
Chemically functionalized phosphorenes and their use in the water splitting reaction
Pratap
Vishnoi
 *,
Aditi
Saraswat
and
C. N. R.
Rao
*
*,
Aditi
Saraswat
and
C. N. R.
Rao
*
New Chemistry Unit, School of Advanced Materials, International Centre for Materials Science, Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur, Bangalore, 560064, India. E-mail: pvishnoi@jncasr.ac.in; cnrrao@jncasr.ac.in
First published on 6th June 2022
Abstract
In the last 7 years, phosphorene (or few layer black phosphorus) has emerged as not only a superior optoelectronic material, but also a potential catalyst for the hydrogen evolution reaction (HER) from water splitting owing to its thickness dependent bandgap, broad spectrum light absorption, high charge-carrier mobility, and high density of surface-active sites. However, pristine phosphorene produces trace amounts of H2 primarily due to its poor ambient stability and a large positive change in the Gibbs free energy of hydrogen (H*) adsorption/desorption (ΔGH* > 0). Due to the recent surge of interest in metal-free HER catalysts, there have been many successful efforts on enhancing the stability as well as the catalytic activity of phosphorene through chemical functionalization, metal doping, and 2D heterocomposites. In this perspective, we present different types of interactions, including covalent, coordination, electrostatic, van der Waals, charge-transfer, and interfacial, which have been utilized in preparing modified phosphorenes. Then, we assess the noteworthy properties of phosphorene, which make it an efficient HER catalyst and discuss the developments in photocatalytic, electrocatalytic, and photo-electrocatalytic means of H2 production using phosphorene based catalysts. We conclude the perspective with suggestions for exciting future developments.
 Pratap Vishnoi | Pratap Vishnoi is a Ramanujan faculty fellow at the Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Bangalore. He received his Ph. D. in chemistry from the Indian Institute of Technology Bombay (IIT Bombay) under the supervision of Prof. R. Murugavel. He then worked as a post-doctoral fellow at the JNCASR with Prof. C. N. R. Rao and subsequently at the University of California Santa Barbara (UCSB) with Prof. Ram Seshadri and Prof. Sir Anthony K. Cheetham. His current research interests are in multifunctional hybrid organic-inorganic materials, halide perovskites and layered nanomaterials. |
 C. N. R. Rao | C. N. R. Rao is the Linus Pauling Research Professor at the JNCASR, Bangalore and Honorary Professor at the Indian Institute of Science, Bangalore. He is the member of several scientific academies, including the Royal Society and the US National Academy of Sciences. He is the recipient of several prestigious awards, including the Royal Medal of the Royal Society, the Dan David Prize, Illy Trieste Science Prize, the August von Wilhelm Hoffmann medal of the German Chemical Society, the von Hippel Award of the Materials Research Society, Eni International Award for Research in Energy Frontiers and Sheikh Saud International Prize for Materials Science. His research interests are in the chemistry of materials, especially 2D materials and their chemical functionalization, reduction of water and CO2 and aliovalent substitution. |
1. Introduction
Heterogeneous catalysis of the hydrogen evolution reaction (HER) by water splitting (H2O → H2 + ½O2; ΔGo = +237 kJ mol−1) is deemed to be a prospective solution for the energy crisis and detrimental environmental effects caused by burning fossil fuels. Among numerous materials, which have been identified as the potential HER catalysts after decades of research, state-of- the-art Pt and Pt group metals have remained the best performing catalyst.1,2 However, the applicability of Pt based systems is limited by their high cost, limited availability, and lack of tolerance to catalyst poisoning.3 To reduce and hopefully eliminate the dependence on metal-based catalysts, there is a surge of interest in catalysts comprising non-metal and earth abundant elements, such as B, C, N, and P. For example, carbon nitride,4 doped graphene, and graphene hybrids,4,5 borocarbonitrides,3 and phosphorene (few layer black phosphorus)6 hold promise for efficient HER catalysts.Phosphorene is the isolated monolayer of black phosphorus (BP), in a similar way as graphene is the isolated monolayer of graphite. It was first obtained in 2014 from the exfoliation of BP crystals.7 During the last 7 years, it has witnessed a major renaissance in a range of applications, including in transistor,8 photodetector,9 gas sensor,10 and catalysis.11 Perhaps, the HER catalysis is one of the most intriguing applications of phosphorene. Phosphorene has a thickness dependent bandgap of 0.3 eV (bulk) to 2.0 eV (monolayer), wide spectrum light absorption capability, high charge carrier mobility, efficient electron–hole separation, and high density of exposed active sites.7,12 Its conduction band lies at an appropriate energy level for the HER, i.e. water reduction.13 However, its inherent HER activity is rather poor, mainly due to ambient instability and a large positive change in the Gibbs free energy of adsorption/desorption for hydrogen intermediate state (ΔGH* > 0).14 The instability is caused by the reactive nature of non-bonding lone pair electrons (LPEs) in phosphorus. The other side of the coin is that the LPEs can be utilized non-destructively in chemical functionalization, which not only protects phosphorene against ambient decomposition but also reduces its ΔGH*. Furthermore, phosphorene based composites have been identified as potential photocatalysts for CO2 reduction.15,16 Among the emerging CO2 conversion techniques, semiconductor based photocatalysis gives rise advantages of utilizing renewable solar energy as well as forming value added chemicals including CO, CH4, and CH3OH.17
Few-layer black phosphorus/phosphorene and its composites have emerged as leading metal-free HER catalysts ever since the first report in 2016.18 The number of publications has remarkably increased (Fig. 1). In recent reviews, the HER application of phosphorene has been summarized along with vastly different topics, including synthetic methods, fundamental properties, and diverse applications.19–21 In this perspective, we provide the highlights of phosphorene based electrocatalysis, photocatalysis, and photo-electrocatalysis of H2 evolution. We start with a brief overview of the fundamental properties of phosphorene and present the key developments on its ambient stabilization through chemical functionalization and 2D-heterostructures. Then, we discuss the developments in H2 production by phosphorene-based catalysts and highlight how they show superior activities, in some cases even close to that of state-of-the-art Pt and Pt group metals. We also provide suggestions for future opportunities.
 | ||
| Fig. 1 (a) Number of research publications on few-layer phosphorus/phosphorene based H2 evolution catalysts. The publications were obtained from web of science on May 06, 2022, with the search keywords combinations of either “phosphorene” and “hydrogen”, “black phosphorus” and “hydrogen” or “black phosphorene” and “hydrogen”. Only those research articles have been included which show experimental reports on H2 evolution from water splitting, whilst purely theoretical reports have been excluded. Only those review articles have been included in which experimental results on HER have been summarized. (b) A timeline of the key milestone developments of few-layer black phosphorus/phosphorene based HER catalysts. | ||
2. History and renaissance
The history of phosphorene traces back to 1914 when Bridgman synthesized BP, whilst studying high pressure phase transition in white phosphorus.22 For a century, between 1914 and 2014, the work on BP progressed steadily on its structural,23 electric transport,24 and superconductivity aspects.25 Inspired by the discovery of graphene from graphite in 2004,26 and its extraordinary transport properties revealed afterwards, the semiconductor research community has explored the exfoliation of many other existing layered materials into their nanosheets. In 2014, Liu et al.7 isolated monolayer phosphorene from scotch tape exfoliation of BP crystal, which captured wide research attention. Since then, phosphorene has seen a major renaissance in nanoscience and nanotechnology. This has also led to a revival of the interest in the bulk properties of BP.27,283. Crystal structure
BP has three pressure dependent polymorphs at room temperature:27 (1) semiconducting orthorhombic phase (A17, Cmca) at the ambient pressure, (2) semimetallic rhombohedral phase (A7, R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) at 5 < P < 11 GPa, and (3) metallic simple cubic phase (sc, Pmm) at ≥ 11 GPa. The orthorhombic phase is the thermodynamically most stable form among all the allotropes of phosphorus. It has a buckled layered structure with covalent P–P bond within the layer and weak van der Waals interaction between the layers (Fig. 2a).29 The layers stack along the b-axis at a distance of 5.23 Å. Each monolayer is a bilayer of two parallel atomic planes comprising sp3 hybridized P atoms. Due to sp3 hybridization, phosphorene adopts a puckered structure, unlike graphene, which is an atomically flat layer of sp2 carbon. The puckered arrangement of atoms makes phosphorene sheet strongly anisotropic along the armchair and the zigzag directions, corresponding to the x-direction (c-axis) and y-direction (a-axis), respectively (Fig. 2b).30 Three of the five valence electrons in phosphorus form the covalent P–P bonds, whilst two remain as the non-bonding lone pair (Fig. 2c and d). Due to puckering, the sheet shows structural anisotropy with smaller P–P distances and ∠P–P–P angles along the zigzag directions compared to those along the armchair direction. Many of the properties of phosphorene, including optical absorption,31 emission,32 thermal conductivity,33 and electric conductivity7 differ along these two directions. Further, the interlayer P–P distance of 3.60 Å suggests that there is a weak van der Waals interaction between the layers, which allows easy exfoliation of BP crystals into thin nanosheets.12 BP crystals can be exfoliated by liquid,34 mechanical,7 and electrochemical methods.35
m) at 5 < P < 11 GPa, and (3) metallic simple cubic phase (sc, Pmm) at ≥ 11 GPa. The orthorhombic phase is the thermodynamically most stable form among all the allotropes of phosphorus. It has a buckled layered structure with covalent P–P bond within the layer and weak van der Waals interaction between the layers (Fig. 2a).29 The layers stack along the b-axis at a distance of 5.23 Å. Each monolayer is a bilayer of two parallel atomic planes comprising sp3 hybridized P atoms. Due to sp3 hybridization, phosphorene adopts a puckered structure, unlike graphene, which is an atomically flat layer of sp2 carbon. The puckered arrangement of atoms makes phosphorene sheet strongly anisotropic along the armchair and the zigzag directions, corresponding to the x-direction (c-axis) and y-direction (a-axis), respectively (Fig. 2b).30 Three of the five valence electrons in phosphorus form the covalent P–P bonds, whilst two remain as the non-bonding lone pair (Fig. 2c and d). Due to puckering, the sheet shows structural anisotropy with smaller P–P distances and ∠P–P–P angles along the zigzag directions compared to those along the armchair direction. Many of the properties of phosphorene, including optical absorption,31 emission,32 thermal conductivity,33 and electric conductivity7 differ along these two directions. Further, the interlayer P–P distance of 3.60 Å suggests that there is a weak van der Waals interaction between the layers, which allows easy exfoliation of BP crystals into thin nanosheets.12 BP crystals can be exfoliated by liquid,34 mechanical,7 and electrochemical methods.35
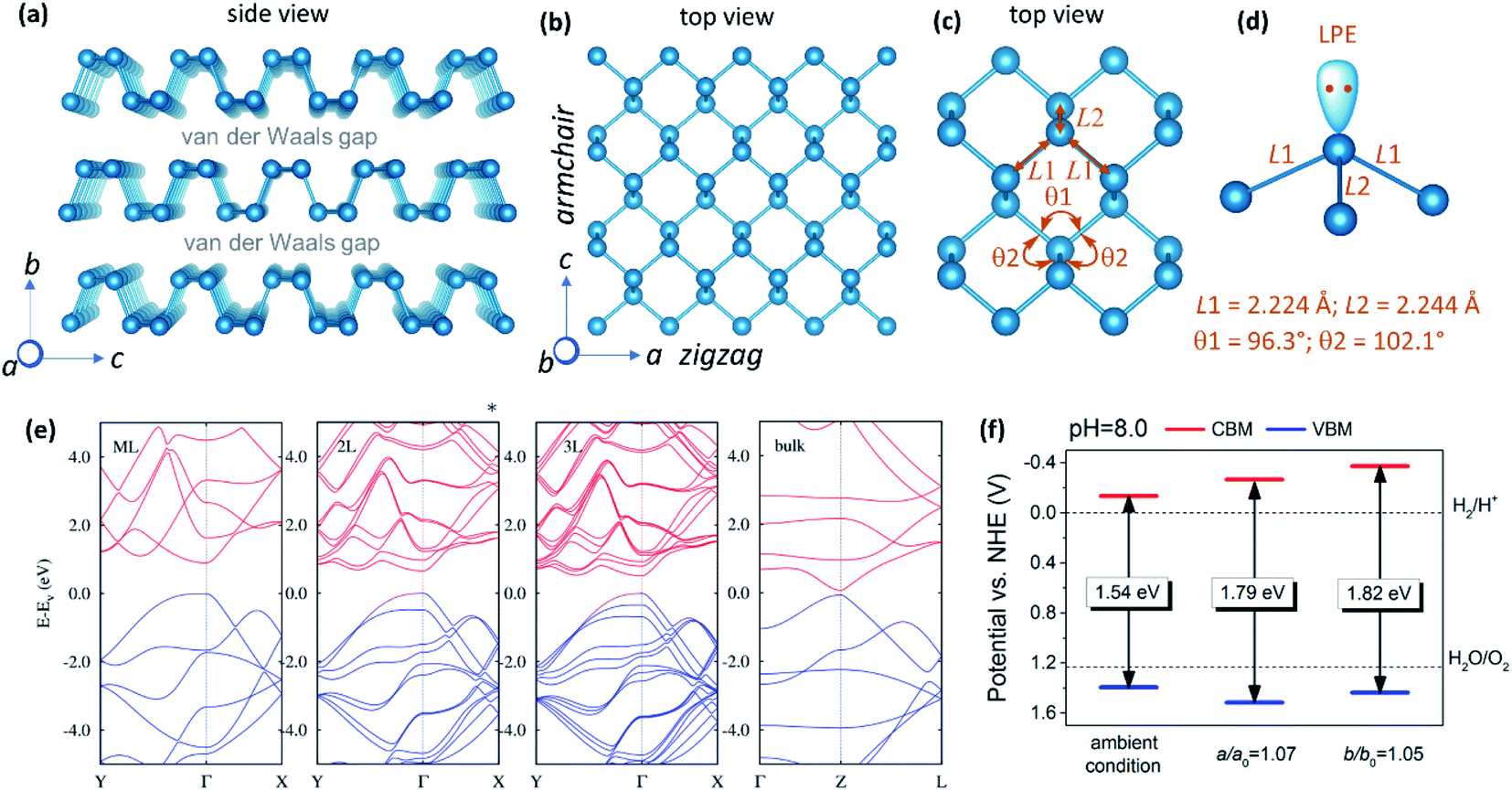 | ||
| Fig. 2 (a) Crystal structure of BP drawn from the crystal information file (CIF) reported elsewhere (orthorhombic Cmca space group; a = 3.3136 Å, b = 10.4788 Å and c = 4.3763 Å; Z = 8).29 (b) Structure of phosphorene sheet showing armchair and zigzag directions. (c) A portion of phosphorene sheet showing P–P bonds and ∠P–P–P angles. (d) Phosphorus atom showing three P–P bonds and a non-bonding lone pair electron. (e) Calculated band structures of monolayer (ML), bilayer (2L), trilayer (3L), and bulk BP. The occupied and unoccupied bands are shown in blue and red, respectively. (f) Calculated (using HSE06 functional) relative energy levels of the conduction and the valance bands of phosphorene under 5% tensile strain along the armchair direction and under 7% tensile strain along the zigzag direction at pH 8.0. Panel (e) is reproduced with permission from ref. 37, © 2014 American Physical Society. Panel (f) is reproduced with permission from ref. 13, © 2014 American Chemical Society. | ||
4. Electronic band structure
The electronic structure and properties of phosphorene have been extensively studied by optical absorption,34 photoluminescence,7,36 and DFT calculations.37 It exhibits a direct bandgap regardless of the thickness, unlike indirect to direct band crossover in transition metal dichalcogenides when exfoliating their bulk crystals into monolayer materials.38 The direct band nature of bulk BP has been experimentally confirmed from the angle-resolved photoemission spectroscopy (ARPES) measurements.8 On increasing the number of layers, the bandgap gradually decreases from ∼2.0 eV to ∼0.30 eV (Fig. 2e).37 It has been suggested that the interlayer van der Waals interaction plays an important role in the layer-dependence of the bandgap. Stronger interaction causes a wider band dispersion and a narrower bandgap with concomitant splitting of the bands as reflected in the band structure of 2L and 3L phosphorene (Fig. 2e).37 The band dispersion is anisotropic along the Γ–X (corresponding to the armchair direction) and Γ–Y (corresponding to the zigzag direction) directions.31 Wide bandgap tuneability along with the anisotropic behaviour of electrons, photons, and phonons make phosphorene a promising material for optoelectronics and related applications. Below are the unique aspects of the electronic structure of phosphorene that favour the HER catalysis: (i) Thickness dependent bandgap: the thickness tuneable bandgap makes phosphorene an excellent material for absorbing photons in the visible and the near-infrared regions of the electromagnetic spectrum to effectively generate excited electrons and holes. (ii) High carrier mobility: conductivity is a prerequisite for photocatalysis and electrocatalysis. Due to high charge carrier mobility, phosphorene exhibits effective separation of charge carriers, and their migration to the catalyst surface. (iii) Band position: calculations have suggested that conduction band minima (CBM) and valence band maxima (VBM) of unstrained phosphorene lie above the required potential for the water reduction reaction (2H+ + 2e−→ H2; E0 = 0.0 V vs. NHE at pH = 7) as well as the water oxidation reaction (H2O → ½O2 + 2e−; E0 = +1.23 V vs. NHE at pH = 7),13,39 (NHE = normal hydrogen electrode), making it suitable for HER catalysis in the presence of a hole scavenger. The utility of phosphorene as a photocatalytic oxygen evolution reaction (OER) is limited. However, at a slightly high pH of 8.0 and intraplanar strain, CBM and VBM of phosphorene straddle the water reduction and oxidation potentials (Fig. 2f),13 thereby making it suitable for overall water splitting.5. Ambient degradation and protection
Although phosphorene is thermodynamically stable, it is chemically unstable due to the reactive nature of LPEs. Under ambient conditions, phosphorene can form P–O and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moieties on its surface by reacting with oxygen.40–42 These surface moieties then absorb water molecules and eventually lead to the oxidation of the phosphorene sheet. In oxygenated water, phosphorene decomposes into water soluble acids, including H3PO2, H3PO3, and H3PO4 as the major products, and the decomposition preferentially occurs on the edges.43 Furthermore, visible light irradiation promotes the decomposition process.44,45 Ambient decomposition suppresses the electronic and the physical properties of phosphorene, which is a major bottleneck for its real-time applications, including HER. Regardless of the instability, the LPEs in phosphorus provide plenty of opportunities to non-destructively interact with a range of electron accepting functionalities through covalent and noncovalent interactions. The functional groups are expected to reduce the LPE density on the phosphorene surface and suppress its reactivity towards oxygen. Furthermore, the functional groups provide shielding against direct contact with reactive species in the ambience and enhance the stability as well as many of the useful properties of phosphorene. Fig. 3 shows various types of interactions, which have been employed for phosphorene functionalization.
O moieties on its surface by reacting with oxygen.40–42 These surface moieties then absorb water molecules and eventually lead to the oxidation of the phosphorene sheet. In oxygenated water, phosphorene decomposes into water soluble acids, including H3PO2, H3PO3, and H3PO4 as the major products, and the decomposition preferentially occurs on the edges.43 Furthermore, visible light irradiation promotes the decomposition process.44,45 Ambient decomposition suppresses the electronic and the physical properties of phosphorene, which is a major bottleneck for its real-time applications, including HER. Regardless of the instability, the LPEs in phosphorus provide plenty of opportunities to non-destructively interact with a range of electron accepting functionalities through covalent and noncovalent interactions. The functional groups are expected to reduce the LPE density on the phosphorene surface and suppress its reactivity towards oxygen. Furthermore, the functional groups provide shielding against direct contact with reactive species in the ambience and enhance the stability as well as many of the useful properties of phosphorene. Fig. 3 shows various types of interactions, which have been employed for phosphorene functionalization.
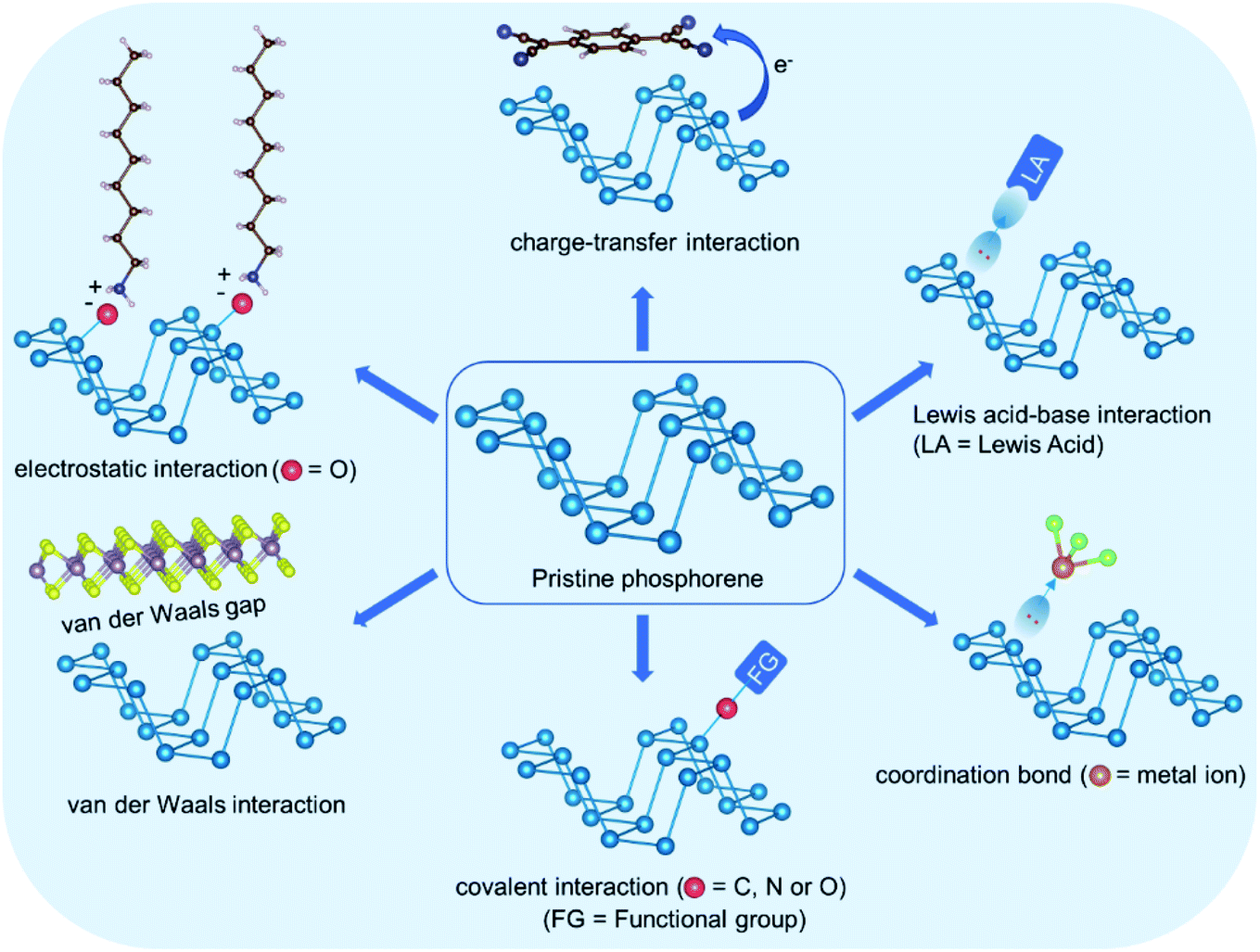 | ||
| Fig. 3 Schematic overview of various types of interactions employed in phosphorene functionalization. | ||
5.1. By covalent interaction
The LPE can form covalent bonds, P–X (X = C, Si, N or O), with appropriate functional groups. Ryder et al.46 have reacted BP nanosheets of ∼10 nm thickness with aryl diazonium salts to attach aryl groups through P–C bonds, which not only stabilized BP under the ambient conditions, but also improved the carrier mobility through p-type doping. The functionalized phosphorus adopts four-coordinate bonding, in a similar way as molecular phosphines form phosphonium compounds with aryl diazonium salts.47 Rao and coworkers48 have synthesized phosphorene ylide from the reaction of liquid exfoliated phosphorene with benzyl bromide. The resultant ylide has been reacted with an aldehyde to form alkene via a Wittig type of reaction. Although P–C bonds firmly hold the functional groups on phosphorene, the four-coordinate phosphorus remains coordinatively unsaturated, due to which an optimal degree of functionalization still cannot be reached. Under ultraviolet (UV) light irradiation, phosphorene has been functionalized with organic azides (R–N3) through PN bonds wherein the functionalized phosphorus atoms are five-coordinated.49,50 The UV light induces the formation of reactive nitrene species from azide, which then attach to phosphorene sheets through PN bonds. Due to five-coordinate bonding, the reactivity of phosphorene towards oxygen reduces significantly, leading to much better ambient stability. Amino (–NH2) functionalized BP nanosheets have been synthesised by ball milling BP crystals with urea, which simultaneously enhances the stability and the HER activity.51 Other studies have shown that hydroxyl groups formed on partially oxidized phosphorene can react with alkyl alcohol (R–OH)52 and trimethylsilyl chloride (TMS–Cl)53 to form P–OR and P–OSi bonds, respectively. The covalent interaction has also been adopted as an efficient strategy to form 2D heterostructures of phosphorene with other important layered materials, such as MoX2 (X = S or Se),48 and g-C3N4.54
5.2. By forming Lewis acid–base complex
Phosphorene acts as a Lewis base due to the LPEs, and it can, therefore, interact with Lewis acid (LA), just like molecular phosphines.55 The Lewis acid–base interaction is expected to make LPEs inaccessible to oxygen.56 Rao and coworkers have reported the adducts of phosphorene with B(C6F5)3 and InCl3. B(C6F5)3 gives rise to better protection owing to stronger electrophilicity and sterical hindrance.11 A recent study on a series of LAs including BBr3, B(C6F5)3, BPh3, Al(OCOR)3, Al(OsBu)3, AlCl3, AlBr3, and GaCl3 has revealed that electrophilicity, steric hindrance, and Pearson hard/soft-ness can be used as the key parameters to qualitatively access the stability of the functionalized phosphorene.57 Highly electrophilic AlCl3, AlBr3, and GaCl3 provide better protection and also cause p-type doping of phosphorene.575.3. By forming coordinating bonds
Metals with empty or partially filled d- or f-orbitals show strong electron affinity and can, therefore, coordinatively bind with phosphorene LPEs. For example, coordinatively unsaturated TiL4 (L = p-toluenesulfonate),58 LnL3 (Ln = Tb, Eu or Nd; L = trifluoromethanesulfonate)59 have been reported to coordinatively interact with phosphorene. The functionalized phosphorenes show excellent stability in air and water. In addition, the lanthanide tagged phosphorenes can potentially be used as contrast agents in magnetic resonance imaging (MRI). Furthermore, the sulfonate ligands strengthen the coordinating ability of metal ions. Ir-complex60 and cisplatin61 have also been attached to phosphorene, and the resultant phosphorenes have been examined as drug delivery agents.Further, phosphorene has been functionalized with chemically adsorbed metal atoms or ions, called adatom. Using sputtered deposition, Cu atoms have been deposited on phosphorene surface and interstitial.62 At both locations, each Cu transfers one electron to phosphorene and oxidizes into a more stable Cu+ state (d10). Thus, the Cu modified phosphorene behaves as an n-type semiconductor, with a remarkable increase in electron mobility from 380 cm2 V−1 s−1 at 300 K to 2140 cm2 V−1 s−1 at 7 K. While there is no experimental report on direct deposition of Cu+ on phosphorene, but Ag+ adatom has been achieved via cation–π interactions, which causes p-type doping of phosphorene with the room temperature hole mobility increasing from 796 cm2 V−1 s−1 to 1593 cm2 V−1 s−1.63 It was proposed that Ag+ ion interacts with three distinct LPEs through η3-coordination. Noble metals such as dipallada unit (Pd2) have been sandwiched between phosphorene sheets.64 The extended X-ray absorption fine structure (EXAFS) analysis and the DFT modelling have highlighted that the coordination of Pd2 occurs between the layer, bridging two phosphorene sheets.
5.4. By charge transfer interaction
Electron withdrawing organic molecules, such as 7,7,8,8-tetracyano-p-quinodimethane (TCNQ)65 and tetracyanoethylene (TCNE),66 accept electrons from phosphorene and form charge transfer (CT) interaction. It has been found that the Raman scattering of phosphorene is sensitive to charge doping.66 It has three Raman modes (i.e., A1g, B2g and A2g) and all of which show softening and broadening on hole and electron doping, with the former causing stronger effects. TCNQ and TCNE have been utilized for hole doping, whilst tetrathiafulvalene (TTF) for electron doping.66 Further, the interactions of TCNQ, TCNE, and TTF with phosphorene have been studied through quenching the photoluminescence of phosphorene quantum dots (PQDs).67 Among these molecules, TCNQ most efficiently quenches the photoluminescence as indicated by the Stern–Volmer constants (Ksv = 7.0 × 104 M−1, 1.0 × 104 M−1, and 6.6 × 103 M−1 for TCNQ, TCNE, and TTF, respectively67). This trend is consistent with their electron withdrawing nature as has been confirmed by the Bader charge-population analysis.68 Furthermore, the electron withdrawing molecules not only reduce the electron density on the phosphorene surface, but also trap the photogenerated electrons, which otherwise cause oxidative decomposition of phosphorene.695.5. By electrostatic interaction
The phosphorene electron density can interact with cationic organic molecules through electrostatic interaction. For example, ionic liquids (1-butyl-3-methylimidazolium tetrafluoroborate),40 surfactants with long hydrophobic chains and small hydrophilic headgroups (cetyltrimethylammonium bromide),70 and long chain alkyl ammonium cation could be effectively utilized.71 In addition, the vertical packing of long alkyl chains creates an additional physical barrier on the phosphorene surface for oxygen and water.5.6. By van der Waals interaction
Organic molecules with extended π-electrons cloud, such as 3,4,9,10-perylene-tetracarboxylic-dianhydride (PTCDA)72 and anthraquinone (AQ)73 show very small charge-transfer to/from phosphorene and adsorb mostly via van der Waals interaction. van der Waals interaction has also been exploited in 2D/2D heterostructures of phosphorene with other important layered materials such as MoS2.74Covalent functionalization is no doubt an effective approach to functionalize and generate different materials with improved properties, such as covalently cross-linked 2D/2D heterostructures. However, in the case of phosphorene, sometimes the covalent functionalization suffers from a few drawbacks: (i) low degree of functionalization due to low reactivity of neutral phosphorene,75,76 and (ii) P–P bond breaking as a consequence of covalent bond formation with the functional groups, which has detrimental effects on the properties.75 On the other hand, non-covalent functionalization protects phosphorene with little (or no) distortion of the lattice, provides a relatively higher degree of functionalization, and preserves most of its properties. Many of the phosphorene-based composite HER catalysts utilize the non-covalent interactions to effectively tune interfacial charge transfer and charge carrier separation.
6. HER catalysts based on phosphorene
6.1. Mechanism of water splitting
Water splitting is an uphill reaction with a positive change in the Gibbs free energy (ΔGo) of +237 kJ mol−1 (equivalent to a potential difference of 1.23 V between anode and cathode or the energy demand of 1.23 eV per electron–hole pair generated). In the electrochemical process, HER occurs at the cathode through the reduction half reaction (2H+ + 2e−→ H2; E0 = 0.0 V vs. NHE at pH = 7), while the oxidation half reaction (H2O → ½O2 + 2e−; E0 = +1.23 V vs. NHE at pH = 7) occurs at the anode (Fig. 4a). Experimentally, the potential requirement for electrolysis of water is >1.23 V. This excess potential is required to account for the overpotential caused by the large activation barrier. A semiconductor with a bandgap of more than 1.23 eV can absorb radiation of wavelength less than 1000 nm and becomes photo excited (Fig. 4b). The photogenerated electrons (e−) can reduce water into H2 if the conduction band minima (CBM) of the semiconductor lies at a more negative potential than water reduction potential, whilst the holes (h+) can oxidize water into O2 if the valence band maxima (VBM) of semiconductor lies at a more positive potential than the water oxidation potential. Experimentally, the energy requirement for the photocatalytic process is much more than 1.23 eV and is, generally, in the range of 1.6–2.4 eV per electron–hole pair generated.77 This excess energy accounts for the additional energy required to overcome the activation energy barrier.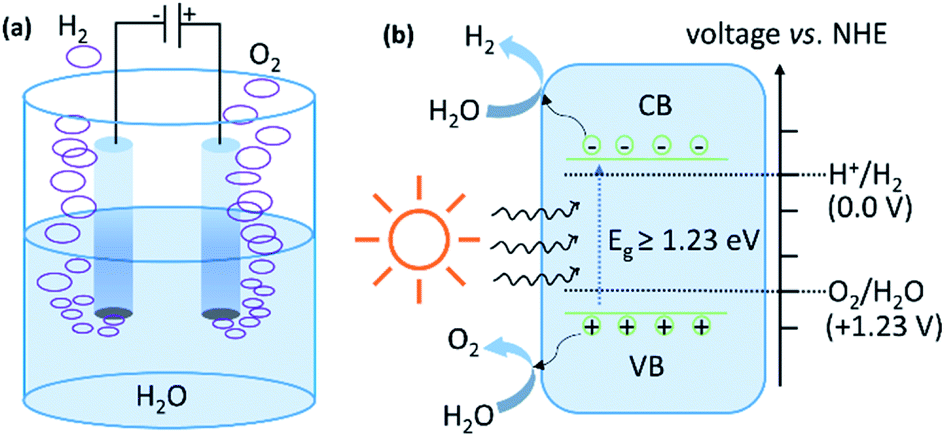 | ||
| Fig. 4 Schematic illustration of the mechanism of water splitting by (a) electrocatalytic (b) photocatalytic routes. | ||
A good HER catalyst reduces the activation barrier by enabling adsorption and desorption of the intermediate adsorbed state (H*) on its active sites while keeping the overall process thermoneutral (|ΔGH*| = 0).78 DFT calculations have suggested that the basal plane of a perfect phosphorene nanosheet has a large positive ΔGH* of 1.25 eV,79 which makes it inherently poor HER catalyst. However, its activity can be boosted by introducing point defects such as Stone–Wales and vacancy defects.14,79 The phosphorus atoms present at the odd size vacancies (i.e. monoatomic or triatomic) as well as at the edge sites are much more HER active when compared to those present at the even size vacancies (i.e. divacancy).14 The atomic vacancies induce a strain field and bond deformation around the defect core, which enhances HER activity through effective hydrogen–phosphorus interaction. There exists a few experimental reports, which suggest that HER activity of phosphorene is indeed higher at its defect and edge sites.18,79
6.2. Electrocatalysis
Phosphorene based HER electrocatalysts have shown activities ranging from very low to high, and in some cases even surpassing the commercial Pt/C catalyst. Experimentally, the electrochemical HER figure of merit is estimated by the overpotential with respect to a reversible hydrogen electrode (RHE), Tafel slope, and charge transfer resistance relative to state-of-the-art Pt/C catalyst.80 Lower the values of these parameters, better the catalyst. We summarize these parameters of phosphorene based electrocatalysts in Table 1, which clearly suggest that phosphorene modified with functional groups, metals, and other important 2D materials show better activity. Generally, it has been found that the phosphorene composites show selective and stable H2 production. Some of the commonly explored electrocatalysts are discussed below.| Catalysts | Overpotential at 10 mA cm−2vs. RHE | Tafel slope (mV dec−1) | Electrolyte | Ref. |
|---|---|---|---|---|
| a Overpotential reported at 100 mA cm−2; NA = not available; PBS = phosphate-buffered saline. | ||||
| Pristine phosphorene | 690 mV | 370 | 0.1 M NaOH | 48 |
| Phosphorene–MoS2 | 226 mV | 144 | 0.1 M NaOH | 48 |
| Phosphorene–MoSe2 | 330 mV | NA | 0.1 M NaOH | 48 |
| BP nanosheets/MoS2 nanoflakes | 85 mV | 68 | 0.5 M H2SO4 | 74 |
| BP nanosheets/MoS2 nanosheets | 126 mV | 68 | 0.5 M H2SO4 | 82 |
| BP nanosheets/MoS2 nanosheets | 237 mV | 99 | 1.0 M KOH | 82 |
| BP nanosheets/MoS2 nanosheets | 258 mV | 154 | 1.0 M PBS | 82 |
| BP nanosheets/MoSe2 | 380 mV | 97 | 0.5 M H2SO4 | 83 |
BP nanosheet/N-doped graphene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) :4) |
191 mV | 76 | 1.0 M KOH | 6 |
| BP nanosheets/Ni2P | 185 mV | 81 | 0.5 MH2SO4 |
84 |
| BP nanosheets/Ni2P | 107 mV | 38.6 | 0.5 M H2SO4 | 85 |
| BP nanosheets/Co2P | 340 mVa | 62 | 0.5 M H2SO4 | 86 |
| BP nanosheets/Co2P | 336 mVa | 72 | 1.0 M KOH | 86 |
| BP nanosheets/CoP nanodots | 105 mV | 92 | 0.5 M H2SO4 | 87 |
| BP nanosheets/CoP nanodots | 118 mV | 79 | 1.0 M KOH | 87 |
| BP nanosheets/Pt-GR | 21 mV | 46.9 | 1.0 M KOH | 88 |
| Phosphorene quantum dots/MoS2 | 600 mV | 162 | 0.1 M KOH | 89 |
| BP nanosheet/Co2P | 305 mV | 65 | 0.5 M H2SO4 | 90 |
| BP nanosheet/W | 500 mV | 112 | 0.5 M H2SO4 | 90 |
| Phosphorene/Ni | 691 mV | 116 | 0.5 M H2SO4 | 91 |
| Phosphorene/Mo | 522 mV | 112 | 0.5 M H2SO4 | 91 |
| Phosphorene/Co | 294 mV | 107 | 0.5 M H2SO4 | 91 |
| Phosphorene/Cu | 550 mV | 139 | 0.5 M H2SO4 | 92 |
| BP quantum dots/Ti3C2TX | 190 mV | 83 | 1.0 M KOH | 93 |
| BP nanosheet/PtRu | 22 mV | 19 | 1.0 M KOH | 94 |
| BP nanosheets/Ir nanoparticles | 1.98 mV | 91 | 1.0 M KOH | 95 |
| BP nanosheets/Ir nanoparticles | 25 mV | 30.9 | 0.5 M H2SO4 | 95 |
| BP nanosheets/Ir nanoparticles | 329 mV | 160 | 1.0 M PBS | 95 |
| Phosphorene–NH2 | 290 mV | 63 | 1.0 M KOH | 51 |
| Phosphorene/B-dopped graphene | 385.9 mV | 110 | 0.5 M H2SO4 | 96 |
| BP nanosheets/Pd-1T–MoS2 (in dark) | 152 mV | 86 | 0.5 M H2SO4 | 97 |
| BP nanosheets/Pd-1T–MoS2 (in light; >420 nm) | 97 mV | 66 | 0.5 M H2SO4 | 97 |
| BP nanosheets/NiCoSe | 206 mV | 77 | 0.5 M H2SO4 | 98 |
| BP nanosheets/NiCoSe | 287 mV | 134 | 1.0 M KOH | 98 |
| BP nanosheets/(NiCoSe|S) | 167 mV | 90 | 0.5 M H2SO4 | 98 |
| BP nanosheets/(NiCoSe|S) | 172 mV | 128 | 1.0 M KOH | 98 |
Sofer et al.18 have investigated the inherent electrochemical HER activity of pristine BP electrode in an acidic medium. The edge-sites have been found to be much more active than the basal sites owing to a higher electron transfer rate at the edge sites when compared to the basal sites. The defect/edge site P atoms show higher reactivity due to the higher density of dangling bonds as well as lone pair electrons. The HER onset potentials at edge-sites and basal-sites have been found to be −0.55 V and −1.13 V,18 respectively, which is consistent with their metallic and semiconducting nature, respectively.81 Further, the DFT calculations have revealed that the basal plane exhibits a negligible surface energy of 9 mJ m−2 compared to that of the edge plane energy of 194 mJ m−2.18 Thus, one of the strategies to enhance the HER activity of phosphorene is to increase the density of defect/edge sites by making nanosheets with reduced size.99
Considering low ΔGH* values of many of the transition metal phosphides (TMPs), doping phosphorene with transition metals is a promising strategy for efficient H2 evolution. To make use of higher reactivity of edge sites of phosphorene, Wang et al.86 have decorated few layer BP nanosheets with dicobalt phosphide (Co2P) from in situ reduction of Co2+ ion under an anhydrous and oxygen-free sealed reaction at high-temperatures (Fig. 5a–e). The composite exhibits low overpotential, low Tafel slope, reduced charge transfer resistance as well as enhanced ambient stability compared to bare BP nanosheets (Fig. 5f–h). Similarly, a composite of Ni2P and BP nanosheets has been synthesized from in situ reduction of Ni2+ on the BP surface.85 The Ni2P particles form uniformly on the BP surface, unlike Co2P. It is not clear from these studies that why does Ni2P form on both edge sites and basal sites, while Co2P form preferentially on the edge sites. A recent report has shown that under ultrahigh vacuum (UHV) conditions, initial deposition of Co metal occurs at the edge-sites of BP nanosheet, but subsequently, it deposits more on the basal-sites.90 The Co atoms present on the edge plane transform to the CoPOx species, whilst those on the basal plane transform to Co2P(112) nanoparticles of 2–3 nm.90 This composite shows electrocatalytic HER activity comparable to that of the edge-site functionalized Co2P/BP composite (see Fig. 5).86 It has been proposed that the increase in HER performance of this composite is mainly due to Co2P(112) owing to its very low ΔGH*. Liu et al.91 have doped phosphorene with Co, Ni, and Mo metals by using a H-type electrochemical setup comprising platinum as the anode and a bulk BP crystal as the cathode in TBA+Br− (TBA = tetrabutylammonium) and metal halide electrolyte (CoCl2, NiCl2 or MoCl3). Nafion 211 membrane has been used as a separator between the anode and the cathode. This setup enables the exfoliation of BP crystals into few-layer phosphorene by TBA+ cation intercalation followed by deposition of metal ions on the resultant nanosheets. The metal ions then undergo in situ reduction to form homogeneously distributed metal nanoparticles. The HER activities follow the order, pristine BP < BP(Ni) < BP(Mo) < BP(Co), which is consistent with their ΔGH* trends. The ΔGH* of BP(Co) is close to that of thermoneutral Pt catalyst (ΔGH* = ∼0 eV). The enhancement in electrocatalytic HER activity of these composites is attributed to the enhanced conductivity, optimal electronic interactions at the metal-phosphorene interface, and the stability of BP nanosheets in alkaline solution. Surprisingly, it has been demonstrated that BP activates state-of-the-art Pt catalysts through strong synergistic interaction in alkaline conditions.88 For this purpose, a BP/Pt/graphite composite has been prepared by sonicating solvent exfoliated BP nanosheet with Pt loaded graphite. The composite forms through strong Pt–P bonds, which exhibits 6.1-fold higher HER activity than that of the Pt/C (20% Pt) catalyst. The density of states (DOS) calculations have suggested that the Pt–P bonding causes a down-shift of the Pt d-band centre, bringing more anti-bonding states below the Fermi level, giving rise to an optimal ΔGH*. Through a similar approach, PtRh nanoclusters have been immobilized on the BP nanosheet.94 The PtRu/BP composite of Pt1.0Ru1.54 stoichiometry exhibits HER activity by an order of magnitude higher than that of the commercial Pt/C (20% Pt) catalyst in alkaline medium. Mei et al.95 have prepared two different pH universal BP-Ir electrocatalysts on carbon cloth (CC) substrate; (1) BPIr_sur: by immersion of a 2 × 2 cm2 CC piece into a dispersion of BP nanosheets and vacuum-drying followed by dipping into a dispersion of Ir nanoparticle and (2) BPIr_be: by immersion of a 2 × 2 cm2 CC piece into a dispersion of Ir nanoparticles and vacuum-drying followed by dipping into a dispersion of BP nanosheets. In 1.0 M KOH aqueous electrolyte, BPIr_be exhibits an overpotential of only 1.98 mV at a current density of 10 mA cm−2, which is superior to the BPIr_sur and BP nanosheet (105 mV, and 420 mV, respectively at a current density of 10 mA cm−2) and far surpassing the Ir nanoparticles (57 mV) as well as the commercial Pt/C (76 mV) catalysts at the same current density. Even at higher current densities of 100 mA cm−2, 200 mA cm−2, and 400 mA cm−2, BPIr_be exhibits small overpotentials of 59 mV, 87 mV, and 125 mV, respectively. The enhanced HER activity of these BPIr composites is attributed to high carrier density around the Fermi level, giving rise to higher electronic conductivity.
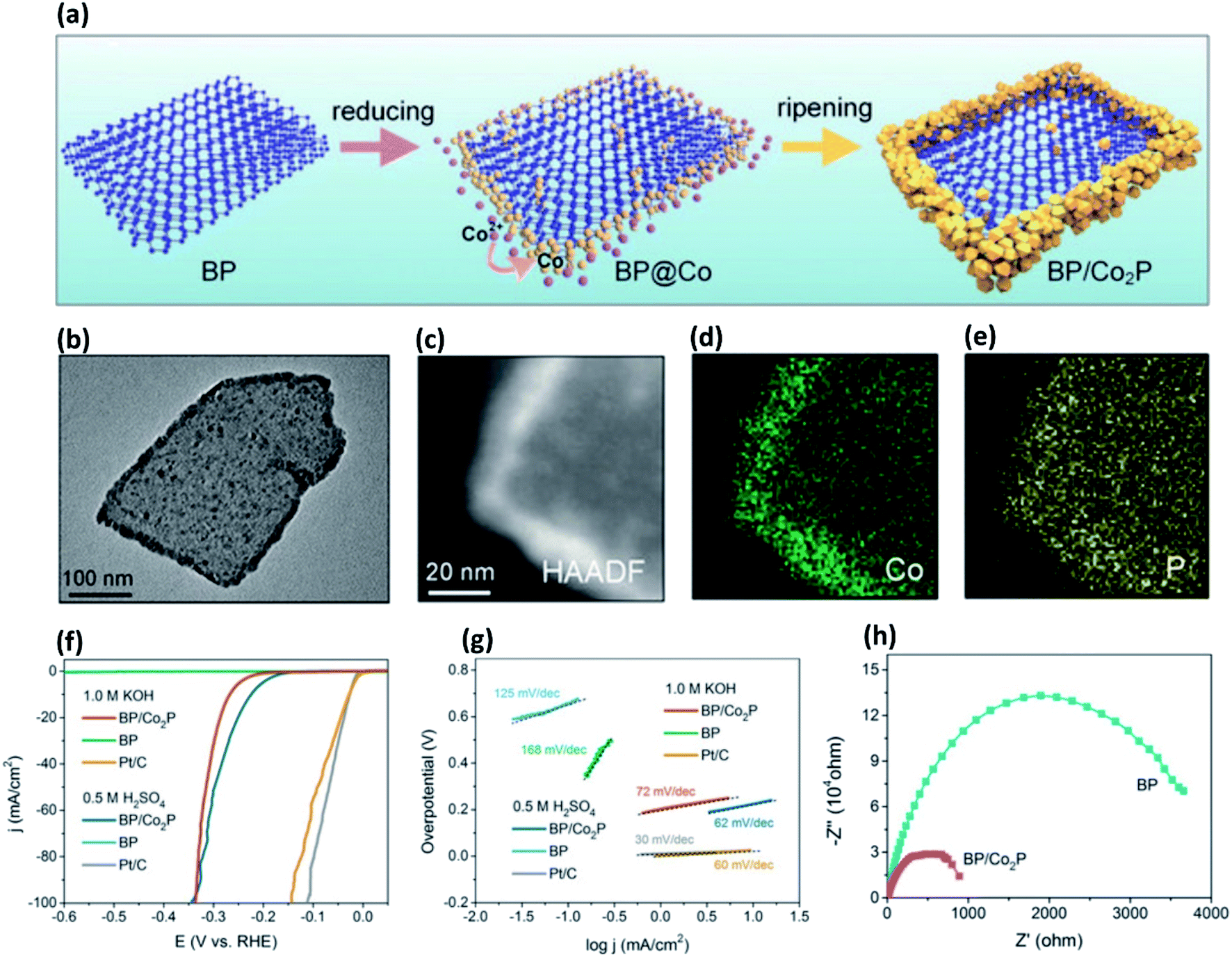 | ||
| Fig. 5 (a) Schematic diagram of BP/Co2P formation through selective interaction of Co2+ ions on phosphorene edge-sites and their in situ reduction into Co2P. (b) Transmission electron microscope (TEM) image of a BP/Co2P nanosheet revealing edge-sites decorated with Co2P. (c) High-angle annular dark-field (HAADF) image of a BP/Co2P nanosheet. (d) and (e) Corresponding energy dispersive spectroscopy (EDS) maps. (f) HER polarization curves of different catalysts in acidic and basic media. (g) Corresponding Tafel slopes. (h) Nyquist plots showing low charge-transfer resistance (Rct) in the case of BP/Co2P. The plots of pristine BP nanosheets are also given for comparison. Reproduced with permission from ref. 86, © 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Although the basal plane of phosphorene is HER inactive, it provides a sufficient contact area for interfacial engineering when integrated into 2D/2D heterocomposites. 2D/2D heterocomposites are expected to show enhanced HER activity due to their large active surface area and facile interfacial charge transfer.100 Semiconducting 2H–MoS2 nanosheet material is considered as a potential HER catalyst, but its inherent activity remains low due to the inertness of its basal plane.101 However, reports have suggested that the MoS2 basal plane can be activated by engineering interfacial charge transfer. He et al.74 have deposited BP nanosheets on MoS2 nanoflakes through van der Waals interaction. The flat-band potential of BP nanosheet (−0.29 V) is more negative than that of the MoS2 nanosheet (- 0.21 V), meaning the Fermi level of BP nanosheet lies at higher energy than that of the MoS2 nanoflake and electrons can, therefore, flow from BP to MoS2. Under acidic conditions, the accumulated electrons on MoS2 effectively promote proton adsorption and reduction processes at the catalyst–electrolyte interface through Volmer [H(aq) + e− → H(ads)] step as the rate-limiting step. The composite exhibits an overpotential of 85 mV at the current density of 10 mA cm−2, which is superior to that of pristine BP as well as MoS2. Another study has revealed that the HER activity of BP/MoS2 van der Waals heterocomposite remains high, even at its large scale prepared from the hydrothermal reaction of electrochemically exfoliated BP nanosheets and (NH4)2MoS4.82 This composite shows electrochemical HER in all pH, but the highest activity has been observed in acidic medium, which is consistent with the electrochemical stability trends of the composite. The van der Waals heterocomposite of BP and MoSe2 exhibits much lower HER activity than that of the BP and MoS2 analogue.83 To strengthen the interfacial charge transfer, Rao and coworkers have developed covalently cross-linked phosphorene–MoS2 and phosphorene–MoSe2 heterostructures (Fig. 6a).48 Carboxylic acid functionalized phosphorene (P–CH2COOH) has been coupled with primary amine functionalized MoS2 or MoSe2 (MoS2–CH2CH2NH2 or MoSe2–CH2CH2NH2). The P–CH2COOH has been obtained from the reaction of phosphorene with bromoacetic acid. The MoS2–CH2CH2NH2 and MoSe2–CH2CH2NH2 have been obtained from the reaction of 2-bromoethylamine hydrobromide with 1T-MoS2 and 1T-MoSe2, respectively, by using a reported protocol.102 The coupling of acid and amine groups forms interlayer amide bonds, which leads to a uniform growth of MoS2 or MoSe2 nanosheets on phosphorene nanosheets. These composites show significantly reduced overpotential, Tafel slope, and charge transfer resistance when compared to those of pristine phosphorene, MoS2, MoSe2 as well as their physical mixtures (Fig. 6b–d). We note that the long-term electrochemical stability of phosphorene–MoSe2 is not as good as phosphorene–MoS2 due to the decomposition of MoSe2 under the electrochemical reaction conditions.
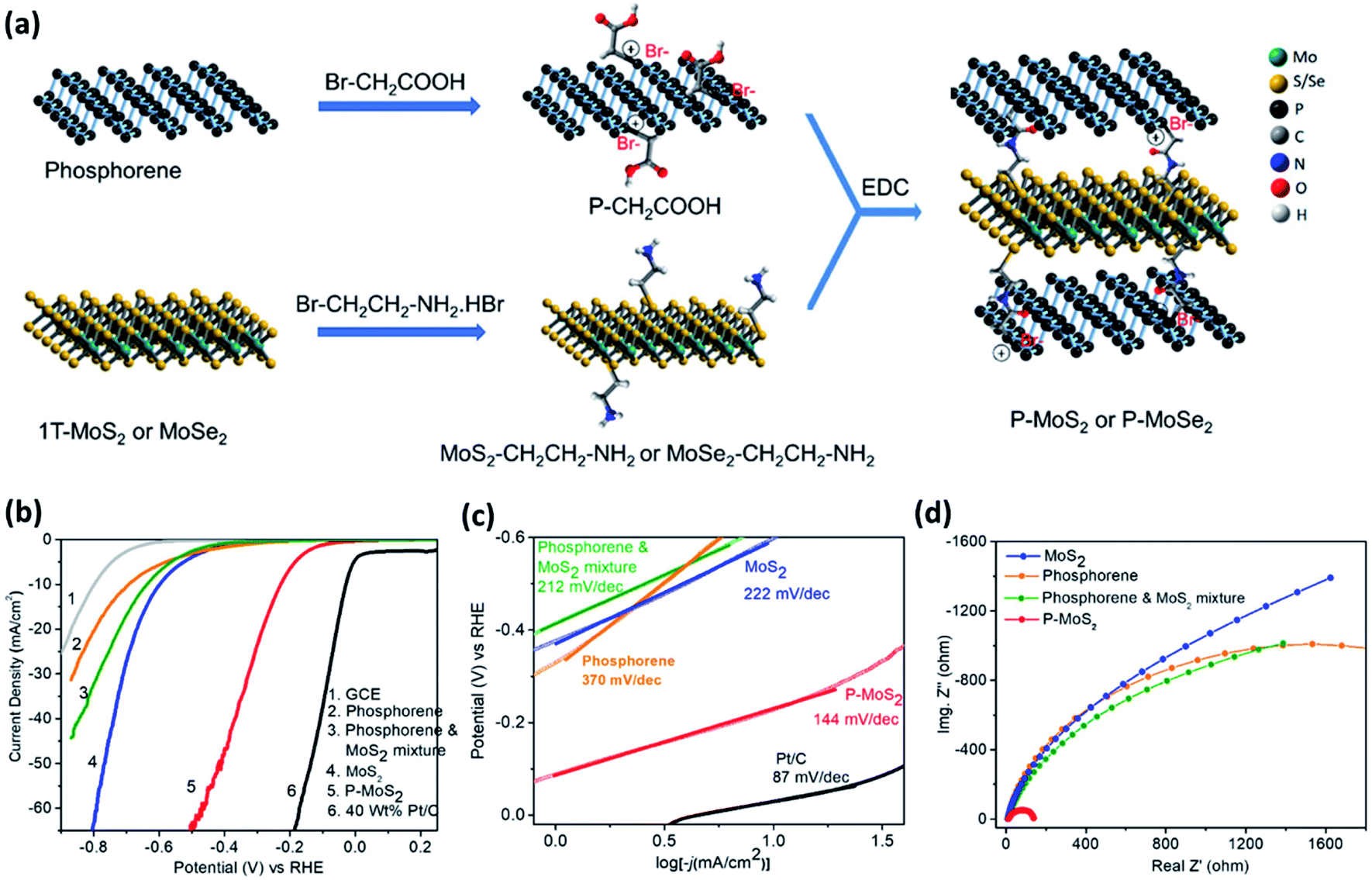 | ||
| Fig. 6 (a) Schematic illustration of the preparation of covalently cross-linked phosphorene–MoS2 and phosphorene–MoSe2 heterocomposites. The amide linkages are formed by the EDC [ethyl-3-(3-dimethylaminopropyl)carbodiimide] coupling between P–CH2COOH and MoS2–CH2CH2–NH2 or MoSe2–CH2CH2–NH2. (b) LSV curves of various catalysts. The curves of 40 wt% Pt/C and glassy carbon electrode (GCE) are also provided for comparison. (c) Corresponding Tafel plots revealing the lowest slope with phosphorene–MoS2. (d) Nyquist plots showing lowest Rct in the case of phosphorene–MoS2. Reproduced with permission from ref. 48, © 2019 American Chemical Society. | ||
Compared to metal-based composite catalysts, metal-free catalysts are preferred in terms of reduced environmental effect and cost. Yuan et al.6 have synthesized a metal free 2D-2D electrocatalyst with exfoliated BP (EBP) and N-doped graphene (NG) (Fig. 7a and b). The Fermi level of NG lies at a higher energy than that of EBP, which makes NG to EBP electron transfer feasible at the heterointerface (Fig. 7c). This composite not only promotes HER on the electron enriched EBP sites, but also promotes OER on the electron depleted NG sites (Fig. 7d). Thus, the EBP@NG composite behaves as an overall water splitting catalyst, with the best performance found at the EBP to NG wt ratio of 1:4 (Fig. 7e and f). Shao et al.51 have obtained ambient stable NH2 functionalized BP nanosheets (NH2-BP) of 2.15–4.87 nm thickness by ball milling BP crystals with urea. The resultant BP nanosheets exhibit abundant –NH2 groups at the edges. The electrocatalytic HER in an alkaline medium indicated that NH2-BP nanosheets need an overpotential of only 290 mV to achieve a current density of −10 mA cm−2, which is ∼3.1-fold less compared to the bulk BP (910 mV) and ∼2.3-fold less compared to milled BP (668 mV) at the same current density. The enhanced HER activity of NH2-BP nanosheets has been attributed to the increased density of electrochemically active surface area (ECSA) as well as feasible charge transfer due to ultra-thinness of functionalized phosphorene. These studies suggest that the density of ECSA can be controlled by morphological engineering.
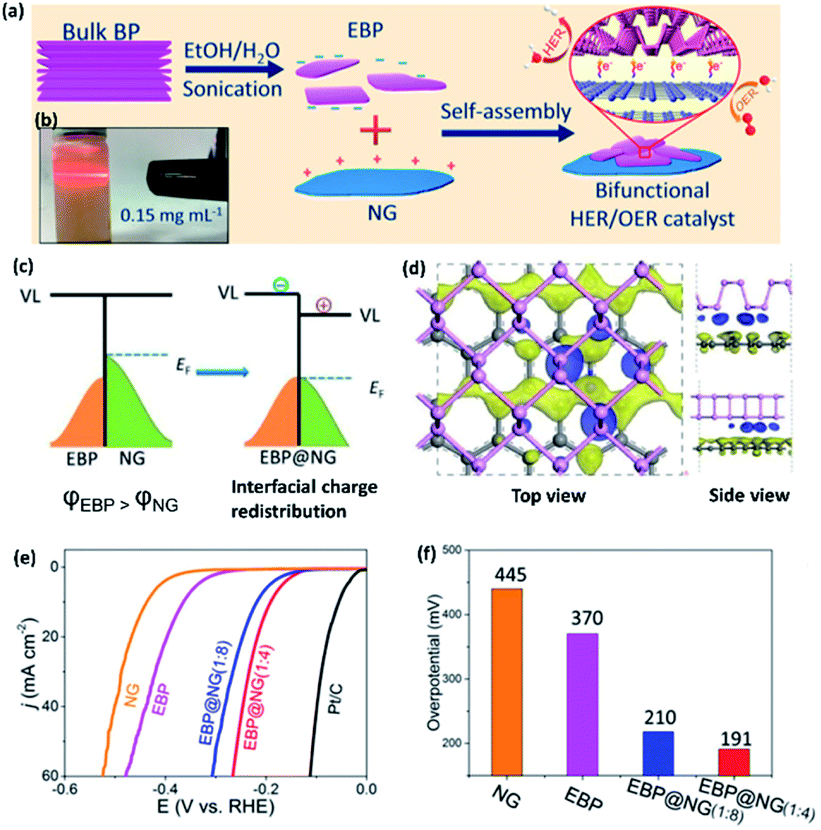 | ||
| Fig. 7 (a) Schematic representation of liquid exfoliation of a BP crystal and preparation of EBP/NG composite (EBP = exfoliated BP; NG = N-doped graphene). (b) Optical image of EBP dispersion showing the Tyndall effect. (c) Illustration showing interfacial charge redistribution between EBP and NG at their Fermi levels. (d) Corresponding differential charge density map with the electron-rich and the hole-rich areas shown by blue and yellow regions, respectively. (e) LSV curves of various catalysts in 1.0 M KOH aqueous solution with the LSV curves of Pt/C and GCE are also given for comparison. (f) Corresponding bar diagram between the HER overpotentials at a current density of 10 mA cm−2 and the mass ratio of EBP to NG in the EBP@NG composite. Reproduced with permission from ref. 6, © 2019 American Chemical Society. | ||
Recently, it has been shown that BP nanosheets can also be utilized as overall water splitting catalysts. A heterostructure composite of nickel-cobalt sulfoselenide/BP (NiCoSe|S/BP) has been fabricated by growing NiCo hydroxide nanosheet on a few-layer BP nanosheet followed by one-step sulfoselenization.98 The composite comprises two redox pairs: Ni2+/Ni3+ and Co3+/Co2+, with the former enhancing HER through electron-donating, whilst the latter enhancing OER through electron-accepting. The composite exhibits excellent electrocatalytic performance for both HER and OER in alkaline conditions with the overpotentials of 172 mV and 285 mV, respectively at a current density of 10 mA cm−2. Stable overall water splitting has been delivered at an overpotential of 1.67 V at a current density of 10 mA cm−2.
6.3. Photocatalysis
Table 2 lists the photocatalytic HER activities of phosphorene-based catalysts comprising single or composite materials. It should be noted that the HER activity depends not only on the nature of the photocatalyst but also on the experimental conditions used in the reaction. Besides, the activity numbers have been reported in a few different ways. It is, therefore, not possible to discuss an accurate comparison of efficiencies of the catalysts reported in different studies. Nevertheless, we have tabulated most of them and discussed important ones found in the commonly investigated studies.| Catalysts | Light source | Sacrificial agent | H2 Evolution rate | AQE (%) | Refs |
|---|---|---|---|---|---|
| a TEOA = triethanolamine; LA = lactic acid; EDTA = ethylenediaminetetraacetic acid; AQE = apparent quantum efficiency; NA = not available. | |||||
| Phosphorene/Pt (50 mg, 3 wt% Pt loading) | 300 W Xe lamp (>420 nm) | CH3OH | 6.9 μmol h−1 | NA | 103 |
| Bare phosphorene (2.2 mg) | 100 W halogen lamp (6 h) | TEOA | 621 μmol h−1 g−1 | 0.8 | 11 |
| Phosphorene·InCl3 (2.2 mg) | 100 W halogen lamp (6 h) | TEOA | 2058 μmol h−1 g−1 | 2.1 | 11 |
| Phosphorene·B(C6F5)3 (2.2 mg) | 100 W halogen lamp (6 h) | TEOA | 6597 μmol h−1 g−1 | 8.4 | 11 |
| Phosphorene·CH–C6H5 (2.2 mg) | 100 W halogen lamp (6 h) | TEOA | 5691 μmol h−1 g−1 | 7.3 | 11 |
| Phosphorene–MoS2 (2.5 mg) | 100 W halogen lamp (6 h) | TEOA | 26.8 mmol h−1 g−1 | NA | 48 |
| Phosphorene–MoSe2 (2.5 mg) | 100 W halogen lamp (6 h) | TEOA | 20.7 mmol h−1 g−1 | NA | 48 |
| Phosphorene–BCN | 100 W halogen lamp (6 h) | TEOA | 6528 μmol h−1 g−1 | NA | 54 |
| Phosphorene–g-C3N4 | 100 W halogen lamp (6 h) | TEOA | 11274 μmol h−1 g−1 |
NA | 54 |
| BP nanosheet/MoS2 (10 mg) | 300 W Xenon lamp (≥420 nm) | Na2S/Na2SO3 | 1286 μmol h−1 g−1 | 1.2 | 114 |
| BP nanoflakes/g-C3N4 nanosheet (1:4) (1.5 mg) |
320 W Xenon lamp (≥420 nm) | Methanol | 427 μmol h−1 g−1 | 3.18 | 108 |
| BP nanoflakes/g-C3N4 nanosheet (1:4) (1.5 mg) |
320 W Xenon lamp (≥780 nm) | Methanol | 101 μmol h−1 g−1 | 1.1 | 108 |
| Phosphorene/g-C3N4 nanosheet (20 mg) | 300 W Xenon lamp (≥420 nm) | LA | 571 μmol h−1 g−1 | 1.2 | 109 |
| BPQDs-g-C3N4 (100 mg) | 200 W Xenon lamp | Methanol | 190 μmol h−1 | NA | 115 |
| BP nanosheet/g-C3N4 (10 mg) | 300 W Xenon (≥420 nm) (24 h) | TEOA | 384.17 μmol g−1 h−1 | NA | 110 |
| Phosphorene/CdS (20 mg) | 300 W Xenon (≥420 nm) (4 h) | LA | 11192 μmol h−1 g−1 |
34.7 | 112 |
| BPQDs/Au nanorod/CdS nanowire (20 mg) | 300 mW cm−2 Xe lamp | Na2S/Na2SO3 | 10.1 mmol h−1 g−1 | 2.3 | 116 |
| BP nanosheets/Co2P (40 mg) | 300 W Xe lamp (≥420 nm) | w/o sacrificial | 29.4 μmol h−1 | 42.55 | 117 |
| BP nanosheet/BiVO4 (5 mg) | 320 W Xe lamp (≥420 nm) (3 h) | w/o sacrificial | 0.80 μmol | 0.89 | 118 |
| BP nanosheet/BiVO4/Co3O4 (5 mg) | 320 W Xe lamp (≥420 nm) (3 h) | w/o sacrificial | 3.9 μmol | NA | 118 |
| BP nanosheet/BiVO4/Co3O4 (5 mg) | 320 W Xe lamp (≥420 nm) (3 h) | EDTA | 7.5 μmol | NA | 118 |
| BP nanosheet/Bi2WO6 (20 mg, 3 wt% H2PtCl6·6H2O loading) | 300 W Xe lamp | TEOA | 21042 μmol g−1 | NA | 119 |
| BP-MoS2/CdS (1.0 mg) | 150 W Xe (5 h) | LA | 183.24 mmol g−1 h−1 | 63.1 | 120 |
| BP/Pt/RGO (BP, 0.2 mg; Pt, 75 μg; RGO, 0.1 mg) | 320 W Xe lamp (4 h) | EDTA | 3.4 mmol g−1 h−1 (>420 nm) | 8.7 | 121 |
| 0.84 mmol g−1 h−1 (>780 nm) | 1.5 | ||||
| BP-Au/La2Ti2O7 (1.5 mg) | 320 W Xe lamp (3 h) | Methanol | 0.74 mmol g−1 h−1 (>420 nm) | NA | 122 |
| 0.30 mmol g−1 h−1 (>780 nm) | NA | ||||
| 1.49 mmol g−1 h−1 (UV-Vis-NIR) | NA | ||||
| BP QDs-CdS-La2Ti2O7 (20 mg) | 300 mW cm−2 Xe lamp | Na2S/Na2SO3 | 0.96 mmol g−1 h−1 (UV-Vis-NIR) | NA | 123 |
| 0.80 mmol g−1 h−1 (≥420 nm) | NA | ||||
| 0.26 mmol g−1 h−1 (≥700 nm) | NA | ||||
| Ni2P@BP/C3N4 (20 mg) | 300 W Xe lamp (≥420 nm) (5 h) | TEOA | 858.2 μmol g−1 h−1 | 2.8 | 107 |
| BP/TiO2 (25 mg) | 300 W Xe lamp (365 nm) (6 h) | TEOA | 122.27 μmol | 18.23 | 124 |
| ZnxCd1−xS/few-layer phosphorene (20 mg) | 300 W Xe lamp (≥420 nm) | LA | 9326 μmol h−1 g−1 | 21.5 | 113 |
| BP/WS2 (0.2 mL dispersion) | 100 mW cm−2 simulated solar (3 h) | EDTA | 9.61 μmol | NA | 125 |
| BP/WS2 (0.2 mL dispersion) | 320 W Xe lamp (3 h) | EDTA | 1.55 μmol (≥808 nm) | NA | 125 |
| 2.49 μmol (≥780 nm) | 2.06 | ||||
| BP/g-C3N4-HKUST-1 (20 mg) | 300 W Xe lamp (≥400 nm) | TEOA | 7380 μmol h−1 g−1 | NA | 126 |
| 0.5%Pt/0.5%BP/ZnIn2S4 (20 mg) | 300 W Xe lamp (≥420 nm) | Na2SO3/Na2S | 1278 μmol h−1 g−1 | 0.25 | 127 |
| BP/Ti3C2/g-C3N4 (10 mg; 2 wt% Pt loading) | 300 W Xe lamp (≥420 nm) | TEOA | 18.42 mmol h−1 g−1 | 17.6 | 111 |
| m-g-CN/BP | Solar simulator 300 W (≥420 nm) | TEOA | 330 μmol g−1 h−1 | NA | 128 |
| m-g-CN/BP-Ni | Solar simulator 300 W (≥420 nm) | TEOA | 442 μmol h−1 g−1 | NA | 128 |
| m-g-CN/BP-Co | Solar simulator 300 W (≥420 nm) | TEOA | 326 μmol h−1 g−1 | NA | 128 |
| m-g-CN/BP-Cu | Solar simulator 300 W (≥420 nm) | TEOA | 223 μmol h−1 g−1 | NA | 128 |
| CdS-BP/Co (20 mg) | 300 W Xe lamp (≥420 nm) | LA | 345.4 μmol h−1 | NA | 129 |
| Pt-BP/CdS (20 mg) | 300 W Xe lamp (≥420 nm) | LA | 24.17 mmol h−1 g−1 |
46.0 | 130 |
Zhao et al.103 and Zhu et al.104 have reported that pristine phosphorene can promote HER under visible light irradiation (λ > 420 nm) in the presence of a sacrificial agent. Pt nanoparticles show improvement in the activity.103 Rao and coworkers11 have reported that the activity of pristine phosphorene can be slightly improved under the visible light illumination using eosin Y as the dye sensitizer and triethanolamine (TEOA) as the sacrificial agent. Furthermore, phosphorene has been chemically functionalized with a benzyl group, InCl3, and B(C6F5)3 (Fig. 8a). The functionalized phosphorenes exhibit several fold enhancements in the H2 evolution rate when compared to pristine phosphorene (Fig. 8b and c). The enhanced activity has been attributed to better ambient stability, enhanced water dispersibility, and slow charge carrier recombination. Indeed, successive theoretical work has predicted that the covalent functionalization significantly reduces the ΔGH* of phosphorene close to 0.0 eV,105 thereby facilitating hydrogen adsorption/desorption and H2 evolution.
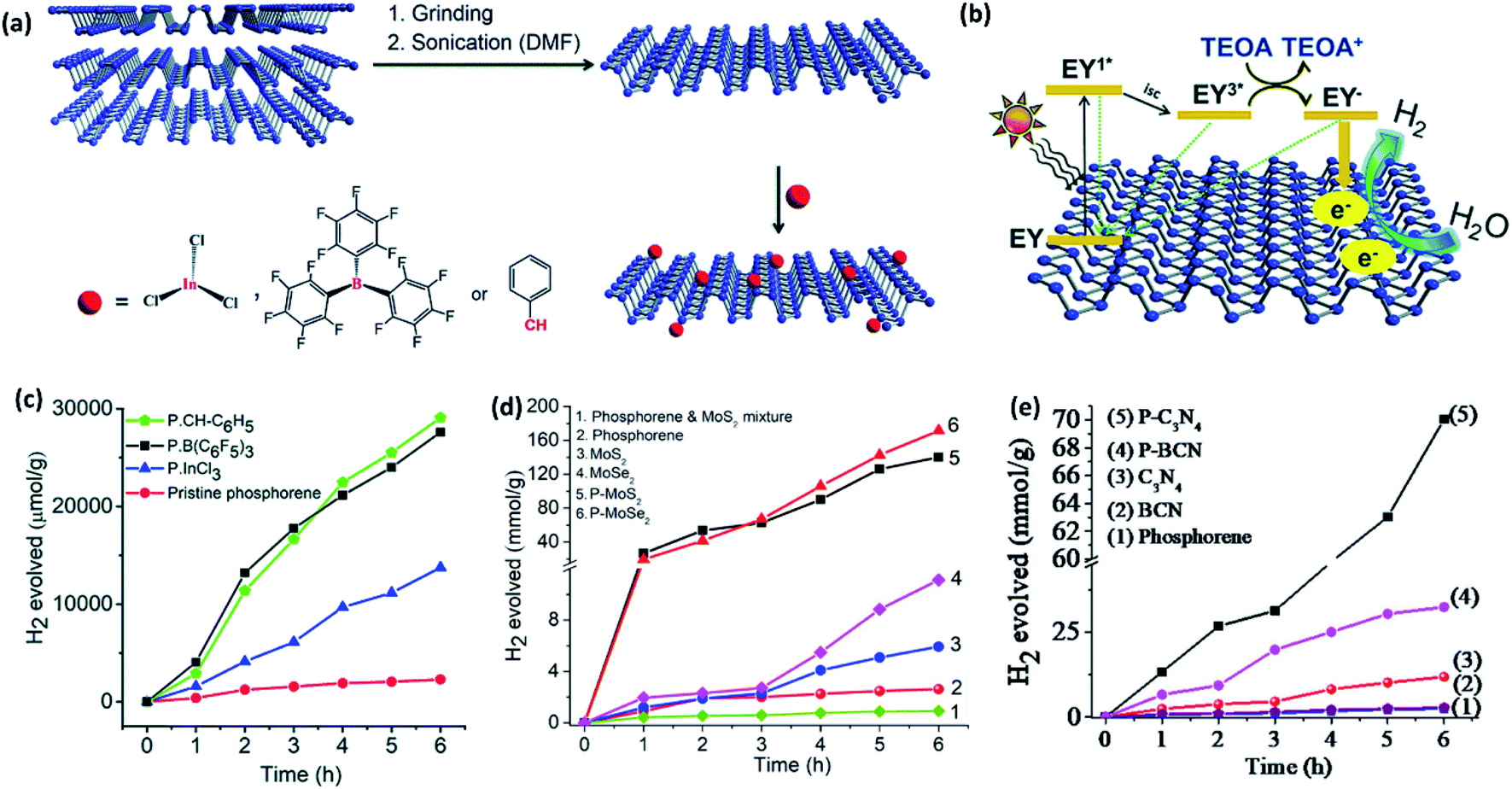 | ||
| Fig. 8 (a) Schematic representation of liquid exfoliation of BP into phosphorene nanosheets and their functionalization with InCl3, B(C6F5)3 and benzyl group. (b) Plausible mechanism of eosin Y sensitised photocatalytic HER in the presence of TEOA as a sacrificial agent. (c) Hydrogen evolved versus time plots of pristine and functionalized phosphorenes. (d) Hydrogen evolved versus time plots of covalently cross-linked phosphorene–MoS2 and phosphorene–MoSe2 composites. (e) Hydrogen evolved versus time plots of metal-free covalently cross-linked phosphorene–C3N4 and phosphorene–BCN composites. Panels (a–c) reproduced with permission from ref. 11, © 2019. The Royal Society of Chemistry. Panel (d) reproduced with permission from ref. 48, © 2019 American Chemical Society. Panel (e) reproduced with permission from ref. 54, ©Author(s) 2020. | ||
Generally, 2D/2D heterostructured photocatalysts show better HER activity owing to broad spectrum light absorption, large interface area, and slow charge carrier recombination.106 In this direction, a ternary composite, Ni2P@BP/g-C3N4, has been obtained by coupling Ni2P@BP nanosheets composite with g-C3N4 nanosheets.107 The composite shows charge transfer from g-C3N4 to Ni2P particles through conducting BP nanosheets, exhibiting HER activity superior to the individual components. Rao and coworkers have synthesized covalently cross-linked binary heterostructures of phosphorene nanosheets with MoS2 and MoSe2 nanosheets through amide linkage (see Fig. 6a for the schematic of phosphorene–MoS2 and phosphorene–MoSe2).48 Further, phosphorene nanosheets have been covalently cross-linked with wide bandgap 2D materials, such as BCN and g-C3N4, to form metal-free 2D heterocomposites.54 These 2D heterocomposites show superior H2 evolution rates when compared to their individual components (Fig. 8d and e). The HER activity of phosphorene–MoS2 composite is as high as 26.8 mmol h−1 g−1, which is ∼30-fold enhanced compared to pristine phosphorene and ∼22-fold compared to few layer MoS2 under similar reaction conditions. The activity of phosphorene–MoS2 is also superior to many other catalysts (Table 2). Similar HER activity trends have been found in the case of phosphorene–MoSe2, phosphorene–BCN, and phosphorene–g-C3N4 composites, but their activities are lower compared to the phosphorene–MoS2 composite.
Zhu et al.108 have synthesized a metal-free 2D/2D heterocomposite of phosphorene with g-C3N4 in different stoichiometries (Fig. 9a). The 1:4 composite shows the highest H2 evolution rate of 427 μmol g−1 h−1 for several days, while pristine phosphorene and g-C3N4 have shown negligible H2 evolution under similar reaction conditions (Fig. 9b and c). The composite forms a type-I heterojunction in which the photoexcited electrons transfer from g-C3N4 to phosphorene, where they are consumed in the proton reduction into H2. The directional electron transfer has been further strengthened by P–N bonds between the exposed P and N atoms on the phosphorene and the g-C3N4 surfaces, respectively. It has been proposed that P–N interactions introduce shallow charge trapping sites, which slow down the charge carrier recombination. Similar trends have been witnessed in a type-I phosphorene/g-C3N4 van der Waals heterostructure (Fig. 9d), exhibiting even higher HER activity than that of Pt loaded g-C3N4 (Fig. 9e).109 The photoexcited electrons migrate from g-C3N4 to phosphorene, creating electron accumulated sites on phosphorene and electron depleted sites on g-C3N4 (Fig. 9f). Another study has revealed that the combination of phosphorene (bandgap of 1.39 eV) and g-C3N4 (bandgap of 2.70 eV) leads to enhanced H2 evolution under broad-spectrum light irradiation.110 The composite exhibits much higher activity under the light of wavelength >420 nm when compared to >475 nm. g-C3N4, being a wide bandgap material, cannot be excited with the light of wavelength >475 nm and the catalysis occurs only on the phosphorene component. A conducting MXene (Ti3C2) layer between phosphorene quantum dots and the g-C3N4 layer enhances the interfacial charge transfer.111 The combination of phosphorene quantum dots and g-C3N4 improves visible light capture, and the MXene nanosheets mediate the migration of photoexcited charge carriers to the catalyst surface.
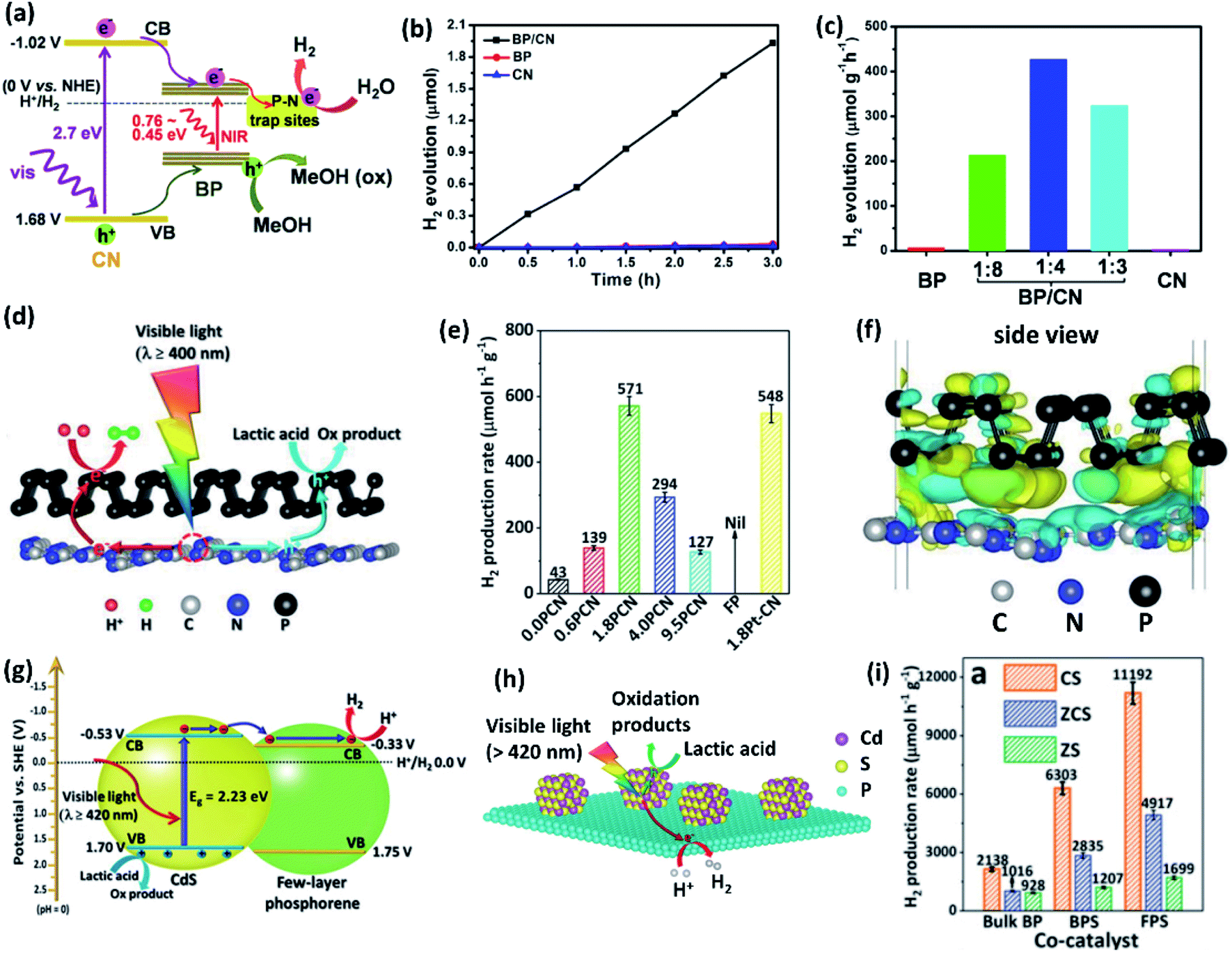 | ||
| Fig. 9 (a) Schematic diagram of visible light photocatalytic HER mechanism of BP/CN composite in the presence of methanol (20 vol%). (b) Corresponding HER activity versus time plots for different catalysts. (c) Effect of BP:CN weight ratio on HER activity under visible light irradiation for 3 h. (d) Schematic of visible light H2 production by the phosphorene/g-C3N4 catalysts in 18 vol% lactic acid aqueous solution. (e) HER activities of phosphorene/g-C3N4 with 0.0, 0.6, 1.8, 4.0 and 9.5 wt% of phosphorene. (f) Differential charge density map of phosphorene/g-C3N4 with the iso-surface value of 0.00015 e Å−3 with the electron accumulation and depletion areas are represented by yellow and blue regions, respectively. (g) and (h) Illustration of type-II band alignment and photocatalytic HER mechanism of few-layer phosphorene/CdS (FPS/CS) composite. (i) Comparison of photocatalytic HER activities of 1.0 wt% bulk BP, BPS (8–11 nm thick BP nanosheet) or FPS (4–5 nm thick BP nanosheet) composites with CdS (CS), Zn0.8Cd0.2S (ZCS) and ZnS (ZS). Panels (a–c) reproduced with permission from ref. 108, © 2017 American Chemical Society. Panels (d–f) reproduced with permission from ref. 109, © 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. Panels (g–i) reproduced with permission from ref. 112, © 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Compared to type-I heterojunction, type-II heterojunction is expected to provide much more efficient charge separation.106 Ran et al.112 have shown that a type-II heterostructure (Fig. 9g and h) of phosphorene nanosheets (4–5 nm thickness) with cadmium sulphide nanoparticles (10–60 nm diameter) exhibits a superior H2 production rate of 11192 μmol h−1 g−1 (Fig. 9i). It is noteworthy that the HER activity reduces due to the formation of type-I heterojunction when the thickness of phosphorene nanosheets is increased to 8–11 nm or beyond. Similar type-II band alignment and HER trends have been observed in the case of ZnxCd1−xS nanoparticles (ca. 15–60 nm diameter) loaded on phosphorene nanosheets of 5–6 nm thickness.113
Most of the above photocatalytic studies have shown the utility of the photoexcited electrons in H2 evolution at the CBM, whilst the photogenerated holes at the VBM are quenched by using sacrificial agents. Zhu et al.118 have prepared a 2D/2D heterostructure of BP (0.2 μm–2.0 μm) and monoclinic bismuth vanadate (BiVO4) (0.1 μm–1.0 μm) nanosheets through electrostatic interaction. The BP/BiVO4 composite catalyzes the overall water splitting reaction through an artificial Z-scheme photocatalytic system (Fig. 10a). The monoclinic BiVO4 is a narrow bandgap (2.4–2.5 eV) layered semiconductor, which has the ability to promote OER. On visible light irradiation, the photogenerated electrons in the CBM of BiVO4 quickly recombine with the photogenerated holes in the VBM of BP owing to their close energies and staggered alignment. In this way, the electrons are utilized in the HER at the CBM of BP, whilst the holes are utilized in the OER at the VBM of BiVO4. The composite shows superior H2 and O2 production rates under visible light irradiation, even in the absence of a sacrificial agent and co-catalyst (Fig. 10b). The HER activity further increases with the assistance of Co3O4 as co-catalyst and EDTA as a sacrificial agent. The optimum HER and OER activities have been observed with 20 wt% BP in the composite, while stoichiometric H2 and O2 evolution has been detected with 40 wt% BP. Similarly, BP nanosheets and monolayer Bi2WO6 have been combined in a Z-scheme photocatalyst, exhibiting HER activity superior to the BP/BiVO4 composite owing to broad spectrum light absorption.119
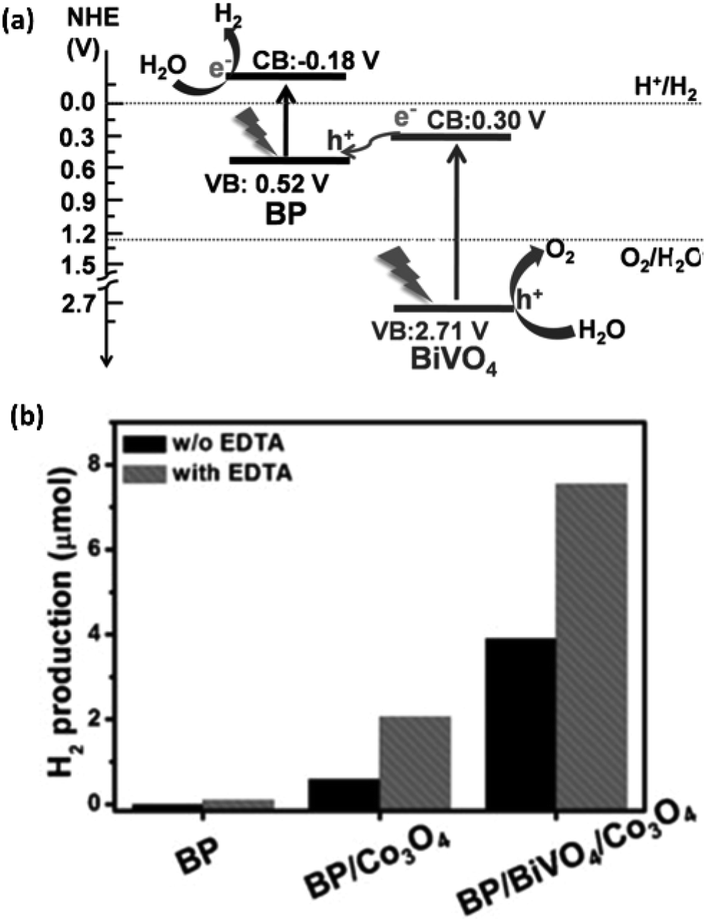 | ||
| Fig. 10 (a) Schematic presentation of BP/BiVO4 composite based Z-scheme photocatalyst for visible and NIR radiation (>420 nm) induced overall water splitting. (b) H2 evolved by different catalysts with and without Co3O4 co-catalyst and EDTA sacrificial agent under >420 nm light irradiation. Reproduced with permission from ref. 118, © 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
6.4. Photo-electrocatalysis (PEC)
Of late, there has been an increase of interest in PEC, which is the combination of photocatalysis and electrocatalysis techniques. Typically, in this method, the HER is carried out with applied potential on a photoelectrode supported by a semiconductor catalyst. It allows more effective separation of photogenerated electron–hole, charge transfer, and enhanced light absorption, thereby increasing the overall energy efficiency of water splitting. Under light irradiation, TiO2 nanorods coated with BP nanosheets show a 140% increase in the incident-photo-current conversion efficiency (IPCE).131 Similarly, enhancement of photocurrent densities of BP/Pt/RGO nanoflakes electrode has been observed under >420 nm or >808 nm light irradiation when compared to dark.121 Recently, it has been reported that van der Waals heterostructure of Pd dopped 1T-MoS2 and BP nanosheet exhibits increased light absorption when compared to Pd-1T-MoS2.97 Under the light irradiation, the composite shows a decrease in the overpotential from 152 mV to 97 mV at the current density of 10 mA cm−2 with a concomitant decrease in the Tafel slope from 86 mV dec−1 to 66 mV dec−1.Furthermore, BP quantum dots (BPQDs) have the capability of harvesting the full visible light spectrum due to size-dependent bandgaps.67,132 The composite of BPQD and g-C3N4 nanosheets has been shown to exhibit enhanced photocatalytic HER activity as well as photocurrent when irradiated with 420 nm laser light.115 The optimal photocurrent of 9.11 μA cm−2 has been obtained on 7 wt% loading of BPQD on g-C3N4 nanosheets. To utilize the broad light absorption capability of BPQDs, Jin et al.133 have fabricated a two-photon E-BiVO4/BPQDs/OL-OEC (E-BiVO4 = etched BiVO4) photoanode passivated with TiO2 overlayer (OL) and coated with NiOOH as oxygen evolution catalyst (OEC). The anode shows a remarkable photocurrent density of 6.2 mA cm−2 at 1.23 V vs. RHE, under AM 1.5 illumination (Fig. 11a). The BPQDs not only enhance the incident photon-to-electron conversion efficiencies (IPCEs), but also extends the light harvesting window up to 800 nm (Fig. 11b). The TiO2 overlayer enhances the ambient stability of BPQD and eliminates the surface trap state. The type-II band alignment (Fig. 11c) of BPQDs and E-BiVO4 promotes efficient separation of charge carriers. NiOOH enhances sluggish O2 evolution kinetics and consequently the E-BiVO4/BPQDs/OL-OEC photoanode exhibits H2 and O2 evolution ∼2:1 molar ratio for over 180 min at a photocurrent density of 6.2 mA cm−2 at 1.23 V vs. RHE (Fig. 11d).
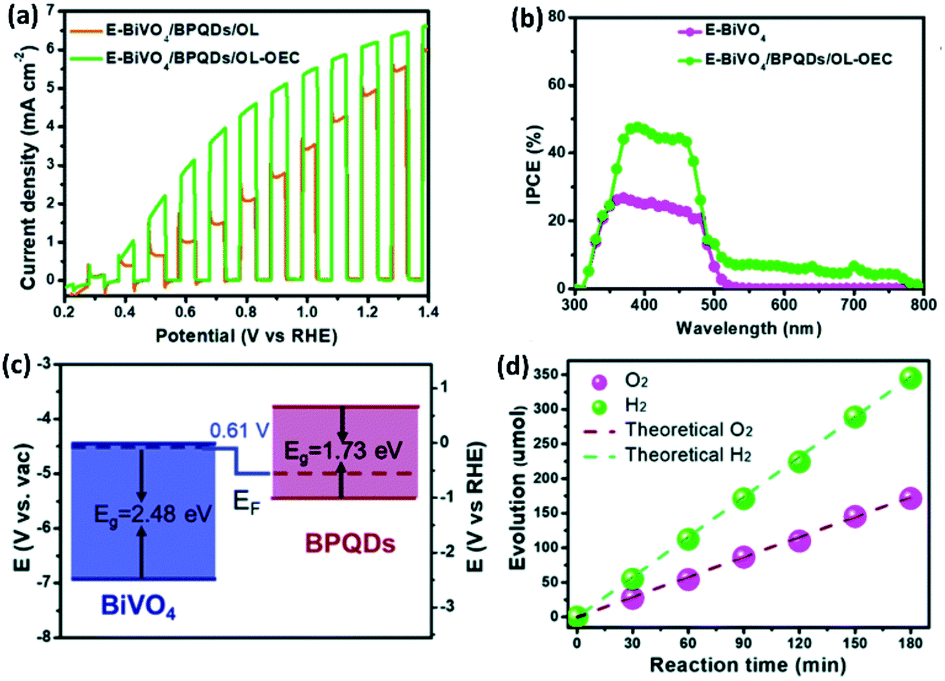 | ||
| Fig. 11 (a) J–V curves of E-BiVO4/BPQDs/OL and E-BiVO4/BPQDs/OL-OEC. (b) IPCE values of E-BiVO4 and E-BiVO4/BPQDs/OL-OEC. (c) Band alignment diagram of E-BiVO4 and BPQDs showing type II heterojunction. (d) HER and OER activities of the E-BiVO4/BPQDs/OL-OEC photoanode at 1.23 V vs. RHE. Reproduced with permission from ref. 133 © 2022 The Royal Society of Chemistry. | ||
7. Conclusions and future opportunities
We have discussed a range of phosphorenes modified with functional groups, atoms/ions, and other catalytically important 2D materials. We have summarized key methods of stabilizing phosphorenes by chemical functionalization as well as interfacial engineering. With appropriate examples, we have discussed diverse interactions (covalent, coordination, electrostatic, van der Waals, charge-transfer, and interfacial) which have been utilized in the functionalization of phosphorene. Although covalent functionalization is an effective approach to functionalize and generate different materials with improved properties, sometimes it leads to a low degree of functionalization as well as P–P bond breaking, affecting the properties of phosphorene. The non-covalent functionalization protects phosphorene with little (or no) distortion of the lattice, provides a relatively higher degree of functionalization, and enhances electronic properties.We have highlighted that phosphorene composites are superior catalysts for H2 production from water splitting reaction and have the potential to replace state-of-the-art Pt metal and Pt group metals. In addition, it is comprised of phosphorus, which is an earth abundant and non-toxic element. However, large-scale practical applications of phosphorene are still limited primarily due to ambient instability and lack of scalable synthesis. We expect that the interest in the chemistry and the applications of phosphorene continue to surge and develop along the following lines:
(1) Understanding chemistry: despite plenty of experimental reports on the stabilization of phosphorene, the long-term stability in oxygen and water is yet to be explored. Further understanding of the chemistry of degradation134 as well as passivation of phosphorene is thus necessary. Quantification of the degree of functionalization remains still a challenge.135 Thus, advanced tools need to be developed to elucidate the degree of functionalization and quality control so that the electronic properties can be appropriately optimized for efficient catalysis and other applications. For example, fluorescence labelling of surface species (FLOSS) can potentially be used as an in situ method.136
(2) Large scale synthesis: although liquid exfoliation has become a significant method of phosphorene synthesis, it has limitations, including uncontrollable lateral size, layer thickness and morphology, solvent contamination, oxidation of sample and formation of structural defects, all of which affect the overall performance. Bottom-up synthetic strategies such as pulsed laser deposition (PLD) have a great potential for high-quality scalable synthesis.137 There is a scope to further developing the top-down approaches such as electrochemical and shear exfoliation.
(3) Metal-free catalysis: p-block elements have the potential to replace the rare and precious noble and other transition metals-based catalysts. Superior HER activities of phosphorene/BCN and phosphorene/N-doped graphene suggest that phosphorene could potentially be used as a metal-free catalyst at a commercial scale. However, the developments of metal-free heterosystems are still at an early stage, and there is considerable scope for future developments. For example, phosphorene–borocarbonitride (BxCyNz) composite is worth exploring where electronic properties can be tuned by varying composition of the latter.
After possible developments of efficient HER catalysts, the realization of the potential of water as a completely renewable hydrogen source could be a viable solution. Solar water splitting reactors need to be developed to ensure large scale production at a low cost. In this direction, the reactors based on photoelectrochemical (PEC) cells have the advantage of using solar as well as electrical energy and simultaneously separating evolved H2 from O2. Bandgap tuneable and high conductivity make phosphorene a promising PEC material for high solar-to-hydrogen efficiency. Besides, strategies for hydrogen storage and transport need to be developed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
PV acknowledges the Science & Engineering Research Board (SERB) of the Govt. of India for Ramanujan Fellowship (Award No. RJF/2020/000106) and the Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR) Bangalore for financial support and research infrastructure.Notes and references
- C. N. R. Rao and S. Dey, Proc. Natl. Acad. Sci., 2017, 114, 13385–13393 CrossRef CAS PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- M. Chhetri, S. Maitra, H. Chakraborty, U. V. Waghmare and C. N. R. Rao, Energy Environ. Sci., 2016, 9, 95–101 RSC.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 CrossRef PubMed.
- M. Latorre-Sánchez, A. Primo and H. García, Angew. Chem., Int. Ed., 2013, 52, 11813–11816 CrossRef PubMed.
- Z. Yuan, J. Li, M. Yang, Z. Fang, J. Jian, D. Yu, X. Chen and L. Dai, J. Am. Chem. Soc., 2019, 141, 4972–4979 CrossRef CAS PubMed.
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tománek and P. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef CAS PubMed.
- Y. Deng, Z. Luo, N. J. Conrad, H. Liu, Y. Gong, S. Najmaei, P. M. Ajayan, J. Lou, X. Xu and P. D. Ye, ACS Nano, 2014, 8, 8292–8299 CrossRef CAS PubMed.
- A. N. Abbas, B. Liu, L. Chen, Y. Ma, S. Cong, N. Aroonyadet, M. Köpf, T. Nilges and C. Zhou, ACS Nano, 2015, 9, 5618–5624 CrossRef CAS PubMed.
- P. Vishnoi, U. Gupta, R. Pandey and C. N. R. Rao, J. Mater. Chem. A, 2019, 7, 6631–6637 RSC.
- M. Z. Rahman, C. W. Kwong, K. Davey and S. Z. Qiao, Energy Environ. Sci., 2016, 9, 709–728 RSC.
- B. Sa, Y.-L. Li, J. Qi, R. Ahuja and Z. Sun, J. Phys. Chem., 2014, 118, 26560–26568 CAS.
- Y. Cai, J. Gao, S. Chen, Q. Ke, G. Zhang and Y.-W. Zhang, Chem. Mater., 2019, 31, 8948–8956 CrossRef CAS.
- J. Li, P. Liu, H. Huang, Y. Li, Y. Tang, D. Mei and C. Zhong, ACS Sustain. Chem. Eng., 2020, 8, 5175–5183 CrossRef CAS.
- X. Zhu, S. Huang, Q. Yu, Y. She, J. Yang, G. Zhou, Q. Li, X. She, J. Deng, H. Li and H. Xu, Appl. Catal. B Environ., 2020, 269, 118760 CrossRef CAS.
- G.-Q. Zhao, J. Hu, X. Long, J. Zou, J.-G. Yu and F.-P. Jiao, Small, 2021, 17, 2102155 CrossRef CAS PubMed.
- Z. Sofer, D. Sedmidubský, Š. Huber, J. Luxa, D. Bouša, C. Boothroyd and M. Pumera, Angew. Chem., Int. Ed., 2016, 55, 3382–3386 CrossRef CAS PubMed.
- S. Li, Y. Zhang and H. Huang, J. Energy Chem., 2022, 67, 745–779 CrossRef.
- Z. Wu, J. Qi, W. Wang, Z. Zeng and Q. He, J. Mater. Chem. A, 2021, 9, 18793–18817 RSC.
- A. M. Kuchkaev, S. Lavate, A. M. Kuchkaev, A. V. Sukhov, R. Srivastava and D. G. Yakhvarov, Energy Technol., 2021, 9, 2100581 CrossRef CAS.
- P. W. Bridgman, J. Am. Chem. Soc., 1914, 36, 1344–1363 CrossRef CAS.
- J. C. Jamieson, Science, 1963, 139, 1291–1292 CrossRef CAS PubMed.
- Y. Akahama, S. Endo and S. Narita, J. Phys. Soc. Jpn., 1983, 52, 2148–2155 CrossRef CAS.
- J. Wittig and B. T. Matthias, Science, 1968, 160, 994–995 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- D. Scelta, A. Baldassarre, M. Serrano-Ruiz, K. Dziubek, A. B. Cairns, M. Peruzzini, R. Bini and M. Ceppatelli, Angew. Chem., Int. Ed., 2017, 56, 14135–14140 CrossRef CAS PubMed.
- X. Li, J. Sun, P. Shahi, M. Gao, A. H. MacDonald, Y. Uwatoko, T. Xiang, J. B. Goodenough, J. Cheng and J. Zhou, Proc. Natl. Acad. Sci., 2018, 115, 9935–9940 CrossRef CAS PubMed.
- Y. Akahama, M. Miyakawa, T. Taniguchi, A. Sano-Furukawa, S. Machida and T. Hattori, J. Chem. Phys., 2020, 153, 14704 CrossRef CAS PubMed.
- P. Vishnoi, K. Pramoda and C. N. R. Rao, ChemNanoMat, 2019, 5, 1062–1091 CrossRef CAS.
- J. Qiao, X. Kong, Z.-X. Hu, F. Yang and W. Ji, Nat. Commun., 2014, 5, 4475 CrossRef CAS PubMed.
- X. Wang, A. M. Jones, K. L. Seyler, V. Tran, Y. Jia, H. Zhao, H. Wang, L. Yang, X. Xu and F. Xia, Nat. Nanotechnol., 2015, 10, 517–521 CrossRef CAS PubMed.
- R. Fei, A. Faghaninia, R. Soklaski, J.-A. Yan, C. Lo and L. Yang, Nano Lett., 2014, 14, 6393–6399 CrossRef CAS PubMed.
- P. Yasaei, B. Kumar, T. Foroozan, C. Wang, M. Asadi, D. Tuschel, J. E. Indacochea, R. F. Klie and A. Salehi-Khojin, Adv. Mater., 2015, 27, 1887–1892 CrossRef CAS PubMed.
- A. Ambrosi, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2017, 56, 10443–10445 CrossRef CAS PubMed.
- J. Yang, R. Xu, J. Pei, Y. W. Myint, F. Wang, Z. Wang, S. Zhang, Z. Yu and Y. Lu, Light: Sci. Appl., 2015, 4, e312 CrossRef CAS.
- T. Low, A. S. Rodin, A. Carvalho, Y. Jiang, H. Wang, F. Xia and A. H. Castro Neto, Phys. Rev. B, 2014, 90, 075434 CrossRef CAS.
- C. N. R. Rao, U. Maitra and U. V. Waghmare, Chem. Phys. Lett., 2014, 609, 172–183 CrossRef CAS.
- W. Hu, L. Lin, R. Zhang, C. Yang and J. Yang, J. Am. Chem. Soc., 2017, 139, 15429–15436 CrossRef CAS PubMed.
- G. Abellán, S. Wild, V. Lloret, N. Scheuschner, R. Gillen, U. Mundloch, J. Maultzsch, M. Varela, F. Hauke and A. Hirsch, J. Am. Chem. Soc., 2017, 139, 10432–10440 CrossRef PubMed.
- Y. Huang, J. Qiao, K. He, S. Bliznakov, E. Sutter, X. Chen, D. Luo, F. Meng, D. Su, J. Decker, W. Ji, R. S. Ruoff and P. Sutter, Chem. Mater., 2016, 28, 8330–8339 CrossRef CAS.
- J. D. Wood, S. A. Wells, D. Jariwala, K.-S. Chen, E. Cho, V. K. Sangwan, X. Liu, L. J. Lauhon, T. J. Marks and M. C. Hersam, Nano Lett., 2014, 14, 6964–6970 CrossRef CAS PubMed.
- T. Zhang, Y. Wan, H. Xie, Y. Mu, P. Du, D. Wang, X. Wu, H. Ji and L. Wan, J. Am. Chem. Soc., 2018, 140, 7561–7567 CrossRef CAS PubMed.
- A. Favron, E. Gaufrès, F. Fossard, A.-L. Phaneuf-ĹHeureux, N. Y. W. Tang, P. L. Lévesque, A. Loiseau, R. Leonelli, S. Francoeur and R. Martel, Nat. Mater., 2015, 14, 826–832 CrossRef CAS PubMed.
- Q. Zhou, Q. Chen, Y. Tong and J. Wang, Angew. Chem., Int. Ed., 2016, 55, 11437–11441 CrossRef CAS PubMed.
- C. R. Ryder, J. D. Wood, S. A. Wells, Y. Yang, D. Jariwala, T. J. Marks, G. C. Schatz and M. C. Hersam, Nat. Chem., 2016, 8, 597–602 CrossRef CAS PubMed.
- S. Yasui, M. Fujii, C. Kawano, Y. Nishimura, K. Shioji and A. Ohno, J. Chem. Soc., Perkin Trans., 1994, 2, 177–183 RSC.
- P. Vishnoi, K. Pramoda, U. Gupta, M. Chhetri, R. G. Balakrishna and C. N. R. Rao, ACS Appl. Mater. Interfaces, 2019, 11, 27780–27787 CrossRef CAS PubMed.
- Y. Liu, P. Gao, T. Zhang, X. Zhu, M. Zhang, M. Chen, P. Du, G.-W. Wang, H. Ji, J. Yang and S. Yang, Angew. Chem., Int. Ed., 2019, 58, 1479–1483 CrossRef CAS PubMed.
- K. L. Walz Mitra, C. H. Chang, M. P. Hanrahan, J. Yang, D. Tofan, W. M. Holden, N. Govind, G. T. Seidler, A. J. Rossini and A. Velian, Angew. Chem., Int. Ed., 2021, 60, 9127–9134 CrossRef CAS PubMed.
- L. Shao, H. Sun, L. Miao, X. Chen, M. Han, J. Sun, S. Liu, L. Li, F. Cheng and J. Chen, J. Mater. Chem. A, 2018, 6, 2494–2499 RSC.
- Z. Sofer, J. Luxa, D. Bouša, D. Sedmidubský, P. Lazar, T. Hartman, H. Hardtdegen and M. Pumera, Angew. Chem., Int. Ed., 2017, 56, 9891–9896 CrossRef CAS PubMed.
- J. Zhang, S. Chen, Y. Ma, D. Wang, J. Zhang, Y. Wang, W. Li, Z. Yu, H. Zhang, F. Yin and Z. Li, J. Mater. Chem. B, 2018, 6, 4065–4070 RSC.
- C. N. R. Rao, K. Pramoda, A. Saraswat, R. Singh, P. Vishnoi, N. Sagar and A. Hezam, APL Mater., 2020, 8, 20902 CrossRef CAS.
- M. Trunk, J. F. Teichert and A. Thomas, J. Am. Chem. Soc., 2017, 139, 3615–3618 CrossRef CAS PubMed.
- A. Ienco, G. Manca, M. Peruzzini and C. Mealli, Dalton Trans., 2018, 47, 17243–17256 RSC.
- D. Tofan, Y. Sakazaki, K. L. Walz Mitra, R. Peng, S. Lee, M. Li and A. Velian, Angew. Chem., Int. Ed., 2021, 60, 8329–8336 CrossRef CAS PubMed.
- Y. Zhao, H. Wang, H. Huang, Q. Xiao, Y. Xu, Z. Guo, H. Xie, J. Shao, Z. Sun, W. Han, X.-F. Yu, P. Li and P. K. Chu, Angew. Chem., Int. Ed., 2016, 55, 5003–5007 CrossRef CAS PubMed.
- L. Wu, J. Wang, J. Lu, D. Liu, N. Yang, H. Huang, P. K. Chu and X.-F. Yu, Small, 2018, 14, 1801405 CrossRef PubMed.
- L. Chan, X. Chen, P. Gao, J. Xie, Z. Zhang, J. Zhao and T. Chen, ACS Nano, 2021, 15, 3047–3060 CrossRef CAS PubMed.
- M. Fojtů, X. Chia, Z. Sofer, M. Masařík and M. Pumera, Adv. Funct. Mater., 2017, 27, 1701955 CrossRef.
- S. P. Koenig, R. A. Doganov, L. Seixas, A. Carvalho, J. Y. Tan, K. Watanabe, T. Taniguchi, N. Yakovlev, A. H. Castro Neto and B. Özyilmaz, Nano Lett., 2016, 16, 2145–2151 CrossRef CAS PubMed.
- Z. Guo, S. Chen, Z. Wang, Z. Yang, F. Liu, Y. Xu, J. Wang, Y. Yi, H. Zhang, L. Liao, P. K. Chu and X.-F. Yu, Adv. Mater., 2017, 29, 1703811 CrossRef PubMed.
- M. Vanni, M. Bellini, S. Borsacchi, L. Calucci, M. Caporali, S. Caporali, F. ďAcapito, M. Geppi, A. Giaccherini, A. Ienco, G. Manca, A. M. Mio, G. Nicotra, W. Oberhauser, M. Serrano-Ruiz, M. Banchelli, F. Vizza and M. Peruzzini, J. Am. Chem. Soc., 2021, 143, 10088–10098 CrossRef CAS PubMed.
- G. Abellán, V. Lloret, U. Mundloch, M. Marcia, C. Neiss, A. Görling, M. Varela, F. Hauke and A. Hirsch, Angew. Chem., Int. Ed., 2016, 55, 14557–14562 CrossRef PubMed.
- P. Vishnoi, S. Rajesh, S. Manjunatha, A. Bandyopadhyay, M. Barua, S. K. Pati and C. N. R. Rao, ChemPhysChem, 2017, 18, 2985–2989 CrossRef CAS PubMed.
- P. Vishnoi, M. Mazumder, M. Barua, S. K. Pati and C. N. R. Rao, Chem. Phys. Lett., 2018, 699, 223–228 CrossRef CAS.
- Y. Jing, Q. Tang, P. He, Z. Zhou and P. Shen, Nanotechnology, 2015, 26, 95201 CrossRef CAS PubMed.
- X. Niu, Y. Li, Y. Zhang, Q. Li, Q. Zhou, J. Zhao and J. Wang, J. Phys. Chem. Lett., 2018, 9, 5034–5039 CrossRef CAS PubMed.
- R. Jain, Y. Singh, S.-Y. Cho, S. P. Sasikala, S. H. Koo, R. Narayan, H.-T. Jung, Y. Jung and S. O. Kim, Chem. Mater., 2019, 31, 2786–2794 CrossRef CAS.
- C. Su, Z. Yin, Q.-B. Yan, Z. Wang, H. Lin, L. Sun, W. Xu, T. Yamada, X. Ji, N. Zettsu, K. Teshima, J. H. Warner, M. Dincă, J. Hu, M. Dong, G. Su, J. Kong and J. Li, Proc. Natl. Acad. Sci., 2019, 116, 20844–20849 CrossRef CAS PubMed.
- C. Wang, D. Niu, B. Liu, S. Wang, X. Wei, Y. Liu, H. Xie and Y. Gao, J. Phys. Chem., 2017, 121, 18084–18094 CAS.
- R. Gusmão, Z. Sofer and M. Pumera, ACS Nano, 2018, 12, 5666–5673 CrossRef PubMed.
- R. He, J. Hua, A. Zhang, C. Wang, J. Peng, W. Chen and J. Zeng, Nano Lett., 2017, 17, 4311–4316 CrossRef CAS PubMed.
- S. Wild, M. Fickert, A. Mitrovic, V. Lloret, C. Neiss, J. A. Vidal-Moya, M. Á. Rivero-Crespo, A. Leyva-Pérez, K. Werbach, H. Peterlik, M. Grabau, H. Wittkämper, C. Papp, H.-P. Steinrück, T. Pichler, A. Görling, F. Hauke, G. Abellán and A. Hirsch, Angew. Chem., Int. Ed., 2019, 58, 5763–5768 CrossRef CAS PubMed.
- A. Mitrović, S. Wild, V. Lloret, M. Fickert, M. Assebban, B. G. Márkus, F. Simon, F. Hauke, G. Abellán and A. Hirsch, Chem. - Eur. J., 2021, 27, 3361–3366 CrossRef PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef.
- J. Lu, X. Zhang, D. Liu, N. Yang, H. Huang, S. Jin, J. Wang, P. K. Chu and X.-F. Yu, ACS Appl. Mater. Interfaces, 2019, 11, 37787–37795 CrossRef CAS PubMed.
- C. N. R. Rao and M. Chhetri, Adv. Mater., 2019, 31, 1803668 CrossRef PubMed.
- S. Fukuoka, T. Taen and T. Osada, J. Phys. Soc. Jpn., 2015, 84, 121004 CrossRef.
- T. Liang, Y. Liu, Y. Cheng, F. Ma and Z. Dai, ChemCatChem, 2020, 12, 2840–2848 CrossRef CAS.
- W. Li, D. Liu, N. Yang, J. Wang, M. Huang, L. Liu, X. Peng, G. Wang, X.-F. Yu and P. K. Chu, Appl. Surf. Sci., 2019, 467–468, 328–334 CrossRef CAS.
- Y. Lin, Y. Pan and J. Zhang, Int. J. Hydrogen Energy, 2017, 42, 7951–7956 CrossRef CAS.
- Z.-Z. Luo, Y. Zhang, C. Zhang, H. T. Tan, Z. Li, A. Abutaha, X.-L. Wu, Q. Xiong, K. A. Khor, K. Hippalgaonkar, J. Xu, H. H. Hng and Q. Yan, Adv. Energy Mater., 2017, 7, 1601285 CrossRef.
- J. Wang, D. Liu, H. Huang, N. Yang, B. Yu, M. Wen, X. Wang, P. K. Chu and X.-F. Yu, Angew. Chem., Int. Ed., 2018, 57, 2600–2604 CrossRef CAS PubMed.
- T. Liang, Y. Liu, P. Zhang, C. Liu, F. Ma, Q. Yan and Z. Dai, Chem. Eng. J., 2020, 395, 124976 CrossRef CAS.
- X. Wang, L. Bai, J. Lu, X. Zhang, D. Liu, H. Yang, J. Wang, P. K. Chu, S. Ramakrishna and X.-F. Yu, Angew. Chem., Int. Ed., 2019, 58, 19060–19066 CrossRef CAS PubMed.
- R. Prasannachandran, T. V. Vineesh, M. B. Lithin, R. Nandakishore and M. M. Shaijumon, Chem. Commun., 2020, 56, 8623–8626 RSC.
- T. Kosmala, L. Bardini, M. Caporali, M. Serrano-Ruiz, F. Sedona, S. Agnoli, M. Peruzzini and G. Granozzi, Inorg. Chem. Front., 2021, 8, 684–692 RSC.
- D. Liu, J. Wang, J. Lu, C. Ma, H. Huang, Z. Wang, L. Wu, Q. Liu, S. Jin, P. K. Chu and X.-F. Yu, Small Methods, 2019, 3, 1900083 CrossRef.
- Y. Zhang, N. Dong, H. Tao, C. Yan, J. Huang, T. Liu, A. W. Robertson, J. Texter, J. Wang and Z. Sun, Chem. Mater., 2017, 29, 6445–6456 CrossRef CAS.
- X.-D. Zhu, Y. Xie and Y.-T. Liu, J. Mater. Chem. A, 2018, 6, 21255–21260 RSC.
- Y. Li, W. Pei, J. He, K. Liu, W. Qi, X. Gao, S. Zhou, H. Xie, K. Yin, Y. Gao, J. He, J. Zhao, J. Hu, T.-S. Chan, Z. Li, G. Zhang and M. Liu, ACS Catal., 2019, 9, 10870–10875 CrossRef CAS.
- J. Mei, T. He, J. Bai, D. Qi, A. Du, T. Liao, G. A. Ayoko, Y. Yamauchi, L. Sun and Z. Sun, Adv. Mater., 2021, 33, 2104638 CrossRef CAS PubMed.
- S. Suragtkhuu, M. Bat-Erdene, A. S. R. Bati, J. G. Shapter, S. Davaasambuu and M. Batmunkh, J. Mater. Chem. A, 2020, 8, 20446–20452 RSC.
- X. Song, B. Li, W. Peng, C. Wang, K. Li, Y. Zhu and Y. Mei, Nanoscale, 2021, 13, 5892–5900 RSC.
- T. Liang, S. Lenus, Y. Liu, Y. Chen, T. Sakthivel, F. Chen, F. Ma and Z. Dai, Energy Environ. Mater., 2022, 1–10 Search PubMed.
- C. C. Mayorga-Martinez, N. Mohamad Latiff, A. Y. S. Eng, Z. Sofer and M. Pumera, Anal. Chem., 2016, 88, 10074–10079 CrossRef CAS PubMed.
- K. Pramoda, U. Gupta, M. Chhetri, A. Bandyopadhyay, S. K. Pati and C. N. R. Rao, ACS Appl. Mater. Interfaces, 2017, 9, 10664–10672 CrossRef CAS PubMed.
- U. Maitra, U. Gupta, M. De, R. Datta, A. Govindaraj and C. N. R. Rao, Angew. Chem., Int. Ed., 2013, 52, 13057–13061 CrossRef CAS PubMed.
- P. Vishnoi, A. Sampath, U. V Waghmare and C. N. R. Rao, Chem. - Eur. J., 2017, 23, 886–895 CrossRef CAS PubMed.
- G. Zhao, T. Wang, Y. Shao, Y. Wu, B. Huang and X. Hao, Small, 2017, 13, 1602243 CrossRef PubMed.
- X. Zhu, T. Zhang, Z. Sun, H. Chen, J. Guan, X. Chen, H. Ji, P. Du and S. Yang, Adv. Mater., 2017, 29, 1605776 CrossRef PubMed.
- W. Zhou, L. Dong, L. Tan and Q. Tang, J. Phys. Chem., 2021, 125, 7581–7589 CrossRef CAS PubMed.
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS.
- R. Boppella, W. Yang, J. Tan, H.-C. Kwon, J. Park and J. Moon, Appl. Catal. B Environ., 2019, 242, 422–430 CrossRef CAS.
- M. Zhu, S. Kim, L. Mao, M. Fujitsuka, J. Zhang, X. Wang and T. Majima, J. Am. Chem. Soc., 2017, 139, 13234–13242 CrossRef CAS PubMed.
- J. Ran, W. Guo, H. Wang, B. Zhu, J. Yu and S.-Z. Qiao, Adv. Mater., 2018, 30, 1800128 CrossRef PubMed.
- Q. Zhang, S. Huang, J. Deng, D. T. Gangadharan, F. Yang, Z. Xu, G. Giorgi, M. Palummo, M. Chaker and D. Ma, Adv. Funct. Mater., 2019, 29, 1902486 CrossRef.
- T. Song, L. Hou, B. Long, A. Ali and G.-J. Deng, J. Colloid Interface Sci., 2021, 584, 474–483 CrossRef CAS PubMed.
- J. Ran, B. Zhu and S.-Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 10373–10377 CrossRef CAS PubMed.
- J. Ran, X. Wang, B. Zhu and S. Z. Qiao, Chem. Commun., 2017, 53, 9882–9885 RSC.
- Y.-J. Yuan, P. Wang, Z. Li, Y. Wu, W. Bai, Y. Su, J. Guan, S. Wu, J. Zhong, Z.-T. Yu and Z. Zou, Appl. Catal. B Environ., 2019, 242, 1–8 CrossRef CAS.
- L. Kong, Y. Ji, Z. Dang, J. Yan, P. Li, Y. Li and S. Frank Liu, Adv. Funct. Mater., 2018, 28, 1800668 CrossRef.
- X. Cai, L. Mao, S. Yang, K. Han and J. Zhang, ACS Energy Lett., 2018, 3, 932–939 CrossRef CAS.
- B. Tian, B. Tian, B. Smith, M. C. Scott, R. Hua, Q. Lei and Y. Tian, Nat. Commun., 2018, 9, 1397 CrossRef PubMed.
- M. Zhu, Z. Sun, M. Fujitsuka and T. Majima, Angew. Chem., Int. Ed., 2018, 57, 2160–2164 CrossRef CAS PubMed.
- J. Hu, D. Chen, Z. Mo, N. Li, Q. Xu, H. Li, J. He, H. Xu and J. Lu, Angew. Chem., Int. Ed., 2019, 58, 2073–2077 CrossRef CAS PubMed.
- D. A. Reddy, E. H. Kim, M. Gopannagari, Y. Kim, D. P. Kumar and T. K. Kim, Appl. Catal. B Environ., 2019, 241, 491–498 CrossRef CAS.
- M. Zhu, Y. Osakada, S. Kim, M. Fujitsuka and T. Majima, Appl. Catal. B Environ., 2017, 217, 285–292 CrossRef CAS.
- M. Zhu, X. Cai, M. Fujitsuka, J. Zhang and T. Majima, Angew. Chem., Int. Ed., 2017, 56, 2064–2068 CrossRef CAS PubMed.
- L. Mao, X. Cai, S. Yang, K. Han and J. Zhang, Appl. Catal. B Environ., 2019, 242, 441–448 CrossRef CAS.
- J. Wu, S. Huang, Z. Jin, J. Chen, L. Hu, Y. Long, J. Lu, S. Ruan and Y.-J. Zeng, J. Mater. Sci., 2018, 53, 16557–16566 CrossRef CAS.
- M. Zhu, C. Zhai, M. Fujitsuka and T. Majima, Appl. Catal. B Environ., 2018, 221, 645–651 CrossRef CAS.
- J. Hu, Y. Ji, Z. Mo, N. Li, Q. Xu, Y. Li, H. Xu, D. Chen and J. Lu, J. Mater. Chem. A, 2019, 7, 4408–4414 RSC.
- Q. Zhang, J. Zhang, L. Zhang, M. Cao, F. Yang and W.-L. Dai, Appl. Surf. Sci., 2020, 504, 144366 CrossRef CAS.
- S. Yılmaz, E. G. Acar, G. Yanalak, E. Aslan, M. Kılıç, İ. Hatay Patır and Ö. Metin, Appl. Surf. Sci., 2022, 593, 153398 CrossRef.
- X. Ren, L. Shi, Y. Li, S. Song, Q. Wang, S. Luo, L. Ren, H. Zhang, Y. Izumi, X. Peng, D. Philo, F. Ichihara and J. Ye, ChemCatChem, 2020, 12, 3870–3879 CrossRef CAS.
- R. Feng, K. Wan, X. Sui, N. Zhao, H. Li, W. Lei, J. Yu, X. Liu, X. Shi, M. Zhai, G. Liu, H. Wang, L. Zheng and M. Liu, Nano Today, 2021, 37, 101080 CrossRef CAS.
- Y. Xu, X. Wang, M. Jin, K. Kempa and L. Shui, ChemElectroChem, 2020, 7, 96–104 CrossRef CAS.
- X. Niu, Y. Li, H. Shu and J. Wang, J. Phys. Chem. Lett., 2016, 7, 370–375 CrossRef CAS PubMed.
- B. Jin, Y. Cho, C. Park, J. Jeong, S. Kim, J. Jin, W. Kim, L. Wang, S. Lu, S. Zhang, S. H. Oh, K. Zhang and J. H. Park, Energy Environ. Sci., 2022, 15, 672–679 RSC.
- W. Li, Z. Wang, F. Zhao, M. Li, X. Gao, Y. Zhao, J. Wang, J. Zhou, Y. Hu, Q. Xiao, X. Cui, M. J. Eslamibidgoli, M. H. Eikerling, R. Li, F. Brandys, R. Divigalpitiya, T.-K. Sham and X. Sun, Chem. Mater., 2020, 32, 1272–1280 CrossRef CAS.
- C. Jellett, J. Plutnar and M. Pumera, ACS Nano, 2020, 14, 7722–7733 CrossRef CAS PubMed.
- M. Barua, M. B. Sreedhara, K. Pramoda and C. N. R. Rao, Chem. Phys. Lett., 2017, 683, 459–466 CrossRef CAS.
- Z. Wu, Y. Lyu, Y. Zhang, R. Ding, B. Zheng, Z. Yang, S. P. Lau, X. H. Chen and J. Hao, Nat. Mater., 2021, 20, 1203–1209 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |