
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterfacial electric fields catalyze Ullmann coupling reactions on gold surfaces†
Ilana B.
Stone‡
a,
Rachel L.
Starr‡
 a,
Norah
Hoffmann‡
a,
Xiao
Wang‡
b,
Austin M.
Evans
a,
Colin
Nuckolls
a,
Tristan H.
Lambert
c,
Michael L.
Steigerwald
a,
Timothy C.
Berkelbach
*ab,
Xavier
Roy
*a and
Latha
Venkataraman
*ad
a,
Norah
Hoffmann‡
a,
Xiao
Wang‡
b,
Austin M.
Evans
a,
Colin
Nuckolls
a,
Tristan H.
Lambert
c,
Michael L.
Steigerwald
a,
Timothy C.
Berkelbach
*ab,
Xavier
Roy
*a and
Latha
Venkataraman
*ad
aDepartment of Chemistry, Columbia University, New York, New York 10027, USA. E-mail: tcb2112@columbia.edu; xr2114@columbia.edu; lv2117@columbia.edu
bCenter for Computational Quantum Physics, Flatiron Institute, New York, New York 10010, USA
cDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA
dDepartment of Applied Physics, Columbia University, New York, New York 10027, USA
First published on 1st September 2022
Abstract
The electric fields created at solid–liquid interfaces are important in heterogeneous catalysis. Here we describe the Ullmann coupling of aryl iodides on rough gold surfaces, which we monitor in situ using the scanning tunneling microscope-based break junction (STM-BJ) and ex situ using mass spectrometry and fluorescence spectroscopy. We find that this Ullmann coupling reaction occurs only on rough gold surfaces in polar solvents, the latter of which implicates interfacial electric fields. These experimental observations are supported by density functional theory calculations that elucidate the roles of surface roughness and local electric fields on the reaction. More broadly, this touchstone study offers a facile method to access and probe in real time an increasingly prominent yet incompletely understood mode of catalysis.
Electric fields are ubiquitous at the interface of a metal and a liquid—for example, in the double layer of electrolyte solutions or in the solvent's reaction field in response to a molecular adsorbate.1–5 These interfacial electric fields can be measured by vibrational Stark shift spectroscopy of probe molecules and are highly variable depending on the ionic concentrations,6 the surface morphology, the nature of adsorbates, and the dielectric properties of the solvent. Such interfacial fields offer new opportunities to promote chemical reactivity without the use of externally applied voltages or other stimuli.1,7–9 In this work, we demonstrate that rough Au surfaces, when submerged in a highly polar solvent, can act as mesoscopic active sites for on-surface aryl iodide dehalogenation, and subsequent aryl–aryl bond formation via an Ullmann-type coupling reaction. The traditional solution-phase Ullmann coupling is a long-standing strategy to form biaryls from aryl halides using stoichiometric elemental Cu in polar solvents.10,11 While still heavily used in traditional solution-phase organic synthesis, the Ullmann coupling has more recently emerged as a stalwart of on-surface chemistry.12–15 In these reactions, aryl bromides or iodides are deposited on single-crystal Cu/Ag/Au surfaces in ultra-high vacuum and heated to cleave the aryl–halide bond and form the biaryl product.16,17 These reactions offer an attractive strategy for the synthesis of covalent two-dimensional polymers, but leave products bound to surfaces, and thus preclude the diversification of reactions and limit the range of accessible chemical substrates that can be probed.18
In this work, we report the Ullmann coupling of aryl iodides on a Au surface submerged in solution under ambient conditions. The surface also serves as a scanning tunneling microscope-based break junction (STM-BJ) substrate, thereby enabling in situ determination of product formation by monitoring single-molecule junction conductance. STM-BJ measurements provide molecule-specific signatures (i.e. the most probable molecular conductance and molecular plateau length) that can be used to identify carbon–carbon bond formation in situ.19 Our observations are consistent with ex situ fluorescence spectroscopy and mass spectrometry measurements that confirm biaryl formation across a range of aryl iodide precursors, including those with strongly coordinating groups such as thiomethyls, pyridines, and amines. Notably, in addition to a Au surface, we find that the Ullmann coupling requires an Au surface that is rough at least on the 10–100 nm scale, as measured by AFM (ESI Fig. S1†), and a highly polar solvent. Furthermore, the reaction works primarily with aryl iodides. Finally, we probe the mechanism of the Ullmann coupling and propose that interfacial electric fields contribute to the reaction catalysis, which is distinct from the mechanisms commonly invoked for solution-based Ullmann couplings.12,16,20–24
We first characterize, in situ, the formation of the homo-coupled dimer (D1, 4,4′-diamino-quaterphenylene) when a solution of 4-iodo-4′-amino-biphenyl (I1) is subject to STM-BJ measurements from a solution deposited on a Au surface (Fig. 1a). A 0.1 mM solution of I1 in propylene carbonate (PC), a highly polar solvent with a dielectric constant of ∼64 at 25 °C,25 is placed on a rough Au STM-BJ substrate prepared by evaporating 70–100 nm of Au on a steel puck (see ESI Fig. S1† for SEM and AFM images). The reaction is monitored by performing STM-BJ measurements over a period of 18 hours using an insulated Au STM tip (see ESI† for details),26 while applying a bias voltage of 100 mV. Fig. 1b presents conductance histograms created over the measurement period showing a molecular conductance peak centered at 3.7 × 10−5G0 (G0 = 2e2/h is the conductance quantum). This conductance value is comparable to previous measurements of tetraphenyl diamine D1 (ESI Fig. S2a†).27 The 2D conductance–displacement histogram shown in Fig. 1c reveals that this conductance peak also features a molecular plateau extending to ∼0.7 nm, which is considerably longer than the length expected for a biphenyl (see Fig. S2† for additional 2D histogram).27 The longer plateau length observed in situ is consistent with that of D1,27 indicating that D1 is formed under these conditions (Fig. 1c). The lower conducting D1 forms via an Ullmann coupling of two I1 molecules, which results in a peak whose height increases over time as is seen in Fig. 1b.
 | ||
| Fig. 1 (a) Molecular structures of aryl iodide I1 and tetraphenyl D1. (b) Logarithmically binned 1D histograms showing a molecular conductance peak for D1 that increases over time in a solution of I1 measured at 100 mV in PC. (c) 2D conductance–displacement histogram created by overlaying all D1 traces taken at 100 mV from the 15–18 h measurement. | ||
We next demonstrate that the coupling reaction happens on the Au surface without performing STM-BJ measurements.28 An aliquot of a solution of I1 and TBAPF6 prepared in PC was deposited on an Au substrate. Samples of the solution were subsequently collected after 2, 5 and 10 hours and measured by fluorescence spectroscopy. If the formation of D1 required the STM measurements, we would not detect any product with this experiment through fluorescence measurements. Fig. 2a and b show fluorescence spectra measured using different substrates and conditions with those of an I1 solution that was not exposed to the Au substrate and a solution of the product D1 synthesized ex situ. These spectra are measured using 284 nm and 340 nm photoexcitation, respectively, with wavelengths chosen based on the electronic absorption spectra of I1 and D1 measured ex situ, (ESI Fig. S3a†). The spectrum reveals that after 2 hours of exposure to the Au substrate in an ionic solution, in addition to the I1 peak at 380 nm, a smaller peak appears at 455 nm corresponding to D1 (Fig. 2a, light blue). Over the course of 10–12 hours, the emission peak consistent with that of D1 increases in intensity (Fig. 2a, dark blue), indicating that dimerization is occurring when I1 is exposed to an interfacial field on a rough Au substrate. Starting with a 100 μM solution of I1, a solution was obtained that has 6.25 μM D1 after 12 hours (Fig. 2b, blue), showing that the conversion is significant.
 | ||
| Fig. 2 (a) Emission spectra (λex = 284 nm) of a solution of I1 placed on a rough Au-coated steel substrate over time (blue traces) overlaid with reference peaks for I1 (yellow trace) and an ex situ synthesized solution of D1 (red trace). These traces are normalized to have the same peak height at 380 nm. (b) Emission spectra (λex = 340 nm) showing a peak for D1 that appears in solution left on a rough Au-coated steel substrate (blue trace) overlaid with reference peaks for I1 (yellow trace) and D1 (red trace). Also shown are data from a solution is left on a smooth Au-coated mica substrate (green trace) and with an added suspension of Au dust (purple trace). (c) Selected ion chromatogram (LC-MS) of the I1 standard (red), the D1 standard (blue), and the solution subject to STM-BJ measurements for 12 hours (green). Masses obtained from these spectra are noted in the legend and shown in ESI Fig. S4.† (d) Molecular structures of aryl iodides studied here I1–I4. | ||
To elucidate the role of the Au substrate in the homocoupling of the aryl iodides, we show, using ex situ fluorescence spectroscopy, that the coupling reaction does not proceed directly on a steel substrate without Au coating (ESI Fig. S3c and d†), indicating that the Au catalyzes the reaction. Similarly, when the rough Au-coated substrate is replaced with an atomically flat Au-coated mica substrate, no dimer formation is observed over 12 hours by fluorescence spectroscopy (Fig. 2b, green trace) or after 12 hours of measurement in the STM-BJ setup (ESI Fig. S3b†). We note here that although the tip and Au-coated mica substrate are locally roughened during the STM-BJ measurement, the number of undercoordinated sites created is likely insufficient to generate a detectible amount of the product. Finally, we also observe dimer formation on an Au-coated glass substrate, which produces a rough gold layer also catalyzes the reaction (ESI Fig. S3b†). Our experiments therefore indicate that a rough Au surface is required and while also eliminating the possibility that steel on its own is catalyzing the reaction. The rough surface thus provides the active catalytic sites, similar to surface-catalyzed reactions under UHV that happen primarily at facets or defects.29–31 These results are further supported by analysis of solutions subjected to STM-BJ measurements via Liquid Chromatography Mass Spectrometry (LC-MS) spectra shown in Fig. 2c. We find that the chromatogram shows a peak at 3.6 minutes when the product D1 is present in the solution while we see a peak at 3.1 minutes when the reactant I1 is present. The reactant and product are identified by their respective masses extracted from these chromatograms and shown in ESI Fig. S4.†
We next evaluate the role of the of the solvent and ions in solution. Although the reaction proceeds in the polar solvent PC and in PC with an added electrolyte (Fig. 2b), when a solution of I1 in the nonpolar solvent trichlorobenzene is deposited on the Au substrate, no peak corresponding to D1 is observed in either STM-BJ measurements or fluorescence spectroscopy (ESI Fig. S5†), demonstrating that the solvent polarity is critical. As shown by Dawlaty and co-workers,1 molecules at the interface of a metal and solvent experience a reaction field whose magnitude is a function of the solvent dielectric constant. Specifically, they show that for a solvent with a dielectric constant over 50 (as is the case with PC), the interfacial fields can be as large as 2 V nm−1. Our experimental observations suggest that such interfacial electric fields are contributors to the catalysis of the Ullmann coupling. We note that identical results are obtained when solutions are created with an added electrolyte (such as tetrabutyl ammonium hexafluorophosphate (TBAPF6) at a 0.1 M concentration) indicating that added ions do not alter the interfacial fields significantly, consistent with previous measurements.32
Finally, when the Au surface is replaced with Au dust (diameter: 500–850 nm) suspended in a reaction mixture which contains I1 dissolved in PC with 0.1 M TBAPF6, the fluorescence spectra (Fig. 2b) indicate no dimer formation. Under these conditions, I1 is exposed to a rough surface comparable in total surface area to the Au substrate, yet no coupling reaction is observed. These results could indicate weaker reaction fields at the interface of a small metallic sphere; however, we cannot rule out other possibilities, including different surface morphologies, fewer undercoordinated sites or inhibited surface diffusion, adsorbed moieties that prevent the reactants from interacting with Au.
The observed homocoupling reaction is generalizable with respect to both the linker and molecular backbone. Three additional aryl iodides (Fig. 2d) were studied under the same conditions as I1, and all appear to homocouple. The coupling of both the thiomethyl- and pyridyl-terminated biphenyl iodides (I2 and I3, respectively) can be probed in situ through STM-BJ measurements because they are equipped with anchoring groups necessary for metallic contact. Similar to the dimerization of I1, when each aryl iodide is measured, a lower molecular conductance peak with a longer molecular plateau appears and continues to grow over time (ESI Fig. S6†). We verify the identity of these products by comparison of the 1D and 2D conductance histograms for D2 and D3 synthesized ex situ and measured independently (Fig. S6†). LC-MS spectra of the extracted solutions also confirms biaryl coupling (ESI Fig. S7†). We note that while these specific iodides were selected for their aurophilic substituents, they demonstrate that both strongly electron withdrawing and electron donating groups are tolerated in this system. Furthermore, even the larger molecule I4 homocouples on the rough Au surface despite its steric encumbrance, as verified by electronic absorption spectroscopy of the extracted solution (ESI Fig. S7†). We note that this particular coupling cannot be probed using the STM-BJ because the large twist angle between adjacent anthracene units in the product decreases its conductance below the measurable limit of our instrument.33,34
We propose a general scheme for the coupling of aryl iodides in an interfacial field (Fig. 3a). We probe this reaction scheme with mechanistic and computational studies described further below. Two aryl iodides undergo an oxidative addition forming two independent C–Au–I constructs at active sites on the rough Au substrate. If two oxidative addition events occur near one another, a small amount of surface migration may occur before a reversible reductive elimination can release the aryl iodides back into solution.35–37 Once the active Au–C bonds are close enough, a C–C bond can form between the activated aryl species, presumably through a mechanism that resembles that of thermally driven reactions on metal surfaces.16,21,38 The resulting tetraphenyl product is then released into solution, likely leaving iodine chemisorbed to the Au surface. We note that this double oxidative addition mechanism differs from the proposed mechanisms for solution-based Ullmann couplings in which the two halides have distinct roles in the coupling process: one aryl iodide free in solution is activated by the metal catalyst, thus forming an organometallic complex with which the second free aryl iodide can interact.39,40
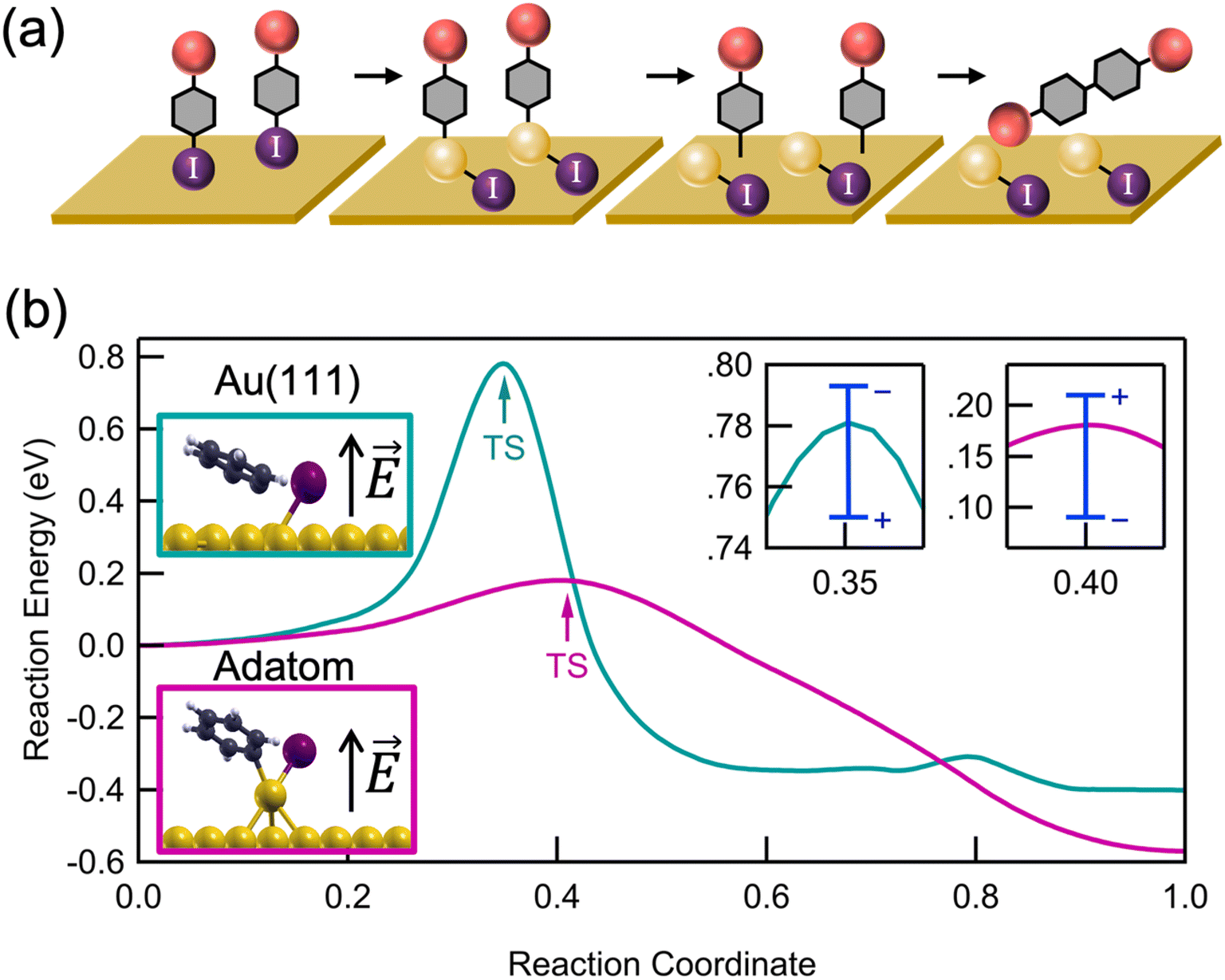 | ||
| Fig. 3 (a) Schematic of the key steps for the proposed Ullmann coupling mechanism on a rough Au surface: (1) binding on the surface, (2) oxidative addition, (3) surface migration and (4) dimerization. The yellow sphere denotes a gold adatom and the red sphere denotes the different substituents used in this study. (b) The DFT reaction energy over the course of the dehalogenation process, from reactant (reaction coordinate = 0) to transition state (TS) to product (reaction coordinate = 1) of iodobenzene. The calculations are performed on an Au(111) surface (green) and with an additional Au adatom (pink). The blue bars at the TS denote the change of the activation energy due to an applied electric field in the range of ±3 V nm−1 (close-up shown in insets at right). Insets at left show the TS geometries and the black arrows indicate the direction of a positive electric field. | ||
Within this reaction scheme, oxidative addition is proposed to be the key step catalyzed by the rough Au surface and the interfacial field. We can probe this step by comparing the reactivity of aryl iodides with that of a preformed Au complex. We synthesized the Au complex Au2 (ESI Fig. S8a†), which has a preformed Au–C bond and a labile PPh3 ligand that readily dissociates in situ.41 This Au complex can be understood as an analog of the product of an oxidative addition of biphenyl iodide I2 to Au, as the dissociation of PPh3 replaces the oxidative addition step such that starting with Au2 instead of an aryl iodide bypasses the oxidative addition step. Indeed, when Au2 is measured in the junction in a non-polar solvent, a molecular conductance peak for D2 appears immediately (ESI Fig. S8b†). Furthermore, when Au2 is left on a smooth mica substrate in polar solvent, D2 is observed in the solution via fluorescence spectroscopy (ESI Fig. S8c†). Together, these results prove that when the oxidative addition step is removed from the reaction scheme altogether, neither a rough surface nor a polar solvent is necessary to the coupling. As such, we conclude that the oxidative addition is the rate limiting step in this transformation and is the key process that is assisted by the rough surface and its interfacial electric field.
For further insights into the oxidative addition process, we turn to electronic structure calculations using density functional theory (DFT); full Computational details are provided in the ESI.† For simplicity, we study the dehalogenation of iodobenzene, whose behavior is representative of other aryl halides. We emphasize that because of the idealized nature of our calculations (especially the surface morphology and absence of explicit solvent), we do not expect quantitative agreement with experimental findings, but rather qualitative insights. Using the nudged elastic band method with 6–12 images, we calculated the minimum energy reaction pathway on an Au(111) surface and on an Au(111) with a single Au adatom (Fig. 3b), as a model for surface roughness. On Au(111), we find an activation energy of 0.76 eV (17.5 kcal mol−1) and a reaction energy of −0.4 eV (−9 kcal mol−1), in good agreement with previous calculations.42 In contrast, in the presence of an Au adatom, which represents an extreme under-coordinated structure, we find a significantly reduced activation energy of 0.18 eV (3.5 kcal mol−1) and more negative reaction energy of −0.6 eV (−13 kcal mol−1).43 The reduced activation energy supports the experimental observation that a rough Au surface, where a fraction of Au atoms on step-edges are undercoordinated (though not as undercoordinated as the adatom shown in Fig. 3b) is essential for catalyzing the Ullmann coupling. The difference in activation energies can be understood by comparing the transition state geometries (Fig. 3b, inset). Compared to Au(111), the Au adatom is more readily inserted into the C–I bond.
Next, we analyze the potential impact of interfacial electric fields on the dehalogenation. In the direction perpendicular to the surface, we apply an electric field of −3 V nm−1 and +3 V nm−1, which is comparable to the interfacial field strengths previously measured1,44 and calculated45 at metal–solvent interfaces. Over this range, we find that the electric field modifies the activation energy by about 0.05–0.1 eV (∼0.5–2 kcal mol−1). We see that a positive electric field (i.e. pointing up from the surface) decreases the activation energy on a pristine surface but increases the activation energy in the presence of an adatom modestly. This contrasting behavior is explained by a difference in the relative dipole moments of the reactant and transition state structures (all dipole moments are directed away from the surface and are thus stabilized by a field pointing in the same direction): on the pristine surface, the dipole moment of the reactant is smaller than that of the transition state, and in the presence of an adatom, the reverse is true. More discussion about this behavior and its implications for a linear free energy relationship can be found in Hoffmann et al.46 These results indicate that both surface roughness and local electric fields are important in determining the kinetics of surface-mediated oxidative addition. We expect an interfacial reaction field to point in the direction of the dipole moment, which is away from the surface.1 Our calculation results show that a field in this direction could either raise or lower the activation energy, depending on the precise local morphology as indicated in Fig. 3b.
To demonstrate that the coupling reaction may involve two independent oxidative additions, we compare the reactivity of a bromide- and triflate-terminated biphenyl (B1 and T1, respectively) (Fig. 4a), which are less prone to oxidative addition than aryl iodides. Indeed, DFT calculations of the dehalogenation of bromobenzene in the presence of an Au adatom predict an activation energy of 0.5 eV (12 kcal mol−1), which is much larger than that for iodobenzene (ESI Fig. S9†). Consistent with our hypothesis of multiple oxidative additions to the Au surface, these less reactive reactants do not homocouple in the junction over the time scale studied. Although these reactants are unlikely to form two C–M–X constructs near each other, when surrounded by reactive iodides, only one such construct is necessary. Based on the hypothesis that two oxidative additions are necessary for aryl–aryl coupling, we suspect an Au-bound intermediate is more likely to be formed by a bromide than a triflate, the latter being less prone to oxidative addition, and thus B1 is more likely to cross-couple with I1 than T1 (Fig. 4a). We note that the methyl group meta to the linker on B1 and T1 is used to distinguish between the cross-coupling product CC1 and the homocoupling product D1.
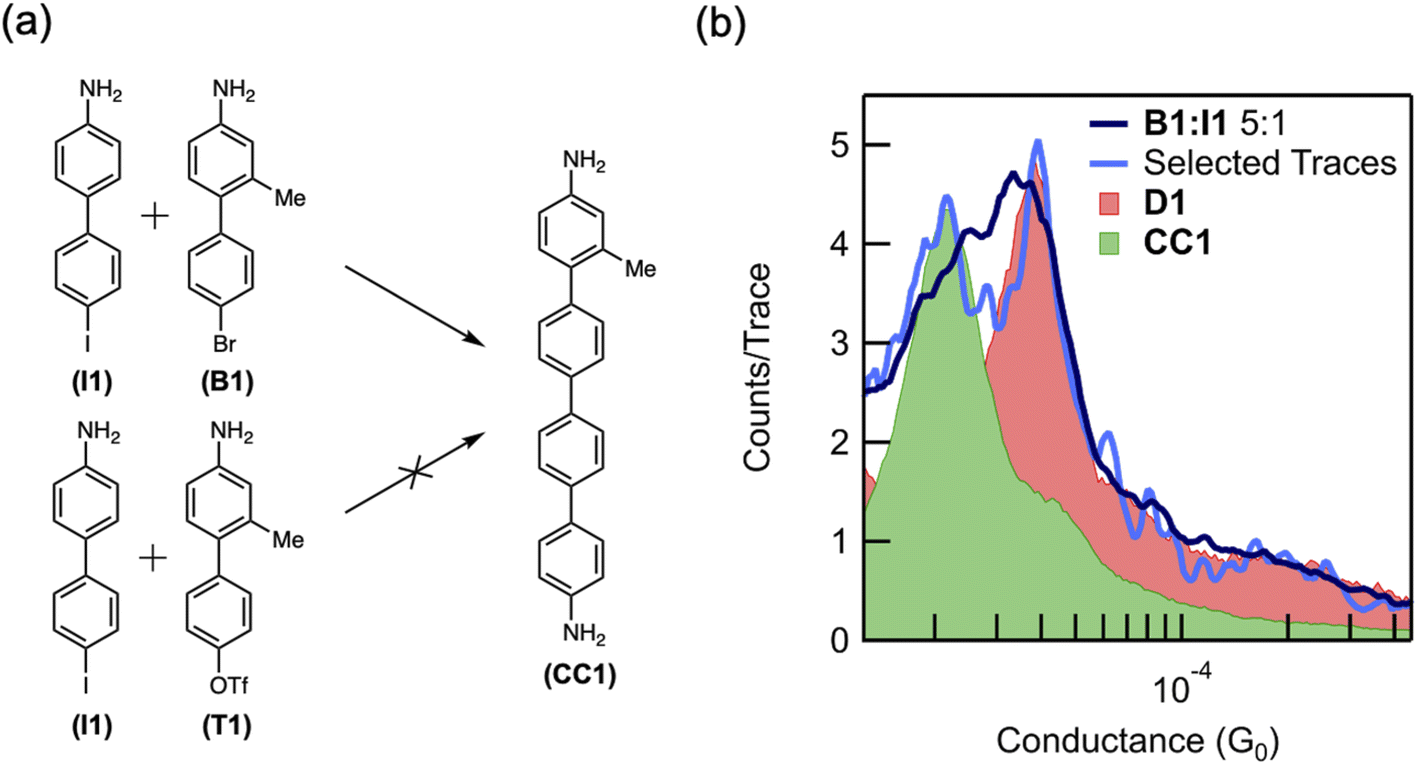 | ||
| Fig. 4 (a) Schematic showing that cross-coupled product CC1 forms from a mixture of I1 and B1 but not from a mixture of I1 and T1. (b) Logarithmically binned 1D histograms showing the in situ cross-coupling of I1 and B1 (dark blue = 1000 traces, light blue = 100 traces), as confirmed by the alignment with the histogram of the product CC1 (green) synthesized ex situ. Also shown in pink is the dimerization product D1 also measured in the same experiment. | ||
When B1 and I1 are exposed to the reaction conditions described above, dimerization is observed. A molecular conductance peak for D1 appears at 3.7 × 10−5G0 in the 1D histogram after 12 hours (Fig. 4b, pink peak). After 18 hours, this peak becomes broader and asymmetric, with a shoulder on the left side (Fig. 4b, dark blue trace). The first 100 traces after the appearance of the shoulder were isolated, and two distinct peaks were resolved at 3.7 × 10−5G0 and 1.9 × 10−5G0 (Fig. 4b, light blue trace). A lower conductance peak is expected for the cross-coupled product, CC1 (Fig. 4a), due to the presence of the methyl group and consequently larger twist angle between the adjacent phenyl rings.34 We therefore assign this new conductance peak to the putative product of the cross-coupling reaction, CC1. To further validate these results, we synthesized CC1ex situ and compared its conductance value (Fig. 4b, green peak) with that of the molecule formed in situ. The alignment of the two peaks supports our hypothesis that the lower conducting peak at 1.9 × 10−5G0 can be assigned to the cross-coupled product (Fig. 4b, green trace). Finally, when we repeat these experiments with T1, no such peak corresponding to CC1 appears (ESI Fig. S10†). The order of reactivity of aryl bromides and sulfonates is highly dependent on the mechanism of the reaction, and aryl bromides are more prone to oxidative addition than aryl sulfonates.47 Although the results with I1 alone do not exclude the possibility of the aryl iodide interacting with C–M–X to form D1, these control measurements indicate strongly that the coupling follows two oxidative additions. This is because T1 should be as reactive (if not more reactive) towards that reaction T1 + C–M–X = CC1 than the aryl iodide. Yet, we do not observe any CC1 product in our mixed measurements with T1 and I1. By contrast, B1 is less reactive towards the same reaction (B1 + C–M–X = CC1), yet we see the CC1 product in the mixed measurements of I1 and B1. However, the reactivity of two oxidative additions forming the product (i.e. two C–M–X forming D1 or CC1) would be highest for I1 and lowest for T1 with B1 having an intermediate reactivity. Our experimental results suggest that in this system, the bromide is more reactive than the triflate, although it is possible that the cross-coupling product with T1 was simply never observed in the six measurements taken. The order of reactivity observed in the cross-coupling experiments is therefore consistent with our hypothesis that two independent oxidative addition steps are involved in the formation of aryl–aryl coupling products.
In this report, we have shown that the Ullmann-type coupling of aryl iodides can be catalyzed by a rough Au surface submerged in a polar solvent under ambient conditions. Through a combination of in situ STM-BJ measurements, ex situ mass spectrometry, fluorescence spectroscopy, and first-principles calculations, we have gained mechanistic insight into these transformations that occur by a double oxidative-addition mechanism. Taken together, this proof-of-principle study also suggests that STM-BJ may provide a valuable method to interrogate surface-based reactivity in real-time. More broadly, this investigation connects interfacial electric fields to chemical reactivity and reiterates the promise of electric-field catalysis that could be leveraged for a wide number of molecular transformations.
Data availability
The data that support the findings of this study are available only on request from the corresponding authors. Prior to making the data available, the data need to be converted from a binary format to a text format, which we are happy to do on request.Author contributions
IBS, RLS, EMA performed all the experiments. NH and XW performed all the calculations. The project was overseen by TCB, XR and LV. IBS, RLS, TCB, XR and LV cowrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported primarily by NSF CHE-2023568 CCI Phase I: Center for Chemistry with Electric Fields. A. M. E. is supported by the Schmidt Science Fellows, in partnership with the Rhodes Trust. I. B. S. was supported in part by NIH R35 GM127135. We thank Lisa Wei for fluorescence measurements taken on an instrument provided by NSF Award 1828491. We thank Ren Wiscons and Boyuan Zhang for SEM and AFM images shown in the ESI† and Claudia Prindle for help with the Mass-Spectra. The Flatiron Institute is a division of the Simons Foundation.References
- S. A. Sorenson, J. G. Patrow and J. M. Dawlaty, Solvation Reaction Field at the Interface Measured by Vibrational Sum Frequency Generation Spectroscopy, J. Am. Chem. Soc., 2017, 139, 2369–2378 CrossRef CAS PubMed.
- V. Oklejas, C. Sjostrom and J. M. Harris, SERS Detection of the Vibrational Stark Effect from Nitrile-Terminated SAMs to Probe Electric Fields in the Diffuse Double-Layer, J. Am. Chem. Soc., 2002, 124, 2408–2409 CrossRef CAS PubMed.
- M. F. Delley, E. M. Nichols and J. M. Mayer, Interfacial Acid–Base Equilibria and Electric Fields Concurrently Probed by In Situ Surface-Enhanced Infrared Spectroscopy, J. Am. Chem. Soc., 2021, 143, 10778–10792 CrossRef CAS PubMed.
- S. N. Steinmann and Z. W. Seh, Understanding electrified interfaces, Nat. Rev. Mater., 2021, 6, 289–291 CrossRef CAS.
- K. Dutta Dubey, T. Stuyver, S. Kalita and S. Shaik, Solvent Organization and Rate Regulation of a Menshutkin Reaction by Oriented External Electric Fields are Revealed by Combined MD and QM/MM Calculations, J. Am. Chem. Soc., 2020, 142, 9955–9965 CrossRef CAS PubMed.
- A. M. Smith, A. A. Lee and S. Perkin, The Electrostatic Screening Length in Concentrated Electrolytes Increases with Concentration, J. Phys. Chem. Lett., 2016, 7, 2157–2163 CrossRef CAS PubMed.
- T. S. Wesley, Y. Román-Leshkov and Y. Surendranath, Spontaneous Electric Fields Play a Key Role in Thermochemical Catalysis at Metal–Liquid Interfaces, ACS Cent. Sci., 2021, 7, 1045–1055 CrossRef CAS PubMed.
- J. Ryu and Y. Surendranath, Tracking Electrical Fields at the Pt/H2O Interface during Hydrogen Catalysis, J. Am. Chem. Soc., 2019, 141, 15524–15531 CrossRef CAS PubMed.
- M. J. Voegtle, T. Pal, A. K. Pennathur, S. Menachekanian, J. G. Patrow, S. Sarkar, Q. Cui and J. M. Dawlaty, Interfacial Polarization and Ionic Structure at the Ionic Liquid–Metal Interface Studied by Vibrational Spectroscopy and Molecular Dynamics Simulations, J. Phys. Chem. B, 2021, 125, 2741–2753 CrossRef CAS PubMed.
- F. Ullmann and J. Bielecki, Ueber Synthesen in der Biphenylreihe, Ber. Dtsch. Chem. Ges., 1901, 34, 2174–2185 CrossRef CAS.
- H. Lin and D. Sun, Recent Synthetic Developments and Applications of the Ullmann Reaction. A Review, Org. Prep. Proced. Int., 2013, 45, 341–394 CrossRef CAS PubMed.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nano-architectures by covalent assembly of molecular building blocks, Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Mullen and R. Fasel, Atomically precise bottom-up fabrication of graphene nanoribbons, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- L. Lafferentz, V. Eberhardt, C. Dri, C. Africh, G. Comelli, F. Esch, S. Hecht and L. Grill, Controlling on-surface polymerization by hierarchical and substrate-directed growth, Nat. Chem., 2012, 4, 215–220 CrossRef CAS PubMed.
- S. Clair and D. G. de Oteyza, Controlling a Chemical Coupling Reaction on a Surface: Tools and Strategies for On-Surface Synthesis, Chem. Rev., 2019, 119, 4717–4776 CrossRef CAS PubMed.
- M. Xi and B. E. Bent, Mechanisms of the Ullmann Coupling Reaction in Adsorbed Monolayers, J. Am. Chem. Soc., 1993, 115, 7426–7433 CrossRef CAS.
- A. Batra, D. Cvetko, G. Kladnik, O. Adak, C. Cardoso, A. Ferretti, D. Prezzi, E. Molinari, A. Morgante and L. Venkataraman, Probing the mechanism for graphene nanoribbon formation on gold surfaces through X-ray spectroscopy, Chem. Sci., 2014, 5, 4419 RSC.
- L. Grill and S. Hecht, Covalent on-surface polymerization, Nat. Chem., 2020, 12, 115–130 CrossRef CAS PubMed.
- M. Kamenetska, M. Koentopp, A. C. Whalley, Y. S. Park, M. L. Steigerwald, C. Nuckolls, M. S. Hybertsen and L. Venkataraman, Formation and Evolution of Single-Molecule Junctions, Phys. Rev. Lett., 2009, 102, 126803 CrossRef CAS PubMed.
- E. A. Lewis, C. J. Murphy, A. Pronschinske, M. L. Liriano and E. C. H. Sykes, Nanoscale insight into C–C coupling on cobalt nanoparticles, Chem. Commun., 2014, 50, 10035–10037 RSC.
- L. Dong, P. N. Liu and N. Lin, Surface-Activated Coupling Reactions Confined on a Surface, Acc. Chem. Res., 2015, 48, 2765–2774 CrossRef CAS PubMed.
- W. Wang, X. Shi, S. Wang, M. A. Van Hove and N. Lin, Single-Molecule Resolution of an Organometallic Intermediate in a Surface-Supported Ullmann Coupling Reaction, J. Am. Chem. Soc., 2011, 133, 13264–13267 CrossRef CAS PubMed.
- A. Monopoli, P. Cotugno, G. Palazzo, N. Ditaranto, B. Mariano, N. Cioffi, F. Ciminale and A. Nacci, Ullmann Homocoupling Catalysed by Gold Nanoparticles in Water and Ionic Liquid, Adv. Synth. Catal., 2012, 354, 2777–2788 CrossRef CAS.
- G. Li, C. Liu, Y. Lei and R. Jin, Au25 nanocluster-catalyzed Ullmann-type homocoupling reaction of aryl iodides, Chem. Commun., 2012, 48, 12005–12007 RSC.
- R. Payne and I. E. Theodorou, Dielectric properties and relaxation in ethylene carbonate and propylene carbonate, J. Phys. Chem., 1972, 76, 2892–2900 CrossRef CAS.
- L. A. Nagahara, T. Thundat and S. M. Lindsay, Preparation and Characterization of STM Tips for Electrochemical Studies, Rev. Sci. Instrum., 1989, 60, 3128–3130 CrossRef CAS.
- Y. Zang, A. Pinkard, Z.-F. Liu, J. B. Neaton, M. L. Steigerwald, X. Roy and L. Venkataraman, Electronically transparent Au-N bonds for molecular junctions, J. Am. Chem. Soc., 2017, 139, 14845–14848 CrossRef CAS PubMed.
- Y. Zang, Q. Zou, T. Fu, F. Ng, B. Fowler, J. Yang, H. Li, M. L. Steigerwald, C. Nuckolls and L. Venkataraman, Directing isomerization reactions of cumulenes with electric fields, Nat. Commun., 2019, 10, 4482 CrossRef PubMed.
- S.-W. Hla, L. Bartels, G. Meyer and K.-H. Rieder, Inducing All Steps of a Chemical Reaction with the Scanning Tunneling Microscope Tip: Towards Single Molecule Engineering, Phys. Rev. Lett., 2000, 85, 2777–2780 CrossRef CAS PubMed.
- G. S. McCarty and P. S. Weiss, Footprints of a Surface Chemical Reaction: Dissociative Chemisorption of p-Diiodobenzene on Cu{111}, J. Phys. Chem. B, 2002, 106, 8005–8008 CrossRef CAS.
- P. S. Weiss, M. M. Kamna, T. M. Graham and S. J. Stranick, Imaging Benzene Molecules and Phenyl Radicals on Cu{111}, Langmuir, 1998, 14, 1284–1289 CrossRef CAS.
- B. Capozzi, Q. Chen, P. Darancet, M. Kotiuga, M. Buzzeo, J. B. Neaton, C. Nuckolls and L. Venkataraman, Tunable Charge Transport in Single-Molecule Junctions via Electrolytic Gating, Nano Lett., 2014, 14, 1400–1404 CrossRef CAS PubMed.
- H. D. Becker, V. Langer, J. Sieler and H. C. Becker, Molecular conformations of 9,9'-bianthryl, di-9-anthrylmethane, and some related twisted anthracene derivatives, J. Org. Chem., 1992, 57, 1883–1887 CrossRef CAS.
- L. Venkataraman, J. E. Klare, C. Nuckolls, M. S. Hybertsen and M. L. Steigerwald, Dependence of single-molecule junction conductance on molecular conformation, Nature, 2006, 442, 904–907 CrossRef CAS PubMed.
- M. Joost, A. Amgoune and D. Bourissou, Reactivity of Gold Complexes towards Elementary Organometallic Reactions, Angew. Chem., Int. Ed., 2015, 54, 15022–15045 CrossRef CAS PubMed.
- B. Huang, M. Hu and F. D. Toste, Homogeneous Gold Redox Chemistry: Organometallics, Catalysis, and Beyond, Trends Chem., 2020, 2, 707–720 CrossRef CAS PubMed.
- L. Rocchigiani and M. Bochmann, Recent Advances in Gold(III) Chemistry: Structure, Bonding, Reactivity, and Role in Homogeneous Catalysis, Chem. Rev., 2021, 121, 8364–8451 CrossRef CAS PubMed.
- M. Lackinger, Surface-assisted Ullmann coupling, Chem. Commun., 2017, 53, 7872–7885 RSC.
- F. Ullmann, Ueber eine neue Bildungsweise von Diphenylaminderivaten, Ber. Dtsch. Chem. Ges., 1903, 36, 2382–2384 CrossRef.
- F. Ullmann, Ueber symmetrische Biphenylderivate, Justus Liebigs Ann. Chem., 1904, 332, 38–81 CrossRef.
- Z. L. Cheng, R. Skouta, H. Vazquez, J. R. Widawsky, S. Schneebeli, W. Chen, M. S. Hybertsen, R. Breslow and L. Venkataraman, In situ formation of highly conducting covalent Au-C contacts for single-molecule junctions, Nat. Nanotechnol., 2011, 6, 353–357 CrossRef CAS PubMed.
- J. Bjork, F. Hanke and S. Stafstrom, Mechanisms of Halogen-Based Covalent Self-Assembly on Metal Surfaces, J. Am. Chem. Soc., 2013, 135, 5768–5775 CrossRef PubMed.
- A. Corma, R. Juárez, M. Boronat, F. Sánchez, M. Iglesias and H. García, Gold catalyzes the Sonogashira coupling reaction without the requirement of palladium impurities, Chem. Commun., 2011, 47, 1446–1448 RSC.
- H. Shi, Z. Cai, J. Patrow, B. Zhao, Y. Wang, Y. Wang, A. Benderskii, J. Dawlaty and S. B. Cronin, Monitoring Local Electric Fields at Electrode Surfaces Using Surface Enhanced Raman Scattering-Based Stark-Shift Spectroscopy during Hydrogen Evolution Reactions, ACS Appl. Mater. Interfaces, 2018, 10, 33678–33683 CrossRef CAS PubMed.
- Z. K. Goldsmith, M. Secor and S. Hammes-Schiffer, Inhomogeneity of Interfacial Electric Fields at Vibrational Probes on Electrode Surfaces, ACS Cent. Sci., 2020, 6, 304–311 CrossRef CAS PubMed.
- N. M. Hoffmann, X. Wang and T. C. Berkelbach, Linear Free Energy Relationships in Electrostatic Catalysis, ACS Catal., 2022, 8237–8241 CrossRef CAS.
- D. F. McMillen and D. M. Golden, Hydrocarbon bond dissociation energies, Annu. Rev. Phys. Chem., 1982, 33, 493–532 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03780g |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |