
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective peptide catalysis in complex environments – from river water to cell lysates†
Tobias
Schnitzer
 ,
Jonas W.
Rackl
and
Helma
Wennemers
*
,
Jonas W.
Rackl
and
Helma
Wennemers
*
Laboratory of Organic Chemistry, ETH Zürich, Vladimir-Prelog-Weg 3, 8093 Zürich, Switzerland. E-mail: Helma.Wennemers@org.chem.ethz.ch
First published on 14th June 2022
Abstract
Many stereoselective peptide catalysts have been established. They consist, like nature's catalysts, of amino acids but have significantly lower molecular weights than enzymes. Whereas enzymes operate with exquisite chemoselectivity in complex biological environments, peptide catalysts are used in pure organic solvents and at higher concentrations. Can a peptide catalyst exhibit chemoselectivity reminiscent of enzymes? Here, we investigated the properties of tripeptide catalysts in complex mixtures in hydrophobic and aqueous solvents. We challenged the catalysts with biomolecules bearing functional groups that could interfere by coordination or reaction with the peptide, the substrates, or intermediates. H-DPro-αMePro-Glu-NHC12H15 emerged through tailoring of the trans/cis ratio of the tertiary amide as a conformationally well-defined tripeptide that catalyzes C–C bond formations with high reactivity and stereoselectivity – regardless of the solvent and compound composition. The chemoselectivity of the tripeptide is so high that it even catalyzes reactions in cell lysates. The findings provoke the question of the potential role of peptide catalysis in nature and during the evolution of enzymes.
Introduction
During the past two decades, peptides have been recognized as potent catalysts for different reactions.1,2 Several of these peptide catalysts feature exquisite levels of stereoselectivity and reactivity. Since they consist, like nature's catalysts, of amino acids but are significantly smaller, peptide catalysts can be viewed as “mini-enzymes”. Yet, whereas enzymes catalyze reactions in aqueous media, most peptidic catalysts operate in organic solvents.‡1–13 A further marked difference is the environment in which catalysis takes place. Enzymes work in highly complex cellular environments – reaction media that require exceptional chemoselectivity – while peptide catalysts are used in well-defined environments consisting only of substrates and products in pure solvents. Moreover, enzymes operate at significantly lower concentrations than peptide catalysts. We became intrigued by the question of whether a peptide catalyst can have a chemoselectivity reminiscent of enzymes. Can stereoselective peptide catalysis occur in water in the presence of compounds abundant in nature, in complex compound mixtures, and possibly even in cell lysates?Herein, we show that the peptide H-DPro-αMePro-Glu-NHC12H15 is so chemoselective that it catalyzes C–C bond formation reactions between aldehydes and nitroolefins with exquisite stereoselectivity in complex mixtures, including cell lysates. Key to the performance of this peptide is the αMePro residue that ensures a high trans/cis ratio of the tertiary amide bond regardless of the solvent and compound composition.
Results & discussion
Peptide catalysis in water in the presence of biomolecules
We used the alkylated tripeptide H-DPro-Pro-Glu-NHC12H151 as a starting point for our studies (Fig. 1). This peptide is a stereoselective catalyst for the conjugate addition reaction of aldehydes to nitroolefins in water and organic solvents.13,14 The catalytic reaction proceeds via the formation of an enamine intermediate between the peptide and the aldehyde followed by a C–C bond formation with the nitroolefin.15–18 Essential for the catalytic efficiency of 1 is a high trans/cis ratio of the DPro-Pro amide bond19 and the CO2H group of glutamate as an intramolecular proton donor.14 In water, the alkyl chain facilitates the formation of an emulsion and thus the solubility of the otherwise water-insoluble substrates.13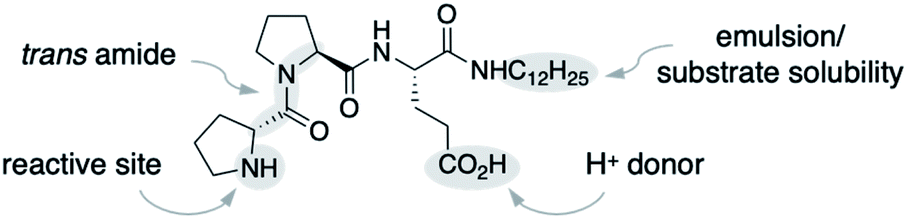 | ||
| Fig. 1 Peptide catalyst 1. | ||
We began by exploring the effect of compounds common in biological systems on the reactivity and stereoselectivity of peptide 1 (Scheme 1). We thus added amino acids (A, B), mono- and disaccharides (C–E), nucleic acid derivatives (F–H), lipids and terpenes (I–K), vitamins (L, M), compounds of the central carbon metabolism (N, O), and glutathione (P) to the peptide-catalyzed conjugate addition reaction between butanal and (E)-nitrostyrene. In the presence of an equimolar amount of additives A–P with respect to 1 (3 mol%), the catalytic reaction proceeded with comparable diastereo- and enantioselectivity (d.r. 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, 91% ee) as in their absence (d.r. 98:2, 91% ee). Even the reactivity of peptide 1 was not affected by these additives (quantitative conversion of starting material to the product after 24 h). The only exception was pyruvic acid (O) which caused a drop to ∼80% conversion. These results are noteworthy since the additives include carboxylic acid and phosphoric acid groups (A, B, H, M–O), metal complexes (M), heteroaromatic groups (B, F–H, M), alcohols (C–E, K–N), and thiols (P). These acidic and basic, H-bond donor and acceptor sites, and the metal complex could interfere by coordination to peptide 1 or the intermediates with the catalytic cycle. Even additives containing carbonyl (C–E), thiol (P), and primary amino groups (A, B, F, G) that could react with the secondary amine catalyst or the aldehyde and nitroolefin substrates do not affect the reaction. The observed deactivation by pyruvic acid is likely due to its acidity (pKa = 2.5), resulting in protonation of the amine, the reactive site of 1. Next, we increased the amount of each additive stepwise from an equimolar amount with respect to the peptide catalyst (3 mol%, 13.2 mM) to 10 mol% (44 mM), 50 mol% (220 mM), and 100 mol% (440 mM; Scheme 1). The latter concentrations are comparable to the overall metabolite concentration inside cells (∼200 mM).20 At the highest concentration (100 mol%, 440 mM), the stoichiometry of the additive and the nitroolefin substrate is 1:1. Even at this concentration, most additives did not affect the catalytic performance of peptide 1 and full conversion; diastereoselectivities between 94:6 and 99:1, and enantioselectivities between 87% ee and 92% ee were observed. Only citric (N) and pyruvic acid (O) reduced the reactivity (<50% conversion), and phenylalanine (B) and glutathione (P) reduced the diastereoselectivity (d.r. ≤80:20). These findings highlight the high chemoselectivity of peptide 1 for the conjugate addition reaction, which proceeds efficiently in water even in the presence of compounds that are abundant in nature and contain functional moieties that could interfere with the catalytic cycle.
:4, 91% ee) as in their absence (d.r. 98:2, 91% ee). Even the reactivity of peptide 1 was not affected by these additives (quantitative conversion of starting material to the product after 24 h). The only exception was pyruvic acid (O) which caused a drop to ∼80% conversion. These results are noteworthy since the additives include carboxylic acid and phosphoric acid groups (A, B, H, M–O), metal complexes (M), heteroaromatic groups (B, F–H, M), alcohols (C–E, K–N), and thiols (P). These acidic and basic, H-bond donor and acceptor sites, and the metal complex could interfere by coordination to peptide 1 or the intermediates with the catalytic cycle. Even additives containing carbonyl (C–E), thiol (P), and primary amino groups (A, B, F, G) that could react with the secondary amine catalyst or the aldehyde and nitroolefin substrates do not affect the reaction. The observed deactivation by pyruvic acid is likely due to its acidity (pKa = 2.5), resulting in protonation of the amine, the reactive site of 1. Next, we increased the amount of each additive stepwise from an equimolar amount with respect to the peptide catalyst (3 mol%, 13.2 mM) to 10 mol% (44 mM), 50 mol% (220 mM), and 100 mol% (440 mM; Scheme 1). The latter concentrations are comparable to the overall metabolite concentration inside cells (∼200 mM).20 At the highest concentration (100 mol%, 440 mM), the stoichiometry of the additive and the nitroolefin substrate is 1:1. Even at this concentration, most additives did not affect the catalytic performance of peptide 1 and full conversion; diastereoselectivities between 94:6 and 99:1, and enantioselectivities between 87% ee and 92% ee were observed. Only citric (N) and pyruvic acid (O) reduced the reactivity (<50% conversion), and phenylalanine (B) and glutathione (P) reduced the diastereoselectivity (d.r. ≤80:20). These findings highlight the high chemoselectivity of peptide 1 for the conjugate addition reaction, which proceeds efficiently in water even in the presence of compounds that are abundant in nature and contain functional moieties that could interfere with the catalytic cycle.
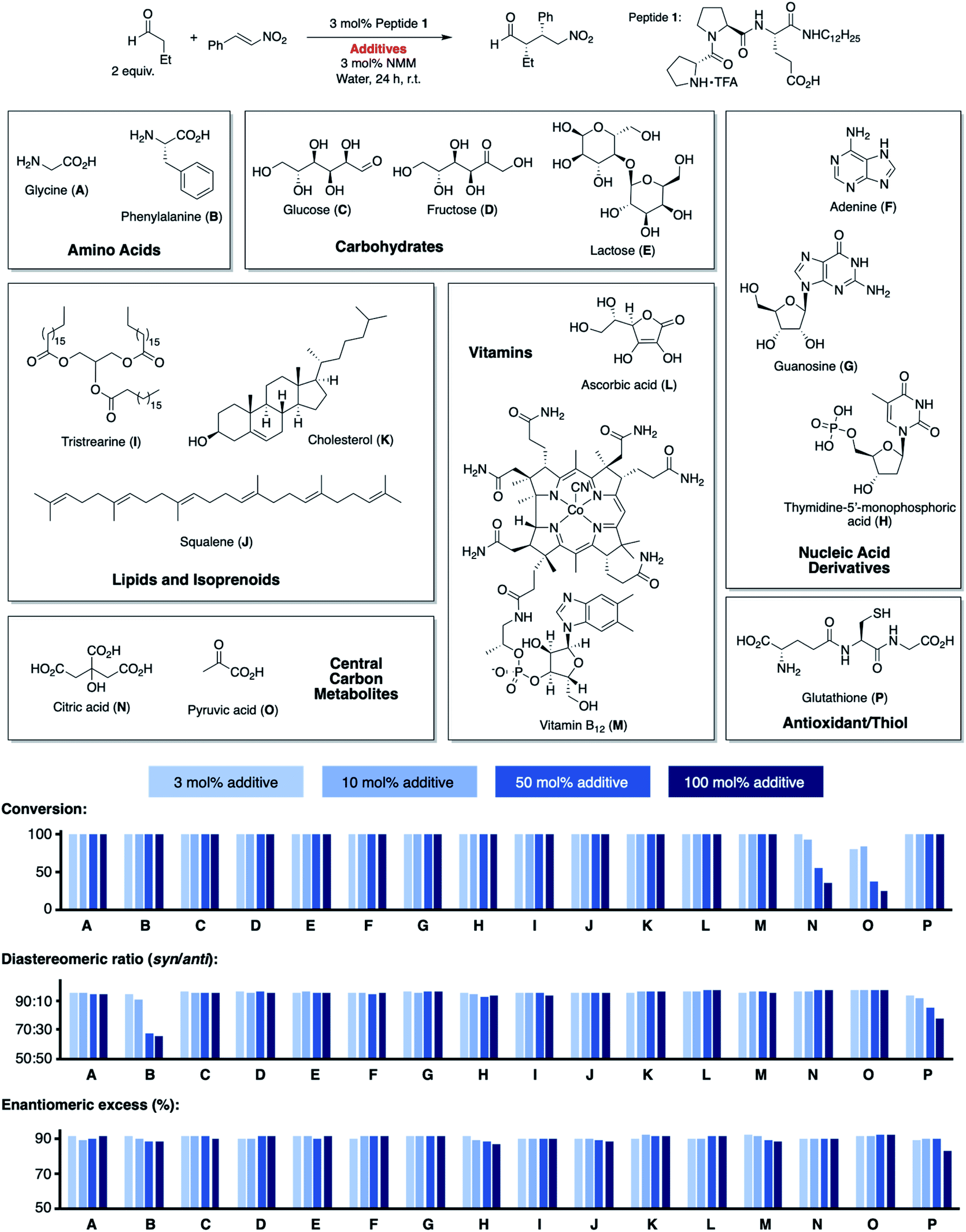 | ||
| Scheme 1 Conjugate addition reaction of butanal to nitrostyrene catalyzed by 1 in the presence of biomolecules A–P. | ||
Peptide catalysis in complex mixtures
We next extended our study to peptide catalysis in multi-component mixtures. Specifically, we performed the catalytic reaction in water from different sources (S1–S4), milk and milk substitutes (S5–S9), fruit and vegetable juices (S10–S15), coffee and tea (S16–S20), honey solution (S21), soft drinks (S22–S25), alcoholics (S26–S33), vinegar (S34–S35) and olive oil (S36; Scheme 2a). These commercial, readily available “solvents” differ significantly from each other in polarity, pH, viscosity, and their ingredients and are, thus, a good platform to probe the effect of different environments on peptide catalysis.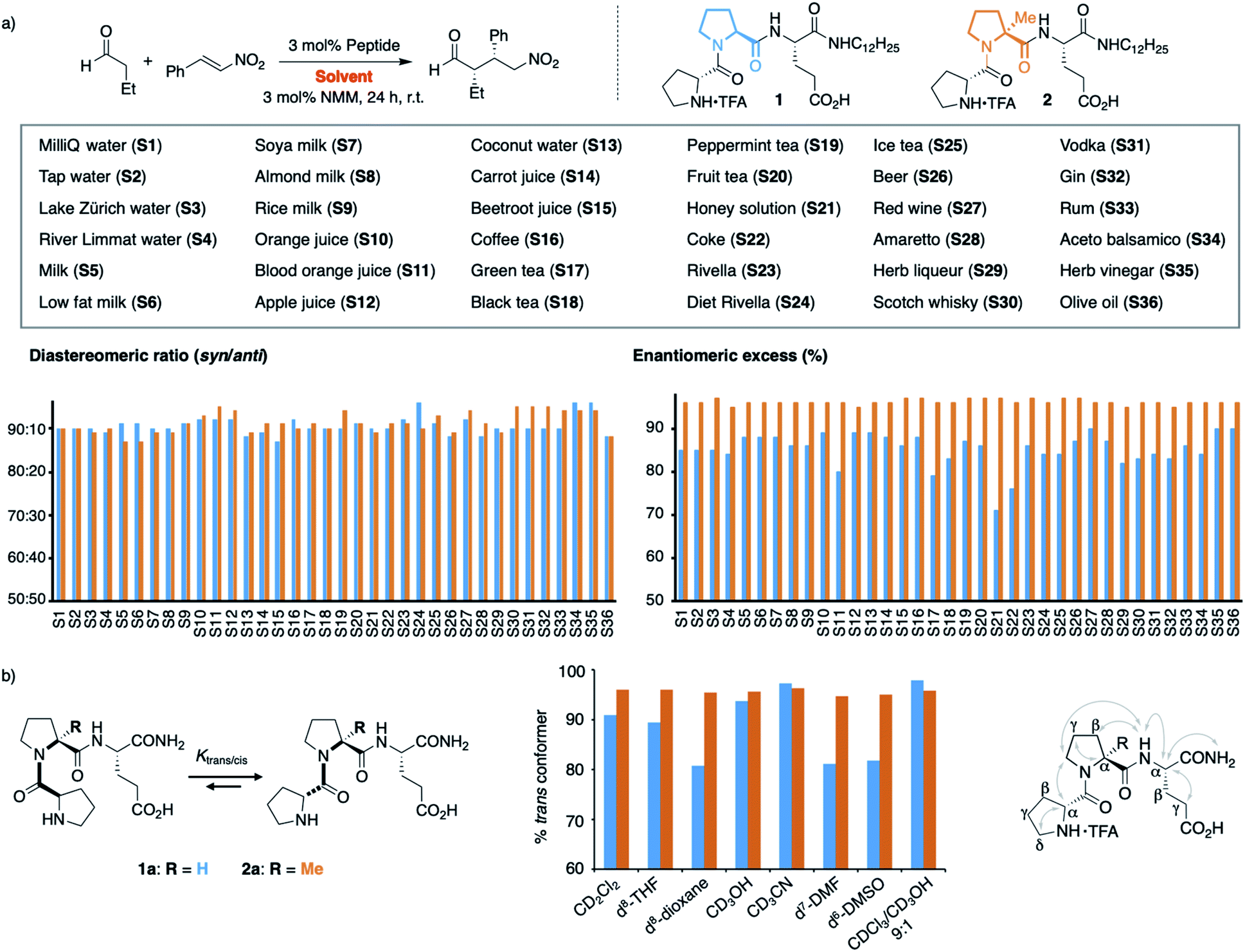 | ||
| Scheme 2 (a) Catalysis in complex mixtures S1–S36 with peptides 1 and 2. (b) Trans/cis ratio of 1a and 2a in different solvents. | ||
Control experiments showed that the aldehyde and nitroolefin do not react with each other in any of these mixtures in the absence of peptide 1. In the presence of 1, the conjugate addition product formed quantitatively in all solvents but with different stereoselectivity. The diastereomeric ratio syn/anti ranged from 93:7 in vodka to 98:2 in green tea, the enantioselectivity from 71% ee in honey solution to 90% ee in olive oil (Scheme 2a, blue bars). Hence, the complex environments affected the enantioselectivity of H-DPro-Pro-Glu-NHC12H251. Whereas these results show that peptide catalysis is possible in complex mixtures, we sought an analog of 1 that would allow for consistently high product formation and stereoselectivity regardless of the environment.
Conformational tailoring of peptide catalyst
We recently found that the stereoselectivity of the parent, non-alkylated peptide H-DPro-Pro-Glu-NH21a correlates with the trans/cis conformer ratio of its DPro-Pro amide bond (Scheme 2b).19 Hence, we suspected that the reduced stereoselectivity in some “solvents” arose from lower trans/cis ratios in these complex environments. Therefore, we sought an analog of 1 with a high trans/cis ratio regardless of the environment. Previously developed catalyst analogs bearing pipecolic acid or γ-substituted Pro derivatives in place of Pro in the middle position have a higher trans/cis ratio and thus higher stereoselectivity than 1a in apolar solvents.19,21 In polar solvents (e.g., H2O, MeOH, DMSO, DMF), these analogs have, however, lower trans/cis ratios and poorer stereoselectivities than 1a.19 Thus, we needed an analog of 1 with a residue in the middle position that “locks” the peptide in the trans conformation. We envisioned that α-L-methylproline (αMePro), a proline derivative known to favor the trans conformer of Xaa–Pro bonds,22–25 could fill this need. Based on the conformational analysis of peptide 1a,16 we reasoned that the additional methyl group at Ca should not affect the conformation of the peptide catalyst. Indeed, a crystal structure of the TFA-salt of H-DPro-αMePro-Glu-NH2 (2a) was almost identical to that of the parent peptide 1a (see ESI† p. S10). Both peptides adopt a β-turn conformation with a H-bond between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of DPro and the C-terminal amide. The similarity of 1a and 2a was further confirmed by NMR spectroscopy, as indicated by the coupling constants and nuclear Overhauser effects (Scheme 2b, right, and ESI† p. S9).
O of DPro and the C-terminal amide. The similarity of 1a and 2a was further confirmed by NMR spectroscopy, as indicated by the coupling constants and nuclear Overhauser effects (Scheme 2b, right, and ESI† p. S9).
Since the trans/cis ratio of 2a is difficult to monitor in complex mixtures, we initially used polar and apolar, protic and aprotic organic solvents to probe the effect of the αMePro residue on the conformation and the catalytic properties of the peptide catalyst (Scheme 2b, middle). Peptide 2a has in all of these solvents a trans/cis ratio of ≥95:1 whereas the trans/cis ratio of peptide 1a varies with the solvent (80–98% of trans conformer). As a result, the stereoselectivity of 2a is higher than that of 1a in all solvents (see ESI† p. S11).
Building on these data, we prepared the alkylated analog H-DPro-αMePro-Glu-NHC12H252 and evaluated its catalytic properties in the different “solvents” S1–S36 (Scheme 2a). Peptide 2 provided the γ-nitroaldehyde with diastereoselectivities of 87:13–95:5 and enantioselectivities of 96% or 97% ee in each of these environments (Scheme 2a, orange bars). The data show that the “locked” trans amide bond of 2 enables peptide catalysis in any environment with uniformly high enantioselectivity. These results are noteworthy since the tested environments have significantly different polarities (e.g., olive oil and milk versus water and alcoholics), viscosities (e.g., honey versus apple juice), pH (e.g., coke and vinegar versus tap water), and contain numerous compounds, including carbohydrates, lipids, peptides, and proteins, that could interfere or inhibit peptide catalysis. The data thus evidence the high chemoselectivity of the peptide catalyst for the conjugate addition reaction.
Peptide catalysis in cell lysate
Encouraged by these data, we challenged peptide 2 to catalyze reactions in cell lysates (Scheme 3). We used lysates with a total protein concentration of 5.3 g mL−1 from human hepatoma cells (HepG2), an immortal cell line that secrets a variety of major plasma proteins. Under standard conditions (880 mM aldehyde, 440 mM nitroolefin, 13.2 mM 2), complete conversion of the nitroolefin was observed, and the conjugate addition product was isolated in 97% yield with a stereoselectivity of d.r. 94:6 and 95% ee. Thus, peptide 2 withstands even the multi-component mixture of a cell lysate with numerous compounds that could either react or coordinate to the catalyst, the substrates, or the reaction intermediates.
 | ||
| Scheme 3 Peptide catalysis in a cell lysate. | ||
Enzyme concentrations range between micro- and millimolar inside cells.20 Therefore, we reduced the reaction concentration by a factor of 10 and 100. Even at the lowest concentration, peptide 2 (132 μM) provided the product with an 81% yield and only slightly lower stereoselectivity (d.r. 85:15, 93% ee). These data show that the peptide is so reactive and chemoselective that stereoselective catalysis takes place in a highly competitive environment at a concentration typical for enzymes.
Conclusion
The study put forth the first examples of stereoselective peptide catalysts that are sufficiently chemoselective to operate in complex mixtures, including cell lysates. The conformationally tailored peptide catalyzes C–C bond formation in both, hydrophobic and aqueous environments with remarkable stereoselectivity, even at concentrations typical for enzyme catalysis. Biomolecules that could interfere through coordination or covalent reaction with the peptide, the catalyst intermediates, or the substrates did not affect the peptide catalysis.Of note, the key to the exquisite performance of the peptide catalyst in any environment was the tuning of the trans/cis amide bond ratio through substituents at proline, a concept that nature uses to regulate protein folding and stability as well as enzyme activity.26–30
The results illustrate that a short-chain peptide can be a potent catalyst in any environment, including those that are hallmarks of naturally evolved enzymes. The findings show the utility of peptide catalysts for organic synthesis and provoke the question of the potential role of small catalytic peptides in the evolution of enzymes. Proteomic analyses have revealed that the cells of microbes as well as those of humans contain a large number of peptides.31 The function of many of these peptides is unknown. The presented data spark the question of whether some of these peptides catalyze biochemical reactions, orchestrating an underworld of clandestine organocatalysis that yet needs to be discovered?
Data availability
Experimental procedures and analytical data are available in the ESI.†Author contributions
H. W. and T. S. conceived the project. T. S. and J. W. R. performed the experiments. All authors contributed to the data analysis and writing of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Fonds der Chemischen Industrie (Germany) for a Kekulé Fellowship for T. S., the Swiss National Science Foundation (grant 200020_188729), and the European Union's Horizon 2020 research and innovation programme (Grant No. 862081) for financial support.Notes and references
- A. J. Metrano, A. J. Chinn, C. R. Shugrue, E. A. Stone, B. Kim and S. J. Miller, Chem. Rev., 2020, 120, 11479 CrossRef CAS PubMed.
- H. Wennemers, Chem. Commun., 2011, 47, 12036 RSC.
- S. Julia, J. Masana and J. C. Vega, Angew. Chem., Int. Ed., 1980, 19, 929 CrossRef.
- S. Julia, J. Guixer, J. Masana, J. Rocas, S. Colonna, R. Annuziata and H. Molinari, J. Chem. Soc., Perkin Trans. 1, 1982, 22, 1317 RSC.
- A. Weyer, D. Díaz, A. Nierth, N. E. Schlörer and A. Berkessel, ChemCatChem, 2012, 4, 337 CrossRef CAS.
- M. Lei, L. Shi, G. Li, S. Chen, W. Fang, Z. Ge, T. Cheng and R. Li, Tetrahedron, 2007, 63, 7892 CrossRef CAS.
- D. K. Romney and S. J. Miller, Org. Lett., 2012, 14, 1138 CrossRef CAS PubMed.
- G. Maayan, M. D. Ward and K. Kirshenbaum, Proc. Natl. Acad. Sci., 2009, 106, 13679 CrossRef CAS PubMed.
- C. Mayer, M. M. Müller, S. H. Gellman and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 6978 CrossRef CAS PubMed.
- K. Akagawa, H. Akabane, S. Sakamoto and K. Kudo, Org. Lett., 2008, 10, 2035 CrossRef CAS PubMed.
- B. Sarkhel, A. Chatterjee and D. Das, J. Am. Chem. Soc., 2020, 142, 4098 CrossRef CAS PubMed.
- C. S. Foden, S. Islam, C. Fernández-García, L. Maugeri, T. D. Sheppard and M. W. Powner, Science, 2020, 370, 865 CrossRef CAS.
- J. Duschmalé, S. Kohrt and H. Wennemers, Chem. Commun., 2014, 50, 8109 RSC.
- J. Duschmalé, J. Wiest, M. Wiesner and H. Wennemers, Chem. Sci., 2013, 4, 1312 RSC.
- M. Wiesner, G. Upert, G. Angelici and H. Wennemers, J. Am. Chem. Soc., 2010, 132, 6 CrossRef CAS PubMed.
- F. Bächle, J. Duschmalé, C. Ebner, A. Pfaltz and H. Wennemers, Angew. Chem., Int. Ed., 2013, 52, 12619 CrossRef PubMed.
- C. Rigling, J. K. Kisunzu, J. Duschmalé, D. Häussinger, M. Wiesner, M. O. Ebert and H. Wennemers, J. Am. Chem. Soc., 2018, 140, 10829 CrossRef CAS PubMed.
- T. Schnitzer and H. Wennemers, J. Org. Chem., 2020, 85, 7633 CrossRef CAS PubMed.
- T. Schnitzer and H. Wennemers, J. Am. Chem. Soc., 2017, 139, 15356 CrossRef CAS PubMed.
- B. D. Bennett, E. H. Kimball, M. Gao, R. Osterhout, S. J. Van Dien and J. D. Rabinowitz, Nat. Chem. Biol., 2009, 5, 593 CrossRef CAS PubMed.
- T. Schnitzer and H. Wennemers, Synthesis, 2018, 50, 4377 CrossRef CAS.
- N. G. Delaney and V. Madison, J. Am. Chem. Soc., 1982, 104, 6635 CrossRef CAS.
- J. H. Welsh, O. Zerbe, W. von Philipsborn and J. A. Robinson, FEBS Lett., 1992, 297, 216 CrossRef CAS PubMed.
- V. Y. Torbeev, E. Fumi, M. O. Ebert, W. B. Schweizer and D. Hilvert, Helv. Chim. Acta, 2012, 95, 2411 CrossRef CAS.
- V. Torbeev, M. O. Ebert, J. Dolenc and D. Hilvert, J. Am. Chem. Soc., 2015, 137, 2524 CrossRef CAS.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929 CrossRef CAS PubMed.
- U. Arnold, M. P. Hinderaker, J. Köditz, R. Golbik, R. Ulbrich-Hofmann and R. T. Raines, J. Am. Chem. Soc., 2003, 125, 7500 CrossRef CAS PubMed.
- S. C. R. Lummis, D. L. Beene, L. W. Lee, H. A. Lester, R. W. Broadhurst and D. A. Dougherty, Nature, 2005, 438, 248 CrossRef CAS PubMed.
- B. Holzberger and A. Marx, J. Am. Chem. Soc., 2010, 132, 15708 CrossRef CAS PubMed.
- S. A. Lieblich, K. Y. Fang, J. K. B. Cahn, J. Rawson, J. LeBon, H. T. Ku and D. A. Tirrell, J. Am. Chem. Soc., 2017, 139, 8384 CrossRef CAS PubMed.
- A. Rathore, T. F. Martinez, Q. Chu and A. Saghatelian, Expert Rev. Proteomics, 2018, 15, 963 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2152664. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02044k |
| ‡ For examples of peptide catalysis in water, or in mixtures of an organic solvent and water, see ref. 3–13. |
| This journal is © The Royal Society of Chemistry 2022 |