
Providing direction for mechanistic inferences in radical cascade cyclization using a Transformer model†
Jiangcheng
Xu‡
 ab,
Yun
Zhang‡
a,
Jiale
Han‡
a,
An
Su
c,
Haoran
Qiao
d,
Chengyun
Zhang
a,
Jing
Tang
a,
Xi
Shen
a,
Bin
Sun
a,
Wenbo
Yu
b,
Silong
Zhai
a,
Xinqiao
Wang
a,
Yejian
Wu
a,
Weike
Su
*a and
Hongliang
Duan
*a
ab,
Yun
Zhang‡
a,
Jiale
Han‡
a,
An
Su
c,
Haoran
Qiao
d,
Chengyun
Zhang
a,
Jing
Tang
a,
Xi
Shen
a,
Bin
Sun
a,
Wenbo
Yu
b,
Silong
Zhai
a,
Xinqiao
Wang
a,
Yejian
Wu
a,
Weike
Su
*a and
Hongliang
Duan
*a
aArtificial Intelligence Aided Drug Discovery Institute, College of Pharmaceutical Sciences; Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, Hangzhou, 310014, P. R. China. E-mail: Pharmlab@zjut.edu.cn; hduan@zjut.edu.cn
bHangzhou Vocational & Technical College, Hangzhou, 310014, P. R. China
cCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, P. R. China
dCollege of Mathematics and Physics, Shanghai University of Electric Power, Shanghai, 201203, P. R. China
First published on 17th March 2022
Abstract
Even in modern organic chemistry, predicting or proposing a reaction mechanism and speculating on reaction intermediates remains challenging. For example, it is challenging to predict the regioselectivity of radical addition in radical cascade cyclization, which finds wide application in life sciences and pharmaceutical industries. In this work, radical cascade cyclization is considered to demonstrate that Transformer, a sequence-to-sequence deep learning model, is capable of predicting the reaction intermediates. A major challenge is that the number of intermediates involved in the different reactions is variable. By defining “key intermediates”, this thorny problem was avoided. We curated a database of 874 chemical equations and corresponding 1748 key intermediates and used the dataset to fine-tune a model pretrained based on the USPTO dataset. The format of the dataset is very different between pretraining and fine-tuning. Correspondingly, the resulting Transformer model achieves remarkable accuracy in predicting the structures and stereochemistry of the key intermediates. The interpretability produced by attention weights of the resulting Transformer model shows a mindset similar to that of an experienced chemist. Hence, our study provides a novel approach to help chemists discover the mechanisms of organic reactions.
Introduction
Reaction mechanisms are crucial to the methodological study of modern organic chemistry.1–4 A comprehensive understanding of chemical reactions can facilitate discovering new chemistry,5,6 predicting reaction products,7,8 designing catalysts,9 and optimizing chemical reactions.2,10,11It is often difficult for us to observe intermediates in chemical reactions. A majority of the mechanistic understanding of organic reactions comes from computational studies6,12 or studies involving both experimental and complementary computational explorations.13–19 Meanwhile, computational research is primarily based on physics-based or physics-inspired models,20 where quantum mechanical (QM) calculations, particularly with density functional theory (DFT), can be applied to real chemical systems studied by experimentalists,3,21,22 or based on a high-accuracy potential energy surface (PES).12,23 However, these methods are computationally intensive and require the involvement of expert chemists.24 Other approaches, such as accurate ab initio methods, are computationally demanding and can only be used to study small systems in the gas phase or regular periodic materials.12
The recent wave of artificial intelligence (AI) within computational explorations has had a major impact on the prediction of small-molecule reaction pathways or understanding of chemical reaction mechanisms.21,24–27 Meanwhile, challenges remain because of computational complexity.12,23,24,28 As an important branch of AI, natural language processing (NLP) models29 have become powerful and effective tools in organic chemistry research, showing promising results in reaction prediction,30–37 reaction condition recommendations,38,39 and retrosynthesis40–45 planning. However, in chemical reaction mechanism speculation, there are negligible related research studies. Here, this study explores the possibility of applying NLP models to intermediate predictions, with the potential to bridge the gap between NLP models and inferences about chemical reaction mechanisms (Fig. 1).
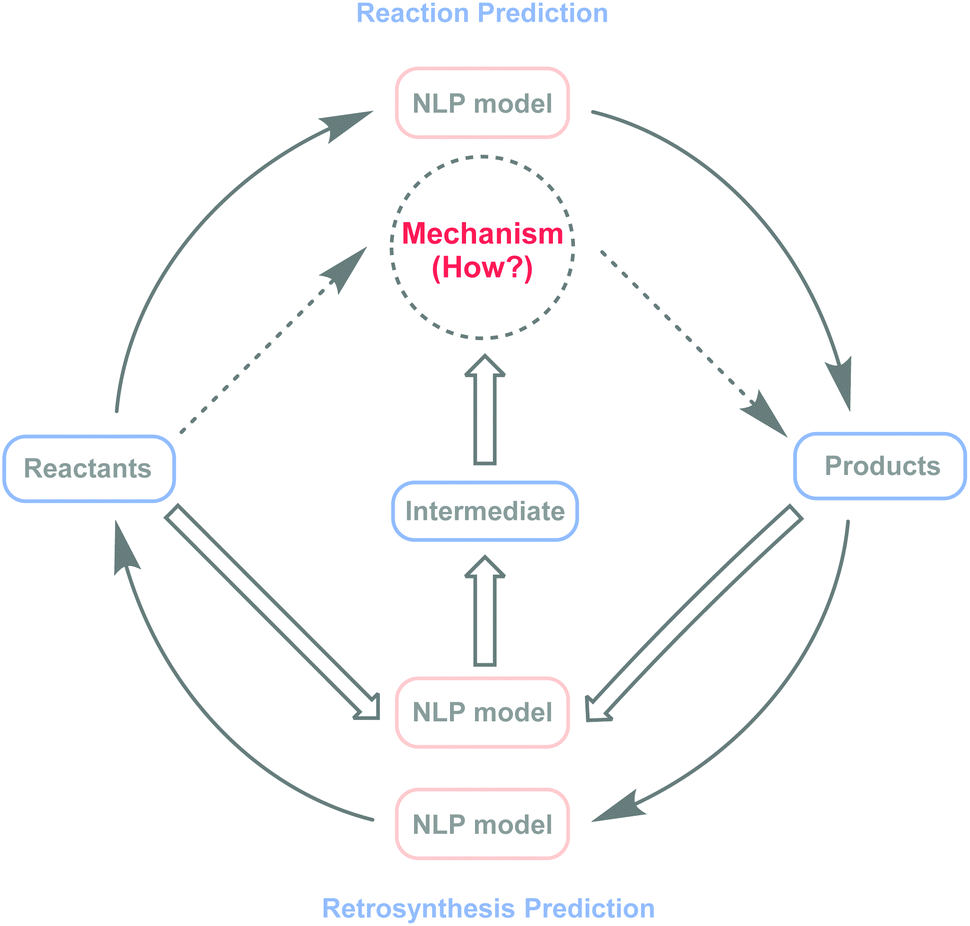 | ||
| Fig. 1 Proof-of-concept method for intermediate prediction. The NLP model has made considerable progress in end-to-end predictions of reaction and retrosynthesis. Determining what happens during a reaction is as important as reaction prediction and retrosynthesis prediction. | ||
In this study, radical cascade cyclization was used as an example to explore the reaction mechanism prediction using an NLP model. Recently, radical chemistry has attracted considerable attention.46 Radical cascade cyclization (Fig. 2) is critical for the preparation of cyclic compounds, as the processes involve only a single step in the generation of carbocycles and heterocycles,46–48 often showing interesting biological properties that are useful in medicinal chemistry.49–51
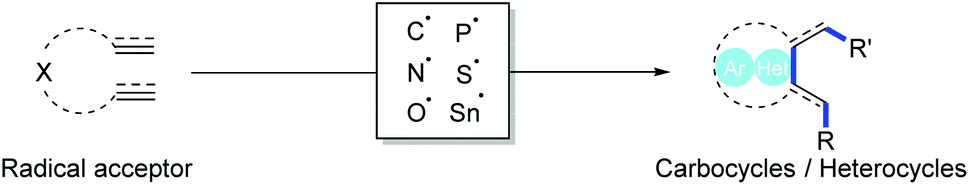 | ||
| Fig. 2 Radical cascade cyclization is promising for the preparation of cyclic compounds because multiple bonds can be formed in a single preparation step. | ||
Radical cascade cyclization was chosen as the research object of this study for two additional reasons. First, it is difficult even for experienced chemists to predict the regioselectivity of radical attack in radical cascade cyclization.46,47,52 Therefore, a simple method for determining the reaction process is urgently needed. Second, “trustworthy” intermediates can be identified because mechanistic studies, such as radical clock experiments, radical inhibition experiments, and radical capture operations, can give detailed insights into the process of radical cascade cyclization.52
The quality and availability of data play a key role in the training of deep learning models. Data on chemical molecules or chemical reactions are available in databases such as PubChem,53 ChEMBL,54 Reaxys,55 and SciFinder.56,57 However, to the best of our knowledge, no data for the intermediates of chemical reactions have been recorded. Therefore, in this study, we curated a validated dataset of reaction mechanisms by collecting the literature manually. Another major challenge is the inconsistent amounts of intermediates in different reactions. By defining the term “key intermediate” (see below for details), one radical cascade cyclization can identify two key intermediates to solve this thorny problem. Based on this definition, a dataset of radical cascade cyclizations containing key intermediates was prepared.
To address the scarcity of training data, a transfer learning strategy was adapted to pretrain the model using the USPTO dataset as we did in previous studies.36,37,41,58 The pre-trained model was then fine-tuned using our curated dataset of reaction intermediates. The results showed that the Transformer model performed satisfactorily in predicting intermediates with an accuracy rate of 93.5%, indicating that it is a useful AI tool for studying chemical reaction mechanisms.
Results and discussion
Construction of a mechanism prediction model
A typical radical cascade cyclization reaction proceeds in four steps:47 (i) radical formation via single-electron transfer (SET); (ii) radical addition via a radical attack on unsaturated bonds to generate radical intermediates; (iii) radical cyclization, resulting in carbon–carbon/heteroatom bonds; and (iv) radical or radical intermediate quenching by another radical donor or by hydrogen abstraction. In this work, the intermediate generated after the first radical addition is called“key intermediate I”, which reflects the regioselectivity of the first radical addition step. If the cascade reaction yields only one carbocycle or heterocycle, the intermediate formed after the cyclization is referred to as“key intermediate II”. For polycyclic formation, the intermediate before the final cyclization step is defined as“key intermediate II”. Key intermediate II can provide valuable information about rearrangements, hydrogen or aryl migrations, and other transformations that occur during the reaction (further details are provided in section 1 of the ESI†).The role of key intermediates in identifying the mechanism of radical cascade cyclization is similar to that of signposts in maze traversal: if there are proper signposts, one can traverse the maze more quickly. Also, if there are multiple potential routes, the signposts can indicate the most plausible route. The “key intermediates” serve as the “signposts” in the mechanism inference methodology.
Data preparation
The state-of-the-art in the development of radical cascade cyclization methodologies, especially the synthesis of carbocycles and heterocycles, was extensively studied by reviewing the existing literature. From these, we analyzed the methods used to infer reaction mechanisms, including experimental and complementary computational exploration. The Simplified Molecular-Input Line-Entry System (SMILES) was chosen to represent the reaction equations and the intermediates (for a comparison of the different representations, see the ESI†). Finally, all the data were canonicalized using RDkit (version 2019.03). The stereo-configuration information on the reactants, products, and intermediates was preserved. In this manner, the dataset for radical cascade cyclization was created manually (see section 4 of the ESI† for more information).The self-built or customized dataset contains a wide range of information, including chemical equations for a total of 874 radical cascade cyclizations and 1748 key intermediates corresponding to the reactions. These reactions were further classified according to the number of rings formed by radical cascade cyclization: 428 of the reactions resulted in a single ring, 321 resulted in two rings, and 125 resulted in three or more rings (Fig. 3a). Another classification was based on the type of radical center: 356 of the reactions had carbon-centered radicals, 266 had sulfur-centered radicals, 104 had phosphorus-centered radicals, 92 had nitrogen-centered radicals, and 56 had tin-centered radicals (Fig. 3b). The molecular mass of the target product follows a normal distribution (Fig. 3c). The formation of radical centers varied because donors can provide radicals with different SETs (Fig. 3d). In the case of radical acceptors, 1,n-diynes, 1,n-enynes, and alkynyl (hetero)arenes are the commonly used varieties (Fig. 3e).
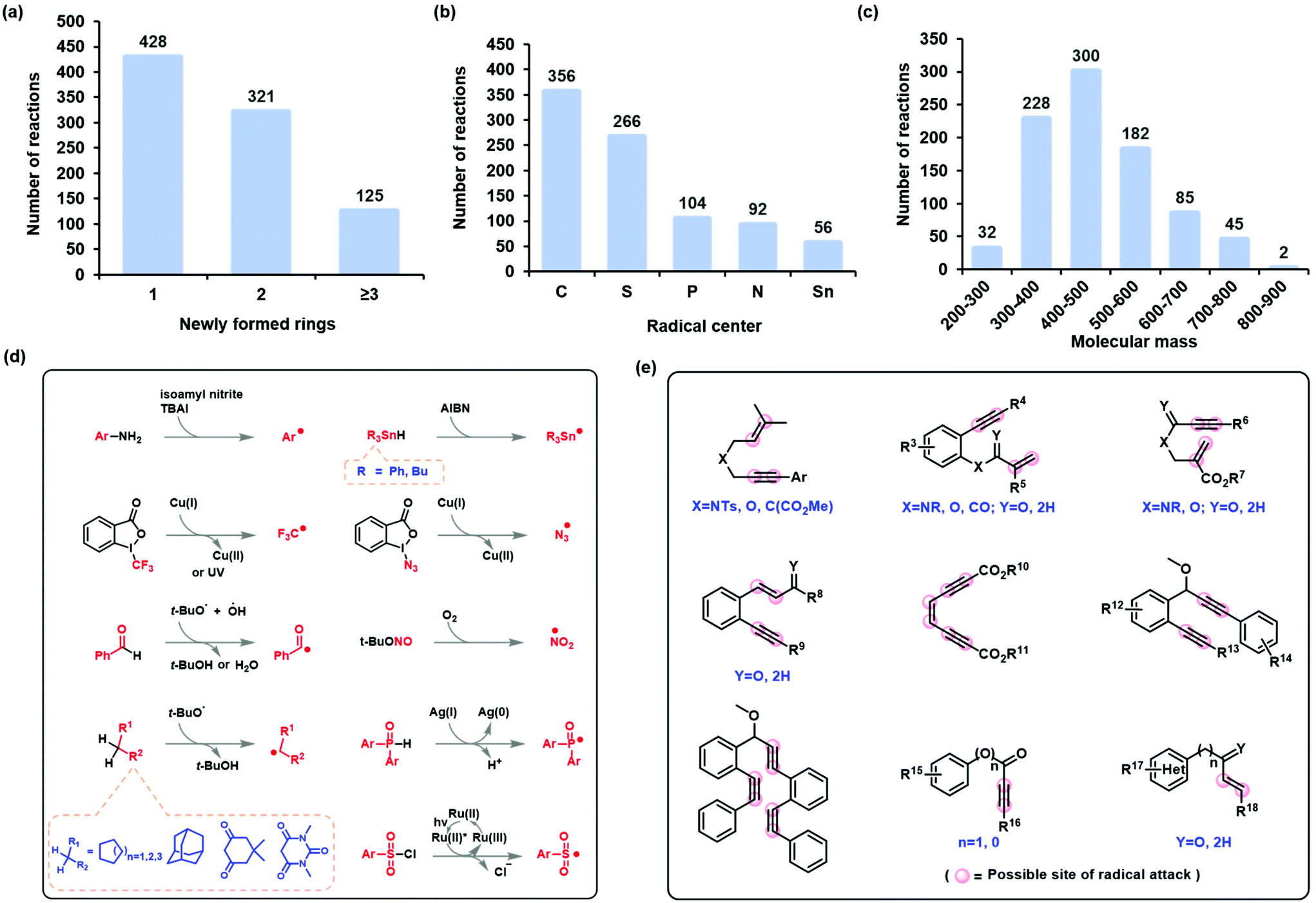 | ||
| Fig. 3 Diverse reactions in the self-built dataset. Distribution of (a) the number of newly formed rings, (b) radical centers, and (c) molecular mass of target products (carbocycle and heterocycle). (d) Typical ways of generating radicals and (e) radical acceptors with possible radical-attack sites. | ||
The pretraining dataset, or the general chemical reaction dataset, contains data regarding approximately 380![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 chemical reactions. These sample reactions were originally from Lowe's dataset,59 extracted from patents filed in the United States Patent and Trademark Office (USPTO), after deleting the reagents, solvent, temperature, and other reaction conditions. Subsequently, the data were filtered to eliminate duplicate, incorrect, and incomplete reactions. Note that each record in the general chemical reaction dataset represents a reaction with no intermediates, whereas the self-built dataset includes radical cascade cyclization and the key intermediates involved. However, if we consider the intermediates as specific types of products that contain radicals, the two datasets are quite similar in form.
000 chemical reactions. These sample reactions were originally from Lowe's dataset,59 extracted from patents filed in the United States Patent and Trademark Office (USPTO), after deleting the reagents, solvent, temperature, and other reaction conditions. Subsequently, the data were filtered to eliminate duplicate, incorrect, and incomplete reactions. Note that each record in the general chemical reaction dataset represents a reaction with no intermediates, whereas the self-built dataset includes radical cascade cyclization and the key intermediates involved. However, if we consider the intermediates as specific types of products that contain radicals, the two datasets are quite similar in form.
Construction of a deep learning model
In this study, we consider the intermediate prediction task as a machine translation problem between the reaction equation and the key intermediates. The Transformer model is a recent development in NLP and is a state-of-the-art model for reaction prediction and retrosynthesis.30,31,34,44 The self-attention mechanism, feed-forward network, and multi-head attention make the Transformer model well-suited for NLP tasks. The Transformer model based on the work from Duan et al.36,37 was applied in this study to predict the reaction intermediates (see section 5 of the ESI †for more information).Transfer learning is often used to adapt established source knowledge to learning tasks in target domains that are weakly labelled or unlabeled.31,34,36,37 In this study, the pretraining using the general chemical reaction dataset corresponded to the initialization or feature extraction tasks in deep learning. The pretraining process uses basic chemical information and features to accomplish the objective task of predicting the key intermediates in a radical cascade cyclization (Fig. 4). Then, the self-built dataset was randomly split into training, validation, and test datasets in the ratio 8:1:1. To verify the improvement in fine-tuning, the performances of the Transformer-baseline model without transfer learning and the model with transfer learning were compared.
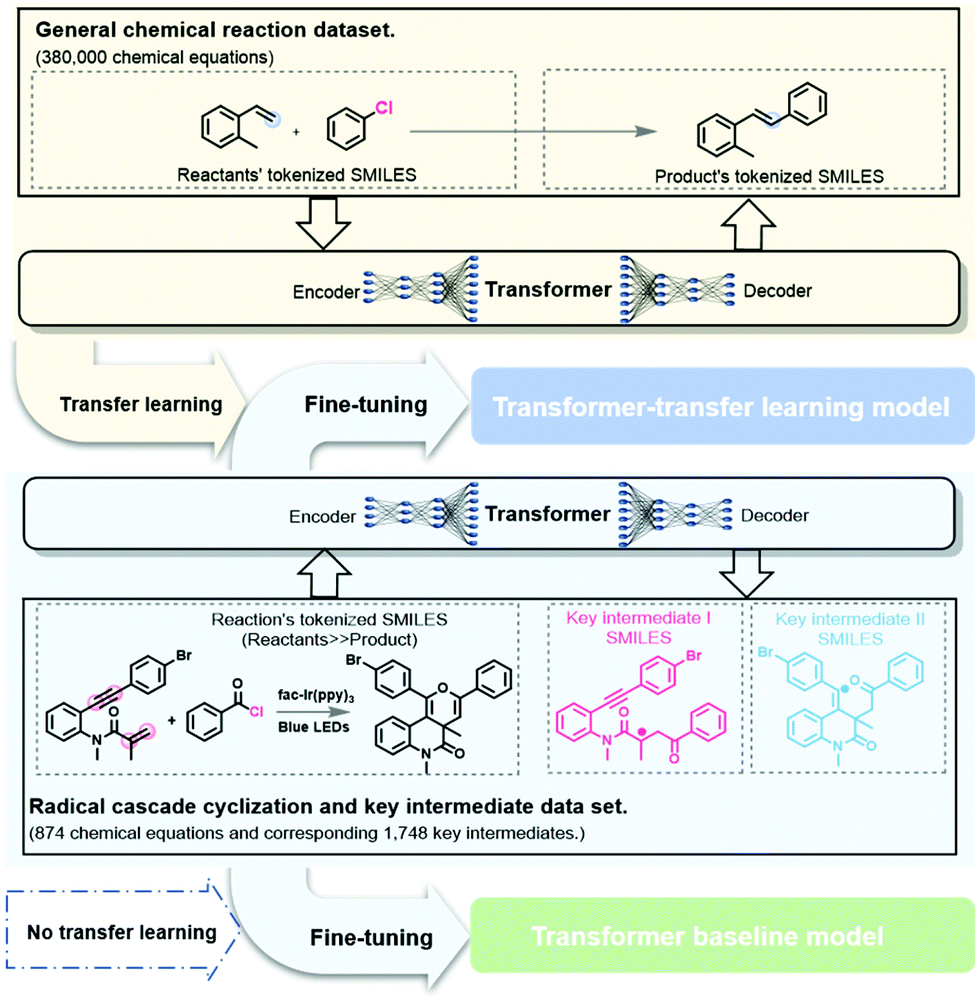 | ||
| Fig. 4 Schematic diagram of the method for the prediction of intermediates in radical cascade cyclizations. The transfer learning model refers to the pretrained version of the Transformer baseline model. The data features in the pretrained dataset and self-built dataset are different. | ||
Performance comparison of the baseline and transfer learning models
The accuracy of the prediction task, which was estimated with 10-fold cross-validation, was used to measure the performance. Fig. 5a–c indicate different perspectives regarding the accuracy of the baseline and transfer learning models for radical cascade cyclization. The Transformer-transfer model has an average accuracy of 94.5% for key intermediate I and 92.5% for key intermediate II, which is much higher than 29.7% and 26.6% associated with the baseline model (Fig. 5c, refer to Tables S15–18 of the ESI† for details). The predictions made by the Transformer-transfer learning model, which were based on the chemical reactivity rules and the knowledge that evolved from pretraining, were sufficiently accurate. This result validates our idealized hypothesis that the intermediates of the radical cascade cyclization are the specific products of the reaction, and this process is consistent with information obtained from the general chemical reaction data.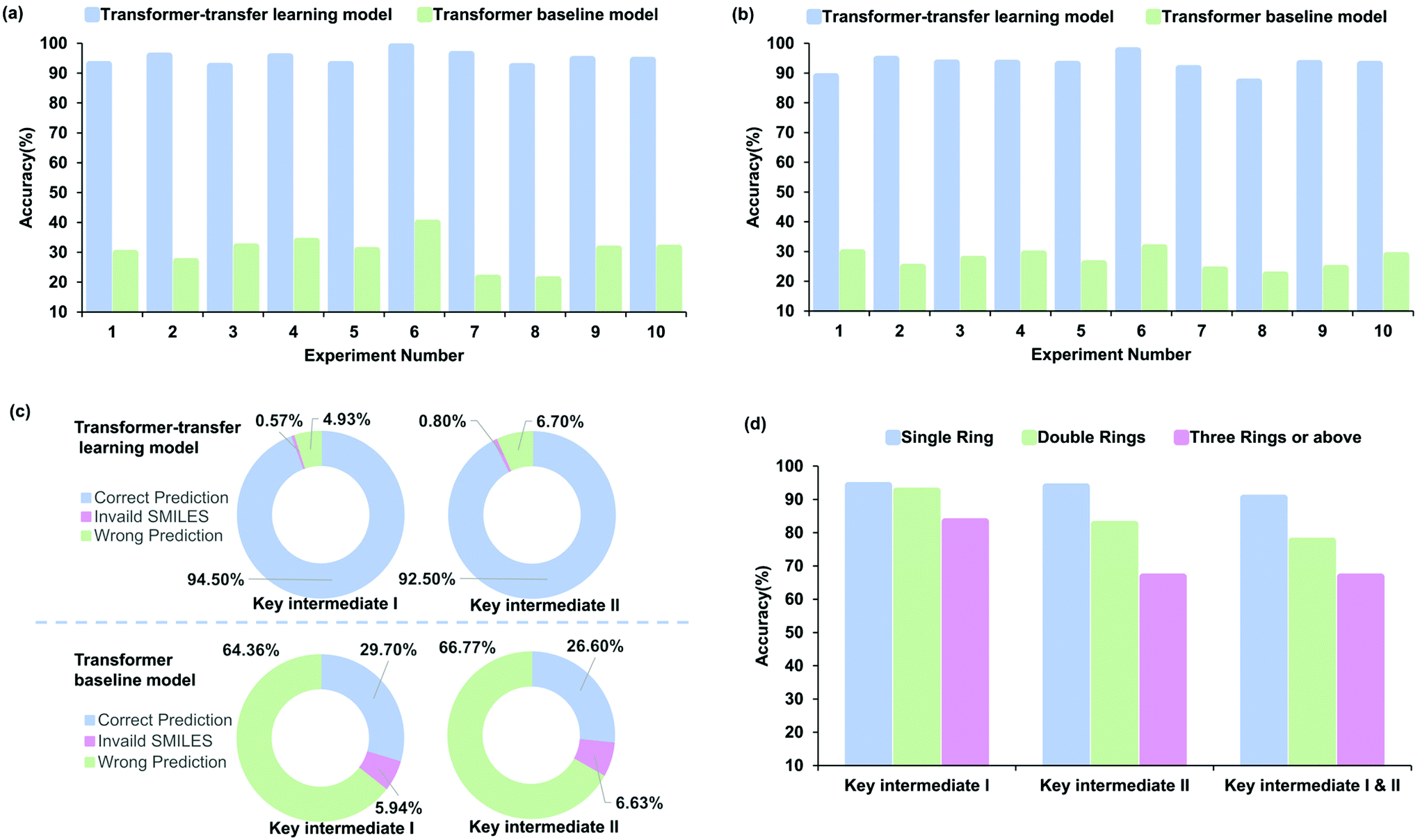 | ||
| Fig. 5 Comparison of baseline and transfer learning models. The results from experiments indicated that the Transformer-transfer learning model has considerable advantages in predicting key intermediate I (a) and key intermediate II (b). (c) Average accuracy of key-intermediate predictions, noting that the Transformer-transfer learning model (upper) does well in avoiding invalid SMILES. (d) Accuracy of the Transformer-transfer learning model for predicting reaction types based on the number of rings. As the number of rings increases, the accuracy decreases. | ||
Performance analysis of different intermediates
To identify the factors that affect the accuracy of intermediate formation predictions, we further explored several types of intermediates. For ease of analysis and understanding, the reactions were classified according to the number of newly formed rings. Experiment 1 (refer to entry 1 in Table S13 of the ESI†) involving 99 reactions was chosen as a representative example for the analysis. The prediction accuracy of different ring numbers obtained with the transfer learning model is shown in Fig. 5d. The number of rings and accuracy are related inversely. For the key intermediate I, the prediction accuracy was 94.2%, 92.5%, and 83.3%, respectively, with an increasing number of rings. To a certain content, the accuracy depends on the complexity of the regio- or stereoselectivity; training data become scarcer for cases with larger numbers of newly constructed rings. The result obtained for key intermediate II was similar, with the accuracy being 93.8%, 82.5%, and 66.7% for increasing ring numbers. The accuracy of key intermediate I was slightly higher than that of II at 92.9% and 88.8%, respectively. The probable reason for this phenomenon is that the key intermediate II is usually more complex than the key intermediate I, which increases the difficulty in identifying the reaction site. We trained the Transformer model to predict the two key intermediates simultaneously. Although the level of difficulty increased considerably, the results showed that there was no significant decrease in the performance, and the prediction accuracy was as high as 81.6%. As expected, the accuracy decreased with the number of rings as 90.4%, 77.5%, and 66.7%, respectively. This result showed the same trend as predicted for key intermediates I and II.Interpretability and analysis of attention weights
According to the Curtin–Hammett principle,60 in a reaction involving a pair of rapidly interconverting reactive intermediates or reactants, the priority for attacking the unsaturated bonds depends primarily on the structure of the radical acceptor. With the 1,n-enynes with non-terminal double bonds, the radical regioselectively attacks the α position of the alkyne initially, presumably because of the subsequent self-sorting kinetic processes via favorable exo-trig radical cyclization (Fig. 6a). In addition, if the radical accepter possesses terminal alkenes, the radicals are selectively added to the activated alkene moiety initially (δ position, Fig. 6b), although there are exceptions.61 This phenomenon may be attributed to the regioselectivity, since alkyne π-bonds are stronger and less reactive than alkene π-bonds, resulting in lower kinetic barriers being associated with the radical addition of double bonds.62,63 Although the rules applicable to complex reactions cannot be generalized easily, they can be indirectly elicited by a deep learning NLP model applied to an appropriate dataset and learned subsequently.28,33,40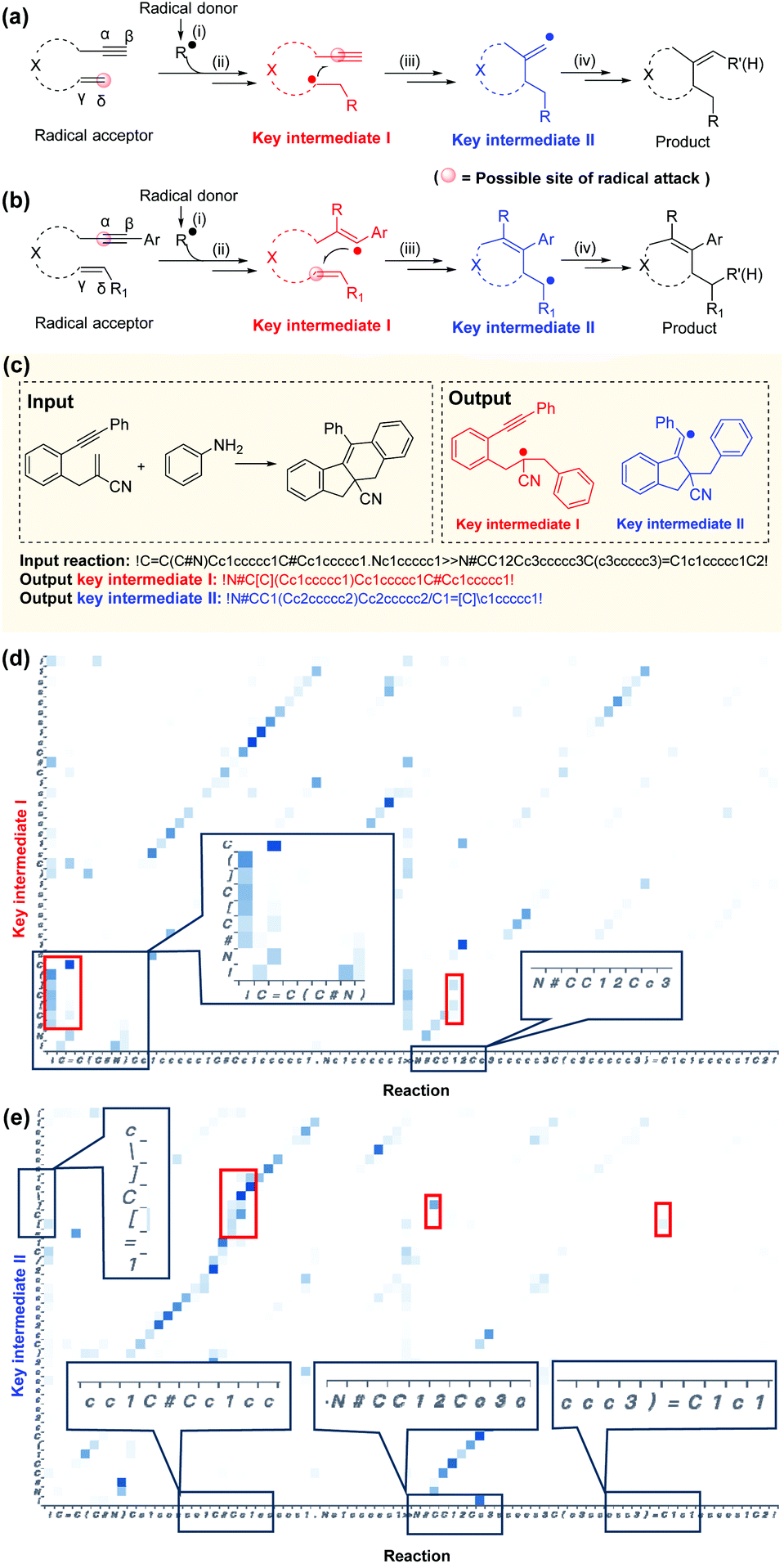 | ||
| Fig. 6 Interpretability and analysis of attention weights. The order of priority for attacking unsaturated bonds depends primarily on the structure of the radical acceptor: for 1,n-enyne radical acceptors with non-terminal double bonds, selective radicals attack the α position of the alkyne (a). For terminal alkenes, radical species (b) are first added regioselectively at the δ position. A random example (c) from the self-built dataset. Visualization of attention weights for key intermediate I (d) and key intermediate II (e) shows that the radical locations are concentrated on the unsaturated bonds of radical attack and the structure and location of functional group structures (circled in red). | ||
Good interpretability is critical for users to understand, trust, and manage powerful AI applications.64–67 Specific functional groups can affect the outcome of a reaction even if they are far from the reaction center in the molecular graph (3D) and the SMILES string (1D). Here, the Transformer-transfer learning model exhibited a mindset similar to that of an experienced chemist. A representative example of correctly predicted intermediates with attention weights is shown in Fig. 5c. Attention is a key factor in considering the complex long-range dependencies between multiple tokens.35 To predict key intermediate I (Fig. 6d), the Transformer model initially focused on the structures of both the reactants and products. Subsequently, the radical location considers the unsaturated bonds attacked by radicals, and the structure and location of nearby functional groups. Particular consideration was given to the position of double bonds, as terminal alkenes and nonterminal alkenes result in different intermediates. In this example, the token “!”, which represents “start” or “end”, placed before “C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C” indicates a terminal alkene, which will be subjected to the radical attack. The noted visualization shows that the radical location of intermediate I (where “[ ]” denotes the radical) is attached to this token, suggesting a similarity to the thought process of an experienced chemist. The observations made during the prediction of key intermediate II are also similar: the structures of the intermediates are based on both the reactants and products, as well as on the functional group near the alkyl group (Fig. 6e). Additional reaction predictions and attention weight details are available in the ESI.†
C” indicates a terminal alkene, which will be subjected to the radical attack. The noted visualization shows that the radical location of intermediate I (where “[ ]” denotes the radical) is attached to this token, suggesting a similarity to the thought process of an experienced chemist. The observations made during the prediction of key intermediate II are also similar: the structures of the intermediates are based on both the reactants and products, as well as on the functional group near the alkyl group (Fig. 6e). Additional reaction predictions and attention weight details are available in the ESI.†
Providing direction for mechanism inference
Successful prediction of the reaction intermediates can provide direction for mechanism inference methodologies. With the self-built dataset, enynes are the most common radical acceptors. There are two unsaturated bonds in enynes involved in radical cascade cyclization, and there are at least four possible sites for radical attacks. The Bu3Sn-mediated radical cascade cyclization of aromatic enynes was presented as a representative example (Fig. 7a) in which the regioselectivity of radical addition is difficult to be determined in the presence of electronically and sterically similar alkenes. However, the Transformer-transfer learning model performed well, even for such complex predictions. After the chemical equation is input, the deep-learning model outputs two key intermediates and indicates that path 2 is the most plausible route for the reaction (Fig. 7b). Alabugin et al.68 reported a plausible reaction pathway identified by computational analysis, and the result was in good agreement with the prediction of the Transformer model. However, our data-driven strategy is more convenient than the complex calculation requirements applicable to other methods.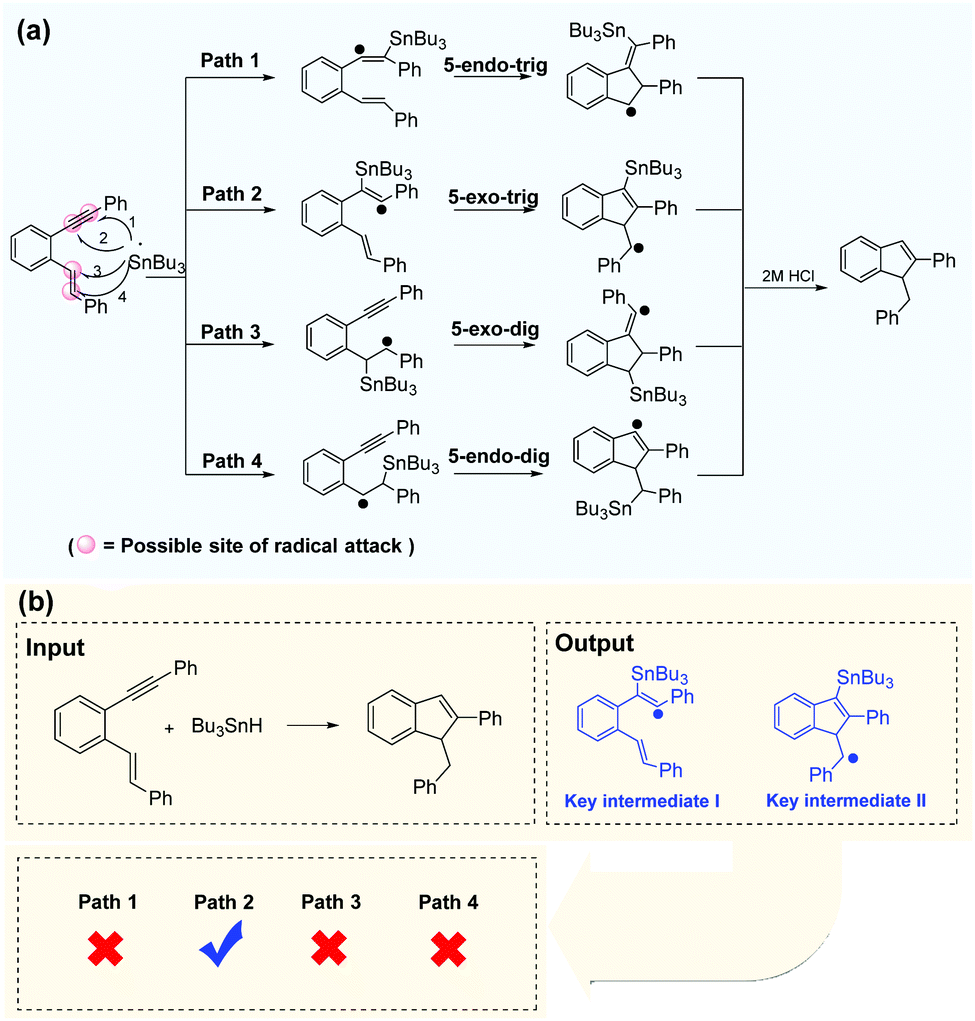 | ||
| Fig. 7 Efficient prediction of reaction pathways. (a) Regioselective attack on a disubstituted alkyne in the presence of electronically and sterically similar alkenes. (b) Path 2 is identified as the most reasonable pathway by the Transformer model, which is consistent with the computational analysis.68 | ||
Li et al.69 reported the radical cascade cyclization of 1,6-enynes with aryl sulfonyl chlorides by visible-light photoredox catalysis (Fig. 8a). A possible mechanism proposed by the authors is shown in Fig. 8b. However, the Transformer model provides additional information by identifying key intermediates I and II, implying that the reaction may have proceeded through a pathway (Fig. 8c) that differs from the mechanism described above. Here, excited [Ru(bpy)3]2+ and sulfonyl radicals are formed by selective cleavage of the S–Cl bond. Subsequent addition of the sulfonyl radicals to the terminal alkene produces tertiary alkyl radicals. The 5-exo-cyclization process leads to the formation of a vinyl radical that further undergoes 1,5-aryl migration and causes subsequent release of SO2 to generate the primary alkyl radical intermediate. The subsequent processing is the same as that inferred by Li et al.69 Further computational studies that we undertook focused on the competition between the two pathways. The DFT studies (Fig. 9, see the ESI† for details) and subsequent literature studies70–74 showed that aryl radicals are not readily generated in the visible-light-induced cyclization of 1,6-enynes with aryl sulfonyl groups, which was reported previously. Furthermore, owing to the difference in reactivity between the alkynes and alkenes, the alkyne π-bonds are stronger and less reactive than the alkene π-bonds.62,63 Thus, the addition of sulfonyl radicals to the terminal activated alkene to provide the tertiary alkyl radical is a more reasonable option (ΔG‡(298 K) = 13.4 vs. 29.9 kcal mol−1, Fig. 9). Hence, in this example, the mechanism indicated by our Transformer model is more reasonable.
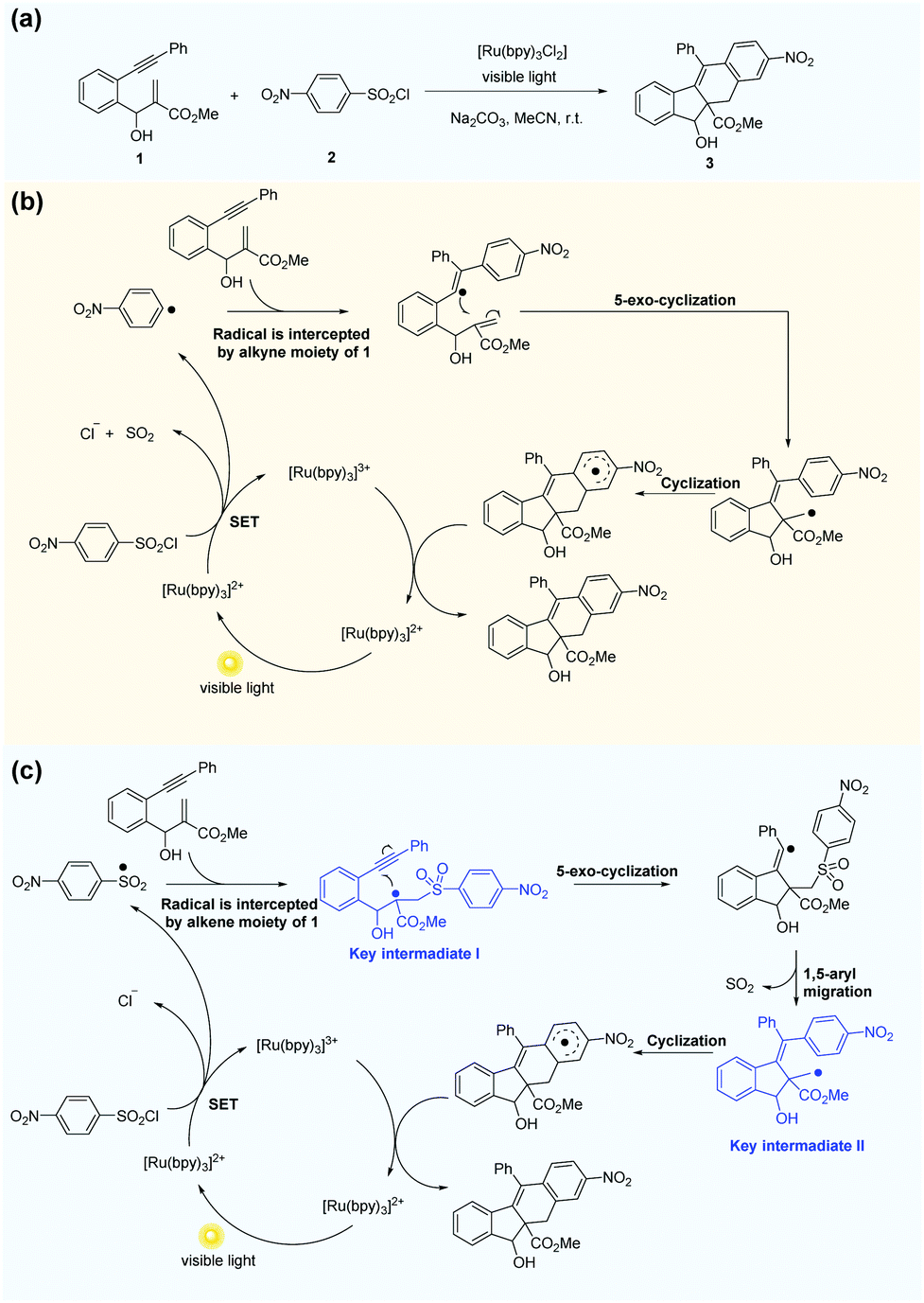 | ||
| Fig. 8 The Transformer model can revise some of the mechanism inference studies in the literature. (a) Radical cascade cyclization of alkynes with alkenes and ArSO2Cl. (b) Possible mechanism inferred by Li et al.69 (c) Possible mechanism proposed by the Transformer model. | ||
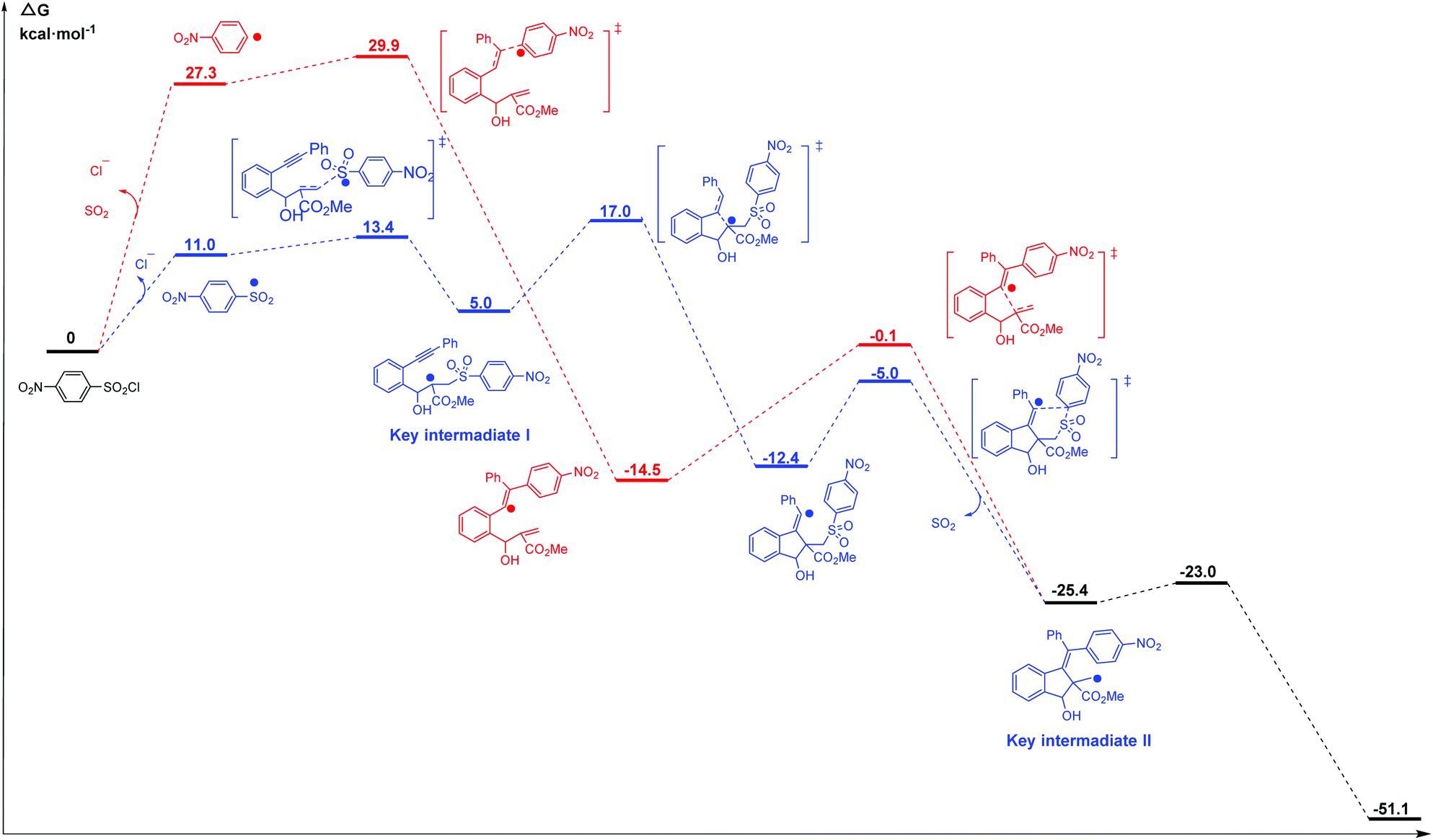 | ||
| Fig. 9 Calculated free energy profiles for the two possible mechanisms (pathway proposed by Li et al. (red) and pathway proposed with the Transformer model (blue)), ΔG values (in kcal mol−1) are calculated at 298 K. | ||
Conclusions
As a proof-of-concept, a Transformer model was applied to predict intermediates, providing direction for the mechanistic inference of organic reactions. By defining key intermediates I and II, a dataset of radical cascade cyclization reactions was curated to train the model. While the dataset format is quite different between pretraining and fine-tuning, transfer learning was used to further overcome the scarcity of training data. The observed improvements demonstrated the potential of integrating transfer learning and Transformer models. Attention weight analysis indicated that the Transformer model exhibited a mindset similar to that of an experienced chemist. The application of the Transformer model to the end-to-end prediction of intermediates in radical cascade cyclization reactions is effective. As data continue to be curated and deep learning algorithms continue to be upgraded, we expect that this work will further encourage the broader chemical community to engage in this emerging field and facilitate AI-accelerated chemistry.Methods
Transformer model
The Transformer model is entirely based on an attention mechanism and avoids the recursion most commonly used in encoder–decoder architectures.75 The Transformer model based on the work from Duan et al.36,37,58 was applied to predict the reaction intermediates. An atom-based tokenization strategy is employed which reduces the vocabulary size of most representations of organic molecules to a relatively small and constant size. Hyperparameters were tuned based on our previous experience.36,37,58 The following hyperparameters were used for key intermediate prediction:Adam optmizer: beta1 = 0.9, beta2 = 0.997.
epsilon = 1e − 9
n_heads = 8
emb_dim = 256
num_layers = 6
FFN_inner_units = 2048
dropout = 0.3
Validation
Canonicalized SMILES of predicted and actual intermediates were compared to assess the accuracy of the model. During testing/validation, there is no significant difference between top-1 and top-k in our experiments due to sharp distribution (high confidence) and small vocabulary size (small search space). Therefore, we use the top-1 accuracy as the metric of our work.Density functional theory calculations
All density functional theory (DFT) calculations were performed using the DMol3 module.76,77 The generalized gradient approximation (GGA) method with the Perdew–Burke–Ernzerhof (PBE) function was performed to simulate the molecules. A global orbital cutoff of 4.5 Å was used. The force and energy convergence criteria were set to 0.002 Ha Å−1 and 10−5 Ha, respectively.Availability of data and materials
The dataset (pretraining and self-built) and the code are available from: https://github.com/hongliangduan/transRCCAuthor contributions
These authors contributed equally: J. X., Y. Z. and J. H. W. S. and H. D. designed the research project. J. X., Y. Z., B. S., J. H., J. T., X. S., S. Z., W. Y., and X. W. collected literature and established a self-built dataset. H. Q., C. Z., and S. Z. designed and trained the models. J. X. and A. S. analysed data and wrote the manuscript. All authors discussed the results and approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the National Natural Science Foundation of China (No. 81903438). We are grateful for the Zhejiang Provincial Key R&D Project (No. 2020C03006 & 2019-ZJ-JS-03) and acknowledge the Natural Science Foundation of Zhejiang Province (LD22H300004).Notes and references
- C. Vallance, Physical chemistry: The finger of reaction mechanism, Nature, 2017, 546, 608–609 CrossRef CAS PubMed.
- T. Deb, J. Tu and R. M. Franzini, Mechanisms and substituent effects of metal-free bioorthogonal reactions, Chem. Rev., 2021, 121, 6850–6914 CrossRef CAS PubMed.
- M. Reiher, N. Wiebe, K. M. Svore, D. Wecker and M. Troyer, Elucidating reaction mechanisms on quantum computers, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7555–7560 CrossRef CAS PubMed.
- V. Gold, Organic reaction mechanisms, Nature, 1977, 267, 471–472 CrossRef.
- L. A. Perego, R. Blieck, A. Groué, F. Monnier, M. Taillefer, I. Ciofini and L. Grimaud, Copper-catalyzed hydroamination of allenes: from mechanistic understanding to methodology development, ACS Catal., 2017, 7, 4253–4264 CrossRef CAS.
- P. H. Cheong, C. Y. Legault, J. M. Um, N. Celebi-Olcum and K. N. Houk, Quantum mechanical investigations of organocatalysis: mechanisms, reactivities, and selectivities, Chem. Rev., 2011, 111, 5042–5137 CrossRef CAS PubMed.
- Q. Peng and R. S. Paton, Catalytic control in cyclizations: From computational mechanistic understanding to selectivity prediction, Acc. Chem. Res., 2016, 49, 1042–1051 CrossRef CAS PubMed.
- Q. Li, Y. Zhao, C. Ling, S. Yuan, Q. Chen and J. Wang, Towards a comprehensive understanding of the reaction mechanisms between defective MoS2 and thiol molecules, Angew. Chem., Int. Ed., 2017, 56, 10501–10505 CrossRef CAS PubMed.
- T. N. Nguyen, J. Guo, A. Sachindran, F. Li, A. Seifitokaldani and C. T. Dinh, Electrochemical CO2 reduction to ethanol: from mechanistic understanding to catalyst design, J. Mater. Chem. A, 2021, 9, 12474–12494 RSC.
- F. Y. Chen, Z. Y. Wu, Z. Adler and H. Wang, Stability challenges of electrocatalytic oxygen evolution reaction: From mechanistic understanding to reactor design, Joule, 2021, 5, 1704–1731 CrossRef CAS.
- T. C. Malig, D. Yu and J. E. Hein, A revised mechanism for the Kinugasa reaction, J. Am. Chem. Soc., 2018, 140, 9167–9173 CrossRef CAS PubMed.
- O. T. Unke, S. Chmiela, H. E. Sauceda, M. Gastegger, I. Poltavsky, K. T. Schutt, A. Tkatchenko and K. R. Muller, Machine learning force fields, Chem. Rev., 2021, 121, 10142–10186 CrossRef CAS PubMed.
- K. Wang, H. Lu, X. Zhu, Y. Lin, M. C. Beard, Y. Yan and X. Chen, Ultrafast reaction mechanisms in perovskite based photocatalytic C–C coupling, ACS Energy Lett., 2020, 5, 566–571 CrossRef CAS.
- K. Wang, Z. Bielan, M. E. Kimura, M. Janczarek, D. Zhang, D. Kowalski, A. Z. Jurek, A. M. Szczupak, B. Ohtani and E. Kowalska, On the mechanism of photocatalytic reactions on CuxO@TiO2 core–shell photocatalysts, J. Mater. Chem. A, 2021, 9, 10135–10145 RSC.
- G. Rózsa, M. Náfrádi, T. Alapi, K. Schrantz, L. Szabó, L. Wojnárovits, E. Takács and A. Tungler, Photocatalytic, photolytic and radiolytic elimination of imidacloprid from aqueous solution: Reaction mechanism, efficiency and economic considerations, Appl. Catal., B, 2019, 250, 429–439 CrossRef.
- M. K. Meadows, X. Sun, I. V. Kolesnichenko, C. M. Hinson, K. A. Johnson and E. V. Anslyn, Mechanistic studies of a “Declick” reaction, Chem. Sci., 2019, 10, 8817–8824 RSC.
- M. Pelckmans, T. Mihaylov, W. Faveere, J. Poissonnier, F. V. Waes, K. Moonen, G. B. Marin, J. W. Thybaut, K. Pierloot and B. F. Sels, Catalytic reductive aminolysis of reducing sugars: Elucidation of reaction mechanism, ACS Catal., 2018, 8, 4201–4212 CrossRef CAS.
- B. G. Stevenson, E. H. Spielvogel, E. A. Loiaconi, V. M. Wambua, R. V. Nakhamiyayev and J. R. Swierk, Mechanistic investigations of an alpha-aminoarylation photoredox reaction, J. Am. Chem. Soc., 2021, 143, 8878–8885 CrossRef CAS PubMed.
- R. Feng, J. A. Smith and K. D. Moeller, Anodic cyclization reactions and the mechanistic strategies that enable optimization, Acc. Chem. Res., 2017, 50, 2346–2352 CrossRef CAS PubMed.
- D. Koner, S. M. Salehi, P. Mondal and M. Meuwly, Non-conventional force fields for applications in spectroscopy and chemical reaction dynamics, J. Chem. Phys., 2020, 153, 010901 CrossRef CAS PubMed.
- K. Jorner, T. Brinck, P. Norrby and D. Buttar, Machine learning meets mechanistic modelling for accurate prediction of experimental activation energies, Chem. Sci., 2021, 12, 1163–1175 RSC.
- F. Planas, M. J. McLeish and F. Himo, Enzymatic stetter reaction: Computational study of the reaction mechanism of MenD, ACS Catal., 2021, 11, 12355–12366 CrossRef CAS.
- S. Manzhos and T. Carrington, Neural network potential energy surfaces for small molecules and reactions, Chem. Rev., 2021, 121, 10187–10217 CrossRef CAS PubMed.
- M. Meuwly, Machine learning for chemical reactions, Chem. Rev., 2021, 121, 10218–10239 CrossRef CAS PubMed.
- G. B. Goh, N. O. Hodas and A. Vishnu, Deep learning for computational chemistry, J. Comput. Chem., 2017, 38, 1291–1307 CrossRef CAS PubMed.
- M. Bragato, G. F. von Rudorff and O. A. von Lilienfeld, Data enhanced Hammett-equation: reaction barriers in chemical space, Chem. Sci., 2020, 11, 11859–11868 RSC.
- S. Heinen, G. F. von Rudorff and O. A. von Lilienfeld, Toward the design of chemical reactions: Machine learning barriers of competing mechanisms in reactant space, J. Chem. Phys., 2021, 155, 064105 CrossRef CAS PubMed.
- J. A. Keith, V. V. Galindo, B. Cheng, S. Chmiela, M. Gastegger, K.-R. Müller and A. Tkatchenko, Combining machine learning and computational chemistry for predictive insights into chemical systems, Chem. Rev., 2021, 121, 9816–9872 CrossRef CAS PubMed.
- H. Li, Deep learning for natural language processing: advantages and challenges, Natl. Sci. Rev., 2018, 5, 22–24 CrossRef.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction, ACS Cent. Sci., 2019, 5, 1572–1583 CrossRef CAS PubMed.
- D. Kreutter, P. Schwaller and J.-L. Reymond, Predicting enzymatic reactions with a molecular transformer, Chem. Sci., 2021, 12, 8648–8659 RSC.
- E. M. Gale and D. J. Durand, Improving reaction prediction, Nat. Chem., 2020, 12, 509–510 CrossRef CAS PubMed.
- P. Schwaller, B. Hoover, J.-L. Reymond, H. Strobelt and T. Laino, Extraction of organic chemistry grammar from unsupervised learning of chemical reactions, Sci. Adv., 2021, 7, eabe4166 CrossRef PubMed.
- G. Pesciullesi, P. Schwaller, T. Laino and J.-L. Reymond, Transfer learning enables the molecular transformer to predict regio-and stereoselective reactions on carbohydrates, Nat. Commun., 2020, 11, 4874 CrossRef CAS PubMed.
- P. Schwaller, T. Gaudin, D. Lanyi, C. Bekas and T. Laino, “Found in Translation”: predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models, Chem. Sci., 2018, 9, 6091–6098 RSC.
- Y. Zhang, L. Wang, X. Wang, C. Zhang, J. Ge, J. Tang, A. Su and H. Duan, Data augmentation and transfer learning strategies for reaction prediction in low chemical data regimes, Org. Chem. Front., 2021, 8, 1415–1423 RSC.
- L. Wang, C. Zhang, R. Bai, J. Li and H. Duan, Heck reaction prediction using a transformer model based on a transfer learning strategy, Chem. Commun., 2020, 56, 9368–9371 RSC.
- A. C. Vaucher, F. Zipoli, J. Geluykens, V. H. Nair, P. Schwaller and T. Laino, Automated extraction of chemical synthesis actions from experimental procedures, Nat. Commun., 2020, 11, 3601 CrossRef PubMed.
- A. C. Vaucher, P. Schwaller, J. Geluykens, V. H. Nair, A. Iuliano and T. Laino, Inferring experimental procedures from text-based representations of chemical reactions, Nat. Commun., 2021, 12, 2573 CrossRef CAS PubMed.
- B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, Retrosynthetic reaction prediction using neural sequence-to-sequence models, ACS Cent. Sci., 2017, 3, 1103–1113 CrossRef CAS PubMed.
- H. Duan, L. Wang, C. Zhang, L. Guo and J. Li, Retrosynthesis with attention-based NMT model and chemical analysis of “wrong” predictions, RSC Adv., 2020, 10, 1371–1378 RSC.
- K. Lin, Y. Xu, J. Pei and L. Lai, Automatic retrosynthetic route planning using template-free models, Chem. Sci., 2020, 11, 3355–3364 RSC.
- P. Schwaller, R. Petraglia, V. Zullo, V. H. Nair, R. A. Haeuselmann, R. Pisoni, C. Bekas, A. Iuliano and T. Laino, Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy, Chem. Sci., 2020, 11, 3316–3325 RSC.
- I. V. Tetko, P. Karpov, R. Van Deursen and G. Godin, State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis, Nat. Commun., 2020, 11, 5575 CrossRef CAS PubMed.
- X. Wang, Y. Li, J. Qiu, G. Chen, H. Liu, B. Liao, C.-Y. Hsieh and X. Yao, RetroPrime: A diverse, plausible and Transformer-based method for Single-Step retrosynthesis predictions, Chem. Eng. J., 2021, 420, 129845 CrossRef CAS.
- J. Xuan and A. Studer, Radical cascade cyclization of 1,n-enynes and diynes for the synthesis of carbocycles and heterocycles, Chem. Soc. Rev., 2017, 46, 4329–4346 RSC.
- J. Liao, X. Yang, L. Ouyang, Y. Lai, J. Huang and R. Luo, Recent advances in cascade radical cyclization of radical acceptors for the synthesis of carbo- and heterocycles, Org. Chem. Front., 2021, 8, 1345–1363 RSC.
- X. Y. Liu and Y. Qin, Indole Alkaloid Synthesis Facilitated by Photoredox Catalytic Radical Cascade Reactions, Acc. Chem. Res., 2019, 52, 1877–1891 CrossRef CAS PubMed.
- J. K. Qiu, B. Jiang, Y. L. Zhu, W. J. Hao, D. C. Wang, J. Sun, P. Wei, S. J. Tu and G. Li, Catalytic dual 1,1-H-abstraction/insertion for domino spirocyclizations, J. Am. Chem. Soc., 2015, 137, 8928–8931 CrossRef CAS PubMed.
- Y. Li, B. Liu, R. J. Song, Q. A. Wang and J. H. Li, Visible light-initiated C(sp3)-Br/C(sp3)-H functionalization of α-carbonyl alkyl bromides through hydride radical shift, Adv. Synth. Catal., 2016, 358, 1219–1228 CrossRef CAS.
- S. Zhou, S. Bommezjin and A. M. John, Formal total synthesis of (±)-vindoline by tandem radical cyclization, Org. Lett., 2002, 4, 443–445 CrossRef CAS PubMed.
- L. Liu, R. M. Ward and J. M. Schomaker, Mechanistic aspects and synthetic applications of radical additions to allenes, Chem. Rev., 2019, 119, 12422–12490 CrossRef CAS PubMed.
- PubChem. https://pubchem.ncbi.nlm.nih.gov, accessed January 28, 2022.
- ChEMBL. https://www.ebi.ac.uk/chembl/, accessed January 28, 2022.
- Reaxys. https://www.reaxys.com/, accessed January 28, 2022.
- SciFinder. https://scifinder.cas.org, accessed January 28, 2022.
- H. Ozturk, A. Ozgur, P. Schwaller, T. Laino and E. Ozkirimli, Exploring chemical space using natural language processing methodologies for drug discovery, Drug Discovery Today, 2020, 25, 689–705 CrossRef PubMed.
- Y. Wu, C. Zhang, L. Wang and H. Duan, A graph-convolutional neural network for addressing small-scale reaction prediction, Chem. Commun., 2021, 57, 4114–4117 RSC.
- D. M. Lowe, Chemical reactions from US patents (1976-Sep2016); https://figshare.com/articles/Chemical_reactions_from_US_patents_1976-Sep2016_/5104873, accessed January 12, 2022.
- T. Foldes, A. Madarasz, A. Revesz, Z. Dobi, S. Varga, A. Hamza, P. R. Nagy, P. M. Pihko and I. Papai, Stereocontrol in diphenylprolinol silyl ether catalyzed michael additions: steric shielding or Curtin−Hammett scenario, J. Am. Chem. Soc., 2017, 139, 17052–17063 CrossRef CAS PubMed.
- R. Fu, W. Hao, Y. Wu, N. Wang, S. Tu, G. Li and B. Jiang, Sulfonyl radical-enabled 6-endo-trig cyclization for regiospecific synthesis of unsymmetrical diaryl sulfones, Org. Chem. Front., 2016, 3, 1452–1456 RSC.
- R. K. Mohamed, S. Mondal, B. Gold, C. J. Evoniuk, T. Banerjee, K. Hanson and I. V. Alabugin, Alkenes as alkyne equivalents in radical cascades terminated by fragmentations: overcoming stereoelectronic restrictions on ring expansions for the preparation of expanded polyaromatics, J. Am. Chem. Soc., 2015, 137, 6335–6349 CrossRef CAS PubMed.
- I. V. Alabugin and E. Gonzalez-Rodriguez, Alkyne origami: Folding oligoalkynes into polyaromatics, Acc. Chem. Res., 2018, 51, 1206–1219 CrossRef CAS PubMed.
- D. Gunning, M. Stefik, J. Choi, T. Miller, S. Stumpf and G. Z. Yang, XAI-Explainable artificial intelligence, Sci. Rob., 2019, 4, eaay7120 CrossRef PubMed.
- J. Stoyanovich, J. J. Van Bavel and T. V. West, The imperative of interpretable machines, Nat. Mach. Intell., 2020, 2, 197–199 CrossRef.
- L. Kohoutova, J. Heo, S. Cha, S. Lee, T. Moon, T. D. Wager and C. W. Woo, Toward a unified framework for interpreting machine-learning models in neuroimaging, Nat. Protoc., 2020, 15, 1399–1435 CrossRef CAS PubMed.
- D. P. Kovacs, W. McCorkindale and A. A. Lee, Quantitative interpretation explains machine learning models for chemical reaction prediction and uncovers bias, Nat. Commun., 2021, 12, 1695 CrossRef CAS PubMed.
- S. Mondal, R. K. Mohamed, M. Manoharan, H. Phan and I. V. Alabugin, Drawing from a pool of radicals for the design of selective enyne cyclizations, Org. Lett., 2013, 15, 5650–5653 CrossRef CAS PubMed.
- G. B. Deng, Z. Q. Wang, J. D. Xia, P. C. Qian, R. J. Song, M. Hu, L. B. Gong and J. H. Li, Tandem cyclizations of 1,6-enynes with arylsulfonyl chlorides by using visible-light photoredox catalysis, Angew. Chem., Int. Ed., 2013, 52, 1535–1538 CrossRef CAS PubMed.
- C. J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. Stephenson, Visible light-mediated atom transfer radical addition via oxidative and reductive quenching of photocatalysts, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed.
- H. Jiang, X. Chen, Y. Zhang and S. Yu, C-H Functionalization of Enamides: Synthesis of b-Amidovinyl Sulfones via Visible-Light Photoredox Catalysis, Adv. Synth. Catal., 2013, 355, 809–813 CrossRef CAS.
- X. Meng, Q. Kang, J. Zhang, Q. Li, W. Wei and W. He, Visible-light-initiated regioselective sulfonylation/cyclization of 1,6-enynes under photocatalyst- and additive-free conditions, Green Chem., 2020, 22, 1388–1392 RSC.
- T. H. Zhu, X. C. Zhang, X. L. Cui, Z. Y. Zhang, H. Jiang, S. S. Sun, L. L. Zhao, K. Zhao and T. P. Loh, Direct C(sp2)-H arylsulfonylation of enamides via iridium(III)-catalyzed insertion of sulfur dioxide with aryldiazonium tetrafluoroborates, Adv. Synth. Catal., 2019, 361, 3593–3598 CrossRef CAS.
- L. Chen, M. Zhou, L. Shen, X. He, X. Li, X. Zhang and Z. Lian, Metal-and base-Free C(sp(2))-H arylsulfonylation of enamides for synthesis of (E)-beta-amidovinyl sulfones via the insertion of sulfur dioxide, Org. Lett., 2021, 23, 4991–4996 CrossRef CAS PubMed.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, L. Kaiser and I. Polosukhin, Attention is all you need, 2017, arXiv:1706.03762.
- B. Delley, From molecules to solids with the DMol3 approach, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- T. Kar and S. Scheiner, Proton and lithium ion transfer between two water molecules with an external restraining force, J. Am. Chem. Soc., 1995, 117, 1344–1351 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed computational methods, full description of deep learning workflow and results, and analysis of “wrong” predictions. See DOI: https://doi.org/10.1039/d2qo00188h |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2022 |