated aging test (60 °C, PBS solution). | ||
Next, ECTs were fabricated according to the vertical geometry introduced by Donahue et al.,34 featuring a channel length of 1 μm. This vertical geometry is beneficial for deposition by electropolymerization as the insulating distance between source and drain contacts is minimized compared to planar geometries. This makes it easier for an electropolymer to “bridge” the source–drain channel distance. This design is convenient for comparing electropolymerized active polymers, but it should be said that due to relatively large, exposed source–drain contact regions the parasitic capacitance makes their modulation speed limited. To deposit PEDOT or G-PEDOT by electrochemical polymerization, both source and drain electrodes were shorted together as a working electrode and a constant amount of current was passed in each case. We found better and more reproducible results with respect to good adhesion and channel forming, using ClO4− as a counter ion as opposed to PF6−. The resultant ECTs were characterized using an AgCl pellet gate electrode (pseudo-reference electrode), and the transfer curves are plotted in Fig. 5, including gate currents and transconductance values. Both polymers provide similar “ON” currents, indicating that the conductivity of both polymers in their doped forms is similar. However, the depletion mode switching off is different. While scanning to positive gate voltages electrochemically de-dopes both materials, by +800 mV the PEDOT channel conductivity declines by roughly a factor of 50. G-PEDOT, in contrast, by +400 mV is clearly completely depleted and conductivity drops to a low value. As a consequence, the on/off ratio of the G-PEDOT ECT is significantly higher (4 orders of magnitude), within a smaller voltage window than PEDOT. This difference in electrochemical doping and dedoping is very evident when comparing the gate current traces. Whereas the G-PEDOT trace features clear, narrow, doping/dedoping peaks, the peaks in the case of PEDOT are wide and poorly defined. This complete depletion of G-PEDOT at relatively low voltages was anticipated already from the CV curve in organic electrolyte shown in Fig. 1d. We verified this difference between PEDOT, PEDOT:PSS and G-PEDOT by measuring CV as well (Fig. S7a and b, ESI‡). The general shape of the recorded voltammograms is very similar and shows a typical pseudo-capacitive response of quasi-rectangular form. On the other hand, finding, the pinch-off mentioned before in G-PEDOT electrochemistry at negative voltages is observed, whereas PEDOT is still doped at these voltages. The G-PEDOT film was fully reduced by applying a potential at least 400 mV less negative than in PEDOT and PEDOT:PSS cases. This correlates with comparing the spectroelectrochemistry between G-PEDOT and PEDOT (see Section 3.1).
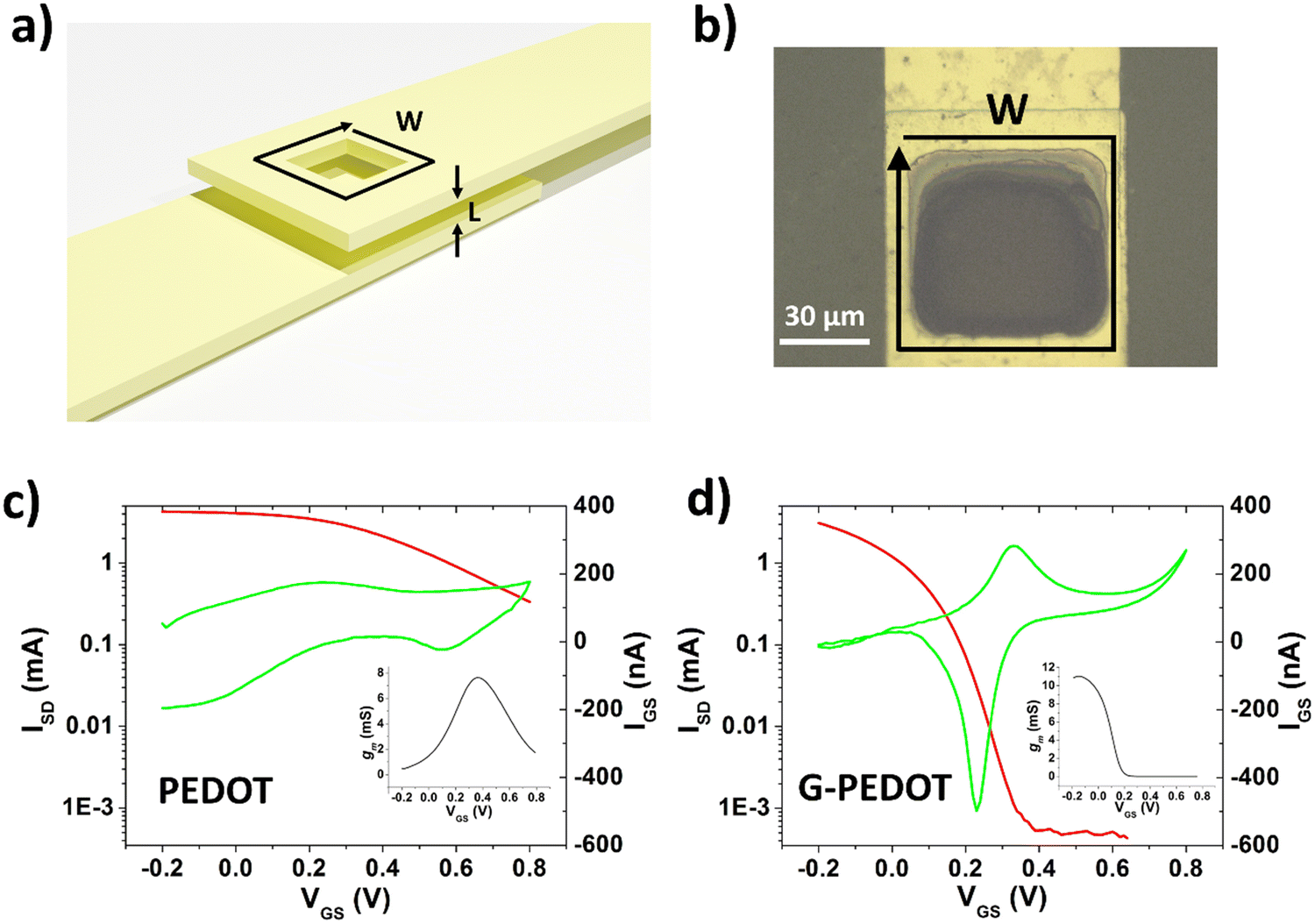 | ||
| Fig. 5 Vertical electrochemical transistors (vECTs). (a) General schematic of a vECT structure, where the channel length is defined by the thickness of an insulator layer (parylene-c, L = 1 μm). (b) Top-view photograph of electropolymerized G-PEDOT vECT. (c and d) transfer curves, VSD = −0.6 V. Red traces are the source–drain current (left y-axis), green represents the gate currents (right y-axis). (c) Electropolymerized PEDOT, compared under the same conditions with electropolymerized G-PEDOT. (d) Three CV cycles were used to complete the channel. | ||
4. Conclusions
Glycolation is a frequently used tool to make conducting polymers which are synthetically manipulatable in organic solvents, yet still hydrophilic enough to function in applications where ionic exchange from water is necessary. In this study, we have endeavored to critically evaluate the effects of glycolation on the archetypical conducting polymer PEDOT. In many respects, G-PEDOT gives properties, such as electrochemical capacitance, very similar to PEDOT. The most significant difference is that electrochemical switching occurs in a smaller window, which is apparent in the ECT application and also in the electrochromic effect. In effect, G-PEDOT is easier to dedope than PEDOT. The higher on/off ratio and smaller voltage window for G-PEDOT ECTs shows that this derivative could be a better choice for some ECT applications than using PEDOT. Nevertheless, our results have been limited to electropolymerization. We have spent considerable effort to conduct bulk electropolymerization and chemical oxidative polymerization, followed by reduction and rinsing of electrolyte or oxidant, respectively, to then re-uptake the polymer in a suitable solvent for solution processing. While this approach works in principle, it is difficult to implement. Our efforts were hampered by very low solubility of these electro- and chemically synthesized and reduced polymers, therefore pure G-PEDOT may be impossible to solution process in a useful way. However, this finding together with the results on interesting electrochemical properties suggest that the glycolated EDOT unit may be a useful component to incorporate into copolymers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for support from the National Science Centre, Poland, within grant 2019/33/B/ST5/01212; funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant agreement No. 949191); funding from the city council of Brno, Czech Republic; and CzechNanoLab Research Infrastructure supported by MEYS CR (LM2018110).References
- J. Rivnay, R. M. Owens and G. G. Malliaras, Chem. Mater., 2014, 26, 679–685 CrossRef CAS.
- D. T. Simon, E. O. Gabrielsson, K. Tybrandt and M. Berggren, Chem. Rev., 2016, 116, 13009–13041 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- A. Giovannitti, D. T. Sbircea, S. Inal, C. B. Nielsen, E. Bandiello, D. A. Hanifi, M. Sessolo, G. G. Malliaras, I. McCulloch and J. Rivnay, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12017–12022 CrossRef CAS PubMed.
- A. Savva, R. Hallani, C. Cendra, J. Surgailis, T. C. Hidalgo, S. Wustoni, R. Sheelamanthula, X. Chen, M. Kirkus, A. Giovannitti, A. Salleo, I. McCulloch and S. Inal, Adv. Funct. Mater., 2020, 30, 1907657 CrossRef CAS.
- D. Moia, A. Giovannitti, A. A. Szumska, I. P. Maria, E. Rezasoltani, M. Sachs, M. Schnurr, P. R. F. Barnes, I. McCulloch and J. Nelson, Energy Environ. Sci., 2019, 12, 1349–1357 RSC.
- A. Giovannitti, I. P. Maria, D. Hanifi, M. J. Donahue, D. Bryant, K. J. Barth, B. E. Makdah, A. Savva, D. Moia, M. Zetek, P. R. F. Barnes, O. G. Reid, S. Inal, G. Rumbles, G. G. Malliaras, J. Nelson, J. Rivnay and I. McCulloch, Chem. Mater., 2018, 30, 2945–2953 CrossRef CAS PubMed.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, 17086 CrossRef CAS.
- R. J. Mortimer, Chem. Soc. Rev., 1997, 26, 147–156 RSC.
- D. Khodagholy, J. Rivnay, M. Sessolo, M. Gurfinkel, P. Leleux, L. H. Jimison, E. Stavrinidou, T. Herve, S. Sanaur, R. M. Owens and G. G. Malliaras, Nat. Commun., 2013, 4, 1–6 Search PubMed.
- R. B. Rashid, X. Ji and J. Rivnay, Biosens. Bioelectron., 2021, 190, 113461 CrossRef CAS PubMed.
- P. Jastrzebska-Perfect, S. Chowdhury, G. D. Spyropoulos, Z. Zhao, C. Cea, J. N. Gelinas, D. Khodagholy, P. Jastrzebska-Perfect, S. Chowdhury, G. D. Spyropoulos, Z. Zhao, C. Cea, J. N. Gelinas and D. Khodagholy, Adv. Funct. Mater., 2020, 30, 1909165 CrossRef CAS.
- X. Chen, A. Marks, B. D. Paulsen, R. Wu, R. B. Rashid, H. Chen, M. Alsufyani, J. Rivnay and I. McCulloch, Angew. Chem., Int. Ed., 2020, 60, 9368 CrossRef PubMed.
- I. P. Maria, B. D. Paulsen, A. Savva, D. Ohayon, R. Wu, R. Hallani, A. Basu, W. Du, T. D. Anthopoulos, S. Inal, J. Rivnay, I. Mcculloch and A. Giovannitti, Adv. Funct. Mater., 2021, 31, 2008718 CrossRef CAS.
- R. B. Rashid, W. Du, S. Griggs, I. P. Maria, I. McCulloch and J. Rivnay, Sci. Adv., 2021, 7, 1–10 Search PubMed.
- S. Akoudad and J. Roncali, Electrochem. Commun., 2000, 2, 72–76 CrossRef CAS.
- F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS PubMed.
- D. K. Hwang, C. Fuentes-Hernandez, M. Fenoll, M. Yun, J. Park, J. W. Shim, K. A. Knauer, A. Dindar, H. Kim, Y. Kim, J. Kim, H. Cheun, M. M. Payne, S. Graham, S. Im, J. E. Anthony and B. Kippelen, ACS Appl. Mater. Interfaces, 2014, 6, 3378–3386 CrossRef CAS PubMed.
- A. Lima, P. Schottland, S. Sadki and C. Chevrot, Synth. Met., 1998, 93, 33–41 CrossRef CAS.
- Q. Zhao, R. Jamal, L. Zhang, M. Wang and T. Abdiryim, Nanoscale Res. Lett., 2014, 9, 557 CrossRef PubMed.
- M. Łapkowski and A. Proń, Synth. Met., 2000, 110, 79 CrossRef.
- M. Dietrich, J. Heinze, G. Heywang and F. Jonas, J. Electroanal. Chem., 1994, 369, 87–92 CrossRef CAS.
- A. Zykwinska, W. Domagala and M. Lapkowski, Electrochem. Commun., 2003, 5, 603–608 CrossRef CAS.
- S. Garreau, J. L. Duvail and G. Louarn, Synth. Met., 2002, 125, 325–329 CrossRef CAS.
- M. Marzocchi, I. Gualandi, M. Calienni, I. Zironi, E. Scavetta, G. Castellani and B. Fraboni, ACS Appl. Mater. Interfaces, 2015, 7, 17993–18003 CrossRef CAS PubMed.
- X. Duan and J. Xu, Int. J. Electrochem. Sci., 2015, 10, 3236–3249 Search PubMed.
- N. Massonnet, A. Carella, O. Jaudouin, P. Rannou, G. Laval, C. Celle and J. P. Simonato, J. Mater. Chem. C, 2014, 2, 1278–1283 RSC.
- V. David, C. Viñas and F. Teixidor, Polymer, 2006, 47, 4694–4702 CrossRef CAS.
- J.-H. Kang, Y.-J. Oh, S.-M. Paek, S.-J. Hwang and J.-H. Choy, Sol. Energy Mater. Sol. Cells, 2009, 93, 2040–2044 CrossRef CAS.
- J. D. Benck, B. A. Pinaud, Y. Gorlin and T. F. Jaramillo, PLoS One, 2014, 9, e107942 CrossRef PubMed.
- C. M. Proctor, J. Rivnay and G. G. Malliaras, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1433–1436 CrossRef CAS.
- C. Boehler, S. Carli, L. Fadiga, T. Stieglitz and M. Asplund, Nat. Protoc., 2020, 15, 3557–3578 CrossRef CAS PubMed.
- G. Nikiforidis, S. Wustoni, C. Routier, A. Hama, A. Koklu, A. Saleh, N. Steiner, V. Druet, H. Fiumelli and S. Inal, Macromol. Biosci., 2020, 20, 1–9 CrossRef PubMed.
- M. J. Donahue, A. Williamson, X. Strakosas, J. T. Friedlein, R. R. McLeod, H. Gleskova and G. G. Malliaras, Adv. Mater., 2018, 30, 1705031 CrossRef PubMed.
Footnotes |
| † This paper is dedicated to Prof. Adam Proń on occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc01448c |
| This journal is © The Royal Society of Chemistry 2022 |