
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic ammonia reforming: alternative routes to net-zero-carbon hydrogen and fuel
Luis C.
Caballero†
 a,
Nicholas E.
Thornburg†‡
*b and
Michael M.
Nigra
*a
a,
Nicholas E.
Thornburg†‡
*b and
Michael M.
Nigra
*a
aDepartment of Chemical Engineering, University of Utah, Salt Lake City, UT, USA. E-mail: Michael.Nigra@utah.edu
bCenter for Integrated Mobility Sciences, National Renewable Energy Laboratory, Golden, CO, USA. E-mail: Nicholas.Thornburg@nrel.gov
First published on 17th October 2022
Abstract
Ammonia is an energy-dense liquid hydrogen carrier and fuel whose accessible dissociation chemistries offer promising alternatives to hydrogen electrolysis, compression and dispensing at scale. Catalytic ammonia reforming has thus emerged as an area of renewed focus within the ammonia and hydrogen energy research & development communities. However, a majority of studies emphasize the discovery of new catalytic materials and their evaluation under idealized laboratory conditions. This Perspective highlights recent advances in ammonia reforming catalysts and their demonstrations in realistic application scenarios. Key knowledge gaps and technical needs for real reformer devices are emphasized and presented alongside enabling catalyst and reaction engineering fundamentals to spur future investigations into catalytic ammonia reforming.
 Luis C. Caballero | Luis C. Caballero is a doctoral candidate in chemical engineering at the University of Utah. He is currently working under Dr Michael Nigra's supervision in the Nanointerface Engineering Laboratory. His research interests are rooted in the design of nanomaterials for heterogeneous catalysts, including their optimization for potential industrial applications. Currently, he is focusing on the design of bifunctional structured Fischer–Tropsch catalysts for the efficient production of hydrocarbons from syngas. Luis has a Bachelor of Science in Chemical Engineering from the University of Alabama. |
 Nicholas E. Thornburg | Dr Nicholas (Nick) E. Thornburg is a chemical reaction engineer in the Fuels and Combustion Science Group within the Center for Integrated Mobility Sciences (CIMS) at the National Renewable Energy Laboratory (NREL). Nick is currently interested in multiphase reaction engineering for on-board fuel reforming and emissions management applications for the renewable synthesis of small-molecule chemicals and fuels, such as ammonia. Nick has a Bachelor of Science in Chemical Engineering from Washington University in St. Louis and a Doctorate in Chemical Engineering from Northwestern University. His doctoral research focused on understanding heterogeneous supported transition metal oxide catalysts for selective oxidation reactions. |
 Michael M. Nigra | Dr Michael M. Nigra is an assistant professor of Chemical Engineering at the University of Utah. He leads the Nanointerface Engineering Laboratory where he and his team design, synthesize, characterize and apply important interfaces in catalysis. He utilizes metal–organic ligand interfaces to enhance the activity, selectivity and stability of catalysts. Michael has Bachelor of Science degrees in Chemical Engineering and Biomedical Engineering from Carnegie Mellon University. He received his PhD at University of California, Berkeley and was a senior research associate at University College London in Chemical Engineering and the Centre for Nature Inspired Engineering before beginning his faculty career. |
Introduction
Ammonia (NH3) is one of the most critical industrial chemicals for sustaining global life, with around 180 million t produced globally every year exclusively via Haber–Bosch-type routes.1 In addition to its current major uses as a fertilizer, chemical intermediate and refrigerant, ammonia is poised to play a crucial role in substituting conventional fossil fuels and hydrogen (H2) resources for future energy and chemical platforms. The U.S. Department of Energy (DOE) has recently identified NH3 as a promising H2 carrier for advanced mobility and storage,2 owing to its high active hydrogen content (17.8 wt%), ease of liquefaction and accessible dissociation chemistries. Although pure NH3 has poor fuel properties itself,3 blended or reformed mixtures of H2 and NH3 are higher performing in internal combustion (IC) engines.4,5 Emerging, renewable-electron-driven NH3 synthesis pathways6,7 enable the molecule's prospect as a net-zero-carbon (NZC) H2 carrier and/or fuel for advanced IC piston and turbine engines and for fuel cells (FCs), broadly applicable to each transportation and stationary energy applications (Scheme 1). | ||
| Scheme 1 Spectrum of NH3-capable internal combustion (IC) and fuel cell (FC) technologies for air, sea and land mobility and for stationary applications as a function of NH3 conversion (i.e., H2 fraction in reformate). Note that additional hydrogen purification is required following catalytic NH3 reforming for FC applications. | ||
Ammonia reforming (i.e., ammonia dissociation or decomposition) occurs in the presence of a heterogeneous catalyst to produce hydrogen, nitrogen (N2) and possibly water (H2O) if oxygen (O2) is present in the feed gas. The extent of NH3 conversion is a strong function of catalyst identity, reforming reaction conditions, participating reforming pathways, reactor geometry, fluid contacting patterns, transport phenomena and more, and different NH3 reforming applications dictate unique performance targets and design constraints. Generally, air-,8,9 sea-10,11 or land-borne12,13 vehicles utilizing ammonia as a liquid fuel and/or carrier will require on-board catalytic reforming reactors, akin to hydrocarbon fuel reforming strategies,14,15 whereas stationary applications for hydrogen storage and dispensing may have lesser spatial constraints but much stricter hydrogen product purity specifications; in particular, ultra-high H2 purities are required for NH3/H2 FC converters when NH3 is used as the carrier16 (Scheme 1). However, unlike high-pressure hydrogen, well-established NH3 infrastructure and supply chains will sustain rapid adoption of new customer end-markets while renewable NH3 generation technologies mature to scale. Ultimately, the future viability of ammonia as a NZC H2 carrier or fuel for IC engines, FC systems and stationary hubs—by air, sea or land—hinges upon the development of efficient, highly integrated catalytic reforming reactors to generate requisite hydrogen energy or fuel at tunable purities for a given end use.
In this Perspective, we review recent advances in heterogeneous catalysis for ammonia reforming alongside enabling reaction engineering principles to motivate future research investigations into this important route to NZC hydrogen and fuel for difficult-to-decarbonize modes of global transportation. We draw parallels across various time and length scales of NH3 reforming to illustrate the unique multidimensionality of this problem, as well as the hurdles that belie its graduation from laboratory to commercial use. While H2O electrolysis and renewable NH3 synthesis systems continue to mature, here we identify key knowledge gaps in catalytic ammonia reforming strategies and technologies to prescribe actionable goals for fundamental researchers and industrial practitioners alike. Successful advances in ammonia reforming will help bridge critical deployment barriers in hydrogen-at-scale, offering flexible, alternative pathways to these NZC energy vectors.
Fundamentals of ammonia reforming catalysis
Catalytic routes to ammonia reforming provide potentially lower pressure and temperature pathways to non-electrolytic hydrogen production. As the reverse reaction of ammonia synthesis, ammonia decomposition is an endothermic reaction, and temperatures of 400 °C are needed to drive the reaction to >99% equilibrium conversion of ammonia. However, reforming applications for IC engine feeds will likely require high-pressure operating conditions that adversely shift equilibrium in favor of the reactants, requiring a new understanding of catalysts in these unfavorable environments.In this section, we will present different catalytic pathways for ammonia reforming focusing on (1) thermal and photocatalytic reforming (i.e., dissociation; eqn (1)), and (2) oxidative reforming (eqn (2)). This Perspective will emphasize thermal and oxidative reforming and provide a brief introduction to photocatalytic reforming. Electrochemical ammonia reforming and ammonia combustion (eqn (3)) will not be discussed. We further note that the terms “reforming”, “dissociation” and “decomposition” may be used interchangeably in the context of these ammonia reactions; however, “cracking” is an inappropriate term to describe the chemistry of this small molecule (just as for methane (CH4)), regardless of pathway or driving force, and should instead be reserved for long-chain hydrocarbon conversion processes.
Thermal reforming
| NH3(g) ⇆ 0.5 N2(g) + 1.5 H2(g), ΔH° = +46 kJ molNH3−1 | (1) |
Oxidative reforming
| NH3(g) + 0.25 O2(g) → 0.5 N2(g) + H2(g) + 0.5 H2O(g), ΔH° = −75 kJ molNH3−1 | (2) |
Stoichiometric combustion
| NH3(g) + 0.75 O2(g) → 0.5 N2(g) + 1.5 H2O(g), ΔH° = −317 kJ molNH3−1 | (3) |
The next three subsections describe thermal reforming, oxidative reforming and photocatalytic reforming catalysts to highlight the current states-of-art as well as historical perspectives in each area. Opportunities for future research directions will also be presented.
Thermal reforming catalysis
Tamaru20 applied a dynamic approach to the study of NH3 decomposition kinetics by elucidating its mechanism over different metal-based catalyst surfaces in 1988. The study first examined the reaction over a tungsten catalyst, determining that the overall reaction order was unity with respect to NH3 at lower ammonia pressures, approaching zero at higher ones. He further concluded that the rate is always zero-order with respect to H2 pressure for this reaction pathway over tungsten. However, mechanistic behavior varies significantly over other transition metals such as iron, where faster hydrogenation of chemisorbed nitrogen dominates. This case typically occurs at lower temperatures and is referred to as the Temkin–Pyzhev mechanism. Conversely, if desorption of nitrogen occurs more rapidly, which is the case at higher temperatures, the tungsten-type behavior is observed. Therefore, the mechanistic pathway depends on the operating temperature, the partial pressure of the reactants and the identity of the catalyst, as stated by Löffler et al.21 in 1976.
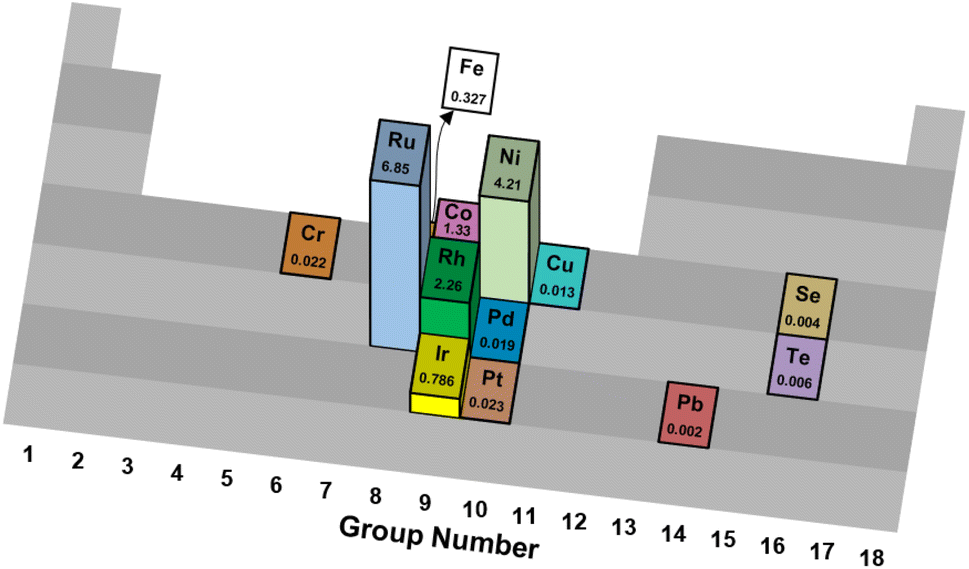 | ||
| Fig. 1 Turnover frequencies (s−1) of various catalytic elements supported on Al2O3 for thermal reforming of NH3.35 | ||
The differences in mechanistic pathways depend on the metal that is used, and the wide range of reaction conditions result in these active metals having clear advantages over one another in particular situations. Ru-based catalysts exhibit high catalytic activity. Supports play a crucial role in these materials, with carbon nanotubes (CNTs),39 MgO,40 and MgAl2O4 (ref. 41) showcasing different catalytic activities. Similarly, different promoters such as Cs, K and Na have been studied with varying results.42 Ru is the most promising metal for ammonia decomposition on the basis of reaction rate. Nonetheless, its major drawback is its higher price compared to Fe and Ni, which serves as a barrier to industrial applications.
Iron-based catalysts are used for ammonia decomposition in hot gas from coal gasification. Fe catalysts have lower activity than Ru catalysts. However, these materials provide valuable insight into the kinetics of the reaction and the interactions with support materials. Some of the studied support materials include CNTs and mesoporous carbon, such as CMK-3. These Fe catalysts are often compared to Ni-based catalysts, which have niche applications in microstructured reactors for hydrogen production. They provide an attractive alternative, as Ni has a lower cost than Ru and displays higher activity than Fe.43 Despite these advantages, ammonia decomposition with Ni as the active metal is highly dependent on structural parameters, such as particle size.44 Such considerations also apply to cobalt-based catalysts. Their activity is a strong function of the physical and chemical properties of the support material.45 Co-based catalysts have become relevant for ammonia decomposition due to their low cost compared to other active components. Metals such as Pt and Rh have also been assessed for their feasibility. Pt-based catalysts have significant drawbacks, as they must be part of a bimetallic system. Pt is not a very active catalyst for ammonia decomposition by itself, as conversions of only 2% are achieved with monometallic Pt catalysts.39 Its advantages lie in its role as an activity enhancer when combined with other metals such as Ni, Fe, or Co.46
Table 1 summarizes results across different studies of catalytic materials for NH3 decomposition. Taken together, these results corroborate Ru being an effective and frequently studied catalytic metal for NH3 thermal reforming.
| Catalytic metal | Metal content (wt%) | Promoter | Metal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :promoter (mol mol−1) :promoter (mol mol−1) |
Support material | Temperature (°C) | Pressureb (atm) | GHSV (mL gcat−1 h−1) | TOF (min−1) | NH3 Conversion (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a All reactions were performed in a fixed-bed flow reactor. b Pressure conditions assumed to be atmospheric if not explicitly stated. | ||||||||||
| Ru | 3.0 | — | — | Ba–ZrO2 | 450 | — | 30000 |
26.6 | 23.6 | 49 |
| Ru | 3.0 | K | 2:1 |
Ba–ZrO2 | 450 | — | 30000 |
36.7 | 32.5 | 49 |
| Ru | 3.0 | Cs | 2:1 |
Ba–ZrO2 | 450 | — | 30000 |
42.8 | 37.8 | 49 |
| Ru | 2.5 | — | — | β-SiC | 400 | 1 | 60000 |
8.4 | 99.3 | 50 |
| Ru | 5.0 | — | — | MgO | 450 | — | 60000 |
21.0 | 30.9 | 51 |
| Ru | 5.0 | — | — | Al2O3 | 450 | — | 60000 |
15.8 | 23.4 | 51 |
| Ru | 5.0 | K | 1:1 |
Carbon nanotubes (CNTs) | 450 | — | 60000 |
65.9 | 97.3 | 51 |
| Ru | 5.0 | — | — | CNTs | 550 | 1 | 30000 |
52.6 | 84.7 | 52 |
| Ru | 5.0 | — | — | Graphitic carbon (GC) | 550 | 1 | 30000 |
58.8 | 95.0 | 52 |
| Ru | 3.2 | — | — | CNFs | 500 | 1 | 6500 | 22.7 | 99.0 | 53 |
| Ru | 11.7 | K | 1:5 |
Graphene aerogel (GA) | 450 | 1 | 30000 |
25.9 | 97.6 | 54 |
| Ru | 5.0 | — | — | Cr2O3 | 600 | — | 30000 |
62.1 | 100 | 55 |
| Ru | 4.8 | — | — | La2O3 | 525 | 1 | 18000 |
25.6 | 90.7 | 56 |
| Co | 10.0 | — | — | Multi-wall CNTs | 500 | 1 | 6000 | 3.0 | 74.6 | 57 |
| Fe | 1.29 | — | — | GC | 600 | — | 6000 | 20.6 | 71.0 | 58 |
| Ir | 10.0 | — | — | SiO2 | 550 | — | 30000 |
18.3 | 30.4 | 59 |
| Ni | 10.0 | — | — | SiO2 | 550 | — | 30000 |
4.0 | 21.6 | 59 |
| Ni | 5.0 | K | 1:2 |
SiO2 (fumed) | 550 | — | 30000 |
16.8 | 42.6 | 60 |
| Ni | 15.0 | — | — | MgAl (6:1) |
550 | — | 30000 |
6.3 | 48.0 | 61 |
| Ni | 10.0 | — | — | La2O3 | 550 | 1 | 30000 |
1.6 | 59.0 | 62 |
| Rh | 4.9 | — | — | CNTs | 400 | — | 30000 |
9.0 | 3.0 | 39 |
| Pt | 9.3 | — | — | CNTs | 400 | — | 30000 |
2.5 | 2.0 | 39 |
| CoMo | 5.0 | — | — | γ-Al2O3 | 600 | — | 36000 |
82.7 | 99.5 | 63 |
| CoFe5 | 5.0 | — | — | CNTs | 600 | — | 36000 |
90.2 | 48.0 | 64 |
| MoS2 | 6.0 | — | — | Laponite | 600 | — | 24000 |
25.9 | 35.0 | 65 |
Oxidative reforming catalysis
Oxidative reforming (eqn (2)) provides an alternative, exothermic pathway for ammonia decomposition. In contrast to combustion (eqn (3)), here a sub-stoichiometric amount of O2 is co-fed to the reactor with NH3 reactant. The catalyst bed is heated through the exothermic combustion of ammonia (−200 kJ molRu−1).78There are a number of important material property considerations when designing an oxidative reforming catalyst: (1) active phase composition, (2) support material, (3) promoters, (4) basicity/acidity, (5) surface area and (6) low amount of electron-withdrawing groups.79 The RDS may be different depending on the composition of the active phase. For example, dehydrogenation of NH3 is rate-determining on noble metal catalysts, whereas the associative desorption of –N atoms is the rate-determining step on non-noble metal catalysts. Further, it is important to choose a catalytic material which does not adsorb CO2 and H2O very well; otherwise, a catalyst pre-treatment step will be necessary. CO2 and H2O may out-compete NH3 and O2 for binding sites on the catalyst, but adsorption behaviors may be mitigated through rational design.
Theory suggests that iridium can produce N2 with 100% selectivity from NH3.80 While environmentally acceptable as a catalytic metal, a main drawback of Ir is its high cost,81 which has motivated continued exploration of ruthenium-based catalysts for oxidative reforming of NH3. It has been proposed that adsorbed ammonia reacts with adsorbed oxygen atom or hydroxyl groups on the active metal surface to remove a hydrogen from ammonia. It is also hypothesized that ammonia dissociation is dependent on co-adsorbed oxygen. The adsorbed oxygen or hydroxyl group also reacts with NH to remove its hydrogen. Co-adsorbed oxygen increases the ammonia desorption on Ir surfaces.80 There are still opportunities for additional studies for further study into the mechanism on Ir and other catalytically active metals. Ru also suffers from deactivation. While the mechanism for deactivation has not been thoroughly studied, it is hypothesized that it is caused by oxide formation, nanoparticle sintering, and diffusion of Ru into the support.82 One of the first studies with Ru/Al2O3 catalysts for oxidative reforming was published in 1967 by Schriber and Parravano with Ru nanoparticles.83 They observed that the rate was dependent on the partial pressures of NH3, O2 and H2O. Higher H2O partial pressures decreased the NH3 oxidation rate, while higher O2 partial pressures increased the NH3 oxidation rate.
One of the recent successful demonstrations of oxidative reforming of NH3 was reported by Nagaoka et al.84 using Ru oxide nanoparticles supported on γ-Al2O3 and La2O3. The catalyst is pre-treated with He to remove CO2 and H2O from the surface, which forms Lewis acid sites in situ to activate NH3. The heat produced from ammonia adsorption and oxidation can in turn be used to drive NH3 decomposition, allowing the reaction to proceed autothermally. In this manner, Ru oxide/γ-Al2O3 is a more active catalyst than Ru oxide/La2O3. The hypothesis for this difference in activity is that La2O3 does not have the Lewis acid sites needed for NH3 adsorption. The quantity of NH3 adsorbed onto the γ-Al2O3-supported sample was 8× higher than that of the La2O3-supported catalyst, contributing to the former support's higher performance within this proof-of-concept study. Another noteworthy example of Ru catalytic activity of NH3 oxidative decomposition is Ru/Ce0.5Zr0.5O2−x by Matsunaga et al.85 Conversion of NH3 was reported to be >96%, with N2 and H2 yields of >96% and 63%, respectively; oxygen conversions were seen to be virtually 100% for this catalyst (Fig. 2). NH3 conversion increased with decreasing NH3/O2 feed ratios. Ru clusters have been immobilized on alkali-exchanged Y zeolites by Cha et al.86 Of the catalysts prepared, Ru/Rb–Y was most active for oxidative reforming, attributed to the low acidity of the Rb–Y and higher electron density around the Ru sites, which allows for more facile N2 desorption.
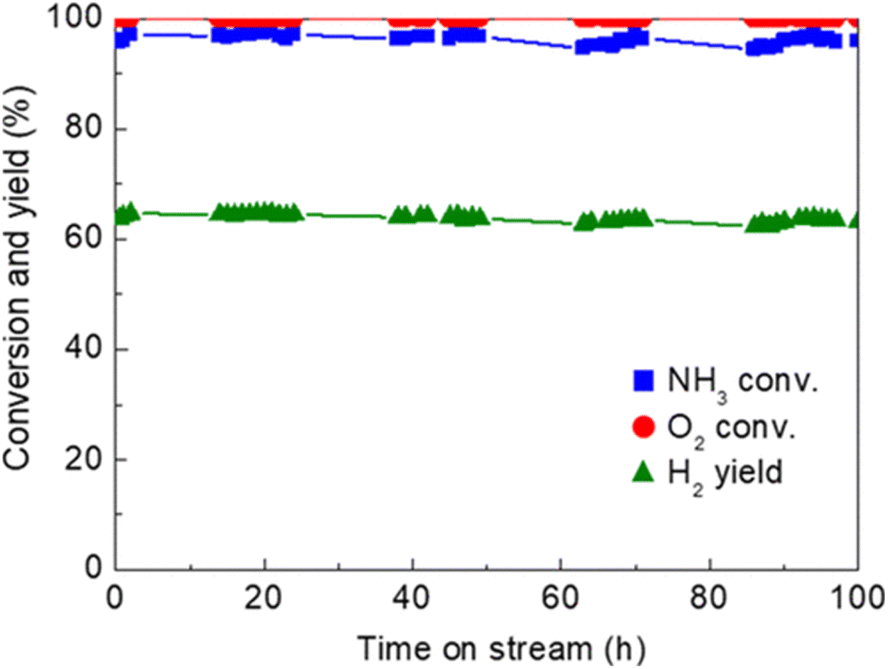 | ||
| Fig. 2 Time-on-stream activity and stability of Ru/Ce0.5Zr0.502 catalyst (1 wt% Ru loading) for oxidative reforming of NH3.85 Reaction conditions:Feed rate 150:37.5:20.8 (NH3:O2:He) mL min−1, 62.5 L h−1 g−1 space velocity, quasi-adiabatic operation at ambient initial temperature. Reproduced with publisher permission from ref. 85. | ||
There have been advances in alloying Ru with other metals to enhance the aggregate catalyst's performance. Experiments performed by Chakraborty et al.87 combined Cu with Ru to study their performance for oxidative reforming at 170 °C under ultra-high vacuum (UHV) conditions. The two metals are immiscible, and the Cu naturally segregates to the surface of the nanoparticles. Ammonia consumption rate enhancements were observed to be 3× greater with respect to the monometallic Ru catalysts and 40× with respect to monometallic Cu catalysts, with the best bimetallic compositions determined to be 0.6–0.8 monolayers of Cu on Ru. All bimetallic compositions exhibited higher activities than the monometallic Cu and Ru.
Future work is needed to discover and understand new catalyst materials for oxidative reforming of NH3. There is much room to further develop alloy catalysts and suitable support materials that enable desirable alloy formation and substrate binding. Opportunities exist to pair electron-donating supports or ligands to these active metals to facilitate nitrogen desorption steps in oxidation mechanisms.
Photocatalytic reforming
Photocatalytic reforming of ammonia shows great promise as a sustainable pathway to produce ammonia-derived hydrogen. There are five processes that underlie photocatalytic ammonia reforming, and to photocatalysis in general. These processes are: (1) generation and (2) separation of photoelectron–hole pairs, (3) absorption of light, (4) adsorption/desorption of reactants and products and (5) redox reactions on the surface of the photocatalysts.88 In recent years, significant advances have been made in the design of heterogeneous photocatalysts for reforming applications, and we highlight some of these advances as well as potential opportunities in the field.In ammonia photocatalytic reforming, a catalytically active metal is typically paired with a photoactive support material; metal nanoparticles are often used to allow for better charge separation.89 Catalytic metals often feature noble metals such as Pt, Pd or Ag, which have significant costs associated with them. Support materials include TiO2, g-C3N4, Ag3PO4 and ZnO as examples. Dopants are often utilized to adjust the band gap to shift the absorption into the visible region.
One of the first reports on using TiO2 as a photocatalyst for NH3 oxidation to produce N2 was published in 1979 by Mozzanega et al. They used TiO2 only as a photocatalyst.90 An early study by Taguchi and Okuhara91 investigated a range of metal nanoparticles such as Pd, Pt, Cu, Ni, Co, Rh and Ru supported on P-25 TiO2 for oxidation of aqueous ammonia. They observed that the N2 production rates trended Pt/TiO2 ≫ Ru/TiO2 > Pd/TiO2 > Rh/TiO2. Altomare and Selli89 showed that Ag/TiO2 had higher NH3 conversion than Pt/TiO2 and Pd/TiO2 but was not as selective to N2 as Pd/TiO2. There have been several successful recent efforts to use more abundant and less expensive metals in photocatalytic materials for ammonia oxidation. Utsunomiya et al.92 tested the following metals supported on TiO2: V, Cr, Ni, Cu, Co, Fe and Mn. Of these metals, Ni/TiO2 exhibited the best N2 selectivity in aqueous solutions. In a departure from using TiO2-based materials, Chen et al. investigate the use of silver phosphate (Ag3PO4) as a photocatalyst,93 which advantageously features the absorption of visible light wavelengths. They compared the activity of Ag3PO4 to that of N-doped TiO2 and g-C3N4 photocatalysts. Ag3PO4 demonstrated activity for NH3 reforming under visible light illumination at ambient conditions. While N-doped TiO2 and g-C3N4 are known to be active as photocatalysts in the visible range, they were not active for NH3 reforming. The authors correlated the formation of ˙OH radicals by Ag3PO4 with the activity in NH3 reforming, as ˙OH radicals were not observed with the other two materials.
There is still much more room for progress in the development of NH3 reforming photocatalysts under oxidative and non-oxidative conditions. Shifting the light absorption range from UV to visible or even near-infrared ranges is desirable and should be further investigated to utilize solar energy inputs more efficiently in such systems. Additionally, utilizing bimetallic nanoparticles such as AuPt nanoparticles94 could provide additional activity and selectivity beyond their monometallic counterparts and should be further investigated in the future.
Reformer reaction engineering and scaling considerations
Beyond catalyst fundamentals, myriad reactor characteristics influence the performance of a given ammonia reforming catalyst. In this section, as a complement to the present literature emphasis on catalyst discovery and evaluation, we discuss important meso- and macroscale aspects of technical catalysts, reactor designs and integration strategies to motivate device development for various end uses. The four main topics of focus are (1) technical catalyst properties, (2) hydrogen product quality, (3) energy (i.e., heat and light) management and (4) exhaust management.Technical catalyst properties
Technical catalysts often vary significantly from laboratory research catalysts in physical form, chemical composition and acceptable performance criteria.95 For ammonia reforming, some primary considerations of a candidate technical catalyst are its metal identity, metal loading, structural formulation and stability.First, trends in sustainable metal sourcing and global availability urge the substitution of platinum-group metals (PGM) with more environmentally benign catalytic metals. While ruthenium exhibits good sustainability metrics,81 its relatively high price has prohibited industrial adoption for Haber–Bosch synthesis;96 the authors predict similar economic constraints for the use of Ru in commercial NH3 reforming applications, although catalyst costs for downscaled devices may be acceptable. However, only a limited number of reports detail metals beyond Ru and Pd to facilitate this chemistry (Table 1 and Fig. 1).23,78,97–102 It has been hypothesized that N2 desorption limits the effective kinetic rates of NH3 decomposition on Fe-, Co- and Ni-based catalysts, while other late transition metals such as Cu, Rh, Pd, Ir and Pt are limited by N–H bond scission kinetics;35 in all cases, Ru is regarded as a highly active metal for NH3 thermal78 and oxidative84,85 reforming. Similarly, low metal contents generally make for more cost-effective materials, but deactivation phenomena may necessitate supra-stoichiometric loadings to prolong catalyst lifetime (vide infra). The authors encourage further research into Earth-abundant, low-cost metals, metal precursors and material preparation methods that yield catalysts with high activity and thermal/oxidative stability for NH3 reforming.
The required form factor of a technical catalyst for ammonia reforming is largely defined by the intended application. While chemical manufacturers frequently employ packed bed reactors, such reactor types and catalyst forms are only appropriate in stationary settings. Indeed, extruded catalyst pellets are not amenable to mobile applications (such as on-board NH3 reforming on an aircraft, maritime vessel or off-road vehicle), where frequent vibrations and vehicle movement would lead to bed unpacking, pellet attrition and/or entrainment of fines.
Instead, monolith reactors are attractive for mobile environments. Monoliths are constructed from metal or ceramic materials that are washcoated to support a catalyst.103 Ubiquitous in automotive exhaust, monoliths feature high mechanical strength, equal flow conditions across all channels, low pressure drop and dynamic, transient operability.104,105 However, catalyst loadings per volume tend to remain low compared to traditional packed beds, which is a disadvantage for any kinetically limited process. These features and limitations imply distinct performance attributes compared to laboratory microreactors operated at differential conversion with powdered catalysts. Indeed, reports on NH3 reforming that employ monoliths are scant in the literature.13,106,107 Plana et al. reported Ni/Al2O3/cordierite monoliths for thermal reforming, exhibiting complete conversion of pure NH3 at 600 °C.106 Compared to packed bed experiments with <200 μm particles of crushed monolith catalyst or of un-supported Ni/Al2O3, the intact cordierite-supported Ni catalyst is capable of 10–20% higher NH3 conversions even when the monolith is operated at 100 °C lower temperatures.106 Clearly, catalyst form factor and construction are critical performance descriptors, suggestive of enhanced mass and/or heat transfer or possible hydrodynamic advantages when NH3 conversion is carried out in real devices. Separately, Wang et al. demonstrated >99.9% conversion at the same temperature and 1100 sccm NH3 feed flowrate over a microfibrous CeO2-promoted Ni/Al2O3 monolith catalyst for 300 h on stream,107 whereas Kane et al. successfully performed on-board NH3 reforming over a FeCrAl monolith-supported 4.7 wt% Ru/Al2O3 catalyst integrated within a John Deere 6400 tractor.13 As a complement to these works, we recommend dedicated research efforts by the ammonia energy community to understand washcoated (vs. powdered) catalyst performance and dynamic monolith cycling strategies for on-board NH3 reforming. Advances in 3D printing of catalyst supports108 and monolith structures109,110 are a further avenue of promising research that may help mitigate effective surface area and volume constraints.
Lastly, a key requirement of technical reforming catalysts is their long-term stability and robustness against deactivation. Deactivation of ammonia reforming catalysts is generally not well understood in the literature47,54,82,98,111 and is worthy of significant attention from the research community. From a reaction engineering perspective, physical pore fouling, active site poisoning, and/or thermal and mass gradients leading to active site restructuring (especially during dynamic and/or oxidative cycling) are the likely deactivation modes of an ammonia reforming catalyst. Intentional fouling/poisoning with known or predicted feed/recycle impurities, accelerated aging112 and extended time-on-stream testing under gradientless conditions113 are urgently needed to advance the viability of NH3 reforming catalyst candidates for a given application. Practitioners should carefully choose reaction conditions for deactivation studies114 that result in incomplete, integral (moderate) conversions in order to observe diminishing catalyst activities over extended spacetimes.
Hydrogen product quality
The requisite hydrogen purity that must exit a catalytic ammonia reformer is specified by the end use. Indeed, for IC piston engines and turbines, only ∼25–30 vol% H2 in unreacted NH3 is required to produce a fungible fuel blend that matches target post-combustion enthalpies for stationary power or vehicle propulsion (Scheme 1). These IC uses necessitate only a single suitable reforming catalyst and reactor. Higher purity applications for FCs, however, require more dedicated strategies to isolate and purify H2.82 Given the equilibrium limitation of the thermal reforming pathway (eqn (1)) and the N2 and H2O (eqn (2) only) co-products, complete conversion to pure hydrogen is not achievable over any sole catalyst—thus requiring creative intensification strategies to simultaneously generate and purify the desired H2 product. Moreover, both IC and FC converters may feed fuels at elevated pressures, requiring auxiliary compressors or catalytic reformers that operate at high pressure despite the unfavorable equilibrium penalty (eqn (1)).Various configurations of a reforming reactor integrated with a hydrogen separator are appropriate strategies to generate high-purity (>99.999%) or ultra-high-purity (>99.9999%) H2 streams for vehicle refueling or distributed FC utility stations. Stationary systems may afford packed bed reactors integrated with downstream and/or interstage sorbent or membrane separators with NH3 recycle loops, whereas mobility applications may require intensified, spatially compact catalytic membrane reactors, with the NH3 dissociation catalysis and H2 removal occurring simultaneously at the same local time and length scales.82,115 Pd-based membranes are well-known for selective H2 separation,116,117 and membrane reactors for NH3 reforming have been reported in the literature.118,119 One recent study has successfully demonstrated the latter for maritime applications up to 86% supra-equilibrium H2 yields at up to 99.998% purity at operating temperatures of ≥425 °C; further, when the permeate was subjected to a vacuum, essentially complete NH3 conversion was achieved by rapid reaction rates at 400 °C, well beyond equilibrium reaction limitations.120 Additional research is needed to identify additional NH3-tolerant membrane, absorbent and adsorbent materials suitable for intensified catalytic reforming strategies.
Energy management
Energy-intensive endothermic reactions such as eqn (1) are challenging to implement in any situation, but particularly so on board of aircraft, maritime vessels or off-road vehicles without auxiliary thermal energy generation systems. Separate challenges arise when light (photons) must be introduced to catalytic reactors outside of laboratory settings.121,122 Effective recovery and transfer of sensible heat from combustion exhaust streams and/or photons and heat derived from practical light sources are therefore crucial for on-board energy management. Energy balances across monoliths include both axial convection and radial conduction terms, complicating interpretation of experimental results and the development of multiscale models.105 Recently, Danilov and Kolb described an autothermal hydrocarbon reforming monolithic reactor using a tanks-in-series model,123 which may be applicable to autothermal ammonia reforming systems. Sensible heat recuperation from exhaust gases has been effectively demonstrated on board a John Deere 6400 tractor that employed a 4.7 wt% Ru/Al2O3 thermal reforming (eqn (1)) catalyst supported on a FeCrAl monolith for a dual-fuel diesel/anhydrous NH3 engine (Fig. 3).13 Specifically, heat exchanger efficiencies of up to 98% were achieved for NH3 flow rates spanning 1.5–12.5 kg h−1 for NH3 conversions of ∼30–50% across various load cycles. While encouraging for an agricultural tractor, such thermal recuperation schemes may be more challenging to implement for an aircraft or a maritime vessel, where higher mass throughputs and heat transfer rates and efficiencies are expected. Ammonia reformers in these contexts pose a steep design challenge of low mass, compact footprint, high effective surface area, low pressure drop and low thermal latency. Further, spatially compact, highly efficient heat exchanger designs are crucially important for endothermic ammonia reformers (i.e., eqn (1) only).124 Finally, light-driven systems are hampered by light attenuation limitations as per the Beer–Lambert Law,122 as well as non-uniform light intensity and photon penetration into densified catalyst forms, constraining their adoption in mobility applications.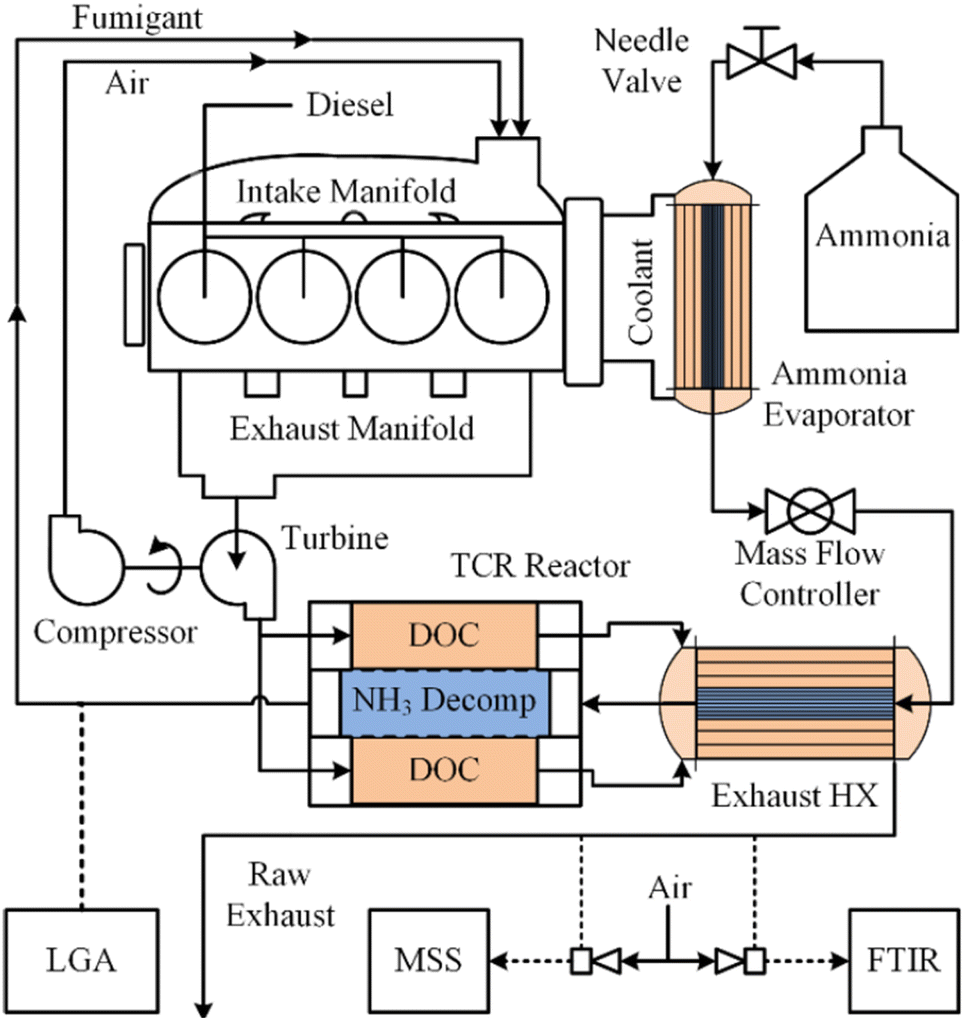 | ||
| Fig. 3 On-board anhydrous NH3 thermal reforming reactor and heat exchangers for a diesel/NH3 dual-fuel tractor engine and exhaust system.13 Reproduced with publisher permission from ref. 13. | ||
Alternatively, drawing inspiration from the chemical industry, endothermic reactions are regularly coupled with exothermic reactions to supply sufficient thermal energy in situ and/or along reactor vessel walls. Perhaps the most ubiquitous commercial practice of enthalpic coupling is steam methane reforming125 (SMR; +206 kJ molCH4−1), which is paired with in situ water-gas shift (−41.2 kJ molCO−1), in situ partial methane oxidation (−35.6 kJ molCH4−1) and/or ex situ reactor wall-side natural gas combustion (−804 kJ molCH4−1). Remarkably, SMR has >4× the enthalpy requirement of eqn (1), suggesting that less demanding thermal coupling approaches exist for ammonia reforming. Such intricately coupled reaction engineering strategies enable energy-efficient endothermic or even autothermal126 steady-state chemical transformations, as well as minimally energy-intensive reactor start-up operations. Chiuta and Bessarabov reported an autothermal microreactor for NH3 decomposition (eqn (1)) that supplied heat via exothermic NH3 oxy-combustion in alternating, countercurrent catalytic plate channels;127 related approaches were modeled previously by Deshmukh and Vlachos.128 Ammonia autothermal reforming (NH3–ATR) combining eqn (1) and (2) has been demonstrated in laboratory reactors84,85 and, promisingly, for a diesel engine employing exhaust gas recirculation (EGR).129 In the latter case, an optimal sub-stoichiometric O2/NH3 feed ratio of 0.04–0.175 was achieved for 2.5–3.2 L minH2−1 over a pelleted 2 wt% Ru/Al2O3 catalyst at a constant NH3 feed rate of 3.0 SLM. H2 and reformer efficiencies were measured up to 80% and 102%, respectively,129 highlighting the viability of on-board NH3–ATR strategies for off-road mobility.
Additional research is recommended to understand cold-start and steady-state phenomena at each the catalyst, reformer and systems levels for different modes of heating (heat exchange, autothermality, Joule heating and combinations thereof). Joule heating is particularly attractive for hybrid vehicle systems that employ batteries and/or FC converters, which could in turn supply renewable electrons to reformer monoliths via local resistive heating; other creative concepts such as induction heating of electromagnetically active catalysts (e.g., core–shell materials130) may also offer promising solutions for next-generation vehicles. Stationary NH3 reforming applications offer significantly more latitude to employ Joule heating by renewable electricity, especially if catalysts and downstream H2 reservoirs are insensitive to thermal cycling. Finally, non-traditional reactor concepts such as electrochemical reactors,131 microreactors127,132 and plasma-driven reactors133 and catalysts134 are also promising approaches for managing thermal energy flows in ammonia reforming. These and other topics are worthy of future investigations.
Exhaust management
Ammonia reforming reactors offer unique integration opportunities with traditional vehicle exhaust systems, many of which already employ NH3 reductant (via urea solutions) for selective catalytic reduction (SCR) of post-combustion pollutants.135 Here, slip streams of ammonia from reformers may be supplied directly to conventional emissions catalysts to reduce harmful NOx gases below compliant thresholds; however, it is still unknown whether existing commercial NOx reduction catalysts will be suitable for the expectedly more concentrated exhaust resulting from combustion of NH3 fuel blends.4 Indeed, steady-state EGR has been demonstrated for thermally recuperated NH3 reforming using gasoline136,137 and diesel129 engines, but creative extensions of NOx exhaust gas recirculation (NOx–EGR) could enable intensified thermal/mass recuperation strategies for simultaneous NH3 reforming and NOx abatement over a dual-functional catalyst bed (Scheme 2). Multiscale and process-level models could aid in determining the dynamic mass and energy balances required for optimal operation, while significant research is yet needed to identify suitable multi-functional catalyst(s) for dual reforming and emissions management. Stationary systems housed in agricultural contexts could further incorporate upstream electrochemical reduction of nitrate contaminants to supply ammonia for on-site or on-board reforming, unlocking inventive concepts in nitrogen atom circularity.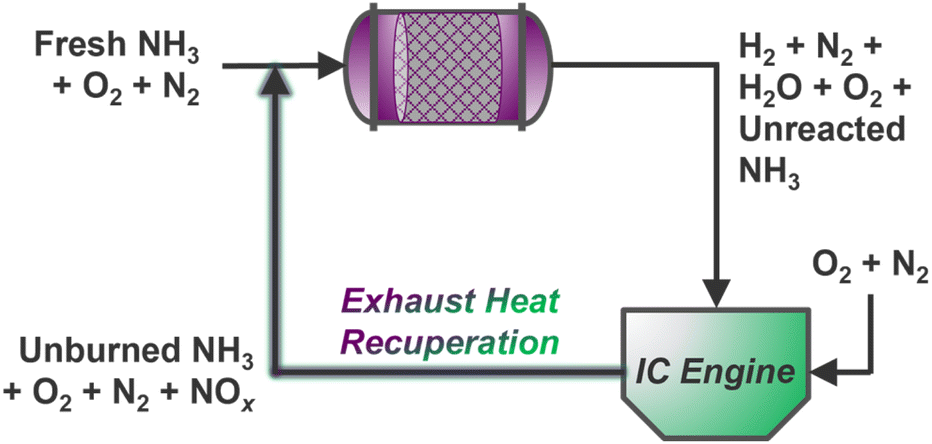 | ||
| Scheme 2 Steady-state intensified NH3–ATR and NOx–EGR concept with continuous mass and heat recycling to an ATR monolith inlet. | ||
Summary and outlook
Catalytic ammonia reforming is an important chemical transformation of broad interest to the hydrogen energy and sustainable fuels communities. Whether the NH3 is partially reformed for IC engines, turbines or solid oxide FCs, or completely reformed with integrated gas separation for ultra-high-purity H2 end uses, the fundamental catalysis and reaction engineering principles underlying such transformations are universal to myriad ammonia and hydrogen energy applications. However, significant knowledge gaps exist to identify and understand catalytic materials capable of dissociating ammonia at low to moderate temperatures, particularly under realistic, dynamic reactor operating conditions. Researchers studying ammonia energy in mobility and/or stationary contexts are recommended to further investigate specific technical hurdles in ammonia reforming, including active site substructures and energetics, catalyst stability, form factor, reactor configuration, and novel strategies for heat management, process intensification and nitrogen atom circularity.Author contributions
N. E. T. and M. M. N. conceived of the article topic and structure, and N. E. T., M. M. N., and L. C. C. wrote the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. C. C. and M. M. N. gratefully acknowledge the Department of Chemical Engineering at the University of Utah for funding this work. This work was authored in part by the National Renewable Energy Laboratory (NREL), operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. This work was supported in part by NREL Institutional Investments. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes. N. E. T. further acknowledges insightful discussions with Drs Robert McCormick, Brad Zigler, Matt Thornton, John Farrell, Bryan Pivovar and Randy Cortright of NREL, with Dr James Barth of Reaction Engines Inc. and with Dr Boris Sheludko of Amogy.References
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- Energy Storage Grand Challenge Roadmap, U.S. Department of Energy, DOE/PA–0022, 2020 Search PubMed.
- O. Elishav, B. Mosevitzky Lis, E. M. Miller, D. J. Arent, A. Valera-Medina, A. Grinberg Dana, G. E. Shter and G. S. Grader, Chem. Rev., 2020, 120, 5352–5436 CrossRef CAS PubMed.
- P. Dimitriou and R. Javaid, Int. J. Hydrogen Energy, 2020, 45, 7098–7118 CrossRef CAS.
- A. Valera-Medina, F. Amer-Hatem, A. K. Azad, I. C. Dedoussi, M. de Joannon, R. X. Fernandes, P. Glarborg, H. Hashemi, X. He, S. Mashruk, J. McGowan, C. Mounaim-Rouselle, A. Ortiz-Prado, A. Ortiz-Valera, I. Rossetti, B. Shu, M. Yehia, H. Xiao and M. Costa, Energy Fuels, 2021, 35, 6964–7029 CrossRef CAS.
- Y. Yang, J. Kim, H. Jo, A. Seong, M. Lee, H.-K. Min, M.-g. Seo, Y. Choi and G. Kim, J. Mater. Chem. A, 2021, 9, 11571–11579 RSC.
- D. Chanda, R. Xing, T. Xu, Q. Liu, Y. Luo, S. Liu, R. A. Tufa, T. H. Dolla, T. Montini and X. Sun, Chem. Commun., 2021, 57, 7335–7349 RSC.
- A. Goldmann, W. Sauter, M. Oettinger, T. Kluge, U. Schröder, J. R. Seume, J. Friedrichs and F. Dinkelacker, Energies, 2018, 11, 392 CrossRef.
- Y. Bicer and I. Dincer, Int. J. Hydrogen Energy, 2017, 42, 10722–10738 CrossRef CAS.
- J. Hansson, S. Brynolf, E. Fridell and M. Lehtveer, Sustainability, 2020, 12, 3265 CrossRef.
- R. H. Dolan, J. E. Anderson and T. J. Wallington, Sustainable Energy Fuels, 2021, 5, 4830–4841 RSC.
- Y. Zhao, B. P. Setzler, J. Wang, J. Nash, T. Wang, B. Xu and Y. Yan, Joule, 2019, 3, 2472–2484 CrossRef CAS.
- S. P. Kane, D. Zarling and W. F. Northrop, ASME 2019 Internal Combustion Engine Division Fall Technical Conference Proceedings, 2019, DOI:10.1115/icef2019-7241.
- D. W. Brookshear, J. A. Pihl and J. P. Szybist, Energy Fuels, 2018, 32, 2267–2281 CrossRef CAS.
- R. K. Ahluwalia, Q. Zhang, D. J. Chmielewski, K. C. Lauzze and M. A. Inbody, Catal. Today, 2005, 99, 271–283 CrossRef CAS.
- J. St-Pierre, Int. J. Hydrogen Energy, 2011, 36, 5527–5535 CrossRef CAS.
- G. Ertl and M. Huber, J. Catal., 1980, 61, 537–539 CrossRef CAS.
- N. Takezawa and I. Toyoshima, J. Phys. Chem., 1966, 70, 594–595 CrossRef CAS.
- R. W. McCabe, J. Catal., 1983, 79, 445–450 CrossRef CAS.
- K. Tamaru, Acc. Chem. Res., 1988, 21, 88–94 CrossRef CAS.
- D. G. Löffler and L. D. Schmidt, J. Catal., 1976, 44, 244–258 CrossRef.
- Z.-P. Hu, C.-C. Weng, C. Chen and Z.-Y. Yuan, Mol. Catal., 2018, 448, 162–170 CrossRef CAS.
- Z.-P. Hu, L. Chen, C. Chen and Z.-Y. Yuan, Mol. Catal., 2018, 455, 14–22 CrossRef CAS.
- A. Jedynak, Z. Kowalczyk, D. Szmigiel, W. Raróg and J. Zieliński, Appl. Catal., A, 2002, 237, 223–226 CrossRef CAS.
- K. Ogasawara, T. Nakao, K. Kishida, T.-N. Ye, Y. Lu, H. Abe, Y. Niwa, M. Sasase, M. Kitano and H. Hosono, ACS Catal., 2021, 11, 11005–11015 CrossRef CAS.
- Z. Wan, Y. Tao, H. You, X. Zhang and J. Shao, ChemCatChem, 2021, 13, 3027–3036 CrossRef CAS.
- C. Zhou, K. Wu, H. Huang, C.-F. Cao, Y. Luo, C.-Q. Chen, L. Lin, C. Au and L. Jiang, ACS Catal., 2021, 11, 10345–10350 CrossRef CAS.
- K. Okura, K. Miyazaki, H. Muroyama, T. Matsui and K. Eguchi, RSC Adv., 2018, 8, 32102–32110 RSC.
- X.-c. Hu, W.-w. Wang, R. Si, C. Ma and C.-J. Jia, Sci. China: Chem., 2019, 62, 1625–1633 CrossRef CAS.
- J. Feng, L. Liu, X. Ju, J. Wang, X. Zhang, T. He and P. Chen, ChemCatChem, 2021, 13, 1552–1558 CrossRef CAS.
- Z. Wang, Z. Cai and Z. Wei, ACS Sustainable Chem. Eng., 2019, 7, 8226–8235 CrossRef CAS.
- G. Novell-Leruth, A. Valcárcel, J. Pérez-Ramírez and J. M. Ricart, J. Phys. Chem. C, 2007, 111, 860–868 CrossRef CAS.
- P. Xie, Y. Yao, Z. Huang, Z. Liu, J. Zhang, T. Li, G. Wang, R. Shahbazian-Yassar, L. Hu and C. Wang, Nat. Commun., 2019, 10, 4011 CrossRef PubMed.
- W. A. Saidi, W. Shadid and G. Veser, J. Phys. Chem. Lett., 2021, 12, 5185–5192 CrossRef CAS PubMed.
- J. C. Ganley, F. S. Thomas, E. G. Seebauer and R. I. Masel, Catal. Lett., 2004, 96, 117–122 CrossRef CAS.
- H. Inokawa, T. Ichikawa and H. Miyaoka, Appl. Catal., A, 2015, 491, 184–188 CrossRef CAS.
- T. A. Le, Q. C. Do, Y. Kim, T.-W. Kim and H.-J. Chae, Korean J. Chem. Eng., 2021, 38, 1087–1103 CrossRef CAS.
- T. E. Bell and L. Torrente-Murciano, Top. Catal., 2016, 59, 1438–1457 CrossRef CAS.
- S.-F. Yin, Q.-H. Zhang, B.-Q. Xu, W.-X. Zhu, C.-F. Ng and C.-T. Au, J. Catal., 2004, 224, 384–396 CrossRef CAS.
- J. Zhang, H. Xu, Q. Ge and W. Li, Catal. Commun., 2006, 7, 148–152 CrossRef CAS.
- D. Szmigiel, W. Raróg-Pilecka, E. Miśkiewicz, Z. Kaszkur and Z. Kowalczyk, Appl. Catal., A, 2004, 264, 59–63 CrossRef CAS.
- A. Klerke, S. K. Klitgaard and R. Fehrmann, Catal. Lett., 2009, 130, 541–546 CrossRef CAS.
- F. Schüth, R. Palkovits, R. Schlögl and D. S. Su, Energy Environ. Sci., 2012, 5, 6278–6289 RSC.
- J. Zhang, H. Xu and W. Li, Appl. Catal., A, 2005, 296, 257–267 CrossRef CAS.
- S. Podila, Y. A. Alhamed, A. A. AlZahrani and L. A. Petrov, Int. J. Hydrogen Energy, 2015, 40, 15411–15422 CrossRef CAS.
- D. A. Hansgen, L. M. Thomanek, J. G. Chen and D. G. Vlachos, J. Chem. Phys., 2011, 134, 184701 CrossRef PubMed.
- B. Lorenzut, T. Montini, C. C. Pavel, M. Comotti, F. Vizza, C. Bianchini and P. Fornasiero, ChemCatChem, 2010, 2, 1096–1106 CrossRef CAS.
- Z.-P. Hu, C. C. Weng, C. Chen and Z. Y. Yuan, Appl. Catal., A, 2018, 49–57 CrossRef CAS.
- Z. Wang, Y.-L. Qu, X. Shen and Z. Cai, Int. J. Hydrogen Energy, 2019, 7300–7307 CrossRef CAS.
- M. Pinzón, A. Romero, A. de Lucas Consuegra, A. R. de la Osa and P. Sánchez, J. Ind. Eng. Chem., 2021, 94, 326–335 CrossRef.
- S.-F. Yin, B.-Q. Xu, C.-F. Ng and C.-T. Au, Appl. Catal., B, 2004, 48, 237–241 CrossRef CAS.
- L. Li, Z. H. Zhu, Z. F. Yan, G. Q. Lu and L. Rintoul, Appl. Catal., A, 2007, 320, 166–172 CrossRef CAS.
- X. Duan, J. Zhou, G. Qian, P. Li, X. Zhou and D. Chen, Chin. J. Catal., 2010, 31, 979–986 CrossRef CAS.
- T. Kocer, F. E. S. Oztuna, S. F. Kurtoğlu, U. Unal and A. Uzun, Appl. Catal., A, 2021, 610, 117969 CrossRef CAS.
- L. Li, Y. Wang, Z. P. Xu and Z. Zhu, Appl. Catal., A, 2013, 467, 246–252 CrossRef CAS.
- C. Huang, Y. Yu, J. Yang, Y. Yan, D. Wang, F. Hu, X. Wang, R. Zhang and G. Feng, Appl. Surf. Sci., 2019, 476, 928–936 CrossRef CAS.
- H. Zhang, Y. A. Alhamed, W. Chu, Z. Ye, A. AlZahrani and L. Petrov, Appl. Catal., A, 2013, 464–465, 156–164 CrossRef CAS.
- L. Li, Q. Meng, W. Ji, J. Shao, Q. Xu and J. Yan, Mol. Catal., 2017, 442, 147–153 CrossRef CAS.
- T. V. Choudhary, C. Sivadinarayana and D. W. Goodman, Catal. Lett., 2001, 72, 197–201 CrossRef CAS.
- X.-K. Li, W.-J. Ji, J. Zhao, S.-J. Wang and C.-T. Au, J. Catal., 2005, 236, 181–189 CrossRef CAS.
- K. Sato, N. Abe, T. Kawagoe, S.-i. Miyahara, K. Honda and K. Nagaoka, Int. J. Hydrogen Energy, 2017, 42, 6610–6617 CrossRef CAS.
- Y. Yu, Y.-M. Gan, C. Huang, Z.-H. Lu, X. Wang, R. Zhang and G. Feng, Int. J. Hydrogen Energy, 2020, 45, 16528–16539 CrossRef CAS.
- J. Ji, X. Duan, G. Qian, X. Zhou, G. Tong and W. Yuan, Int. J. Hydrogen Energy, 2014, 39, 12490–12498 CrossRef CAS.
- J. Zhang, J.-O. Müller, W. Zheng, D. Wang, D. Su and R. Schlögl, Nano Lett., 2008, 8, 2738–2743 CrossRef CAS PubMed.
- P. Santhana Krishnan, M. Neelaveni, P. Tamizhdurai, M. Mythily, S. Krishna Mohan, V. L. Mangesh and K. Shanthi, Int. J. Hydrogen Energy, 2020, 45, 8568–8583 CrossRef CAS.
- J.-L. Cao, Z.-L. Yan, Q.-F. Deng, Z.-Y. Yuan, Y. Wang, G. Sun, X.-D. Wang, B. Hari and Z.-Y. Zhang, Catal. Sci. Technol., 2014, 4, 361–368 RSC.
- Z.-P. Hu, H. Zhao, Z.-M. Gao and Z.-Y. Yuan, RSC Adv., 2016, 6, 94748–94755 RSC.
- L. li, F. Chen, J. Shao, Y. Dai, J. Ding and Z. Tang, Int. J. Hydrogen Energy, 2016, 41, 21157–21165 CrossRef CAS.
- Z.-P. Hu, C.-C. Weng, G.-G. Yuan, X.-W. Lv and Z.-Y. Yuan, Int. J. Hydrogen Energy, 2018, 43, 9663–9676 CrossRef CAS.
- H. Zhang, Q. Gong, S. Ren, M. A. Arshid, W. Chu and C. Chen, Catal. Sci. Technol., 2018, 8, 907–915 RSC.
- H. Inokawa, M. Maeda, S. Nishimoto, Y. Kameshima, M. Miyake, T. Ichikawa, Y. Kojima and H. Miyaoka, Int. J. Hydrogen Energy, 2013, 38, 13579–13586 CrossRef CAS.
- Z.-W. Wu, X. Li, Y.-H. Qin, L. Deng, C.-W. Wang and X. Jiang, Int. J. Hydrogen Energy, 2020, 45, 15263–15269 CrossRef CAS.
- W. I. F. David, J. W. Makepeace, S. K. Callear, H. M. A. Hunter, J. D. Taylor, T. J. Wood and M. O. Jones, J. Am. Chem. Soc., 2014, 136, 13082–13085 CrossRef CAS PubMed.
- F. Chang, H. Wu, R. v. d. Pluijm, J. Guo, P. Ngene and P. E. de Jongh, J. Phys. Chem. C, 2019, 123, 21487–21496 CrossRef CAS PubMed.
- T. J. Wood and J. W. Makepeace, ACS Appl. Energy Mater., 2018, 1, 2657–2663 CrossRef CAS.
- A. Jain, H. Miyaoka and T. Ichikawa, J. Phys. Chem. C, 2016, 120, 27903–27909 CrossRef CAS.
- P. L. Bramwell, S. Lentink, P. Ngene and P. E. de Jongh, J. Phys. Chem. C, 2016, 120, 27212–27220 CrossRef CAS.
- S. Mukherjee, S. V. Devaguptapu, A. Sviripa, C. R. F. Lund and G. Wu, Appl. Catal., B, 2018, 226, 162–181 CrossRef CAS.
- I. Lucentini, X. Garcia, X. Vendrell and J. Llorca, Ind. Eng. Chem. Res., 2021, 60, 18560–18611 CrossRef CAS.
- C.-z. He, H. Wang, L.-y. Huai and J.-y. Liu, J. Phys. Chem. C, 2012, 116, 24035–24045 CrossRef CAS.
- M. Bystrzanowska, P. Petkov and M. Tobiszewski, ACS Sustainable Chem. Eng., 2019, 7, 18434–18443 CrossRef CAS.
- K. E. Lamb, M. D. Dolan and D. F. Kennedy, Int. J. Hydrogen Energy, 2019, 44, 3580–3593 CrossRef CAS.
- T. J. Schriber and G. Parravano, Chem. Eng. Sci., 1967, 22, 1067–1078 CrossRef CAS.
- K. Nagaoka, T. Eboshi, Y. Takeishi, R. Tasaki, K. Honda, K. Imamura and K. Sato, Sci. Adv., 2017, 3, e1602747 CrossRef PubMed.
- T. Matsunaga, S. Matsumoto, R. Tasaki, Y. Ogura, T. Eboshi, Y. Takeishi, K. Honda, K. Sato and K. Nagaoka, ACS Sustainable Chem. Eng., 2020, 8, 13369–13376 CrossRef CAS.
- J. Cha, T. Lee, Y.-J. Lee, H. Jeong, Y. S. Jo, Y. Kim, S. W. Nam, J. Han, K. B. Lee, C. W. Yoon and H. Sohn, Appl. Catal., B, 2021, 283, 119627 CrossRef CAS.
- D. Chakraborty, C. D. Damsgaard, H. Silva, C. Conradsen, J. L. Olsen, H. W. P. Carvalho, B. Mutz, T. Bligaard, M. J. Hoffmann, J.-D. Grunwaldt, F. Studt and I. Chorkendorff, Angew. Chem., Int. Ed., 2017, 56, 8711–8715 CrossRef CAS PubMed.
- S. Zhang, Z. He, X. Li, J. Zhang, Q. Zang and S. Wang, Nanoscale Adv., 2020, 2, 3610–3623 RSC.
- M. Altomare and E. Selli, Catal. Today, 2013, 209, 127–133 CrossRef CAS.
- H. Mozzanega, J. M. Herrmann and P. Pichat, J. Phys. Chem., 1979, 83, 2251–2255 CrossRef CAS.
- J. Taguchi and T. Okuhara, Appl. Catal., A, 2000, 194–195, 89–97 CrossRef CAS.
- A. Utsunomiya, A. Okemoto, Y. Nishino, K. Kitagawa, H. Kobayashi, K. Taniya, Y. Ichihashi and S. Nishiyama, Appl. Catal., B, 2017, 206, 378–383 CrossRef CAS.
- M. Chen, C. Zhang and H. He, Environ. Sci. Technol., 2020, 54, 10544–10550 CrossRef CAS PubMed.
- Y. Shiraishi, S. Toi, S. Ichikawa and T. Hirai, ACS Appl. Nano Mater., 2020, 3, 1612–1620 CrossRef CAS.
- S. Mitchell, N.-L. Michels and J. Pérez-Ramírez, Chem. Soc. Rev., 2013, 42, 6094–6112 RSC.
- J. Humphreys, R. Lan and S. Tao, Adv. Energy Sustain. Res., 2021, 2, 2000043 CrossRef.
- Z.-P. Hu, C.-C. Weng, C. Chen and Z.-Y. Yuan, Appl. Catal., A, 2018, 562, 49–57 CrossRef CAS.
- L. Huo, X. Han, L. Zhang, B. Liu, R. Gao, B. Cao, W.-W. Wang, C.-J. Jia, K. Liu, J. Liu and J. Zhang, Appl. Catal., B, 2021, 294, 120254 CrossRef CAS.
- J. Martinez-Ortigosa, C. W. Lopes, G. Agostini, A. E. Palomares, T. Blasco and F. Rey, Microporous Mesoporous Mater., 2021, 323, 111230 CrossRef CAS.
- G. Xu, Y. Zhang, J. Lin, Y. Wang, X. Shi, Y. Yu and H. He, ACS Catal., 2021, 5506–5516 CrossRef CAS.
- N. Morlanés, S. Sayas, G. Shterk, S. P. Katikaneni, A. Harale, B. Solami and J. Gascon, Catal. Sci. Technol., 2021, 11, 3014–3024 RSC.
- J. W. Makepeace, J. M. Brittain, A. Sukhwani Manghnani, C. A. Murray, T. J. Wood and W. I. F. David, Phys. Chem. Chem. Phys., 2021, 23, 15091–15100 RSC.
- J. L. Williams, Catal. Today, 2001, 69, 3–9 CrossRef CAS.
- R. M. Heck, S. Gulati and R. J. Farrauto, Chem. Eng. J., 2001, 82, 149–156 CrossRef CAS.
- V. Tomašić, Z. Gomzi and S. Zrnčević, Chem. Eng. Technol., 2006, 29, 59–65 CrossRef.
- C. Plana, S. Armenise, A. Monzón and E. García-Bordejé, J. Catal., 2010, 275, 228–235 CrossRef CAS.
- M. Wang, J. Li, L. Chen and Y. Lu, Int. J. Hydrogen Energy, 2009, 34, 1710–1716 CrossRef CAS.
- R. Hubesch, M. Mazur, K. Föger, P. R. Selvakannan and S. K. Bhargava, Chem. Commun., 2021, 57, 9586–9589 RSC.
- X. Li, W. Li, F. Rezaei and A. Rownaghi, Chem. Eng. J., 2018, 333, 545–553 CrossRef CAS.
- M. R. Hartings and Z. Ahmed, Nat. Rev. Chem., 2019, 3, 305–314 CrossRef.
- B. Lin, Y. Guo, J. Lin, J. Ni, J. Lin, L. Jiang and Y. Wang, Appl. Catal., A, 2017, 541, 1–7 CrossRef CAS.
- D. Kunwar, C. Carrillo, H. Xiong, E. Peterson, A. DeLaRiva, A. Ghosh, G. Qi, M. Yang, M. Wiebenga, S. Oh, W. Li and A. K. Datye, Appl. Catal., B, 2020, 266, 118598 CrossRef CAS.
- D. Nazimek and J. Ryczkowski, Adsorpt. Sci. Technol., 1999, 17, 805–811 CrossRef CAS.
- S. L. Scott, ACS Catal., 2018, 8, 8597–8599 CrossRef CAS.
- D. G. Vlachos, A. B. Mhadeshwar and N. S. Kaisare, Comput. Chem. Eng., 2006, 30, 1712–1724 CrossRef CAS.
- S. Yun and S. Ted Oyama, J. Membr. Sci., 2011, 375, 28–45 CrossRef CAS.
- A. Basile, F. Gallucci and S. Tosti, in Membrane Science and Technology, Elsevier, 2008, vol. 13, pp. 255–323 Search PubMed.
- J. P. Collins and J. D. Way, J. Membr. Sci., 1994, 96, 259–274 CrossRef CAS.
- N. Itoh, A. Oshima, E. Suga and T. Sato, Catal. Today, 2014, 236, 70–76 CrossRef CAS.
- V. Cechetto, L. Di Felice, J. A. Medrano, C. Makhloufi, J. Zuniga and F. Gallucci, Fuel Process. Technol., 2021, 216, 106772 CrossRef CAS.
- H. E. Bonfield, T. Knauber, F. Lévesque, E. G. Moschetta, F. Susanne and L. J. Edwards, Nat. Commun., 2020, 11, 804 CrossRef CAS PubMed.
- K. C. Harper, E. G. Moschetta, S. V. Bordawekar and S. J. Wittenberger, ACS Cent. Sci., 2019, 5, 109–115 CrossRef CAS PubMed.
- V. A. Danilov and G. Kolb, Chem. Eng. Sci., 2021, 231, 116269 CrossRef CAS.
- A. Takahashi and T. Fujitani, Chem. Eng. Res. Des., 2021, 165, 333–340 CrossRef CAS.
- S. D. Angeli, G. Monteleone, A. Giaconia and A. A. Lemonidou, Int. J. Hydrogen Energy, 2014, 39, 1979–1997 CrossRef CAS.
- R. C. Brown, Joule, 2020, 4, 2268–2289 CrossRef CAS.
- S. Chiuta and D. G. Bessarabov, Catal. Today, 2018, 310, 187–194 CrossRef CAS.
- S. R. Deshmukh and D. G. Vlachos, Ind. Eng. Chem. Res., 2005, 44, 4982–4992 CrossRef CAS.
- W. Wang, J. M. Herreros, A. Tsolakis and A. P. E. York, Int. J. Hydrogen Energy, 2013, 38, 9907–9917 CrossRef CAS.
- W. Wang, G. Tuci, C. Duong-Viet, Y. Liu, A. Rossin, L. Luconi, J.-M. Nhut, L. Nguyen-Dinh, C. Pham-Huu and G. Giambastiani, ACS Catal., 2019, 9, 7921–7935 CrossRef CAS.
- D.-K. Lim, A. B. Plymill, H. Paik, X. Qian, S. Zecevic, C. R. I. Chisholm and S. M. Haile, Joule, 2020, 4, 2338–2347 CrossRef CAS.
- S. Chiuta, R. C. Everson, H. W. J. P. Neomagus and D. G. Bessarabov, Int. J. Hydrogen Energy, 2015, 40, 2921–2926 CrossRef CAS.
- J. E. Brandenburg, Plasma reactor for cracking ammonia and hydrogen-rich gases to hydrogen, U.S. Pat., 7628 962 B1, University of Central Florida Research Foundation, Inc., U. S. A, 2009 Search PubMed.
- Y. Yi, X. Wang, A. Jafarzadeh, L. Wang, P. Liu, B. He, J. Yan, R. Zhang, H. Zhang, X. Liu, H. Guo, E. C. Neyts and A. Bogaerts, ACS Catal., 2021, 1765–1773 CrossRef CAS.
- L. Han, S. Cai, M. Gao, J.-y. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS PubMed.
- K. Ryu, G. E. Zacharakis-Jutz and S.-C. Kong, Int. J. Hydrogen Energy, 2014, 39, 2390–2398 CrossRef CAS.
- M. Comotti and S. Frigo, Int. J. Hydrogen Energy, 2015, 40, 10673–10686 CrossRef CAS.
Footnotes |
| † These authors contributed equally. |
| ‡ Lead contact. |
| This journal is © The Royal Society of Chemistry 2022 |