
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymer architecture dictates thermoreversible gelation in engineered emulsions stabilised with branched copolymer surfactants†
A.
Rajbanshi
 ac,
M. A.
da Silva
a,
D.
Murnane
a,
L.
Porcar
b,
C. A.
Dreiss
c and
M. T.
Cook
*a
ac,
M. A.
da Silva
a,
D.
Murnane
a,
L.
Porcar
b,
C. A.
Dreiss
c and
M. T.
Cook
*a
aSchool of Life and Medical Sciences, University of Hertfordshire, Hatfield, Hertfordshire AL10 9AB, UK. E-mail: m.cook5@herts.ac.uk
bInstitut Laue Langevin, 71 Avenue des Martyrs, 38042 Grenoble, France
cInstitute of Pharmaceutical Science, King's College London, Franklin-Wilkins Building, 150 Stamford Street, London, SE1 9NH, UK
First published on 26th September 2022
Abstract
The generation of materials that switch from a liquid to gel state upon warming can enable new healthcare technologies with improved functionality, such as in situ gel-forming materials for drug delivery to topical or parenteral sites. The majority of these materials are aqueous polymer solutions, which then suffer from an inability to solubilise hydrophobic drugs. This study investigates the generation of thermoresponsive “engineered emulsions” which are low-viscosity emulsions at low temperature and switch to a gel state upon warming. This is achieved by the synthesis of novel branched copolymer surfactants (BCS) containing di(ethylene glycol) methyl ether methacrylate (DEGMA) as a thermoresponsive component giving a lower critical solution temperature (LCST). The copolymers were employed as emulsifiers to prepare 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dodecane:water emulsion systems. The effect of polymer architecture is shown to be intimately linked to the rheology of these systems, where branching, elevation of molecular weight, and the presence of hydrophobic end groups is demonstrated to be commensurate with gel formation upon heating. Mechanisms of gel formation were probed by small-angle neutron scattering, which demonstrated that the branched copolymer surfactants formed oblate ellipsoids in solution that grew anisotropically with temperature, forming larger disk-like nanoparticles. The formation of these elongated particles leads to thickening of the emulsions, whilst connectivity of the aggregates and BCS at the oil–water interface is required for gel formation to occur. Overall, the study provides design principles for this novel class of thermoresponsive material with great potential in healthcare, cosmetic, and energy applications.
:1 dodecane:water emulsion systems. The effect of polymer architecture is shown to be intimately linked to the rheology of these systems, where branching, elevation of molecular weight, and the presence of hydrophobic end groups is demonstrated to be commensurate with gel formation upon heating. Mechanisms of gel formation were probed by small-angle neutron scattering, which demonstrated that the branched copolymer surfactants formed oblate ellipsoids in solution that grew anisotropically with temperature, forming larger disk-like nanoparticles. The formation of these elongated particles leads to thickening of the emulsions, whilst connectivity of the aggregates and BCS at the oil–water interface is required for gel formation to occur. Overall, the study provides design principles for this novel class of thermoresponsive material with great potential in healthcare, cosmetic, and energy applications.
Introduction
Emulsions are metastable colloids made up of at least two immiscible liquids in which one phase is dispersed into another and forms droplets.1,2 The immiscible fluids in emulsion systems may be kinetically stabilised by lowering interfacial tension,3 traditionally achieved using small-molecule or polymeric surfactants which adsorb at the liquid–liquid interface.1 The use of macromolecules at the interface offers great opportunity in manipulation of chemistry such that in addition to stabilising the emulsion system, they impart stimuli-responsive – so-called “smart” – functionality.4,5 These stimuli-responsive polymeric materials undergo a modification in their physical and chemical properties in response to changes in their environment, such as mechanical stress, pH, light, temperature, and biological stimuli.6–9Thermoresponsive polymers modify their physical properties in response to temperature. Typically this manifests as an alteration of the solubility of the constituent units with temperature, which can be coupled to the intrinsic ability to control polymer properties through modification of architecture and inclusion of co-monomers to lead to complex materials.10 Thermoresponsive materials are being widely employed in healthcare research, enabling novel and improved therapies and diagnostics, amongst a plethora of applications such as drug delivery platforms, additive manufacturing and tissue engineering.11 Polymer solutions that are capable of transforming to a gel state with elevation of temperature to a critical point are referred to as “Thermoreversible gels” or “Thermogelling materials”.11 A sol–gel transition occurs when heating a thermoreversible gel above a critical temperature, affected by an overall increase in hydrophobicity above the lower critical solution temperatures (LCST), triggering self-assembly processes and physical interaction, which increases the viscosity of the system.12 These materials could transition from liquid to gel at body temperature, enabling in situ gelation and thus enhanced retention and therapeutic effect.11 However, the vast majority of these materials are aqueous polymer solutions which often limits the application of the materials to hydrophilic encapsulants.
In 2008, Weaver introduced the term “emulsion engineering”13 to describe a droplet trapping concept in which the droplet surface functionality is designed to kinetically trap the droplets giving a percolating network structure in the material, resulting in solidification. Droplet assembly and dispersion were dependent on inter-droplet hydrogen bonding, controlled by acidic or basic pH conditions.13 Weaver and colleagues achieved these phenomena by stabilising emulsions with branched copolymer surfactants (BCSs) composed of a pH-responsive monomer, a hydrophilic macromonomer for steric stabilisation, a cross-linker for branching, and a chain-transfer agent to introduce hydrophobic alkyl chain ends.14,15 The hydrophobic chain ends of the branched copolymers offer excellent attachment to the surface of the oil droplets and the capacity to alter the stabilisation and surface functionality of the droplets in the continuous aqueous phase.16 These features are the essentials of “engineered emulsions”. This concept was demonstrated with weakly acidic polymeric surfactants that were able to cooperatively form hydrogen-bonds between emulsion droplets in the unionised state, imparting a pH-dependent sol–gel behaviour. As is typical of emulsions, these systems are complex and polymers imparting stimuli-responsive behaviour are required to stabilise emulsions with oils of widely varied polarity, droplet size and oil/water ratio, as well as varied emulsification techniques.3 Thus, the field is both promising, and highly challenging. Our group has recently demonstrated for the first time that thermoresponsive engineered emulsions may be generated from poly(N-isopropylacrylamide) BCSs, in which poly(N-isopropyl acrylamide) exhibits an LCST. These emulsions successfully exhibited thermoreversible gelation, however the gel state was weak (G′ of ca. 30 Pa) and existed over a narrow temperature range.17 The study found that polymer exists at the O/W interface and as nanoscale aggregates in the bulk, with rising temperature inducing changes to nanostructure which triggers gelation. There is a need to generate thermoresponsive engineered emulsions of higher performance, which requires an understanding of how polymer architecture links to rheology.
This study reports the first thermoresponsive engineered emulsions from di(ethylene glycol) methyl ether methacrylate (DEGMA) branched copolymer surfactants. DEGMA, as a member of the highly versatile and biocompatible oligo(ethylene glycol) methacrylate class,18 offers a promising blueprint for the generation of these materials. Polymer architecture will be varied to establish correlations with the temperature-dependent rheological behaviour of these systems, in particular how it is affected by branching degree, hydrophobic chain ends and molecular weight. The nanoscale processes underpinning thermoresponse will be determined by small-angle neutron scattering. This approach is promising to generate novel materials for healthcare applications, along with design principles for these advanced functional materials.
Materials and methods
Materials
Di(ethylene glycol) methyl ether methacrylate (DEGMA, 95%), poly(ethylene glycol) methyl ether methacrylate (PEGMA, Mn 950 gmol−1), ethylene glycol dimethacrylate (EGDMA, 98%), 1-dodecanethiol (DDT, 99%), and dodecane anhydrous (99%) were purchased from Sigma-Aldrich (UK). α, α-azoisobutyronitrile (AIBN, >99%) was obtained from Molekula (UK). 1-hexadedecanethiol (HDT, 97%) and 1-tetradecanethol (TDT, 94%) were purchased from Alfa Aesar (UK). Ethanol and dimethylformamide (DMF) were supplied by VWR (UK). Lithium bromide (99%) and 2-mercaptoethanol (ME, 99%) were purchased from Acros Organics (UK). Dialysis tubing with molecular weight cut off (MWCO) of 14 kg mol−1 was purchased from Sigma Aldrich (UK). GPC EasiVial poly(methyl methacrylate) mixed standards and a poly(methyl methacrylate) single standard (72 kg mol−1) were procured from Agilent (UK). Deionised H2O was employed in all experiments. All chemicals were used as received.Synthesis of PDEGMA-co-PEGMA branched copolymer surfactant by free radical polymerisation
A series of thermoresponsive BCSs were synthesized by free radical polymerisation. In a general synthesis, DEGMA, PEGMA, cross-linker (EGDMA) and chain-transfer agent (DDT/HDT/TDT/ME) were dissolved in 190 mL ethanol and bubbled with nitrogen gas. After 1 h of nitrogen purging, an ethanolic solution of AIBN (10 mL) was added to the solution. The apparatus was set at 70 °C for 48 h for polymerisation to proceed. After 48 h, the synthesised polymer was distilled at 80 °C to remove excess ethanol. The resultant crude polymer was then dissolved in water and transferred to a pre-soaked dialysis bag. The dialysis bag was immersed in a beaker containing de-ionised water for 7 to 10 days and the water was replaced at regular intervals to facilitate the purification process. The resultant polymer solution was subjected to lyophilisation for 48 h to obtain a freeze-dried product. The yields for all the lyophilised polymers were 85 ± 1% (Table S1†).A library of 9 polymers were produced, labelled P1–P9. P1–P3 explored the effect of branching degree by variation of cross-linker in the feed (Table 1) and P4–P6 explored the effect of molecular weight by control of initiator/chain-transfer agent:monomer ratio (Table 1). P7–P9 explored the effect of hydrophobic end group using the feed for P1 (Table 1) but switching DDT for TDT (P7), HDT (P8) and ME (P9).
| Sample ID | DEGMA (mmol) | PEGMA (mmol) | EGDMA (mmol) | DDT (mmol) | AIBN (mmol) |
|---|---|---|---|---|---|
| P1 | 174 | 6 | 12 | 12 | 1.2 |
| P2 | 174 | 6 | 6 | 12 | 1.2 |
| P3 | 174 | 6 | 0 | 12 | 1.2 |
| P4 | 174 | 6 | 12 | 8 | 0.8 |
| P5 | 174 | 6 | 12 | 6 | 0.6 |
| P6 | 174 | 6 | 12 | 3 | 0.3 |
Characterisation of thermoresponsive BCSs
1H NMR was used to characterise the BCS on Bruker Advance AM 600 NMR instrument using CDCl3 as a solvent at ambient temperature. As an internal standard, the residual solvent peak was used. Delta 5.3.1 NMR software was used to process the data.The number average molecular weight (Mn) and polydispersity of synthesised BCSs were characterised using an Agilent 1260 Infinity II GPC equipped with a refractive index (RI) detector. The system was equipped with an Agilent Varian PLGel 5 μm mixed D column. 0.1% w/v lithium bromide in dimethylformamide was used as the eluent, at a flow rate of 0.8 mL min−1 with the column temperature set at 30 °C. All samples were prepared in dimethylformamide at a concentration of 2 mg mL−1 prior to analysis. Dynamic light scattering was conducted on 1 mg mL−1 BCS samples in deionised water without filtration using a Malvern Nano-ZS instrument from 20–60 °C at 1 °C intervals.
Emulsion preparation
A series of 1:1 w/w oil in water emulsions were prepared to study the thermoresponsive behaviour by rheology. 2.5 g polymer solutions of 2.5, 5 and 10 wt% were prepared in ice cold water with stirring in a 30 mL glass vial. 2.5 g of dodecane oil phase was added to the polymer solution which was then homogenised for 2 min at 2400 rpm to obtain a 1:1 w/w oil in water emulsion using a Silverson L4R Heavy Duty Mixer Emulsifier (US). The resulting emulsions were kept undisturbed for 36 h under ambient conditions. Approximately 1.1 g of the water phase separated as the lower phase of the creamed emulsion was withdrawn, and the rheological behaviour of the emulsion cream was studied as a function of temperature. The approximate oil phase volume in the emulsions studied was 0.57.
Rheology of thermoresponsive emulsions
Rheological measurements were performed on an AR 1500ex rheometer (TA instruments, USA) equipped with a Peltier temperature control unit and a 40 mm parallel plate geometry with a specified gap distance of 500–750 μm at an oscillating stress of 1 Pa and frequency of 6.28 rad s−1. The change in storage modulus (G′) and loss modulus (G′′) as a function of temperature was recorded and the loss tangent (tan δ) calculated as G′′/G′. Temperature ramps were performed in the range 20 to 60 °C at 1 °C per minute heating/cooling rate. Frequency sweeps were conducted on emulsions stabilised with 10 wt% P1 at 30 and 50 °C between 0.628 and 100 rad s−1 at a fixed shear strain of 0.1%.SANS studies on BCS solutions with variation of temperature
SANS experiments were performed on the D22 instrument at the Institut Laue–Langevin (Grenoble, France). The neutron wavelength was 6 Å, the sample-detector distances were 2, 5.6, and 17.6 m, and the collimation distances were 2.8, 8, and 17.6 m, respectively. The detector offset was 300 mm. These settings gave a wave vector range 2.7 × 10−3 ≤ q ≤ 0.45 Å−1. Rectangular quartz cuvettes with a thickness of 1 mm were used for all samples. Measurements were performed at 25, 40, and 50 °C with a minimum equilibration time of 15 min prior to sample run. All polymer solutions (P1–6) were prepared at 20 wt% in D2O prior to measurement. Rheological analysis of these samples was also conducted, using the protocols described above. Data stitching was performed on Igor Pro (Wavemetrics, USA)19 and data fitting was conducted using SasView 4.2.2. The following factors were fixed to reduce the number of fitting parameters: scale/volume fraction (0.2) and solvent scattering length density, SLD (6.37 × 10−6 Å−2). The remaining parameters were fitted using form factors and structure factors described elsewhere.20,21 The SLDs of the BCS were calculated from the monomeric unit using the NIST Neutron activation and scattering calculator,22 and left to float, to account for hydration of the polymers.The scattering intensity I(q) can be written as follows:
| I(q) = A(P(q)AS(q)A) + BKG, |
A was set to 0.2, as described above. If more than one scattering object is present, or the objects studied have a hierarchical structure that generates scattering over distinct length scales, the expression can be extended to include further terms:
| I(q) = A(P(q)AS(q)A) + B(P(q)B) + BKG, |
For the BCS solution data, the majority of data were fitted using the an ellipsoid form factor20 with either a hard sphere or sticky hard sphere structure factor.21 When a power law form factor was included, it took the form I(q) = q−4 which is characteristic of Porod scattering arising from a sharp interface.23
Results and discussion
Synthesis of thermoresponsive BCS by free radical polymerisation
A modified one-pot synthesis was employed to generate an initial thermoresponsive branched copolymer surfactant (P1), informed by Weaver and co-worker's studies on pH-responsive Engineered Emulsions.13,14,24 P1 was synthesised using DEGMA as the thermoresponsive unit, PEGMA as the hydrophilic macromonomer, EGDMA as the cross-linker, AIBN as the initiator and DDT as the chain-transfer agent (Fig. 1(i)), in ratios guided by previous publications.25 DEGMA, a monomer which imparts temperature sensitivity with an LCST of 26 °C,26 is expected to transition from a hydrophilic to a relatively hydrophobic state upon warming that was intended to trigger self-assembly processes leading to gelation. PEGMA, a hydrophilic macromonomer for stabilisation, is expected to preferentially be solubilised in the water phase at the liquid–liquid interface and offer steric stabilisation over the measured temperature range. EGDMA is a widely used cross-linker in free radical polymerization that provides branching in the system. DDT was chosen as a chain-transfer agent to interact favourably with the dodecane oil phase due to the hydrophobicity of the C12 alkyl chain, and aimed to improve the thermodynamic stability to the emulsion.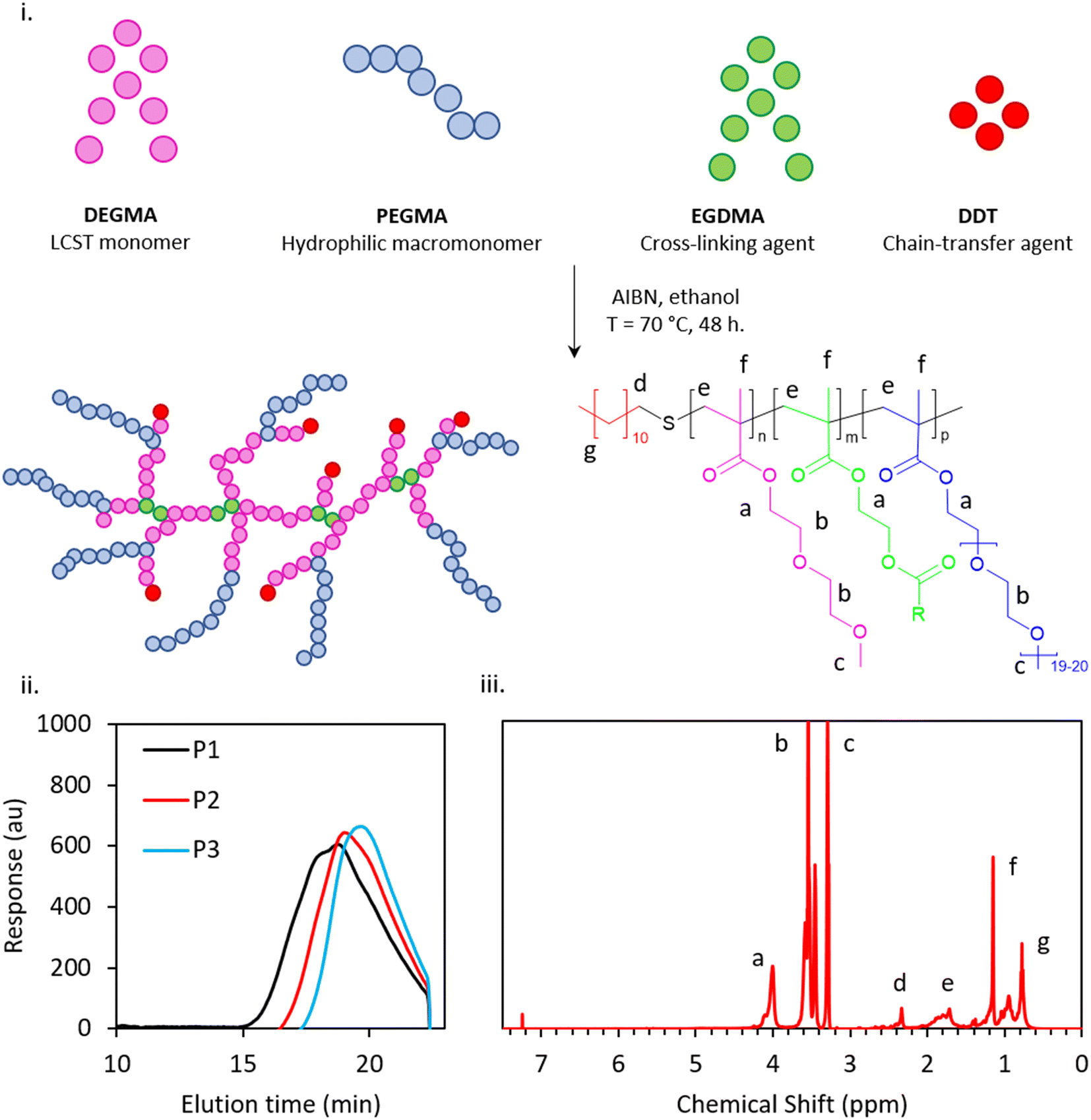 | ||
| Fig. 1 Free radical polymerisation of PDEGMA-PEGMA-EGDMA-DDT and theoretical structure of the branched copolymer surfactant (BCS) (i), exemplar GPC chromatogram of P1–3 (ii) and exemplar 1H NMR spectrum (iii). | ||
Following the synthesis of P1, a library of thermoresponsive BCSs were synthesised by varying composition of cross-linker, molecular weight and chain-transfer agents of longer and shorter carbon chain lengths, relative to P1, with the overall aim of linking polymer architecture to emulsion behaviour (Table 2). The successful synthesis of these copolymers was confirmed by GPC and 1H NMR (Fig. 1(ii and iii)). P2 and P3 were based on the initial synthesis (i.e., P1) with half the quantity of cross-linker (EGDMA) and with no cross-linker, i.e. 6 and 0 mmol respectively, with the intention of exploring the effect, and necessity, of branching. P4, P5 and P6 were variations of P1 with reduced levels of initiator (AIBN) and chain-transfer agent (DDT) at 0.8/8 mmol AIBN/DDT, 0.6/6 mmol AIBN/DDT and 0.4/4 mmol AIBN/DDT, respectively. As chains may be propagated from either AIBN-derived radicals or those transferred to DDT, a reduction in the quantity of these elements whilst retaining their ratio offers control of molecular weight by increasing the average degree of polymerisation per chain. P7, P8 and P9 were synthesised with varying carbon chain length of the chain transfer-agents whilst keeping the composition of DEGMA, PEGMA and EGDMA constant as per P1. P7, P8 and P9 were synthesised with TDT, HDT and mercaptoethanol respectively, giving chain ends of C-14, C-16, and a “zero-length” ethyl alcohol group. This series evaluates the impact of the polymer chain-end hydrophobicity on the branched copolymer's aqueous solution characteristics, as well as exploring behaviour without hydrophobic termini (P9).
| Study | Sample | Composition/variable | M n (kg mol−1) | M w (kg mol−1) | Đ |
|---|---|---|---|---|---|
| Effect of architecture & cross-linker | P1 | Full cross-linker (12 mmol) | 8.2 | 18.5 | 2.3 |
| P2 | Half cross-linker (6 mmol) | 6.5 | 11.3 | 1.7 | |
| P3 | No cross-linker (0 mmol) | 5.4 | 7.9 | 1.5 | |
| Effect of molecular weight | P4 | 0.8/8 mmol AIBN/DDT | 10.8 | 33.0 | 3.1 |
| P5 | 0.6/6 mmol AIBN/DDT | 14.1 | 68.3 | 4.8 | |
| P6 | 0.4/4 mmol AIBN/DDT | 21.3 | 180.9 | 8.5 | |
| Effect of chain-transfer agent (CTA) | P7 | Tetradecanethiol (C14) | 8.9 | 23.0 | 2.6 |
| P8 | Hexadecanethiol (C16) | 9.0 | 23.7 | 2.6 | |
| P9 | Mercaptoethanol (C0) | 15.1 | 28.5 | 1.9 |
The 1H NMR spectra (Fig. 1(iii)) confirmed the polymerisation of branched copolymers with no evidence of vinylic protons in the 6–7 ppm region (P1–P9 spectra given in ESI†). 1H NMR of the crude product indicated that the reactions achieved >98% conversion of monomer in all cases and after purification >85% yield was obtained. The reactivity ratios of DEGMA and PEGMA are ca. 0.3–0.6 and 3.1–1.7, respectively, and a gradient structure in the monomer sequence distribution throughout the BCS is likely.27 Given the chemical similarity of PEGMA, DEGMA, and EGDMA, quantification of branching was not possible by 1H NMR and the BCS are assumed to follow the composition driven by the feed mixture given the high conversions. GPC was employed to determine molecular weights and polydispersity indices (PDIs) of the BCSs, as well as confirming that distributions were monomodal. Indeed, GPC chromatograms (exemplar data in Fig. 1(ii), full data in Fig. S10†) for all the polymers P1 to P9 displayed monomodal distribution of molecular weight. The GPC data recorded for such complex and amphiphilic architectures are likely to deviate from the true values of molecular weight.28 As such, recorded molecular weight parameters are not absolute, but expressed relative to PMMA standards, but they do indicate that the polymerizations were successful, followed expected trends, and allow the determination of polydispersity indices (Đ). The polymers exhibited consistent and systematic compositional variation throughout the series.
P1 to P3 evaluates the impact of cross-linker for the thermoresponsive behaviour of the emulsion systems. GPC analyses for the branched copolymers P1, P2 and P3 revealed number average molecular weights (Mn) of 8.2, 6.5 and 5.4 kg mol−1, respectively. A decrease in Mn and Đ was observed with lowering cross-linker concentration (Table 1) whilst keeping the stoichiometry of the monomer to chain-transfer agent constant. Thus, it is intuitive that P1 with the highest concentration of cross-linker displays the greatest degree of branching and thus the highest molecular weight, with the Mn decreasing as cross-linker is reduced. P4 to P6 demonstrate that Mn may be tailored by AIBN/DDT in the feed. An increase in molecular weight and polydispersity is observed with reduced concentration of chain initiator and chain-transfer agent, allowing clear control over this parameter and granularity between Mn in emulsifiers. P7, P8, P9 are synthesized with varying carbon chain length chain-transfer agents. P7 is synthesized with TDT (C14), P8 with HDT (C16) and P9 with ME (C0). Comparing P9 (i.e., synthesis with ME) with P7 and P8, an apparent increase in Mn is observed, although ME has a shorter carbon chain length than DDT. This is attributed to the hydrophobic P7 and P8 interacting with column packing to increase elution time and reducing apparent Mn.28 GPC analysis of BCS systems likely underestimates molecular weight where branched copolymers of this type are expected to have a compact structure of lower solvodynamic volume relative to their molecular weight than the linear systems.29
All BCS were capable of stabilizing dodecane-in-water emulsions, giving rise to a creamed phase which was isolated and used in all further experiments. P1 demonstrated temperature-induced gelation, which could be macroscopically observed by heating of the sample followed by tilting the vial and allowing gravitational force to act on the material (Fig. 2). When heated, the sample did not flow after vial inversion. Microscopy confirmed the presence of dodecane droplets within the emulsions at a high internal oil phase volume (φ = 0.57). Droplet size analysis by light microscopy (Fig. S11†) revealed mean droplet radii between 7.5 and 11 μm, which gave rise to comparable levels of creaming between emulsions.
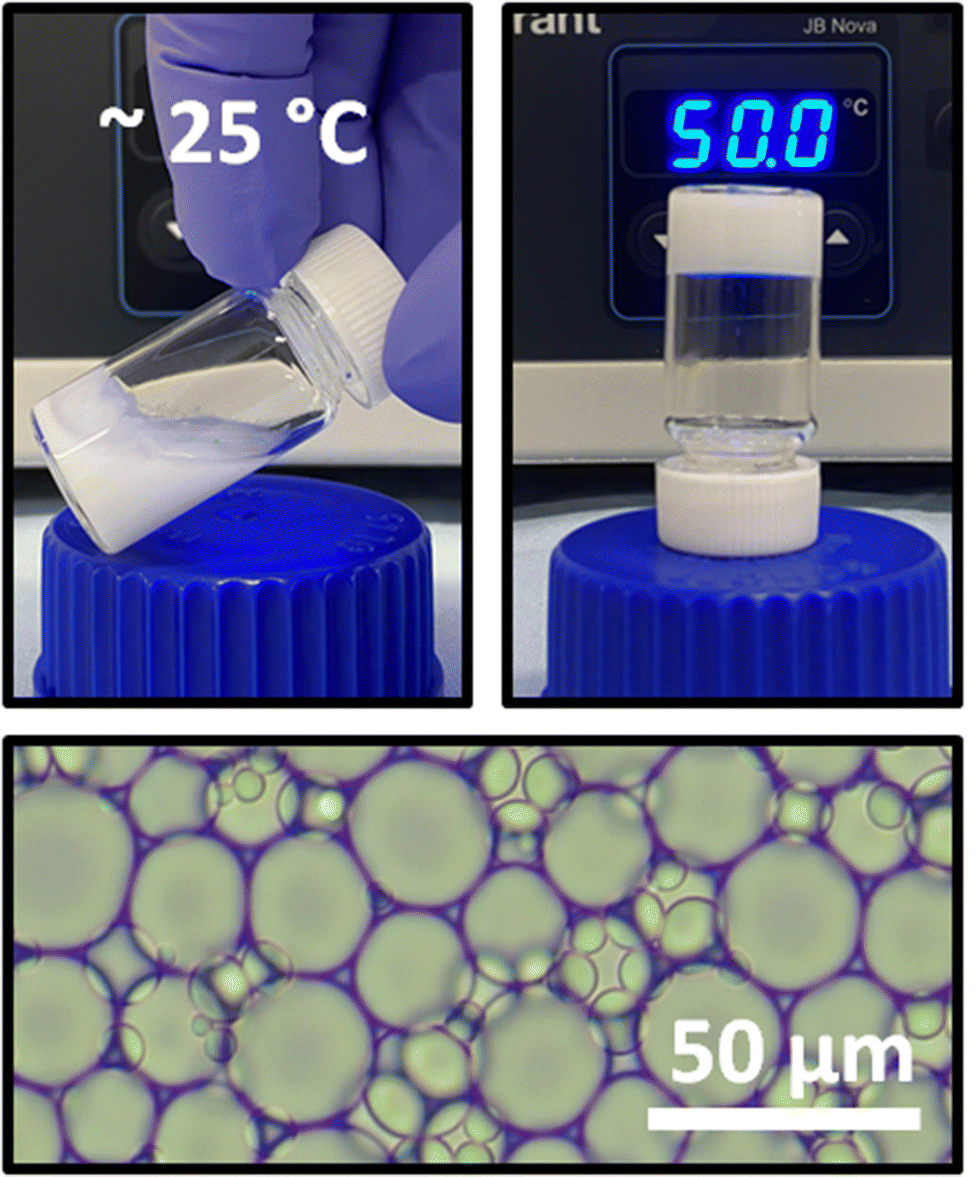 | ||
| Fig. 2 Dodecane-in-water emulsions stabilized by P1 BCS flow under the effect of gravity at room temperature but transition to a rigid gel state upon heating (top). Microscopy confirms the presence of oil droplets (bottom). | ||
Effect of polymer branching on thermoresponse of BCS-stabilised emulsions
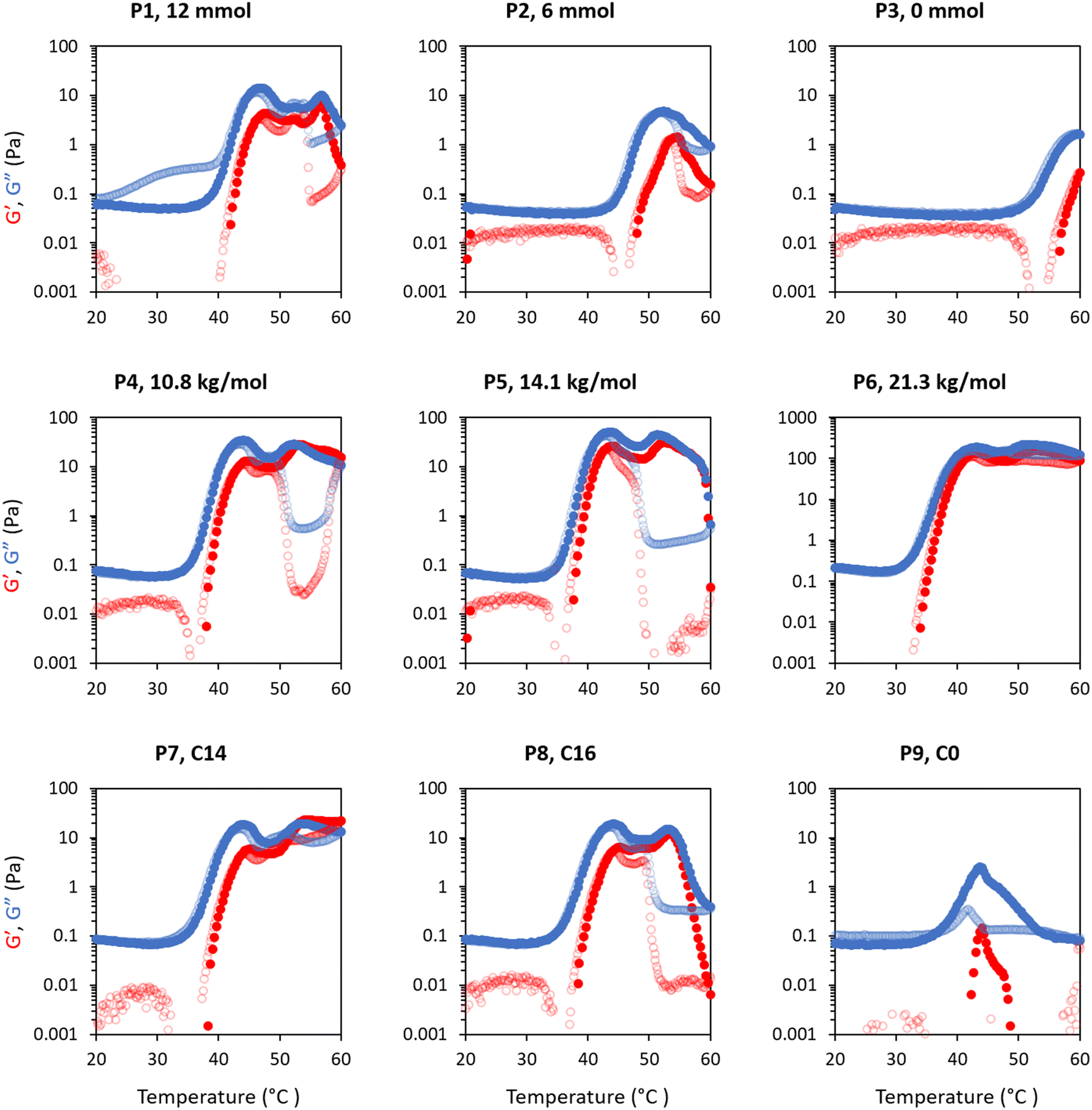
Rheology was used to measure temperature-induced changes in viscoelasticity of the creamed phase of BCS-stabilised emulsions. Viscoelasticity of the emulsions is characterised by the storage (or “elastic”) modulus (G′) and loss (or “viscous”) modulus (G′′) with temperature. The emulsions behave as “solid-like” when G′>G′′ and “liquid-like” when G′ < G′′. The ratio G′′/G′ is defined as tan δ and allows quantification of the relative contributions of these factors to the overall resistance to deformation. In this work, the intersection of the G′ and G′′, giving tan δ < 1, is considered as the gel point.BCS concentration had a large effect on emulsion behaviour with temperature (Fig. 3). For the P1 system with the highest branching degree, all emulsions showed evidence of thermoresponse. At 2.5 wt% polymer concentration, a slight rise in G′ and G′′ was observed at 45 °C, which represents an overall increase in viscosity, and was termed “thermo-thickening” behaviour. However, gelation, defined as a point where G′ exceeds G′′, did not occur. A similar behaviour was observed with a polymer concentration of 5 wt% where the temperature-induced thickening behaviour started at approximately 40 °C, which increased with temperature up to 55 °C, however the emulsion again remained predominantly liquid-like at all temperatures (G′′ > G′). The magnitude of G′ and G′′ increase with temperature was greater in the 5 wt% emulsion than in the 2.5 wt% system. For emulsion systems with 10 wt% polymer concentration, a significant rise in G′ and G′′ was observed at 40 °C followed by a cross-over point (i.e., when solid-like behaviour was first observed, as G′ > G′′) at ca. 48 °C, defined as the gelation temperature (Tgel). This Tgel is believed to correspond to the establishment of an inter-droplet percolating gel-network, as hypothesised in prior studies on responsive emulsions.13,14,17 As aggregation progresses, both G′ and G′′ increased up to 55 °C, which coincided with a minima for tan δ, indicating a point of maximum relative elasticity (Fig. S13†). Beyond this temperature, the network giving rise to the gel state appeared to break down, due to a reduction in both G′ and G′′ and elevation of tan δ. Thus the region 40–55 °C is associated with the formation of an elastic structure due to an LCST-induced alteration in BCS hydrophilicity, whereas the region >55 °C is associated with the loss of elastically-active interactions.
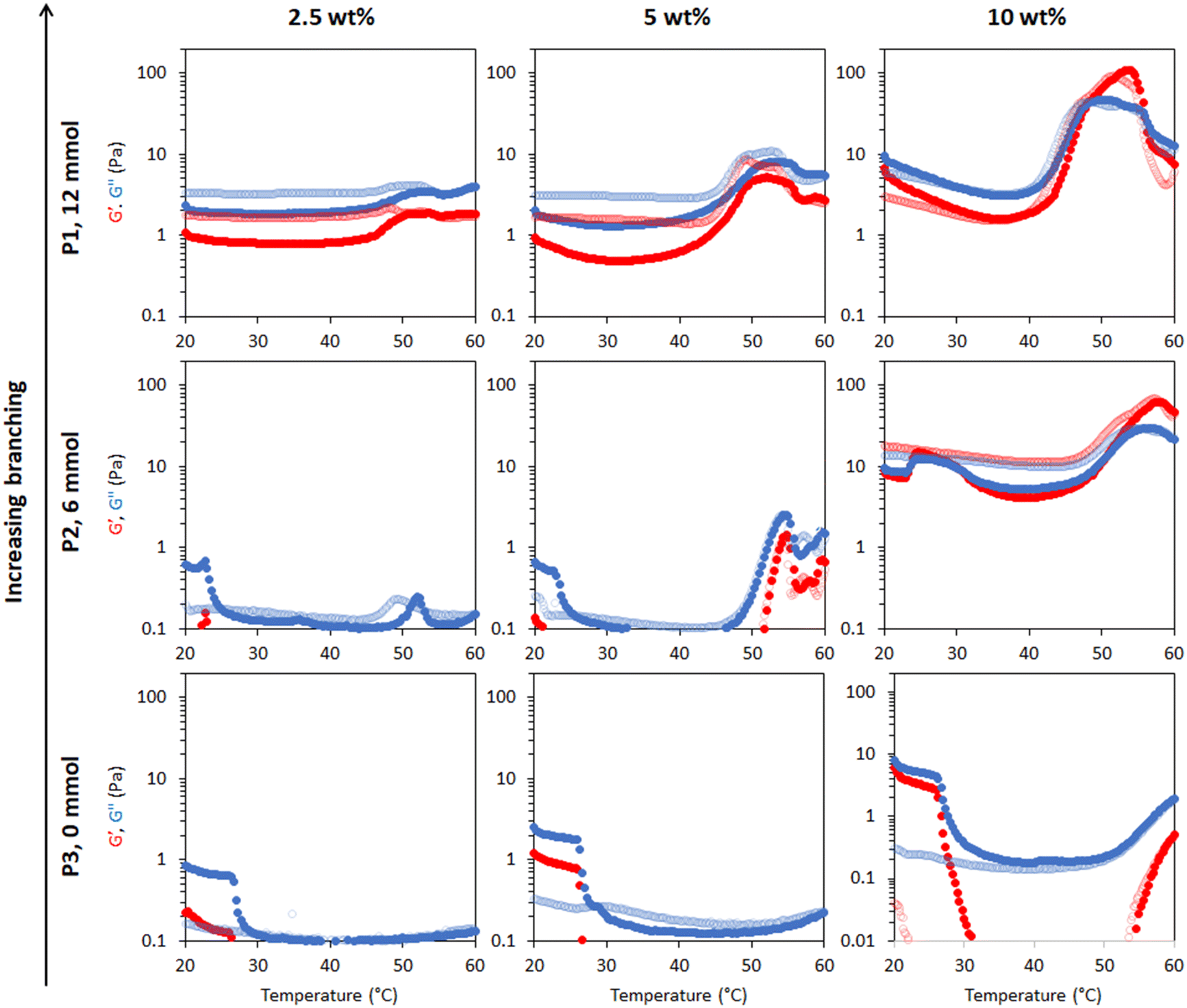 | ||
| Fig. 3 Rheological behaviour of emulsions stabilised by thermoresponsive BCSs where P1 is the sample with “full cross-linker”, P2 is with “half cross-linker” and P3 has no cross-linker. Emulsions were explored at 2.5, 5 and 10 wt% polymer concentration. G′ is shown in red and G′′ is shown in blue. Dark colours show the ‘up’ heating ramp whilst light colours show the subsequent ‘down’ cooling ramp. The Y-axis scales were chosen to enable better comparison of the different emulsion systems. Full rheograms showing the Y-axis region < 0.1 Pa are shown in Fig. S12.† | ||
This study showed that the polymer concentration is an important factor that could be manipulated in an emulsion system to increase the thermal response and/or trigger thermo-gelation. Indeed, the high concentrations required for gelation suggest that the polymer is likely to be spatially distributed both at the droplet interface and in the bulk in order to enable the formation of a percolating network. All systems demonstrated reversibility of the rheological transitions, which is desirable to retain functionality after heating/cooling, barring the small increase in viscosity (G′′) noticeable in the “down” curve of the P1 emulsions at 2.5 and 5% w/w. Overall, the results obtained with P1-emulsions demonstrate that the polymerisation of DEGMA, which presents an LCST, and the hydrophilic macromonomer PEGMA in the BCS architecture produce the desired thermoreversible gelation behaviour.
The viscoelastic nature of the emulsions stabilised with 10 wt% P1 was further probed by rheology frequency sweeps both sides of the transition, at 30 and 50 °C (Fig. 4). At 30 °C, the system behaved as a viscoelastic liquid exhibiting Maxwell-type behaviour with a cross-over at 16 rad s−1, giving a relaxation time of ca. 0.06 s. When heated above Tgel to 50 °C the materials exhibited classic gel-like behaviour with G′ > G′′ over the range of frequencies measured.30
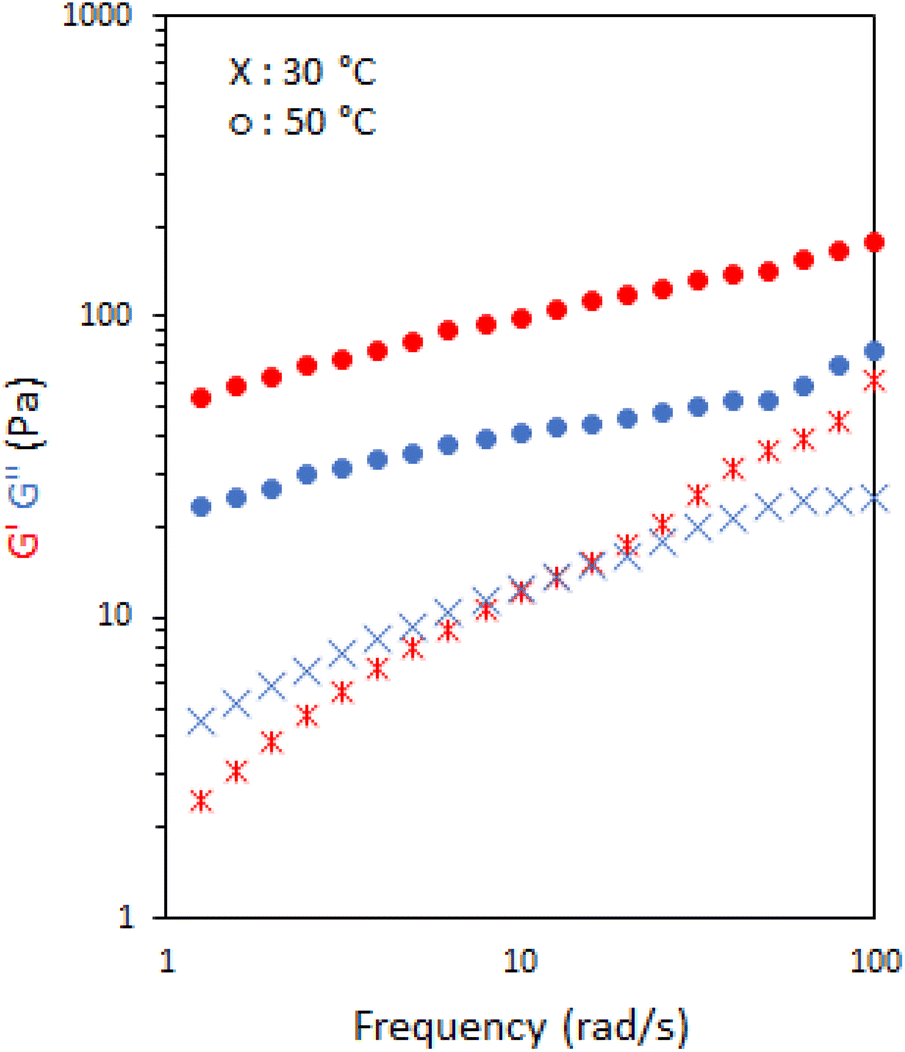 | ||
| Fig. 4 Frequency sweeps of emulsions stabilised with 10 wt% P1. G′ is shown in red and G′′ is shown in blue. | ||
Variation of EGDMA in the monomer feed allowed control of branching and its exploration as a factor in emulsion thermoresponse. P1 is the most highly branched, followed by P2, while P3 has no branching (Table 1). In different systems, the cross-linker to monomer ratio allowed tuning of the LCST, polymer size and colloidal stability,31,32 and thus was hypothesized to affect BCS performance. As discussed above, thermoresponsive emulsions stabilized by P1 (“full cross-linker”) showed thermo-thickening behaviour at 2.5 and 5 wt% and thermoreversible gelation at a concentration of 10 wt%. The rheology of the emulsions stabilised by the BCS synthesised with half the quantity of cross-linker (P2) was assessed at the same polymer concentrations (Fig. 3). With 2.5 wt% BCS, emulsion thinning was observed at 23 °C which was stable up to 47 °C and a small rise in G′′ was then observed at 50 °C. A similar behaviour was observed with 5 wt% polymer, however, the increase seen in G′ and G′′ observed at 47 °C was greater in magnitude. Beyond this point, both moduli collapsed. Emulsions stabilised at 10 wt% polymer concentration showed two thermal events, which occurred at 25 and 50 °C. A second event of thermo-thickening was observed with a rise in temperature to 40 °C, with Tgel occurring at 50 °C. The gel state was then stable up to 60 °C, the highest temperature assessed.
The P3 construct contains no cross-linker and is expected to have a linear structure of poly(DEGMA-ran-PEGMA) with dodecyl chain ends. Temperature-induced thinning was observed at 25 °C and no sign of thickening behaviour was observed up to 60 °C. A gradual increase in polymer concentration from 2.5 to 5 and 10 wt% did not lead to any significant change to the thermal response. Considering the evolution of tan δ with temperature shows that whilst P1 and P2 achieved a transition to a predominantly elastic state, P3 reduced its elasticity during a heating cycle (Fig. S13†). This indicates that the presence of cross-linker to form the BCSs, and therefore a branched structure, is essential to produce a thermal response of the emulsions. This finding is consistent with Weaver and co-workers’ study of engineered emulsions, who attributed the pH-triggered gelation to the presence of multiple hydrophobic DDT domains per polymer chain in the branched systems, allowing stronger tethering to the oil phase, and ultimately the ability to form elastically active bridges between emulsion droplets.14,33
Small-angle neutron scattering measurements were performed on BCS solutions to explore the thermal behaviour of the polymers, aiming to shed light on the mechanisms underlying the thermoresponsive behaviour of the engineered emulsion systems. Three temperatures were investigated to capture the behavior across the gelation process, namely: 25, 40 and 50 °C, where 25 °C lies below the LCST, 40 °C around the expected onset of transition and 50 °C above the transition. 20 wt% solutions were explored for each temperature, which is equivalent to the aqueous phase used in the generation of 10 wt% emulsion systems. The data were fitted with the combination of form factors, P(q), and a structure factor, S(q). The form factor describes the morphology of the scattering objects and the structure factor accounts for interactions between the particles. Form factors used to fit the data were ellipsoids (El), associated with a power law (PL) where needed, whilst a hard sphere (HS) or a sticky hard sphere (SHS) structure factor were required to account for interactions.
Initially, the BCS series exploring branching density (P1, P2 and P3) was examined by SANS. This series had cross-linker feed at 12, 6, and 0 mmol, for P1, P2, and P3, respectively (Fig. 5). At 25 °C (below the LCST), the 20% w/v solutions were best described by an ellipsoid form factor with a polar radius greater than equatorial radius, indicating an oblate spheroid (Fig. 6). Fitting was attempted with spherical, prolate, and tri-axial spheroid models, but adequate fits could not be achieved. Cylinder form factors resulted in disk-like objects of large radii and short length, giving poorer fits than the oblate spheroids. The fitting required a contribution from a hard-sphere structure factor in most instances. A power-law form factor was also required to fit the low q region, and attributed to large aggregates or clusters of polymer, as observed in many polymer systems, for example PEO homopolymers.34 The precise nature of these objects cannot be determined reasonably within this q range but fit to a q−4 decay, indicative of a sharp interface. Upon warming to 40 and 50 °C, the oblate ellipsoid model gave strong fits to the data but the power law contribution was greatly reduced and was no longer required to fit the data in the branched systems (P1 and P2), which can be interpreted as a reduced fraction of polymer chains in solution giving rise to this clustering.
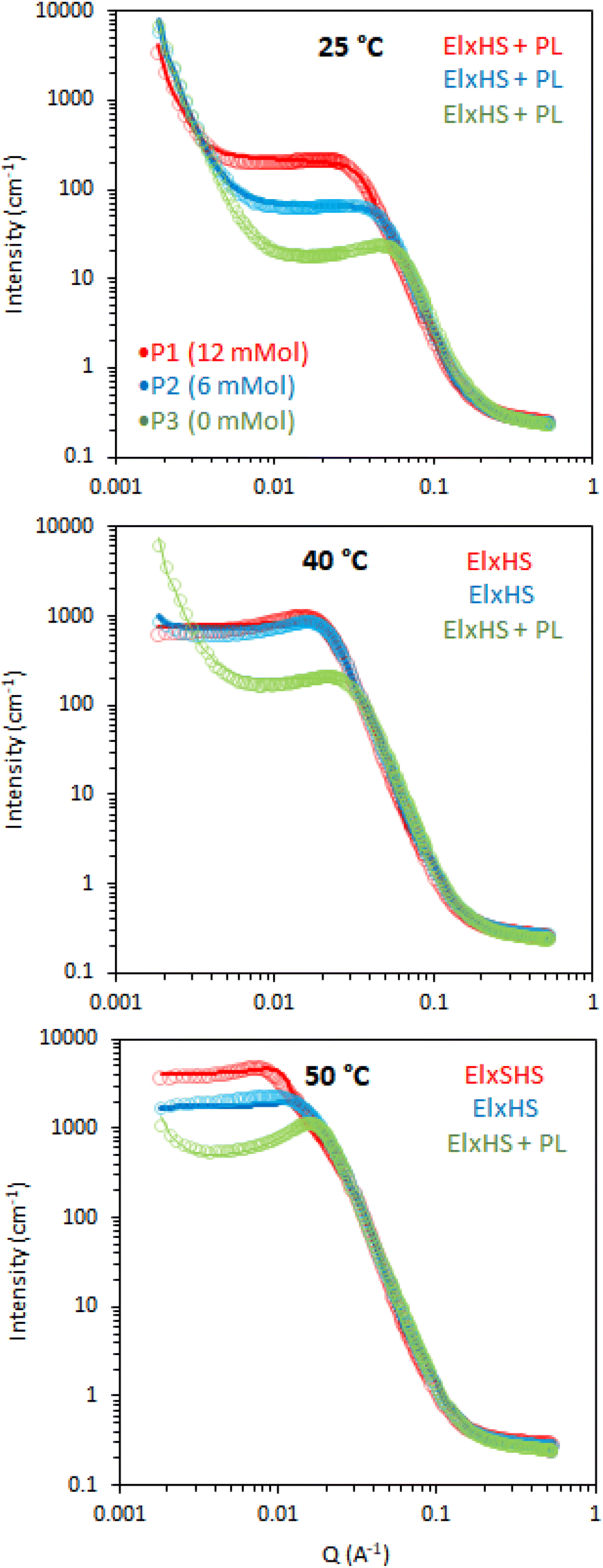 | ||
| Fig. 5 SANS data (circles) with fits (lines) of 20 wt% BCS solutions in D2O with variation of cross-linking and temperature. Legend inset shows sample ID and cross-linker feed. Models used to fit data are inserted at the top right. El is an ellipsoid form factor, HS and SHS are hard sphere and sticky hard sphere structure factors, respectively, whilst × and + are mathematical operators. PL is a power law with a q−4 decay. | ||
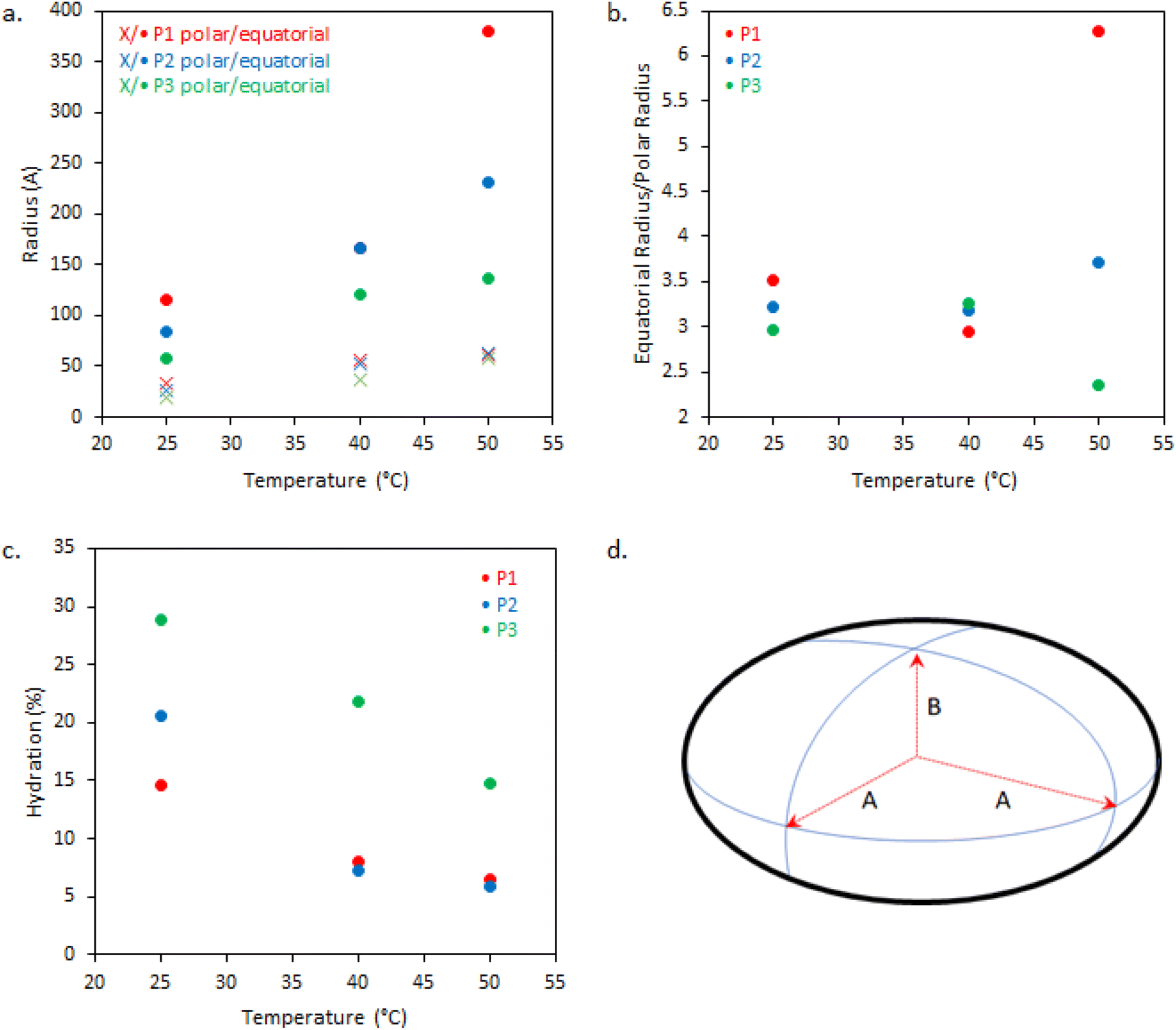 | ||
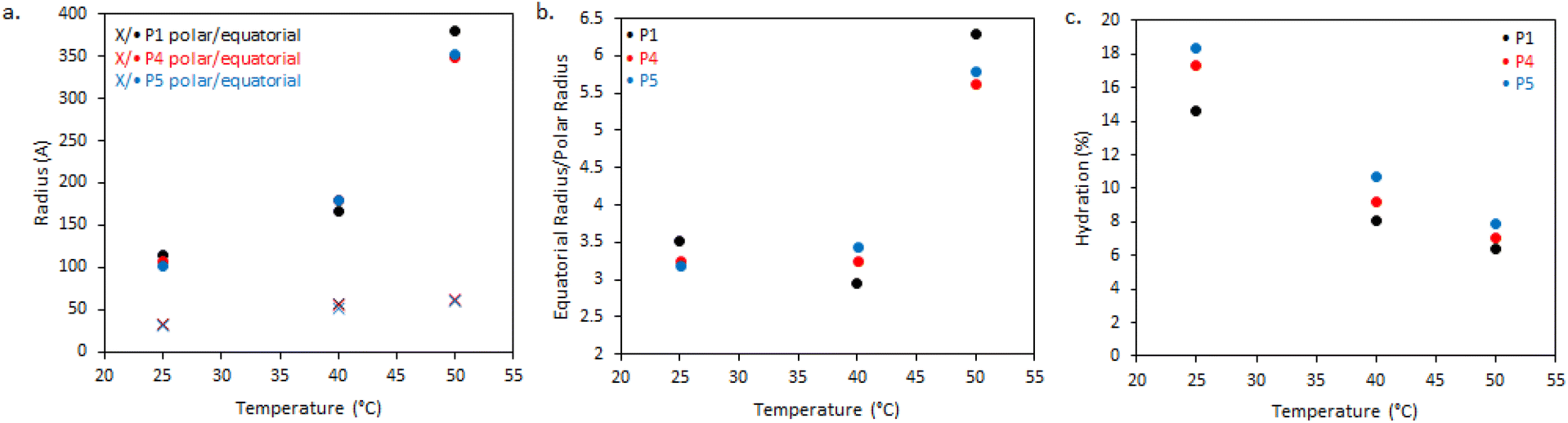
| Fig. 6 (a) Dimensions of BCS ellipsoidal aggregates with variation of cross-linking and temperature, derived from SANS fitting, (b) the ratio of equatorial to polar radii, and (c) degree of hydration of BCS particles estimated from SLD values obtained from the fits. (d) Shows the geometric structure of an ellipsoid where A and B are the equatorial and polar radii, respectively. When A > B the ellipsoid is oblate. | ||
In the absence of steric constraints, surfactant molecules tend to form spherical micelles, however it is known that depending on surfactant structure other micellar shapes with lower curvature can be attained.35 In particular, when the tail-group of the surfactant is large compared the head-group, the surfactant will tend to form anisotropic structures such as cylinders, ellipsoids, or lamellae, based on the “critical packing parameter”.36 This theory has also been applied with great success to block copolymers.37 In the BCS reported here, it may be hypothesized that, particularly above the LCST, the volume of the “hydrophobic” moieties (DEGMA above the LCST and DDT chain-ends) will be large relative to the PEG headgroups, given the excess DEGMA in the feed, which may prevent the formation of spherical micelles. Furthermore, given the presence of multiple DDT chain ends per BCS and steric constraints from the branched structure, it is plausible that the macromolecules cannot take a spherical conformation and instead tend towards objects of lower curvature, such as these oblate ellipsoids.
The dimensions of the oblate ellipsoidal (equatorial radius > polar radius) aggregates formed by P1–P3 solutions are given in Fig. 6. Strikingly, all ellipsoids increase in dimensions with heating and P1 becomes increasingly oblate. For example, P1 has an elliptical form which upon heating increases approximately 11.5-fold in volume and becomes more disk-like in shape, with the ratio of equatorial to polar radius increasing from 3.5 to 6.3 (Fig. 6b). Whilst P1–P3 have similar polar radii, the equatorial radii are greatly increased with branching, indicating an increased oblate character in the order P1 > P2 > P3. This is likely the result of the greater restrictions on acquiring high curvature as branching is introduced. For example, P3 at 25 °C has the lowest degree of ellipsoidal character and could be fitted as a polydisperse sphere, but was kept as an ellipsoid for consistency.
The systems appear to become increasingly dehydrated as they are heated. Hydration of the BCS aggregates was estimated using SLD values from the fits against SLD values calculated for pure DEGMA (0.620 × 10−6 Å−2)22 and D2O (6.37 × 10−6 Å−2). Fig. 6 shows that all BCS aggregates become increasingly dehydrated with heating. This is consistent with the thermoresponsive DEGMA component undergoing a desolvation when heated above the LCST and expelling D2O. Given the potentially counterintuitive combination of particle growth and desolvation, the hypothesized process occurring during heating is the association of ellipsoidal species as DEGMA desolvates, resulting in growth. P1 and P2 show a lower extent of hydration than P3, which is attributed to the dense branching structure hindering solvation of the inner moieties of the BCS.
Overall, the BCS formed ellipsoids which became less solvated with temperature. The nature of thermothickening (P1 and P2) vs. thermothinning (P3) (Fig. 3) from this data may be prescribed to the differing nature of aggregates, with P1 and P2 forming larger, more oblate, aggregates facilitating jamming and connectivity in the system. Indeed, P1 at 50 °C, which gave the strongest gel-like response, was fitted to a sticky hard-sphere structure factor, indicative of inter-ellipsoid attractions. Obviously, an additional key factor in gel formation, observed when emulsions are formulated with the BCSs which cannot be observed from SANS of the polymer solutions, is the interfacial behaviour of the BCS at the oil/water interface. It has previously been postulated that pH-responsive BCS exhibited multiple tethering points to oil droplets when a branched structure with multiple dodecyl-chain ends was present.25 This strength of tethering to the oil phase could be essential to enabling droplet-droplet connectivity to achieve gel formation, providing a rationale for the thermothickening behavior of the branched (P1 and P2) vs. non-branched (P3) polymers, given that the branched (P1 and P2) systems have multiple hydrophobic chain ends per macromolecule.
Effect of polymer molecular weight on the rheology of BCS-stabilised emulsions
The effect of molecular weight on the rheological behaviour of thermoresponsive emulsions was then studied. Fig. 7 shows the rheology of thermoresponsive emulsions stabilised with P1, P4, P5 and P6 (increasing Mn, Table 1) at concentrations 2.5, 5 and 10 wt%. The rheological behaviour of P1 was discussed above, and this data is included as a reference point. At 2.5 wt%, the rheological behaviour of emulsions stabilised with P4, P5 and P6 did not show clear differences with P1, with weak thermal response. The thermo-thickening was enhanced relative to P1 at 5 wt% for emulsions stabilised with P4, however, gelation did not occur (G′ < G′′). Emulsions stabilised with 5 wt% P5 demonstrated a Tgel at ca. 45 °C. However, this gel was not stable over a broad range of temperatures, breaking down at 50 °C. Similarly, the emulsion stabilised with the highest molecular weight polymer, P6, at 5 wt% exhibited a Tgel at 45 °C, with the gel phase extending up to 55 °C. Thus, larger molecular weights appear to favour gel formation, shifting the concentrations required for gelation to lower values, as well as displaying higher moduli. This effect may be attributed in part to longer polymer chains being more likely to form physical connections with other polymer chains. With an increase in polymer concentration to 10 wt%, the emulsions stabilised with P4, P5, and P6 exhibited a Tgel at 46, 45, and 40 °C and showed a similar thermo-reversible behaviour while cooling. Moreover, the gel region extended up to the highest temperature assayed, 60 °C. This effect is clearly seen in the value of tan δ with temperature (Fig. S13†). The storage moduli (G′) of P5 and P6 is ca. ten-fold greater in the gel state (up to ca. 300 Pa) than the previously reported PNIPAM BCS systems (ca. 30 Pa).17 Furthermore, the onset of thickening for these two constructs was ca. 35 °C, such that hardening could be triggered upon exposure to the body's heat, which is advantageous for drug delivery.38 Thus, this new blueprint for BCS is able to generate materials with much more appropriate thermoresponsive properties for applications requiring resistance to shear, such as drug delivery to topical sites or depot injections.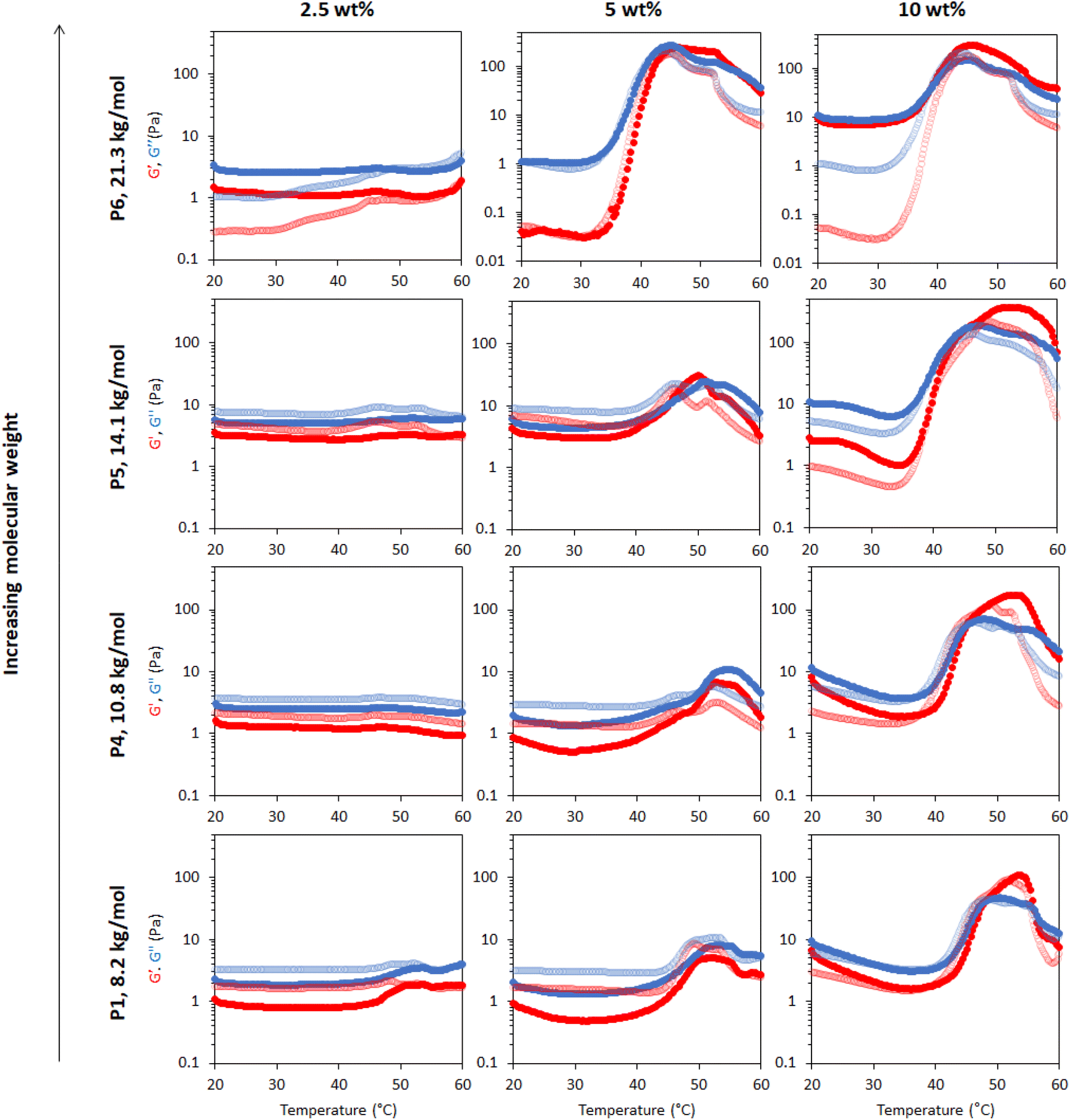 | ||
| Fig. 7 Effect of molecular weight and polymer concentration on thermoresponse of emulsions stabilized with BCSs. The molecular weight of BCSs follows: P1 < P4 < P5 < P6. G′ is shown in red and G′′ is shown in blue. Dark colours show the ‘up’ heating ramp whilst light colours show the subsequent ‘down’ cooling ramp. Full rheograms showing the Y-axis region < 0.1 Pa for P6 at 5 and 10 wt% are shown in Fig. S14.† | ||
The effect of molecular weight of BCSs on the nanostructures they form in solution was then evaluated by SANS (Fig. 8). This series explored Mn at 8.2, 10.8, 14.1, and 21.3 for P1, P4, P5, and P6, respectively. It is worth noting that polydispersity also increased with Mn (Table 1). At 25 °C, the P4 data could again be fitted to ellipsoids with a power law contribution, however the power law was not needed with P5. Once samples were heated to 40 and 50 °C, SANS data for P1, P4 and P5 showed only subtle variations in structure, in-line with their comparable rheology at these temperatures. Parameters extracted from the fits reflect this, with only small deviations in hydration and aggregate dimensions (Fig. 9). SANS analysis of P6 solutions instead showed vastly different scattering profiles, which were tentatively assigned to multiple ellipsoids at 25 and 40 °C, before forming an ellipsoid of equivalent dimensions to the other Mn samples at 50 °C, without the need however for a structure factor. The presence of multiple objects in P6 solutions is likely a result of its high polydispersity (Đ = 8.8). Overall, these SANS experiments with varying Mn BCS reproducibly demonstrate the presence of oblate ellipsoids at high temperatures, amenable to a gel state.
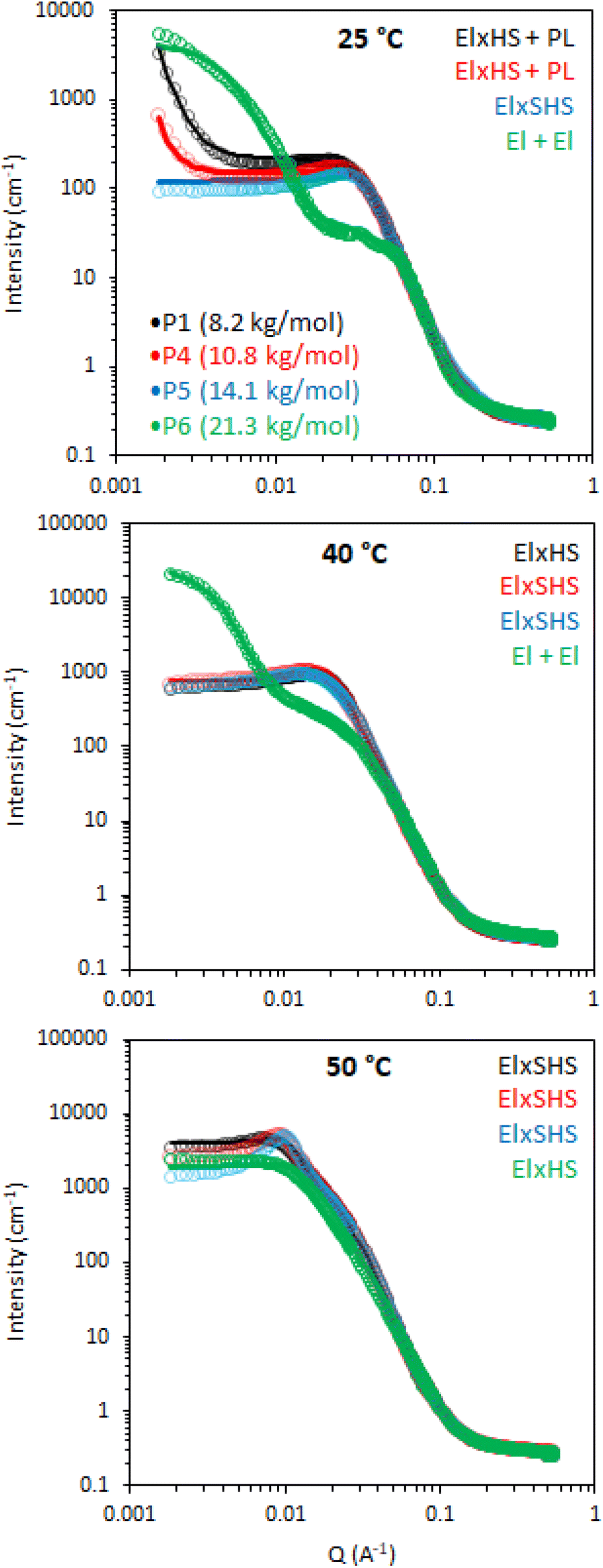 | ||
| Fig. 8 SANS data (circles) with fits (lines) of 20 wt% BCS solutions in D2O with constructs of varying molecular weight and temperature. Legend inset with sample id and Mn determined by GPC. Models used to fit data are inserted top right. El is an ellipsoid form factor, HS and SHS are hard sphere and sticky hard sphere structure factors, respectively, whilst × and + are mathematical operators. PL is a power law with a q−4 decay. | ||
 | ||
| Fig. 9 (a) Dimensions of BCS ellipsoidal aggregates with variation of Mn and temperature, derived from SANS fitting, (b) ratio of equatorial to polar radii, and (c) degree of hydration of BCS particles estimated from the values of the SLDs returned by the fits. The parameters for P6 are omitted due to the different approach to fitting used and the presence of multiple species in solution. | ||
Effect of chain-transfer agent (CTA)
The effect of the polymer chain end hydrophobicity on the thermoresponsive behaviour of the emulsions was studied. P1, P7 and P8 were synthesised with mercaptoalkanes comprising of C12, C14 and C16 carbon chain lengths, leading to hydrophobic tails on the BCS. BCS P9 was synthesised with mercaptoethanol as a CTA to generate a “zero-length” chain end with 2-hydroxyethyl-functionality, which is expected to be hydrophilic. Emulsions prepared with hydrophilic and hydrophobic thiols as CTAs at different polymer concentrations provided an insight on the thermoresponsive activity as shown in Fig. 10.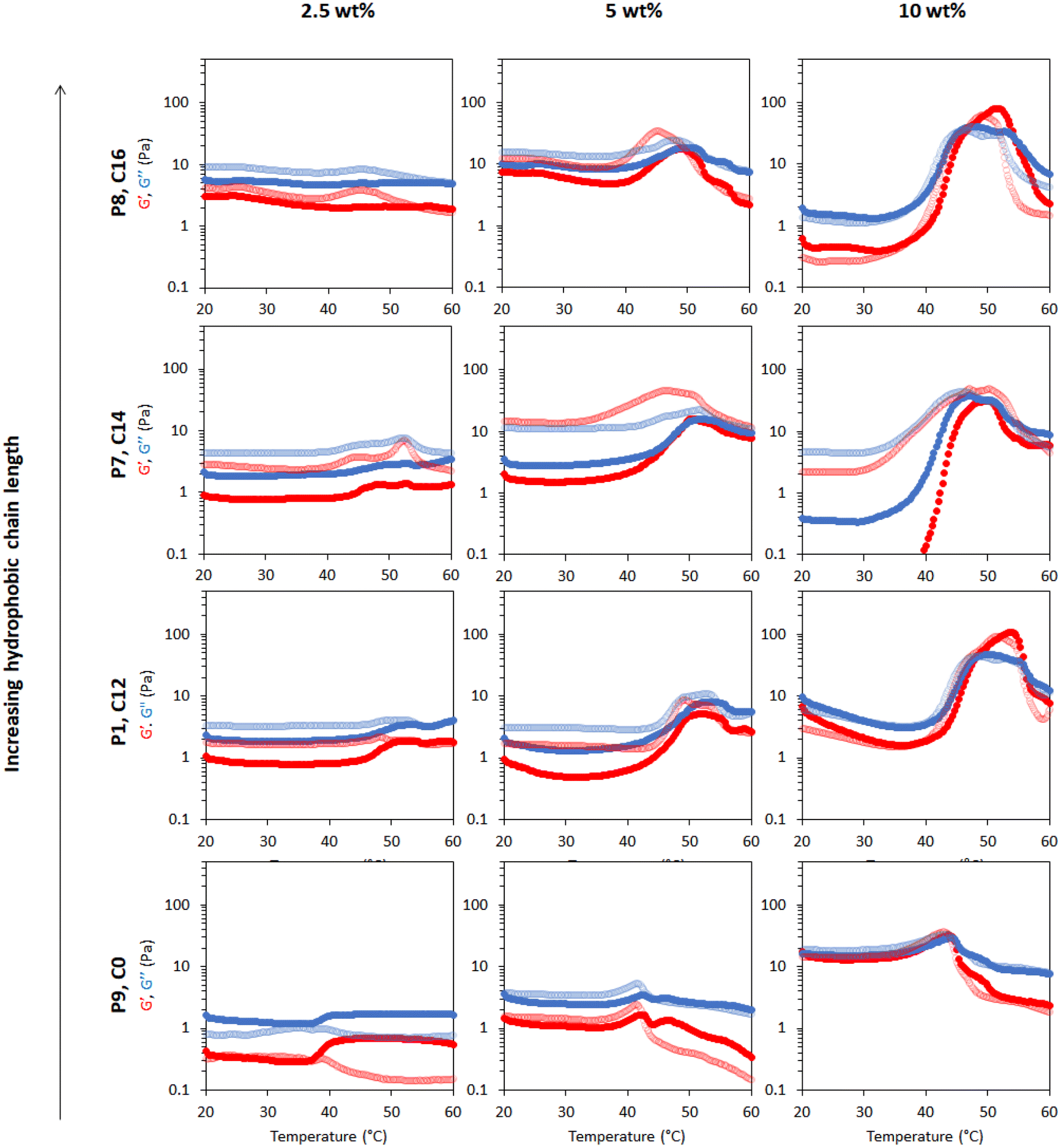 | ||
| Fig. 10 Effect of hydrophobic alkyl tail length on thermo-gelation activity of emulsions as determined by a rheology temperature ramp. P9 is synthesized with mercaptoethnol, P1 with dodecanethiol, P7 with tetradecanethiol and P8 with hexadecanethiol, leading to alkyl chain lengths of 0, 12, 14, and 16 C atoms, respectively. G′ is shown in red and G′′ is shown in blue. Full rheograms showing the Y-axis region < 0.1 Pa for P7 at 10 wt% are shown in Fig. S15.† | ||
The thermoresponsive activity of emulsions at 2.5 wt% polymer concentration P1, P7, P8 and P9 showed a very weak response and were comparable. At 5 wt% polymeric concentration, thermo-thickening was enhanced with P7 and P8 compared to P1. Tgel occurred at 50 °C, however this gel state was only stable over a narrow temperature range. Similarly, emulsions stabilised with 10 wt% P7 showed thermo-thickening but no gelation. The thermoresponsive behaviour of emulsions stabilised with P8 resembled P1 with gelation occurring at ca. 47 °C, however, the gel phase was unstable beyond 55 °C. The rheology of the emulsions stabilised with P9 (Fig. 4a) demonstrated that hydrophobic CTA is necessary for thermoresponsive activity. It was observed that P9 stabilised the emulsions despite a lack of hydrophobic tails, forming emulsions at various polymer concentrations of 2.5, 5 and 10 wt%, but demonstrating a weak thermoresponse. Analysis of tan δ indicated that the material had some degree of elasticity at low temperatures (tan δ ≈ 1), but became increasingly liquid-like when heated-up. The rheology data suggested that the CTAs exert control over the thermo-thickening behaviour of the emulsions because these hydrophobic alkyl groups with strong interaction with the oil phase are required for gelation to occur.
Dynamic light scattering (Fig. 11) revealed further differences in self-assembly between the BCS with hydrophobic alkyl groups (P7) and those terminated with –CH2CH2–OH (P9). P9 (C0) showed a single, sharp transition at 29 °C, with a sharp increase in scattering, and a large DH and PDI (270 nm, 0.27 PDI). P7 (C14) scattered light below 25 °C, associated with clusters of polymer (169 nm diameter, 0.09 PDI), before transitioning to smaller aggregates (ca. 34 nm, 0.12 PDI, at 30 °C) in the region 27–35 °C. A second transition then occurred above 35 °C, with aggregates of higher DH (51 nm, 0.07 PDI). In line with SANS, the DLS of P7 evidenced larger aggregates at low temperatures, followed by the formation of well-defined aggregates above the LCST of PDEGMA, which grew with temperature. Without the presence of hydrophobic alkyl groups, P9 (C0) simply underwent a transition from polymer in solution to heterogeneous aggregates.
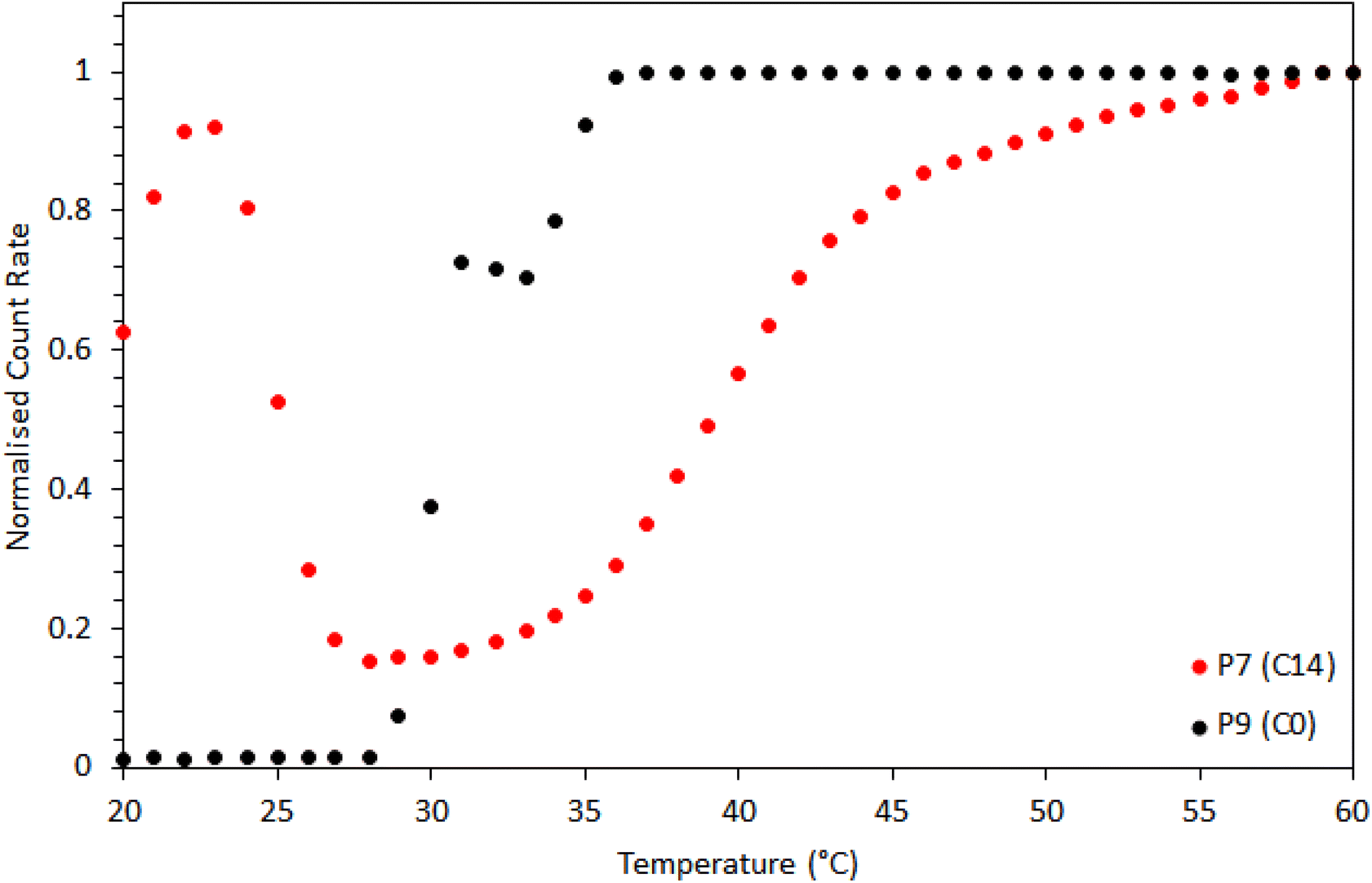 | ||
| Fig. 11 Dynamic light scattering (DLS) of P7 (C14) and P9 (C0) solutions showing normalized scattered count rates with temperature. | ||
The rheology of BCS solutions (20 wt%) in D2O was also studied to probe whether structuration in SANS is associated with rheological changes and if the presence of emulsion droplets is required for network formation (Fig. 12). This concentration is equivalent to the concentration of polymer expected in the aqueous phase of 10 wt% BCS-stabilised emulsions, assuming all the polymer is in the continuous phase (which is, therefore, an over-estimation since a large amount is likely to be at the interface). All BCS solutions retained thermal transitions which were in accordance with the emulsions' rheology, showing sharp thickening with temperature, except for P9 which has no hydrophobic chain ends and showed a weaker response. However, a notable difference is in the overall liquid-like behavior of the systems, in which G′ never exceeds G′′, showing overall a predominantly liquid-like response. Thus, the presence of emulsion droplets is crucial for the formation of a gel network. This connectivity could arise from the interactions of BCS at the oil–water interface with the polymer aggregates in the bulk, acting as junctions to connect droplets, or from interface–interface interactions between droplets, as hypothesized by Weaver et al. in their pH-responsive engineered emulsions.15 The rheology of emulsions does not, however, show thermal gelation at low BCS concentrations (2.5 wt%) which are however sufficient to stabilize the emulsions. In this 2.5 wt% system it is expected that concentration of polymer in the bulk water is low and that presence of the BCS at the O/W interface alone is not sufficient to enable gel formation.
 | ||
| Fig. 12 Temperature ramp rheology of 20 wt% BCS solutions in D2O. Filled and open circles show the heating and cooling cycles, respectively. | ||
Conclusions
This research demonstrated that thermoresponsive engineered emulsions may be generated from branched copolymers of polyDEGMA and PEGMA that act as emulsifiers and trigger gelation. The materials exhibit a sol–gel transition upon warming across a range of concentrations and architectures, generating materials up to ten-times stronger than the previously reported thermoresponsive engineered emulsions.17 Variation of cross-linker density proves for the first time that a branched structure is required for an effective thermal transition to the gel state, as the phenomenon is “turned off” when non-branched polymers are used to stabilize the emulsions. Increasing the molecular weight of the BCS improves the gel strength (as assessed by the elastic modulus G′) and shifts the onset of thickening into a physiologically relevant temperature range (ca. 35 °C). Variation of alkyl length on the BCS chain ends shows that the architecture reliably exhibits a thermoreversible gelation across C12–C16, but that the phenomenon is lost when this hydrophobic character is removed. This demonstrates that effective tethering to the emulsion droplets with this hydrophobic anchor is required to form a gel phase, which is likely to require both BCS-BCS interaction in the bulk and at the droplet interface. Bulk behavior of the BCS was explored by SANS and revealed that the materials form oblate ellipsoids in solution which grow anisotropically with temperature, accompanying the thermothickening/thermoreversible gelation events. This builds-up a hierarchical picture of self-assembly. At low temperatures, the polymer is present at the oil/water interface, tethered by hydrophobic end groups into the oil phase, and in the bulk as a mixture of ellipsoidal aggregates and larger clusters. Upon heating, the aggregates increase in size, with growth along the equatorial radius exceeding growth of the polar radius. It is expected that connectivity of these aggregates in solution and polymer adsorbed at the interface leads to the formation of a percolating network. This new class of BCS allows the effective generation of thermoresponsive engineered emulsions and establishes some principles for the design of advanced materials for exploitation in a wide range of fields, particularly in pharmaceuticals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView also contains code developed with funding from the European Union's Horizon 2020 research and innovation programme under the SINE2020 project, grant agreement No. 654000. The authors thank the Institut Laue Langevin for the provision of neutron beam time on D22 instrument (10.5291/ILL-DATA.9-11-2028). The Engineering and Physical Sciences Research Council (EP/T00813X/1) and Royal Society of Chemistry (RF17-9915) are acknowledged for funding the research. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising. Data is available on reasonable request to the authors.References
- H. Shinohara, Rep. Prog. Phys., 2001, 64, 297–381 CrossRef.
- Q. Chen, X. Cao, H. Liu, W. Zhou, L. Qin and Z. An, Polym. Chem., 2013, 4, 4092–4102 RSC.
- W. Richtering, Langmuir, 2012, 28, 17218–17229 CrossRef CAS.
- M. E. Aulton, Aulton's Pharmaceutics: The Design and Manufacture of Medicines, Elsevier Limited, Philadelphia, 3rd edn, 2007 Search PubMed.
- R. I. Mahato, Pharmaceutica Dosage Forms and Drug Delivery, CRC Press LLC, Boca Raton, FL, 2007 Search PubMed.
- B. Brugger and W. Richtering, Langmuir, 2008, 24, 7769–7777 CrossRef CAS PubMed.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435 RSC.
- L. Wang, W. Huang, S. Wang, Y. Cui, P. Yang, X. Yang and J. V. M. Weaver, J. Appl. Polym. Sci., 2015, 132, 42183 Search PubMed.
- C. Zhao, J. Tan, W. Li, K. Tong, J. Xu and D. Sun, Langmuir, 2013, 29, 14421–14428 CrossRef CAS PubMed.
- E. Cabane, X. Zhang, K. Langowska, C. G. Palivan and W. Meier, Biointerphases, 2012, 7, 1–27 CrossRef PubMed.
- M. T. Cook, P. Haddow, S. B. Kirton and W. J. McAuley, Adv. Funct. Mater., 2021, 31, 2008123 CrossRef CAS.
- T. Arai, T. Joki, M. Akiyama, M. Agawa, Y. Mori, H. Yoshioka and T. Abe, J. Neuro-Oncol., 2006, 77, 9–15 CrossRef CAS PubMed.
- J. V. M. Weaver, S. P. Rannard and A. I. Cooper, Angew. Chem., 2009, 121, 2165–2168 CrossRef.
- J. V. M. Weaver, R. T. Williams, B. J. L. Royles, P. H. Findlay, A. I. Cooper and S. P. Rannard, Soft Matter, 2008, 4, 985–992 RSC.
- J. V. M. Weaver, S. P. Rannard and A. I. Cooper, Angew. Chem., Int. Ed., 2009, 48, 2131–2134 CrossRef CAS PubMed.
- R. T. Woodward, L. Chen, D. J. Adams and J. V. M. Weaver, J. Mater. Chem., 2010, 20, 5228–5234 RSC.
- M. A. Silva, A. Rajbanshi, D. Okopu-Achampong and M. T. Cook, Macromol. Mater. Eng., 2022, 2200321, DOI:10.1002/mame.202200321.
- J. F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- S. R. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- L. A. Feigin and D. I. Svergun, Structure Analysis by Small-Angle X-Ray and Neutron Scattering, Springer New York LLC, 1st edn, 1987 Search PubMed.
- M. Kotlarchyk and S. H. Chen, J. Chem. Phys., 1983, 79, 2461–2469 CrossRef CAS.
- NIST SLD Calculator, https://www.ncnr.nist.gov/resources/sldcalc.html, (accessed 20 April 2021).
- S. M. King, in Modern Techniques for Polymer Characterisation, ed. R. A. Pethrick and J. Dawkins, John Wiley & Sons, Ltd, 1999, pp. 171–232 Search PubMed.
- N. O'Brien, A. McKee, D. C. Sherrington, A. T. Slark and A. Titterton, Polymer, 2000, 41, 6027–6031 CrossRef.
- J. V. M. Weaver, S. P. Rannard and A. I. Cooper, Angew. Chem., Int. Ed., 2009, 48, 2131–2134 CrossRef CAS.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- C. Pietsch, M. W. M. Fijten, H. M. L. Lambermont-thijs, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2811–2820 CrossRef CAS.
- K. Philipps, T. Junkers and J. J. Michels, Polym. Chem., 2021, 2522–2531 RSC.
- I. Koichi, Y. Tomi and S. Kawaguchi, Macromolecules, 1992, 25, 1534–1538 CrossRef.
- P. Biais, M. Engel, O. Colombani, T. Nicolai, F. Stoffelbach and J. Rieger, Polym. Chem., 2021, 12, 1040–1049 RSC.
- N. Welsch and A. L. Lyon, PLoS One, 2017, 12(7), e0181369, DOI:10.1371/journal.pone.0181369.
- N. Nun, S. Hinrichs, M. A. Schroer, D. Sheyfer, G. Grübel and B. Fischer, Gels, 2017, 3, 34 CrossRef PubMed.
- R. T. Woodward and J. V. M. Weaver, Polym. Chem., 2011, 2, 403–410 RSC.
- B. Hammouda, D. L. Ho and S. Kline, Macromolecules, 2004, 37, 6932–6937 CrossRef CAS.
- R. Nagarajan, Langmuir, 2002, 18, 31–38 CrossRef CAS.
- J. Israelachvili, Intermolecular and Surface Forces, Academic Press, London, 2nd edn, 1992 Search PubMed.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- P. Haddow, W. J. McAuley, S. B. Kirton and M. T. Cook, Mater. Adv., 2020, 1, 371–386 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00876a |
| This journal is © The Royal Society of Chemistry 2022 |