
Highly efficient electrochemical CO2 reduction over crystalline–amorphous In2O3–CeOx heterostructures†
Cuifeng
Wang
,
Zhaohui
Wu
,
Guihao
Liu
,
Sha
Bai
,
Lin
Guo
,
Lei
He
 * and
Yu-Fei
Song
*
* and
Yu-Fei
Song
*
State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: helei@mail.buct.edu.cn; songyf@mail.buct.edu.cn; Fax: +86 10 64431832; Tel: +86 10 64431832
First published on 30th September 2022
Abstract
The electrochemical reduction reaction of CO2 (CO2RR) to fuels and chemicals is a promising approach to consume greenhouse gases and mitigate the dependence on fossil fuels. Herein, we synthesized an effective crystalline–amorphous In2O3–CeOx heterostructure, which exhibited high catalytic performance for CO2-to-formate conversion. The maximum faradaic efficiency (FE) of 94.8% was achieved, and above 90% FE can be maintained in a wide potential range from −0.8 to −1.2 V vs. RHE. Detailed studies showed that In2O3 functioned as the active site for CO2 activation and formate formation, and the amorphous CeOx was beneficial for the electron transfer, leading to the electronic structure reconfiguration of In2O3. Hence, the In2O3–CeOx heterostructure enhanced the adsorption of *OCHO intermediates and lowered the energy barrier forming *HCOOH from *OCHO.
Introduction
The electrochemical reduction reaction of CO2 (CO2RR) into value-added chemicals and fuels is a promising approach to consume excess industrial CO2 emissions and address the current ongoing consumption of non-renewable fossil fuels.1–4 Nevertheless, there are still problems restricting the practical application of the CO2RR. For example, CO2 gas molecules are thermodynamically stable, and the complexity of the reaction pathway as well as the competitive hydrogen evolution reaction (HER) leads to diverse products and low selectivity. Considering the cost of electricity and the product's market price, the two-electron transfer products formate and CO are the most promising target products among the numerous CO2 reduction products.5 Formate is one of the fundamental starting materials for industrial production, which is widely used in the fields of pesticides, dyes, medicine, rubber and tanning industries.6 As such, efficient catalysts need to be developed to lower the energy barrier and improve the selectivity for converting CO2 to formate.The p-block metals such as In, Bi, Pb, and Sn are efficient catalysts for CO2 electroreduction due to their high overpotential for the HER.7–10 Among them, low-toxicity and environmentally friendly In-based materials show high selectivity to formate.11 Various strategies on In-based catalysts have been proposed such as morphology design,12 defect engineering,13 doping,14 and alloying15 to reinforce the catalytic activities. Although these strategies are ingenious in design, they still suffer from cumbersome synthetic procedures, and the electrochemical performance still needs to be further improved. As such, the development of novel In-based catalysts is promising for electroreduction of CO2 to formate.
Constructing heterostructures is an effective strategy to enhance the electrocatalytic performance due to their efficient charge transfer at the interface and the sufficiently exposed catalytically active sites.16–18 In particular, heterostructures can direct high selectivity toward a specific product in complicated CO2RR reaction routes via adjusting the binding strength with key intermediates.19,20 CeO2-based materials possess unique transformation characteristics between Ce3+ and Ce4+ states, which are beneficial for the gain and loss of electrons,21 and have been proved to significantly strengthen the adsorption and promote the activation of CO2 molecules. These properties make CeO2 suitable for constructing heterostructures to enhance the CO2 conversion process. For example, Bao et al.22 have demonstrated that the CO2RR can be significantly improved by the interface of Au–CeOx heterostructures. Gong et al.23 have revealed that high Ce3+ concentration is beneficial for CO2 activation. Notably, in comparison with their crystalline–crystalline counterparts, crystalline–amorphous heterostructures can inherit the merits of amorphous structures, such as abundant active sites and defects, high flexibility, and better corrosion resistance.24,25 Therefore, rational design of crystalline–amorphous heterostructures is an effective method to achieve the purpose of improving CO2RR performance.
Herein, crystalline–amorphous In2O3–CeOx heterostructures were fabricated for the first time and were used as an electrocatalyst for the CO2RR. It was discovered that the maximum faradaic efficiency (FE) can reach 94.8% at −0.9 V vs. RHE over In2O3–CeOx, which was obviously more efficient than those of a pure In2O3 catalyst and a crystalline–crystalline In2O3–CeO2 heterostructure. The improved performance of the In2O3–CeOx catalyst was attributed to the electronic reconfiguration of indium by strengthened electron transfer between In2O3 and amorphous CeOx, which significantly enhanced the adsorption of *OCHO intermediates and lowered the energy barrier of *OCHO → *HCOOH, leading to the high activity and selectivity for the CO2RR to formate.
Results and discussion
The In2O3–CeOx catalyst was synthesized via two steps (Fig. S1a†). Firstly, an aqueous solution containing indium nitrate, sodium carbonate and as-prepared CeOx was stirred to form an In(OH)3–CeOx precipitate for one hour at room temperature. Secondly, the precursor In(OH)3–CeOx was annealed under an Ar/H2 flow for one hour to obtain the In2O3–CeOx catalyst. The structures of the samples (e.g., In2O3–CeOx, In2O3–CeO2, the individual CeOx, CeO2 and In2O3) were determined using X-ray diffraction (XRD) patterns. As shown in Fig. 1a, no obvious peak was observed for CeOx, suggesting that the amorphous structure was successfully achieved. In addition, no obvious peak of In2O3 was observed in the In2O3–CeOx sample, which was caused by the small particle size and uniform dispersion of In2O3. In contrast, CeO2 displayed the characteristic peaks of the crystal structure, which were consistent with those reported in the literature (Fig. 1b).26 The peaks of In2O3–CeO2 matched well with the standard patterns of CeO2 and In2O3, as shown in Fig. 1b. The high-resolution transmission electron microscopy (HR-TEM) images showed the crystalline–amorphous heterostructure interface of In2O3–CeOx and crystalline–crystalline heterostructure interface of In2O3–CeO2 (Fig. 1c–f). As shown in Fig. 1d, the lattice fringe of In2O3–CeOx could be assigned to the (222) plane of In2O3 while the amorphous CeOx did not show any lattice stripes. In contrast, the HR-TEM image of In2O3–CeO2 clearly presented the lattice fringes for the (222) facet of In2O3 and (111) facet of CeO2 (Fig. 1f). Furthermore, energy dispersive X-ray spectroscopy (EDX) elemental mapping of the samples showed that In, Ce, and O were evenly distributed (Fig. 1g–j and S2†), confirming the successful preparation of In2O3–CeOx and In2O3–CeO2 heterostructures.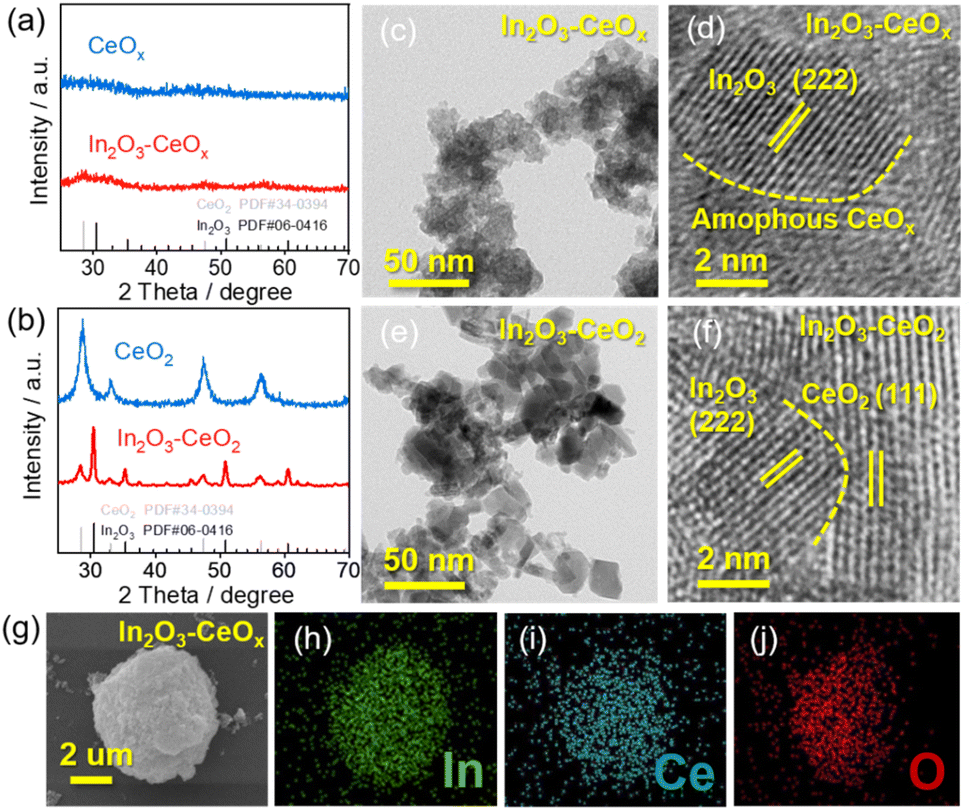 | ||
| Fig. 1 (a) XRD patterns of CeOx and In2O3–CeOx, (b) XRD patterns of CeO2 and In2O3–CeO2, (c and d) HR-TEM image of In2O3–CeOx and (e and f) HR-TEM image of In2O3–CeO2. (g–j) The corresponding EDX elemental mapping of In2O3CeOx. | ||
X-ray photoelectron spectroscopy (XPS) was utilized to investigate the electronic properties and chemical compositions of the samples. The Ce 3d XPS spectra of CeOx showed the presence of both Ce4+ and Ce3+ peaks (Fig. 2a), where Ce3+ included one pair of peaks (V and V′), whilst Ce4+ had two pairs of peaks (U1, U2, U′1, and U′2). Notably, the proportion of Ce3+ in In2O3–CeOx was increased to 48.3% (Fig. 2b) compared to the individual CeOx (43.2%). In contrast, the proportion of Ce3+ in In2O3–CeO2 showed no obvious variation compared to that in CeO2 (Fig. S3a and b†). Additionally, the In 3d spectra of In2O3 showed two peaks at 451.7 (In 3d3/2) and 444.2 eV (In 3d5/2), while the binding energy of In 3d in In2O3–CeOx shifted positively about 1 eV compared to that of In2O3 (Fig. 2c). The results demonstrated the strong interaction between In2O3 and CeOx, and electrons transferred from the crystalline In2O3 to amorphous CeOx, which led to an increase in the proportion of Ce3+ in In2O3–CeOx. Moreover, the linear sweep voltammetry (LSV) curves of In2O3 showed an obvious reduction peak at −0.487 V (vs. RHE), corresponding to a typical In3+/In0 reduction peak (Fig. S4†).27
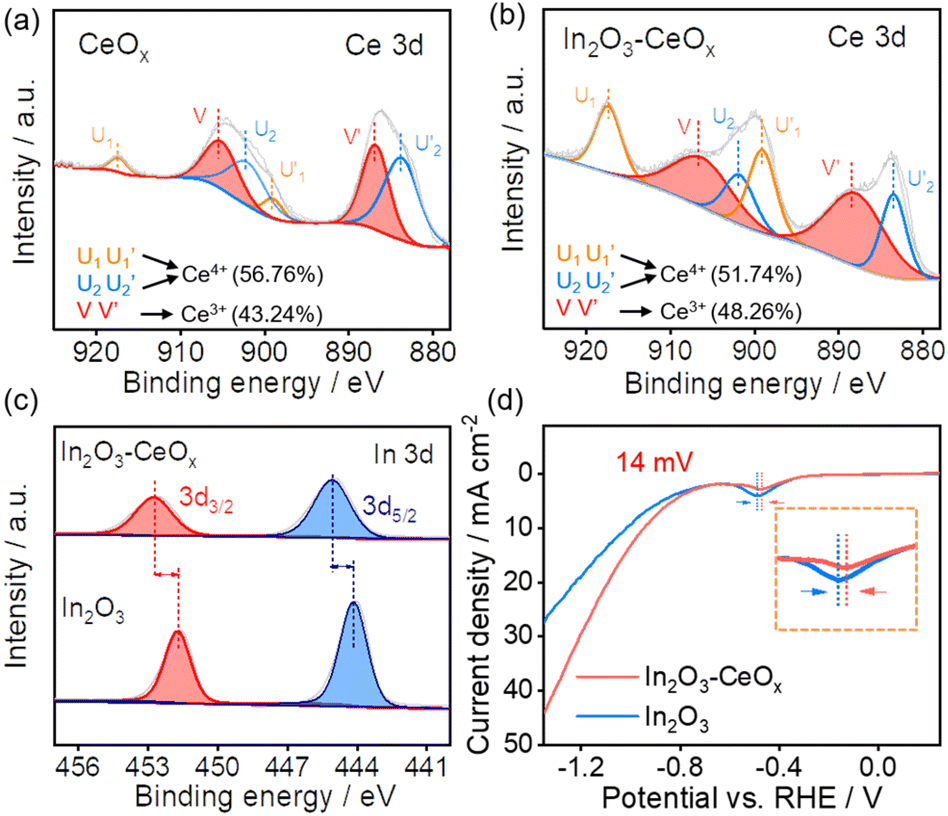 | ||
| Fig. 2 Ce 3d XPS spectra of (a) CeOx and (b) In2O3–CeOx; (c) In 3d XPS spectra of In2O3–CeOx and In2O3; and (d) LSVs of In2O3 and In2O3–CeOx in CO2-saturated 0.5 M KHCO3 at a scan rate of 50 mV s−1. | ||
It should be noted that the reduction peak potential of In2O3–CeOx positively shifted by 14 mV (appearing at −0.473 V) (Fig. 2d). The lower reduction potential of In2O3–CeOx meant an enhanced electrochemical redox activity.28 In contrast, no obvious change in the reduction peak potential was detected for In2O3–CeO2 compared to that of In2O3 (Fig. S3c†). This result was consistent with the XPS data, revealing that the crystalline–amorphous heterostructure of In2O3–CeOx led to stronger interactions between its individual components.
The electrocatalytic CO2RR performances of In2O3, In2O3–CeOx, and In2O3–CeO2 were tested in a three-electrode system, with CO2 saturated 0.1 M KHCO3 as an electrolyte. Linear sweep voltammetry (LSV) curves were obtained at a scan rate of 50 mV s−1 between −1.38 V and −0.62 V (vs. RHE, all potentials used were vs. RHE unless otherwise stated). As depicted by the linear sweep voltammetry (LSV) curves of all catalysts (Fig. 3a and S5†), significantly higher current density was observed in CO2 than that under an Ar atmosphere, indicating that the CO2 reduction reaction occurred over these catalysts. Meanwhile, the current density of In2O3–CeOx was larger than those of In2O3–CeO2 and In2O3 in a CO2-saturated electrolyte, indicating that a crystalline–amorphous In2O3–CeOx heterostructure shows the best CO2RR performance.
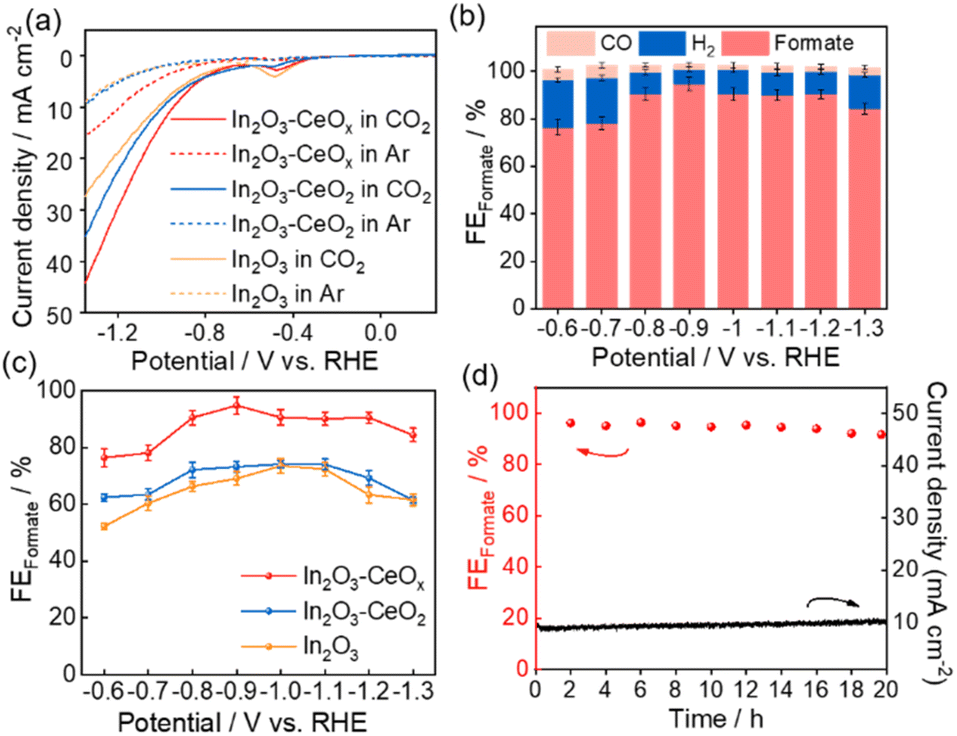 | ||
| Fig. 3 CO2 electroreduction in a 0.5 M KHCO3 electrolyte. (a) LSV curves of In2O3–CeOx, In2O3–CeO2 and In2O3 electrodes in a CO2-saturated electrolyte. (b) The FEformate, FECO and FEH2 of In2O3–CeOx at different applied potentials, respectively. (c) FEformate of In2O3–CeOx, In2O3–CeO2 and In2O3 at different applied potentials, respectively. (d) Long-term stability of the CO2 electroreduction for In2O3–CeOx at −0.9 V (vs. RHE). | ||
To further analyze the electroreduction products, controlled potential electrolysis of CO2 was performed at a constant working potential ranging from −0.6 V to −1.3 V. The gaseous and liquid products were quantified by gas chromatography (GC) and nuclear magnetic resonance (NMR) spectroscopy, respectively. For In2O3, In2O3–CeO2 and In2O3–CeOx catalysts, CO and H2 were detected as gas products, and formate was the unique liquid product (Fig. S6†). The formate faradaic efficiency (FEformate) of the three catalysts all exhibited a volcanic-like pattern over the range of applied operating potentials, among which, the In2O3–CeOx electrode exhibited higher selectivity toward formate against H2 and CO (Fig. 3b and c). More remarkably, the FEformate on the In2O3–CeOx electrode remained above 90% over a wide electrochemical window from −0.8 V to −1.2 V. The maximum FEformate reached 94.8% with a current density of 7 mA cm−2 at −0.9 V (Fig. 3c). By comparison, the maximum FEformate for In2O3–CeO2 and In2O3 electrodes was only 74.2% and 73.6%, respectively, under −1.0 V (Fig. S7a and b†). The optimal electrolysis potential of In2O3–CeOx was more positive than that of In2O3 and In2O3–CeO2, which suggested that lower electrical energy was required to cross the energy barrier and realize the CO2RR by the In2O3–CeOx catalyst. Notably, H2 was the only product of the individual CeOx and CeO2 (Fig. S7c and d†), indicating that the amorphous and crystalline ceria contributed to the formate formation via functions rather than being the catalyst.
The stability of In2O3–CeOx was tested by continuous electrolysis at −0.9 V. As shown in Fig. 3d, no evident decay of current density and FEformate was observed after 20 h-electrolysis, demonstrating the good stability of the prepared In2O3–CeOx electrode. Moreover, the LSV curves before and after long-term test showed no obvious difference (Fig. S8†), which further manifested the good stability of In2O3–CeOx. After the test, the element mapping of In2O3–CeOx showed that In, Ce, and O were uniformly distributed (Fig. S9†). In addition, the HRTEM image of In2O3–CeOx after electrolysis still showed the amorphous morphology of CeOx and the (222) plane of In2O3 (Fig. S9a and b†), indicating that the crystalline–amorphous heterostructure of In2O3–CeOx can be retained during the electrolytic process.
To gain a deeper understanding of the kinetics of CO2 electroreduction, we fitted the overpotentials of the three catalysts as a function of the bias current density for formate production to obtain the corresponding Tafel slopes (Fig. 4a). In2O3–CeOx presented a smaller calculated Tafel slope (176 mV dec−1) compared to In2O3–CeO2 (186 mV dec−1) and pure In2O3 (375 mV dec−1). The smallest Tafel slope of In2O3–CeOx suggested the largest increment in the CO2 reduction rate with the increasing overpotential. The electron transfer properties were further investigated by an electrochemical impedance spectroscopy (EIS) test. As shown in Fig. 4b, In2O3–CeOx showed smaller charge transfer resistance (Rct) compared to In2O3–CeO2 and In2O3. The results suggested that the crystalline–amorphous heterostructure rendered faster charge transfer than the bare In2O3 and crystalline–crystalline heterostructure, and the interaction of electrochemically active In2O3 with the amorphous CeOx led to a more efficient electron transfer. The electrochemically active surface area (ECSA) was evaluated by the electrochemical double layer capacitance (Cdl) due to their positive proportion relationship (Fig. 4c).2,29,30 The ECSA of In2O3–CeOx (13.75 cm−2) was higher than those of In2O3–CeO2 (11.50 cm−2) and In2O3 (9.50 cm−2), which can promote the electrocatalytic performance. In addition, the partial current density of formate was normalized by the ECSA (Fig. S11†).2 Similar to the geometric current density, the normalized current density of In2O3–CeOx was the highest among the three samples. The rough surface of the CeOx substrate promoted the adsorption and dispersion of In2O3, thereby increasing the active surface area. Furthermore, to verify the phenomenon of electron transfer between In2O3 and CeOx in In2O3–CeOx, we performed Mott–Schottky and valence band XPS experiments (Fig. S12†). As shown in Fig. 4d, the flat band potentials of In2O3 and CeOx were located at −0.87 V and −0.47 V (vs. NHE), respectively. Such a band alignment can effectively facilitate the interfacial electrons transferring from the key component In2O3 to CeOx. However, the flat band and valence band potentials of CeO2 and In2O3 were not significantly different (Fig. S13†), and thus electron transfer hardly occurred between CeO2 and In2O3.
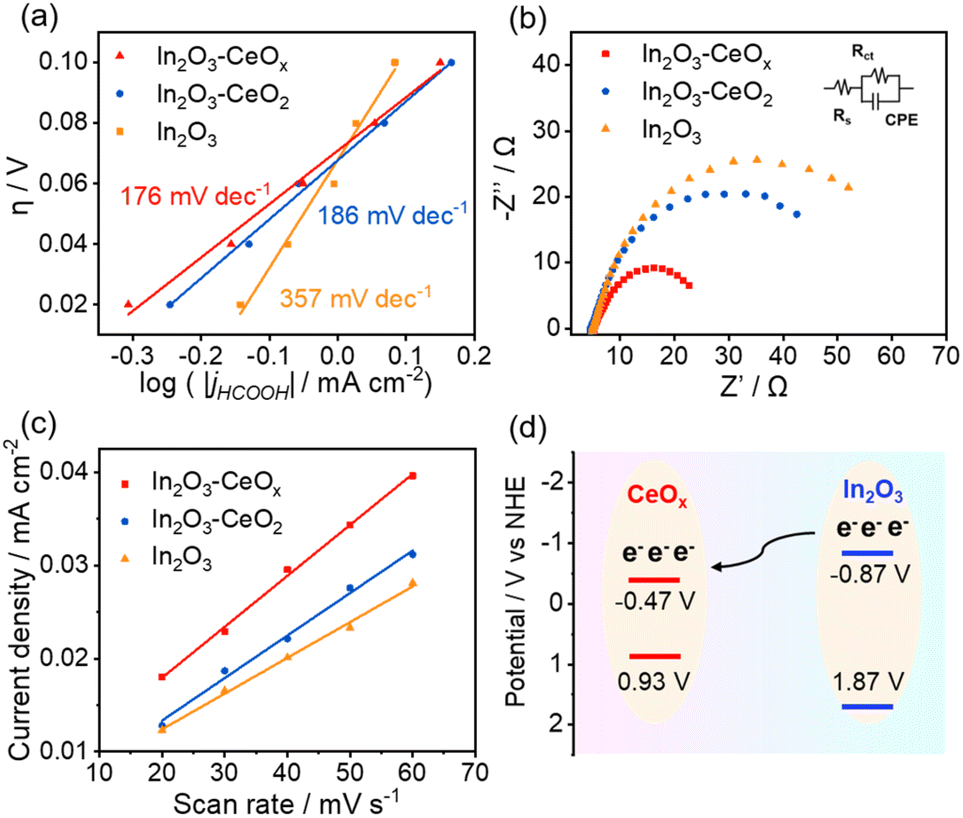 | ||
| Fig. 4 Advantages of In2O3–CeOx for CO2 reduction. (a) Tafel plots, (b) Nyquist plots (inset: equivalent circuit), (c) linear fitting of double-layer capacitive currents versus scan rates to estimate the ECSA of In2O3–CeOx, In2O3–CeO2 and In2O3, and (d) schematic illustration of the charge transfer in In2O3–CeOx. | ||
Density functional theory (DFT) calculations were carried out to shed light on the origin of the enhanced activity of In2O3–CeOx for the CO2RR (Fig. S14–S16†). The charge density difference and charge displacement curves of In2O3–CeOx and In2O3–CeO2 were obtained to study the transfer of electrons.31–33 As shown in Fig. 5a, the positive and negative signals of the charge displacement curve represented electron accumulation and depletion, respectively. The In2O3–CeOx heterostructure displayed a significant charge rearrangement around the crystalline–amorphous heterostructure interface, and electron transfer took place from In2O3 to CeOx, which was in good agreement with the XPS results. Moreover, the reaction pathways for the generation of formate were considered. According to the calculations, the Gibbs free energy for the rate-determining step (RDS) on In2O3–CeOx is lower than that on In2O3–CeO2 and In2O3 for the CO2-to-formate process (Fig. 5b), suggesting that In2O3–CeOx was thermodynamically more favourable for CO2-to-formate conversion. The adsorption energies of the *OCHO intermediates on In2O3–CeOx and In2O3–CeO2 were further explained by the projected density of states (p-DOS). As shown in Fig. 5c, the centre of the p-band for In2O3–CeOx–*OCHO was closer to the Fermi level compared with that of In2O3–CeO2–*OCHO, which verified that the antibonding state filling of the In-5p orbital of In2O3–CeOx is lower than that of In2O3–CeO2, thereby enhancing the adsorption strength of *OCHO.14 In addition, the Gibbs free energy for the dominant competitive HER was also explored (Fig. 5d). The Gibbs free energy for generating *H over In2O3–CeOx was much larger than that for In2O3–CeO2 and In2O3, indicating that the HER was more difficult to occur on In2O3–CeOx. Therefore, the crystalline–amorphous In2O3–CeOx heterostructure accelerated the electron transfer, enhanced the adsorption to *OCHO, lowered the energy barrier for the process from OCHO* to *HCOOH, and finally promoted the formation of the formate product. Furthermore, the In2O3–CeOx catalyst significantly suppressed the HER competition reaction compared with In2O3 and In2O3–CeO2.
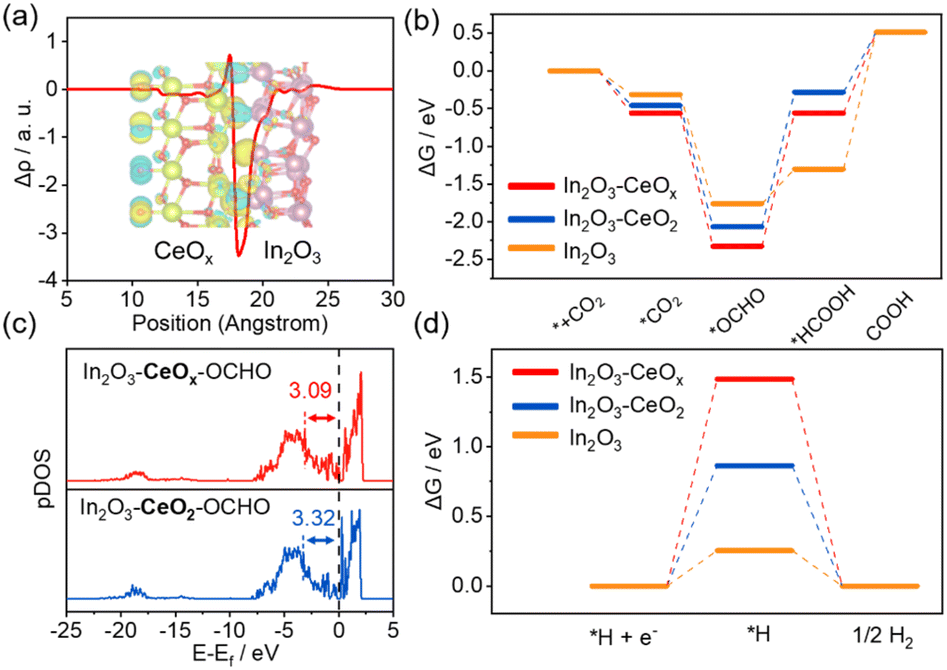 | ||
| Fig. 5 DFT calculations. (a) Charge density difference and charge displacement curves of In2O3–CeOx; purple, yellow, and red spheres represent In, Ce, and O atoms, respectively. (b) Gibbs free-energy diagrams of CO2-to-formate. (c) In-5p p-DOS of In2O3–CeOx and In2O3–CeO2 after adsorbing *OCHO. (d) Gibbs free-energy diagrams of the HER. | ||
Conclusion
In conclusion, we described a facile strategy to fabricate the crystalline–amorphous In2O3–CeOx heterostructure and studied its performance for the CO2RR. The experimental results showed that In2O3–CeOx achieved better catalytic performance than the crystalline–crystalline In2O3–CeO2 heterostructure and In2O3 alone. The maximum FE of 94.8% was achieved on the In2O3–CeOx catalyst, and the FE remained above 90% over a wide potential range of −0.8 to −1.2 V vs. RHE. Detailed studies have shown that there was a remarkable electron transfer from In2O3 to amorphous CeOx on the In2O3–CeOx heterostructure compared to the crystalline–crystalline In2O3–CeO2 heterostructure, which decreased the interfacial charge transfer resistance. The In2O3–CeOx heterostructure promoted the adsorption for *OCHO intermediates and thereby the energy barrier of *OCHO → *HCOOH was reduced, which led to the high selectivity of the CO2RR to formate. We believe that the work may provide an effective strategy for catalysing CO2 conversion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22007004, 22178019, 21625101 and 21521005), the National Key Research and Development Program of China (2017YFB0307303) and the Fundamental Research Funds for the Central Universities (buctrc202010, XK1802-6, XK1803-05, XK1902 and 12060093063).Notes and references
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- C. Chen, X. Sun, X. Yan, Y. Wu, H. Liu, Q. Zhu, B. B. A. Bediako and B. Han, Boosting CO2 electroreduction on N,P-Co-doped carbon aerogels, Angew. Chem., Int. Ed., 2020, 59, 11123–11129 CrossRef CAS PubMed.
- W. Guo, X. Tan, J. Bi, L. Xu, D. Yang, C. Chen, Q. Zhu, J. Ma, A. Tayal, J. Ma, Y. Huang, X. Sun, S. Liu and B. Han, Atomic indium catalysts for switching CO2 electroreduction products from formate to CO, J. Am. Chem. Soc., 2021, 143, 6877–6885 CrossRef CAS PubMed.
- Z. Wang, Y. Zhou, C. Xia, W. Guo, B. You and B. Y. Xia, Efficient electroconversion of carbon dioxide to formate by a reconstructed amino-functionalized indium-organic framework electrocatalyst, Angew. Chem., Int. Ed., 2021, 60, 19107–19112 CrossRef CAS PubMed.
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Promises of main group metal-based nanostructured materials for electrochemical CO2 reduction to formate, Adv. Energy Mater., 2020, 10, 1902338 CrossRef CAS.
- F. Valentini, V. Kozell, C. Petrucci, A. Marrocchi, Y. Gu, D. Gelman and L. Vaccaro, Formic acid, a biomass-derived source of energy and hydrogen for biomass upgrading, Energy Environ. Sci., 2019, 12, 2646–2664 RSC.
- W. Guo, S. Liu, X. Tan, R. Wu, X. Yan, C. Chen, Q. Zhu, L. Zheng, J. Ma, J. Zhang, Y. Huang, X. Sun and B. Han, Highly efficient CO2 electroreduction to methanol through atomically dispersed Sn coupled with defective CuO catalysts, Angew. Chem., Int. Ed., 2021, 60, 21979–21987 CrossRef CAS PubMed.
- L. Lu, W. Guo, C. Chen, Q. Zhu, J. Ma, H. Wu, D. Yang, G. Yang, X. Sun and B. Han, Synthesis of Sn4P3/reduced graphene oxide nanocomposites as highly efficient electrocatalysts for CO2 reduction, Green Chem., 2020, 22, 6804–6808 RSC.
- D. Yang, Q. Zhu, X. Sun, C. Chen, W. Guo, G. Yang and B. Han, Electrosynthesis of a defective indium selenide with 3D structure on a substrate for tunable CO2 electroreduction to syngas, Angew. Chem., Int. Ed., 2020, 59, 2354–2359 CrossRef CAS PubMed.
- J. E. Pander, M. F. Baruch and A. B. Bocarsly, Probing the mechanism of aqueous CO2 reduction on post-transition-metal electrodes using ATR-IR spectroelectrochemistry, ACS Catal., 2016, 6, 7824–7833 CrossRef CAS.
- J. Li, M. Zhu and Y.-F. Han, Recent advances in electrochemical CO2 reduction on indium-based catalysts, ChemCatChem, 2021, 13, 514–531 CrossRef CAS.
- Y. Huang, X. Mao, G. Yuan, D. Zhang, B. Pan, J. Deng, Y. Shi, N. Han, C. Li, L. Zhang, L. Wang, L. He, Y. Li and Y. Li, Size-dependent selectivity of electrochemical CO2 reduction on converted In2O3 nanocrystals, Angew. Chem., Int. Ed., 2021, 60, 15844–15848 CrossRef CAS PubMed.
- J. Zhang, R. Yin, Q. Shao, T. Zhu and X. Huang, Oxygen vacancies in amorphous InOx nanoribbons enhance CO2 adsorption and activation for CO2 electroreduction, Angew. Chem., Int. Ed., 2019, 58, 5609–5613 CrossRef CAS PubMed.
- Z. Chen, G. Yu, B. Li, X. Zhang, M. Jiao, N. Wang, X. Zhang and L. Liu, In situ carbon encapsulation confined nickel-doped indium oxide nanocrystals for boosting CO2 electroreduction to the industrial level, ACS Catal., 2021, 11, 14596–14604 CrossRef CAS.
- W. Luo, W. Xie, R. Mutschler, E. Oveisi, G. L. De Gregorio, R. Buonsanti and A. Züttel, Selective and stable electroreduction of CO2 to CO at the copper/indium interface, ACS Catal., 2018, 8, 6571–6581 CrossRef CAS.
- M. A. Ahsan, T. He, J. C. Noveron, K. Reuter, A. R. Puente-Santiago and R. Luque, Low-dimensional heterostructures for advanced electrocatalysis: an experimental and computational perspective, Chem. Soc. Rev., 2022, 51, 812–828 RSC.
- P. Prabhu, V. Jose and J.-M. Lee, Heterostructured catalysts for electrocatalytic and photocatalytic carbon dioxide reduction, Adv. Funct. Mater., 2020, 30, 1910768 CrossRef CAS.
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S.-Z. Qiao, Surface and interface engineering in copper-based bimetallic materials for selective CO2 electroreduction, Chem, 2018, 4, 1809–1831 CAS.
- P.-F. Sui, C. Xu, M.-N. Zhu, S. Liu, Q. Liu and J.-L. Luo, Interface-induced electrocatalytic enhancement of CO2-to-formate conversion on heterostructured bismuth-based catalysts, Small, 2022, 18, 2105682 CrossRef CAS PubMed.
- Z. Zhang, F. Ahmad, W. Zhao, W. Yan, W. Zhang, H. Huang, C. Ma and J. Zeng, Enhanced electrocatalytic reduction of CO2 via chemical coupling between indium oxide and reduced graphene oxide, Nano Lett., 2019, 19, 4029–4034 CrossRef CAS PubMed.
- J. Wang, X. Xiao, Y. Liu, K. Pan, H. Pang and S. Wei, The application of CeO2-based materials in electrocatalysis, J. Mater. Chem. A, 2019, 7, 17675–17702 RSC.
- D. Gao, Y. Zhang, Z. Zhou, F. Cai, X. Zhao, W. Huang, Y. Li, J. Zhu, P. Liu, F. Yang, G. Wang and X. Bao, Enhancing CO2 electroreduction with the metal–oxide interface, J. Am. Chem. Soc., 2017, 139, 5652–5655 CrossRef CAS PubMed.
- H. Dong, L. Zhang, L. Li, W. Deng, C. Hu, Z.-J. Zhao and J. Gong, Abundant Ce3+ ions in Au-CeOx nanosheets to enhance CO2 electroreduction performance, Small, 2019, 15, 1900289 CrossRef PubMed.
- S. Shen, Z. Wang, Z. Lin, K. Song, Q. Zhang, F. Meng, L. Gu and W. Zhong, Crystalline-amorphous interfaces coupling of CoSe2/CoP with optimized d-band center and boosted electrocatalytic hydrogen evolution, Adv. Mater., 2022, 34, 2110631 CrossRef CAS PubMed.
- M. Yang, M. Zhao, J. Yuan, J. Luo, J. Zhang, Z. Lu, D. Chen, X. Fu, L. Wang and C. Liu, Oxygen vacancies and interface engineering on amorphous/crystalline CrOx-Ni3N heterostructures toward high-durability and kinetically accelerated water splitting, Small, 2022, 18, 2106554 CrossRef CAS PubMed.
- Y. X. Duan, Y. T. Zhou, Z. Yu, D. X. Liu, Z. Wen, J. M. Yan and Q. Jiang, Boosting Production of HCOOH from CO2 Electroreduction via Bi/CeOx, Angew. Chem., Int. Ed., 2021, 60, 8798–8802 CrossRef CAS PubMed.
- Z. M. Detweiler, J. L. White, S. L. Bernasek and A. B. Bocarsly, Anodized indium metal electrodes for enhanced carbon dioxide reduction in aqueous electrolyte, Langmuir, 2014, 30, 7593–7600 CrossRef CAS PubMed.
- F. Yang, X. Bao, P. Li, X. Wang, G. Cheng, S. Chen and W. Luo, Boosting hydrogen oxidation activity of Ni in alkaline media through oxygen-vacancy-Rich CeO2/Ni heterostructures, Angew. Chem., Int. Ed., 2019, 58, 14179–14183 CrossRef CAS PubMed.
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef CAS PubMed.
- C. Zhang, Y. Huang, Y. Yu, J. Zhang, S. Zhuo and B. Zhang, Sub-1.1 nm ultrathin porous CoP nanosheets with dominant reactive {200} facets: a high mass activity and efficient electrocatalyst for the hydrogen evolution reaction, Chem. Sci., 2017, 8, 2769–2775 RSC.
- G. Ciancaleoni, F. Nunzi and L. Belpassi, Charge displacement analysis-A tool to theoretically characterize the charge transfer contribution of halogen bonds, Molecules, 2020, 25, 300–316 CrossRef CAS PubMed.
- Y. Zhou, Y. Yao, R. Zhao, X. Wang, Z. Fu, D. Wang, H. Wang, L. Zhao, W. Ni, Z. Yang and Y. M. Yan, Stabilization of Cu+via strong electronic interaction for selective and stable CO2 electroreduction, Angew. Chem., Int. Ed., 2022, 61, e202205832 CAS.
- D. Sorbelli, E. Rossi, R. W. A. Havenith, J. Klein, L. Belpassi and P. Belanzoni, Gold-aluminyl and gold-diarylboryl complexes: bonding and reactivity with carbon dioxide, Inorg. Chem., 2022, 61, 7327–7337 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qi01646j |
| This journal is © the Partner Organisations 2022 |